
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidation of the acid reactivity of polyhedral orthoformates for the synthesis of carbasugar derivatives†
Kazuki Usuguchi ,
Akira Takagi,
Ippei Takashima‡
and
Kensuke Okuda*
,
Akira Takagi,
Ippei Takashima‡
and
Kensuke Okuda*
Laboratory of Bioorganic & Natural Products Chemistry, Kobe Pharmaceutical University, 4-19-1, Motoyamakita, Higashinada, Kobe, Hyogo 658-8558, Japan. E-mail: okuda@kobepharma-u.ac.jp
First published on 19th June 2025
Abstract
Carbasugar-containing natural products such as uvaridacol L have a variety of bioactivities, motivating chemists to develop methods for their synthesis. The conversion of myo-inositol is one of the most efficient methods for the synthesis of carbasugars. However, selective conversion of myo-inositol derivatives remains to be explored. In our synthesis of uvaridacol L derivatives, we found that the methoxy olefin derivatives of orthoester-protected myo-inositols, the key synthetic intermediates of our study, exhibit differing reaction selectivities depending on their geometric isomerism and substituents. Here we present new insights that contribute to the synthesis of carbasugar-type derivatives by elucidating the mechanism of the selectivity using density functional theory (DFT) calculations.
1 Introduction
Carbasugar-containing natural products1–6 such as gabosines, streptol, uvamalol, and valienamine have a variety of biological activities, including antimicrobial7 and antitumor activities8 (Fig. 1). Since carbasugars have roles as pseudosugars, they are at times incorporated into pharmaceuticals, the diabetes drug voglibose being a notable example. On the other hand, (−)-uvaridacol L (1), a carbasugar analogue isolated from the leaves of Uvaria dac, is known to exhibit selective cytotoxicity (50% preferential cytotoxicity, PC50 = 20.1 μM) toward the PANC-1 human pancreatic cancer cell line in nutrient deprived medium (NDM).9 Some types of cancer cells, including PANC-1, adapt to nutrient starvation – a characteristic of solid tumors – and resistance to existing conventional anticancer drugs such as gemcitabine.10 Therefore, uvaridacol L is attracting attention as a seed compound for novel drug discovery targeting the cancer-specific low nutrition environment. | ||
| Fig. 1 Carbasugar-type natural products and their derivatives. | ||
Carbasugars have characteristic structures which consist of highly oxygenated cyclohex-1-en-1-ylmethanol and successive chiral centers.11 Also, hydroxy groups of carbasugar-type natural products are often functionalized by acyl groups such as benzoyl and/or acetyl moieties. Several synthetic methods are currently employed to synthesize carbasugars with these characteristics. One is the construction of new skeletons of carbasugars from sugars such as glucose.11–15 This strategy has some advantages such as the preexisting chiral hydroxy groups that are found in the starting material. However, the hemiacetal oxygen ether functionality must be replaced with a methylene group by a carbocyclization such as the Horner–Wadsworth–Emmons reaction with a suitable protection–deprotection strategy, which requires a multistep synthesis. The other is the semisynthetic approach by using shikimic acid that possesses the cyclohexene moiety and quinic acid (the hydrated form of shikimic acid) as starting materials.12,13 These starting materials derived from natural products already have similar chirality corresponding to the target carbasugars with a cyclohexene core, but they require the introduction of a hydroxy group at the 6-position with a possible need for stereo inversion of the 3,4,5-hydroxy groups. One of the other approaches uses myo-inositol.14–16 Although requiring exo-homologation at the 1-potision, the construction of carbasugars from myo-inositol provides many advantages particularly in regard to the stereochemistry at the 3,4,5,6-position hydroxy groups (all-trans) which perfectly match that of the target compound. Leveraging this advantage, we previously reported the first total synthesis of (±)-uvaridacol L (1) from myo-inositol in seven steps.17 We also showed that the benzoyl moiety on the primary hydroxy group is essential for the antiausterity property of (±)-1,17 and the potency of racemic (±)-1 is almost the same as that of natural (−)-uvaridacol L (1).10 This implies that the racemic compound can be used for structure activity relationship (SAR) studies.
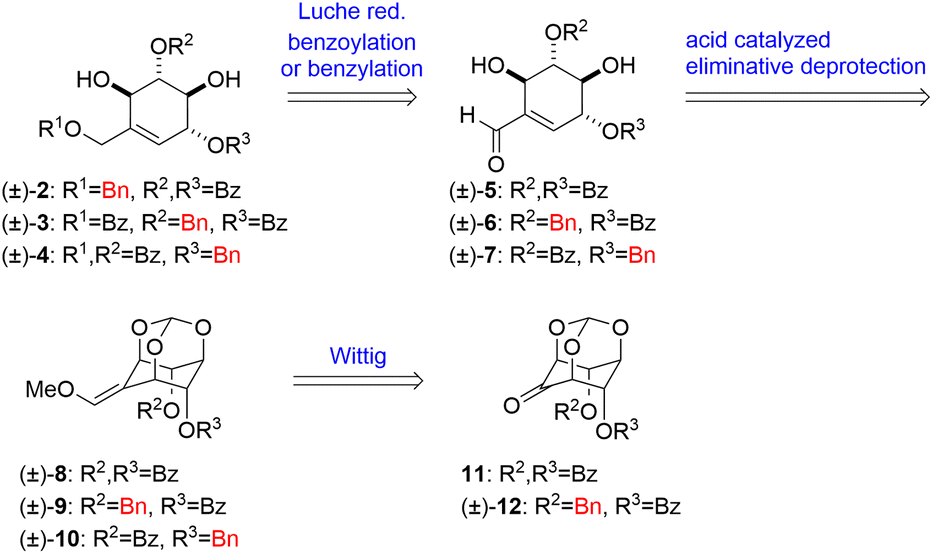
For further SAR investigations, we were interested in how the ester carbonyl moiety affects the antiausterity properties. To explore the pharmacophore, deoxygenated derivatives ((±)-2–4, i.e. the benzoyloxy moieties of (±)-uvaridacol L substituted with benzyloxy groups) are necessary (Fig. 1). In this study, we aimed to establish a new synthetic route for and evaluate the antiausterity activities of uvaridacol L derivatives ((±)-2–4). Furthermore, we found that the direction of double bond migration in the ring-opening reaction of orthoesters for methoxy olefins changed depending on the substituent properties and we investigated the mechanism of this preference using a theoretical study.
2 Results and discussion
To begin the study, we established an alternative synthetic route for the synthesis of (±)-2–4. Retrosynthetic analysis is as follows (Scheme 1). (±)-2–4 are accessible by Luche reduction of conjugated aldehydes (±)-5–7 followed by benzoylation or benzylation of the primary hydroxy group. The cyclohexene skeleton of (±)-5–7 can be constructed by Brønsted acid catalyzed eliminative deprotection of orthoesters (±)-8, (±)-9 and (±)-10. The methoxy olefin moieties of (±)-8, (±)-9 and (±)-10 can be obtained by Wittig reaction from the key intermediates 11 and (±)-12, which are accessible from myo-inositol orthoformate (13) in several steps. E/Z-selectivity of this Wittig reaction governs the yield of asymmetric (±)-9 and (±)-10, which also affects (±)-6 and (±)-7. We have previously shown the preparation of (±)-5 through (±)-8 and 11.17 To prepare the other key intermediates (±)-6 and (±)-7, first, using myo-inositol orthoformate 13 as the starting material, the two hydroxy groups at the axial positions of 13 were protected with benzyl groups to afford the dibenzyl product 14 in 71% yield. The remaining hydroxy group of 14 was oxidized by 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) to give ketone 15 in 88% yield. One of the benzyl groups of 15 was removed by Pd/C-catalyzed reduction to afford the monobenzyl product (±)-16 in 61% yield, and the resulting hydroxy group was benzoylated to afford the benzoylate (±)-12 in 75% yield. Then Wittig reaction of (±)-12 gave methoxy E-olefin (±)-9 and methoxy Z-olefin (±)-10 (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 7:10) in 81% total yield (Scheme 2).
:Z = 7:10) in 81% total yield (Scheme 2).
 | ||
| Scheme 1 Retrosynthetic analysis of deoxygenated derivatives of (±)-uvaridacol L (2–4). | ||
 | ||
| Scheme 2 Synthesis of (±)-methoxy olefins (9 and 10). | ||
The obtained (±)-9 and (±)-10 by further purification were treated with acid for the construction of the cyclohexene ring followed by cleavage of the orthoester, respectively (Scheme 3). Upon treating (±)-9 and (±)-10 with 1:1 1 M aq. HCl and THF, (±)-9 provided (±)-6 with the double bond formed on the benzoyloxy group side and (±)-7 with the double bond formed on the benzyloxy group side in 40% and 36% yields, respectively, while (±)-10 provided (±)-7 in 70% yield as a single product. These differences in selectivity are considered to be due to a match-mismatch between the two orientations of the substituents on the hydroxy group and the electronic effect of the methoxy group.
 | ||
| Scheme 3 The ring-opening reaction of (±)-methoxy olefins (9 and 10). | ||
The obtained (±)-6 and (±)-7 were converted to the respective derivatives by the same synthetic process (Scheme 4). Luche reduction of (±)-6 and (±)-7 gave allylic alcohols (±)-17 and (±)-18 in 96% and 98% yields respectively. The deoxygenated derivatives of uvaridacol L ((±)-3 and (±)-4) were synthesized by benzoylation of the resulting primary alcohols with 2,4,6-collidine in 83% and 71% yields, respectively. Finally, allylic alcohol (±)-1917 was converted to benzyl ether (±)-2 in 17% yield by acidic benzylation with 2,4,6-tris(benzyloxy)-1,3,5-triazine (TriBOT).18
 | ||
| Scheme 4 Synthesis of deoxygenated derivatives of (±)-uvaridacol L (2–4). | ||
During these transformations, we were interested in understanding the mechanism that determines the regioselectivity of the construction of the cyclohexene skeleton ((±)-6 and (±)-7) from methoxy olefin orthoformates ((±)-9 and (±)-10). The direction of the cleavage is influenced by the type of substituent and the geometry of the methoxy olefin (Scheme 3). Controlling the regioselectivity of this cleavage would lead to the development of the selective synthesis of carbasugars using myo-inositol and we aimed to investigate the mechanism of this selectivity by computational chemistry. In a previous study, Sureshan and co-workers reported experiments using orthovalerate ((±)-20, Scheme 5), which gave a stable valerate ester after ring-opening rather than the acid-labile formate ester, as a substrate in the cleavage reaction of orthoesters to confirm the selectivity of the position activated by protonation.14 They confirmed that the ring-opening of the orthovalerate quantitatively provided a 5-O-acylated product ((±)-21). One can expect that the reaction mechanism is as follows: first, protonation at the orthoester O atom (intermediate (IM)1) of (±)-10 (starting material (SM)) leads to C–O bond cleavage to give IM2 followed by hydrolysis to yield (±)-7 (Scheme S1†). Also, one can assume that the protonation of each orthoester oxygen atom of (±)-9 and (±)-10 is reversible; however, putative density functional theory (DFT) calculations showed that every protonation triggered spontaneous generation of H-IM1 with more than 120 kcal mol−1 of stabilization compared to the SM, which suggests that the protonation is irreversible (Scheme S2†). Therefore, we considered this mechanism implausible. To continue our investigation of the mechanism, we then assumed that the reaction proceeds through a two-step transition state (Scheme 6).
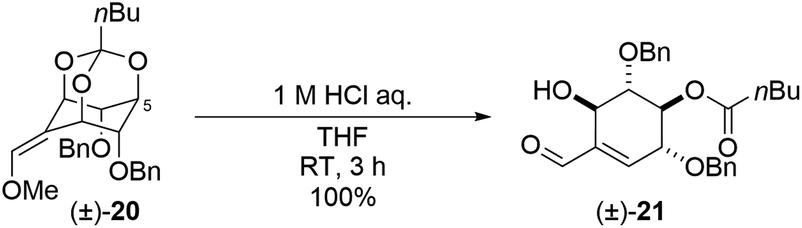 | ||
| Scheme 5 Related substrate (±)-20 for (±)-9 and (±)-10 and its product under acidic condition ((±)-21). | ||
 | ||
| Scheme 6 Mechanism of ring-opening reaction for (±)-10. | ||
First, the orthoester oxygen is activated by coordination to a proton, and the neighboring C–O bond is cleaved (transition state (TS)1). Next, electron donation from the lone pair of the methoxy group leads to olefin transfer, cleaving the C–O bond to form a formyl group (TS2). Based on the assumption that the reaction passes through these transition states, we investigated the mechanism of cleavage and the factors involved in the selectivity by using DFT calculations. All DFT optimization and transition state calculations were performed at the B3LYP/6-31G** level and in water using the SMD (solvation model based on density) model.19 Single-point energy calculations were performed at the B3LYP/def2-TZVP level and in water using the SMD model.
To analyze more detailed modelling of the protonation step, we calculated the structural stabilization using a model in which the hydronium ion was added. As a result, the structures of E isomer (±)-9 with 1-O, 3-O and 5-O coordinating to the hydrogen atoms of the oxonium ion were destabilized by 1.6 kcal mol−1 (1-O: IM1A-E), 2.0 kcal mol−1 (3-O: IM1B-E), and 2.5 kcal mol−1 (5-O: IM1C-E), respectively (Fig. 2). Then, the activation energies of the transition state (TS1) in the C–O bond cleavages of the orthoester were calculated from each hydrated oxonium ion adduct as the ground state. The activation energies of each transition state, TS1A-E, TS1B-E, and TS1C-E, were 3.5 kcal mol−1, 2.8 kcal mol−1, and 1.7 kcal mol−1, respectively. From these energy differences, it can be assumed that the proton transfer between the orthoester oxygen of the hydrated oxonium ion is reversible and that the route of C–O bond cleavage via TS1C-E, which requires the lowest activation energy, is the major pathway to producing the ring-opened products (±)-6 and (±)-7, followed by the route of C–O bond cleavage via TS1B-E. The double bond transfer and resulting orthoester cleavage at the 1-O and 3-O positions compete during the conversion through TS2C-E from IM2C-E to the product (PC). The activation energies (ΔΔG‡) of the transition states providing the respective products were 4.5 kcal mol−1 for ring opening in the 1-O direction (TS2Ca-E) and 5.2 kcal mol−1 in the 3-O direction (TS2Cb-E). The difference between the two activation energies is 0.7 kcal mol−1 which suggests that (±)-6, resulting from PCa, is preferred slightly as the product compared to (±)-7 which is derived from PCb by this pathway. On the other hand, the route via IM2B-E, which requires the second lowest activation energy after that of IM2C-E, produces only PB to lead to (±)-6. These results suggest that the reaction with (±)-9 as the substrate has poor selectivity. This result corresponds to the experimental results in which the two products ((±)-6 and (±)-7) were obtained as a mixture.
 | ||
| Fig. 2 Energy profile for ring-opening reaction of E-isomer (±)-9 calculated by DFT (B3LYP/def2-TZVP//B3LYP/6-31G** [SMD = water]). | ||
The same calculation was applied for the Z isomer (±)-10, where the protonated forms (IM1A-Z, IM1B-Z, and IM1C-Z) were in equilibrium, and the two lower TS1 activation energies were observed when 5-O (TS1C-Z: ΔΔG‡ = 2.3 kcal mol−1) or 1-O was protonated (TS1A-Z: ΔΔG‡ = 2.8 kcal mol−1) (Fig. 3). The activation energies of each transition state giving products from IM2C-Z were estimated to be 6.1 kcal mol−1 for the ring-opening reaction in the 1-O direction (TS2Ca-Z) and 3.4 kcal mol−1 for the ring-opening reaction in the 3-O direction (TS2Cb-Z). The difference between the two activation energies was 2.7 kcal mol−1, suggesting that the reaction via TS2Cb-Z gives PCb to lead to (±)-7 as the more favorable product from (±)-10. Additionally, the route via TS1A-Z produces only PA to lead to (±)-7. These considerations of the mechanism correspond well with the experimental result that the reaction of (±)-10 gave exclusively (±)-7 in 70% yield.
 | ||
| Fig. 3 Energy profile for ring-opening reaction of Z-isomer (±)-10 calculated by DFT (B3LYP/def2-TZVP//B3LYP/6-31G** [SMD = water]). | ||
From another perspective of the reactions converting (±)-9 and (±)-10 to (±)-6 and (±)-7, the sites of double bond formation tended to be on the side trans to the methoxy group with less preference for the side adjacent to the benzoyl moiety. To elucidate the mechanism of regioselectivity further, the electrostatic interactions which are important for the reactions of the dibenzoylated compound (±)-8 and dibenzylated compound (±)-22, in addition to (±)-9 and (±)-10, were calculated using second-order perturbation theory analysis of the Fock matrices in the natural bond orbital (NBO) analyses (Table 1).
|
|||
|---|---|---|---|
| NBO | Donor (BO) | Acceptor (ABO) | E (kcal mol−1) |
| E-Isomer (±)-9 | C1–O | C6–O | 2.1 |
| C3–O | C4–O | 1.9 | |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (p) C (p) |
C1–O (trans) | 5.5 | |
| CC (p) |
C3–O | 4.8 | |
| Z-Isomer (±)-10 | C1–O | C6–O | 2.1 |
| C3–O | C4–O | 1.8 | |
| CC (p) |
C1–O | 5.1 | |
| CC (p) |
C3–O (trans) | 5.6 | |
| BzBz (±)-8 | C1–O | C6–O | 2.1 |
| C3–O | C4–O | 1.9 | |
| CC (p) |
C1–O (trans) | 5.5 | |
| CC (p) |
C3–O | 4.8 | |
| BnBn (±)-22 | C1–O | C6–O | 2.0 |
| C3–O | C4–O | 1.6 | |
| CC (p) |
C1–O (trans) | 5.4 | |
| CC (p) |
C3–O | 4.7 | |

The greater the electron donation from the bonding orbital (BO) of the orthoester C–O bond to the antibonding orbital (ABO) of the neighbouring C–O bond, the greater the dissociation energy, making it more difficult to cleave the bond. Comparing the donation of electrons from the C1–O or C3–O BO to the C6–O or C4–O ABO in (±)-9 and (±)-10, the stabilization energy from the donation of energy to the ABO on the benzoyloxy group side is higher than that to the ABO on the benzyloxy group side. This result may be attributed to the increased electron acceptability of the antibonding orbital due to the electron-withdrawing property of the benzoyl group, which generates unfavorable effects on the C–O bond cleavage at the neighboring site. In addition, the donation of electrons from the π–electrons of the methoxy olefin double bond to the ABOs of the C1–O and C3–O bond may contribute to the advantageous delocalization of electrons.
To evaluate the methoxy olefin orientation effect simply, stabilization by electron donation from the π electrons of (±)-8 and (±)-22, which are protected by the same protecting group, to the ABO of the C1–O (trans) bond and C3–O (cis) bond were calculated, respectively. Protection with both benzoyl ((±)-8) or benzyl ((±)-22) groups showed strong preference for the π–electron donating orientation toward the trans position of the methoxy group. From these electrostatic effects, the cleavage of orthoester (±)-9 resulted in a mixture of (±)-6 and (±)-7 due to the competing substituent effects of the orientation of enol ether and ether/ester protection, whereas the reaction of (±)-10 resulted in (±)-7 as a single product due to collaborative substituent effects. We expect that these insights into the selectivity of the orthoester cleavage directed by the characteristics of the neighboring substituents will be useful for the molecular design and syntheses of other carbasugar derivatives from myo-inositol.
Finally, synthesized deoxygenated uvaridacol L derivatives (±)-2–4 were evaluated for their preferential cytotoxicity toward PANC-1 cells under nutrient deprived conditions by WST-8 assay. As (±)-2–4 are racemic mixtures, their stereochemistry may affect their potency. Therefore, we first evaluated the cytotoxicity of the (±)-uvaridacol L (1) racemic mixture, the natural enantiomer (−)-uvaridacol L (1), and the unnatural enantiomer (+)-uvaridacol L (1) in NDM compared to standard low glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (+FBS) as well as FBS-free DMEM (−FBS) and FBS, sodium pyruvate, and glucose-free DMEM (−FBS, −SP & −Glc) over 24 h (Fig. S1,† and Table 2).
| Compound | +FBS | −FBS | −FBS, −SP, & −Glc | NDM |
|---|---|---|---|---|
| a We have previously reported the cytotoxicity of (−)-1.10 | ||||
| (±)-1 | 98.5 ± 10.6 | 51.1 ± 14.8 | 32.3 ± 1.2 | 33.5 ± 1.8 |
| (−)-1a | 81.8 ± 10.0 | 30.4 ± 2.0 | 24.6 ± 5.0 | 14.2 ± 1.1 |
| (+)-1 | 102.9 ± 22.9 | 31.1 ± 1.9 | 26.7 ± 3.8 | 20.1 ± 8.3 |
| (±)-2 | 108.8 ± 26.4 | 40.8 ± 17.9 | 27.7 ± 8.3 | 22.3 ± 11.7 |
| (±)-3 | 138.5 ± 29.3 | 60.4 ± 34.4 | 43.9 ± 25.2 | 33.7 ± 8.4 |
| (±)-4 | 141.4 ± 12.8 | 48.8 ± 20.7 | 35.6 ± 10.9 | 44.8 ± 25.8 |
We did not observe much difference between the enantiomers and the racemic mixture in each medium and they all showed preferential cytotoxicity in NDM. These data imply that the stereochemistries of 2–4, which are quite similar to 1, also have little effect on their potency. Then the evaluation of racemic (±)-2–4 was conducted to observe their cytotoxicities. As (±)-2–4 are derivatives of (±)-1, where one of the three benzoyl esters was converted to a benzyl ether, these results implied that the contribution of the ester carbonyl oxygen to their potency is small, contrary to the mono debenzoylated derivative (±)-19 which was found to be significantly less potent than (±)-1.17 These datapoints are useful for the design of more fine-tuned derivatives of uvaridacol L (1) as antiausterity compounds.
To explore the general preferential cytotoxicity of 1–4 in NDM toward cancer cells that have resistance to conventional anticancer drugs, we also evaluated the activity of (+)-1, (−)-1 and (±)-1–4 toward HT-29 colorectal cancer cells which, like PANC-1 cells, are also viable under nutrient starved conditions and resistant to gemcitabine.10 The WST-8 assay was performed in RPMI-1640 supplemented with 10% FBS (+FBS), FBS free RPMI-1640 medium (−FBS), FBS, sodium pyruvate, and glucose free RPMI-1640 medium (−FBS, −SP, and −Glc), and NDM. The trend with HT-29 cells (Fig. S2,† and Table 3) matched that of PANC-1 cells (Fig. S1,† and Table 2), giving evidence that 1–4 may act more generally on cancer cells under nutrient deprived conditions than currently known.
| Compound | +FBS | −FBS | −FBS, −SP, & −Glc | NDM |
|---|---|---|---|---|
| a We have previously reported the cytotoxicity of (−)-1.10 | ||||
| (±)-1 | 67.9 ± 7.0 | 25.3 ± 11.5 | 28.7 ± 11.0 | 17.7 ± 5.6 |
| (−)-1a | 66.0 ± 12.0 | 23.3 ± 9.4 | 19.2 ± 6.3 | 13.3 ± 3.7 |
| (+)-1 | 70.2 ± 11.3 | 43.0 ± 32.1 | 42.1 ± 34.7 | 17.4 ± 6.8 |
| (±)-2 | 49.5 ± 23.7 | 17.1 ± 2.4 | 15.6 ± 2.2 | 13.0 ± 1.7 |
| (±)-3 | 138.6 ± 50.5 | 89.2 ± 35.5 | 28.0 ± 6.3 | 19.8 ± 8.6 |
| (±)-4 | 122.3 ± 23.3 | 46.4 ± 17.4 | 30.9 ± 3.4 | 28.5 ± 5.0 |
3 Conclusions
In summary, we have synthesized deoxygenated derivatives of uvaridacol L (±)-2–4 by building a cyclohex-1-en-1-ylmethanol skeleton from myo-inositol. Moreover, we discovered that the direction of acid-catalyzed double bond migration from methoxy olefin to a key intermediate in this synthesis depends on the type of adjacent substituent (ester or ether) and the geometric isomer of the intermediate methoxy olefin. We have found that the regioselectivity of migration of the double-bond depends on the electronic interactions of the BO and the ABO by using DFT calculations. These results provide useful guidance for the synthesis of carbasugars using myo-inositol as the starting material. SAR of antiausterity agent uvaridacol L (1) was also performed using (±)-2–4 to show that the ester carbonyl oxygen of uvaridacol L (1) is not essential for its antiausterity properties.4 Experimental
4.1 General method
All reactions were carried out under an ambient atmosphere in a round bottom flask containing a stir-bar with a rubber septum except as described otherwise. Anhydrous tetrahydrofuran (THF) and CH2Cl2 were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) and used without further purification. All other reagents were purchased from Tokyo Chemical Industry Co. (Tokyo, Japan), Kishida Chemical Co. (Osaka, Japan), Nacalai Tesque Inc. (Kyoto, Japan), or FUJIFILM Wako Pure Chemical Corporation and used without further purification. Silicycle Inc. (Quebec, Canada) silica gel (SiliaFlash® F60, 40–63 μm, #R10030B) or Chromatorex PSQ60B (Fuji Silysia Chemical Ltd, Kasugai, Japan) was used for flash column chromatography.4.2 Analytical methods
All reactions were monitored by thin-layer chromatography with E. Merck silica gel 60 F254 pre-coated plates (0.25 mm, 1.05715.0001) and were visualized by UV (254 nm). 1H and 13C NMR spectra were recorded on a JEOL ECZ400S spectrometer (1H: 400 MHz, 13C: 100 MHz) instrument. Chemical shifts in 1H NMR are reported in ppm relative to the residual protons of deuterated solvents (acetone-d6: 2.04 ppm) or the internal standard tetramethylsilane (CDCl3: 0.00 ppm for 1H). Chemical shifts in 13C NMR are reported in ppm relative to the carbon of deuterated solvent (CDCl3: 77.0 ppm, acetone-d6: 29.8 ppm). The mass spectra were measured on a Thermo Fisher Scientific LTQ Orbitrap Discovery. Melting points were determined by Yanaco micro melting point apparatus MP-J3. Specific rotations were measured with JASCO DIP-370 digital polarimeter using the sodium D line and are reported as follows: [α]tD (c = 10 mg mL−1, solvent). Yield refers to isolated yields of compounds greater than 95% purity as determined by 1H NMR analysis. New compounds ((±)-16, (±)-12, (±)-17, (±)-3, (±)-18, (±)-4, and (±)-2) were characterized by 1H NMR, 13C NMR, and HRMS. E or Z-isomers ((±)-9 and (±)-10) were identified unambiguously by 1H NMR, 13C NMR, COSY, and NOESY. (±)-6 and (±)-7 were identified unambiguously by 1H NMR, 13C NMR, COSY, HMBC, and HMQC. Known compounds (14,20 15,20 and (±)-1917) were synthesized according to the literature. We have previously reported preparation of (+)-uvaridacol L ((+)-1)10 and its [α]25D was +89.3 (c = 0.3, CHCl3).
4.3 Experimental procedure for synthesis
:1 three times, and hexane/EtOAc = 3:1 two times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (CHCl3) to afford 14 (264 mg, 71%) as a colorless solid. Rf: 0.58 (hexane/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3) δ: 3.00 (1H, d, J = 11.2 Hz), 4.19–4.24 (3H, m), 4.37 (2H, t, J = 3.6 Hz), 4.45–4.48 (1H, m), 4.59 (2H, d, J = 11.2 Hz), 4.67 (2H, d, J = 11.2 Hz), 5.47 (s, 1H), 7.28–7.30 (m, 10H). 1H NMR spectrum of 14 is consistent with the reported data.21:1). 1H NMR (400 MHz, CDCl3) δ: 4.44–4.45 (2H, m), 4.55–4.59 (3H, m), 4.59 (2H, d, J = 12.0 Hz), 4.61 (2H, d, J = 12.0 Hz), 5.65 (1H, s), 7.23–7.30 (10H, m). 1H NMR spectrum of 15 is consistent with the reported data.21:1 to 1:1) to afford (±)-16 and its corresponding geminal diol. The afforded oil was dissolved in toluene and refluxed with Dean–Stark apparatus overnight. After cooling to room temperature, the solution was evaporated under reduced pressure to afford (±)-16 (67.3 mg, 61%) as a pale yellow oil. Rf: 0.63 (CHCl3/MeOH = 20:1). 1H NMR (400 MHz, CDCl3) δ: 3.59 (1H, d, J = 11.2 Hz), 4.41 (1H, dt, J = 4.4, 2.0 Hz), 4.45 (1H, tt, J = 3.2, 2.0 Hz), 4.49 (1H, dt, J = 4.4, 2.0 Hz), 4.56 (1H, d, J = 11.6 Hz), 4.63 (1H, dd, J = 7.2, 3.2 Hz), 4.68 (1H, tt, J = 7.2, 3.2 Hz), 4.73 (1H, d, J = 11.6 Hz), 5.66 (1H, s), 7.28–7.30 (2H, m), 7.36–7.41 (3H, m); 13C NMR (100 MHz, CDCl3) δ: 68.5, 71.5, 72.7, 76.8, 77.4, 80.7, 102.1, 128.3 (2C), 128.9 (2C), 129.0, 135.2, 200.3; HR-ESI-MS m/z 301.0684 (calcd for C14H14O6Na [M + Na+]: 301.0683).:1) to afford (±)-12 (382 mg, 75%) as a colorless solid. Rf: 0.33 (hexane/EtOAc = 3:1). Mp: 114–116 °C. 1H NMR (400 MHz, acetone-d6) δ: 4.42 (1H, dt, J = 4.0, 2.0 Hz), 4.50 (1H, d, J = 11.6 Hz), 4.56 (1H, dt, J = 4.0, 2.0 Hz), 4.62 (1H, d, J = 11.6 Hz), 4.65 (1H, td, J = 3.6, 2.8 Hz), 4.79 (1H, tt, J = 3.6, 2.0 Hz), 5.74 (1H, td, J = 3.6, 2.8 Hz), 5.79 (1H, s), 7.09–7.13 (5H, m), 7.17 (2H, t, J = 8.0 Hz), 7.43 (1H, tt, J = 8.0, 1.6 Hz), 7.67 (2H, d, J = 8.0 Hz); 13C NMR (100 MHz, acetone-d6) δ: 68.7, 72.0, 72.7, 77.7, 77.9, 78.4, 103.4, 128.6, 128.7 (2C), 129.1 (2C), 129.3 (2C), 129.8, 130.5 (2C), 134.3, 138.1, 165.5, 200.0; HR-ESI-MS m/z 405.0948 (calcd for C21H18O7Na [M + Na+]: 405.0945).:1) to afford isomeric mixture of (±)-9 and (±)-10 (100 mg, 81%) as a colorless solid. The isomeric mixture was separated by column chromatography (hexane/EtOAc = 5:1) to afford (±)-9 (30 mg, 24%) and (±)-10 (48 mg, 39%) as a colorless solid.
4.3.5.1 (±)-9 (E-isomer). Rf: 0.50 (hexane/EtOAc = 2
:1). Mp: 86–89 °C. 1H NMR (400 MHz, CDCl3) δ: 3.69 (3H, s), 4.38 (1H, td, J = 3.6, 0.8 Hz), 4.48 (1H, dt, J = 3.6, 2.0 Hz), 4.52 (1H, d, J = 11.2 Hz), 4.64 (1H, tt, J = 2.0, 3.6 Hz), 4.67 (1H, d, J = 11.2 Hz), 5.24–5.25 (1H, m), 5.59 (1H, td, J = 3.6, 0.8 Hz), 5.72 (1H, s), 6.26 (1H, s), 7.17 (2H, t, J = 8.0 Hz), 7.25–7.28 (5H, m), 7.45 (1H, tt, J = 8.0, 1.2 Hz), 7.84 (2H, dd, J = 8.0, 1.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 60.3, 66.4, 68.0, 69.1, 70.5, 71.5, 73.4, 104.7, 106.3, 127.7, 127.9 (2C), 128.17 (2C), 128.25 (2C), 129.5, 129.8 (2C), 133.0, 137.5, 145.7, 165.5; HR-ESI-MS m/z 433.1259 (calcd for C23H22O7Na [M + Na+]: 433.1258).
4.3.5.2 (±)-10 (Z-isomer). Rf: 0.47 (hexane/EtOAc = 2
:1). Mp: 89–91 °C. 1H NMR (400 MHz, CDCl3) δ: 3.57 (3H, s), 4.30 (1H, t, J = 3.2 Hz), 4.40 (1H, dt, J = 3.2, 1.6 Hz), 4.52 (1H, d, J = 12.0 Hz), 4.56 (1H, d, J = 12.0 Hz), 4.74 (1H, tt, J = 3.2, 1.6 Hz), 5.14 (1H, dd, J = 3.6, 1.6 Hz), 5.62 (1H, t, J = 3.6 Hz), 5.70 (1H, s), 6.23 (1H, s), 7.16–7.30 (7H, m), 7.50 (1H, t, J = 7.2 Hz), 7.94 (2H, d, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 60.2, 65.9, 67.8, 68.5, 71.4, 71.7, 73.9, 104.6, 106.0, 127.7 (2C), 127.8, 128.2 (2C), 128.3 (2C), 129.6, 129.9 (2C), 133.0, 137.5, 145.7, 165.6; HR-ESI-MS m/z 433.1260 (calcd for C23H22O7Na [M + Na+]: 433.1258).
:Z = 36:64) in THF (3.8 mL), 1 M aqueous HCl (3.8 mL) was added and stirred at 40 °C for 25 h. After cooling to room temperature, the reaction mixture was diluted with H2O and extracted with EtOAc three times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc = 1:1) to afford (±)-6 (38.6 mg, 28%) and (±)-7 (83.2 mg, 60%) as a colorless solid.(±)-6 Rf: 0.67 (hexane/EtOAc = 1:1). Mp: 140–143 °C. 1H NMR (400 MHz, CDCl3) δ: 2.84 (1H, d, J = 1.6 Hz), 3.66 (1H, d, J = 2.4 Hz), 3.76 (1H, dd, J = 10.0, 6.8 Hz), 3.99 (1H, tt, J = 10.0, 1.6 Hz), 4.81 (1H, d, J = 11.6 Hz), 4.83–4.86 (1H, m), 5.13 (1H, d, J = 11.6 Hz), 5.90 (1H, dt, J = 8.0, 2.4 Hz), 6.67 (1H, s), 7.31–7.42 (5H, m), 7.46 (2H, t, J = 8.0 Hz), 7.60 (1H, tt, J = 8.0, 1.6 Hz), 8.07 (2H, dd, J = 8.0, 1.6 Hz), 9.53 (1H, s); 13C NMR (100 MHz, CDCl3) δ: 70.9, 71.1, 73.1, 75.1, 81.9, 128.1, 128.2 (2C), 128.5 (2C), 128.6 (2C), 129.1, 129.9 (2C), 133.6, 137.9, 140.6, 145.2, 166.0, 194.1; HR-ESI-MS m/z: 391.1150 (calcd for C21H20O6Na [M + Na+]: 391.1152).
(±)-7 Rf: 0.47 (hexane/EtOAc = 1:1). Mp: 121–123 °C. 1H NMR (400 MHz, CDCl3) δ: 2.65 (1H, d, J = 4.4 Hz), 3.57 (1H, d, J = 2.0 Hz), 4.02 (1H, ddd, J = 10.0, 7.6, 4.0 Hz), 4.41 (1H, dt, J = 7.6, 2.0 Hz), 4.85 (1H, d, J = 11.6 Hz), 4.87 (1H, d, J = 11.6 Hz), 4.85–4.88 (1H, m), 5.39 (1H, dd, J = 10.0, 7.6 Hz), 6.70–6.71 (1H, m), 7.32–7.42 (5H, m), 7.45 (2H, t, J = 7.6 Hz), 7.59 (1H, tt, J = 8.0, 1.2 Hz), 8.09 (2H, dd, J = 8.0, 1.2 Hz), 9.53 (1H, s); 13C NMR (100 MHz, CDCl3) δ: 68.4, 72.7, 73.5, 76.1, 78.3, 128.1 (2C), 128.3, 128.4 (2C), 128.7 (2C), 129.4, 130.0 (2C), 133.4, 137.2, 139.2, 147.0, 166.9, 193.6; HR-ESI-MS m/z: 391.1151 (calcd for C21H20O6Na [M + Na+]: 391.1152).
4.3.7.1 The reaction of (±)-9 (E-isomer). To a solution of (±)-9 (31.0 mg, 0.076 mmol) in THF (0.7 mL), 1 M aqueous HCl (0.7 mL) was added and stirred at 40 °C for 22.5 h. After cooling to room temperature, the reaction mixture was diluted with H2O and extracted with EtOAc three times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (CHCl3) to afford (±)-6 (11 mg, 40%) and (±)-7 (10 mg, 36%) as a colorless solid.
4.3.7.2 The reaction of (±)-10 (Z-isomer). To a solution of (±)-10 (48.0 mg, 0.12 mmol) in THF (0.7 mL), 1 M aqueous HCl (0.7 mL) was added and stirred at 40 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with H2O and extracted with EtOAc three times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (CHCl3) to afford (±)-7 (30 mg, 70%) as a colorless solid.
:1) to afford (±)-17 (28 mg, 96%) as a colorless solid. Rf: 0.57 (CHCl3/MeOH = 9:1). Mp: 135–137 °C. 1H NMR (400 MHz, CDCl3) δ: 2.14 (1H, t, J = 2.0 Hz), 2.65 (1H, d, J = 4.0 Hz), 2.98 (1H, d, J = 2.4 Hz), 3.67 (1H, dd, J = 10.0, 8.0 Hz), 4.03 (1H, ddd, J = 10.0, 7.2, 2.4 Hz), 4.27 (2H, d, J = 4.0 Hz), 4.50 (1H, dd, J = 7.2, 2.0 Hz), 4.88 (1H, d, J = 12.0 Hz), 4.99 (1H, d, J = 12.0 Hz), 5.67 (1H, s), 5.68–5.70 (1H, m), 7.31–7.42 (5H, m), 7.45 (2H, t, J = 7.6 Hz), 7.58 (1H, tt, J = 7.6, 1.2 Hz), 8.06 (2H, dd, J = 7.6, 1.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 64.0, 72.6, 73.5, 75.2, 75.4, 83.8, 121.9, 128.1 (2C), 128.2, 128.4 (2C), 128.8 (2C), 129.6, 129.8 (2C), 133.3, 138.2, 140.3, 166.8; HR-ESI-MS: m/z 393.1314 (calcd for C21H22O6Na [M + Na+]: 393.1309).:1). Mp: 142–144 °C. 1H NMR (400 MHz, CDCl3) δ: 2.89 (1H, d, J = 2.4 Hz), 3.15 (1H, d, J = 4.8 Hz), 3.70 (1H, dd, J = 10.0, 7.6 Hz), 4.03 (1H, ddd, J = 10.0, 8.0, 2.4 Hz), 4.40–4.44 (1H, m), 4.70 (1H, d, J = 13.2 Hz), 4.93 (1H, d, J = 11.6 Hz), 4.99 (1H, d, J = 11.6 Hz), 5.22 (1H, d, J = 13.2 Hz), 5.70–5.72 (1H, m), 5.83 (1H, s), 7.30–7.42 (5H, m), 7.44 (2H, t, J = 8.0 Hz), 7.45 (2H, t, J = 7.6 Hz), 7.57 (1H, t, J = 7.6 Hz), 7.59 (1H, d, J = 8.0 Hz), 8.05 (2H, dd, J = 7.6, 1.2 Hz), 8.06 (2H, dd, J = 8.0, 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 64.2, 71.4, 72.9, 74.5, 75.3, 83.4, 124.9, 128.1 (3C), 128.4 (2C), 128.5 (2C), 128.7 (2C), 129.5, 129.6, 129.8 (2C), 129.9 (2C), 133.3, 133.4, 137.0, 138.1, 166.5, 166.9; HR-ESI-MS: m/z 497.1575 (calcd for C28H26O7Na [M + Na+]: 497.1571).:1) to afford (±)-18 (68 mg, 98%) as a hygroscopic colorless solid. Rf: 0.50 (CHCl3/MeOH = 9:1). 1H NMR (400 MHz, CDCl3) δ: 2.52 (1H, br s), 2.68 (1H, br s), 3.59 (1H, br s), 3.99 (1H, t, J = 8.8 Hz), 4.17 (1H, d, J = 8.0 Hz), 4.22 (2H, s), 4.56 (1H, d, J = 8.0 Hz), 4.71 (1H, d, J = 11.6 Hz), 4.76 (1H, d, J = 11.6 Hz), 5.19 (1H, dd, J = 11.6 Hz), 5.78 (1H, s), 7.28–7.39 (5H, m), 7.44 (2H, t, J = 8.0 Hz), 7.59 (1H, tt, J = 8.0, 1.2 Hz), 8.07 (2H, dd, J = 8.0, 1.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 64.0, 72.0, 72.4, 73.0, 78.7, 78.8, 123.9, 127.9 (2C), 128.0, 128.49 (2C), 128.54 (2C), 129.3, 129.9 (2C), 133.6, 137.8, 138.2, 167.8; HR-ESI-MS: m/z 393.1312 (calcd for C21H22O6Na [M + Na+]: 393.1309).:1) to afford (±)-4 (22.6 mg, 71%) as a colorless solid. Rf: 0.63 (hexane/EtOAc = 1:1). Mp: 41–44 °C. 1H NMR (400 MHz, CDCl3) δ: 2.47 (1H, d, J = 2.8 Hz), 3.33 (1H, d, J = 5.2 Hz), 4.06 (1H, ddd, J = 10.0, 8.0, 2.0 Hz), 4.22 (1H, d, J = 8.0 Hz), 4.52–4.56 (1H, m), 4.77 (2H, s), 4.79 (1H, d, J = 13.2 Hz), 5.15 (1H, d, J = 13.2 Hz), 5.26 (1H, dd, J = 10.0, 8.0 Hz), 5.93 (1H, s), 7.29–7.38 (5H, m), 7.46 (4H, t, J = 8.0 Hz), 7.59 (1H, t, J = 8.0 Hz), 7.60 (1H, t, J = 8.0 Hz), 8.05 (2H, dd, J = 8.0, 1.6 Hz), 8.10 (2H, dd, J = 8.0, 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 64.2, 70.6, 72.6, 73.1, 78.3, 78.8, 126.5, 127.96 (2C), 127.99, 128.5 (4C), 128.6 (2C), 129.4, 129.7, 129.8 (2C), 130.0 (2C), 133.3, 133.5, 135.0, 137.8, 166.6, 167.6; HR-ESI-MS: m/z 497.1568 (calcd for C28H26O7Na [M + Na+]: 497.1571).17 (230 mg, 0.60 mmol) and TfOH (20 μL, 0.23 mmol) in 1,4-dioxane (6 mL), TriBOT (96.0 mg, 0.24 mmol) was added and stirred at room temperature for 6 h. The reaction mixture was quenched by the addition of saturated aqueous NaHCO3 and extracted with CHCl3 three times. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc = 2:1) to afford (±)-2 (48 mg, 17%) as a colorless solid. Rf: 0.27 (hexane/EtOAc = 2:1). Mp: 42–44 °C. 1H NMR (400 MHz, CDCl3) δ: 3.06 (1H, d, J = 5.6 Hz), 3.23 (1H, d, J = 5.2 Hz), 4.14 (1H, d, J = 12.0 Hz), 4.24 (1H, dt, J = 9.2, 5.6 Hz), 4.28 (1H, d, J = 12.0 Hz), 4.56 (1H, d, J = 12.0 Hz), 4.59 (1H, d, J = 12.0 Hz), 4.59–4.62 (1H, m), 5.40 (1H, dd, J = 9.2, 6.8 Hz), 5.74 (1H, ddt, J = 6.8, 4.8, 2.0 Hz), 5.86–5.87 (1H, m), 7.28–7.35 (5H, m), 7.38 (2H, t, J = 8.0 Hz), 7.43 (2H, t, J = 8.0 Hz), 7.55 (1H, tt, J = 8.0, 1.2 Hz), 7.58 (1H, tt, J = 8.0, 1.2 Hz), 8.00 (2H, dd, J = 8.0, 1.2 Hz), 8.08 (2H, dd, J = 8.0, 1.2 Hz); 13C NMR (100 MHz, acetone-d6) δ: 70.1, 70.8, 72.0, 73.1, 76.1, 78.6, 121.9, 128.2, 128.5 (2C), 129.0 (2C), 129.2 (2C), 129.3 (2C), 130.3 (2C), 130.5 (2C), 131.2, 131.7, 133.7, 134.0, 139.6, 140.7, 166.4, 166.5; HR-ESI-MS: m/z 497.1571 (calcd for C28H26O7Na [M + Na+]: 497.1571).4.4 Procedure of biological assay
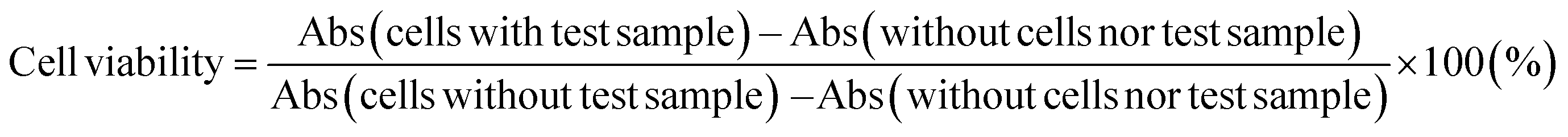 | (1) |
| Abs = Abs450 − Abs650 |
Each experiment was performed in duplicate and repeated independently. The WST-8 assay on HT-29 cells was also performed according to the method on PANC-1 cells except that the medium was changed to RPMI-1640 medium.
Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
KO conceived the project. All authors designed the experiments. KU, AT and KO performed the experiments and analyzed the data. All authors contributed to manuscript preparation and revision.Conflicts of interest
There are no conflicts of interest to declare.Note added after first publication
This article replaces the version published on 19 June 2025, which included an incorrectly placed equation. The text of the article remains unchanged.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 19K07035 (K. O.), 22K06570 (K. O.), the Vehicle Racing Commemorative Foundation Grant Number 6110, 6231, 6297 (K. O.), Chugai Foundation for Innovative Drug Discovery Science Grant Number 21-B1-2 (A. T.), and SHIONO WELLNESS FOUNDATION Grant Number R4-4 (A. T.). K. U. is grateful for Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan, Scholarship Grants from Chugai Foundation for Innovative Drug Discovery Science, and Scholarship Grants from Suntory Foundation for Life Sciences. We are thankful to Ms. Miho Miyake, Ms. Chiemi Tsuda, Ms. Yuka Iio, and Ms. Nana Urakami (Laboratory of Bioorganic & Natural Products Chemistry, Kobe Pharmaceutical University) for performing the WST-8 assay experiment.Notes and references
- T. K. M. Shing and H. M. Cheng, Synlett, 2010, 2010, 142–144 Search PubMed.
- C.-H. Wang, H. T. Wu, H. M. Cheng, T.-J. Yen, I-H. Lu, H. C. Chang, S.-C. Jao, T. K. M. Shing and W.-S. Li, J. Med. Chem., 2011, 54, 8574–8581 Search PubMed.
- T. K. M. Shing, Y. Chen and W. L. Ng, Synlett, 2011, 2011, 1318–1320 CrossRef.
- Q. R. Li, S. I. Kim, S. J. Park, H. R. Yang, A. R. Baek, I. S. Kim and Y. H. Jung, Tetrahedron, 2013, 69, 10384–10390 CrossRef CAS.
- P. Yuan, X. Liu, X. Yang, Y. Zhang and X. Chen, J. Org. Chem., 2017, 82, 3692–3701 Search PubMed.
- S. Roscales and J. Plumet, Int. J. Carbohydr. Chem., 2016, 2016, 4760548 Search PubMed.
- T. W. Miller, B. H. Arison and G. Albers-Schonberg, Biotechnol. Bioeng., 1973, 15, 1075–1080 CrossRef CAS PubMed.
- T. Yamada, M. Iritani, H. Ohishi, K. Tanaka, K. Minoura, M. Doi and A. Numata, Org. Biomol. Chem., 2007, 5, 3979–3986 RSC.
- S. Awale, A. M. Tawila, D. F. Dibwe, J. Ueda, S. Sun, S. Athikomkulchai, C. Balachandran, I. Saiki, K. Matsumoto and H. Esumi, Bioorg. Med. Chem. Lett., 2017, 27, 1967–1971 CrossRef CAS PubMed.
- A. Takagi, K. Usuguchi, I. Takashima and K. Okuda, Chem. Pharm. Bull., 2023, 71, 641–649 CrossRef CAS PubMed.
- P. Banachowicz and S. Buda, RSC Adv., 2019, 9, 12928–12935 Search PubMed.
- X.-L. Zhu, Y.-Q. Luo, L. Wang, Y.-K. Huang, Y.-G. He, W.-J. Xie, S.-L. Liu and X.-X. Shi, ACS Omega, 2021, 6, 17103–17112 CrossRef CAS PubMed.
- T. K. M. Shing, C. S. K. Kwong, A. W. C. Cheung, S. H.-L. Kok, Z. Yu, J. Li and C. H. K. Cheng, J. Am. Chem. Soc., 2004, 126, 15990–15992 CrossRef CAS PubMed.
- S. Mondal, A. Prathap and K. M. Sureshan, J. Org. Chem., 2013, 78, 7690–7700 CrossRef CAS PubMed.
- S. Mondal and K. M. Sureshan, Org. Biomol. Chem., 2014, 12, 7279–7289 Search PubMed.
- S. Mondal and K. M. Sureshan, J. Org. Chem., 2016, 81, 11635–11645 Search PubMed.
- A. Takagi, K. Usuguchi, I. Takashima and K. Okuda, Org. Lett., 2021, 23, 4083–4087 Search PubMed.
- H. Fujita, N. Hayakawa and M. Kunishima, J. Org. Chem., 2015, 80, 11200–11205 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Stewart Campbell and G. R. J. Thatcher, Tetrahedron Lett., 1991, 32, 2207–2210 CrossRef.
- S. Pasari, S. M. Ismail, M. R. Wenk and M. J. Lear, Tetrahedron Lett., 2015, 56, 2597–2601 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary schemes, figures, copies of 1H NMR, 13C NMR, COSY, NOESY, HMBC, and HMQC spectra of the compounds, Cartesian coordinates and total energies for the DFT calculated structures, and results of second order perturbation theory analysis of the Fock matrices in the NBO analyses for the DFT calculated structures. See DOI: https://doi.org/10.1039/d5ra01049g |
| ‡ Present address: National Institute of Technology, Hakodate College; 14-1, Tokurachou, Hakodate, Hokkaido, 042-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2025 |