DOI:
10.1039/D5RA01038A
(Paper)
RSC Adv., 2025,
15, 15164-15177
Novel C-3 and C-20 derived analogs of betulinic acid as potent cytotoxic agents: design, synthesis, in vitro and in silico studies†
Received
12th February 2025
, Accepted 29th April 2025
First published on 8th May 2025
Abstract
In this report, novel derivatives of betulinic acid were designed and synthesized by targeting the C-3-OH group and C-20 olefinic bond in an endeavour to develop potent antitumor agents. These analogs were screened for their anticancer activity against six different human cancer cell lines including breast cancer MCF-7, lung cancer A549, colon cancer HCT-116, leukemia MOLT-4, prostate carcinoma cell PC-3 and pancreatic cancer cell Miapaca-2 by MTT assay. Many derivatives displayed better cytotoxicity than the parent compound BA. More significantly compounds 9b, 9e, 10 and 11a were found to have more promising activity than BA. Compound 11a was the most potent analog with IC50 values of 7.15 (MCF-7), 8.0 (A549), 3.13 (HCT-116), 13.88 (MOLT-4), 8.0 (PC-3) and 6.96 (MiaPaCa-2) μM. In addition to experimental investigations, in silico aspects were evaluated for the parent compound, BA and 11a derivative based on its potential bioactive behaviour. The representative compounds were optimized structurally using density functional theory (DFT). GaussView 6.1 graphical interface associated GAUSSIAN 09 (Revision C.01) software package was used for the calculations under 6-311g(d,p)/B3LYP formalism using under a SMD model (water as solvent) for the parent compound BA and 11a to explain the respective bioactive behaviour. This was followed by molecular docking studies suggesting that compound 11a binds efficiently with all the three proteins with the docking score of −7.2 kcal mol−1 in the case of matrix metalloproteinase-2 (PDB ID: 1HOV) and poly[ADP-ribose] polymerase-1 (PDB ID: 1UK0) and −6.7 kcal mol−1 in the case of TRAF2 (PDB ID: 2X7F). Further, molecular dynamics studies between 11a and the three proteins were carried out using Desmond Maestro v11.3 to study protein–ligand interactions and protein stability.
Introduction
Cancer is a leading cause of death worldwide. According to a WHO report, in 2020, ten million deaths were contributed by cancer alone, and the most common cancers responsible for this mortality include lung, colon, liver, stomach and breast.1 Colon cancer was found to be the third most common cancer worldwide and is the second leading cause of cancer related deaths. By the end of 2040, it is expected that the colon cancer burden will increase to 3.2 million new cancer cases and 1.6 million deaths each year.2 Available treatments for colon cancer including surgery, chemotherapy, radiation and targeted therapy have certain limitations associated with them such as adverse side effects, multidrug resistance and limited availability. There is an urgent need for the development of novel anti-colon cancer agents with fewer side effects and greater efficacy.3 Natural products have played a highly significant role as a source of lead molecules for anticancer drug discovery. In the timeframe of 39 years, from 1981 to 2019, out of 185 small molecules as anticancer drugs, 75 were related to natural products and their derivatives.4 Among the natural products, triterpenoids of lupane-, oleanane-, ursane- and cucurbitane-type and their structural derivatives have been reported for their potent in vitro and in vivo anticancer effects.5
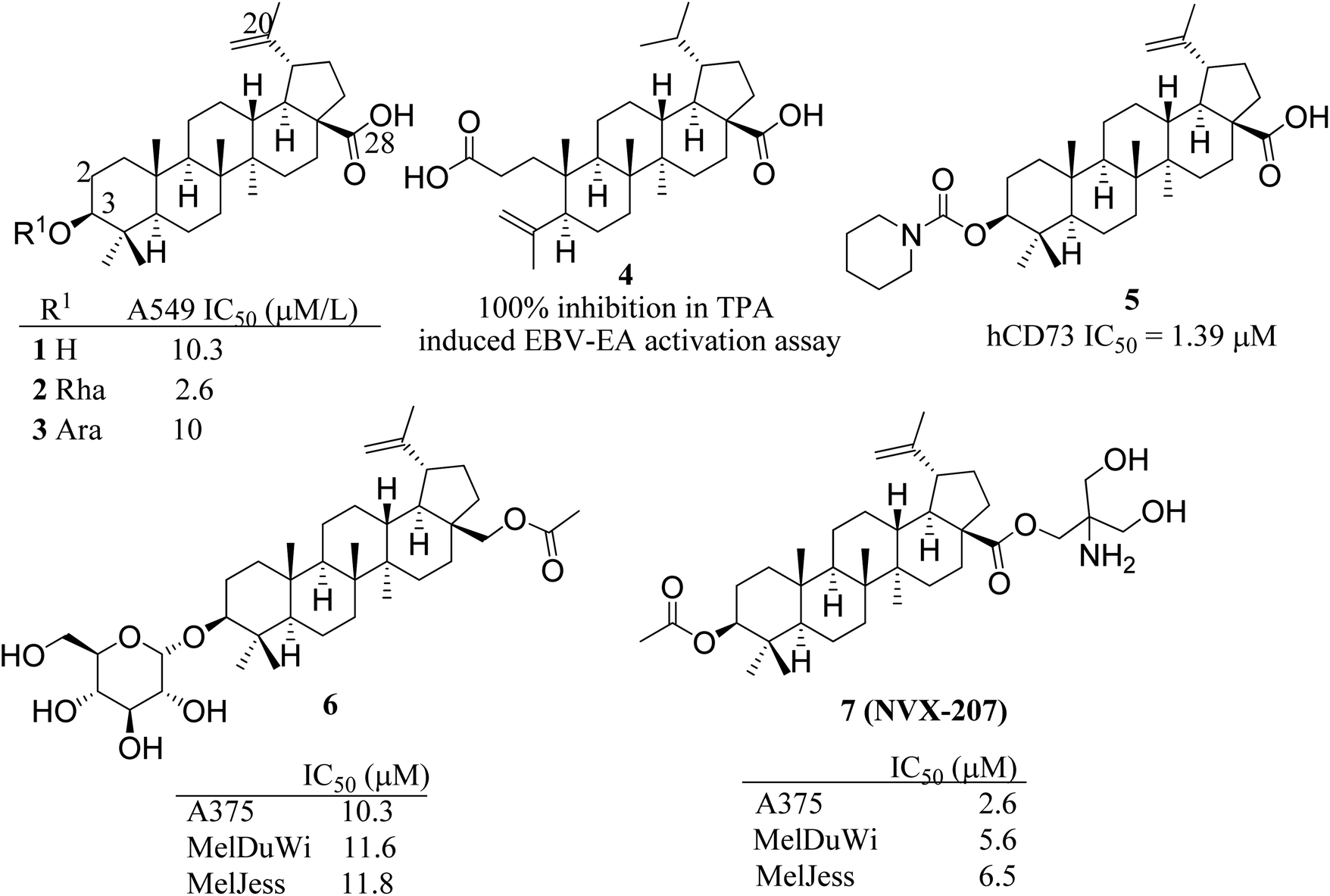
Betulinic acid (BA), a pentacyclic lupane-type triterpenoid, exhibits anticancer effects with various mode of action including triggering the apoptosis in cancer cells via activating mitochondrial pathway, regulating the cell cycle and angiogenesis via different factors such as specificity protein transcription factors, cyclin D1 and epidermal growth factor receptor, inhibition of signal transducer and activator of transcription 3 and NFκB signalling pathways. It also prevents the invasion and metastasis of tumor cells, and affects the topoisomerase I, p53 and lamin B1 expression.6 BA is a pharmacologically active scaffold but possess low water solubility, poor bioavailability and less efficacy.7 As such a number of analogs have been reported by exploiting the key positions C-2, C-3, C-20 and C-28 available on the BA (Fig. 1). Several BA modified derivatives having polar sugar moiety at C-3 (2, 3) and 3,4-seco (4) derivatives have been reported in literature for improved water solubility as well as promising cytotoxicity against cancer cell lines.8 Miao et al. reported the betulinic acid carbamate derivative 5 at C-3 as human CD73 inhibitor and promising immuno-oncology target.9 NVX-207, a C-28 modified BA derivative reported to possess cytotoxicity on several cancer cell lines. Paschke et al. reported the anticancer effects of BA and its two C-3 modified derivatives (6 & 7) including NVX-207 for antiproliferative effects on equine and human melanoma cell lines. NVX-207 beside having the improved water solubility was found to be the potent antiproliferative agent among all three compounds.10 Some selected C-3 modified derivatives of betulinic acid are shown in Fig. 1.
 |
| | Fig. 1 Some selected C-3 modified betulinic acid derivatives (2–7) as anticancer agents. | |
In continuation to our previous communications,11–13 in the present paper, we report the synthesis of new analogs by targeting less explored C-3 and C-20 positions of BA. The new derivatives were evaluated against six different human cancer cell lines including MCF-7, A549, HCT-116, MOLT-4, PC-3 and Miapaca-2. In addition to experimental studies, DFT calculations were done using reliable level of theory to speculate optimized structure and various other physicochemical properties. Molecular docking studies were carried out against three targets involved in pathophysiology of colon cancer including matrix metalloproteinase-2 (PDB ID: 1HOV), poly[ADP-ribose] polymerase-1 (PDB ID: 1UK0) and TRAF2 (PDB ID: 2X7F). The molecular dynamics simulation studies of lead compound and above-mentioned proteins were evaluated to determine ligand protein interactions and stability.
Material and methods
All the chemicals and reagents for synthesis were purchased from Sigma Aldrich. The chemical reactions monitored using silica gel F254 plates using ceric ammonium sulphate solution as spraying reagent for detection of spots. The synthesized products were purified via column chromatography using silica gel 60–120 mesh as stationary phase. NMR spectra of all the compounds recorded on Bruker DPX 400 and DPX 500 instrument using CDCl3 solvent. The chemical shifts are expressed in δ and coupling constant in Hertz. High Resolution Mass Spectra (HRMS) were recorded on Agilent Technologies 6540 instrument.
Isolation of betulinic acid (1)
Betulinic acid (1) was isolated from the stem bark of Platanus orientalis. Shade dried and course powdered plant material (1.6 kg) was extracted with DCM–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature for 24 h, and filtered. The marc was re-extracted twice with same solvent proportions. The filtrate was concentrated on rotary evaporator at 40 °C under reduced pressure and afforded about 120 grams of DCM–methanol extract (extractive value (EV) = 7.5%). The desired compound was purified by column chromatography over silica gel 60–120 mesh and further purified by repeated crystallization. The NP was characterized using various spectroscopic techniques and comparison of data with that reported in the literature.12
:1) at room temperature for 24 h, and filtered. The marc was re-extracted twice with same solvent proportions. The filtrate was concentrated on rotary evaporator at 40 °C under reduced pressure and afforded about 120 grams of DCM–methanol extract (extractive value (EV) = 7.5%). The desired compound was purified by column chromatography over silica gel 60–120 mesh and further purified by repeated crystallization. The NP was characterized using various spectroscopic techniques and comparison of data with that reported in the literature.12
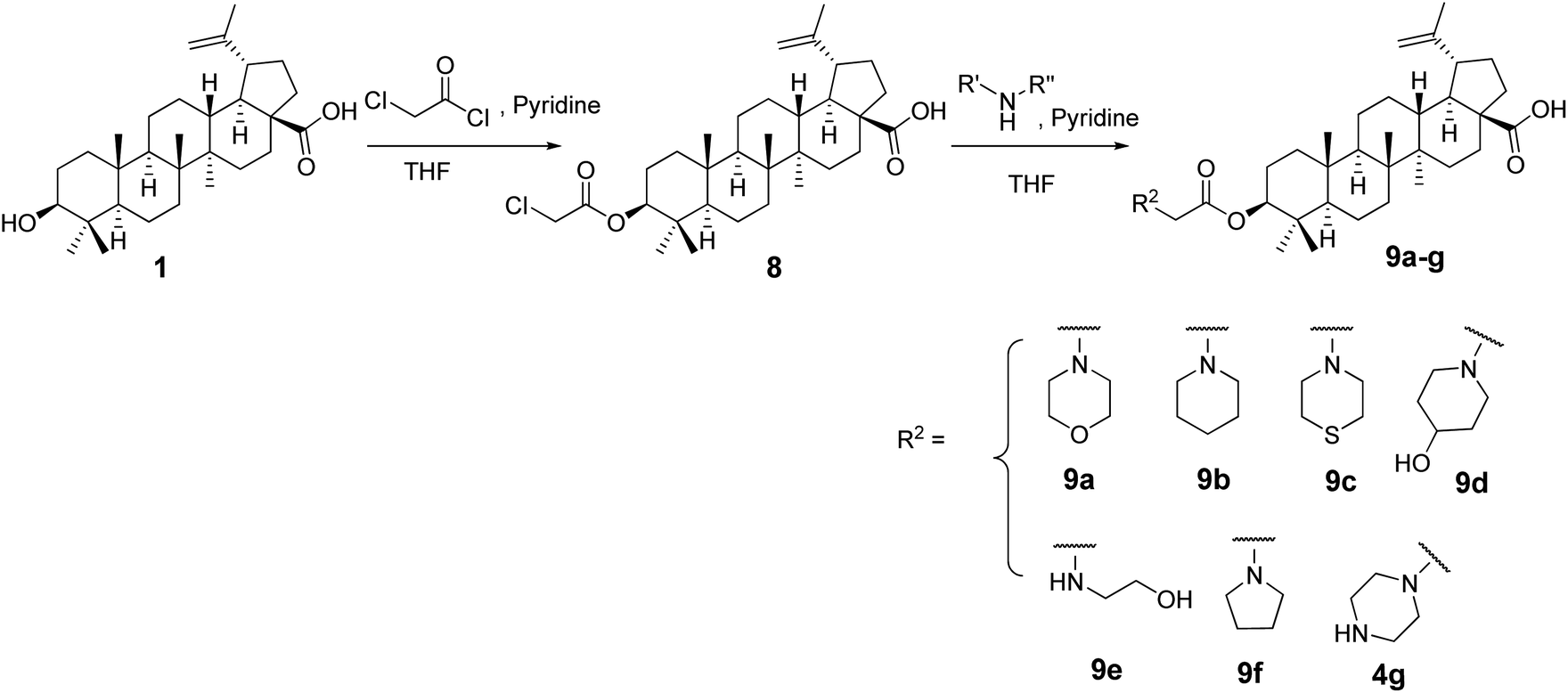
Preparation of 3-chloroacetyl betulinic acid (8)
To compound 1 (1.0 g, 2.2 mmol) dissolved in dry THF (20 mL) was added pyridine (3.3 mmol) and stirred at 0 °C for 5–10 minutes. Chloroacetyl chloride (0.31 mL, 4.4 mmol) was then added dropwise while stirring at 0 °C. After addition, the reaction mixture was allowed to react at room temperature till completion for 2–3 h monitored by TLC. To the reaction mixture was then added 50 mL of ice-cold water and allow the crude product to precipitate and then recrystallized in methanol and DCM furnished 2 as white solid (96% yield). 1H NMR (CDCl3, 400 MHz): δ 4.71 and 4.61 (1H, each, s, H-29), 4.56 (1H, t, J = 8.8 Hz, H-3), 4.05 (2H, d, J = 1.9 Hz, COCH2Cl), 3.00 (1H, m, H-19), 2.26 and 1.66 (1H, each, m, H-22), 2.19 and 1.51 (1H each, m, H-16), 1.97 (2H, m, H-2), 1.69 (3H, s, H-30), 1.66 (1H, m, H-13), 1.62 (1H, m, H-18), 1.63 and 1.39 (1H each, m, H-6), 1.52 and 1.25 (1H each, m, H-15), 1.55 and 1.41 (1H each, m, H-7), 1.42 and 1.17 (1H each, m, H-21), 1.38 and 1.25 (1H each, m, H-12),1.38 (1H, m, H-9), 1.06 and 1.16 (1H each, m, H-11), 1.61 and 1.10 (1H each, m, H-1), 0.97 (3H, s, H-26), 0.93 and 0.86 (3H, each, s, H-27 and H-23), 0.85 (3H, s, H-24), 0.85(3H, s, H-25), 0.80 (1H, m, H-5); 13C NMR (125 MHz, CDCl3) δ 182.92, 167.21, 150.37, 109.80, 83.38, 56.43, 55.34, 50.34, 49.22, 46.96, 42.42, 41.29, 40.66, 38.40, 38.27, 38.02, 38.01, 37.09, 34.1, 32.14, 30.56, 29.68, 27.94, 25.39, 23.55, 20.85, 19.36, 18.11, 16.42, 16.17, 16.03, 14.67. HRMS m/z calcd for C32H49ClO4 [M + H]+ 533.0230, found 533.0236.
General procedure for the preparation of amino derivatives (9a–g)
The title compounds 9a–g were prepared by dissolving compound 8 (0.2 mmol) in THF and was added appropriate amine (0.3 mmol) and pyridine (0.2 mmol) and stirred at room temperature. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured onto 50 mL of water and partitioned with ethyl acetate (3 × 20 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography/by recrystallization to afford the desired products in excellent yields. The spectral data of the compounds is given in the ESI.†
Preparation of 3-azidoacetyl betulinic acid (10)
The title compound was synthesised by reaction of 8 (0.2 mmol) with NaN3 (0.4 mmol) in THF:water (2:1) at room temperature. After completion of the reaction, the reaction mixture was poured in ice cold water to allow the crude product to precipitate. The precipitate was finally washed with methanol to furnish 10 (yield 85%).1H NMR (CDCl3, 400 MHz): δ 4.74 and 4.62 (1H, each, s, H-29), 4.57 (1H, m, H-3), 3.49 (2H, s, COCH2N3), 3.00 (1H, m, H-19), 2.27 and 1.66 (1H, each, m, H-22), 2.19 and 1.51 (1H each, m, H-16), 1.97 (2H, m, H-2), 1.69 (3H, s, H-30), 1.66 (1H, m, H-13), 1.62 (1H, m, H-18), 1.63 and 1.39 (1H each, m, H-6), 1.52 and 1.25 (1H each, m, H-15), 1.55 and 1.41 (1H each, m, H-7), 1.42 and 1.17 (1H each, m, H-21), 1.38 and 1.25 (1H each, m, H-12),1.38 (1H, m, H-9), 1.06 and 1.16 (1H each, m, H-11), 1.61 and 1.10 (1H each, m, H-1), 0.97 (3H, s, H-26), 0.93 and 0.85(3H, each, s, H-27 and H-23), 0.87 (3H, s, H-24), 0.85 (3H, s, H-25), 0.79 (1H, m, H-5). HRMS m/z calcd for C32H49N3O4 [M + H]+ 540.0124, found 540.0125.
Synthesis of phosphoramidate 11a
The compound 10 (0.2 mmol) dissolved in THF and to this added trimethylphosphite (0.3 mmol), and the reaction was stirred at room temperature till completion. The reaction mixture was poured onto 30 mL of water and partitioned with ethyl acetate (3 × 20 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 5% methanol/dichloromethane as eluent to afford the desired product 5a in 58% yield.1H NMR (CDCl3, 400 MHz): δ 4.72 and 4.59 (1H, each, s, H-29), 4.53 (1H, t, J = 8.0 Hz, H-3), 3.73 and 3.70 (6H, each, s, 2× OCH3), 3.67 (2H, m, COCH2NH), 3.25 (1H, m, NH), 2.98 (1H, m, H-19), 2.26 and 1.65 (1H, each, m, H-22), 2.18 and 1.47 (1H each, m, H-16), 1.95 (2H, m, H-2), 1.68(3H, s, H-30), 1.64 (1H, m, H-13), 1.61 (1H, m, H-18), 1.59 and 1.39 (1H each, m, H-6), 1.52 and 1.25 (1H each, m, H-15), 1.54 and 1.41 (1H each, m, H-7), 1.41 and 1.17 (1H each, m, H-21), 1.37 and 1.23 (1H each, m, H-12),1.34(1H, m, H-9), 1.15 and 1.07 (1H each, m, H-11), 1.62 and 1.02 (1H each, m, H-1), 0.95 (3H, s, H-26), 0.91 (3H, s, H-24), 0.82 (6H, each, s, H-27 and H-23), 0.80(3H, s, H-25), 0.76 (1H, m, H-5). 13C NMR (125 MHz, CDCl3) δ 181.39, 170.64, 150.55, 109.65, 82.57, 56.32, 55.39, 53.33, 53.32, 50.39, 49.20, 46.91, 42.97, 42.42, 40.68, 38.31, 37.91, 37.09, 34.19, 32.20, 30.58, 29.68, 27.95, 25.42, 23.68, 20.86, 19.36, 18.13, 16.45, 16.18, 15.98, 14.65, 14.14. HRMS m/z calcd for C34H56NO7P [M + H]+ 622.2654, found 622.2656.
Synthesis of phosphoramidate 11b
The compound 10 (0.2 mmol) dissolved in THF was added triethylphosphite (0.3 mmol). The reaction mixture was stirred at room temperature till completion. The reaction mixture was poured onto 30 mL of water and partitioned with DCM (3 × 20 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 5% methanol dichloromethane as eluent to afford the desired product 11b in 60% yield. 1H NMR (CDCl3, 400 MHz): δ 4.72 and 4.60 (1H, each, s, H-29), 4.56 (1H, t, J = 8.4 Hz, H-3), 3.68 (2H, m, COCH2NH), 3.70 (4H, m, 2× OCH2CH3), 3.39 (1H, m, NH), 3.00 (1H, m, H-19), 2.24 and 1.63 (1H, each, m, H-22), 2.17 and 1.48 (1H each, m, H-16), 1.97 (2H, m, H-2), 1.68 (3H, s, H-30), 1.65 (1H, m, H-13), 1.62 (1H, m, H-18), 1.63 and 1.39 (1H each, m, H-6), 1.52 and 1.25 (1H each, m, H-15), 1.55 and 1.41 (1H each, m, H-7), 1.42 and 1.17 (1H each, m, H-21), 1.37 and 1.23 (1H each, m, H-12),1.35 (1H, m, H-9), 1.15 and 1.07 (1H each, m, H-11), 1.61 and 1.10 (1H each, m, H-1), 1.31 (6H, m, 2× CH2CH3), 0.96 (3H, s, H-26), 0.92 (3H, s, H-24), 0.83 (6H, each, s, H-27 and H-23), 0.81(3H, s, H-25), 0.78 (1H, m, H-5). 13C NMR (125 MHz, CDCl3) δ 181.33, 170.66, 150.55, 109.65, 82.53, 62.67, 62.79, 56.32, 55.39, 53.32, 53.18, 53.29, 50.39, 49.21, 46.91, 43.00, 42.42, 40.68, 38.30, 37.91, 37.09, 34.20, 32.20, 30.59, 29.68, 27.96, 25.43, 23.69, 20.87, 19.36, 18.14,16.45, 16.18, 16.13, 15.99, 14.65. HRMS m/z calcd for C36H60NO7P [M + H]+ 650.2005, found 650.2007.
Synthesis of compound 12
To a solution of compound 1 (1.0 mmol) in dry THF taken in an oven dried and air tight RB was added N-bromosuccinimide (1.25 mmol). The heterogeneous reaction mixture was stirred at room temperature till completion monitored by TLC. The reaction mixture was poured onto 100 mL of water and partitioned with DCM (3 × 50 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 12% ethyl acetate/hexane as eluent to afford the desired product 12 in 76% yield. 1H NMR (CDCl3, 400 MHz): δ 5.14 and 5.05 (1H, each, s, H-29), 4.0 (2H, s, H-23), 3.15 (1H, t, J = 7.6 Hz, H-3), 3.05 (1H, ddd, J = 4.8, 4.3, 4.4 Hz, H-19), 2.29 and 1.67 (1H, each, m, H-22), 2.26 and 1.47 (1H each, m, H-16), 1.93 (2H, m, H-2), 1.73 (3H, s, H-30), 1.69 (1H, m, H-13), 1.67 (1H, m, H-18), 1.63 and 1.42 (1H each, m, H-6), 1.58 and 1.37 (1H each, m, H-15), 1.56 and 1.42 (1H each, m, H-7), 1.41 and 1.17 (1H each, m, H-21), 1.38 and 1.23 (1H each, m, H-12),1.35 (1H, m, H-9), 1.15 and 1.07 (1H each, m, H-11), 1.62 and 1.02 (1H each, m, H-1), 0.96 (3H, s, H-26), 0.95 (3H, s, H-24), 0.82 (3H, each, s, H-27), 0.75 (3H, s, H-25), 0.68 (1H, m, H-5); 13C NMR (125 MHz, CDCl3) δ 178.87, 151.49, 146.08, 113.14, 101.07, 78.62, 56.20, 55.33, 50.68, 50.47, 47.63, 42.23 40.58, 38.75, 38.24, 37.08, 36.77, 34.30, 33.05, 32.08, 31.22, 30.01, 27.74, 26.80, 20.94, 18.22, 15.98, 15.77, 15.25, 14.53. HRMS m/z calcd for C30H47BrO3 [M + H]+ 535.2300, found 535.2301.
Synthesis of compound 7
To a solution of compound 13 (0.6 mmol) in THF:water (1:1) was added NaN3 (0.9 mmol) and stirred at room temperature till completion of the reaction. The reaction mixture was poured onto 50 mL of water and partitioned with DCM (3 × 30 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 15% ethyl acetate hexane as eluent to afford the desired product 13 in 70% yield. 1H NMR (CDCl3, 400 MHz): δ 5.04 and 4.96 (1H, each, s, H-29), 3.76 (2H, s, H-23), 3.19 (1H, dd, J = 5.2, 4.8 Hz, H-3), 2.95 (1H, ddd, J = 4.4, 4.3, 4.4 Hz, H-19), 2.29 and 1.67 (1H, each, m, H-22), 2.27 and 1.47 (1H each, m, H-16), 1.93 (2H, m, H-2), 1.73 (3H, s, H-30), 1.69 (1H, m, H-13), 1.67 (1H, m, H-18), 1.63 and 1.42 (1H each, m, H-6), 1.58 and 1.37 (1H each, m, H-15), 1.56 and 1.42 (1H each, m, H-7), 1.41 and 1.17 (1H each, m, H-21), 1.38 and 1.23 (1H each, m, H-12),1.35 (1H, m, H-9), 1.19 and 1.07 (1H each, m, H-11), 1.62 and 0.95 (1H each, m, H-1), 0.96 (3H, s, H-26), 0.93 (3H, s, H-24), 0.82 (3H, each, s, H-27), 0.75 (3H, s, H-25), 0.69 (1H, m, H-5). 13C NMR (125 MHz, CDCl3) 13C NMR (126 MHz, CDCl3) δ 178.83, 149.09, 111.21, 78.76, 56.16, 55.40, 55.29, 50.44, 50.22, 48.85, 43.52, 42.32, 40.63, 38.73, 38.15, 37.09, 36.77, 34.28, 32.05, 31.89, 29.58, 27.80, 26.93, 26.78, 20.93, 18.20, 16.00, 15.81, 15.25, 14.54. HRMS m/z calcd for C30H47N3O3 [M + H]+ 498.0125, found 498.0129.
Synthesis of compound 14a
The compound 13 (0.2 mmol) dissolved in THF and was added trimethylphosphite (0.3 mmol) and stirred at room temperature till completion. The reaction mixture was poured onto 30 mL of water and partitioned with DCM (3 × 20 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 5% methanol/dichloromethane as eluent to afford the desired product 14a in 61% yield.1H NMR (CDCl3, 400 MHz): δ 4.98 and 4.89 (1H, each, s, H-29), 4.07 (4H, m, OCH2CH3), 3.45 (3H, m, H-23, NH), 3.19 (1H, dd, J = 5.2, 4.8 Hz, H-3), 2.88 (1H, ddd, J = 4.4, 4.3, 4.4 Hz, H-19), 2.28 and 1.67 (1H, each, m, H-22), 2.27 and 1.47 (1H each, m, H-16), 1.93 (2H, m, H-2), 1.73 (3H, s, H-30), 1.69 (1H, m, H-13), 1.67 (1H, m, H-18), 1.63 and 1.42 (1H each, m, H-6), 1.58 and 1.37 (1H each, m, H-15), 1.56 and 1.42 (1H each, m, H-7), 1.41 and 1.17 (1H each, m, H-21), 1.38 and 1.23 (1H each, m, H-12),1.35 (1H, m, H-9), 1.31 (6H, m, OCH2CH3), 1.19 and 1.07 (1H each, m, H-11), 1.62 and 0.95 (1H each, m, H-1), 0.96 (3H, s, H-26), 0.91 (3H, s, H-24), 0.81 (3H, each, s, H-27), 0.75 (3H, s, H-25), 0.67 (1H, m, H-5). 13C NMR (125 MHz, CDCl3) δ 180.39, 153.01, 106.98, 78.93, 62.31 (2× CH2), 56.20, 55.30, 50.44, 50.14, 45.21, 43.25, 42.34, 40.66, 38.85, 38.67, 38.22, 37.16, 36.82, 34.30, 32.35, 31.95, 29.39, 28.01, 27.26, 26.81, 22.72, 21.00, 18.28, 16.23, 16.18, 16.02, 15.43, 14.70, 14.17. HRMS m/z calcd for C32H54NO6P [M + H]+ 580.0417, found 580.0419.
Synthesis of compound 14b
To a solution of compound 13 (0.2 mmol) in THF and was added triethylphosphite (0.3 mmol) and stirred at room temperature. After completion, the reaction mixture was poured onto 30 mL of water and partitioned with DCM (3 × 20 mL). The organic layer was dried over Na2SO4 and purified via silica gel column chromatography in 5% methanol/dichloromethane as eluent to afford the desired product 14b in 68% yield. 1H NMR (CDCl3, 400 MHz): δ 4.98 and 4.89 (1H, each, s, H-29), 4.07 (4H, m, OCH2CH3), 3.45 (3H, m, H-23, NH), 3.19 (1H, dd, J = 5.2, 4.8 Hz, H-3), 2.88 (1H, ddd, J = 4.4, 4.3, 4.4 Hz, H-19), 2.28 and 1.67 (1H, each, m, H-22), 2.27 and 1.47 (1H each, m, H-16), 1.93 (2H, m, H-2), 1.73 (3H, s, H-30), 1.69 (1H, m, H-13), 1.67 (1H, m, H-18), 1.63 and 1.42 (1H each, m, H-6), 1.58 and 1.37 (1H each, m, H-15), 1.56 and 1.42 (1H each, m, H-7), 1.41 and 1.17 (1H each, m, H-21), 1.38 and 1.23 (1H each, m, H-12),1.35 (1H, m, H-9), 1.31 (6H, m, OCH2CH3), 1.19 and 1.07 (1H each, m, H-11), 1.62 and 0.95 (1H each, m, H-1), 0.96 (3H, s, H-26), 0.91 (3H, s, H-24), 0.81 (3H, each, s, H-27), 0.75 (3H, s, H-25), 0.67 (1H, m, H-5). 13C NMR (125 MHz, CDCl3) δ 180.39, 153.01, 106.98, 78.93, 62.31 (2× CH2), 56.20, 55.30, 50.44, 50.14, 45.21, 43.25, 42.34, 40.66, 38.85, 38.67, 38.22, 37.16, 36.82, 34.30, 32.35, 31.95, 29.39, 28.01, 27.26, 26.81, 22.72, 21.00, 18.28, 16.23, 16.18, 16.02, 15.43, 14.70, 14.17. HRMS m/z calcd for C34H58NO6P [M + H]+ 608.2145, found 608.2147.
Biological assay
Cell culture and treatment. The human cancer cell lines including breast cancer cell MCF-7, lung cancer A549, colon cancer HCT-116, leukemia MOLT-4, prostate carcinoma PC-3 and pancreatic cancer Miapaca-2 were purchased from ECACC, England. Cancer cells were grown in RPMI/MEM/McCOY/DMEM growth media containing 10% FCS, 100 U penicillin G, and 100 μg streptomycin per mL. Cells were grown in a CO2 incubator (Thermocon Electron Corporation, Texas, USA) at 37 °C with 95% humidity. Cells were treated with compounds dissolved in DMSO while the untreated control cultures received only DMSO with concentration <0.2%.
Cell proliferation assay. Cells were seeded in 96 well plates. When at 70–75% confluency after 4 h, cells were treated with given compounds and placed in incubator for 48 h. MTT dye was added to the plates 4 h prior to experiment termination at concentration of 2.5 mg mL−1, dissolved in media. MTT formazon crystals formed were dissolved in DMSO and absorbance at 570 nm was recorded.12
DFT evaluation. The energy minimal state or optimized structural conformation of the target compounds, was obtained by optimizing at the DFT level 6-311g(d,p) basis set along with B3LYP functional. GaussView 6.1 graphical interface associated GAUSSIAN 09 (Revision C.01) was the software-package used for the different calculations and visualization of results. BA and 11a in their carboxylate forms have been computed and compared to explain the bioactive behaviour. Various systematic algorithms under combined non-planar group symmetry and theoretical force constants generate useful conclusions from the calculation. Molecular orbital analysis, and other electron density mapping14 were further analysed to describe various physicochemical properties linked with the study.15,16
Molecular docking and dynamic studies
Protein preparation. The 3-dimensional structures of proteins were retrieved from RCSB PDB (Research Collaboratory for Structural Bioinformatics PDB). Matrix metalloproteinase-2 bound to N-[4-[[(2R)-1-(hydroxyamino)-3-methyl-1-oxo-butan-2-yl]-(2-morpholin-4-ylethyl)sulfamoyl]phenyl]-4-pentyl-benzamide (ligand id: SC-74020 or I52) (PDB id: 1HOV), structure of catalytic domain of human poly(ADP-ribose) polymerase in complex with 2-[3-[(1R)-4-(4-fluorophenyl)-3,6-dihydro-2H-pyridin-1-yl]propyl]-8-methyl-3H-quinazolin-4-one (ligand id: FRM) (PDB id: 1UK0) and structure of the Kinase Domain of Human Traf2- and Nck complexed with a selective inhibitor, 9-hydroxy-4-phenyl-6H-pyrrolo[3,4-c]carbazole-1,3-dione (ligand id: 824) (PDB id: 2X7F) were downloaded in the .pdb format. The proteins were converted from pdb to pdbqt using the PyRX0.8.
Ligand preparation. The bound ligands were removed and redocked with the respective proteins to obtain the vina score to validate the docking tool and to obtain the interaction score. The ligand molecules were drawn using ACD ChemSketch and saved as a .mol file. The OPENBABEL program available inside the PyRX0.8 was used to convert all the molecules into PDB format, and universal force field (uff) was used for energy minimization with the number of runs kept to 2500.
Molecular docking. For docking studies, AutoDock vina program17 was used. It is an efficient tool for prediction of protein–ligand binding. AutoGrid17 was used to define docking area for proteins. The grid box size of 78 × 42 × 42 Å and centred at x, y, z coordinates of 8.78, 17.43, 26.21 for 1HOV, grid box size of 78, 71, 49 and grid center of 5.46, 0.09, 34.96 for 1UK0 and grid box size of 21, 0.31, 52 and grid center of 77.75, 57.64, 42.79 for 2X7F for all the ligands including reference to the target proteins. The AutoDock Vina exhaustiveness of 8 was applied using PyRx 0.8, a Graphical User Interface for docking studies (Wolf, 2009). VinaLigGen was used to generate LigPlot for each conformation.18–20
Molecular dynamics simulation. MD simulations were performed on all three proteins by considering their APO form and the protein–ligand complex identified after the docking result using Desmond Maestro v11.3.21 Initially, the protein preparation wizard was used to pre-process the protein and the system builder utility for preparing the system for simulation. The protein was then individually placed in an orthorhombic simulation box with a distance of 10 Å away from the edges. The system was solvated by pre-equilibrated simple point charge (SPC) water model and 0.15 M salt (NaCl) concentration was added along with Na+ and Cl− ions to neutralize the system. Prior to MD simulations, the model system was relaxed using the standard Desmond protocol. This a six-step relaxation protocol which includes an initial short simulation of 12 ps in NVT ensemble at T = 10 K with restraints on solute heavy atoms, a 12 ps simulation in NPT ensemble with T = 10 K and pressure (P) = 1 atm restraining the solute heavy atoms, a 12 ps simulation in NPT ensemble with restraints on nonhydrogen solute atoms, and a 24 ps simulation in NPT ensemble without restraints. The pressure and temperature conditions were stabilized with the isobaric and isothermal ensemble using the Martyna–Tobias–Klein barostat and Nosé–Hoover thermostat which are the default protocols of Desmond.Finally, OPLS3 (ref. 22) force field was applied to the protein and ligand. System preparation included solvation, neutralization, minimization, and equilibration before a 100 ns NPT ensemble (T = 300 K and P = 1 atm) simulation and trajectory was recorded at intervals of 100 ps by generating approximately 1000 frames.
Result and discussion
Chemistry
The C-3-hydroxyl and C-20 double bond positions in BA are comparatively less explored than position C-28 with carboxylic acid. In this study we explored C-3 and C-20 positions and envisaged that introduction of nitrogen containing compounds (amine; oxime, phosphoramidate) could enhance the cytotoxic activity and even water solubility.23,24 Additionally, BA derivatives containing an acyl moiety at C-3 position are known to display higher cytotoxicity.23 To achieve this, we synthesised C-3 derived amine analogs with intact acyl group by treating compound 1 with chloroacetyl chloride in dry THF in presence of dry pyridine to synthesize intermediate 8 which was used as a starting material to synthesize target compounds. In order to keep the acyl group intact, compound 8 was subjected to react with different amines to afford desired products (9a–g) in excellent yields (Scheme 1).
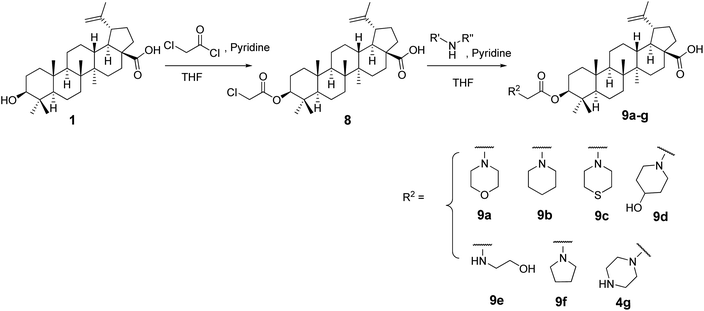 |
| | Scheme 1 Synthetic route to 9a–g. | |
The phosphoramidate moiety is known to improve the antitumor activity of the parent molecules.25,26 Therefore, it was feasible to targeted C-3 and C-20 positions of betulinic acid. To synthesis C-3 derived phosphoramidate analogs, compound 2 with NaN3 to furnish azide 10 with intact acetyl group. Compound 10 was treated with appropriate trialkyl phosphite in THF to produce the desired products 11a and 11b (Scheme 2).
 |
| | Scheme 2 Synthetic route to 10, 11a and 11b. | |
To synthesize phosphoramidate at C-20 position by exploiting the olefinic bond, compound 1 was treated with N-bromosuccinimide (NBS) to obtain 12 which when treated with NaN3 afforded azide 13 that was finally treated with appropriate trialkyl phosphite to afford desired phosphoramidates 14a and 14b (Scheme 3). The structures of all the synthesized derivatives were confirmed using spectroscopic techniques.
 |
| | Scheme 3 Synthetic route to 14a and 14b. | |
Biology
All the derivatives including parent compound 1 were evaluated in vitro for anti-tumor activity on six different human cancer cell lines: breast cancer MCF-7, lung cancer A549, colon cancer HCT-116, leukaemia MOLT-4, prostate carcinoma cell PC-3 and pancreatic cancer cell Miapaca-2by MTT assay. The IC50 values are summarized in Table 1 and given as the average value of the triplicate analysis. As shown in Table 1, conversion of 1 to chloroacetyl chloride derivative 8, slightly improved the activity. Among amine derivatives 9a–g designed to evaluate the role played by amino moiety in influencing the anti-cancer activity 9b was effective against MCF-7, A549, MOLT-4 and PC-3 cell lines with IC50 values 13.0, 12.59, 18.0 and 12.0 μM respectively and two to three times more active than 1 (BA). Compound 9d was found active against A549, MOLT-4 and PC-3 cell lines with IC50 values between 9.98, 8.35 and 12.36 μM. 9e was the most active compound among the amine derivatives having IC50 values 9.48 (MCF-7), 7.67 (A549), 6.78 (HCT-116), 11.70 (MOLT-4), 7.48 (PC-3) and 6.62 (Miapaca-2) μM. Interestingly, both of these compounds (9d and 9e) have a free hydroxyl group in the amino moiety. Compound 10 was effective against HCT-116 and Miapaca-2 cell lines, IC50 values 5.25 and 8.34 μM respectively. Among the phosphoramidate analogs, 11a and 11b, 11a was two to four-fold more active than parent 1, and found to be most active analog among all the synthesized derivatives, with IC50 values 7.15 (MCF-7), 8.00 (A549), 3.13 (HCT-116), 13.88 (MOLT-4), 8.00 (PC-3) and 6.96 (Miapaca-2) μM respectively on all the six cancer cell lines tested.
Table 1 IC50 values of BA and its derivatives against various cancer cell lines
| Compound |
Breast MCF-7 |
Lung A549 |
Colon HCT-116 |
Leukemia MOLT-4 |
Prostate PC-3 |
Pancreatic MiaPaca-2 |
| 1 |
22.86 |
24.74 |
12.23 |
44.30 |
15.55 |
16.72 |
| 8 |
19.13 |
18.15 |
11.15 |
>50 |
12.11 |
15.23 |
| 9a |
19.24 |
>50 |
22.50 |
>50 |
>50 |
>50 |
| 9b |
13.00 |
12.59 |
18.42 |
18.02 |
12.00 |
34.01 |
| 9c |
>50 |
9.21 |
11.44 |
>50 |
12.83 |
29.00 |
| 9d |
16.00 |
9.98 |
13.99 |
8.35 |
12.36 |
14.00 |
| 9e |
9.48 |
7.67 |
6.78 |
11.70 |
7.48 |
6.62 |
| 9f |
>50 |
10.87 |
9.78 |
>50 |
16.70 |
16.00 |
| 9g |
>50 |
14.79 |
12.50 |
>50 |
15.36 |
45.00 |
| 10 |
13.12 |
10.26 |
5.25 |
14.04 |
12.68 |
8.34 |
| 11a |
7.15 |
8.00 |
3.13 |
13.88 |
8.00 |
6.96 |
| 11b |
18.00 |
12.00 |
13.00 |
16.00 |
18.10 |
11.00 |
| 12 |
>50 |
30.21 |
6.82 |
>50 |
13.12 |
>50 |
| 13 |
15.10 |
18.16 |
8.25 |
18.00 |
12.79 |
10.34 |
| 14a |
>50 |
46.71 |
12.00 |
>50 |
41.14 |
23.00 |
| 14b |
>50 |
>50 |
11.55 |
>50 |
42.16 |
32.00 |
| Paclitaxel |
<0.01 |
<0.01 |
0.09 |
0.05 |
0.03 |
0.07 |
Compounds 12 and 13 were effective against HCT-116 cell line. In compounds 14a and 14b, the activity was significantly reduced compared to the parent 1 except for HCT-116 for which the activity was comparable to that of 1. These results indicate that cytotoxicity profile of BA analogs may be susceptible to the size and the electrostatic sensitivity of the substituents present at C-20 position. In general, the above results demonstrated that C-3 position was a favourable site to carry out structural modification to enhance the anti-tumor potential of BA. However, modification at C-20 resulted in the loss of activity indicated that C-20 position of BA was not a favourable site to derivatize to enhance cytotoxicity against cancer cell lines.
DFT-based global reactive descriptors
Geometry optimization is the first step of configuration/conformational analysis to arrive at a minimal energy state of a molecular system, provided no negative frequency is obtained in the calculation. Both molecular structures in their anionic forms were optimized at the relevant level of theory given in the computational methodology. Different optimization parameters, including bond lengths, bond angles, and dihedral angles of the parent compound, betulinic acid, and the potent derivative 11a in their carboxylate form, can be visualized as given in Fig. 2. The frontier orbitals of both structures are shown in Fig. 3 and 4, respectively, for BA and 11a.27–30 The frontier molecular orbitals (FMO) play an important role in describing the reactivity of a molecule in association with global reactive descriptors given in Table 2. From the calculated reactive descriptors, it is clear that BA has more kinetic stability than 11a due to a higher HOMO–LUMO gap, and the latter by global softness, global hardness, and dipole moment. These factors are hence responsible for the fascinating bioactivity exhibited by 11a.
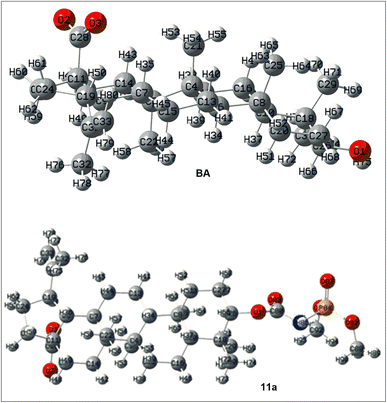 |
| | Fig. 2 Optimized structures of the representative compounds in their carboxylate form. | |
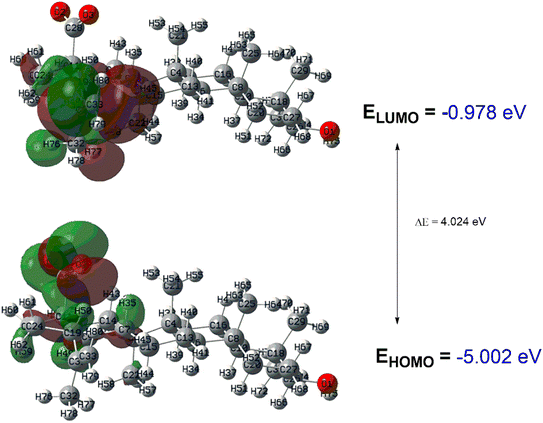 |
| | Fig. 3 FMO diagram of BA with the respective HOMO–LUMO gap. | |
 |
| | Fig. 4 FMO diagram of the 11a with the respective HOMO–LUMO gap. | |
Table 2 Reactive descriptors of the target compounds
| Descriptor |
BA |
11a |
| EHOMO |
−0.978 eV |
−0.773 eV |
| ELUMO |
−5.002 eV |
−5.088 |
| HOMO–LUMO gap |
4.024 eV |
4.315 eV |
| χ |
−2.990 |
−2.930 |
| σ |
0.497 |
0.464 |
| η |
2.012 |
2.157 |
| μ |
22.72 |
29.77 D |
| Pi |
2.990 |
2.930 |
| ω |
2.222 |
1.989 |
Molecular docking
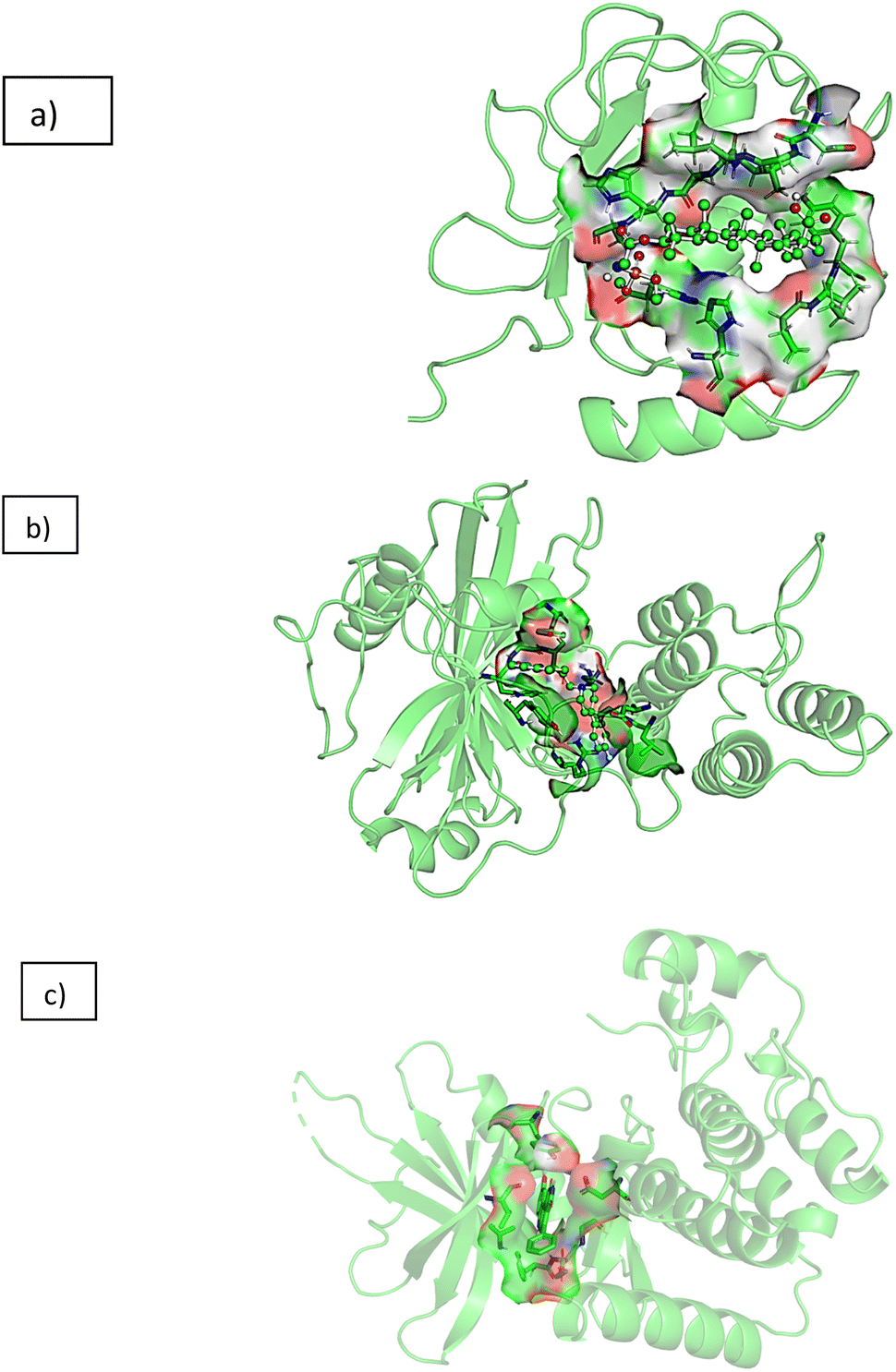
From the in vitro studies, compound 11a was found to be the lead compound with maximum activity against colon cancer cell line HCT-116 (IC50 value 3.13 μM), hence taken for molecular docking studies to get insights into the molecular mode of action against colon cancer. As mentioned in our recent publication three targets involved in pathophysiology of colon cancer including matrix metalloproteinase-2 (PDB ID: 1HOV), poly[ADP-ribose] polymerase-1 (PDB ID: 1UK0) and TRAF2 (PDB ID: 2X7F) were chosen to elucidate the mechanism of action against colon cancer.31 Fig. 5 represents the 3D interactions of 11a with all the three proteins mentioned above. Table 3 represent the data obtained from Molecular Docking analysis of all the synthesized compounds 1, 8, 9a–g, 10, 11a and 11b, 12, 13, 14a and 14b along with the parent molecule 1. Post docking analysis like hydrogen bond and hydrophobic interactions between each conformation of ligand with protein was performed using VinaLigGenand. The ligands which showed better vina score along with interaction were further considered for dynamics simulation. It was observed that the reference ligands I52 (PDB 1HOV) formed five hydrogen bonds with Ala86, Phe87, His124, His120 and His130 with vina score of −6.5, co-crystallised inhibitor FRM (PDB 1UK0) forms one hydrogen bond with Gly202 with vina score of −11.6 and co-crystallised inhibitor 824 (PDB 2X7F) forms one hydrogen bond with Tyr36 with the vina score of −9.7. All the interactions matched with the interactions available at the PdbSum to validate the docking tool and to consider the vina score as reference.32–34
 |
| | Fig. 5 3D interaction diagram of 11a with (a) matrix metalloproteinase-2 (1HOV) (b) poly(ADP-ribose) polymerase (1UK0) (c) kinase domain of human Traf2- and Nck (2X7F). | |
Table 3 Vina score and hydrogen bonds for the ligand interacting with three proteins
| Protein |
Ligand |
Vina score |
Hydrogen bonds |
| 1HOV |
1 |
−6.8 |
HIS55|ARG53 |
| 8 |
−7.5 |
— |
| 9a |
−7.7 |
— |
| 9b |
−8.1 |
— |
| 9c |
−8.2 |
ALA86 |
| 9d |
−8.3 |
PRO75 |
| 9e |
−7.3 |
LEU128|HIS124|ALA86 |
| 9f |
−7.8 |
ALA86 |
| 9g |
−7.6 |
— |
| 10 |
−7 |
ARG38|ARG38 |
| 11a |
−7.2 |
ALA86|HIS85 |
| 11b |
−7.5 |
ALA86|HIS85 |
| 12 |
−6.6 |
HIS55|ARG53 |
| 13 |
−6.8 |
GLU30|ARG53 |
| 14a |
−7.3 |
— |
| 14b |
−7.3 |
— |
| i52 |
−6.5 |
ALA86| PHE87|HIS124|HIS120|HIS130 |
| 1UK0 |
1 |
−10 |
ASN207|GLU327 |
| 8 |
−8.1 |
PRO281|LEU280|GLY283 |
| 9a |
−8.3 |
ASN207|SER203 |
| 9b |
−7.7 |
— |
| 9c |
−10.5 |
SER203|ASP109|HIS201 |
| 9d |
−8 |
GLN46|SER213|ARG180 |
| 9e |
−9.9 |
SER203|HIS201|HIS201 |
| 9f |
−9.1 |
GLU102 |
| 9g |
−8.4 |
GLN98|ARG217 |
| 10 |
−8.6 |
ASP105|SER322 |
| 11a |
−7.2 |
LEU117|SER121|GLU19 |
| 11b |
−9.8 |
HIS201|HIS201 |
| 12 |
−8.9 |
— |
| 13 |
−7.5 |
GLY283|GLN335|TYR331|HIS285 |
| 14a |
−7.8 |
PRO189|PRO281|ASP332 |
| 14b |
−8.9 |
GLU327|HIS248 |
| FRM |
−11.6 |
GLY202 |
| 2X7F |
1 |
−6.9 |
GLY109|PHE107 |
| 8 |
−6.8 |
GLY190 |
| 9a |
−6.8 |
LEU50|LEU50|GLN49 |
| 9b |
−7.1 |
— |
| 9c |
−6.9 |
ASN164 |
| 9d |
−6.9 |
LEU50|LEU50 |
| 9e |
−6.6 |
THR84 |
| 9f |
−6.8 |
LEU50|LEU50 |
| 9g |
−6.8 |
LEU50|LEU50 |
| 10 |
−6.9 |
LYS168|GLU106|THR84 |
| 11a |
−6.7 |
TYR86|PHE107|ARG80|ARG80 |
| 11b |
−6.2 |
GLU128|GLU127 |
| 12 |
−6.8 |
TYR86 |
| 13 |
−6.6 |
LEU50|LYS168|GLU106|LYS168|GLU106|THR84 |
| 14a |
−6.6 |
GLU163|TYR86|THR47 |
| 14b |
−6.3 |
LEU50|LYS168|TYR86 |
| 824 |
−9.7 |
TYR36 |
Based on the vina score, interactions and the in vitro studies the ligand 11a exhibited the vina score of −7.2 with 1HOV protein. In the case of 1UK0, it formed three hydrogen bonds with Leu117, Ser121, Glu19, yielding a vina score of −7.2. Ligand displayed the vina score of −6.7 by forming four hydrogen bonds with Tyr86, Phe107 and Arg80 for 2X7F. These complexes were subsequently chosen for dynamics simulation.
Molecular dynamics simulation
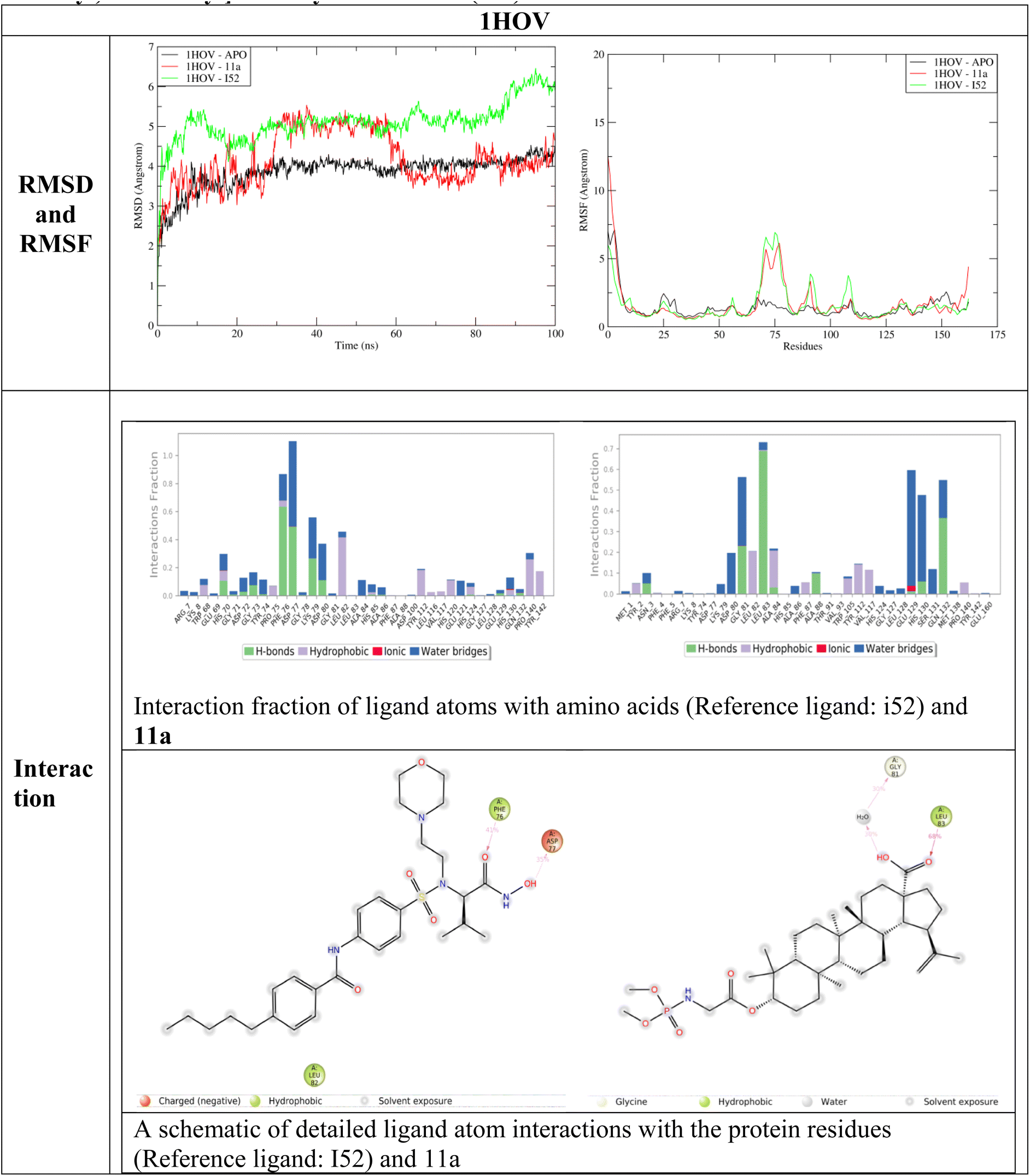
Molecular dynamics simulation studies provide a detailed account of binding interactions and interaction stability related to protein and ligand. The two terms RMSD provide details about structure deviation and RMSF detailed about the protein fluctuations due to ligand interactions.27 Fig. 6–8 represent the RMSD, RMSF, interaction fraction and ligand protein contacts between 11a and three proteins 1HOV, 1UK0 and 2X7F respectively.
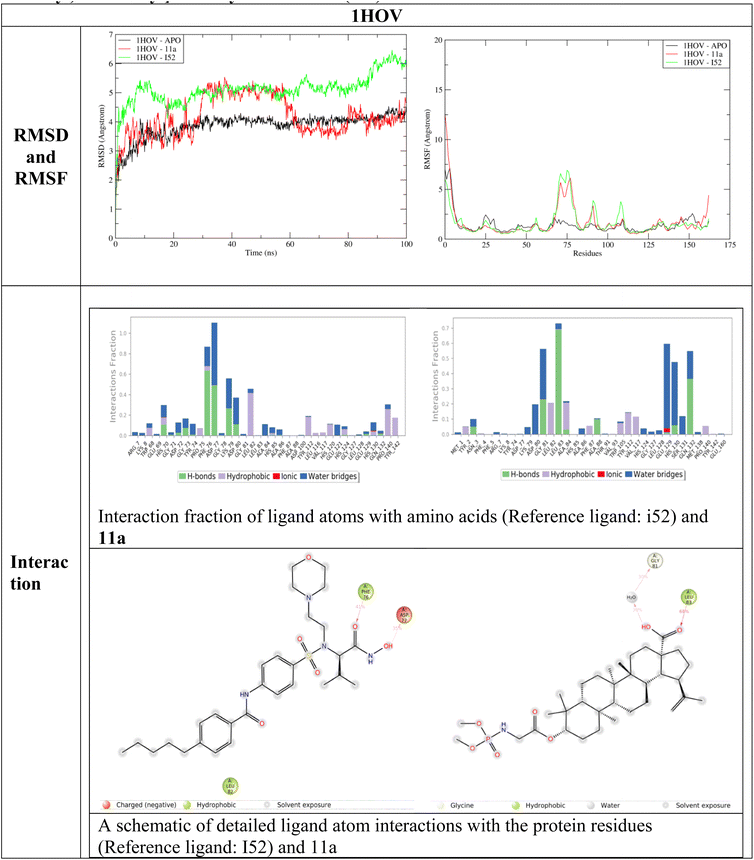 |
| | Fig. 6 RMSD, RMSF, interaction fraction and ligand–protein contacts between 11a and reference ligand N-[[3-[4-[[(2S)-1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl]sulfamoyl]phenyl]phenyl]methyl]-4-oxo-3H-quinazoline-2-carboxamide (I52) with 1HOV. | |
 |
| | Fig. 7 RMSD, RMSF, Interaction fraction and ligand–protein contacts between 11a and reference 2-{3-[4-(4-fluorophenyl)-3,6-dihydro-1(2H)-pyridinyl]propyl}-8-methyl-4(3H)-quinazolinone (RFM) with 1UK0. | |
 |
| | Fig. 8 RMSD, RMSF, interaction fraction and ligand–protein contacts between 11a and reference 9-hydroxy-4-phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione[9-hydroxy-4-phenyl-6H-pyrrolo[3,4-c]carbazole-1,3-dione] (824) with 2X7F. | |
During the interaction study with 1HOV, the RMSD plot indicates that the ligand-bound form shows structural deviations throughout the simulation time of 100 ns. The unbound form of the protein maintains stability. The deviation in the complex form (both reference ligand I52 and 11a) of the protein due to fluctuations in amino acid position 60–85. There is a significant secondary structural change around the residues 82–85. In the APO form, beta-sheet is formed while in the complex the beta sheet is completely lost throughout the simulation time of 100 ns bringing more deviation to the protein complex. Interaction analysis of reference ligand I52 shows hydrophobic interaction with Phe76, and Leu82, hydrogen bond with Asp77. Ligand 11a formed hydrophobic interaction with fraction values near 0.7 with Leu83 and formed water bridge with Gly81. These residues constitute the binding site of the protein as observed in PDBSum (1HOV – Sc-74020 complex). Overall, ligand 11a exhibits deviation around 30 ns and 60 ns and stability after the simulation time of 60 ns.
The RMSD analysis of 11a with 1UK0 indicates that binding of ligand 11a reduces overall deviation compared to the APO form when compared with reference ligand FRM. Notably, there is a significant reduction in fluctuation around positions 50–60, 110–130 and 150–260 supporting in deviation reduction. The reference ligand formed hydrogen bond with Gly202 and Asn207. Hydrophobic interaction with Leu108, His201, Tyr235, Tyr246. Ligand 11a formed a weak interaction between ligand and multiple residues Lys13, Asp17, Leu117, Gly119 and Asn132 was observed. Since interaction fraction less than 0.4, residues were not observed between the ligand–protein contact analysis.
From the RMSD analysis, both reference and 11a demonstrated stability with 2X7F. However, compared to reference 824, the overall deviation slightly decreases in the 11a complex after 35 ns of simulation time. Reduction in fluctuation is observed in the initial positions, 220 to 260, contributing to a significant deviation compared to the APO form. A hydrogen bond with Phe107 was observed for both the ligands and water bridge formation with Met105 for 11a. The protein–ligand complex shows a stable interaction is the active site.
Conclusion
In this paper, different synthetic strategies were used to exploit C-3 and C-20 positions of betulinic acid to design and synthesized novel derivatives to evaluate their effect on anticancer activity. All the analogs were tested in vitro for anti-tumor activity on six different human cancer cell lines including breast cancer MCF-7, lung cancer A549, colon cancer HCT-116, leukaemia MOLT-4, prostate carcinoma PC-3 and pancreatic cancer Miapaca-2 by MTT assay. Many derivatives displayed better cytotoxicity profile than the parent compound. More significantly compounds 9b, 9e, 10 and 11a were found to have promising activity profile than BA. Compound 11a was found as the most potent and lead compound, with maximum activity against colon cancer HCT-116 cell line (IC50 values 3.13 μM). DFT calculation also supports the observed data followed by the molecular docking and dynamic studies of compound 11a showing more stable interactions with protein 2X7F. Compound 11a exhibited the potential to be developed as potent anticancer agent against colon cancer and should be further explored for in vitro and in vivo mechanism of action studies against colon cancer.
Data availability
The associated data with this article can be found in ESI.†
Conflicts of interest
There is no conflict of interest associated with this article.
Author contributions
Nisar A. Dangroo: conceptualization, data interpretation, writing – original draft, Ziad Moussa: funding acquisition/review drafting/data interpretation, Mustafa S. Alluhaibi: in silico/software, Abdulrahman A. Alsimaree: in silico/software, Mohammed B. Hawsawi: docking, Reem I. Alsantali: methodology investigation, Jasvinder Singh: methodology, investigation, bioassays, Nidhi Gupta: manuscript writing, editing and MD, Basavarajaiah S. M.: software Md simulations, molecular docking, Prashantha Karunakar: molecular docking and MD study, J. M. Mir: Gaussian based DFT calculations, Manzoor A. Rather: formal analysis and characterization, and Saleh A. Ahmed: supervision, review, editing and fund acquisition.
Acknowledgements
The authors extend their appreciation to Taif university, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-86). Ziad Moussa is grateful to the United Arab Emirates University (UAEU) and to the Research Office for supporting the research developed in his laboratory and reported herein (UPAR grant code G00004605). NAD sincerely thanks the Department of Science and Technology-Science and Engineering Research Board (DST-SERB) New Delhi, India for providing fellowship under File No. PDF/20l7/002716CS.
References
- https://www.who.int/news-room/fact-sheets/detail/cancer, accessed on 18.06.2024.
- https://www.who.int/news-room/fact-sheets/detail/colorectal-cancer, accessed on 18.06.2024.
- https://www.cancer.gov/types/colorectal/patient/colon-treatment-pdq, accessed on 18.06.2024.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- Y. Ren and A. D. Kinghorn, Planta Med., 2019, 85, 802–814 CrossRef CAS.
- X. Zhang, J. Hu and Y. Chen, Mol. Med. Rep., 2016, 14, 4489–4495 CrossRef CAS PubMed.
- Y. Zhong, N. Liang and M.-S. CHENG, Chin. J. Nat. Med., 2021, 19, 641–647 CAS.
- R. Csuk, Expert Opin. Ther. Pat., 2014, 24, 913–923 CrossRef CAS.
- Y. Zhang, S. Ye, Y. Wang, C. Wang, Y. Zhu, Y. Wu, Y. Zhang, H. Zhang and Z. Miao, Bioorg. Med. Chem., 2022, 59, 116672 CrossRef CAS PubMed.
- G. Liebscher, K. Vanchangiri, T. Mueller, K. Feige, J.-M. Cavalleri and R. Paschke, Chem.-Biol. Interact., 2016, 246, 20–29 CrossRef CAS.
- N. Gupta, S. K. Rath, J. Singh, A. Qayum, S. Singh and P. L. Sangwan, Eur. J. Med. Chem., 2017, 135, 517–530 CrossRef CAS PubMed.
- N. A. Dangroo, J. Singh, S. K. Rath, N. Gupta, A. Qayum, S. Singh and P. L. Sangwan, Steroids, 2017, 123, 1–12 CrossRef CAS PubMed.
- I. Khan, S. K. Guru, S. K. Rath, P. K. Chinthakindi, B. Singh, S. Koul, S. Bhushan and P. L. Sangwan, Eur. J. Med. Chem., 2016, 108, 104–116 CrossRef CAS.
- C. J. Brabec and N. S. Sarici, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, GAUSSIAN 09 (Revision C.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- S. Thandivel, P. Rajan, T. Gunasekar, A. Arjunan, S. Khute, S. R. Kareti and S. Paranthaman, Heliyon, 2024, 10, e27880 CrossRef CAS PubMed.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., Des. Sel., 1995, 8, 127–134 CrossRef CAS PubMed.
- S. Dallakyan and A. J. Olson, in Chemical Biology, ed. J. E. Hempel, C. H. Williams and C. C. Hong, Springer New York, New York, NY, 2015, vol. 1263, pp. 243–250 Search PubMed.
- K. J. Bowers, F. D. Sacerdoti, J. K. Salmon, Y. Shan, D. E. Shaw, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary and M. A. Moraes, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing SC '06, ACM Press, Tampa, Florida, 2006, p. 84 Search PubMed.
- J. Wiemann, L. Heller, V. Perl, R. Kluge, D. Ströhl and R. Csuk, Eur. J. Med. Chem., 2015, 106, 194–210 CrossRef CAS PubMed.
- M. Willmann, V. Wacheck, J. Buckley, K. Nagy, J. Thalhammer, R. Paschke, T. Triche, B. Jansen and E. Selzer, Eur. J. Clin. Invest., 2009, 39, 384–394 CrossRef CAS.
- F. M. Oliveira, L. C. Barbosa and F. M. Ismail, RSC Adv., 2014, 4, 18998–19012 RSC.
- C. R. Wagner, V. V. Iyer and E. J. McIntee, Med. Res. Rev., 2000, 20, 417–451 CrossRef CAS.
- P. J. Stephens, D. M. McCann, F. J. Devlin and A. B. Smith, J. Nat. Prod., 2006, 69, 1055–1064 CrossRef CAS.
- T. Erdogan, J. Mol. Struct., 2021, 1242, 130733 CrossRef CAS.
- D. F. Tegegn, H. Z. Belachew and A. O. Salau, Sci. Rep., 2024, 14, 8146 CrossRef CAS.
- C. F. Matta, J. Comput. Chem., 2014, 35, 1165–1198 CrossRef CAS.
- C.-H. Lai, C.-C. Chang, Y.-L. Weng and T.-H. Chuang, Molecules, 2018, 23, 3170 CrossRef.
- E. O. Akintemi, K. K. Govender and T. Singh, Comput. Theor. Chem., 2022, 1210, 113658 CrossRef CAS.
- N. Gupta, G. Singh, A. Qayum, M. Ovais Dar, S. Singh, M. Katoch and P. L. Sangwan, ChemistrySelect, 2024, 9, e202400637 CrossRef CAS.
- S. M. Basavarajaiah, J. Badiger, N. G. Yernale, N. Gupta, P. Karunakar, B. T. Sridhar, M. Javeed, K. S. Kiran and B. Rakesh, Bioorg. Chem., 2023, 137, 106598 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ziad Moussa
*a,
Ziad Moussa