
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of a novel interlayer expanded zeolite with a 12 × 12-ring structure†
Boting Yang *a,
Feijie Wanga,
Yutong Zhaoa,
Mengmeng Suna,
Jiayang Lia,
Yong-Qing Qiub and
Chun-Guang Liua
*a,
Feijie Wanga,
Yutong Zhaoa,
Mengmeng Suna,
Jiayang Lia,
Yong-Qing Qiub and
Chun-Guang Liua
aCollege of Science, Beihua University, Jilin 132013, P. R. China. E-mail: ybt199@126.com
bInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China
First published on 27th February 2025
Abstract
In this work, the lamellar precursor PLS-1 was successfully modified by acid treatment, intercalation and interlayer silylation. The layered precursor PLS-1 with CDO topological structure was synthesized using H-magadiite as silicon source and tetramethylammonium hydroxide (TMAOH) as template agent. After acid treatment to remove interlayer organic molecules, various OSDA were introduced between the layers, and the interlayer expanded intercalated materials were obtained. Intercalation can not only improve the crystallization degree of the sample, but also introduce larger organic molecules between the layers to increase the interlayer distance. 1,3-Dichlorotetramethyldisiloxane was used to silanize the intercalated material. The silanization conditions were investigated and the best conditions were obtained. Through the construction of the theoretical model, the experimental data were compared to show that the new molecular sieve PLS-1-2Si material has a 12 × 12-R interlayer expanded structure.
1. Introduction
Zeolites are an important class of multi-phase catalysts because they not only have excellent activity and high specific surface area, but also have certain shape and size selectivity.1 Due to their special pore structure, zeolites are widely used in a variety of applications such as catalysts, ion exchangers, detergents, and adsorbents,2–5 and the scope of their applications is still expanding. Based on the great application potential of zeolites, the preparation and development of new zeolite structures by different methods is one of the research hot-points in the field of zeolites.One of the most common method for synthesizing new zeolites is the formation of three-dimensional zeolites with different structures from two-dimensional layered zeolite by interlayer condensation, and the results of these researches have been extensively reviewed.6–11 Lamellar zeolites have an unique structural property that their layers are connected by hydrogen bonding between terminal silica hydroxyl groups. The hydrogen bonding of the interlayer connection is weak and suitable for post-treatment modification. Post-treatments include swelling, pillaring, intercalation, or interlayer silylation,12–15 and a variety of layered zeolite can be modified by post-treatments to derive more open structures. The synthesis of new zeolite catalysts from layered zeolites has attracted great interest due to the excellent catalytic performance of layered catalysts in many promising processes.16–20
PLS-1 is a layered silicate prepared by Takuji Ikeda's group, it has a five-ring basic structural unit,21 and will experience the interlayer dehydration and condensation after calcination to formed a new zeolite CDS-1 with CDO topology. CDS-1 has a two-dimensional eight-ring (8-R) pore system (4.7 × 3.1 Å and 4.2 × 2.5 Å). Fethi Kooli's group prepared Na-magadiite and its plasmonic derivatives and synthesized PLS-1.22 The influence of factors such as hydrothermal treatment temperature and time water content on the effect of hydrothermal treatment was investigated, and the synthesis time of PLS-1 was shortened from 10 to 3 days, which greatly improved the synthesis efficiency. Yoshihiro Sugi group realized the application of solid base catalysts by using TMAOH molecules between the layer of PLS-1.23
In the later reports, some groups have conducted studies related to the introduction of hetero-atoms into PLS-1 structures. For example, gallium silicate molecular sieve [Ga]CDS-1 zeolite with CDO topology was successfully prepared from layered precursor [Ga]PLS-1 by dehydration condensation,24 and this hetero-atom zeolite could be used to catalyze the methanol-to-olefin (MTO) reaction. Some groups also succeeded in introducing B, Ge, and Ti hetero-atoms into PLS-1 and synthesized the corresponding hetero-atomic CDS-1 zeolite.25–27 Takashi Tatsumi's group developed a one-step interlayer silylation reaction of the layered zeolite precursor PLS-1 for the preparation of interlayer-expanded microporous crystals, IEZ-1, and investigated its adsorption and catalytic properties.28 These studies indicate that PLS-1 has a large potential for further study and application.
Mochizuki developed a new method to construct two- and three-dimensional network molecules to form ordered silica nanostructures,29 where two-dimensional layered silicates formed new pore structures by silylation of dioxydichlorosilanes, and new three-dimensional interlayer-expanded zeolites were prepared. And then, various layered precursors with different topologies, such as MWW, FER, CDO, and MCM-47 were silylated and constructed a novel zeolite structure with enlarged interlayer pores. In a later study, they used DEDMS silanes to silylate the layered precursors PLS-4, Nu-6(1), and PLS-3,30–34 and the results showed that the silylation effectively enlarged the interlayer pore entrances and the specific surface area, and exhibited enhanced activity in terms of porosity and catalytic properties. The interlayer silylation reaction of layered precursors with silane reagents proved to be a versatile technique for post-synthesis of novel zeolites with larger porosity.
In this study, we chose the layered zeolite PLS-1 for preparation and post-treatment modification. We need to perform acid treatment and intercalation treatment on the parent material PLS-1 before silylation because the interlayer spacing of PLS-1 is not large enough to accommodate the insertion of silane reagent 1,3-dichlorotetramethyldisiloxane between the layers if we start from PLS-1 directly. Therefore, we used acid treatment to partially remove the organic molecules between the layers of PLS-1, and then intercalated the larger organic molecules into the material after acid treatment, so as to obtain the intercalated material with the same layer lamina as PLS-1 but the larger interlayer spacing than PLS-1. We tested different organic molecules and finally found that the material intercalated with tetrabutylammonium hydroxide (TBAOH) has a sufficiently large interlayer spacing, which achieve the insertion of silane reagent 1,3-dichlorotetramethyldisiloxane, and finally obtained the product PLS-1-2Si. Thus we adopted an acid treatment to partially remove TMAOH between the layers, and then utilized organic structure-directing agents (OSDAs) with larger size to intercalate the acid-treated samples to obtain the layers with an increased interlayer spacing, and then finally subjected them to silylization with 1,3-dichlorotetramethyldisiloxane to obtain the pore-size-increased zeolite. The resulting material PLS-1-2Si has –O–Si–O–Si–O– units silylated between the layers and possesses a novel structure with 12 × 12-R pore system.
2. Experimental section
2.1 Materials
The chemicals used were of analytical reagent grade: Ludox HS-40 (colloidal SiO2), solid NaOH, TMAOH solution (tetramethylammonium hydroxide 25% in water), TEAOH solution (tetraethylammonium hydroxide 25% in water), TPAOH solution (tetrapropylammonium hydroxide 25% in water), TBAOH solution (tetrabutylammonium hydroxide 25% in water), TEMP (4-amino-2,2,6,6-tetramethylpiperidine 98%), anhydrous ethanol, HCl (31%), dimethyldichlorosilane (99%), 1,3-dichlorotetramethyldisiloxane (95%).2.2 Characterization techniques
The structure and crystallinity of the zeolite were characterized by powder X-ray diffraction (XRD). The XRD spectra were acquired on a DX-2700 diffractometer using Cu Kα radiation with a tube voltage of 30 kV, a tube current of 40 mA, and a 2θ angle in the range of 5–50° with a step angle of 0.02°. Infrared analysis was carried out using a Fourier transform infrared spectrometer (SHIMADZU, IRAFFINITY-1S). The samples were mixed and ground with KBr crystals and tested in the range of 400–4000 cm−1 after conventional pressing to analyze the form of functional groups present in the zeolite. This was performed using a TG/DTA apparatus (Beijing Jingyi Gaoke Instrument Co., Ltd, ZCT-A). The crystal morphology of the solid product was observed by thermal field emission high resolution scanning electron microscope (Zeiss, SUPRA55, Germany). The 29Si MAS NMR spectra were measured on a VARIAN VNMRS 400WB NMR spectrometer using the single-pulse method with a 4 mm MAS BB/BB probe at a frequency of 119.29 MHz, a rotational rate of 3 kHz, and a recovery delay of 60 s. The chemical shifts are those of the sodium salt of 2,2-dimethyl-2-silapentane-5-sulfonate ((CH3)3Si(CH2)3SO3Na).2.3 Material preparation
2.3.5.1 Preparation of PLS-1-TEAOH. PLS-1-acid-x-y-z was immersed in tetraethylammonium hydroxide (TEAOH) solution at a molar ratio of SiO2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEAOH:H2O = 1:0.4:30 and reacted at 443 K for 8 h. The product was filtered and washed and dried at 333 K overnight.
:TEAOH:H2O = 1:0.4:30 and reacted at 443 K for 8 h. The product was filtered and washed and dried at 333 K overnight.
2.3.5.2 Preparation of PLS-1-TPAOH. PLS-1-acid-x-y-z was immersed in tetrapropylammonium hydroxide (TPAOH) solution at a molar ratio of SiO2
:TPAOH:H2O = 1:0.2–0.4:30, and reacted at 373 K for 8 h. The product was filtered and washed, and dried at 333 K overnight.
2.3.5.3 Preparation of PLS-1-TBAOH. PLS-1 acid treatment was immersed in tetrabutylammonium hydroxide (TBAOH) solution at a molar ratio of SiO2
:TBAOH:H2O = 1:0.2–0.4:30, reacted at 373 K for 8 h, the product was filtered and washed, and dried at 333 K overnight.
2.3.6.1 Preparation of PLS-1-Si. Using 1,3-dichlorodimethylsilane (DCDMS) as the silylation reagent, 1.0 g of solid powder was added to a mixture of 0.23 g of silylation reagent and 30 mL of 1.0 M HCl–EtOH solution and stirred for 30 min at room temperature. The resulting mixture was then stored statically at 443 K for 20 h. The mixture was then dried at 333 K for 1 h. The mixture was then dried at 333 K for 1 h. The mixture was then stored at 333 K for 1 h. The mixture was then stored at 333 K for 2 h. After silicification, the solids were recovered by filtration, washed with distilled water, and then dried at 333 K. The material was then dried at 333 K for 30 min. The resulting material was named PLS-1-Si.
2.3.6.2 Preparation of PLS-1-2Si. The interlayer expanded material was subjected to insertion of silicon using 1,3-dichlorotetramethyldisiloxane as a silylation reagent. 1.0 g of the interlayer-expanded material was added to a mixture of 0.23 g of the silylation reagent and 30 mL of 2.0 M HCl solution. The resulting mixture was then stored statically at 443 K for 20 hours. After silylation, the solid was recovered by filtration, washed with distilled water, and then dried at 333 K. The material was then dried at 333 K for 3 hours. The resulting material was named PLS-1-2Si.
3. Results and discussion
3.1 Exploration of the intercalation of PLS-1 under different acid treatment conditions
PLS-1 is a layered silicate with the presence of the organic substance TMAOH between the layer, and the introduction of organics with larger size and volume into the interlayer can result in an interlayer-expanded silicate material. Considering the limited amount of organic matter that can be accommodated between the layer, in this study, TMAOH was partially removed by acid treatment to leave empty space for OSDA with larger volume. During the experiments, we explored the influence of different acid treatment conditions on the resulted intercalated materials using TEAOH. The changes of the material layer spacing and the crystallinity of the resulting samples can be seen by XRD. TG analysis was performed to compare the amount of TMAOH removed between the layers of the PLS-1 matrix and the acid-treated samples to derive the optimal acid treatment conditions that would enable the intercalation.Fig. 1 gives the XRD plots of PLS-1 and its samples after treatment with 0.1 M HCl acid for 0.5–2 h at 443 K and after intercalation using TEAOH. From the figure, it can be seen that PLS-1 has good crystallinity and phase purity (Fig. 1a), with 2θ = 8.46° for the laminar characteristic peaks (marked with arrow), while the sample after calcination is CDS-1 (Fig. 1b), which satisfies the CDO topology, with 2θ = 9.62° for the laminar characteristic peaks. After calcination, the silica hydroxyl groups at the end of the laminates were dehydrated and condensed, the layer spacing was reduced, and the position of the layered characteristic peak was shifted back. After a short high-temperature acid treatment, the position of the laminar characteristic peaks of the product remained essentially unchanged. Observation of the XRD images showed that the layer spacing did not increase significantly, indicating that the acid treatment conditions did not have a significant effect on the removal of TMAOH from the interlayer of PLS-1. Therefore, we changed the conditions of acid treatment to further explore.
 | ||
| Fig. 1 XRD plots of PLS-1 parent and various post-treated materials: PLS-1 (a), PLS-1-cal (b), PLS-1-acid-0.1H-443-0.5 (c), PLS-1-acid-0.1H-443-0.5-TEAOH (d), PLS-1-acid-0.1H-443-1 (e), PLS-1-acid-0.1H-443-1-TEAOH (f), PLS-1-acid-0.1H-443-2 (g), PLS-1-acid-0.1H-443-2-TEAOH (h). | ||
In Fig. 2, a lower-temperature, longer-time acid treatment of the PLS-1 layered precursor was carried out using a 0.1 M HCl solution, and the sample was acid treated for 4 h at room temperature, 333 K, and 353 K. As can be seen from the Fig. 2, the position of the layered characteristic peaks remained essentially unchanged for the 4 h of acid treatment at different temperatures. At room temperature, the XRD of the acid-treated samples was consistent with that of the matrices, and the XRD peaks changed slightly at 20–30° after increasing the temperature. The position of the characteristic peaks of the layers remained unchanged after the use of TEAOH intercalation, indicating that the TMAOH between the layer during the acid treatment process was not effectively removed, which limited the access of TEAOH to the interlayer. In the later experiments, the present study extended the acid treatment time to compare the acid treatment and the intercalation effect.
 | ||
| Fig. 2 XRD plots of PLS-1 parent and various post-treated materials: PLS-1 (a), PLS-1-cal (b), PLS-1-acid-0.1H-RT-4 (c), PLS-1-acid-0.1H-RT-4-TEAOH (d), PLS-1-acid-0.1H-333-4 (e), PLS-1-acid-0.1H-333-4-TEAOH (f), PLS-1-acid-0.1H-353-4 (g), PLS-1-acid-0.1H-353-4-TEAOH (h). | ||
Subsequently, the time of acid treatment was increased from 4 h to 6 h. As can be seen from Fig. S1,† the XRD curves of the intercalated layer after 6 h of acid treatment using 0.1 M HCl solution did not undergo any significant changes compared with the XRD curves of the intercalated samples after 4 h of acid treatment in Fig. 2, and the laminar characteristic peaks of the intercalated samples did not move forward, which indicates that the acid treatment for 6 h in did not produce any effect. Based on the above results, we can conclude that in the 0.1 M HCl system, changing the temperature and time of acid treatment has no significant effect on the removal of TMAOH. We guessed that the acid concentration was too low, so we increased the acid concentration to 1.0 M for experimental investigation in this study.
After increasing the acid concentration, we first tried the high-temperature short-term acid treatment for PLS-1 at 443 K for 0.5–1 h. From Fig. S2,† it can be seen that the XRD peaks of the samples changed somewhat at 20–30° after the acid treatment, but the laminar characteristic peaks remained undisplaced after the intercalation treatment. Meanwhile, the positions of the XRD peaks of the samples (e, h, k) and the parent calcined sample (b) are basically the same after the intercalation calcination, which indicates that the topology has not been changed. This result suggests that after increasing the acid concentration, TEAOH did not enter the interlayer successfully even under high-temperature short-duration acid treatment conditions. Therefore, we tried the low-temperature long-time acid treatment condition to observe the effect of acid treatment and TEAOH intercalation.
The samples were acid treated with 1 M HCl solution at room temperature, 333 K and 353 K for 4 h. After filtration and washing, the samples were intercalated with TEAOH solution and then the intercalated samples were calcined. As can be seen in Fig. S3,† comparing with the original sample, after the low-temperature and long-time acid treatment and the intercalation of PLS-1 using 1 M HCl solution, the characteristic peaks of the layers remained undisplaced, and TEAOH did not replace TMAOH into the interlayer. It indicates that the hydrochloric acid solution has limited effect on the acid treatment of PLS-1 matrices, therefore, in this study, the system of the acid treatment process solution was adjusted to use ethanol hydrochloric acid solution for the acid treatment.
We firstly used 0.1 M HCl–EtOH solution for short-time high-temperature acid treatment of PLS-1 parent, as can be seen in Fig. 3, when PLS-1 was acid treated at 423 K for 0.5–1 h, the XRD characteristic peaks of the acid-treated samples changed only slightly compared with those of PLS-1 parent, and the laminar characteristic peaks of the acid-treated samples after intercalation still remained at 8.46° and did not move forward, indicating that TEAOH did not enter the interlayer. It is worth noting that the XRD of the sample changed greatly at 20–30° after 2 h of acid treatment, and the 2θ of the characteristic peak of the layer was 7.7° after the insertion of the layer, which indicates that the spacing between the layers of the sample increased after the insertion of the layer according to the Bragg equation. This indicates that the acid treatment in ethanol hydrochloric acid solution for 2 h can partially remove the TMAOH from the interlayer of PLS-1, and leave enough empty space to enable TEAOH to enter the interlayer smoothly during the intercalation process. In order to further investigate the removal of TMAOH from the interlayer after acid treatment, we selected 423 K acid-treated samples for 1 h and 2 h for TG analysis.
 | ||
| Fig. 3 XRD plots of PLS-1 parent and various post-treatment materials: PLS-1 (a), PLS-1-cal (b), PLS-1-acid-0.1HE-423-0.5 (c), PLS-1-acid-0.1HE-423-0.5-TEAOH (d), PLS-1-acid-0.1HE-423-0.75 (e), PLS-1-acid-0.1HE-423-0.75-TEAOH (f), PLS-1-acid-0.1HE-423-1 (g), PLS-1-acid-0.1HE-423-1-TEAOH (h), PLS-1-acid-0.1HE-423-2 (i), PLS-1-acid-0.1HE-423-2-TEAOH (j). | ||
From the TG curve in Fig. 4A, it can be seen that the total weight loss of PLS-1 parent (a) is 18.92%, there are two heat-absorbing processes between 100–320 °C, the first stage of mass loss is the desorption of water physically adsorbed in the PLS-1 interlayer that occurs as well as interlayer silica-hydroxyl dehydration and condensation (about 9.54%), and it is an exothermic process between 320–600 °C, and the second stage of mass loss occurred with interlayer condensation and removal of organic matter (about 9.38%). As can be seen from the figure, the mass loss of the acid-treated samples (b and c) was significantly reduced. The total weight loss of sample b for 1 h of acid treatment was 8.94% and the total weight loss of sample (c) for 2 h of acid treatment was 3.65%. It can be seen that the time of acid treatment in hydrochloric acid ethanol solution was extended, and the weight loss rate of the organic matter decreased. When acid treatment was carried out for 1 h, the amount of TMAOH remaining between the layer was large, and the interlayer vacancies were not enough to allow TEAOH to be inserted into the interlayer smoothly. When the time was extended to 2 h, about 60% of the TMAOH between the layer was removed, leaving enough space between the layer to accommodate the TEAOH molecules, and the insertion process was carried out smoothly, indicating that organic matter with larger sizes could be successfully inserted into the interlayer only when the removal rate of the organic matter between the layer had reached a certain level. In the subsequent study, we mainly investigated the temperature and time of the acid treatment in the hydrochloric acid–ethanol system to find out the optimal acid treatment conditions to accomplish the TEAOH intercalation.
 | ||
| Fig. 4 TG (A) and DTG (B) curves for PLS-1 parent (a), PLS-1-acid-0.1HE-423-1 (b), PLS-1-acid-0.1HE-423-2 (c). | ||
After the exploration of short-time high-temperature acid treatment, we further investigated the acid treatment of PLS-1 matrices using 0.1 M HCl–EtOH under low-temperature and long-time conditions, and it can be seen from Fig. S4† that at 4 h of acid treatment, after the acid-treated samples were intercalated by TEAOH at room temperature (d) and 313 K (g) temperature, the XRD at 2θ = 7.58 and 2θ = 8.4 two peaks appeared, which corresponded to the characteristic peaks of the layers of the parent material and TEAOH intercalation, indicating that the intercalation process was not carried out thoroughly, and a part of the crystals still maintained the original structure. The position of the laminar characteristic peaks after the intercalation of the samples treated with acid at 333 K (j) and 353 K (m) temperatures is consistent with that of sample j in Fig. 3, indicating that TEAOH enters into the interlayer and the interlayer spacing increases. As can be seen from the figure, the intercalated (j) and intercalation-calcined (k) samples after acid treatment at 333 K have better crystallinity and larger peak intensities in XRD. Meanwhile, several intercalated samples were CDO topology after calcination, indicating that the topology of the samples was not changed after intercalation. In conclusion, 333 K was chosen as the temperature of acid treatment in this study, and the time of acid treatment was explored.
We acid treated PLS-1 with 0.1 M HCl–EtOH solution at 333 K for 4 h, 6 h, 8 h, and 10 h. As can be seen in Fig. S5,† intercalation of the samples after acid treatment at 333 K all yielded interlayer-expanded materials with increased layer spacings (d, g, j, and m), and the resulting intercalated samples became more crystalline with increasing acid treatment time The crystallinity of the resulting intercalated samples was better with increasing acid treatment time. However, the interlayer-expanded materials with 8 h of acid treatment showed better characteristic peaks of CDO topology after calcination than those with 10 h of acid treatment. In summary, the optimal acid treatment condition explored in this study was 8 h of PLS-1 acid treatment at 333 K in 0.1 M ethanol hydrochloride system.
We selected samples that were able to achieve TEAOH intercalation after acid treatment for thermogravimetric analysis to further compare the interlayer TMAOH removal under these acid treatment conditions. As shown in Fig. S6,† these samples that were able to successfully intercalate, the total weight loss of the samples ranged from 2.95% to 4.92%, and the weight loss of these samples was smaller than that of sample b (PLS-1-acid-0.1HE-423-1) in Fig. 4, which indicates that after the acid treatment, when the interlayer organics removal reached a certain level, in other words, when sufficient vacancies were generated between the layer, the size of the bigger of OSDA can enter the interlayer smoothly, thus widening the interlayer spacing.
3.2 Exploration of different OSDA interpolation conditions
After selecting the optimal acid treatment condition, in the later study, we used the acid-treated samples with this condition for the investigation of the intercalation conditions, and the size of the interlayer spacing of the resulting interlayer swelling materials after intercalation with various OSDAs was calculated and compared.The intercalation process was probed for the time of TEAOH intercalation using a sample of PLS-1-acid-0.1HE-333-8 at a temperature of 443 K. As can be seen in Fig. 5, the intercalated samples have consistent XRD shapes with a 2θ = 7.62° for the lamellar characteristic peak. It is worth noting that the crystallinity of the intercalated samples increased with the increase of reaction time until 8 h; the samples intercalated for 10 h showed a decrease in crystallinity compared to 8 h. The crystallinity of the intercalated samples increased with the increase of reaction time. Based on the experimental results, 8 h was used as the reaction condition for intercalation in this study.
 | ||
| Fig. 5 XRD plots of PLS-1 intercalated using TEAOH, at different times 2 h (a), 4 h (b), 6 h (c), 8 h (d), 10 h (e). | ||
Then, we referred to the optimal conditions for TPAOH intercalation, which was reacted for 8 h at 443 K. However, at the end of the reaction, all the solids were dissolved, and we guessed that it might be caused by the enhanced alkalinity of TPAOH. Therefore, we lowered the reaction temperature to 373 K and found that TPAOH was able to intercalate into the interlayer with 2θ = 6.72° for the characteristic peak of the layer (Fig. S7a†). Due to the enhanced basicity of TPAOH, we also tried to reduce its content for exploration. During the experiment, the amount of TPAOH was halved, and it can be seen from the figure that at n(TMAOH)/n(TPAOH) = 0.5, a better crystallinity of the interlayer swelling material was obtained (Fig. S7d†). In this study, the dosage of 0.5 TPAOH was chosen and the reaction temperature was explored at low temperatures, 333 K (Fig. S7b†), 353 K (Fig. S7c†) and 373 K (Fig. S7d†) were chosen for the experiments, and it can be seen from Fig. S7† that the best crystallinity of the intercalation product was obtained at 373 K. We conclude that the optimum reaction condition for intercalation using TPAOH is SiO2:TPAOH:H2O = 1:0.2:30 at 373 K for 8 h.
The effect of TBAOH dosage on the intercalation was explored according to the optimal intercalation conditions of TPAOH. During the experiments, 373 K and 8 h were used as the reaction conditions, and the intercalation effects of n(TBAOH)/n(TPAOH) = 1 and 0.5 were compared, as shown in Fig. 6, after the TBAOH intercalation, the 2θ of the characteristic peaks of the lamina = 6.3°, which is lower than that of the TPAOH, indicated the larger interlayer distance. And the interlayer expansion material obtained when n(TBAOH)/n(TPAOH) = 0.5 with similar crystallinity and interlayer distance with using TBAOH for intercalation only.
 | ||
| Fig. 6 XRD plots of PLS-1 intercalation using TBAOH under different conditions PLS-1-0.5TBAOH 373 K 8 h (a), PLS-1-TBAOH 373 K 8 h (b). | ||
In later studies, we also tried to use 4-amino-2,2,6,6-tetramethylpiperidine (TEMP), a cyclic molecule, as well as a mixture of the two OSDAs for intercalation. We can see the XRD in Fig. S8,† when using TEAOH, TPAOH, TBAOH and TEMP for intercalation, the characteristic peaks of the layers all shifted to lower degree indicated the space between the layers increased. Using TBAOH for intercalation can produce the intercalation material with the lowest lamina diffraction peak which diffraction angle is 2θ = 6.3°, the diffraction angle for TPAOH and TEMP are similar and slightly higher, TEAOH is the highest (2θ = 7.6°), but the gap between TBAOH and TPAOH is not big.
We calculated the interlayer spacing using Bragg's equation for the parent body and various interlayer expansion materials. As can be seen from Table 1, the diffraction angles of the characteristic peaks of the layers during the interpolation of PLS-1 after acid treatment were all reduced, and the layer spacing was also increased to different degrees, and the interlayer spacing of the interlayer swelling material obtained by interpolation using TBAOH was the largest, which was 14.02 Å. These data provide a reference for us to build the theoretical model in the following as well.
| No. | Sample | 2θ (°) | d (Å) |
|---|---|---|---|
| 1 | PLS-1 | 8.46 | 10.44 |
| 2 | PLS-1-TEAOH | 7.6 | 11.62 |
| 3 | PLS-1-TPAOH | 6.62 | 13.34 |
| 4 | PLS-1-TBAOH | 6.3 | 14.02 |
| 5 | PLS-1-TEMP | 6.72 | 13.14 |
| 6 | PLS-1-TPAOH+TEMP | 6.62 | 13.34 |
| 7 | PLS-1-TBAOH+TEMP | 6.46 | 13.67 |
In order to further accurately compare the content of interlayer organics in the layers of these several interlayer-expanded materials, we performed thermogravimetric analysis on the materials of various OSDA intercalations. As can be seen from Fig. S9,† the weight loss of various intercalation products after high-temperature calcination ranged from 9.42% to 16.87%, which was slightly lower than that of the original PLS-1 sample, which was 18.92% (Fig. 4a); the weight loss of the sample after intercalation was significantly higher than that of the sample after the acid treatment (Fig. S6†), indicating that organics with larger sizes succeeded in getting into the interlayer of the molecular sieve after intercalation, and the lamellar spacing was subsequently increased.
3.3 Investigation of silicon insertion conditions after organic structure-directing agent insertion
In a previous report, Takashi Tatsumi's group used dichlorodimethylsilane for PLS-1 can be directly interpolated with silicon.28 In this study, we compared the use of dichlorodimethylsilane for silicone insertion of PLS-1 matrices and interlayer swelling materials after insertion using TEAOH. As can be seen in Fig. 7, the silica insertion reaction proceeded smoothly before and after PLS-1 intercalation; it is noteworthy that the crystallinity of the material was relatively improved by inserting silica after intercalation, indicating that the organic structure-directing agent for intercalation has a certain promotion effect on the silica insertion reaction of molecular sieves. Meanwhile, the XRD of several calcined materials were observed, and both PLS-1 parent and PLS-1-TEAOH calcined were in CDO topology. However, the diffraction angle of the lamina characteristic peaks of the samples after calcination did not decrease after insertion of silicon, indicating that after insertion of silicon we obtained a molecular sieve structure with increased pore size. Since we have obtained a series of intercalated materials with increasing interlayer spacing in this work, we began to try to use larger silane reagents with Si–O–Si structure for silylation to get novel interlayer expanded structure.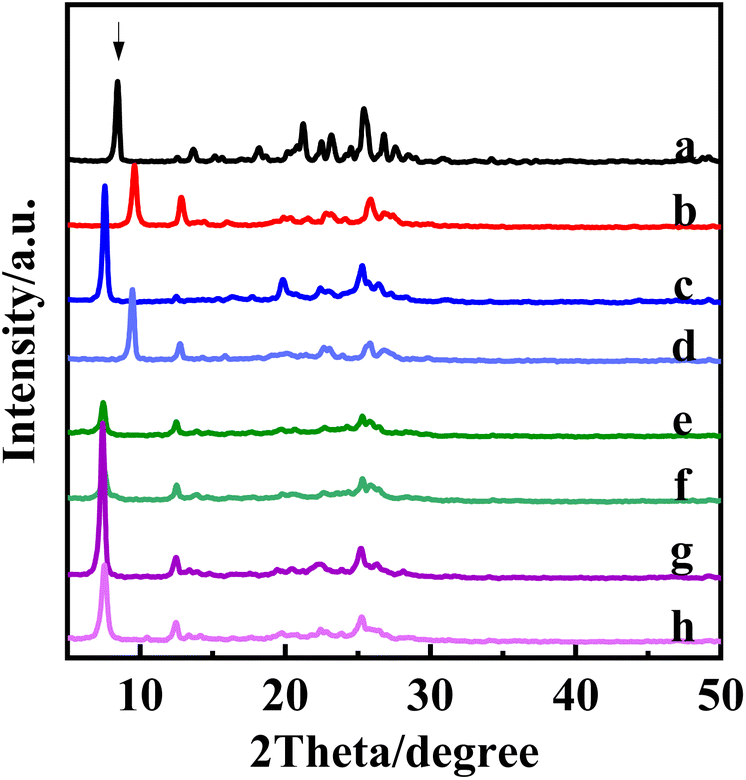 | ||
| Fig. 7 XRD plots of the parent and various post-treated materials: PLS-1 (a), PLS-1-cal (b), PLS-1-TEAOH intercalation (c), PLS-1-TEAOH-cal (d), PLS-1 direct insertion of silicon (e), PLS-1 direct insertion of silicon calcining (f), PLS-1-TEAOH intercalation of silicon (g), PLS-1-TEAOH intercalation with silicon calcining (h). | ||
It can be seen from the SEM diagram in Fig. 8 that several samples show a lamellar structure, and the morphology does not change after modification, indicating that the post-treatment process does not belong to secondary crystallization. We noticed that after acid treatment, intercalation and silicon insertion, some small particles appeared on the surface of the sample. It can be seen from the SEM image that small particles appeared from the step of acid treatment, and it can be reasonably guessed that a part of silicon fragment was generated during the pickling process, which had no effect on the crystal structure of the material.
 | ||
| Fig. 8 SEM image of PLS-1 (a), PLS-1-acid-0.1HE-333-8 (b), PLS-1-0.5TBAOH (c), PLS-1-2Si (d). | ||
After the intercalation of PLS-1-acid-0.1HE-333-8 with TEAOH, the intercalation distance increases, and the intercalation sample is silylated with 1 M HCl–EtOH as solvent and 1,3-dichlorotetramethyldisiloxane as silanization reagent. It can be seen from Fig. S10.† Under this reaction condition, the diffraction angle of the XRD layered characteristic peak inserted into the two Si is 2θ = 7.44° and does not move to lower degree, indicating that the Si–O–Si bond is not successfully inserted into the interlayer. This result indicated that the spacing of TEAOH intercalated PLS-1 is enough for accommodate one silica unit –Si–, but not two silica unit Si–O–Si. The reason is the spacing between the layers is not enough to accommodate the Si–O–Si bond. Therefore, we tried other larger molecular organic compounds intercalated materials to conduct silylation. According to Table 1, we choose sample No. 3 and No. 4 for test as a representative since samples No. 5–7 possessed almost similar interlayer distance and crystallinity with them.
As can be seen from Fig. S11,† after intercalation with TPAOH, the Si–O–Si bond still failed to be successfully inserted. Considering that the Si–O bond length in 1,3-dichlorotetramethylsilane is 10 Å, the layer spacing after intercalation with TBAOH is sufficient to accommodate the molecule theoretically. Actually, as shown in Fig. 9, the Si–O–Si bond was successfully inserted using a hydrochloric acid system, and the 2θ of the layered characteristic peak = 6.2°, the condition for silylation is 2.0 M HCI solution at 443 K for 20 h, and reduce the concentration of hydrochloric acid will affect the result of silylation (see Fig. 9a). It can be seen from the figure that the materials after TBAOH intercalation can successfully insert Si–O–Si bonds and have good crystallinity (see Fig. 9b) and this material is named as PLS-1-2Si to be conducted further characterization.
 | ||
| Fig. 9 XRD patterns of interlayer expanded materials using TBAOH-intercalated material as parent with different conditions, PLS-1-2Si-1M HE-443-20 (a), PLS-1-2Si-2M HCl-443-20 (b). | ||
Fig. 10 shows the infrared spectrum of PLS-1-2Si. It can be seen from the figure that there is a strong absorption peak at 1085 cm−1, which is caused by the anti-symmetric stretching vibration of Si–O–Si bond, and the absorption peak around 810 cm−1 is the symmetric stretching vibration absorption peak of Si–O–Si bond. The appearance of Si–CH3 asymmetric stretching vibrations at 2970 and 850 cm−1 after silanization confirms that (CH3)2Si is partially incorporated into the zeolite. The infrared spectra further confirmed that the Si–O–Si bond was successfully inserted between layers. After insertion of silicon, a new 12 × 12-R pore system is formed between the layers.
 | ||
| Fig. 10 FT-IR spectrum of PLS-1-2Si in different wavenumber ranges, 2500–3500 cm−1 (a), 700–1200 cm−1 (b). | ||
29Si MAS NMR was used to characterize the change of silicon hydroxyl group in the material during treatment. Fig. 11 shows solid 29Si MAS NMR spectra of PLS-1, PLS-1-acid-0.1HE-333-8, PLS-1-0.5TBAOH and PLS-1-2Si samples. The silanization of Si(CH3)2OSi(CH3) 2 groups into the layered precursor was further studied. The characteristic peaks at −110 to −120 ppm in the figure correspond to a silicon species Si(OSi)4 (Q4) coordinated with four silicones, while the NMR peaks at −101 to −110 ppm correspond to a silicon species (OH)Si(OSi)3 (Q3) attached to a hydroxyl group. As can be seen from the Fig. 11, the spectrum of PLS-1 shows the signal peaks of Q3 and Q4, with Q3 accounting for 47.18% and Q4 accounting for 52.82%, while Q3 of PLS-1-acid-0.1HE-333-8 decreases (33.90%) and Q4 increases slightly (66.10%). The Q3 of PLS-1-0.5TBAOH is about 35.48% and the Q4 is about 64.52%. PLS-1-2Si presents a new signal peak at −16.32 ppm, which is the characteristic peak of silanizing reagent group Si (CH3)2OSi(CH3) 2 inserted between the CDO layers in the form of D2. At the same time, the content of Q3 decreased significantly by 14.10% and Q4 increased to 72.24%. These experimental results show that silanization occurs through the reaction of silyl groups with the silicon hydroxyl group on the surface of the layer.
 | ||
| Fig. 11 29Si MAS NMR of PLS-1 (a), PLS-1-acid-0.1HE-333-8 (b), PLS-1-0.5TBAOH (c), PLS-1-2Si (d). | ||
The layers of PLS-1-2Si contain organic compounds in the preparation process, mainly residue TMAOH and TBAOH after silylation and –CH3 from silane agent, which occupy the new generated 12 × 12-R pores formed between the layers, limiting the adsorption and catalytic properties of the material. Therefore, we used the calcining method to remove organic matter between layers. First of all, after calcining for 6 h at 723 K, it can be seen from the Fig. S12† that the structure of the sample after calcining partially collapsed and the layer spacing narrowed, indicating that the thermal stability of PLS-1-2Si is not ideal. After reducing the calcining temperature, the structure was maintained at 523 K. In order to analyze the variation of organic matter between layers, thermogravimetric analysis was performed on samples calcined at 523 K.
Fig. 12 is the TG-DTA diagram of the sample calcined at 523 K by PLS-1-2Si. It can be seen that the sample still has a weight loss rate of 5.83% after calcining at 523 K for 6 h, indicating that organic matter remains between the layer. The organic matter between the layer cannot be completely removed after calcining at a lower temperature. The residual organic part occupies the position in the pore, which has a certain limitation on the characterization and application of PLS-1-2Si.
 | ||
| Fig. 12 TG-DTA diagram of PLS-1-2Si samples calcined at 523 K. | ||
3.4 Simulation of crystal structure of interlayer expanded material
Based on the XRD obtained in the experiment, the theoretical layer spacing was calculated. The theoretical model of PLS-1-2Si was built using Materials Studio software, and its structure was optimized using Vasp. After the structure was optimized and reasonable, the theoretical XRD was derived using Materials Studio software. The theoretical XRD was compared with the experimental XRD, and it was found that the positions of the XRD peaks were highly consistent in the Fig. 13. It further confirmed that our experimental process is reasonable and successful.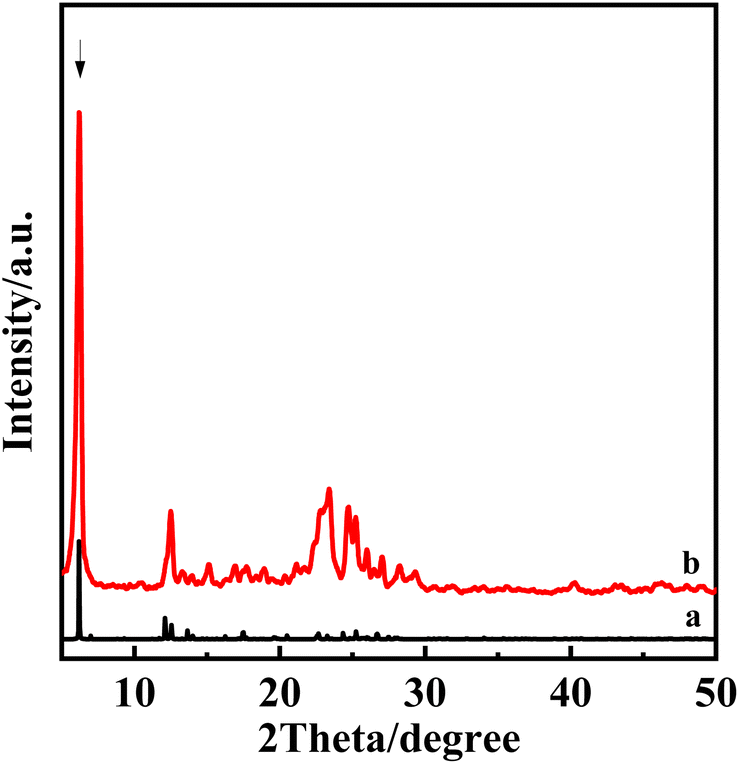 | ||
| Fig. 13 Theoretical and experimental XRD patterns of PLS-1-2Si, theoretical PLS-1-2Si (a) and experimental PLS-1-2Si (b). | ||
Based on the above characterization results, Fig. 14 shows the schematic diagram of the whole post-processing process. The lamellar precursor PLS-1 was treated with hydrochloric ethanol solution at 333 K for 6 h, and the interlamellar template agent TMAOH was partially removed and the disordered PLS-1-acid structure was formed. Then, starting from PLS-1-acid, TBAOH intercalation is carried out. During the intercalation process, macromolecular organic compounds enter the PLS-1-acid interlayer and bring about the order of lamellar deposition, and finally form a new layered molecular sieve PLS-1-TBAOH. 1,3-Dichlorotetramethyldisiloxane was used as silanizing reagent to silylate PLS-1-TBAOH. 1,3-Dichlorotetramethyldisiloxane molecules bonded with the silicon hydroxyl group on the PLS-1-TBAOH layer and inserted into the molecular sieve layer to form a new molecular sieve with 12 × 12-R interlayer expanded structure.
 | ||
| Fig. 14 Schematic diagram of the structural transformation of the layered precursor PLS-1 to PLS-1-2Si. | ||
4. Conclusions
In this study, we successfully prepared the layered molecular sieve PLS-1 and modified it after treatment. After removing the organic matter between the layer by acid treatment, various kinds of OSDA were introduced into the interlayer, and various interlayer expansion materials with enlarged interlayer spacing were obtained. Later, experiments were conducted to compare the crystallinity of the samples with PLS-1 directly and after intercalation, and it was found that intercalation could not only improve the crystallinity of the samples, but also introduce large functional groups into the interlayer and increase the pore size of the molecular sieve. Based on this idea, we investigated whether the layer spacing of various interlayer expansion materials could accommodate Si–O–Si. After using TBAOH intercalation, the reaming material PLS-1-2Si was successfully prepared in 2 M HCl system, and the optimal intercalation conditions were obtained.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Joint Funds of Jilin Natural Science Foundation, Free Exploration Project, Central Government to Guide Local Science and Technology Development Funds, Grant No. YDZJ202101ZYTS170.Notes and references
- B. Yilmaz, U. Müller, B. Tijsebaert, D. D. Vos, B. Xie, F. S. Xiao, H. Gies, W. P. Zhang, X. H. Bao, H. Imaig and T. Tatsumi, Chem. Commun., 2011, 47, 1812–1814 RSC.
- L. Z. Yang, Q. L. Liu, R. H, K. X. Fu, Y. Su, Y. F. Zheng, X. Q. Wu, C. F. Song, N. Ji, X. B. Lu and D. G. Ma, Appl. Catal., B, 2022, 309, 121224 CrossRef CAS.
- Y. S. Tong, D. H. Yuan, W. N. Zhang, Y. X. Wei, Z. M. Liu and Y. P. Xu, J. Energy Chem., 2021, 58, 41–47 CrossRef CAS.
- A. M. Cardoso, M. B. Horn, L. S. Ferret, C. M. N. Azevedo and M. Pires, J. Hazard. Mater., 2015, 287, 69–77 CrossRef CAS PubMed.
- J. Mokrzycki, M. Fedyna, M. Marzec, J. Szerement, R. Panek, A. Klimek, T. Bajda and M. M-Hersztek, J. Environ. Chem. Eng., 2022, 10, 108567 CrossRef CAS.
- B. Marler and H. Gies, Eur. J. Mineral., 2012, 24, 405–428 CrossRef CAS.
- F. S. O. Ramos, M. K. de Pietre and H. O. Pastor, RSC Adv., 2013, 3, 2084–2111 RSC.
- J. Přech, P. Pizarro, D. P. Serrano and J. Čejka, Chem. Soc. Rev., 2018, 47, 8263–8306 RSC.
- E. Schulman, W. Wu and D. X. Liu, Materials, 2020, 13, 1822 CrossRef CAS PubMed.
- M. Shamzhy, B. Gil, M. Opanasenko, W. Roth and J. Čejka, ACS Catal., 2021, 11, 2366–2396 CrossRef CAS.
- L. Xu and P. Wu, New J. Chem., 2016, 40, 3968–3981 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- L. Wei, K. Song, W. Wu, S. Holdren, G. Zhu, E. Shulman, W. Shang, H. Chen, M. R. Zachariah and D. X. Liu, J. Am. Chem. Soc., 2019, 141, 8712–8716 CrossRef CAS PubMed.
- A. Corma, V. Fornes, S. B. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353–356 CrossRef CAS.
- P. Wu, J. F. Ruan, L. L. Wang, L. L. Wu, Y. Wang, Y. M. Liu, W. B. Fan, M. Y. He, O. Terasaki and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 8178–8187 CrossRef CAS PubMed.
- L. Q. Meng, B. Mezari, M. G. Goesten, W. Wannapakdee, R. Pestman, L. Gao, J. Wiesfeld and E. J. M. Hensen, Catal. Sci. Technol., 2017, 7, 4520–4533 RSC.
- A. Feliczak-Guzik, Microporous Mesoporous Mater., 2018, 259, 33–45 CrossRef CAS.
- S. H. Cha, J. Lee, J. Shin and S. B. Hong, Top. Catal., 2015, 58, 537–544 CrossRef CAS.
- Y. J. Zhang and S. N. Che, Angew. Chem., Int. Ed., 2020, 59, 50–60 CrossRef CAS PubMed.
- J. X. Zhang, S. C. Bo, W. P. Liao, K. X. Yang, T. Su, H. Y. Lü and Z. G. Zhu, Fuel, 2024, 357, 129662 CrossRef CAS.
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892–4896 CrossRef CAS PubMed.
- F. Kooli, J. Plevert, Y. Liu, K. Hbaieb and R. Al-Faze, Clay Miner., 2016, 51, 781–791 CrossRef CAS.
- K. Komura, T. Kawamura and Y. Sugi, Catal. Commun., 2007, 8, 644–648 CrossRef CAS.
- H. Suzuoki, S. Takegawa and K. Komura, J. Jpn. Pet. Inst., 2014, 57, 184–191 CrossRef CAS.
- K. Komura, T. Murase, Y. Sugi and M. Koketsu, Chem. Lett., 2010, 39, 948–949 CrossRef CAS.
- T. Murase and K. Komura, J. Porous Mater., 2016, 23, 11–17 CrossRef CAS.
- T. Murase and K. Komura, J. Porous Mater., 2017, 24, 203–209 CrossRef CAS.
- I. S. Inagaki, T. Yokoi, Y. Kubota and T. Tatsumi, Chem. Commun., 2007, 48, 5188–5190 RSC.
- D. Mochizuki, A. Shimojima and T. Imagawa, J. Am. Chem. Soc., 2005, 127, 7183–7191 CrossRef CAS PubMed.
- H. Xu, B. T. Yang, J. G. Jiang, L. L. Jia, M. Y. He and P. Wu, Microporous Mesoporous Mater., 2013, 169, 88–96 CrossRef CAS.
- J. G. Jiang, L. L. Jia, B. T. Yang, H. Xu and P. Wu, Chem. Mater., 2013, 25, 4710–4718 CrossRef CAS.
- B. T. Yang, H. H. Wu and P. Wu, J. Phys. Chem. C, 2014, 118, 24662–24669 CrossRef CAS.
- B. T. Yang, J. G. Jiang, H. Xu, H. H. Wu and P. Wu, Chin. J. Chem., 2018, 36, 227 CrossRef CAS.
- B. T. Yang, J. G. Jiang, H. Xu, H. H. Wu, M. Y. He and P. Wu, Angew. Chem., Int. Ed., 2018, 57, 9515 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01027f |
| This journal is © The Royal Society of Chemistry 2025 |