
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple self-assembly aptasensor for ultrasensitive detection of kanamycin based on carbon dots and Ti3C2 MXene nanocomposite†
Fang Li *a,
Shuyue Xiongb,
Yijun Tanb,
Mingming Luob and
Zijian Wu*b
*a,
Shuyue Xiongb,
Yijun Tanb,
Mingming Luob and
Zijian Wu*b
aSchool of Information Engineering, Tianjin University of Commerce, Tianjin, China. E-mail: lifang_6952@163.com; Tel: +86-15522412909
bTianjin Key Laboratory of Food Biotechnology, College of Biotechnology and Food Science, Tianjin University of Commerce, Tianjin, China. E-mail: wzjian@tjcu.edu.cn; Tel: +86-18622219597
First published on 9th April 2025
Abstract
Antibiotics play an excellent role in preventing and treating animal diseases, but their improper use poses a potential threat to public health. Designing a sensing platform to detect trace amounts of antibiotic residues in actual samples is a significant challenge. To achieve this objective, a self-assembly aptasensor based on a two-round signal amplification strategy for kanamycin (KANA) trace detection was demonstrated by employing carbon dot (CD) decorated Ti3C2 MXene as electrode modification material and the complex of double-stranded DNA (dsDNA) and methylene blue (MB) as probes, where dsDNA was composed of complementary DNA (cDNA) and aptamers. The CDs can effectively suppress the self-stacking of Ti3C2 MXene to promote electron transfer and provide plenty of exposed active sites for the aptamer to capture KANA precisely. The MB inserted into dsDNA would be liberated upon interaction with KANA due to a competitive process that occurs among cDNA and KANA, reducing electrical signal. Under the optimal conditions, the constructed aptasensor exhibited a good linear relationship between the output signal and the logarithm of KANA concentration in the range of 1 fM–1 mM, and the limit of detection is 0.892 fM. The satisfactory selectivity, stability, and reproducibility suggested that the prepared aptasensor can be a universal platform to detect other antibiotic residues by anchoring corresponding aptamers. Furthermore, it has been successfully applied to determine KANA in milk samples (recovery rates ranged from 101.01% to 107.21%, and the RSD was below 5%), demonstrating potential application prospects in the food-safety analysis field.
1. Introduction
Kanamycin (KANA) is derived from Streptomyces bacteria and belongs to the class of aminoglycoside antibiotics.1 It is widely used in medical and agricultural fields, especially dairy farming, to target bacterial infections due to its inexpensive cost and favorable bacteriostatic effect.2,3 However, KANA could accumulate in the body through the food chain, resulting in many side effects, such as ototoxicity, nephrotoxicity, and allergic reactions.4 Therefore, effective identification and detection of KANA residues are crucial for food safety regulation and health protection. So far, many methods have been reported to determine KANA, such as enzyme-linked immunosorbent assay, high-performance liquid chromatography, capillary electrophoresis, and liquid chromatography-mass spectrometry.5–8 Although these methods possess certain reliability, they also inevitably have drawbacks, including cumbersome operation, high cost, and a time-consuming and laborious process. In recent years, some sensing technologies, such as surface plasmon resonance, colorimetric or fluorescence measurements, and electrochemical analytical methods, have received more and more attention, primarily due to their expedient response times, affordability, user-friendliness, and exceptional sensitivity.9–11 In particular, the electrochemical sensor has received widespread attention because of its feasible operation, simplicity, and ease of miniaturization.12With the rapid development of nanomaterials, electrochemical sensors using nanomaterials as electrode material have opened up the way for detecting various biomolecules because of the enhanced performance. Among the numerous nanomaterials, Ti3C2 MXene nanosheets, as an emerging two-dimensional material (2D) with merits of high electrical conductivity, unique surface properties, and outstanding biocompatibility, have been considered as an ideal sensing element for constructing electrochemical sensors. However, only Ti3C2 MXene is inevitably prone to self-stacking due to the hydrogen bonding and van der Waals force between layers,13 which could weaken its electrical conductivity to a certain extent. Moreover, it is easily oxidized during the interface forming process, affecting the electrochemical analysis performance. To suppress the self-stacking and oxidation, decorating and functionalizing the Ti3C2 MXene surface opens new avenues, enhancing the sensing performance. Carbon dots (CDs), as zero-dimensional (0D) carbon-based nanomaterials, have been extensively utilized in electrochemical sensors due to their unique properties, such as excellent electrical conductivity, high surface area, and abundant surface functional groups.14,15 For example, Huang et al. constructed CDs based electrochemical aptasensor for Staphylococcus aureus detection, in which the CDs enhanced the sensitivity of the electrochemical aptasensor due to their excellent conductivity and provided stability through their abundant carboxyl groups;16 CDs, due to their small size and functional groups, can intercalate between the layers of Ti3C2 MXene nanosheets. This creates physical spacing between the layers, preventing them from restacking and maintaining a high surface area. Moreover, the surface charges of CDs and Ti3C2 MXene can create electrostatic repulsion between the nanosheets, further preventing their aggregation in solution, facilitating efficient electron transport and enhancing aptasensor sensitivity.17 CDs can also act as a nanocarrier to provide plenty of efficiently exposed sites for anchoring probes, further improving the sensitivity. However, few articles exist on applying CDs and Ti3C2 MXene hybrids in electrochemical sensors. Ti3C2 MXene coupled with CDs by electrostatic self-assembly to form a free-standing has opened up the possibility of creating novel technology for antibiotic residue determination.
The rational design and application of electrode materials are essential factors in enhancing sensing performance. Sensing strategies also play a crucial role in the electrochemical sensors, which can be divided into three categories: (i) the direct measurement of electron transfer between targets and electrode materials; (ii) anchoring the aptamer (as capture probe) on the electrode materials to recognize targets by the conformation change of the aptamer specifically; (iii) the hybridization of aptamer with its complementary DNA (cDNA) for targets detection through competitive strategy. Compared to direct measurement, aptamer-based sensing strategies have better specificity due to the specific recognition ability of aptamers for target molecules.18 However, the aptamer-specific recognition of the target generates a minute change in the electrochemical signal compared to the only aptamer-modified electrode, which limits its wider application. Electrochemical signal tags like ferrocene (Fc), methylene blue (MB), and thionin (Thi) are typically labeled on aptamer to enhance the signal output.19–22 The target binding-induced conformational change causes the difference in distance from tags to the electrode surface, leading to changes in signal output. The signal tag is usually placed on the terminal of the aptamer, which may affect the process of target binding and result in poor sensing performance. So, label-free emerged as the times required labeling the signal tags on DNA or electrode materials. Among these signal tags, MB as an electrochemical indicator is commonly employed in electrochemical sensors because of its significant difference in the ability to bind single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA).23 It is reported that MB is more inclined to interact with dsDNA compared to ssDNA, and C–G base pairs are the preferred binding sites for MB.24 Therefore, utilizing dsDNA and MB as probes, where dsDNA is composed of cDNA and aptamers, is considered an excellent auxiliary means for constructing label-free electrochemical sensors and enhancing their sensing performance.
In this paper, a self-assembly aptasensor for ultrasensitive KANA detection was constructed by 0D/2D nanocomposites of Ti3C2 MXene decorated with CDs and DNA-MB complex. In which, Ti3C2 MXene exhibitd metallic conductivity, a large surface area, hydrophilicity, and surface functional groups. These properties significantly enhanced electron transfer kinetics, provided ample surface area for aptamer immobilization, and allowed for easy modification and covalent bonding with aptamers in electrochemical sensors, leading to higher sensitivity and selectivity, while the CDs can effectively suppress the self-stacking of Ti3C2 MXene to promote electron transfer and provide plenty of exposed active sites for aptamer to capture the KANA specifically. The aminohydroxylation with carboxyl groups on CDs immobilized the aminated cDNA on the electrode first. Then, aptamer was hybridized with cDNA to form a dsDNA structure and interacted with MB subsequently. In the presence of KANA, the aptamer binds explicitly to KANA, and MB would be liberated due to a competitive process among cDNA and KANA, resulting in the output signal variation (Scheme 1). The proposed method enhanced performance from two perspectives: electrode surface modification and sensing strategy. Compared with other reported KANA detection methods, the proposed method demonstrated more substantial capabilities in sensitivity and detection range, indicating that it has potential application prospects in the food safety analysis field.
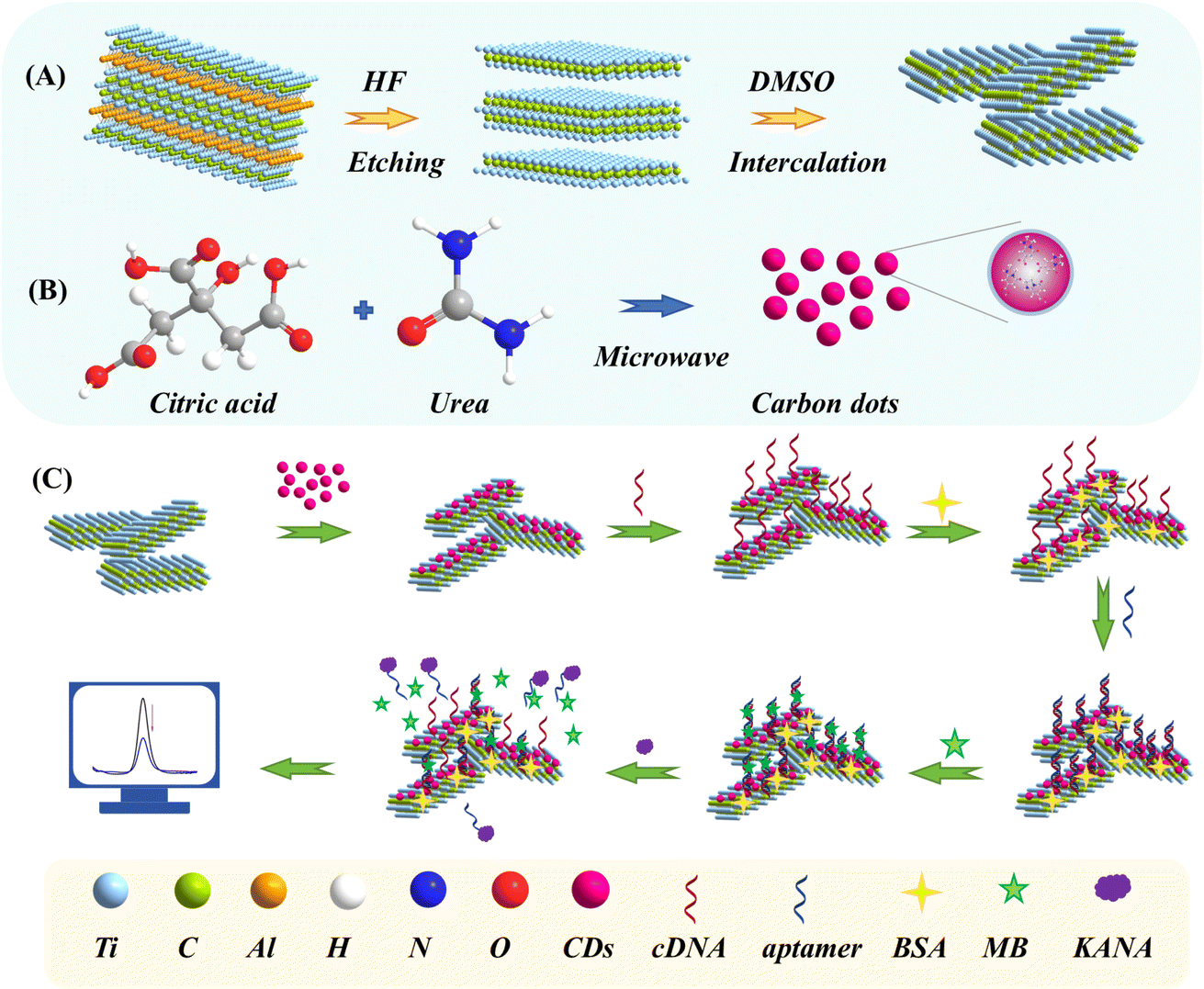 | ||
| Scheme 1 Schematic illustration of the preparation of (A) Ti3C2 MXene and (B) CDs; (C) fabrication procedures for the self-assembly aptasensor. | ||
2. Materials and methods
2.1. Materials
Citric acid was purchased from Tianjin Winda Rare Chemical Reagent Factory. Urea and bovine serum albumin (BSA) were purchased from Beijing Solarbio Science&Technology Co., Ltd. Ti3AlC2 and hydrofluoric acid (HF) were purchased from Beijing Beike New Materials Technology Co., Ltd. Dimethylsulfoxide (DMSO) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. KANA, Chloramphenicol (CAP), doxycycline hydrochloride (DOX), oxytetracycline (OTC), streptomycin (STR), and neomycin (NEO) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Tetracycline (TET) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. MB was purchased from Weifang Xingbo Biotechnology Co., Ltd. Phosphate buffered saline (PBS, 0.1 M, pH 7.4) was purchased from Quanzhou Yida Technology Co., Ltd. KCl, K4[Fe(CN)6]·3H2O, and K3 [Fe(CN)6] were bought from Shanghai Sinopharm Group Chemical Reagent Co., Ltd. Different brands of milk (milk 1–3) were purchased from CR Vanguard, the raw milk and milk that has not undergone high-temperature processing (non-UHT milk) were provided by Tianjin Mengniu Dairy Co., Ltd. Aminated-cDNA. Aptamer was synthesized by Shanghai Shengong Bioengineering Co., Ltd, with the following sequences:
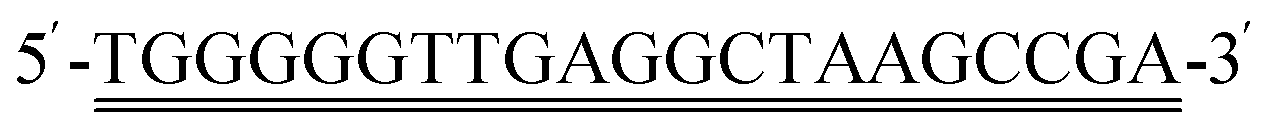
2.2. Instruments
The CHI-660E electrochemical workstation (Chenhua Instrument, Shanghai, China) was applied to electrochemical characterizations with a three-electrode system, in which the working electrode (WE) was a glassy carbon electrode (GCE), the counter electrode (CE) was platinum (Pt), and the reference electrode (RE) was a saturated silver/silver chloride (Ag/AgCl) electrode. The morphological characterizations of the CDs and Ti3C2 MXene were observed using scanning electron microscopy (SEM, Hitachi, Ltd), the atomic force microscope (AFM, Bruck), and a transmission electron microscope (TEM, JEOL). X-ray photoelectron spectroscopy (XPS) was conducted (Kratos Analytical Ltd) to analyze the electrode surface's elemental composition and chemical states. Electrochemical impedance spectroscopy (EIS) was conducted in a solution of 5 mM K3Fe(CN)6/K4Fe(CN)6 (0.1 M KCl) under the applied voltage of 0.2 V and a frequency range of 10−1 Hz to 105 Hz. Differential pulse voltammetry (DPV) experiments were conducted in a 5 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl under the voltage range of −0.4 to 0.8 V to detect the different concentrations of KANA (pulse period was 0.2 s, pulse amplitude was 50 mV, and potential increment was 4 mV). The cyclic voltammetry (CV) was recorded within the voltage range of −0.4 to 0.8 V in a 5 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl for optimizing the experimental conditions and electrochemical characterization of aptasensor.2.3. Preparation of CDs
CDs were synthesized according to the methods reported in previous literature.25,26 First, 0.5 g citric acid and 0.5 g urea were added into 5 mL deionized water (DIW) and stirred until completely dissolved. Then, the obtained aqueous solution was treated by microwave oven at 500 W for 3 min. After cooling the reaction product to room temperature, it was poured into 20 mL DIW and subjected to ultrasonic treatment. Finally, the CDs solution was obtained through a syringe filter with a pore size of 0.45 μm, as illustrated in Scheme 1A.2.4. Preparation of Ti3C2 MXene
Ti3C2 MXene was prepared according to the reported methods.27 Firstly, 2 g Ti3AlC2 was added to 20 mL 40% HF solution and stirred at 40 °C for 12 h. Secondly, the mixture was centrifuged 5–6 times at 5000 rpm for 5 min until the pH reached 6, and then centrifugation was stopped, and the upper liquid layer was poured out. Thirdly, a 25 mL DMSO intercalator was added to the leftover sediment and stirred for 12 h. After that, the obtained product was washed 3 times by centrifugation at 5000 rpm for 5 min. Finally, the obtained supernatant was Ti3C2 MXene dispersion (Scheme 1B).2.5. Preparation of CDs@Ti3C2 MXene nanocomposite
The CDs were added to the Ti3C2 MXene solution and continuously stirred magnetically for 10 min to prepare a composite with different volume ratios (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, 2:1, 1:5, and 1:10), and then ultrasonic treatment was performed for 10 min. Finally, the obtained mixed solution was CDs@Ti3C2 MXene dispersion.
:1, 5:1, 2:1, 1:5, and 1:10), and then ultrasonic treatment was performed for 10 min. Finally, the obtained mixed solution was CDs@Ti3C2 MXene dispersion.
2.6. Construction of the self-assembly aptasensor
The fabrication procedures of aptasensor were shown in Scheme 1C. Firstly, the GCE was polished successively with 0.3 μm and 50 nm Al2O3 powder and then thoroughly cleaned with ethanol and DIW. Secondly, the CDs@Ti3C2 MXene/GCE was prepared by dropping 8 μL homogeneous CDs@Ti3C2 MXene suspension onto the surface of GCE, and gently rinsed with DIW to remove the unbounded particles. Thirdly, the CDs@Ti3C2 MXene/GCE was incubated with cDNA solution for 160 min at room temperature (represented by cDNA/CDs@Ti3C2 MXene/GCE) and rinsed with DIW. Then, the electrode surface was incubated with BSA solution (1%, 10 μL) for 1 h to avoid nonspecific adsorption (represented by BSA/cDNA/CDs@Ti3C2 MXene/GCE). Subsequently, the aptamer (0.2 μM, 10 μL) was dropped onto the BSA/cDNA/CDs@Ti3C2 MXene modified electrode for 120 min (represented by aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE). Finally, the electrode was applied to 3.5 μM MB solution for 1 h (represented by MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE).2.7. Samples pretreatment
A standard addition method was employed to evaluate milk samples and verify the aptasensor's practicality. Different concentrations of KANA were added to the milk samples. The milk samples were centrifuged at 12000 rpm at 4 °C for 10 min and then allowed to stand for 10 min to remove the upper layer of fat. The lower clear liquid was filtered through a 0.22 μm filter membrane and diluted 10 times with 0.01 M PBS solution (pH 7.4). Finally, the content of KANA was determined using the proposed aptasensor, and the detection value was obtained to calculate the recovery rates.
3. Results and discussion
3.1. Morphological characterization of nanomaterials
The surface morphology of Ti3C2 MXene, CDs, and CDs@Ti3C2 MXene were studied by SEM, TEM, and AFM. Fig. 1A displayed the layered sheet-like structures of Ti3C2 MXene as well as the curled edges. In contrast, after the addition of CDs, many small pores appeared on the surface, and the layered structure became more regular, as shown in Fig. 1B. The TEM image in Fig. 1C showed a single flake of intercalated Ti3C2 MXene, with lateral sizes up to a few hundred nanometers. In Fig. 1D, CDs distributed on the surface of Ti3C2 MXene can be observed. AFM images (Fig. 1E and F) provided a more intuitive demonstration of the impact of CDs on the structure of Ti3C2 MXene. At the same time, the surface roughness can be estimated by NanoScope® Analysis software. The roughness of the etched Ti3C2 MXene was approximately 1.71 nm (Fig. 1E), while the roughness of the prepared CDs was 1.22 nm (Fig. 1F). The AFM results for the synthesized CDs@Ti3C2 MXene nanocomposite indicated a roughness of 2.88 nm (Fig. 1G), which was close to the sum of the roughness values of CDs and Ti3C2 MXene. It suggested that CDs might be embedded between the layers of Ti3C2 MXene, thereby preventing its self-stacking.28 | ||
| Fig. 1 SEM images of (A) Ti3C2 MXene and (B) CDs@Ti3C2 MXene; TEM images of (C) Ti3C2 MXene and (D) CDs@Ti3C2 MXene (inset: a cropped section of the image); AFM images of (E) Ti3C2 MXene, (F) CDs, and (G) CDs@Ti3C2 MXene. | ||
The XPS analysis was performed to investigate the chemical composition of each step modification. The broad spectra can be employed to observe the variation of elements, as shown in Fig. 2A. The simultaneous appearance of N and Ti elements in the XPS spectra of CDs@Ti3C2 MXene, provides preliminary evidence for the successful synthesis of CDs@Ti3C2 MXene. The XPS scan of cDNA/CDs@Ti3C2 MXene exhibited an additional peak of P 2p, suggesting that cDNA had successfully been modified on CDs@Ti3C2 MXene. Compared to cDNA/CDs@Ti3C2 MXene, the percentage content of P element in aptamer/cDNA/CDs@Ti3C2 MXene increased from 3.07% to 3.42%, which is attributed to the complementary pairing between the aptamer and cDNA, leading to an increase in the P element content on the electrode surface.
 | ||
| Fig. 2 (A) XPS wide spectra of Ti3C2 MXene, CDs, CDs@Ti3C2 MXene, CDs@Ti3C2 MXene/cDNA, CDs@Ti3C2 MXene/cDNA/aptamer; the N 1s core spectra (B) and C 1s core spectra (C) of CDs; the C 1s core spectra (D) and Ti 2p core spectra (E) of Ti3C2 MXene; the N 1s core spectra (F) and C 1s core spectra (G) of CDs@Ti3C2 MXene; (H) the N 1s core spectra of cDNA/CDs@Ti3C2 MXene. | ||
The specific chemical components were analyzed to verify each step modification's accomplishment. The CDs' C 1s and N 1s core spectra were analyzed, shown in Fig. 2B and C. The four peaks at 284.77 eV, 285.70 eV, 286.90 eV, and 288.36 eV were attributed to C–C, C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO, respectively.29,30 The peaks in the N 1s spectra of CDs at 399.78 eV and 400.98 eV were related to the C–N–C and N–H, respectively, indicating that the N element may be in a porphyrin ring-like structure that helped to form π–π stacking on the film of Ti3C2 MXene.31 The C 1s and Ti 2p core spectra of Ti3C2 MXene were analyzed and presented in Fig. 2D and E, the core spectra of C 1s could be broken up into four sub-peaks at 282.34 eV, 284.71 eV, 286.36 eV, and 288.91 eV, which corresponded to the C–Ti, C–C, C–O, and O–CO/C–F bonds, respectively.29,32 The Ti 2p core spectrum of Ti3C2 MXene could be divided into six peaks, which are located at 455.35 eV, 456.08 eV, 457.18 eV, 458.94 eV, 461.87 eV, and 464.79 eV, respectively. These peaks correspond to Ti, Ti2+, Ti3+, TiO2, Ti–C, and Ti3+ satellite peaks, which were consistent with reports in the literature.33 The N 1s spectra could offer additional evidence to enhance the comprehension of the molecular structure of CDs@Ti3C2 MXene. As displayed in Fig. 2F, the fitted peaks at about 399.91 eV and 401.23 eV, which belonged to C–N–C and N–H bonds, respectively, were consistent with the peak splitting results of CDs, indicating that CDs had been successfully combined with Ti3C2 MXene. The C 1s spectrum of CDs@Ti3C2 MXene (Fig. 2G) could be divided into five sub-peaks, which were 281.91 eV, 282.4 eV, 284.76 eV, 286.29 eV, and 288.57 eV, corresponding to C–H, C–Ti, C–C, CO/C–N, and O–CO/C–F. Notably, the appearance of the CO/C–N can be attributed to the CDs, further proving the successful preparation of the CDs@Ti3C2 MXene hybrid.34 As shown in Fig. 2H, the appearance of additional N 1s peak (C–N) compared to CDs@Ti3C2 MXene, which was attributed to the formation of amide bonds between the amino groups on cDNA and the OH groups on Ti3C2 MXene, further proving the successful anchoring of cDNA on the electrode surface.
O, and O–CO, respectively.29,30 The peaks in the N 1s spectra of CDs at 399.78 eV and 400.98 eV were related to the C–N–C and N–H, respectively, indicating that the N element may be in a porphyrin ring-like structure that helped to form π–π stacking on the film of Ti3C2 MXene.31 The C 1s and Ti 2p core spectra of Ti3C2 MXene were analyzed and presented in Fig. 2D and E, the core spectra of C 1s could be broken up into four sub-peaks at 282.34 eV, 284.71 eV, 286.36 eV, and 288.91 eV, which corresponded to the C–Ti, C–C, C–O, and O–CO/C–F bonds, respectively.29,32 The Ti 2p core spectrum of Ti3C2 MXene could be divided into six peaks, which are located at 455.35 eV, 456.08 eV, 457.18 eV, 458.94 eV, 461.87 eV, and 464.79 eV, respectively. These peaks correspond to Ti, Ti2+, Ti3+, TiO2, Ti–C, and Ti3+ satellite peaks, which were consistent with reports in the literature.33 The N 1s spectra could offer additional evidence to enhance the comprehension of the molecular structure of CDs@Ti3C2 MXene. As displayed in Fig. 2F, the fitted peaks at about 399.91 eV and 401.23 eV, which belonged to C–N–C and N–H bonds, respectively, were consistent with the peak splitting results of CDs, indicating that CDs had been successfully combined with Ti3C2 MXene. The C 1s spectrum of CDs@Ti3C2 MXene (Fig. 2G) could be divided into five sub-peaks, which were 281.91 eV, 282.4 eV, 284.76 eV, 286.29 eV, and 288.57 eV, corresponding to C–H, C–Ti, C–C, CO/C–N, and O–CO/C–F. Notably, the appearance of the CO/C–N can be attributed to the CDs, further proving the successful preparation of the CDs@Ti3C2 MXene hybrid.34 As shown in Fig. 2H, the appearance of additional N 1s peak (C–N) compared to CDs@Ti3C2 MXene, which was attributed to the formation of amide bonds between the amino groups on cDNA and the OH groups on Ti3C2 MXene, further proving the successful anchoring of cDNA on the electrode surface.
3.2. Electrochemical characterization of aptasensor
CV and EIS were utilized as powerful methods to elucidate the incremental construction process of the self-assembly aptasensor. As shown in Fig. 3A, the bare GCE electrode (15.43 μA, blank line) showed distinct reversible redox peaks. The peak current significantly increased after being modified by CDs@Ti3C2 MXene (48.63 μA, red line), indicating that CDs@Ti3C2 MXene can effectively improve the conductivity of the electrode. When the cDNA was assembled on CDs@Ti3C2 MXene/GCE, the peak current was found to decrease (26.29 μA, blue line), which was possibly due to electrostatic repulsion generated by the negatively charged phosphate skeleton in the cDNA chain with [Fe(CN)6]3−/4−. After treating the electrode with BSA to block the unbound sites, the electron transfer was hindered, and the current was further decreased due to the non-electroactivity of BSA (22.78 μA, green line). The aptamer was then anchored to the electrode surface through complementary pairs with cDNA bases. The peak current was observed to continue decreasing, mainly because the addition of the aptamer increased the steric hindrance of the electrode, impeding the transfer of electrons (12.90 μA, orange line). When the MB was modified on the electrode surface, it was found that the current had a clear upward trend, which was due to the electrochemical activity of MB (25.89 μA, purple line). In the presence of KANA, the aptamer had a higher affinity for KANA compared to cDNA, resulting in the untangling of the double chain and causing MB to lose the attachment site on the electrode, which reduced the electron transfer on the electrode surface and thus resulted in a decrease in the peak current (17.43 μA, cyan line). | ||
| Fig. 3 (A) CV and (B) EIS curves for each step of the aptasensor's construction process. (a) Bare GCE; (b) CDs@Ti3C2 MXene/GCE; (c) cDNA/CDs@Ti3C2 MXene/GCE; (d) BSA/cDNA/CDs@Ti3C2 MXene/GCE; (e) aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE; (f) MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE; (g) KANA/MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE; (C) (I): left (ΔI = Ip, Ti3C2 MXene/GCE − Ip, GCE), right (ΔI = Ip, CDs@Ti3C2 MXene/GCE − Ip, GCE); (II): left (ΔI = Ip, KANA/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE − Ip, aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE), right (ΔI = Ip, KANA/MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE − Ip, MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE); (III): left (ΔI = Ip, KANA/BSA/aptamer/Ti3C2 MXene/GCE − Ip, Ti3C2 MXene/GCE), right (ΔI = Ip, KANA/MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE − Ip, CDs@Ti3C2 MXene/GCE); the CV response of (D) GCE, (E) Ti3C2 MXene/GCE and (F) CDs@Ti3C2 MXene/GCE at scan rates from 20 mV s−1 to 200 mV s−1; calibration curves of redox peak current response against the square root of scan rate for (G) GCE, (H) Ti3C2 MXene/GCE and (I) CDs@Ti3C2 MXene/GCE. | ||
Simultaneously, EIS measurements were also explored to characterize the development procedure of aptasensor, as shown in Fig. 3B. The high-frequency semicircle observed in EIS represents the electron transfer-limited process, with its diameter directly reflecting the electron transfer resistance.35 The bare GCE exhibited a typical Nyquist plot (28.96 kΩ, curve a). The semicircular diameter of CDs@Ti3C2 MXene/GCE (6.46 kΩ, curve b) was significantly smaller than that of GCE, owing to the excellent conductivity of the prepared CDs@Ti3C2 MXene composite, which facilitated electron transfer on the electrode surface. After cDNA was modified onto CDs@Ti3C2 MXene/GCE, the negatively charged phosphate backbone of the DNA chain obstructed the electron transfer of [Fe(CN)6]3−/4−, resulting in an increase in impedance on the electrode surface (11.03 kΩ, curve c). Upon modification with BSA, the resistance increased due to its non-electroactive nature (16.70 kΩ, curve d). When the aptamer was modified onto BSA/cDNA/CDs@Ti3C2 MXene/GCE, the steric hindrance increased due to the electrostatic repulsion between the negatively charged phosphate groups in the aptamer and [Fe(CN)6]3−/4− (29.87 kΩ, curve e). Further modification with MB led to a decrease in resistance due to the electroactive properties of MB (14.85 kΩ, curve f). When KANA was modified onto the surface of MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE, the specific binding between KANA and aptamer caused the double strand to separate and MB to be released, increasing impedance (18.46 kΩ, curve g). These results demonstrated that EIS curves were consistent with CV measurements.
The electrochemical performance of the constructed aptasensor was enhanced in two key areas: electrode surface modification and sensing strategy, as shown in Fig. 3C. Specifically, (I) the surface conductivity of the electrode modified with CDs@Ti3C2 MXene has significantly increased compared to that of the electrode modified solely with Ti3C2 MXene. The peak current (ΔI) increase observed with Ti3C2 MXene modified GCE was 3.53 μA (Fig. S1A†) and CDs modified GCE was 4.19 μA (Fig. S1B†), whereas the ΔI for a GCE modified with CDs@Ti3C2 MXene was 33.2 μA. This was sufficient evidence to demonstrate that CDs@Ti3C2 MXene composites play a crucial role in enhancing the conductivity of the electrode; (II) utilizing the different binding affinities of MB to ssDNA or dsDNA, the signal has been further amplified. The comparison of electrode surface with (8.46 μA) or without MB (Fig. S1C†) (1.74 μA), demonstrating that the synergistic effect of MB and DNA molecules contributed to signal enhancement; (III) compared to our previous work (ΔI = 9.16 μA),36 the sensing performance of aptasensor has been enhanced (ΔI = 31.2 μA) in terms of both electrode materials and sensing strategies.
Fig. 3D–F exhibited CV response curves of GCE, Ti3C2 MXene/GCE, and CDs@Ti3C2 MXene/GCE electrodes at different scan rates ranging from 20 mV s−1 to 200 mV s−1. The redox peak current was proportional to the scan rate, and the slight shift of the redox peak potential was due to the rapid transfer of electrons. The linear plots of the redox peak current versus the square root of the scan rate were obtained (Fig. 3G–I). The regression equations of bare GCE electrode were determined to be Ipa (μA) = 37.745v1/2 + 0.392, R2 = 0.991 (anodic peak), and Ipc (μA) = −26.322v1/2 − 1.506, R2 = 0.992 (cathodic peak), the regression equations of Ti3C2 MXene/GCE electrode were determined to be Ipa (μA) = 34.650v1/2 + 0.919, R2 = 0.991 (anodic peak) and Ipc (μA) = −26.140v1/2 − 0.859, R2 = 0.991 (cathodic peak), and the regression equations of CDs@Ti3C2 MXene/GCE electrode were determined to be Ipa (μA) = 82.985v1/2 + 4.884, R2 = 0.992 (anodic peak) and Ipc (μA) = −75.307v1/2 − 9.719, R2 = 0.992 (cathodic peak), indicating the occurrence of the diffusion-controlled electron transfer process on the surface of bare GCE, Ti3C2 MXene/GCE and CDs@Ti3C2 MXene/GCE. The electroactive specific surface area (ECSA) of GCE, Ti3C2 MXene/GCE, and CDs@Ti3C2 MXene/GCE can be calculated by the Randles–Sevcik equation:37
| Ip = (2.69 × 105)n3/2AD1/2Cv1/2 |
3.3. Optimization of experimental conditions
To improve sensing performance, several important experimental parameters, such as the volume ratio of Ti3C2 MXene to CDs, cDNA incubation time, cDNA concentration, aptamer binding time, aptamer concentration, and KANA reaction time, were optimized by CV. The influence of different volume ratios of Ti3C2 MXene to CDs (10:1, 5:1, 2:1, 1:5, and 1:10) on sensing performance was investigated under the same conditions shown in Fig. 4A. It is found that the output peak current increased with the increasing amount of CDs when its volume ratio to Ti3C2 MXene is 1:5. However, more CDs led to the gradual decrease of the current signal. This can be explained by the fact that fewer CDs cannot avoid the aggregation of Ti3C2 MXene, but more CDs would weaken the conductivity of Ti3C2 MXene. Therefore, 5:1 was selected as the volume ratio of Ti3C2 MXene to CDs in subsequent experiments.
 | ||
| Fig. 4 Effect of (A) the volume ratio of Ti3C2 MXene to CDs; (B) the cDNA incubation time; (C) the cDNA concentration; (D) the aptamer binding time; (E) the aptamer concentration; (F) the KANA reaction time. The shift of peak current (ΔIp) was obtained by subtracting the current of the previous modification step. The error bars represented the standard deviation for three repeated measurements. | ||
The experimental results of different cDNA incubation times (40, 80, 120, 160, and 200 min) were shown in Fig. 4B, which revealed that the current increased as the incubation time of cDNA from 40 min to 160 min. When the incubation time of cDNA was more than 160 min, the current decreased remarkably. So, the incubation time of cDNA was fixed at 160 min. The peak current of different cDNA concentrations (0.05, 0.1, 0.25, 0.5, and 1 μM) modified electrode was recorded and shown in Fig. 4C. The current increased with the increase of cDNA concentration from 0.05 to 0.25 μM and decreased subsequently, which indicated that the cDNA anchored on CDs@Ti3C2 MXene had reached saturation state. Thus, 0.25 μM was chosen as the optimal concentration for cDNA.
The optimal time for the binding of aptamers to cDNA (40, 80, 120, 160, and 200 min) was studied as shown in Fig. 4D, the current increased from 40 min to 120 min and then tended to stabilize. So, it can be determined that the optimal incubation time for aptamer was 120 min. The best aptamer concentration (0.05, 0.1, 0.2, 0.4, and 0.5 μM) for hybridization with cDNA was investigated, as shown in Fig. 4E, the current gradually increased with the increase of aptamer concentration and then decreased until the aptamer concentration was 0.2 μM, indicating that excessive aptamer absorbed on the electrode surface would hinder electron transport. Thus, the aptamer concentration was selected as 0.2 μM for subsequent experiments. The effect of KANA reaction time (5 min, 10 min, 15 min, 20 min, and 25 min) on the output current was studied and recorded, as shown in Fig. 4F. The current remained constant when the incubation time of KANA reached 15 min. Therefore, the optimal incubation time for KANA was 15 min.
3.4. The performance analysis of aptasensor
Under the optimal conditions, the DPV was applied to investigate the electrochemical responses of different concentrations of KANA (0.01 fM–1 mM), as shown in Fig. 5A. It can be seen that the peak current of DPV regularly decreased with the increase of KANA concentrations. It indicated that the aptamer was more inclined to bind with KANA, leaving cDNA alone on the electrode surface, thus resulting in the signal molecule MB losing its attachment site and disengaging from the electrode. The calibration curve exhibited a good linear relationship between the DPV peak current and the logarithm values of KANA concentrations varying from 1 fM to 1 mM. The linear equation was fitted to Ip (μA) = −0.95lgCKANA + 12.98 (Pearson's r = 0.9857) (Fig. 5B). The limit of detection (LOD) was estimated to be 0.892 fM with the signal to noise ratio being 3. The sensing performance of the constructed aptasensor exhibited a wider linear range and lower LOD compared to previously reported KANA detection strategies, as shown in Table 1. The enhanced performance should be attributed to the synergistic effect of CDs, Ti3C2 MXene and electron mediating of MB intercalated into dsDNA structures.
 | ||
| Fig. 5 (A) DPV responses of the sensing interface toward different concentrations of KANA in a solution containing 5 mM [Fe(CN)6]3−/4− and 0.1 M KCl. From 0.01 fM to 1 mM, the KANA concentration corresponds to 0.01 fM, 0.1 fM, 1 fM, 10 fM, 100 fM, 1 pM, 10 pM, 100 pM, 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM, and 1 mM. (B) Linear relationship between the peak current and the concentration of KANA. The error bars represent standard deviation for three repeated measurements. | ||
| Materials | Detection range | LOD | Real sample | Ref. |
|---|---|---|---|---|
| a Abbreviated definition: Co/Ni-Bio-MOF_an adenine-based Co/NiMOF; ZrO2@Au_noble metal Au-doped coral-like ZrO2; PEI-MoS2-MWCNTs_polyethyleneimine-molybdenum disulfide-multi-walled carbon nanotubes; g-C3N4–COOH/ZnSe_carboxylated g-C3N4 was combined with ZnSe quantum dots (QDs) through amide reaction; Dy-MOF_dysprosium metal–organic framework; GdPc (acac)_a new Gd(III) complex GdPc(acac) (Pc = phthalocyanine, acac = acetylacetonate); DNA-AgNCs_DNA stabilized silver nanoclusters; AuNPs_gold nanoparticles; GR-TH_thionine functionalized graphene; HNP-PtCu_hierarchical nanoporous-PtCu; MWCNTs-IL_multi-walled carbon nanotubes, ionic liquid of 1-hexyl-3-methylimidazolium hexafluorophosphate; NP-PtTi_nanoporous PtTi. | ||||
| Co/Ni-Bio-MOF, ZrO2@Au | 10–1000 μM | 0.037 μM | Milk | 38 |
| PEI-MoS2-MWCNTs | 1 × 10−3–1 × 103 ng mL−1 | 0.21 pg mL−1 | Milk | 39 |
| Au@FeP | 2.4–150 nM | 0.98 nM | Milk; tap water | 40 |
| g-C3N4–COOH/ZnSe | 1.0 nM–100 μM | 0.7982 nM | Milk | 41 |
| Dy-MOF, GdPc | 0.001 pg mL−1–1000 ng mL−1 | 0.3 fg mL−1 | Milk | 42 |
| DNA-AgNCs, AuNPs | 100–1100 nM | 22.6 nM | Milk | 43 |
| GR-TH, HNP-PtCu | 5 × 10−7–5 × 10−2 μg mL−1 | 0.42 pg mL−1 | Pork meat; chicken; liver | 44 |
| MWCNTs-IL, NP-PtTi | 0.05–100 ng mL−1 | 3.7 pg mL−1 | Milk | 45 |
| CDs@Ti3C2 MXene | 1 fM–1 mM | 0.892 fM | Milk | This work |
3.5. Specificity, stability, and reproducibility of the sensing interface
The specificity, stability, and repeatability of the aptasensor were vital factors for its practical application. To evaluate the specificity of the constructed aptasensor, several common antibiotics like DOX, OTC, STR, NEO, CAP, and TET with 100 nM concentration were measured and shown in Fig. 6A. Compared with the evident changes in the electrochemical signal response of KANA, the signal response of other interfering substances was insignificant, indicating that the proposed aptasensor had excellent specificity towards KANA. The coexistence interference of different antibiotics can be negligible.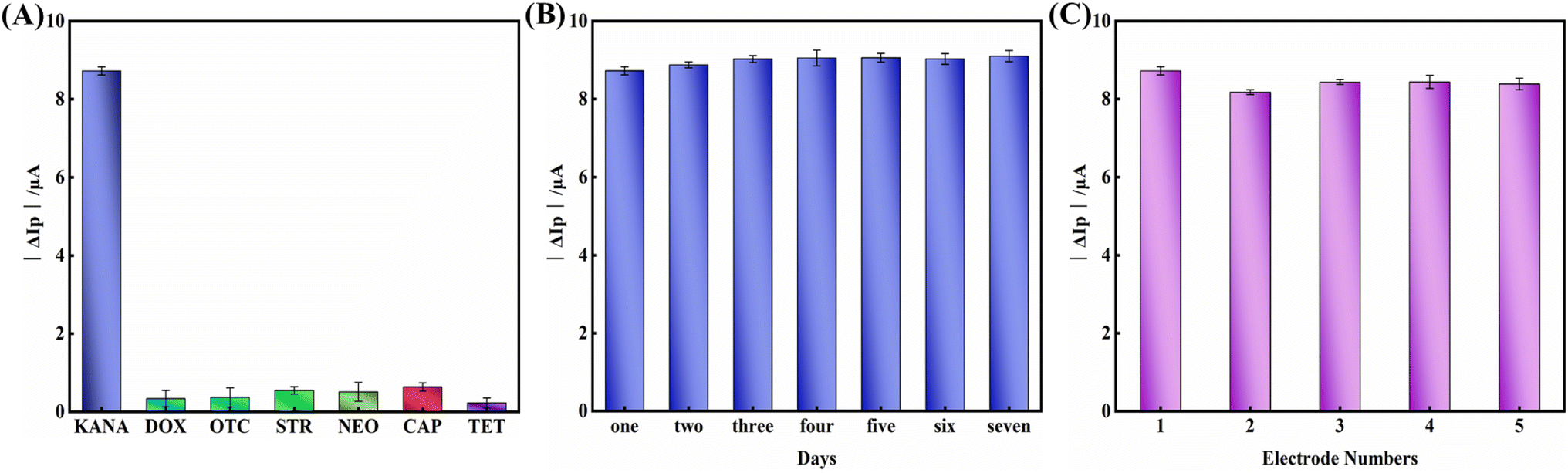 | ||
| Fig. 6 (A) The anti-interference detection of the proposed aptasensor (the concentration of KANA and interferent was set 100 nM). (B) Stability and (C) reproducibility of the proposed aptasensor for detecting KANA with the concentration of 100 nM, ΔIP = IP, KANA/MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE − IP, MB/aptamer/BSA/cDNA/CDs@Ti3C2 MXene/GCE. Error bars represent the standard deviations of three independent measurements. | ||
The stability of the proposed aptasensor was investigated to determine whether it can be used for long-term detection. The aptasensor was stored at 4 °C and measured every day for 7 days. As shown in Fig. 6B, 95.8% of the original electrochemical response could remain within one week of storage, indicating that the stability of the aptasensor was acceptable.
Meanwhile, five independent electrodes prepared with the same method were used for KANA detection to explore the reproducibility of the prepared aptasensor (Fig. 6C). The relative standard deviation (RSD) of the five electrodes was as low as 1.276%, confirming the aptasensor's good reproducibility.
3.6. Analysis of milk samples
To evaluate the application potential of the electrochemical aptasensor, different concentrations of KANA (i.e., 10 pM, 100 pM, 1 nM, 10 nM, and 100 nM) were spiked into real milk samples. The detection was performed on spiked real samples with different processing treatments, including raw milk, non-UHT milk (pasteurized milk), as well as various UHT milk brands. The recovery rates for raw milk ranged from 93.89% to 112.79%, with relative standard deviation (RSD) values between 0.99% and 4.39%. For non-UHT milk, the recovery rates varied from 88.59% to 116.68%, with RSD values ranging from 1.38% to 3.94%. As for the UHT milk samples from different brands, the recovery rates for milk 1, milk 2, and milk 3 were in the ranges of 101.01% to 107.21%, 91.57% to 118.49%, and 92.39% to 111.34%, respectively, while the RSD values ranged from 2.23% to 4.81%, 1.64% to 4.14%, and 1.02% to 5.05%, respectively. As summarized in Table 2, all recovery rates for the actual samples fell within 90% to 118%, with RSD values predominantly below 5%. These results demonstrated that the constructed aptasensor exhibited robust stability and excellent reproducibility. Furthermore, the satisfactory recovery rates confirmed the practical feasibility of this electrochemical aptasensor for detecting KANA in real milk samples.| Sample | Sample no. | Added | Found | Recovery (%) | RSD (%, n = 3) |
|---|---|---|---|---|---|
| Raw milk | 1 | 10 pM | 11.081 pM | 110.80 | 2.19 |
| 2 | 100 pM | 93.893 pM | 93.89 | 2.14 | |
| 3 | 1 nM | 1.127 nM | 112.79 | 0.99 | |
| 4 | 10 nM | 9.864 nM | 98.64 | 3.18 | |
| 5 | 100 nM | 111.254 nM | 111.25 | 4.39 | |
| Non-UHT milk | 1 | 10 pM | 11.668 pM | 116.68 | 3.94 |
| 2 | 100 pM | 112.975 pM | 112.97 | 1.63 | |
| 3 | 1 nM | 1.103 nM | 110.36 | 2.09 | |
| 4 | 10 nM | 8.859 nM | 88.59 | 1.38 | |
| 5 | 100 nM | 108.853 nM | 108.85 | 2.36 | |
| Milk 1 | 1 | 10 pM | 10.494 pM | 104.94 | 4.38 |
| 2 | 100 pM | 104.489 pM | 104.49 | 2.23 | |
| 3 | 1 nM | 1.043 nM | 104.3 | 4.71 | |
| 4 | 10 nM | 10.100 nM | 101.01 | 4.81 | |
| 5 | 100 nM | 107.209 nM | 107.21 | 4.03 | |
| Milk 2 | 1 | 10 pM | 11.107 pM | 111.07 | 2.07 |
| 2 | 100 pM | 118.491 pM | 118.49 | 4.14 | |
| 3 | 1 nM | 1.059 nM | 105.90 | 1.64 | |
| 4 | 10 nM | 9.157 nM | 91.57 | 2.28 | |
| 5 | 100 nM | 106.677 nM | 106.68 | 2.53 | |
| Milk 3 | 1 | 10 pM | 11.134 pM | 111.34 | 3.75 |
| 2 | 100 pM | 101.629 pM | 101.63 | 1.02 | |
| 3 | 1 nM | 1.097 nM | 109.74 | 2.93 | |
| 4 | 10 nM | 9.291 nM | 92.91 | 2.61 | |
| 5 | 100 nM | 92.388 nM | 92.39 | 5.05 |
4. Conclusion
In summary, an electrochemical aptasensor with enhanced performance was developed for the ultrasensitive detection of KANA. This sensing platform was improved in two key areas: electrode surface modification and sensing strategy. Specifically, CDs decorated Ti3C2 MXene were utilized as the electrode material, while MB-intercalated dsDNA served as the probe. The CDs can effectively prevent the self-stacking of Ti3C2 MXene and facilitate electron transfer. In the presence of KANA, the high affinity of the aptamer for KANA led to the denaturation of dsDNA into ssDNA and the subsequent release of MB, resulting in a significant decrease in output current. This unique strategy exhibited substantial advantages due to the synergistic effect among the different components. Compared to other reported methods, it demonstrated a lower limit of detection (LOD) of 0.892 fM and a wider linear range (1 fM to 1 mM). Furthermore, the aptasensor exhibited exceptional specificity, reproducibility, and stability, with a recovery rate in milk samples ranging from 101.01% to 107.21%, underscoring its practical applicability and reliability. Therefore, this aptasensor showed great potential for application in food safety analysis and provided a novel and valuable detection platform for other fields.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Fang Li: conceptualization, methodology, supervision, project administration, writing – review & editing, validation, resources. Shuyue Xiong: conceptualization, methodology, data curation, formal analysis, writing – original draft, validation, software, resources. Yijun Tan: data curation, formal analysis, software, resources. Mingming Luo: data curation, formal analysis, software, resources. Zijian Wu: funding acquisition, supervision, writing – review & editing, validation, resources.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was funded by the Tianjin Municipal Education Commission Scientific Research Project [grant number 2024KJ076].References
- J. Tang and X. Tao, Microchem. J., 2023, 194, 109313 CrossRef CAS.
- Y. Jin, Y. Zhang, H. Xu, X. Lu, Y. Yuan and W. Zhang, Food Control, 2024, 161, 110390 CrossRef CAS.
- W. Cui, C. Hu, R. Zhu, D. Qiu, R. Gong, R. Wang, Q. Li, T. Yan, C. Li, M. Qiao and S. Xu, J. Food Compos. Anal., 2024, 131, 106261 CrossRef CAS.
- X. Gao, Z. Sun, X. Wang, W. Zhang, Y. Wang, J. Han, X. Sun, Y. Guo, F. Li and S. Xu, Sens. Actuators, B., 2022, 373, 132706 CrossRef CAS.
- A. Guironnet, C. Sanchez-Cid, T. M. Vogel, L. Wiest and E. Vulliet, J. Chromatogr. A, 2021, 1651, 462133 CrossRef CAS PubMed.
- E. Loomans, J. Van Wiltenburg, M. Koets and A. Van Amerongen, J. Agric. Food Chem., 2003, 51, 587–593 CrossRef CAS PubMed.
- C.-Z. Yu, Y.-Z. He, G.-N. Fu, H.-Y. Xie and W.-E. Gan, J. Chromatogr. B, 2009, 877, 333–338 CrossRef CAS PubMed.
- X. P. Zhang, J. J. Wang, Q. H. Wu, L. Li, Y. Wang and H. L. Yang, Molecules, 2019, 24(10), 1902 CrossRef CAS PubMed.
- X. Wang, M. Zou, X. Xu, R. Lei, K. Li and N. Li, Anal. Bioanal. Chem., 2009, 395, 2397–2403 CrossRef CAS PubMed.
- X. Wang, X. Yuwen, S. Lai, X. Li and G. Lai, Anal. Chim. Acta, 2024, 1287, 342139 CrossRef CAS PubMed.
- L. Tian, J. Zhang, H. Fan, Y. Zhang, Z. Wang, O. Oderinde, Y. Wang and J. Cui, Anal. Biochem., 2023, 663, 115028 CrossRef CAS PubMed.
- S. M. Taghdisi, N. M. Danesh, M. Ramezani, M. Alibolandi, M. A. Nameghi, G. Gerayelou and K. Abnous, Talanta, 2021, 223, 121705 Search PubMed.
- Y. Feng, K. Wu, S. Wu, Y. Guo, M. He and M. Xue, ACS Appl. Mater. Interfaces, 2023, 15, 3077–3088 CrossRef CAS PubMed.
- C. Liu, X. Lin, J. Liao, M. Yang, M. Jiang, Y. Huang, Z. Du, L. Chen, S. Fan and Q. Huang, Chin. Chem. Lett., 2024, 35, 109598 CrossRef CAS.
- C. Liu, Y. Mei, Q. Lei, X. Ma, X. Nan, Y. Zhu, J. Liao, Y. Xu, Y. Luo, H. Zhang, M. Yang, X. Lin and Q. Huang, Chem. Eng. J., 2024, 499, 156434 CrossRef CAS.
- X. Lin, C. Liu, Q. Lei, X. Nan, Y. Zhu, J. Liao, Z. Du, C. Ye, Y. Xiong, M. Yang, X. Fang, Y. Luo and Q. Huang, J. Hazard. Mater., 2025, 485, 136845 CrossRef CAS PubMed.
- Z. Tan, W. Wang, M. Zhu, Y. Liu, Y. Yang, X. Ji and Z. He, Desalination, 2023, 548, 116267 CrossRef CAS.
- S. Cheng, H. Zhang, J. Huang, R. Xu, X. Sun and Y. Guo, Sci. Total Environ., 2020, 737, 139785 CrossRef CAS PubMed.
- H. Morsalpour, H. R. Zare, Z. Shekari and M. Mirbagheri-Firousabad, Microchem. J., 2024, 207, 111940 CrossRef CAS.
- B. He and X. Dong, Sens. Actuators, B., 2019, 294, 192–198 CrossRef CAS.
- C. Y. Deng, X. M. Pi, P. Qian, X. Q. Chen, W. M. Wu and J. Xiang, Anal. Chem., 2017, 89, 966–973 CrossRef CAS PubMed.
- X. Cui, K. Zhao, Z. Qu, X. Chao, L. Xie, H. Chen, B. He and B. Zhang, Bioelectrochem., 2025, 163, 108881 CrossRef CAS PubMed.
- D. Pan, X. L. Zuo, Y. Wan, L. H. Wang, J. Zhang, S. P. Song and C. H. Fan, Sensors, 2007, 7, 2671–2680 CrossRef CAS PubMed.
- Z. Y. Wang, J. Zhao, Z. J. Li, J. C. Bao and Z. H. Dai, Anal. Chem., 2017, 89, 6815–6820 CrossRef CAS PubMed.
- R. Yuan, B. D. Yan, C. Y. Lai, X. H. Wang, Y. Cao, J. C. Tu, Y. Li and Q. Wu, ACS Omega, 2023, 8, 22099–22107 CrossRef CAS PubMed.
- P. Y. Sun, X. Li, B. H. Kong, Y. A. Zhu, M. H. Wang, H. Wang and Q. Liu, Int. J. Biol. Macromol., 2023, 253, 127130 CrossRef CAS PubMed.
- W. H. Wang, S. Xiao, Z. J. Jia, H. Z. Xie, T. H. Li, Q. Q. Wang and N. Gan, Sens. Actuators, B., 2022, 351, 130839 CrossRef CAS.
- J. H. Ding, H. R. Zhao and H. B. Yu, Chem. Eng. J., 2022, 430, 132838 CrossRef CAS.
- Y. Zhang, X. Du, M. Wei, X. Shan, W. Wang, D. Jiang, H. Shiigi and Z. Chen, Anal. Chim. Acta, 2023, 1238, 340645 CrossRef CAS PubMed.
- C. Li, N. Li, L. Yang, L. Liu and D. Zhang, Spectrochim. Acta, Part A., 2024, 309, 123824 CrossRef CAS PubMed.
- F. Li, J. Z. Zhang, S. H. Hu and Y. F. Jia, ACS Sensors, 2021, 6, 1218–1227 CrossRef CAS PubMed.
- D. Jiang, M. Wei, X. J. Du, M. Qin, X. L. Shan, W. C. Wang and Z. D. Chen, Biosens. Bioelectron., 2022, 200, 113917 CrossRef CAS PubMed.
- H. S. Peng, Y. Y. Hui, L. Zhang, F. X. Zhang, Y. F. Liu, J. B. Zheng, R. Jia, Y. X. Song and B. N. Wang, Sens. Actuators B Chem., 2022, 368, 132119 CrossRef CAS.
- L. Lu, G. Guan, J. Wang, W. Meng, S. Li, Y. Zhang and F. Guo, Chem. Eng. J., 2024, 480, 147999 CrossRef CAS.
- J. Yang, W. Zhong, Q. Yu, J. Zou, Y. S. Gao, S. W. Liu, S. B. Zhang, X. Q. Wang and L. M. Lu, Molecules, 2022, 27, 1871 CrossRef CAS PubMed.
- S. Xiong, Z. Wu, F. Li, P. Zhao, H. Li, H. Bao and X. Yang, LWT, 2024, 200, 116186 CrossRef CAS.
- T. Sha, J. Liu, M. Sun, L. Li, J. Bai, Z. Hu and M. Zhou, Talanta, 2019, 200, 300–306 CrossRef CAS PubMed.
- M. Yin, L. Zhang, X. Wei, Y. Sun, S. Qi, Y. Chen, X. Tian, J. Qiu and D. Xu, J. Electroanal. Chem., 2022, 920, 116647 CrossRef CAS.
- S. Cheng, R. Xu, F. Yang, J. Huang, X. Sun, X. Huang, H. Li, F. Li, Y. Guo, M. Hasanzadeh and Y. Zhu, Bioelectrochemistry, 2022, 147, 108174 CrossRef CAS PubMed.
- X. Qi, L. Zhang, X. Wang, S. Chen and X. Wang, Food Control, 2023, 149, 109700 CrossRef CAS.
- X.-P. Liu, J.-L. Cheng, C.-J. Mao, M.-Z. Wu, J.-S. Chen and B. Kang Jin, Microchem. J., 2022, 172, 106928 CrossRef CAS.
- X. Duan, N. Zhang, Z. Li, L. Zhang, F. Sun, Z. Zhou, H. Liu, Y. Guo, X. Sun, J. Jiang and D. Zhang, J. Colloid Interface Sci., 2023, 632, 171–178 CrossRef CAS PubMed.
- X. Wang, W. Zhang, X. Gao, Z. Sun, X. Sun, Y. Guo, F. Li and N. E. Boboriko, Sens. Actuators B Chem., 2022, 360, 131665 CrossRef CAS.
- X. Qin, Y. Yin, H. Yu, W. Guo and M. Pei, Biosens. Bioelectron., 2016, 77, 752–758 CrossRef CAS PubMed.
- W. Guo, N. Sun, X. Qin, M. Pei and L. Wang, Biosens. Bioelectron., 2015, 74, 691–697 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01006c |
| This journal is © The Royal Society of Chemistry 2025 |