
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the multifaceted nature of terpyridine-based metal complexes: a synergistic study of their photophysical and electrochemical behavior with computational insights†
Nafeesa Naeem a,
Ehsan Ullah Mughal*a,
Ebru Bozkurt*bc,
Ishtiaq Ahmedd,
Muhammad Naveed Zafare,
Ali Raza Ayubf,
Amina Sadiqg,
Abdulrahman A. Alsimareeh,
Reem I. Alsantalii and
Saleh A. Ahmedj
a,
Ehsan Ullah Mughal*a,
Ebru Bozkurt*bc,
Ishtiaq Ahmedd,
Muhammad Naveed Zafare,
Ali Raza Ayubf,
Amina Sadiqg,
Abdulrahman A. Alsimareeh,
Reem I. Alsantalii and
Saleh A. Ahmedj
aDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk; ebrubozkurt@atauni.edu.tr
bProgram of Occupational Health and Safety, Vocational College of Technical Sciences, Atatürk University, 25240 Erzurum, Turkey
cDepartment of Nanoscience and Nanoengineering, Graduate School of Natural and Applied Sciences, Atatürk University, 25240, Erzurum, Turkey
dDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, UK
eDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
fKey Laboratory of Clusters Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, PR China
gDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
hDepartment of Chemistry, College of Science and Humanities, Shaqra University, Shaqra, Saudi Arabia
iDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P. O. Box 11099, Taif 21944, Saudi Arabia
jDepartment of Chemistry, Faculty of Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
First published on 7th April 2025
Abstract
This manuscript provides a comprehensive overview of the synthesis, characterization, and photophysical and electrochemical properties of terpyridine-based metal complexes (C1–C20). The synthesis of these terpyridine (TPY) complexes involves the coordination of TPY ligands (L1–L11) with transition metal ions, leading to a variety of novel structural and electronic configurations. The characterization of TPY ligands and their complexes is carried out using various techniques, including UV-Vis spectroscopy, NMR, FTIR and mass spectrometry. To the best of our knowledge, for the first time, we comprehensively investigate the photophysical, solvatochromic, electrochemical, and computational properties of an extensive series of TPY-based metal complexes (C1–C20) within a single framework. The solvatochromic behavior of the synthesized complexes (C1–C20) is explored, revealing their sensitivity to solvent polarity, which is a key factor influencing their photophysical properties. The TPY-based complexes (C1–C20) exhibited solvent-dependent fluorescence behavior, with distinct ILCT and MLCT mechanisms, and enhanced fluorescence in specific solvents, particularly for Zn(II) and Cu(II) complexes. The absorption and emission characteristics of the complexes are studied in dilute solutions to explore their structure–property relationships. Additionally, the electrochemical properties of the TPY-based metal complexes (C1–C20) are investigated, highlighting their redox activity and potential for use in energy storage and conversion applications. Density functional theory (DFT) calculations are employed to provide detailed insights into the electronic structure and reactivity of these complexes, supporting the experimental observations. The correlation of electronic band gaps with photophysical and electrochemical behaviors showed compounds as promising candidates with efficient charge transfer and strong fluorescence. The integrated analyses reveal the exceptional potential of this scaffold for advanced materials applications, highlighting its versatility and significance in cutting-edge research.
1. Introduction
Terpyridine-based metal complexes (Fig. 1) have attracted considerable attention in recent years due to their versatile structural framework and unique electronic properties, which make them promising candidates for various applications in photophysics, electrochemistry, and molecular sensing.1 The terpyridine (TPY) ligand (Fig. 1), characterized by its tridentate coordination mode, allows for robust metal coordination, resulting in stable complexes that exhibit fascinating electronic transitions and photophysical properties.2,3 These complexes are not only easy to synthesize but also offer extensive tunability, which can be strategically modulated by altering the metal center or substituent groups on the TPY ligand, thus enabling fine control over their optical and electronic characteristics.4,5 | ||
| Fig. 1 General structure of TPY ligand (a) and complex (b). | ||
The photophysical properties of TPY-based metal complexes are particularly significant, as they display strong absorption in the UV-visible region and often exhibit metal-to-ligand charge transfer (MLCT) bands.6–8 These properties make them excellent candidates for applications in light-emitting devices, photovoltaic cells, and sensors. One prominent phenomenon observed in TPY metal complexes is solvatochromism—the dependence of a compound's color or photophysical response on the polarity of the solvent.9,10 This property is attributed to the reorganization of electronic distribution in different solvent environments, making TPY complexes sensitive to their surroundings and, therefore, useful as molecular probes or chemosensors.11,12 Understanding and controlling solvatochromic behavior in such complexes can provide insights into their electronic transitions, enhancing their application potential in chemical sensing.13
The absorption and emission spectra of TPY-based metal complexes offer crucial insights into their electronic transitions and excited-state dynamics.11,14 Analyzing the absorption spectra allows for identification of specific wavelengths where these dyes effectively absorb light, which is essential for optimizing light-harvesting and energy transfer applications.7,15 In contrast, emission spectra reveal the wavelengths of light emitted by the dyes as they relax from the excited to the ground state, providing valuable data for fluorescence-based applications, such as imaging and sensing.9,16
A key photophysical parameter, the quantum yield (Φ), indicates the efficiency with which a dye converts absorbed photons into emitted photons.17,18 Measuring the quantum yield of TPY-based metal complexes helps assess their suitability for use in fluorescence-based devices and sensors.12,19 A detailed examination of factors affecting quantum yield, including molecular structure and the influence of substituents, can support the design of dyes with improved performance and higher efficiency.20,21
Furthermore, solvent choice plays a significant role in shaping the photophysical behavior of TPY-based metal complexes.22,23 Understanding solvent effects is essential for adapting these dyes for specific applications and maximizing their efficiency across various environments. A comprehensive exploration of these influences is key to tailoring the properties of TPY-based metal complexes for desired applications.24,25
In addition to their photophysical features, TPY-based metal complexes often possess intriguing electrochemical properties that can be exploited in sensor development.26,27 Electrochemical studies provide valuable information on the redox behavior and electronic structure of these complexes, as well as their stability under various conditions.28–30 These electrochemical characteristics not only complement the photophysical properties but also enable TPY complexes to serve as redox-active sensors, facilitating the detection of various analytes based on their oxidation or reduction responses.6,31,32
This manuscript focuses on the design, synthesis, and characterization of TPY-based metal complexes with an emphasis on their photophysical, solvatochromic, and electrochemical properties. The findings from this work aim to advance our understanding of TPY metal complexes and their application in photophysical and sensory technologies, highlighting their multifunctional capabilities in modern scientific applications, multifunctional dyes for use in optoelectronics, dye-sensitized solar cells, and biological imaging applications. Additionally, computational studies will be employed to investigate their electronic structures and further elucidate the relationships between their structural features and observed properties. By integrating synthetic, spectroscopic, electrochemical, and computational approaches, this work seeks to contribute to the expanding knowledge of TPY-based systems and their potential applications in advanced materials and technologies.
2. Materials and methods
All reagents used in this study were obtained from Merck (Germany) and were employed as received, without further purification. Melting points were measured using an uncalibrated electrothermal melting point apparatus. Infrared (IR) spectra were recorded on a Bio-Rad spectrophotometer, while Nuclear Magnetic Resonance (NMR) spectra were obtained using a Bruker DRX 600 instrument, operating at 500 MHz and 600 MHz for 1H-NMR and 126 MHz and 151 MHz for 13C-NMR, respectively. Reaction progress was monitored by thin-layer chromatography (TLC) on silica gel pre-coated plates, with spots visualized under UV light.All solvents (Sigma and Merck), quinine sulfate (Fluka), and sulfuric acid (Sigma) were purchased and used without further purification. Stock solutions of the compounds were prepared in ethanol. Fresh probe samples in varying solutions were obtained from these stock solutions by evaporating the solvent. For all measurements, the concentrations of the compounds were maintained at 10 μM. All experiments were carried out at room temperature.
2.1. General procedure for syntheses of TPY ligands (L1–L11) and metal complexes (C1–C20)33
A solution of 2-acetylpyridine (2.43 g, 20.0 mmol) in methanol (20 mL) was mixed with a substituted aryl aldehyde (10.0 mmol). Potassium hydroxide (KOH) pellets (1.54 g, 24.0 mmol) and 35% aqueous ammonia solution (40 mL) were subsequently added to the mixture. The reaction mixture was refluxed for 4–6 hours, with its progress monitored using thin-layer chromatography (TLC). Upon completion, the solvent was evaporated under reduced pressure, and the resulting solid was collected by filtration. The precipitate was washed extensively with distilled water to remove residual base, followed by ice-cold ethanol until the washings were neutral. The purified product was obtained by recrystallization from ethanol after air drying the solid.33To prepare the metal complex, a hot methanolic solution (20 mL) of the metal salt (0.5 mmol) was added dropwise to a dichloromethane solution (20 mL) containing the substituted TPY ligand (0.28 g, 1.0 mmol) under continuous stirring. The solution's color changed immediately, indicating complex formation. The reaction mixture was stirred at room temperature for 2 hours, and the addition of excess NH4PF6 caused the product to precipitate. The precipitate was filtered and washed sequentially with cold methanol (5 mL) and diethyl ether (15 mL). Recrystallization from CH3CN, CH3OH, or a combination of the two provided the analytically pure complex.33
2.2. UV-vis absorption and fluorescence spectroscopy
The UV-vis absorption and fluorescence spectra of the samples were obtained using a Shimadzu UV-1800 spectrophotometer and an Agilent Technologies Cary Eclipse fluorescence spectrophotometer, respectively. Steady-state fluorescence measurements were performed by exciting the samples at 300 nm and 350 nm, and the fluorescence emission intensities were recorded over the wavelength ranges of 310–600 nm and 350–650 nm. The fluorescence quantum yields of the compounds were determined following the method described previously34 using the Parker–Rees equation (eqn (1)).
 | (1) |
2.3. FTIR measurements
The infrared (IR) spectra of the solid samples were measured using a Nicolet FT-IR Impact 400D spectrometer over the range of 400–4000 cm−1. The samples were prepared by finely grinding them with spectroscopic-grade potassium bromide (KBr) powder and pressing the mixture into 1 mm pellets. A blank KBr disk served as the reference for the background. Spectral smoothing and baseline correction were carried out automatically using the integrated software, Spectra Manager™ II (JASCO Corporation).352.4. Cyclic voltammetry
Electrochemical analyses were performed using a CHI620 C electrochemical analyzer in a standard three-electrode setup. The system comprised a platinum wire as the counter electrode, a glassy carbon electrode as the working electrode, and an Ag/AgCl (3 M KCl) electrode as the reference. Measurements were conducted in CH3CN containing tetrabutylammonium bromide (TBAB) as the supporting electrolyte. Sample solutions were prepared at a concentration of 0.1 mM in CH3CN. Cyclic voltammograms were recorded over a potential window ranging from −2.0 V to +1.0 V at scan rates of 40, 100, and 200 mV s−1. Scanning was initiated from the most negative potential (∼−2.0 V) and proceeded towards the positive potential (∼+1.0 V). All CV measurements were carried out in aqueous medium, ensuring that the choice of the Ag/AgCl reference electrode is appropriate for our study. All experiments were carried out at ambient temperature.62.5. Computational (DFT) calculations
In this study, DFT quantum analyses were performed to ascertain the geometrical configurations, photophysical properties, FMO analysis and molecular electrostatic potential analysis of our molecules by using the Gaussian 09W36 for calculations and GaussView 6.0 (ref. 37) for the visualized ChemDraw 12.0 (ref. 38) as well. Our synthesized molecules were optimized using the method B3LYP/LAN2DZ39 for the geometrical analysis, frontier molecular orbitals, and molecular electrostatic potentials to evaluate the best molecules for our further investigations. Additionally, we utilized the B3LYP/def2-TZVP basis set for light elements, including carbon (C), hydrogen (H), nitrogen (N), and oxygen (O).3. Results and discussion
3.1. Chemistry
The synthesis of TPY ligands and their complexes was performed in accordance with a previously reported procedure33 (as outlined in Scheme 1 and Table 1). The structural characterization of the newly synthesized ligands and metal complexes was accomplished using FTIR, UV-Vis, 1H NMR, and 13C NMR spectroscopy and mass spectrometry. | ||
| Scheme 1 Synthesis of TPY ligands (L1–L11) and their metal complexes (C1–C20). | ||
| Compound no. | Chemical structure and IUPAC names |
|---|---|
| L1 |  |
| L2 |  |
| L3 |  |
| L4 | 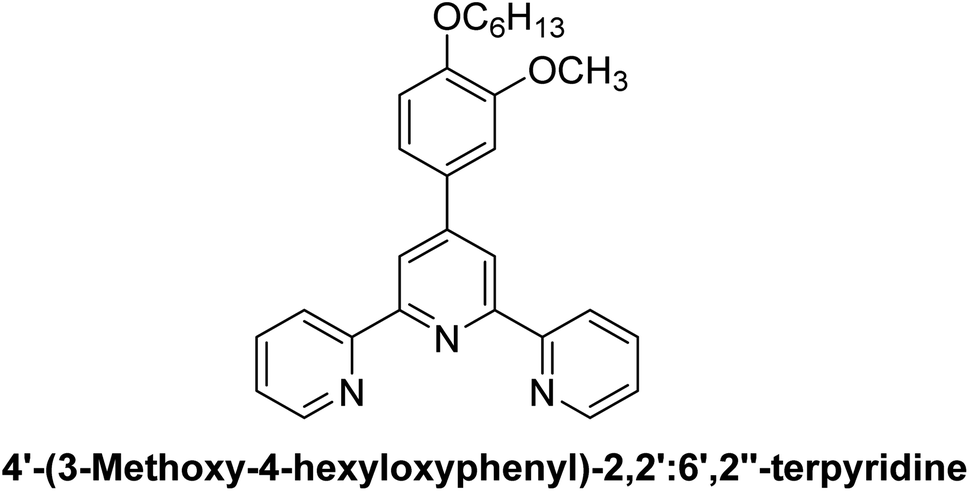 |
| L5 |  |
| L6 | 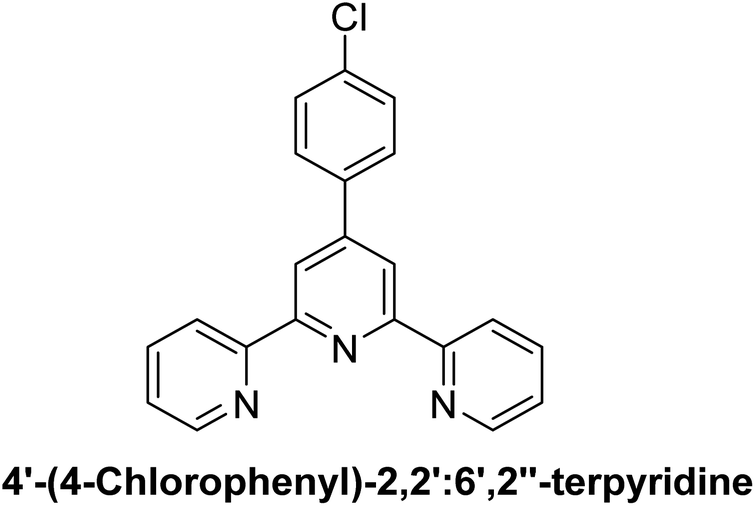 |
| L7 |  |
| L8 |  |
| L9 |  |
| L10 |  |
| L11 |  |
| C1 |  |
| C2 |  |
| C3 |  |
| C4 |  |
| C5 |  |
| C6 | 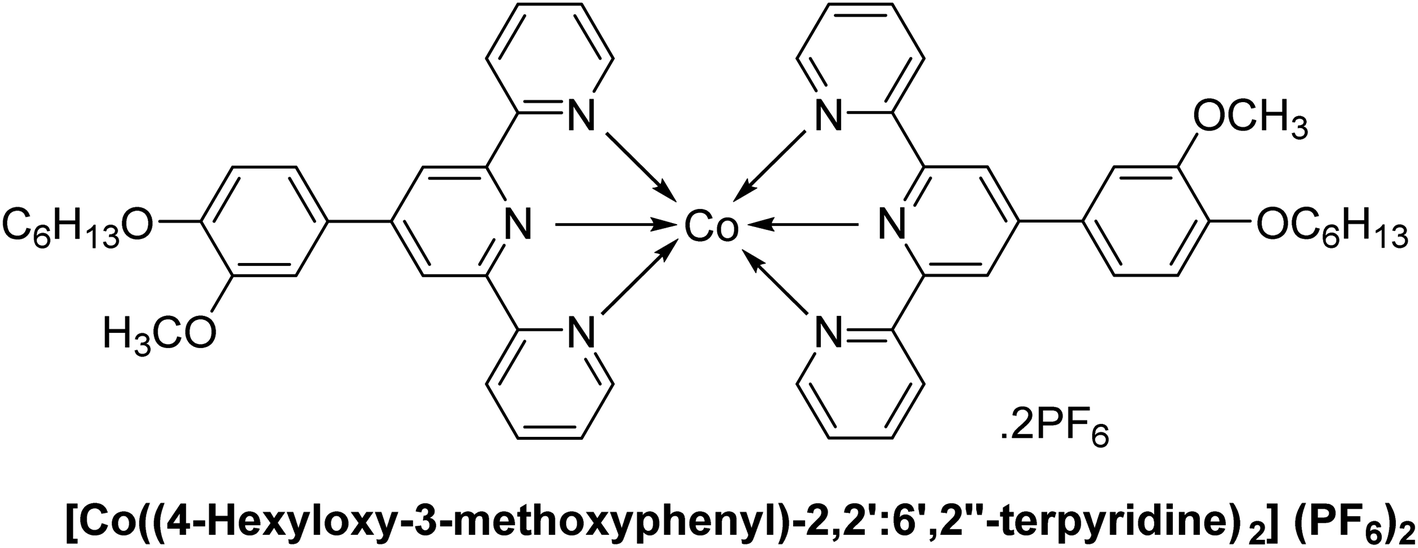 |
| C7 | 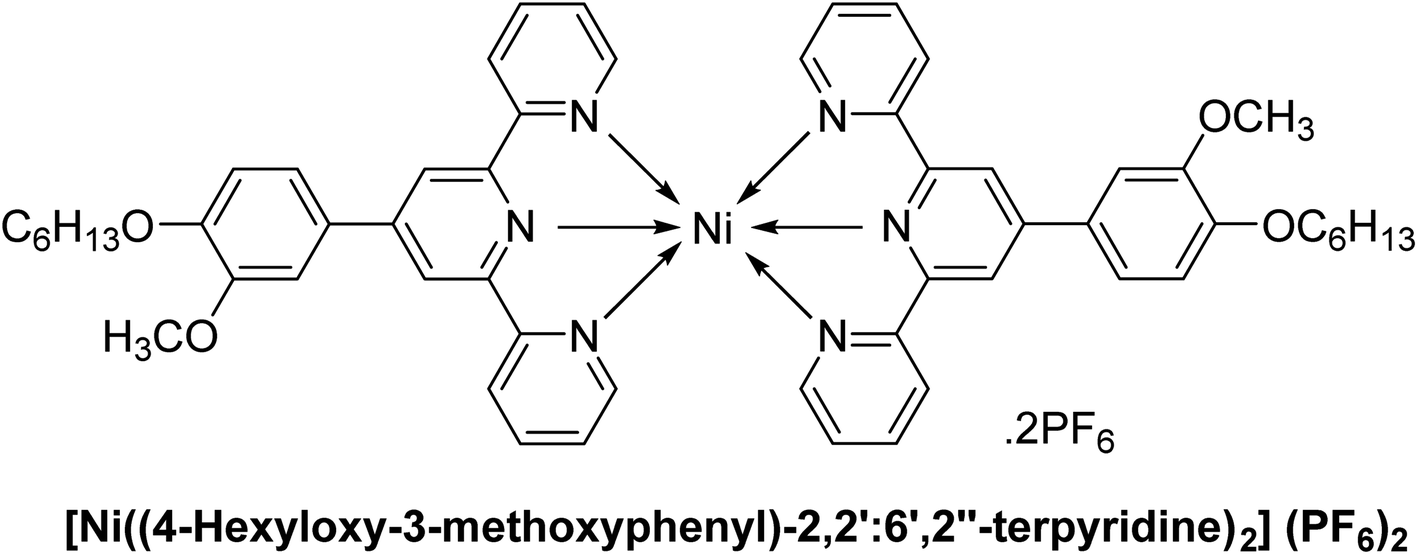 |
| C8 |  |
| C9 |  |
| C10 |  |
| C11 |  |
| C12 |  |
| C13 |  |
| C14 |  |
| C15 |  |
| C16 |  |
| C17 |  |
| C18 |  |
| C19 |  |
| C20 |  |
The 1H-NMR spectrum of the substituted 2,2′:6′,2′′-TPY ligand (Fig. 1) typically shows distinct signals corresponding to the aromatic protons of the terpyridine framework ranging from δ = 7.0–9.0 ppm, consistent with the deshielded nature of the aromatic environment. Signals arising from the pyridine protons in the ortho positions to the nitrogen atoms are often observed as doublets or doublet of doublets in the downfield region (δ = 8.5–9.0 ppm) due to the electron-withdrawing effects of the nitrogen atoms and spin–spin coupling.
The central pyridine ring protons typically show a characteristic singlet around δ = 7.6–8.2 ppm owing to chemically and magnetically equivalent protons. Substituents on the TPY scaffold cause additional shifts; for instance, EDGs may shift signals upfield, while EWGs result in downfield shifts. The presence of these substituents is further evidenced by singlets, doublets, or triplets depending on their specific placement and influence on coupling patterns.
Upon complexation with a metal cation, significant changes occur in the 1H-NMR spectrum. Signals corresponding to the protons near the coordination sites (particularly those ortho to the pyridine nitrogen atoms) typically shift downfield (δ = 8.8–9.5 ppm), indicating deshielding due to metal–ligand interactions. The protons of the central pyridine ring may show further splitting or broadening, often appearing as unresolved multiplets, depending on the coordination geometry and the electronic nature of the metal center.
In some cases, the formation of the metal complex results in signal broadening across the spectrum, particularly if the complex exhibits dynamic behavior in solution or if the metal ion is paramagnetic. For 1H-NMR spectra of diamagnetic complexes, distinct splitting patterns and downfield shifts provide evidence for successful coordination. For example, singlets or doublets corresponding to substituent protons may shift slightly or remain unchanged, depending on their proximity to the coordination site.
The 13C-NMR spectrum of the free TPY ligand typically displays peaks in the range of 120–160 ppm, attributed to the aromatic carbons of the terpyridine framework. Coordination to the metal center often causes downfield shifts for the carbons directly bonded to the pyridine nitrogens (δ = 150–165 ppm), reflecting the influence of electron density redistribution upon complexation.
The spectroscopic data of all the previously synthesized ligands (L1, L2, L3, L6 and L7) and complexes (C3, C9 and C10) are given in literature.4,33 In addition, the complete synthesis and spectroscopic characterization of two newly synthesized ligands (L8 and L11) are available in another protocol,35 while the spectral data of other newly synthesized ligands (L4, L5, L9, L10) and complexes (C1, C2, C4, C5, C6, C7, C8, C11–C20) are given below:
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2CH2CH2CH2CH2CH3), 3.92 (s, 3H, –OMe), 1.78–1.73 (m, 2H, –OCH2C2CH2CH2CH2CH3), 1.47–1.43 (m, 2H, –OCH2CH2C2CH2CH2CH3), 1.35–1.30 (m, 4H, –OCH2CH2CH2C2C2CH3), 0.91–0.89 (m, 3H, –OCH2CH2CH2CH2CH2C3); 13C NMR (151 MHz, DMSO-d6): δ 156.0, 150.07, 150.01, 149.98, 149.96, 149.8, 138.0, 130.4, 125.0, 121.4, 120.1, 118.1, 113.7, 110.8, 68.7, 56.2, 31.4, 29.1, 25.6, 22.5, 14.4 (remaining carbons are isochronous); accurate mass (ESI) of [M + H]+: calculated for C28H30N3O2 440.2338; found 440.2328.2CH3), 1.42 (t, J = 5.0 Hz, 3H, –OCH2C3); 13C NMR (126 MHz, DMSO-d6): δ 160.2, 156.0, 155.5, 149.7, 149.5, 137.8, 129.8, 129.6, 128.6, 125.0, 121.3, 117.7, 116.0, 115.6, 114.6, 63.7, 15.07; accurate mass (ESI) of [M + H]+: calculated for C23H20N3O5SZn 514.0415; found 514.0410.2CH3), 1.38–1.33 (bm, 3H, –OCH2C3); 13C NMR (126 MHz, DMSO-d6): δ 160.1, 156.1, 156.0, 155.5, 149.7, 149.4, 138.0, 129.8, 128.6, 124.9, 124.8, 121.2, 117.7, 115.6, 63.7, 15.0; accurate mass (ESI) of [M + Na]+: calculated for C46H38CoN6O2Na 788.2285; found 788.2273.
2CH2CH2CH2CH2CH3), 3.92 (s, 3H, –OMe), 1.78–1.73 (m, 2H, –OCH2C2CH2CH2CH2CH3), 1.47–1.43 (m, 2H, –OCH2CH2C2CH2CH2CH3), 1.35–1.30 (m, 4H, –OCH2CH2CH2C2C2CH3), 0.91–0.89 (m, 3H, –OCH2CH2CH2CH2CH2C3); 13C NMR (151 MHz, DMSO-d6): δ 156.0, 150.07, 150.01, 149.98, 149.96, 149.8, 138.0, 130.4, 125.0, 121.4, 120.1, 118.1, 113.7, 110.8, 68.7, 56.2, 31.4, 29.1, 25.6, 22.5, 14.4 (remaining carbons are isochronous); accurate mass (ESI) of [M + H]+: calculated for C28H30N3O2 440.2338; found 440.2328.2CH3), 1.42 (t, J = 5.0 Hz, 3H, –OCH2C3); 13C NMR (126 MHz, DMSO-d6): δ 160.2, 156.0, 155.5, 149.7, 149.5, 137.8, 129.8, 129.6, 128.6, 125.0, 121.3, 117.7, 116.0, 115.6, 114.6, 63.7, 15.07; accurate mass (ESI) of [M + H]+: calculated for C23H20N3O5SZn 514.0415; found 514.0410.2CH3), 1.38–1.33 (bm, 3H, –OCH2C3); 13C NMR (126 MHz, DMSO-d6): δ 160.1, 156.1, 156.0, 155.5, 149.7, 149.4, 138.0, 129.8, 128.6, 124.9, 124.8, 121.2, 117.7, 115.6, 63.7, 15.0; accurate mass (ESI) of [M + Na]+: calculated for C46H38CoN6O2Na 788.2285; found 788.2273.3.2. Photophysical studies of all the synthesized TPY complexes (C1–C20)
The UV-Vis absorption and fluorescence spectra of all the new TPY complexes (C1–C20) were recorded in thirteen different solvents such as dichloromethane (DCM), diethyl ether, dimethyl formamide (DMF), tetrahydrofuran (THF), ethyl acetate, toluene, dimethyl sulfoxide (DMSO), acetonitrile (ACN), chloroform, isopropanol (i-PrOH), ethanol (EtOH), methanol (MeOH) and water at room temperature. As can be seen in Fig. 1a–20a in ESI,† all compounds exhibited absorption peaks within the ranges of 250–300 nm and 300–400 nm across nearly all selected solvents. While the bands at 250–300 nm originate from the n–π* and π–π* transitions of the aromatic rings, the bands appearing in the 300–400 nm range correspond to the intra-ligand π–π* and n–π* transitions. The peaks around 570–580 nm observed only in the C3 complex correspond to the metal–ligand charge transfer band (MLCT). However, MLCT bands were observed as a shoulder of the π–π* transition bands, which are generally most blue-shifted at around 400 nm in other complexes. The reason why the MLCT band, which originates from weak d–d transitions that should be in the visible region of the spectrum (∼500 nm), was not observed in other complexes was that the charge transfer transition band tails were dominant.6,41,42 Moreover, oxidation of the d6 metal complexes is generally referred to occur on the metal center while reduction is centered on the ligands. In accordance with this, the lowest visible light energy absorption process is considered as MLCT.40,43 This is also another reason why the MLCT band is observed only in the Fe complex. In addition, although the aromatic ring transitions and MLCT transitions were not affected by solvent polarity, the bands of intra-ligand transitions were observed to be broader in highly polar solvents.To understand the effects of intra-ligand and MLCT on the fluorescence properties of molecules, fluorescence measurements of all new TPY derivative compounds were also taken at room temperature at both 300 nm and 350 nm excitation wavelengths. While Co(II) complexes generally showed fluorescence in DMF at 300 nm excitation wavelength, they showed high fluorescence intensity especially in apolar solvents at 350 nm excitation wavelength (Fig. 1b, c, 6b, c, 3b, c, 8b and c in ESI†). However, C12, C15 and C19 compounds had very high fluorescence properties in most of the solvents studied at 300 nm excitation wavelength, and it was observed that C19 compound exhibited fluorescence properties in all solvents except water at an excitation wavelength of 350 nm (Fig. 12b, c, 15b, c, 19b and c in ESI†). It was observed that while Ni(II) complexes exhibited low fluorescence intensities at 300 nm excitation wavelength in all solvents, they exhibited high fluorescence intensities at 350 nm excitation wavelength, especially in apolar solvents (Fig. 2b, c, 4b, c, 5b, c, 7b and c in ESI†). It was determined that the Fe(II) complex C3 compound showed fluorescence only in polar aprotic solvents at an excitation wavelength of 300 nm and had high fluorescence intensity in apolar solvents when excited at 350 nm (Fig. 3b and c in ESI†). While Zn(II) complexes exhibited fluorescence in polar solvents at 300 nm excitation wavelength (except C10), they displayed better fluorescence properties in apolar solvents when excited at 350 nm (Fig. 10b, c, 11b, c, 14b, c, 17b, c, 18b and c in ESI†). Moreover, C18 compound had a single fluorescence peak (Fig. 2a–c) while three peaks are observed in apolar solvents at 350 nm excitation wavelength for all compounds. Among the Cu(II) complexes, C13 exhibited good fluorescence in polar solvents, especially in water, when excited at 300 nm, while C16 and C20 showed very good fluorescence properties in almost all solvents (Fig. 13b, 16b and 20b in ESI†). When the excitation wavelength was 350 nm, C13 and C16 showed fluorescence in apolar solvents, while C20 had three fluorescence peaks in apolar solvents and a single fluorescence peak in polar solvents (Fig. 13c, 16c and 20c in ESI†). While the fluorescence observed at the excitation wavelength of 300 nm was due to the intra-ligand charge transfer from the aromatic ring to the ligand, the fluorescence at the excitation wavelength of 350 nm was attributed to the MLCT. A significant quenching of the MLCT transitions was observed in highly polar solvents, as evident from the fluorescence quantum yields in all compounds (Table 2).
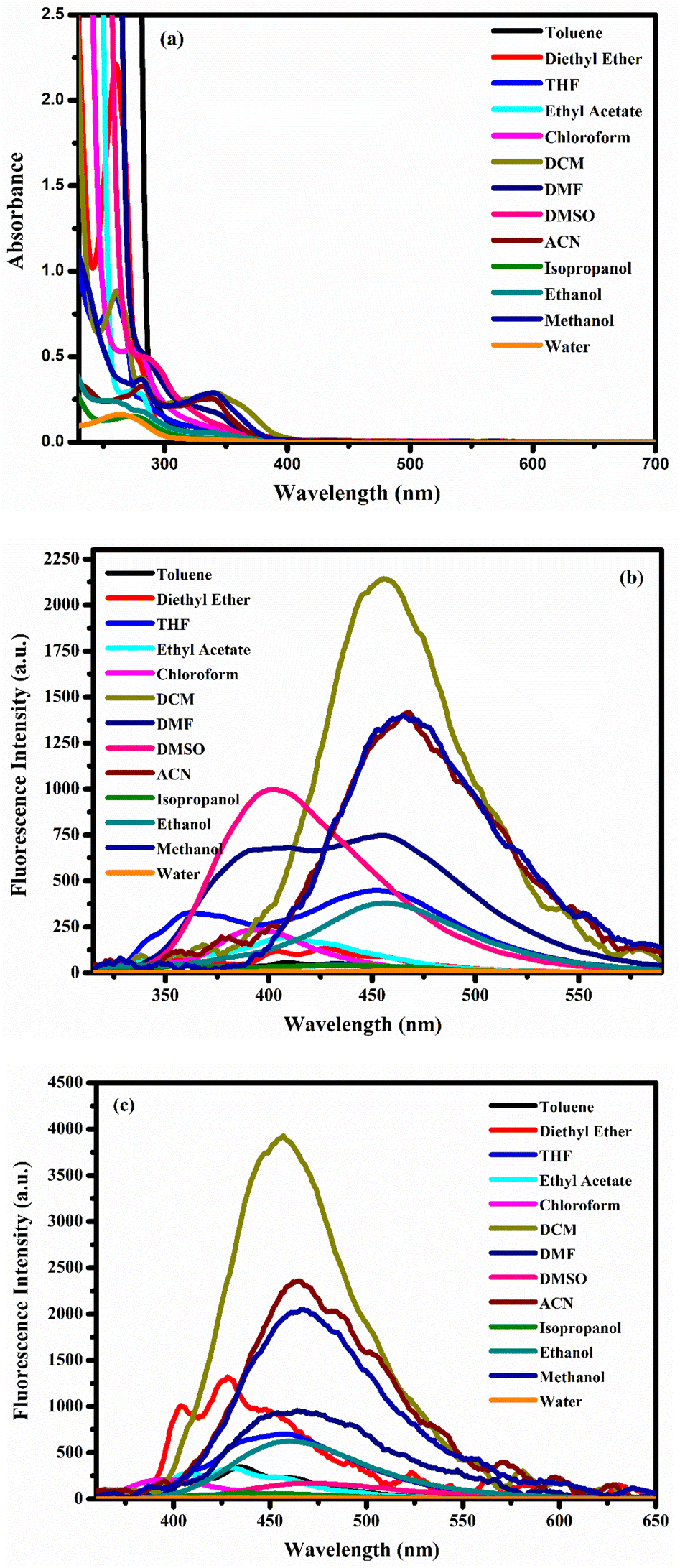 | ||
| Fig. 2 (a) The UV-vis absorption and (b) λexc = 300 nm fluorescence spectra and (c) λexc = 350 nm fluorescence spectra of compound C18 (10 μM) in different solvents. | ||
| No. | Solvent | λabs (nm) | ε ×104 (M−1 cm−1) | λem (nm) (λexc = 300 nm) | Φ (λexc = 300 nm) | λem (nm) (λexc = 350 nm) | Φ (λexc = 350 nm) |
|---|---|---|---|---|---|---|---|
| C1 | Toluene | 289 | 2.27 | 401 | 0.02 | 410/433/457 | 0.05 |
| Diethyl ether | 262 | 2.32 | 403 | 0.01 | 403/428/450 | 0.01 | |
| THF | 268 | 3.86 | 406 | 0.02 | 408/433/457 | 0.04 | |
| Ethyl acetate | 276 | 3.11 | 408 | 0.02 | 406/430/452 | 0.05 | |
| Chloroform | 278 | 4.66 | 436 | 0.01 | 435 | 0.01 | |
| DCM | 265 | 3.39 | 446 | 0.01 | 411/436/463 | 0.02 | |
| DMF | 287 | 4.47 | 438 | 0.08 | 416/440/461 | 0.01 | |
| DMSO | 287 | 4.06 | 448 | 0.03 | — | — | |
| ACN | 283 | 3.15 | — | — | — | — | |
| i-PrOH | 283 | 1.67 | — | — | 410/433/458 | 0.01 | |
| EtOH | 284 | 2.94 | — | — | — | — | |
| MeOH | 283 | 3.55 | — | — | — | — | |
| Water | 283 | 1.03 | — | — | — | — | |
| C2 | Toluene | 289 | 2.13 | — | — | 410/433/458 | 0.05 |
| Diethyl ether | 263 | 1.89 | — | — | 403/428/451 | 0.02 | |
| THF | 269 | 2.90 | 408 | 0.01 | 408/433/460 | 0.05 | |
| Ethyl acetate | 275 | 1.95 | 346/418 | 0.01 | 405/426/455 | 0.08 | |
| Chloroform | 278 | 3.55 | — | — | — | — | |
| DCM | 265 | 2.49 | 413 | 0.02 | 411/435/455 | 0.03 | |
| DMF | 276 | 2.73 | — | — | — | — | |
| DMSO | 270 | 2.92 | — | — | — | — | |
| ACN | 273 | 2.08 | — | — | — | — | |
| i-PrOH | 278 | 1.62 | — | — | 410/435/458 | 0.01 | |
| EtOH | 276 | 2.08 | — | — | — | — | |
| MeOH | 275 | 2.57 | — | — | — | — | |
| Water | 281 | 1.23 | — | — | — | — | |
| C3 | Toluene | 289 | 2.00 | 396 | 0.01 | 410/433/458 | 0.04 |
| Diethyl ether | 263 | 1.13 | 356/416 | 0.01 | 403/428/448 | 0.02 | |
| THF | 268 | 3.16 | 408 | 0.04 | 408/433/454 | 0.06 | |
| Ethyl acetate | 276 | 1.26 | 410 | 0.04 | 406/431/451 | 0.19 | |
| Chloroform | 277 | 4.22 | — | — | — | — | |
| DCM | 264/576 | 4.03 | 438 | 0.01 | 411/436/461 | 0.02 | |
| DMF | 270/576 | 4.37 | 438 | 0.03 | 411/436/457 | 0.01 | |
| DMSO | 290/580 | 4.26 | 448 | 0.02 | — | — | |
| ACN | 274/571 | 3.50 | — | — | — | — | |
| i-PrOH | 283/572 | 1.04 | — | — | 41/433/452 | 0.02 | |
| EtOH | 284/574 | 2.49 | — | — | — | — | |
| MeOH | 284/570 | 2.76 | — | — | — | — | |
| Water | 279/570 | 1.84 | — | — | — | — | |
| C4 | Toluene | 289 | 1.78 | 338/428 | 0.01 | 408/433/455 | 0.09 |
| Diethyl ether | 261 | 9.00 | 337/418 | 0.01 | 403/426/451 | 0.06 | |
| THF | 268 | 2.75 | — | — | 408/431/452 | 0.05 | |
| Ethyl acetate | 273 | 1.22 | 342/423 | 0.01 | 406/430/455 | 0.13 | |
| Chloroform | 278 | 3.79 | — | — | — | — | |
| DCM | 265 | 2.57 | — | — | 413/435 | 0.03 | |
| DMF | 276 | 2.60 | — | — | — | — | |
| DMSO | 270 | 3.44 | 438 | 0.01 | — | — | |
| ACN | 273 | 2.29 | — | — | — | — | |
| i-PrOH | 271 | 7.70 | — | — | 408/433 | 0.03 | |
| EtOH | 269 | 1.21 | 337 | 0.01 | — | — | |
| MeOH | 276 | 3.00 | — | — | — | — | |
| Water | 275 | 4.10 | — | — | — | — | |
| C5 | Toluene | 289 | 1.52 | 343/426 | 0.01 | 410/435/457 | 0.13 |
| Diethyl ether | 260 | 5.30 | 337/426 | 0.01 | 403/428/451 | 0.11 | |
| THF | 266 | 1.68 | 441 | 0.01 | 408/431/458 | 0.10 | |
| Ethyl acetate | 274 | 1.00 | 343/428 | 0.01 | 406/430/452 | 0.15 | |
| Chloroform | 277 | 3.57 | — | — | — | — | |
| DCM | 264 | 2.56 | — | — | 411/436/454 | 0.03 | |
| DMF | 276 | 3.06 | — | — | — | — | |
| DMSO | 270 | 3.06 | — | — | — | — | |
| ACN | 275 | 2.65 | — | — | — | — | |
| i-PrOH | 277 | 5.00 | — | — | 410/433/455 | 0.06 | |
| EtOH | 270 | 1.73 | 336/451 | 0.01 | — | — | |
| MeOH | 275 | 2.40 | — | — | — | — | |
| Water | 270 | 4.70 | — | — | — | — | |
| C6 | Toluene | 289 | 1.71 | 343/423 | 0.01 | 408/433/457 | 0.17 |
| Diethyl ether | 289 | 0.72 | 338/423 | 0.01 | 403/428/451 | 0.17 | |
| THF | 261 | 1.57 | 413 | 0.02 | 408/431/454 | 0.11 | |
| Ethyl acetate | 277 | 1.00 | 416 | 0.02 | 406/430/458 | 0.17 | |
| Chloroform | 279 | 2.73 | — | — | 441 | 0.01 | |
| DCM | 261 | 2.22 | — | — | 411/436/460 | 0.04 | |
| DMF | 276 | 2.75 | 445 | 0.07 | 435 | 0.02 | |
| DMSO | 289 | 2.87 | 454 | 0.03 | — | — | |
| ACN | 281 | 1.03 | — | — | 428 | 0.01 | |
| i-PrOH | 276 | 0.41 | — | — | 411/430 | 0.04 | |
| EtOH | 261 | 0.71 | 335 | 0.01 | — | — | |
| MeOH | 282 | 1.77 | — | — | — | — | |
| Water | 269 | 0.29 | — | — | — | — | |
| C7 | Toluene | 289 | 1.84 | 340/423 | 0.01 | 408/433/460 | 0.06 |
| Diethyl ether | 261 | 0.85 | 335/425 | 0.01 | 403/428/446 | 0.12 | |
| THF | 262 | 2.41 | 344/448 | 0.01 | 408/433/455 | 0.05 | |
| Ethyl acetate | 277 | 1.17 | 344/423 | 0.01 | 405/430/450 | 0.09 | |
| Chloroform | 277 | 3.81 | — | — | — | — | |
| DCM | 262 | 2.52 | — | — | 411/436 | 0.03 | |
| DMF | 276 | 2.92 | — | — | — | — | |
| DMSO | 289 | 2.97 | — | — | — | — | |
| ACN | 274/362 | 2.04 | — | — | — | — | |
| i-PrOH | 276 | 0.67 | — | — | 408/433/457 | 0.02 | |
| EtOH | 269 | 1.12 | 335 | 0.01 | — | — | |
| MeOH | 277 | 2.58 | — | — | — | — | |
| Water | 269 | 0.44 | — | — | — | — | |
| C8 | Toluene | 289 | 1.80 | 352/428 | 0.01 | 408/433/461 | 0.13 |
| Diethyl ether | 261 | 0.71 | 354/411 | 0.01 | 403/428/448 | 0.20 | |
| THF | 262 | 1.90 | — | — | 408/403/451 | 0.12 | |
| Ethyl acetate | 276 | 1.10 | 353/425 | 0.01 | 406/430/451 | 0.13 | |
| Chloroform | 278 | 2.33 | — | — | — | — | |
| DCM | 262 | 1.77 | — | — | 411/435/460 | 0.07 | |
| DMF | 276 | 2.39 | 363 | 0.02 | 411/436 | 0.01 | |
| DMSO | 288 | 2.33 | — | — | — | — | |
| ACN | 278/334 | 1.56 | — | — | — | — | |
| i-PrOH | 271 | 0.93 | — | — | 408/433/455 | 0.02 | |
| EtOH | 283 | 1.72 | — | — | — | — | |
| MeOH | 278/334 | 1.71 | — | — | — | — | |
| Water | 281/329 | 0.93 | — | — | — | — | |
| C9 | Toluene | 289 | 1.81 | 348/425 | 0.01 | 410/435/455 | 0.16 |
| Diethyl ether | 260 | 0.80 | 352/416 | 0.01 | 403/428/446 | 0.16 | |
| THF | 261 | 1.46 | 356 | 0.01 | 408/433/451 | 0.15 | |
| Ethyl acetate | 281 | 0.97 | 343/425 | 0.01 | 406/431/452 | 0.17 | |
| Chloroform | 278 | 2.23 | — | — | — | — | |
| DCM | 260 | 1.55 | — | — | 411/435/461 | 0.11 | |
| DMF | 276 | 1.67 | 357 | 0.01 | — | — | |
| DMSO | 270 | 1.99 | — | — | — | — | |
| ACN | 283 | 1.18 | — | — | — | — | |
| i-PrOH | 275 | 0.47 | — | — | 408/433 | 0.04 | |
| EtOH | 262 | 1.02 | 334 | 0.01 | — | — | |
| MeOH | 281 | 1.52 | — | — | — | — | |
| Water | 269 | 0.28 | — | — | — | — | |
| C10 | Toluene | 289 | 1.61 | 341/433 | 0.01 | 410/435/458 | 0.20 |
| Diethyl ether | 262 | 0.84 | 335/416 | 0.01 | 403/428/446 | 0.16 | |
| THF | 262 | 1.43 | — | — | 408/431/460 | 0.16 | |
| Ethyl acetate | 276 | 1.06 | 342/452 | 0.01 | 406/430/451 | 0.18 | |
| Chloroform | 278 | 2.26 | — | — | 438 | 0.02 | |
| DCM | 262 | 0.81 | 342/441 | 0.01 | 411/436 | 0.17 | |
| DMF | 276 | 1.33 | — | — | 413/436 | 0.02 | |
| DMSO | 270 | 1.63 | — | — | — | — | |
| ACN | 274 | 0.55 | — | — | 435 | 0.02 | |
| i-PrOH | 276 | 0.58 | — | — | 408/435 | 0.05 | |
| EtOH | 257 | 0.70 | 335 | 0.01 | — | — | |
| MeOH | 276 | 0.73 | 340 | 0.01 | — | — | |
| Water | 267 | 0.32 | — | — | — | — | |
| C11 | Toluene | 289 | 1.87 | — | — | 410/435/460 | 0.18 |
| Diethyl ether | 260 | 1.14 | — | — | 408/433/460 | 0.20 | |
| THF | 262 | 2.55 | 358 | 0.11 | 408/433/461 | 0.15 | |
| Ethyl acetate | 276 | 1.85 | 355 | 0.07 | 406/430/454 | 0.16 | |
| Chloroform | 278 | 2.49 | 352/363 | 0.02 | — | — | |
| DCM | 261/338 | 2.24 | 352/365 | 0.18 | 411/436/460 | 0.18 | |
| DMF | 290 | 1.82 | 357 | 0.03 | — | — | |
| DMSO | 289 | 2.05 | — | — | — | — | |
| ACN | 284/327 | 2.00 | 322/363 | 0.16 | — | — | |
| i-PrOH | 284 | 1.02 | 353/366 | 0.12 | — | — | |
| EtOH | 284 | 1.96 | 356/366 | 0.15 | — | — | |
| MeOH | 283/336 | 1.75 | 353/366 | 0.14 | — | — | |
| Water | 283/336 | 0.77 | 353/366 | 0.24 | — | — | |
| C12 | Toluene | 300 | 3.21 | 359 | 0.12 | 410/435/458 | 0.20 |
| Diethyl ether | 260 | 2.38 | 355 | 0.14 | 403/428/451 | 0.20 | |
| THF | 259 | 3.26 | 356 | 0.08 | 411/436/463 | 0.13 | |
| Ethyl acetate | 275 | 2.39 | 355 | 0.11 | 410/435/463 | 0.14 | |
| Chloroform | 278 | 4.13 | 358 | 0.09 | — | — | |
| DCM | 260 | 3.15 | 357 | 0.13 | 413/440/465 | 0.13 | |
| DMF | 290 | 3.92 | 359 | 0.08 | — | — | |
| DMSO | 286 | 3.45 | 361 | 0.01 | — | — | |
| ACN | 250/279 | 2.29 | 357 | 0.09 | — | — | |
| i-PrOH | 248/280 | 1.93 | 355 | 0.18 | 411/438 | 0.06 | |
| EtOH | 249/283 | 2.34 | 356 | 0.13 | — | — | |
| MeOH | 281 | 3.18 | 358 | 0.06 | — | — | |
| Water | 267 | 0.45 | 361 | 0.05 | — | — | |
| C13 | Toluene | 289 | 1.41 | 359 | 0.01 | 411/435 | 0.05 |
| Diethyl ether | 261 | 0.79 | 355/403/426 | 0.02 | 405/428/451 | 0.08 | |
| THF | 262 | 2.15 | 356/408/433 | 0.02 | 408/433/455 | 0.13 | |
| Ethyl acetate | 276 | 1.30 | 355 | 0.02 | 405/431/455 | 0.01 | |
| Chloroform | 281 | 2.33 | — | — | — | — | |
| DCM | 262 | 2.06 | 355/411/436 | 0.02 | 411/436/463 | 0.12 | |
| DMF | 290 | 1.86 | 358 | 0.02 | 411/436/461 | 0.02 | |
| DMSO | 290 | 1.85 | — | — | — | — | |
| ACN | 281 | 1.18 | 356 | 0.01 | 408/433 | 0.01 | |
| i-PrOH | 283 | 0.85 | 354 | 0.01 | 410/433/451 | 0.04 | |
| EtOH | 282 | 1.60 | 355 | 0.01 | — | — | |
| MeOH | 285 | 1.63 | 355 | 0.01 | — | — | |
| Water | 282 | 1.29 | 355/365 | 0.04 | — | — | |
| C14 | Toluene | 289 | 1.32 | 346 | 0.01 | 410/433/463 | 0.06 |
| Diethyl ether | 262 | 0.59 | 353 | 0.02 | 403/428/451 | 0.11 | |
| THF | 260 | 1.89 | 357 | 0.05 | 408/433/460 | 0.18 | |
| Ethyl acetate | 281 | 0.98 | 353 | 0.01 | 405/430 | 0.02 | |
| Chloroform | 281 | 2.05 | 352/365 | 0.01 | — | — | |
| DCM | 262 | 1.46 | 353 | 0.11 | 411/436/461 | 0.21 | |
| DMF | 288 | 2.00 | 357 | 0.02 | 415/435/466 | 0.02 | |
| DMSO | 288 | 1.71 | — | — | — | — | |
| ACN | 283 | 1.15 | 355 | 0.19 | — | — | |
| i-PrOH | 280 | 0.52 | 355/366 | 0.03 | 411/433 | 0.06 | |
| EtOH | 285 | 0.91 | 355 | 0.09 | — | — | |
| MeOH | 284 | 1.29 | 353 | 0.17 | — | — | |
| Water | 267 | 0.29 | 353 | 0.02 | — | — | |
| C15 | Toluene | 299 | 3.24 | 359 | 0.12 | 410/435/454 | 0.07 |
| Diethyl ether | 253 | 1.94 | 351 | 0.13 | 403/426/454 | 0.12 | |
| THF | 251 | 2.80 | 358 | 0.07 | 408/433/458 | 0.05 | |
| Ethyl acetate | 276 | 2.41 | 352 | 0.09 | 406/428 | 0.02 | |
| Chloroform | 278 | 4.46 | 352 | 0.05 | — | — | |
| DCM | 253 | 2.40 | 358 | 0.14 | 411/436 | 0.05 | |
| DMF | 289 | 2.80 | 356 | 0.06 | — | — | |
| DMSO | 287 | 3.16 | 358 | 0.01 | — | — | |
| ACN | 249/277 | 2.01 | 355 | 0.10 | — | — | |
| i-PrOH | 250/278 | 2.08 | 354 | 0.09 | — | — | |
| EtOH | 250/280 | 2.36 | 354 | 0.08 | — | — | |
| MeOH | 283 | 2.46 | 354 | 0.05 | — | — | |
| Water | 248/278 | 1.21 | 381 | 0.03 | — | — | |
| C16 | Toluene | 296 | 2.56 | 357 | 0.10 | 410/435/458 | 0.12 |
| Diethyl ether | 260 | 2.04 | 354 | 0.09 | 403/428/450 | 0.17 | |
| THF | 260 | 1.96 | 355 | 0.07 | 408/433/455 | 0.07 | |
| Ethyl acetate | 278 | 1.80 | 354 | 0.07 | 406/430 | 0.09 | |
| Chloroform | 281 | 3.41 | 355 | 0.05 | — | — | |
| DCM | 260 | 2.82 | 357 | 0.10 | 411/436/463 | 0.06 | |
| DMF | 290 | 3.18 | 357 | 0.04 | — | — | |
| DMSO | 285 | 3.53 | 360 | 0.01 | — | — | |
| ACN | 248/278 | 2.45 | 355 | 0.04 | — | — | |
| i-PrOH | 248/278 | 1.73 | 355 | 0.07 | — | — | |
| EtOH | 251/281 | 1.87 | 354 | 0.07 | — | — | |
| MeOH | 283 | 3.36 | 356 | 0.03 | — | — | |
| Water | 257 | 1.24 | 365 | 0.03 | — | — | |
| C17 | Toluene | 294 | 1.35 | 341/411/433 | 0.01 | 410/435/458 | 0.12 |
| Diethyl ether | 262 | 0.74 | 341/405/426/454 | 0.03 | 403/428/450 | 0.17 | |
| THF | 260 | 1.33 | 354/406/431 | 0.01 | 408/431/457 | 0.07 | |
| Ethyl acetate | 276 | 1.09 | 354/406/428 | 0.01 | 406/430/452 | 0.09 | |
| Chloroform | 278 | 2.59 | 358/366 | 0.01 | — | — | |
| DCM | 264 | 0.95 | 363/411/435 | 0.02 | 411/436/460 | 0.12 | |
| DMF | 285 | 1.67 | 363 | 0.01 | — | — | |
| DMSO | 280 | 1.42 | — | — | — | — | |
| ACN | 272 | 0.73 | 363 | 0.01 | — | — | |
| i-PrOH | 276 | 0.57 | 358 | 0.02 | — | — | |
| EtOH | 262 | 0.85 | 358 | 0.02 | — | — | |
| MeOH | 285 | 2.04 | 359 | 0.02 | — | — | |
| Water | 283 | 0.84 | 363 | 0.02 | — | — | |
| C18 | Toluene | 289 | 1.43 | 410/433 | 0.02 | 410/435/457 | 0.12 |
| Diethyl ether | 260 | 0.94 | 403/428/448 | 0.04 | 403/428/446 | 0.31 | |
| THF | 262 | 1.47 | 359/451 | 0.12 | 458 | 0.17 | |
| Ethyl acetate | 281 | 0.86 | 406 | 0.06 | 406/428/455 | 0.12 | |
| Chloroform | 281 | 2.26 | 394 | 0.02 | 396 | 0.04 | |
| DCM | 260/343 | 2.24 | 455 | 0.28 | 457 | 0.26 | |
| DMF | 289 | 3.26 | 391/455 | 0.12 | 463 | 0.13 | |
| DMSO | 292 | 3.82 | 401 | 0.10 | 471 | 0.04 | |
| ACN | 281/341 | 2.19 | 468 | 0.19 | 465 | 0.21 | |
| i-PrOH | 276 | 0.63 | 357/439 | 0.02 | 436/452 | 0.05 | |
| EtOH | 257 | 1.00 | 455 | 0.11 | 560 | 0.19 | |
| MeOH | 281/341 | 2.11 | 465 | 0.19 | 466 | 0.14 | |
| Water | 264 | 0.36 | — | — | — | — | |
| C19 | Toluene | 299 | 6.81 | 360 | 0.10 | 410/433/458 | 0.13 |
| Diethyl ether | 283 | 5.33 | 342/355 | 0.09 | 401/425 | 0.07 | |
| THF | 262/291 | 5.99 | 358 | 0.09 | 408/431 | 0.09 | |
| Ethyl acetate | 281 | 5.46 | 358 | 0.10 | 406/428 | 0.11 | |
| Chloroform | 281 | 7.22 | 374 | 0.08 | 401 | 0.03 | |
| DCM | 264/288 | 6.83 | 375 | 0.09 | 411/436 | 0.08 | |
| DMF | 288 | 6.46 | 392 | 0.10 | 431 | 0.04 | |
| DMSO | 285 | 7.00 | 405 | 0.10 | 418/433 | 0.03 | |
| ACN | 250/284 | 5.10 | 393 | 0.12 | 440 | 0.04 | |
| i-PrOH | 251/285 | 5.38 | 390 | 0.11 | 415/430 | 0.06 | |
| EtOH | 251/285 | 5.72 | 397 | 0.13 | 433 | 0.04 | |
| MeOH | 281 | 5.66 | 411 | 0.03 | 458 | 0.03 | |
| Water | 269 | 1.04 | 451 | 0.03 | — | — | |
| C20 | Toluene | 297 | 6.01 | 360 | 0.11 | 408/433/455 | 0.13 |
| Diethyl ether | 281 | 5.18 | 354 | 0.08 | 401/426/448 | 0.07 | |
| THF | 260/291 | 4.98 | 361 | 0.09 | 408/433 | 0.10 | |
| Ethyl acetate | 281 | 4.68 | 357 | 0.10 | 406/430/457 | 0.11 | |
| Chloroform | 281 | 6.81 | 370 | 0.08 | 397 | 0.03 | |
| DCM | 263/292 | 6.60 | 374 | 0.10 | 411/436 | 0.04 | |
| DMF | 292 | 6.59 | 394 | 0.09 | 446 | 0.02 | |
| DMSO | 290 | 7.07 | 405 | 0.12 | 410 | 0.01 | |
| ACN | 253/285 | 5.13 | 398 | 0.09 | 453 | 0.01 | |
| i-PrOH | 251/285 | 4.80 | 393 | 0.12 | 435 | 0.07 | |
| EtOH | 251/285 | 5.00 | 400 | 0.10 | 457 | 0.11 | |
| MeOH | 282/349 | 5.49 | 415 | 0.03 | 463 | 0.02 | |
| Water | 277 | 1.32 | 396 | 0.05 | 400 | 0.01 |
Additionally, in highly polar solvents, the electronic environment around the complex changes due to hydrogen bonds and dipole–dipole interactions. This change affects the energy levels of intra-ligand transitions and creates a broadening of the spectrum.44 No significant shifts (either blue or red) were observed in the fluorescence spectra of the compounds with respect to solvent polarity. However, when the substituent effect was considered in all complexes, it was observed that the fluorescence intensity was generally high in the presence of electron-withdrawing groups (EWGs), as shown by the fluorescence quantum yields (Table 2).
3.3. FTIR study
Vibrational analysis was conducted to identify the molecular modes contributing to the bands observed in the FTIR spectra. A thorough examination of the band shapes, positions, and intensities enabled accurate assignment of the vibrational modes. The FTIR spectra of the synthesized derivatives (C1–C20) exhibited distinct bands corresponding to aromatic and aliphatic C–H and C–O stretching vibrations, with absorption in the ranges of 1145–1020 cm−1 and 1370–1215 cm−1, respectively. Comparative spectral analysis confirmed successful coordination of the substituted TPY ligands with Zn(II), Co(II), Ni(II), Cu(II), and Fe(II). Evidence for chelation through the TPY ligands was observed by the shifts in specific ligand bands following complexation. Remarkably, the ν(C–N) stretching band, associated with nitrogen coordination, shifted to lower frequencies in the spectra of the metal complexes, confirming the coordination of the TPY nitrogen atoms. The band corresponding to the azomethine group, initially located at 1600–1580 cm−1 in the free ligands, shifted to lower frequencies (1520–1425 cm−1) upon complexation, indicating the involvement of the azomethine nitrogen in coordination. A characteristic band observed above 554 cm−1 further supported the coordination of TPY nitrogen atoms with the metal ions. Additionally, a new band in the range of 655–556 cm−1 was detected, confirming the formation of Zn–N, Co–N, Ni–N, Cu–N, and Fe–N bonds in the complexes. This detailed vibrational analysis offers valuable insights into the structural characteristics and coordination behavior of the synthesized metal complexes.3.4. Cyclic voltammetric analysis
The electrochemical behavior of TPY complexes (C1–C20) was investigated using cyclic voltammetry (CV) over a potential range of −2.0 to +1.0 V at room temperature. The experiments were conducted at scan rates of 40, 100 and 200 mV s−1 (Table 3 and Fig. 3a–e). To confirm solvent purity, an initial CV scan was conducted using CH3CN as the blank solution across the predetermined potential range (−2.0 to +1.0 V). The absence of any peaks in both the forward and reverse scans indicated a contaminant-free medium.| Compound no. | Epasa (V) | Epcsb (V) | ΔEc (V) | Ipad![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ipce (μA) :Ipce (μA) |
E½ (V) |
|---|---|---|---|---|---|
| a Epa = anodic peak potential.b Epc = cathodic peak potential.c ΔE = difference of Epa & Epc.d Ipa = anodic peak current.e Ipc = cathodic peak current. | |||||
| C1 | −0.5, −0.3, 0.7 | −0.8, −1.5 | 0.3 | 30, 20, 10 | −0.60 |
| 150, 200 | |||||
| C2 | −0.6, 0.4, 0.8 | −0.75, −1.4 | 0.15 | 20, 10, 50 | −0.52 |
| 150, 210 | |||||
| C3 | −0.35, —, 0.50 | −0.75, −1.6 | 0.4 | 40, —, 30 | −0.55 |
| 130 | |||||
| C4 | −0.30, —, 0.60 | −0.70, −1.5 | 0.4 | 25, —, 100 | −0.50 |
| 125, 240 | |||||
| C5 | −0.45, —, 0.70 | −0.80, −1.3 | 0.35 | 40, —, 30 | −0.62 |
| 130 | |||||
| C6 | −0.5, −0.15, 0.7 | −0.80, −1.4 | 0.30 | 30, 35, 80 | −0.65 |
| 210, 120 | |||||
| C7 | −0.3, —, 1.3 | −0.80 | 0.50 | 70, 180 | −0.55 |
| C8 | −0.5, −0.14, 0.30 | −0.7, −1.3 | 0.20 | 40, 30, 140 | −0.60 |
| 215, 130 | |||||
| C9 | −0.4, −0.3, 0.8 | −0.75, −1.2 | 0.35 | 20, 70, 90 | −0.57 |
| 210, 120 | |||||
| C10 | −0.4, —, 0.65 | −0.75, −1.6 | 0.35 | 40, —, 35 | −0.57 |
| 130 | |||||
| C11 | −0.35, 0.45, 0.6 | −0.75 | 0.4 | 33, 30, 35 | −0.55 |
| 130 | |||||
| C12 | −0.25, 0.50, 0.75 | −0.80, −1.5 | 0.55 | 33, 30, 35 | −0.52 |
| 130 | |||||
| C13 | −0.3, −0.2, 0.5 | −0.75, −1.4 | 0.45 | 30, 35, 80 | −0.52 |
| 210, 120 | |||||
| C14 | −0.5, −0.3, 0.6 | −0.70, −1.4 | 0.20 | 20, 30, 90 | −0.60 |
| 100, 180 | |||||
| C15 | −0.5, —, 0.65 | −0.75 | 0.25 | 40, 50 | −0.62 |
| 150 | |||||
| C16 | −0.5, 0.4, 0.7 | −0.80, −1.2 | 0.3 | 30, 50, 20 | −0.65 |
| 150 | |||||
| C17 | −0.30, —, 0.55 | −0.70, −1.6 | 0.4 | 40, —, 30 | −0.50 |
| 130 | |||||
| C18 | −0.3, 0.2, 0.65 | −0.80, −1.5 | 0.5 | 20, 10, 50 | −0.55 |
| 150, 210 | |||||
| C19 | −0.5, 0.3, 0.55 | −0.85, −1.4 | 0.35 | 25, 20, 10 | −0.67 |
| 90, 70 | |||||
| C20 | −0.3, 0.2, 0.65 | −0.80, −1.5 | 0.50 | 30, 15, 10 | −0.55 |
| 90, 110 | |||||
 | ||
| Fig. 3 Comparative cyclic voltammograms based on metal centers (Co, Ni, Fe, Zn and Cu) of all the synthesized TPY-complexes (C1–C20) recorded at a scan rate of 200 mV s−1. (a) CV of Co-based TPY complexes. (b) CV of Ni-based TPY complexes. (c) CV of Fe-based TPY complex. (d) CV of Zn-based TPY complexes. (e) CV of Cu-based TPY complexes. | ||
The redox properties of TPY-based metal complexes/organic dyes (C1–C20) were analyzed to assess their electron-transfer potential. Electrochemical analysis was performed using cyclic voltammetry (CV) with a 0.1 mM solution of the synthesized complexes. The experiments were conducted at a platinum (Pt) electrode in CH3CN, with tetrabutylammonium bromide (TBAB) as the supporting electrolyte at a concentration of 0.1 M. The scan rate applied during the measurements was 200 mV s−1. Additionally, the oxidation peaks at more positive potentials are indicative of metal-centered redox transitions, while the lower-potential reductions correspond to ligand-based processes, consistent with reported electrochemical studies of similar systems.
Fig. 3 illustrates three quasi-reversible oxidation peaks for the TPY complexes. The observation of one or two cathodic peaks suggests that the electron transfer process involves multiple electrons, typically more than two. Following this, two additional physically adsorbed, condensed films appear, each corresponding to an adsorption process at notably negative potentials.
Furthermore, peak currents (Ipc, Ipa) and their respective peak potentials (Epc, Epa) were evaluated based on specific parameters (as shown in Fig. 3). These include the anodic-to-cathodic peak current ratio (Ipa/Ipc) and the peak potential separation (ΔE = Epa − Epc), which are reported in Table 3. The data presented in Table 3 indicate that the redox behavior of the metal-based compounds follows quasi-reversible processes, as evidenced by potential separations greater than 0.3 V, with the Ipa/Ipc ratio generally below 1. Furthermore, the E1/2 values (eqn (2)) for both quasi-reversible and reversible reactions were determined using the provided equation and are shown in Table 3.
| E1/2 = Epa − ΔE/2 | (2) |
CV was used to evaluate the peak potential and current for the electrooxidation of various metal-based TPY complexes. By analyzing the relationship between Ip and v1/2, as well as lnIp and ln(v), insights were gained into the reversibility of the reactions and whether they are governed by adsorption or diffusion processes. The effect of scan rates on the electrooxidation of a 0.1 mM solution of TPY-based complexes was investigated at scan rates of 40, 100, and 200 mV s−1, as detailed in Table S3 of the ESI.† The quasi-reversible nature of the process was verified by the shift in redox peaks with varying scan rates. Electrooxidation was challenging for derivatives with EWGs but was facilitated by EDGs, aligning well with previously reported findings on similar derivatives.
The recurring oxidation signals are attributed to the ‘TPY’ unit and its specific substituents, whereas the reduction signals are linked to the terpyridyl connectivity. The peak potential positions are influenced by the surrounding environment, as evidenced by a signal at +0.5 V confirming the TPY unit. The data also indicate that the TPY derivatives undergo a multi-electron transfer process, highlighting their rich electroactivity, with multiple electroactive centers that make them intriguing scaffolds for further electrochemical characterization.
4. Structure–property relationship
The compounds C1–C20 exhibit a strong absorption band attributed to π–π* transitions within the conjugated framework, with maximum absorption (λmax) observed in the range of 235–590 nm. Furthermore, due to the presence of a similar central TPY core across all derivatives, the UV-Vis absorption spectra display comparable spectral shapes and profiles. The observed variations in absorption intensity and wavelength are primarily attributed to the effect of the 4-alkylphenyl substituents at the 4′-position of the 2,2′:6′′,2-TPY scaffold. The phenyl groups at the p-position of the TPY structure create a conjugated backbone that significantly contributes to photon absorption.The fluorescence properties of Zn, Co, Ni, Cu, and Fe-based TPY complexes (C1–C20) were investigated in different solvents. Fluorescence studies revealed that complexes incorporating EWGs exhibit enhanced fluorescence intensity and higher quantum yields compared to those with EDGs. This behavior is attributed to the ability of EWGs to stabilize the excited state, thereby improving the emission efficiency of the complexes.
Electrochemical properties were analyzed using CV, revealing that the complexes are electrochemically stable and display quasi-reversible behavior. The cathodic and anodic peak values for each molecule remained consistent across three repeated cycles at scan rates of 40, 100, and 200 mV s−1. The position and nature of substituents on the TPY scaffold were found to influence the redox behavior and current. Moreover, EDGs increase electron density on the terpyridine ligand, especially at the pyridine rings. This typically lowers the reduction potential (makes it easier to reduce) because the ligand's LUMO is stabilized. EDGs can also slightly destabilize the metal center in metal complexes by increasing electron density, affecting the oxidation potential. Enhanced donating ability generally leads to stronger π-backbonding interactions in metal complexes. Although varying substituents did not significantly alter the cathodic or anodic peak positions, increasing the scan rate resulted in an increased current magnitude, indicating a reversible electrochemical redox process. Furthermore, EWGs reduce electron density on the terpyridine ligand, making it more electrophilic. This increases the reduction potential (makes it harder to reduce) by destabilizing the LUMO. In metal complexes, EWGs can stabilize the metal center by withdrawing electron density, thereby increasing the oxidation potential. Enhanced withdrawing ability weakens π-backbonding interactions but can stabilize high oxidation states of the metal.
The oxidation peak potential of the synthesized scaffolds shifted to more positive values. EDGs on the p-position of the TPY moiety enhanced the redox activity, whereas EWGs reduced it. The study underscores the critical role of substituent type, position, and number on the aryl rings in modulating both the physicochemical and electrochemical properties of the derivatives (C1–C20) (Fig. 4).
 | ||
| Fig. 4 Structure–property relationship of the TPY-based metal complexes. | ||
5. DFT studies
5.1. Optimization
The synthesized compounds (C1, C4, C5, C6, C8, C9, C11, C16, C18, and C19) were optimized using the B3LYP/LanL2DZ level of theory, without imposing any symmetry requirements. Molecular structure significantly influences a substance's electronic and optical characteristics. As a result of structural optimization, all molecules possess twisted structures. All the investigated molecules (C1, C4, C5, C6, C8, C9, C11, C16, C18, and C19) undergo computational calculations to ensure their minimized potential energy surfaces. The optimized structures of all the molecules are shown in Fig. 5. | ||
| Fig. 5 Optimized structures of few targeted molecules C1, C4, C5, C6, C8, C9, C11, C16, C18, and C19. | ||
5.2. HOMO–LUMO analysis
HOMO & LUMO is a crucial analysis for investigating the hole-transporting materials, to facilitate the hole extraction at the interface and increase the VOC of the materials. The HOMO and LUMO energy bandgaps of the studied molecules were investigated from their ground-optimized structures. As shown in Table 4, the calculated HOMO values of all the synthesized molecules are −2.87, −2.53, −2.57, −3.00, −3.09, −3.11, −3.54, −3.18, −3.44, and −2.70 eV, and their LUMO values are −1.65, −1.90, −1.96, −1.72, −2.45, −1.97, −2.54, −2.12, −2.59, and −2.05 eV. The results have shown that our synthesized molecules have enough HOMO values that would be comparative, which would be enhanced the VOC in solar cells by facilitating hole extraction at the interface. As displayed in Fig. 6, the highest occupied molecular orbitals density is delocalized across the donor-based units whereas the lowest unoccupied molecular orbitals density is mainly distributed on the accepting fragments that make them un-symmetrical charge transport.| Molecules | EHOMO (eV) | ELUMO (eV) | Band gap (EHOMO − ELUMO) (eV) |
|---|---|---|---|
| C1 | −2.87 | −1.65 | 1.22 |
| C4 | −2.53 | −1.90 | 0.63 |
| C5 | −2.57 | −1.96 | 0.61 |
| C6 | −3.00 | −1.72 | 1.28 |
| C8 | −3.09 | −2.45 | 0.64 |
| C9 | −3.11 | −1.97 | 1.14 |
| C11 | −3.54 | −2.54 | 1.00 |
| C16 | −3.18 | −2.12 | 1.06 |
| C18 | −3.44 | −2.59 | 0.85 |
| C19 | −2.70 | −2.05 | 0.65 |
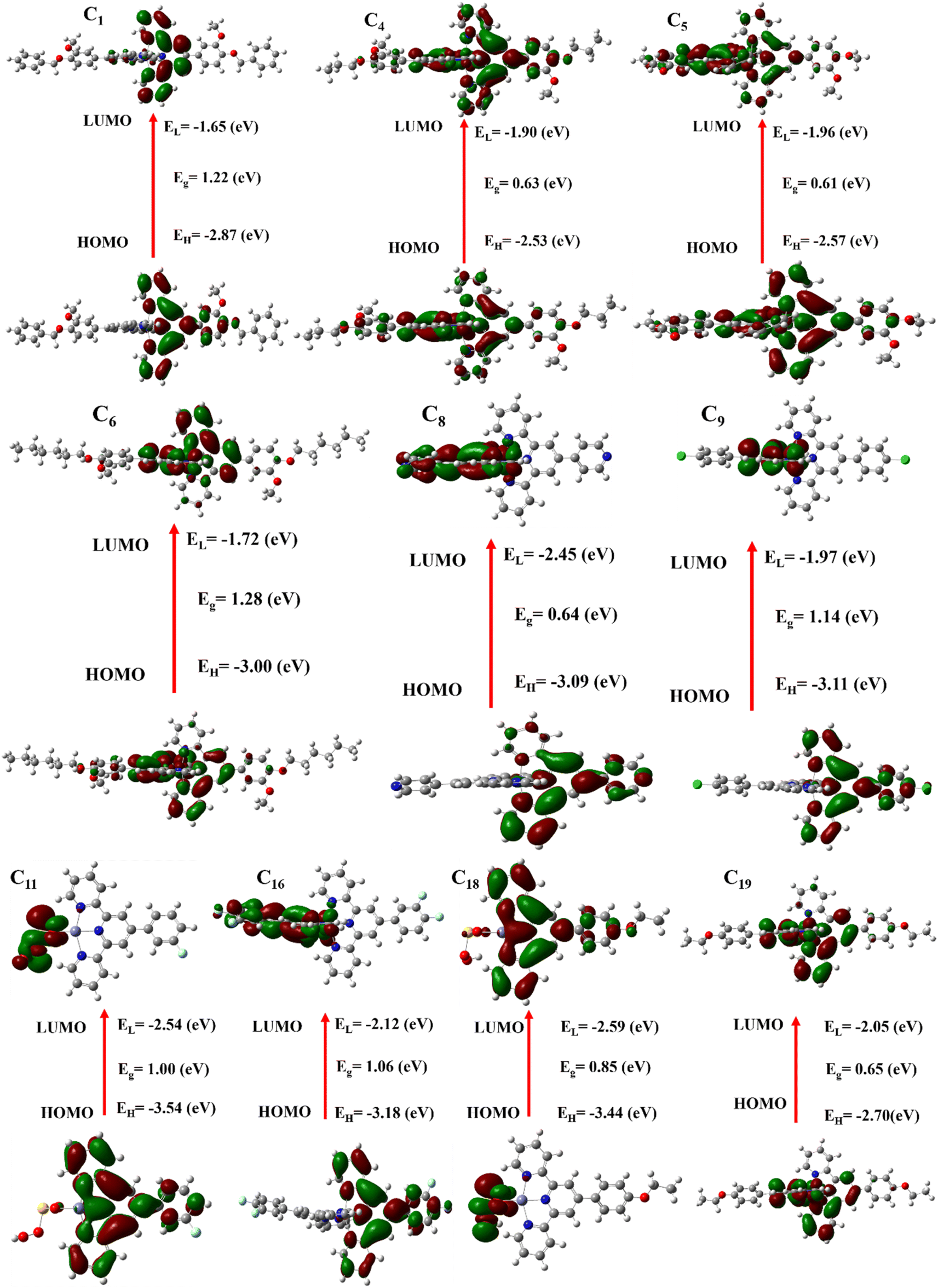 | ||
| Fig. 6 HOMO & LUMO energy values of few synthesized molecule with concerned energy gap. | ||
The energy levels of synthesized molecules are calculated using eqn (3) and their results are illustrated in Table 4. The energy levels of our synthesized molecules are 1.22, 0.63, 0.61, 1.28, 0.64, 1.14, 1.00, 1.06, 0.85, and 0.65 eV. The synthesized molecules possess a lower energy gap and their values are displayed in Table 4.
| Eg = EHOMO − ELUMO | (3) |
5.3. Physical parameters
Different chemical properties were calculated to assess the investigated molecules' kinetic stability and chemical reactivity, and the outcomes are displayed in Table S4 in ESI file.† Chemical potential (μ), found using eqn (4), illustrates the ability to escape an electronic cloud. The chemical potential calculations of the synthesized molecules (C1, C4, C5, C6, C8, C9, C11, C16, C18, and C19) are −2.26, −2.22, −2.27, −2.36, −2.77, −2.54, −3.04, −2.65, −3.015, and −2.38 eV. The synthesized molecules except C4 other molecules possess greater negative chemical potential than C4 with the highly reactive molecules.| μ = ½(EHOMO + ELUMO) | (4) |
The absolute hardness and softness describe the chemical reactivity and stability of the synthesized molecules, which would be better for device manufacturing. PCE will be enhanced by making designed molecules less rigid. The eqn (5) and (6) can be used to assess the softness and hardness, illustrated in Table S4 in ESI file.†
 | (5) |
| Hardness (η) = ½(ELUMO − EHOMO) | (6) |
Chemical hardness for our synthesized molecules is given, 0.61, 0.315, 0.305, 0.64, 0.32, 0.57, 0.5, 0.53, 0.425, and 0.325 eV with the order of hardness our synthesized molecules are C5 < C4 < C8 < C19 < C18 < C11 < C16 < C9 < C1 < C6 which represented that our synthesized molecules are comparatively stable and less rigid as well. The softness of the synthesized molecules is 1.646, 3.17, 3.28, 1.56, 3.13, 1.75, 2.00, 1.89, 2.35, and 3.08 but expressed in orders are C6 < C1 < C9 < C16 < C11 < C18 < C19 < C8 < C4 < C5 which represented that our synthesized molecules are reactive.
The electronegativity and electrophilicity index of the synthesized molecules were determined using eqn (7) and (8). Electronegativity serves as an indicator of the synthesized molecules' ability to attract electrons. By modulating electronegativity, it is possible to enhance the charge transfer capacity (eqn (9)) and reduce energy losses. Additionally, the electrophilicity index provides insight into the molecules' capacity to accept electrons; a higher electrophilicity index value facilitates more efficient electron transfer. Together, the electronegativity and electrophilicity index quantitatively characterize the electron-accepting capabilities of the synthesized molecules.
 | (7) |
 | (8) |
 | (9) |
5.4. Molecular electrostatic potential surfaces (MEP)
The MEP surface investigation gives highly sufficient information related to the reactivity of the molecules. Fig. 7 illustrates the MEP of the molecules in the studied molecules. The blue and red regions on the surface represent positive and negative charge densities, respectively. The negative charge density was redistributed near the electrophile associated with our molecules. Furthermore, the observed reduction in negative potential scattering in our synthesized compounds suggests the presence of diminished oxidation sites, which contributes to the enhanced chemical stability of the molecules. | ||
| Fig. 7 MEPs of our synthesized molecules. | ||
6. Conclusions
This study explores the synthesis, characterization, and detailed investigation of the photophysical and electrochemical properties of a series of Zn(II), Cu(II), Fe(II), Co(II), and Ni(II) complexes (C1–C20) synthesized from a range of substituted 2,2′:6′,2′′-TPY ligands (L1–L11). This study uniquely integrates photophysical and electrochemical analyses with computational investigations, providing a holistic understanding of their properties within a single protocol. Such an approach not only highlights the unique characteristics of these complexes but also underscores their potential as versatile candidates for advanced materials and functional applications. EWGs were observed to enhance fluorescence intensities, as confirmed by quantum yield measurements. Under 300 nm excitation, fluorescence predominantly arose from ILCT, with Co(II), Zn(II), and Cu(II) complexes exhibiting pronounced fluorescence in polar solvents. Among these, complexes C12, C15, and C19 demonstrated consistent fluorescence across various solvents. In contrast, Zn(II) and Cu(II) complexes such as C16, C18, and C20 exhibited notable fluorescence in both polar and nonpolar environments. Excitation at 350 nm shifted the dominant process to MLCT, with Co(II), Ni(II), and Fe(II) complexes showing enhanced fluorescence primarily in nonpolar solvents.Electrochemical studies using CV highlighted the distinct redox behaviors of the complexes, influenced by both the metal centers and ligand substituents. Multiple oxidation and reduction peaks were observed, signifying intra-ligand and metal-centered charge transfer processes. Complexes C1, C2, C6, and C16 exhibited three distinct oxidation peaks with ΔEa values of approximately 0.30–0.35 V, indicative of efficient electron transfer dynamics. Complexes with higher ΔEa values, such as C12, C18, and C20 (∼0.50–0.55 V), demonstrated improved redox stability. Moderate ΔEa values (∼0.35–0.40 V) were observed for C3, C5, and C10, reflecting balanced redox processes. Furthermore, C11, C13, and C18 showed steady redox behavior with moderate ΔEa values and consistent current densities, while C6, C8, and C19 displayed superior electrochemical performance, characterized by reversible redox peaks and solvent-dependent potential shifts. Quasi-reversible redox behavior was a consistent feature across all complexes, underscoring their electroactive nature.
A relationship between photophysical and electrochemical properties and electronic features such as band gaps was established. Such compounds with narrower band gaps, for example, C5 (0.61 eV) and C8 (0.64 eV), showed very efficient electron transfer processes. The latter showed improved electrochemical reversibility, in addition to fluorescence emission that is more appreciable, primarily in non-polar solvents. These compounds also exhibited a high charge transfer capacity, as evidenced by their respective ΔNmax values of 7.43 and 8.66. Particularly, complexes with large band gaps, such as C1 (1.22 eV) and C11 (1.00 eV), were stable at redox states but only medium fluorescence intensities, indicating good electronic stability. The softness values for complexes like C4, C5, and C8 are increased; this leads to better redox performance. On the other hand, compounds having a higher hardness value, like C6 with η = 0.64 eV, displayed more stable redox behavior with larger potential ranges.
Detailed photophysical and electrochemical studies have identified C5, C6, and C8 as the most promising candidates for optoelectronic applications and catalytic processes because of their excellent electronic, redox, and photophysical properties. This paper discusses the potential of metal complexes derived from TPY as flexible materials in the field of redox-active compounds with variable photophysical properties, which advances material science and related applications.
Data availability
All the data is provided in the manuscript and ESI file.†Author contributions
Nafeesa Naeem: experimental work performance, data analysis and collection, first-draft preparation; software, CV analysis; Ehsan Ullah Mughal: supervision, main idea, reviewing and editing, final writing the manuscript; Ebru Bozkurt: performed photophysical and solvatochromic studies; Ishtiaq Ahmed: performed NMR and mass analyses; Muhammad Naveed Zafar: formal analysis; Ali Raza Ayub: DFT studies; Amina Sadiq: formal analysis; Abdulrahman A. Alsimaree: formal analysis, reviewing the final manuscript; Reem I. Alsantali: formal analysis, reviewing the final manuscript; Saleh A. Ahmed: formal analysis, reviewing and editing the final manuscript.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors extend their appreciation to Umm Al-Qura University, Saudi Arabia for funding this research work through grant number: 25UQU4320545GSSR04. This research work was funded by Umm Al-Qura University, Saudi Arabia under grant number: 25UQU4320545GSSR04.References
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 Search PubMed.
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS.
- B. Z. Momeni, N. Davarzani, J. Janczak, N. Ma and A. S. Abd-El-Aziz, Coord. Chem. Rev., 2024, 506, 215619 CAS.
- E. U. Mughal, S. F. Kainat, N. Naeem, M. Imran, A. Javaid, A. Sadiq, A. Y. A. Alzahrani, S. B. Moussa and S. A. Ahmed, Dyes Pigm., 2024, 112254 CAS.
- O. S. Taniya, D. S. Kopchuk, A. F. Khasanov, I. S. Kovalev, S. Santra, G. V. Zyryanov, A. Majee, V. N. Charushin and O. N. Chupakhin, Coord. Chem. Rev., 2021, 442, 213980 CAS.
- E. U. Mughal, R. J. Obaid, A. Sadiq, M. A. Alsharif, N. Naeem, S. Kausar, A. A. Altaf, R. S. Jassas, S. Ahmed and R. I. Alsantali, Dyes Pigm., 2022, 201, 110248 CAS.
- L. Zhu, M. Bai, S. Xiao, Y. Liu, Q. Zhu, Z. Wang, J. Zhao, W. Zhang and D. Chen, Talanta, 2024, 279, 126559 CAS.
- J. Shi, S. Yin, H. Li, X. Yi, H. Wu, T. Cao, X. Fan, J. Liu and F. Zhou, Carbon, 2025, 120089 CAS.
- N. E. Guesmi, J. Fluoresc., 2023, 33, 2315–2326 CAS.
- Z. Zhang, M. Zhang, X. Li, K. Li, X. Lu, Y. Wang and X. Zhu, ACS Sustain. Chem. Eng., 2018, 6, 11614–11623 CAS.
- A. Szlapa-Kula, M. Małecka and B. Machura, Dyes Pigm., 2020, 180, 108480 Search PubMed.
- Y. Shi, L. Chen, L. Xiong, X. Wang, Y. Yu and M. Yang, Chem. Eng. J., 2025, 160393 CAS.
- C. Bhaumik, D. Maity, S. Das and S. Baitalik, RSC Adv., 2012, 2, 2581–2594 CAS.
- S. Vaidya, C. Johnson, X.-Y. Wang and R. H. Schmehl, J. Photochem. Photobiol., A, 2007, 187, 258–262 Search PubMed.
- X. Chen, Q. Zhou, Y. Cheng, Y. Geng, D. Ma, Z. Xie and L. Wang, J. Lumin., 2007, 126, 81–90 Search PubMed.
- P. Pal, T. Ganguly, A. Sahoo and S. Baitalik, Inorg. Chem., 2021, 60, 4869–4882 Search PubMed.
- S. R. Barzegar Kiadehi and H. Golchoubian, Struct. Chem., 2024, 1–14 Search PubMed.
- Y. Zhang, Z. Liu, J. Wang, H. Du, Q. Sun, R. Gao and Z. Xu, Waste Manage., 2025, 198, 95–105 CrossRef CAS PubMed.
- J. Palion-Gazda, A. Kwiecień, K. Choroba, M. Penkala, K. Erfurt and B. Machura, Inorg. Chem., 2024, 29(13), 3074–3094 CAS.
- X. Tian, J. Li, C. Gu, G. Yin, Y. Zhang, J. Zhao and X. Cao, New J. Chem., 2023, 47, 4245–4251 RSC.
- N. An, T. Chen, J. Zhang, G. Wang, M. Yan and S. Yang, Small Methods, 2024, 8, 2300910 CrossRef CAS PubMed.
- S. Bhattacharya, P. Pal and S. Baitalik, Dalton Trans., 2024, 53, 1307–1321 RSC.
- D. Cai, Z. Yang, R. Tong, H. Huang, C. Zhang and Y. Xia, Small, 2024, 20, 2305778 Search PubMed.
- T. Klemens, A. Świtlicka-Olszewska, B. Machura, M. Grucela, E. Schab-Balcerzak, K. Smolarek, S. Mackowski, A. Szlapa, S. Kula and S. Krompiec, Dalton Trans., 2016, 45, 1746–1762 RSC.
- D. Toledo, F. Brovelli, J. Soto-Delgado, O. Pena, J.-Y. Pivan and Y. Moreno, J. Mol. Struct., 2018, 1153, 282–291 Search PubMed.
- B. Samrudhi, A. Narayanan, D. Devadiga, A. Idrissi, S. Bouzakraoui, M. Padaki and T. Ahipa, J. Mol. Liq., 2024, 125259 CrossRef CAS.
- A. Maroń, S. Kula, A. Szlapa-Kula, A. Świtlicka, B. Machura, S. Krompiec, J. G. Małecki, R. Kruszyński, A. Chrobok and E. Schab-Balcerzak, Eur. J. Org Chem., 2017, 2017, 2730–2745 CrossRef.
- J. Klein, A. Stuckmann, S. Sobottka, L. Suntrup, M. van der Meer, P. Hommes, H. U. Reissig and B. Sarkar, Chem.–Eur. J., 2017, 23, 12314–12325 CrossRef CAS PubMed.
- Sonia, S. Khatua, S. Samanta and B. N. Mongal, Polycyclic Aromat. Compd., 2024, 44, 3408–3418 Search PubMed.
- T. Ezhilarasu and S. Balasubramanian, ChemistrySelect, 2018, 3, 12039–12049 Search PubMed.
- N. Mateyise, M. Conradie and J. Conradie, Polyhedron, 2024, 117075 Search PubMed.
- X. Hao, W. Song, Y. Wang, J. Qin and Z. Jiang, Small, 2025, 21, 2408624 CrossRef PubMed.
- E. U. Mughal, M. Mirzaei, A. Sadiq, S. Fatima, A. Naseem, N. Naeem, N. Fatima, S. Kausar, A. A. Altaf and M. N. Zafar, R. Soc. Open Sci., 2020, 7, 201208 Search PubMed.
- E. Bozkurt, ChemistrySelect, 2022, 7, e202203777 CAS.
- E. U. Mughal, S. F. Kainat, N. Naeem, M. Imran, A. Javaid, A. Y. A. Alzahrani, S. B. Moussa and S. A. Ahmed, Dyes Pigm., 2024, 228, 112254–112262 Search PubMed.
- B. A. Mohammed, R. O. Kareem, O. A. Hamad and H. Kebiroglu, S. Afr. J. Chem., 2024, 78, 85–94 CAS.
- S. Silvarajoo, U. M. Osman, K. H. Kamarudin, M. H. Razali, H. M. Yusoff, I. U. H. Bhat, M. Z. H. Rozaini and Y. Juahir, Data Brief, 2020, 32, 106299 Search PubMed.
- F. A. Muslikh, R. R. Samudra, B. Ma'arif, Z. S. Ulhaq, S. Hardjono and M. Agil, Borneo J. Pharm., 2022, 5, 357–366 Search PubMed.
- A. Ismail, H. Ezzat, A. Menazea and M. Ibrahim, J. Electron. Mater., 2021, 50, 4741–4751 Search PubMed.
- A. R. Guerroudj, E. U. Mughal, N. Naeem, A. Sadiq, J. H. Al-Fahemi, B. H. Asghar, N. Boukabcha, A. Chouaih and S. A. Ahmed, Spectrochim. Acta, Part A, 2024, 313, 124093 CrossRef PubMed.
- K. Tummalapalli, C. Vasavi, P. Munusami, M. Pathak and M. Balamurali, Int. J. Biol. Macromol., 2017, 95, 1254–1266 CrossRef CAS PubMed.
- Q. Wang, F. Xiong, F. Morlet-Savary, S. Li, Y. Li, J.-P. Fouassier and G. Yang, J. Photochem. Photobiol., A, 2008, 194, 230–237 CrossRef CAS.
- E. M. Johansson, M. Odelius, S. Plogmaker, M. Gorgoi, S. Svensson, H. Siegbahn and H. Rensmo, J. Phys. Chem. C, 2010, 114, 10314–10322 CrossRef CAS.
- B. N. Ghosh, F. Topić, P. K. Sahoo, P. Mal, J. Linnera, E. Kalenius, H. M. Tuononen and K. Rissanen, Dalton Trans., 2015, 44, 254–267 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01005e |
| This journal is © The Royal Society of Chemistry 2025 |