DOI:
10.1039/D5RA00962F
(Review Article)
RSC Adv., 2025,
15, 9676-9755
Recent advances in metal-free catalysts for the synthesis of N-heterocyclic frameworks focusing on 5- and 6-membered rings: a review
Received
9th February 2025
, Accepted 5th March 2025
First published on 31st March 2025
Abstract
The tremendous potential of transition metal-free multi-component reactions (MCR) in the synthesis of N-heterocyclic frameworks is examined in this review, offering a complete overview of this subject matter. The discussion on the mechanistic rationale of the reaction routes and intermediates provides profound insights into the underlying changes, encouraging deeper investigation into various molecular frameworks. This review serves as a doorway to study the practicality of exploiting these reactions for the efficient and uncomplicated synthesis of specific nitrogen heterocycles. Specifically, we reveal the potential of transition metal-free catalysts in this field. Because of their extensive scope and diversity, these reactions enable the synthesis of various heterocycles that contain nitrogen, which include 5-membered (carbazole, pyrimidines, and pyrroles) and 6-membered rings (piperidine, pyridine, quinoline, diazinane, pyrazine, quinoxaline, and 1,2,3-triazine). In addition, the compatibility of transition metal-free catalysts with various functional groups and substrates not only increases the synthetic value of these compounds but also broadens their relevance in the domains of medical chemistry, materials science, and other relevant areas of study. To motivate future study and development in this field, the successful examples described in this review highlight the potential of transition metal-free catalysts as powerful instruments for the quick and efficient synthesis of nitrogen heterocycles. In general, this review provides a thorough and insightful examination of transition metal-free catalysts, highlighting the relevance of these compounds in contemporary organic synthesis and their potential to revolutionize the field of nitrogen heterocycle synthesis.
 Hai Truong Nguyen | Hai Truong Nguyen is currently a Lecturer at the University of Science, Vietnam National University Ho Chi Minh City (VNUHCM-US). He received his PhD in Organic Chemistry from VNUHCM-US in 2023. His research is focused on developing new green catalysts, such as ILs, deep eutectic solvents, eutectogels, amorphous carbon, zeolite, graphene oxide and carbon materials, and the synthesis and evaluation of biological activities of heterocyclic compounds. He has published over 35 papers and 2 book chapters. |
 The Thai Nguyen | The Thai Nguyen was born in Binh Thuan province, Vietnam. He obtained his B.S. degree from Ho Chi Minh City University of Education (HCMUE) in 2015. Later, he received his M.S. degree from the University of Science, Vietnam National University–Ho Chi Minh City (VNUHCM-US) in 2019. He is currently pursuing his PhD degree at the University of Science, Vietnam National University–Ho Chi Minh City under the supervision of Assoc. Prof. Phuong Hoang Tran (VNUHCM-US) and Assoc. Prof. Cong Tien Nguyen (HCMUE). His current research interests focus on the synthesis of heterocyclic compounds with green catalysts. |
 Vinh Thanh Chau Doan | Doan Chau Thanh Vinh is currently a Teaching Assistant at the University of Science, Vietnam National University, Ho Chi Minh City, Vietnam (VNUHCM-US). He also received his Bachelor's degree with a Chemistry major from this university in 2022. Now, he is a PhD student in Organic Chemistry at VNUHCM-US. His research is focused on carbonaceous materials, which can be applied as catalysts for biomass conversion to valuable compounds and for the synthesis of furfural derivatives. He has published 5 papers. |
 Trinh Hao Nguyen | Trinh Hao Nguyen is a Researcher, working at the Faculty of Interdisciplinary Science, University of Science, Ho Chi Minh City, Vietnam. He also received his Bachelor's degree with a Chemistry major from this university in 2022. He is currently studying for his PhD degree under the supervision of Assoc. Prof. Phuong Hoang Tran (VNUHCM-US). His research is focused on green catalysts, including carbonaceous materials (amorphous carbon, graphite carbon, and carbon nanotube) and ionic liquids, and their application in the synthesis of furan derivatives from biomass and heterocyclic compounds under green reaction conditions. |
 Minh Hai Tran | Minh Hai Tran is a student at the Faculty of Chemistry, University of Science, Vietnam National University Ho Chi Minh City (VNUHCM-US). He is conducting research under the supervision of Dr Hai Truong Nguyen focusing on the synthesis and evaluation of biological activities of heterocyclic compounds, utilizing green catalysts such as graphene oxide and other carbon-based materials. |
1 Introduction
The study of heterocyclic compounds holds great significance within the realm of organic chemistry, and it has garnered considerable interest from the scientific community. The appeal of heterocyclic compounds stems from their pronounced electron-donating properties and robust coordination capabilities, making them highly bioactive.
Recent advancements in synthetic methodologies have provided rapid access to a variety of functionalized heterocyclic compounds, which are essential for medicinal chemists.1–3 This is due to the capacity to broaden the accessible drug-like chemical space and promote more effective drug development programs.4 As a result, medicinal chemists are now able to discover new drugs more quickly. Additionally, the discovery of dependable synthetic pathways capable of producing large amounts of the required molecule contributes to speeding up the drug development processes.5 During a drug discovery program, it is a common practice to utilize established synthetic methodologies. Nevertheless, the pharmaceutical industry has witnessed significant advantages as a result of the development of novel heterocyclic syntheses that enable alternative methods of bond formation. This review examines the application of recent advancements in C–H activation, photoredox chemistry, hydrogen-borrowing catalysis, MCR, regioselective and stereoselective synthesis, and other innovative methodologies for the generation of ring structures in actual project delivery.6 In addition, emphasis is placed on the significance and the value of collaborative efforts between the pharmaceutical industry and academic institutions in helping to shape the advancement of novel synthetic methodologies for the functionalization of heterocycles, garnering significant attention from the pharmaceutical sector.7
The creation of novel compounds and composites has received much research attention in heterocyclic chemistry, which is a significant and distinctive class among the applied areas of organic chemistry.8–10 Over the past two decades, these compounds have attracted increasing interest. They have helped in the creation of many different organic synthesis procedures and widely used in chemical sciences.
One of the most basic classes of organic compounds is known as N-heterocycles. These molecules contain nitrogen atoms in their ring structures. Their relevance stems from the fact that they are found in a broad variety of physiologically active compounds, medicines, agrochemicals, and functional materials.11 As a result of the wide range of chemical characteristics that N-heterocycles possess, they have been used as essential building blocks in organic synthesis processes. This makes it possible to create novel pharmaceuticals, catalysts, and electronic materials.12,13 Consequently, researchers have extensively investigated a wide range of synthetic techniques for the purpose of effectively constructing N-heterocycles, with particular emphasis on enhancing the reaction efficiency, selectivity, and preservation of the environment. Herein, we provide an overview of various compounds belonging to the N-heterocyclic class, encompassing both 5- and 6-ring systems synthesized using metal-free catalysts under green reaction conditions.
2. 5-Membered rings
2.1. Five-membered heterocycle containing one nitrogen atom
2.1.1. Pyrrolidine. Pyrrolidine is a saturated, five-membered heterocycle containing nitrogen, which plays an essential role in several biological and pharmacological applications.14 It functions as a structural core in several natural alkaloids, including nicotine, hygrine, and kakuol, which demonstrate a variety of biological actions. Pyrrolidine derivatives have been extensively used in medicinal chemistry owing their capacity to improve pharmacokinetic parameters and influence biological targets (such as nicotine, L-proline, and hygrine) (Scheme 1).15–18
 |
| | Scheme 1 Chemical structures of bioactive compounds containing a pyrrolidine framework. | |
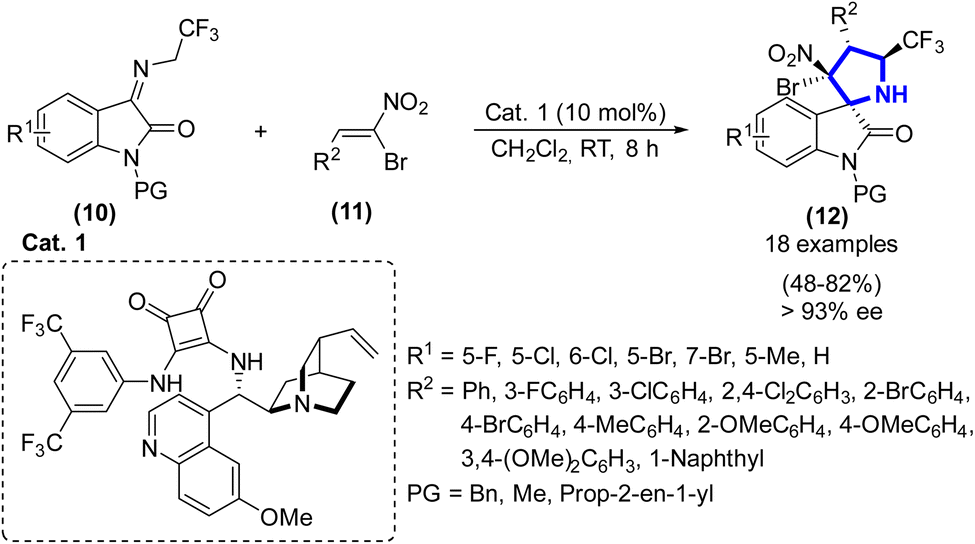
F.-Y. Chen et al. (2020) synthesized pyrrolidinyl spirooxindoles (3) using cinchonidine-derived squaramide (10 mol%) as the catalyst from isatin-derived ketamine and (Z)-α-bromonitroalkene in the presence of CH2Cl2 (2 mL) at r.t. in 8 h (Scheme 2).19 The researchers analyzed the unrefined reaction mixture using 1H NMR spectroscopy, resulting in the determination of the dr value, which exceeded 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The determination of the ee value was conducted using chiral HPLC analysis, specifically using a Chiralpak AD-H column with a mobile phase consisting of 20% 2-propanol and n-hexane at a flow rate of 1 mL min−1. A wavelength of 254 nm was used for the UV measurement. The analysis focused on the primary diastereoisomer, which exhibited an ee value exceeding 93%. Additionally, the efficacy of the catalyst was successfully exhibited in relation to the quantity of product in grams, resulting in a yield of 70%.
:1. The determination of the ee value was conducted using chiral HPLC analysis, specifically using a Chiralpak AD-H column with a mobile phase consisting of 20% 2-propanol and n-hexane at a flow rate of 1 mL min−1. A wavelength of 254 nm was used for the UV measurement. The analysis focused on the primary diastereoisomer, which exhibited an ee value exceeding 93%. Additionally, the efficacy of the catalyst was successfully exhibited in relation to the quantity of product in grams, resulting in a yield of 70%.
 |
| | Scheme 2 Synthesis of pyrrolidinyl spirooxindoles catalyzed by Cat. 1. | |
In the study by X. Zhang et al. (2019), they employed acetic acid as a catalyst to facilitate the synthesis of tetracyclic compounds with pyrrolidine rings. The synthesis was accomplished by exploiting oxazolidin-5-ones as the initial substrates and leveraging the 1,3-dipolar cycloaddition process of azomethine ylides (Scheme 3).20 A new and effective one-pot five-component method was described in this study for the selective synthesis of N-bridged bicyclic pyrrolidines with different types of side chains. Also, CO2 and H2O were produced as by-products, which were seen as the highlight of this reaction process.
 |
| | Scheme 3 Synthesis of pyrrolidine-containing tetracyclic compounds catalyzed by AcOH/EtOH. | |
In a recent study conducted by Z.-J. Wu et al. (2018), a novel organocatalyzed electrochemical approach was devised for the efficient synthesis of pyrrolidine frameworks. In this process, N-allyl amides are dehydrogenated, and then joined with 1,3-dicarbonyl compounds (Scheme 4).21 This method was demonstrated using an RVC-anode (100 PPI, 1 cm × 1 cm × 1.2 cm) and Pt-cathode at 7.5 mA (janode ∼0.1 mA cm−2), in 4.4 h under argon.
 |
| | Scheme 4 Synthesis of pyrrolidine and tetrahydropyridine derivatives catalyzed by Cat. 2. | |
In a recent study conducted by D. Kowalczyk et al. (2017), they investigated the methodology employed for synthesizing pyrrolidine derivatives that incorporate a benzofuran-3(2H)-one framework (Scheme 5).22 The [3 + 2]-cycloaddition process was conducted using 2-arylidene-benzofuran-3(2H)-ones and imines produced from salicylaldehyde and diethyl aminomalonates. The experiment was conducted with the inclusion of cinchona alkaloid quinine as the organocatalyst, with a loading of 10 mol%. The identification of the primary products was accomplished through 1H NMR analysis conducted on the unrefined reaction mixture. The er value of the primary products was determined using chiral stationary phase HPLC.
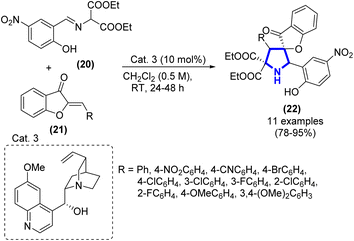 |
| | Scheme 5 Synthesis of pyrrolidine derivatives catalyzed by Cat. 3. | |
In a recent study conducted by Azomethine Ylides Z. Huang et al. (2017), they focused on the utilization of phosphine as a catalyst for the [3 + 2]-cycloaddition reaction between allenoates and o-hydroxyaryl. Their objective was to synthesize functionalized 4-methylenepyrrolidine derivatives using this catalytic process (Scheme 6).23 In this research, triphenylphosphine (20 mol%) acted as an efficient catalyst in the presence of dichloromethane solvent (2 mL), and a high yield of the major products was recorded of over 78%. Moreover, the reaction could be performed on a gram scale with 76% yield. Also, the functionalized 4-methylenepyrrolidines were used as precursors to synthesize other derivatives for pharmaceutical applications.
 |
| | Scheme 6 Synthesis of 4-methylenepyrrolidine derivatives catalyzed by Cat. 4. | |
The use of cyclic ethers as alkylation reagents for the N-alkylation of sulphonamides to synthesize pyrrolidine was reported by W. Shi et al. (2014) (Scheme 7).24 The reaction was conducted with trifluoromethanesulfonic acid (TfOH) as the catalyst and toluene as the solvent at 120 °C for 18–42 h. The predominant product yields were evidenced using 1H-NMR spectroscopy, exhibiting favorable to outstanding outcomes.
 |
| | Scheme 7 Synthesis of pyrrolidine catalyzed by TfOH. | |
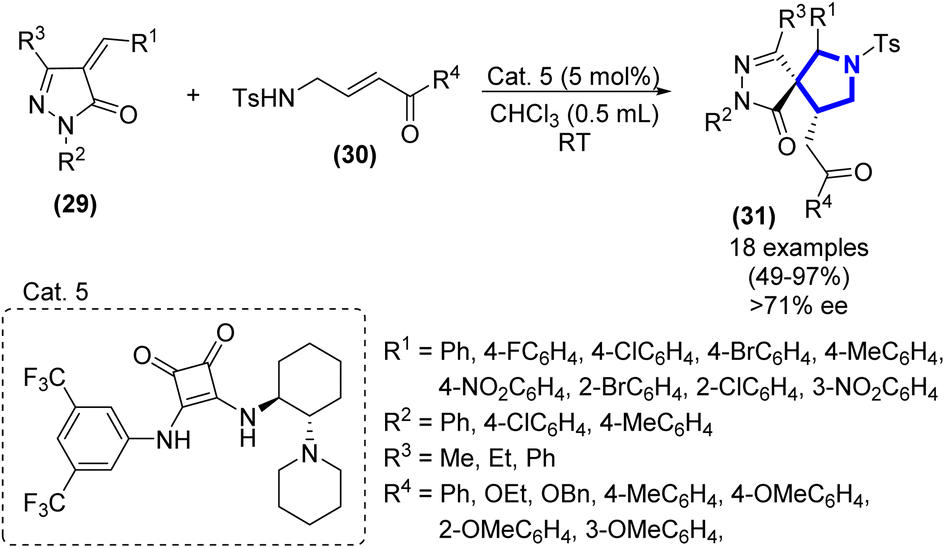
A study focusing on the diastereo- and enantio-selective synthesis of spiro-pyrrolidine-pyrazolones via the aza-Michael/Michael addition process was introduced by J.-H. Li et al. (2016) (Scheme 8).25 The process was performed between pyrazolone and tosyl aminomethyl enone with 5 mol% catalyst Cat. 5. The required product was achieved in good to exceptional yield (49–97%) when dichloromethane was utilized at r.t. together with excellent enantioselectivities (over 71% ee, based on chiral HPLC analysis). This method demonstrated efficacy during a gram-scale trial, which resulted in a yield of 85% of the starting material.
 |
| | Scheme 8 Synthesis of spiro-pyrrolidine-pyrazolones catalyzed by Cat. 5. | |
E. Da et al. (2014) published a method for the synthesis of 2,4-disubstituted pyrrolidines from N-allylic-substituted α-amino nitriles via intramolecular Michael reactions (Scheme 9).26 The 5-endo-trig cyclization reaction afforded the best results when carried out using chloroform as the solvent in the presence of Cat. 6 (5 mol%) at r.t. The investigation involved the examination of the reactions using both electron-donating and electron-withdrawing groups. The determination of significant product yields was accomplished through 1H NMR measurement, reaching up to 96%.
 |
| | Scheme 9 Synthesis of 2,4-disubstituted pyrrolidines catalyzed by Cat. 6. | |
G. Talavera et al. (2013) introduced the process of synthesizing α-amino-δ-keto esters or β-substituted δ-oxoamides from α,β-unsaturated aldehydes through the Michael reaction under iminium activation (Scheme 10).27 The process was demonstrated with a chiral secondary amine catalyst in toluene. In the second oxidation step of the process, the reaction was carried out in pyridinium chlorochromate (PCC) and dichloromethane at r.t. after 16 h. The cis/trans ratio was measured via NMR analysis, while the ee value was determined via HPLC analysis.
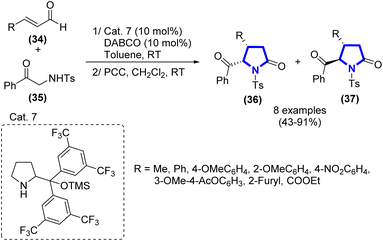 |
| | Scheme 10 Synthesis of α-amino-δ-keto esters or β-substituted δ-oxoamides catalyzed by Cat. 7. | |
C. Guo et al. (2013) invented a technique for the synthesis of pyrrolidine frameworks from β-ketoesters and benzylamine via a three-component reaction (Scheme 11).28 In the presence of Cat. 7 (10 mol%), a yield of pyrrolidine was obtained at 50 °C in 8 days. The principal chemicals were identified via HPLC and acquired with yields ranging from moderate to exceptional (up to 90%).
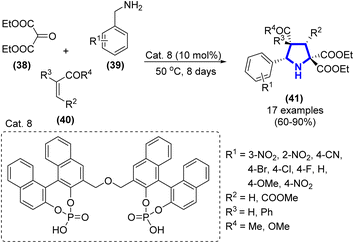 |
| | Scheme 11 Synthesis of pyrrolidine derivatives catalyzed by Cat. 8. | |
In the study conducted by E. Gomez-Torres et al. (2011), they reported the synthesis of pyrrolidine frameworks using the process of adding 1,3-diketones, β-ketoesters, and malonates to maleimide and N-substituted maleimides (Scheme 12).29 At r.t. and in toluene as the solvent, the application of organocatalyst Cat. 9 at a concentration of 10 mol% was shown to produce the best results. The pyrrolidines of interest were synthesized with satisfactory to outstanding efficiency. The catalytic activity of the chiral 2-aminoben-zimidazole-derived organocatalyst was evaluated on the gram scale.
 |
| | Scheme 12 Synthesis of pyrrolidines catalyzed by Cat. 9. | |
In a study conducted by Yu et al. (2010), they successfully demonstrated the application of Michael addition reaction for the conjugate addition of ketones to maleimides. This reaction was catalyzed by a bifunctional monosulfonyl DPEN salt (Scheme 13).30 The experiment was conducted using a catalyst, specifically 10 mol% of Cat. 10, and benzoic acid in the same concentration. The reaction took place at r.t. in a toluene solution with a volume of 0.5 mL. The reaction was permitted to continue for 48 h. The initial products were subjected to a screening process, which led to substantial yields (reaching up to 99%) and remarkable enantioselectivities (reaching up to 99%).
 |
| | Scheme 13 Synthesis of pyrrolidines catalyzed by Cat. 10. | |
Ankita Chaudhary et al. (2018) developed a catalyst-free three-component domino reaction characterized and applied for synthesis of pyrimidinetriones. The compounds were synthesized in aqueous medium with chemoselectivity. This technology is distinguished by its rapid reaction time, high yield, operational simplicity, and absence of column chromatographic purification (Scheme 14).31
 |
| | Scheme 14 Catalyst-free chemoselective synthesis of pyrrolidine derivatives. | |
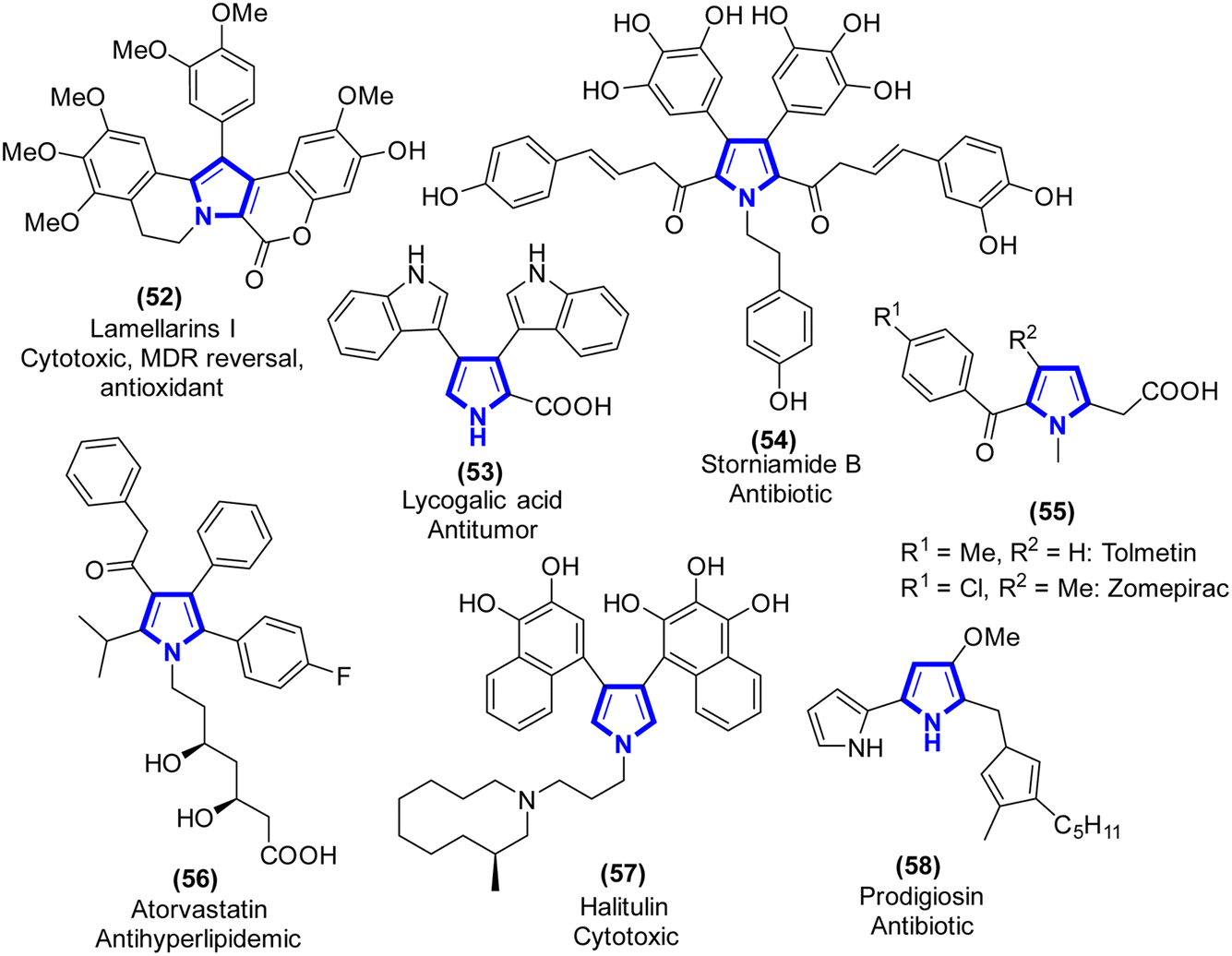
2.1.2. Pyrrole. Pyrrole is a substance belonging to a group of compounds called the heterocycles. Specifically, it is a heterocyclic aromatic organic molecule possessing a ring structure consisting of four carbon atoms and one nitrogen atom.32 Proline and hydroxyproline are examples of amino acids that have a pyrrole ring structure. In medicinal chemistry, one of the most pressing challenges is the development of new synthetic molecules having drug-like characteristics. The majority of pharmaceutically useful compounds have their origins in natural products, and these compounds are typically based on N-heterocyclic motifs. Many pharmacological features of commercially available medications with pyrrole rings are known, including antipsychotic,33 anticancer,34 antifungal,35 antiprotozoal,36 and antimalarial (Scheme 15).37
 |
| | Scheme 15 Chemical structures of bioactive compounds containing a pyrrole framework. | |
A synthetic technique for producing poly-substituted pyrroles from 1,3-dicarbonyl compounds and primary amines was disclosed by M. Xiong et al. (2021) (Scheme 16).38 The experiment was conducted using electro-oxidative intermolecular annulation. This was accomplished by employing a graphite rod as the anode and a platinum plate as the cathode, with a steady current of 6 mA. The reaction was performed in nBu4NBF4 and CF3SO3H, and a volume of 10 mL iPrOH and EtOH in a 4:1 ratio v/v was introduced in the reaction system. The reaction proceeded for a duration of 5 h at a temperature of 55 °C under argon. The major products were obtained in moderate yields via intermolecular cyclization.
 |
| | Scheme 16 Synthesis of poly-substituted pyrroles using electro-oxidative intermolecular annulation. | |
A study focusing on the synthesis of novel tricyclic pyrrolo[2,1-a]isoquinolines was published by M. Ghandi et al. (2021) (Scheme 17).39 The experimental procedure involved the utilization of ethanol in an intramolecular acid-catalyzed cyclization process. The duration of the reaction was 3 h. The target products were obtained in yields ranging from modest to excellent.
 |
| | Scheme 17 Synthesis of tricyclic pyrrolo[2,1-a]isoquinolines in ethanol. | |
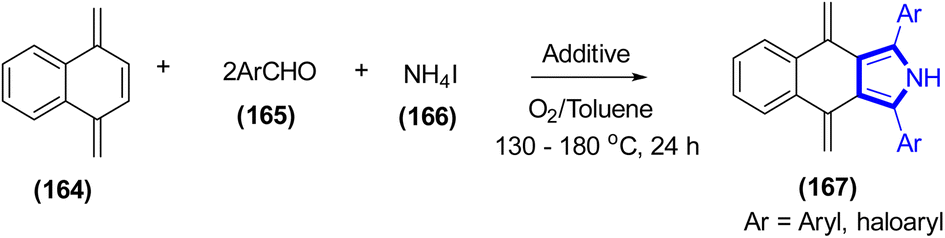
D. Chang et al. (2021) pioneered a novel approach for the preparation of multiaryl-substituted pyrrole frames from arylketones, amines, and nitrovinylarenes (Scheme 18).40 The experiment was conducted for a duration of 24 h at 160 °C, utilizing ammonium iodide as the catalyst and toluene as the solvent. The principal compounds were obtained in favorable to excellent yields (up to 94%). Also, 2,3,5-triaryl-substituted pyrroles were recorded in the absence of nitrovinylarenes. The yield of the reaction was about 77% under the optimal conditions.
 |
| | Scheme 18 Synthesis of multiaryl-substituted pyrrole catalyzed by ammonium iodide. | |
A novel procedure was developed by Q.-X. Zi et al. (2020) to show the preparation of 2-amino-4-coumarinyl-5-arylpyrrole derivatives (Scheme 19).41 The oxidation reaction of arylglyoxal monohydrates, 1,1-enediamines, and 4-hydroxy-2H-chromen-2-ones was conducted in ethanol under reflux conditions for a duration of 6 h. The primary products were synthesized in the absence of metal catalysts, resulting in yields varying from satisfactory to exceptional, exceeding 87%.
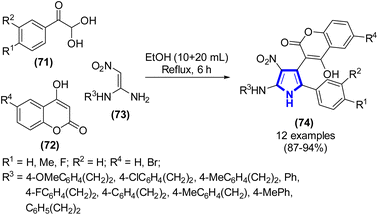 |
| | Scheme 19 Synthesis of 2-amino-4-coumarinyl-5-arylpyrroles in ethanol. | |
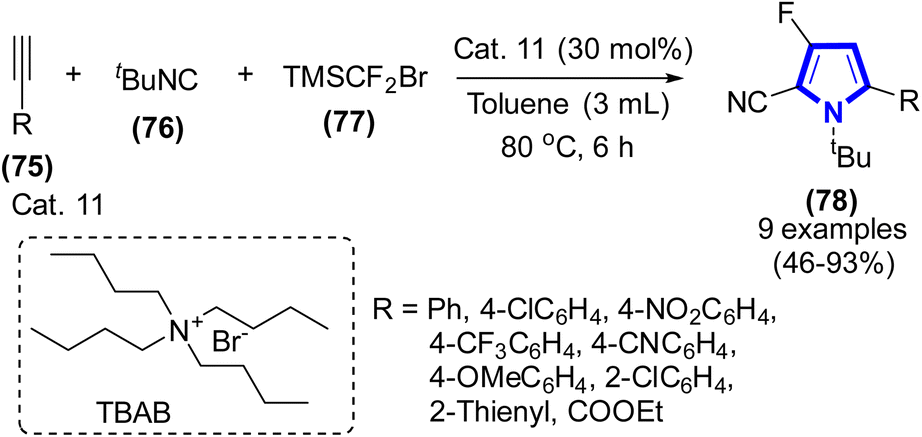
In the presence of TBAB (Cat. 11), the reaction of alkynes, 2-isocyano-2-methylpropane, and (bromodifluoromethyl)trimethyl-silane was stirred at 80 °C for 6 h to form mono-fluorinated pyrrole derivatives, which was demonstrated by R. Zhang et al. (2020) (Scheme 20).42 The mono-fluorinated pyrroles of interest were successfully synthesized by a [3 + 2]-cycloaddition reaction. The reaction proceeded under an N2 environment, with toluene serving as the solvent. The yields achieved ranged from moderate to good.
 |
| | Scheme 20 Synthesis of mono-fluorinated pyrroles catalyzed by Cat. 11. | |
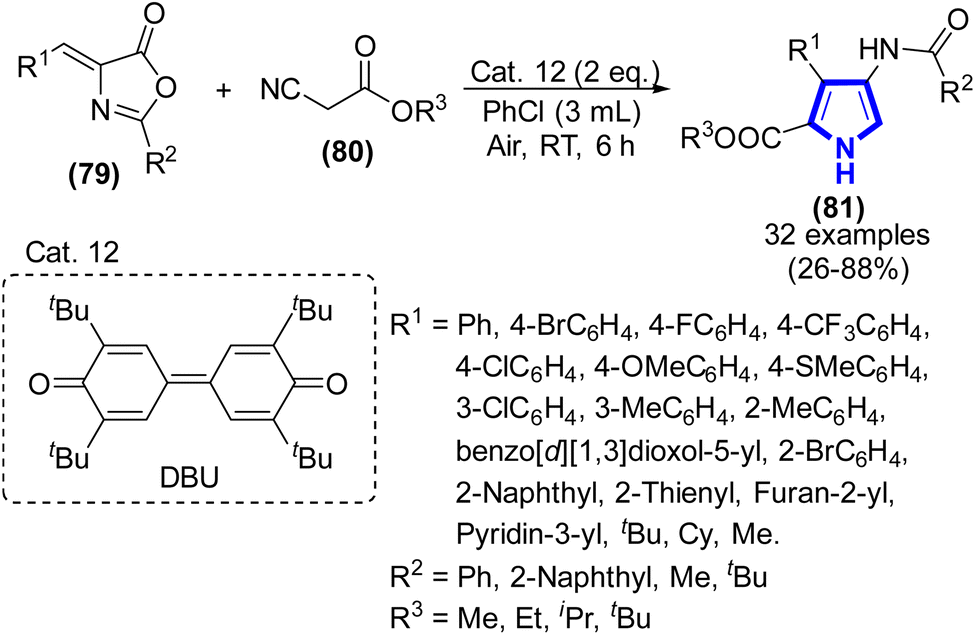
In the study by Zhang et al. (2020), they successfully synthesized 2,3,4-trisubstituted pyrrole frameworks via a [3 + 2]-reaction. The process of cyclization involving activated methylene isocyanides with 4-(arylidene) compounds was considered. The compounds referred to are oxazol-5(4H)-ones having a substituent at the 2-position (Scheme 21).43 The [3 + 2]-cyclization reaction occurred at r.t. to form 2,3,4-trisubstituted pyrroles in the presence of DBU (Cat. 12) and chlorobenzene for 6 h. The intended product could be achieved in yields ranging from 26% to 88%, depending on the substrate scope.
 |
| | Scheme 21 Preparation of 2,3,4-trisubstituted pyrroles catalyzed by Cat. 12. | |
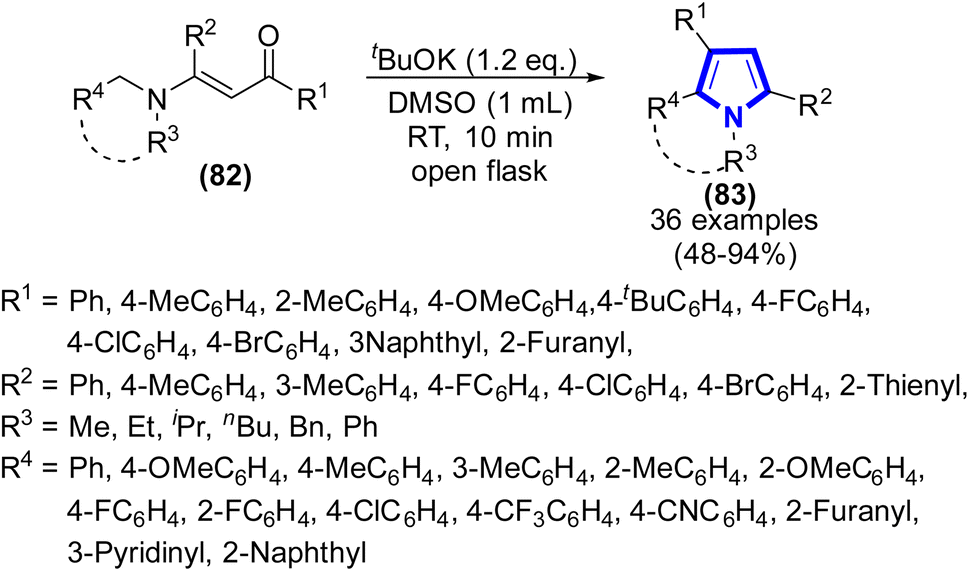
L. Xu et al. (2020) successfully devised a method utilizing a superbase catalyst to facilitate the N–α-sp3C–H functionalization of tertiary enaminones, enabling the synthesis of pyrroles with various substituents (Scheme 22).44 The experimental procedure was conducted at ambient temperature with the addition of tBuOK and DMSO. It was observed that this method was highly effective for the synthesis of substituted pyrrole derivatives. This study concentrated on evaluating the impact of various substituents on the substrate, resulting in a very favorable yield of the desired product, which accounted for about 48–94%.
 |
| | Scheme 22 Synthesis of substituted pyrroles catalyzed by tBuOK. | |
L. Wei et al. (2020) devised a methodology for the synthesis of functionalized pyrroles by a sequential process involving condensation and intramolecular cyclization (Scheme 23).45 This reaction process was employed to synthesize pyrrole frames from α-amino acid esters hydrochloride and alkynals in the presence of triethylamine and dichloromethane at ambient temperature for 24 h. To validate the efficacy of this approach, the reaction was conducted on a significant scale, utilizing a quantity of 10 mmol.
 |
| | Scheme 23 Synthesis of pyrroles catalyzed by triethylamine. | |
L. Su et al. (2020) presented a new method for synthesizing 5-amino-1H-pyrrole-2-carboxylates. This was achieved using the [3 + 2]-cycloaddition process involving benzylidenemalononitriles and ethyl glycinate hydrochloride, utilizing iodine and DMF as catalysts. The reaction was conducted at 120 °C and prolonged reaction times of 18–36 h (Scheme 24).46 The geometric structure of the target substance was determined through X-ray crystallographic analysis.
 |
| | Scheme 24 Synthesis of 5-amino-1H-pyrrole-2-carboxylates catalyzed by iodine. | |
In a recent publication by R. Shrestha et al. (2020), a novel and highly efficient one-pot methodology for the synthesis of polyfunctionalized pyrroles was elucidated (Scheme 25).47 The reaction was carried out by a series of chemical transformations, namely Knoevenagel condensation, intramolecular Michael addition, and ring opening, which occurred as a result of N-annulation of 3-formylchromones and α-amino ketones. This reaction was conducted in the presence of Cat. 13. The employed methodology shows notable benefits, including its ability to accommodate diverse functional groups and its utilization of moderate reaction circumstances. The reaction was conducted using acetonitrile as the solvent, resulting in a high yield of main products after a duration of 7 in the range of 52–85%.
 |
| | Scheme 25 Synthesis of polyfunctionalized pyrroles catalyzed by Cat. 13. | |
The preparation of pyrrole-3-carboxaldehydes from aldehydes, amines, and 1,4-ketoaldehydes through chemoselective Mannich reaction-cyclization, which was published by N. A. Mir et al. (2020) (Scheme 26).48 This reaction was performed in two steps, where a mixture of aldehydes and amines was reacted with each other in DMSO for 2 h, in the presence of L-proline (Cat. 14), and then 1,4-ketoaldehydes was added to form pyrrole-3-carboxaldehyde products for 24 h at r.t. The principal products were evaluated for their in vitro antibacterial and antifungal characteristics, with the most positive results noted at an MIC of 16 μg mL−1, particularly for bacterial strains.
 |
| | Scheme 26 Synthesis of pyrrole-3-carboxaldehydes catalyzed by Cat. 14. | |
Three-component domino reactions to synthesize multi-functionalized pyrroles were established by V. A. Mamedov et al. (2020) (Scheme 27).49 The experimental procedure involved the combination of 3-aroylquinoxalin-2-ones, malononitrile, secondary cyclic amine (or primary alcohols), and an alcohol solution in acetic acid. The mixture was subjected to heating for a duration of 7 h. The quinoxalinone-benzimidazolone rearrangement was employed to initiate the pathways, resulting in the formation of pyrrolecarbonitriles in significant yields. The yield reached about 25–83%, depending on the substrate scope.
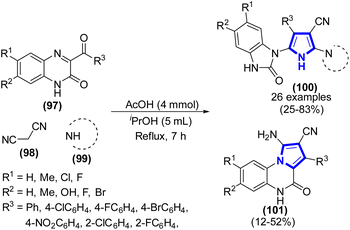 |
| | Scheme 27 Synthesis of pyrroles catalyzed by acetic acid. | |
In the recent study by H. Jia et al. (2020), they devised a highly effective approach for synthesizing substituted dihydropyrrole and pyrrole frameworks by the process of [3 + 2]-cyclization and subsequent aromatization (Scheme 28).50 The cyclization of β-chloro-vinyl dithianes with imines was carried out under the reaction conditions of an argon atmosphere using tBuOK and THF as the solvent at ambient temperature for a duration of 30 min. The pure products were acquired in satisfactory yields.
 |
| | Scheme 28 Synthesis of pyrrole scaffolds catalyzed by tBuOK. | |
Q.-W. Gui et al. (2020) discovered a sustainable method for the synthesis of multi-substituted pyrroles using a metal- and solvent-free approach (Scheme 29).51 This investigation involved the utilization of alkynes, TMSCN, and N,N-disubstituted formamides in the presence of iodine at a concentration of 20 mol%. Under the ideal reaction conditions, the major compounds were determined in good yields (up to 98%). Importantly, in the gram-scale preparation of pyrroles (under 8 products), the targeted compounds were separated in outstanding yields (64–98%).
 |
| | Scheme 29 Synthesis of multi-substituted pyrroles catalyzed by iodine. | |
A methodology for synthesizing poly-substituted pyrroles by the reaction of 1,2-disubstituted 2-imidazolines with alkynes was executed by Golantsov et al. (2020) (Scheme 30).52 The aza-Claisen rearrangement or/and transannular nucleophilic addition ring-opening reactions were performed in two steps to form the major products. During the initial stage, the reaction involved the combination of 2-imidazolines and alkynes in acetonitrile, which was maintained at ambient temperature for a duration of 4 h. Subsequently, poly-substituted pyrroles were synthesized in xylene medium.
 |
| | Scheme 30 Synthesis of polysubstituted pyrroles. | |
P. Gao et al. (2020) carried out a study on a metal-free methodology for the synthesis of 2,3-disubstituted pyrroles using N-hydroxyalkyl enamines (Scheme 31).53 The primary products were obtained with good selectivity through the conversion of N-hydroxyalkyl enamines via oxidative cyclization using Cat. 15 (IBX) in THF. The experiment was conducted at a temperature of 120 °C for a duration of 12 h.
 |
| | Scheme 31 Synthesis of 2,3-disubstituted pyrroles catalyzed by Cat. 15. | |
A novel approach for the preparation of 2,4-disubstituted pyrroles from isocyanides was reported by I. V. Efimov et al. (2020) (Scheme 32).54 The authors demonstrated the [3 + 2] cycloaddition reaction of isocyanides and enamines and/or enaminones to form 2,4-disubstituted pyrroles in the presence of tBuOK and DMF in air at 0 °C. The major products were obtained in moderate yields (up to 96%).
 |
| | Scheme 32 Synthesis of 2,4-disubstituted pyrroles catalyzed by tBuOK. | |
A study proposed an environmentally friendly method for the synthesis of chromenopyrrole frameworks. The reaction is facilitated by the presence of a piperidine catalyst, as described by E. Dhanasekar et al. (2020) (Scheme 33).55 The experimental procedure was conducted in an aqueous solution using a domino Knoevenagel–Michael addition–cyclization–rearrangement sequence. This approach exhibits several advantages, including a rapid reaction time, simple of operation, and high yield. The reaction was investigated on a gram scale with the objective of synthesizing the desired molecule in high yield.
 |
| | Scheme 33 Synthesis of chromenopyrrole catalyzed by Cat. 16. | |
The synthesis of iodopyrrole-3-carboxaldehydes using succinaldehyde and imines was investigated by S. Choudhary et al. (2020) using the aforementioned methods (Scheme 34).56 The straightforward method was demonstrated to form 4- or 5-iodopyrrole-3-carboxaldehydes using Cat. 14 in the presence of I2 through Mannich reaction-cyclization.
 |
| | Scheme 34 Synthesis of iodopyrrole-3-carboxaldehydes catalyzed by Cat. 14. | |
T. Yang et al. (2019) introduced a highly effective approach to produce polysubstituted pyrroles using α,β-unsaturated ynones and N-substituted ethyl glycine ethyl ester hydrochlorides (Scheme 35).57 Cat. 12 was employed as the catalyst in a two-step process for the synthesis of pyrrole frameworks. The initial procedure involved the reaction between a combination of α,β-unsaturated ynones and N-substituted ethyl glycine ethyl ester hydrochlorides. This reaction took place in the presence of Cat. 12 under ambient conditions for a duration of 12 h, followed by the removal of ethanol after the reaction. Subsequently, dimethyl sulfoxide (DMSO) was introduced in the solution, and the resulting mixture was subjected to stirring at 140 °C for a duration of 4 h. The primary products were acquired in favorable yields. This approach exhibits several advantages, including the utilization of a non-metallic catalyst and the employment of mild reaction conditions.
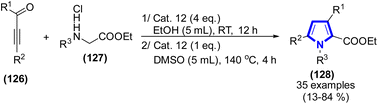 |
| | Scheme 35 Synthesis of polysubstituted pyrroles catalyzed by Cat. 12. | |
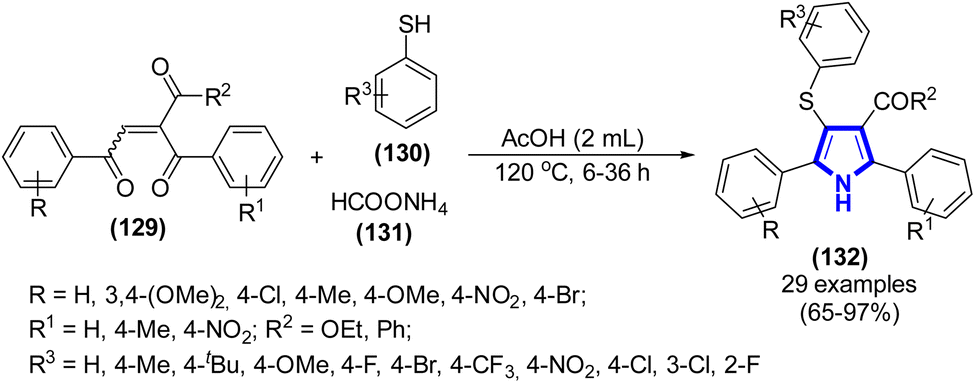
In the study by V. Rajeshkumar et al. (2019), they devised a novel methodology for the synthesis of 4-thioarylpyrroles using a one-pot three-component approach (Scheme 36).58 The experimental procedure involved the utilization of 1,4-enediones, thiols, and ammonium formate in a consecutive thiol-Michael/Paal–Knorr reaction for the purpose of synthesizing C–S and C–N bonds.
 |
| | Scheme 36 Synthesis of 4-thioarylpyrroles catalyzed by ammonium formate. | |
An organocatalytic technique for synthesizing functionalized pyrroles by the cycloisomerization of Z-1-iodo-4-N-methylbenzenesulfonyl-1,6-enynes was described by L. Meng et al. (2019) (Scheme 37).59 The chemical reactions were performed in the presence of organomolecule Cat. 17 and tBuOK for a period of 10 min.
 |
| | Scheme 37 Synthesis of functionalized pyrroles catalyzed by Cat. 17. | |
An effective procedure for the synthesis of tetrahydrocyclopenta[b]pyrroles was provided by J. A. Malone et al. (2019) (Scheme 38).60 The reaction was conducted using α′-hydroxy silylenol ether and a silylenol ether generated from acetophenone. This reaction was performed via the [2 + 2 + 1] annulation reaction mechanism, employing a Brønsted acid as a catalyst. Utilizing this method, they could successfully produce high yields of these biologically important N-heterocycles. The application of this method to access heterocycles and complex molecules that are structurally related is currently underway in the laboratory.
 |
| | Scheme 38 Synthesis of tetrahydrocyclopenta[b]pyrroles catalyzed by Py TfOH. | |
V. Kumar et al. (2019) described a unique one-pot method for the synthesis of 1,2,4-trisubstituted pyrroles (Scheme 39).61 This reaction proceeds via a chain reaction involving aziridines and β-bromo-β-nitrostyrenes. Initially, N-substituted-aziridine-2-carboxylates are produced using dibrominated-compounds and ethanolamine as the starting materials. Similarly, β-bromo-β-nitrostyrene can be derived from β-nitrostyrene via synthesis. The desired product was obtained in satisfactory to outstanding yields. The protocol for the formal preparation of ningalin B was delineated, encompassing the utilization of the established approach.
 |
| | Scheme 39 Synthesis of 1,2,4-trisubstituted pyrroles. | |
According to B. S. Karki et al. (2019), under the conditions of visible light photoredox catalyzed reactions, a formal [3 + 2] cycloaddition reaction involving 2H-azirines and nitroalkenes led to the metal-free synthesis of trisubstituted pyrroles (Scheme 40).62 This technique involved a base-mediated denitration reaction, followed by the Michael addition of the 2H-azaallenyl radical to the β-nitrostyrenes. The direction of the nitro group influence regulates the regiochemistry of the reaction. The notable characteristics of the reaction include its perfect regioselectivity, and in the majority of situations, excellent product yields.
 |
| | Scheme 40 Synthesis of trisubstituted pyrroles via a visible-light photoredox-catalyzed reaction. | |
By using electrooxidation annihilation to create polypotent pyrroles from amines and aldehydes or ketones, a generic and useful method was developed by X. Gao et al. (2019) (Scheme 41).63 Arylacetaldehyde and primary amines may favorably engage in this metabolism to produce β-substituted pyrroles without the need for extra oxidants. Using imines as a substrate was a highly effective way to acquire tetrasubstituted pyrroles. This reaction was tolerant to functional groups under moderate conditions and may be scaled up to large quantities.
 |
| | Scheme 41 Synthesis of polypotent pyrroles via electrooxidation. | |
A mild and efficient three-component domino-catalyzed Cat. 19 was developed by X. Chang et al. (2019) for the synthesis of highly functional NH-pyrroles from arylglyoxal monohydrates, enzymatic esters, and 1,3-dicarbonyl cyclic compounds in 1,4-dioxane at r.t. for 0.5 h (Scheme 42).64 The current work used a domino-catalyzed method to synthesize a variety of substituted NH-pyrroles, resulting in moderate to favorable yields, of 54–80%.
 |
| | Scheme 42 Synthesis of tetrasubstituted NH-pyrroles catalyzed by Cat. 20. | |
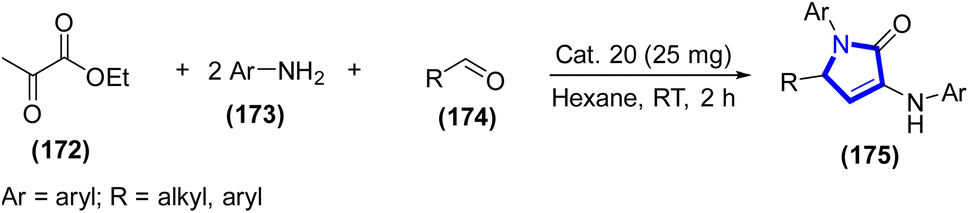
Tetrasubstituted pyrroles were synthesized utilizing one-pot, three-component methodologies and an exceptionally efficient, transition metal-free Brønsted acidic IL, which was introduced by A. Balu Atar et al. (2019) (Scheme 43).65 The present approach uses Cat. 20 for the solvent-free, efficient synthesis of amines, dialkyl acetylenedicarboxylates, and β-nitrostyrenes. The tetrasubstituted pyrrole was synthesized using Cat. 20. Many functionalized tetrasubstituted pyrrole derivatives could be synthesized in high yields under transition metal- and solvent-free conditions. Furthermore, the reuse of Cat. 20 in successive processes did not decrease the product yield. In one pot, this eco-friendly approach produces a variety of tetrasubstituted pyrroles with various therapeutic uses.66
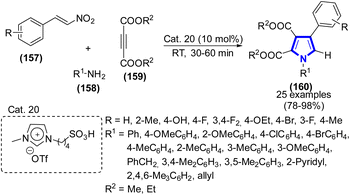 |
| | Scheme 43 Synthesis of tetrasubstituted pyrroles catalyzed by Cat. 20. | |
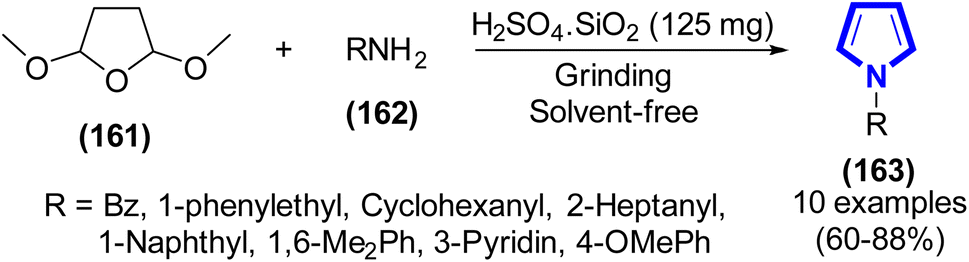
Recently, A. J. Khammas et al. (2019) developed a method for the production of N-substituted pyrrole derivatives, where a sulfuric acid catalyst immobilized on silica gel (SSA) was found to be extremely efficient (Scheme 44).67 They established the optimized reaction conditions to produce a high yield, including simple grinding of the reactants, absence of a solvent, and short reaction time. Additionally, this reaction satisfies the majority of green chemistry criteria.
 |
| | Scheme 44 Synthesis of N-substituted pyrroles catalyzed by H2SO4 SiO2 (SSA). | |
C. Zhou et al. (2023) documented the establishment of a four-component reaction for the synthesis of tetrasubstituted pyrroles under metal-free conditions. The pyrrole ring was synthesized in a single vessel via [2 + 1 + 1 + 1] condensation with ammonium salt as the nitrogen source. Substituted benzo[f]isoindole-4,9-diones and pyrrolo[3,4-c]pyrrole-1,3-diones were synthesized from 1,4-naphthoquinones and maleimides as versatile C2 fragments. This study employed ammonium salt as a nitrogen source, utilized readily available starting materials, and synthesized two carbon–carbon and two carbon–nitrogen bonds in a single process (Scheme 45).68
 |
| | Scheme 45 Synthesis of tetrasubstituted pyrroles. | |
In the study reported by K. Afratis et al. (2023), they aimed to synthesized substituted fused pyrroles through MCR among thioamides, aldehydes, and ammonium acetate. Intercepting the cyclic intermediate with compounds in the required oxidation state eliminated the requirement of harmful oxidizing agents. This reaction is advantageous due to several diversification points that may be readily used, given the accessibility of commercially available aldehydes and anilines, and may have extensive applications in the synthesis of physiologically significant pyrrole-containing molecules (Scheme 46).69
 |
| | Scheme 46 Synthesis of fully substituted fused pyrroles through an MCR. | |
Tajbakhsh et al. (2023) established an efficient method for the acid-catalyzed synthesis of 1H-pyrrol-2(5H)-ones. This was achieved using a unique N-sulfonic acid modified poly(styrene-diethylenetriamine) via a three-component reaction including aldehydes, amines, and ethyl pyruvate. The reaction was conducted within a short, producing elevated yield promptly (Scheme 47).70
 |
| | Scheme 47 Synthesis of 1H-pyrrol-2(5H)-ones. | |
Kalegowda Shivashankar and colleagues (2018) published a study on the development of a novel I2-catalyzed one-pot multi-component approach for the synthesis of a broad variety of elusive benzofuro[2,3-b]pyrroles. To date, there has been no investigation on the possibility of cyclization occurring in one pot among alkanones, hydrazine, and 2-bromobenzofuran or 2-bromobenzothiophene. Considering this, the described single-step procedure offers a flexible alternative to the current pathways for access to relevant benzofuro/thieno[2,3-b]pyrroles (Scheme 48).71
 |
| | Scheme 48 Synthesis of benzofuro/thieno[2,3-b]pyrroles. | |
Borghs et al. (2019) reported the detailed synthesis of around thirty-five substituted pyrrole derivatives using multicomponent reactions under distinctive base metal-catalyzed conditions (Scheme 49). PNP (N,N′-bis(diphenylphosphine)-2,6-diaminopyridine) served as a ligand integrated into the metal catalyst, which was a magnesium complex characterized by air stability and cost-effectiveness. A substantial quantity of water and hydrogen gas was generated as by-products of the operation. The computational analysis indicated the participation of an acceptor dehydrogenation hydrogen auto-transfer mechanism in this process. The metal–ligand combination has a dual function in the dehydrogenation of ethylene glycol by the acceptor, followed by an unusual hydrogen auto-transfer mechanism.72
 |
| | Scheme 49 Manganese-catalysed multicomponent synthesis of pyrroles. | |
Under moderate reaction conditions, L. Kumar S and colleagues (2024) developed an ultrasound-assisted, one-pot tandem MCR assay for various tri- and tetra-substituted pyrroles (Scheme 50). In addition to having excellent chemo- and regio-selectivity, their process does not involve the use of metals or solvents. Additionally, the reaction makes it possible to produce a wide range of substrates with a high tolerance for functional groups, resulting in excellent to exceptional yields. In this context, iodine serves as both a catalyst and an oxidant, facilitating a significant reduction in chemical waste. In comparison to traditional heating, this method, together with ultrasonic irradiation, enhanced the reaction efficiency and speed, while concurrently reducing the occurrence of side reactions. This approach enables the synthesis of several polysubstituted pyrrolecarbonitriles, many of which are challenging to obtain using conventional procedures.73
 |
| | Scheme 50 Synthesis of pyrroles using ultrasound-assisted method. | |
In 2017, Xiang Li and colleagues developed a strategy for synthesizing pyrrole analogs via a one-pot method, yielding disulfide-tethered pyrrole (Scheme 51). This reaction involves a sequence of transformations, including sequential Knoevenagel condensation, Michael addition, N-/O-cyclization, ring opening, double tautomerization, and oxidative coupling. These reactions provide two new pyrrole rings and nine chemical bonds. The published approach enables the synthesis of products with high formation efficiency and atom economy, while also exhibiting notable attributes including the absence of transition metals, simple reaction conditions, shorter reaction time, and uncomplicated synthesis process. This technique is an extension of a feasible method for synthesizing disulfides linked to pyrroles.74
 |
| | Scheme 51 Synthesis of pyrrole analogs using Cat. 19. | |
Thangavel Pavithra and colleagues (2023) successfully synthesized tetrasubstituted pyrroles without the need for any solvent or catalyst, at ambient temperature and under the influence of white light. Overall, the product was successfully obtained via intermolecular cyclization and aromatization. According to previous reports, this method is also incredibly affordable and exceptionally beneficial to the environment. Additionally, early in vitro cytotoxic experiments were carried out on compounds against hepatocellular carcinoma cells (HepG2) to determine their potential cytotoxicity. One of the compounds showed significant activity, which had an IC50 value of 17.82 M (Scheme 52).75
 |
| | Scheme 52 Three-component synthesis of tetra-substituted pyrroles without solvents and catalysts. | |
The group led by Sridhar Pervaram (2020) aimed to produce poly-substituted pyrrolidinone derivatives via a green multicomponent reaction. Polyethylene glycol-400 was employed as the solvent, and 1-butyl-3-methylimidazolium tetrafluoroborate as an IL. Because ILs are inexpensive and can be reused, they are regarded as superior catalysts for phase transfer given that they do not cause toxicity. Consequently, an approach that is simple, very effective, and environmentally friendly was developed (Scheme 53).76
 |
| | Scheme 53 Synthesis of pyrrolidinone derivatives using PEG-400 and [bmin]BF4. | |
Tongyan Yu and colleagues (2021) successfully designed a method employing enaminone as a substrate for the precise capture of carbocation intermediates. This allowed them to produce highly functionalized pyrroles and pyrrole-fused piperidine-4-ones in yields ranging from modest to good. This four-component reaction, mediated by BF3·Et2O, has several benefits, including gentle reaction conditions, absence of metals, and availability of practically relevant pathways to create highly functionalized pyrroles, making it an scheme appealing (Scheme 54).77
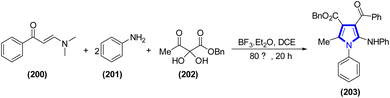 |
| | Scheme 54 Synthesis of pyrroles via a four-component reaction. | |
The group of researchers led by Xiangqing Chang (2019) devised a three-component reaction AS an effective and innovative multicomponent reaction. To produce highly functionalized NH-pyrroles, the reaction was supported by a solvent composed of 1,4-dioxane and catalyzed with the assistance of Cat. 19 at the appropriate temperature. The reaction tolerated various functional groups, which resulted in moderate to excellent yields of different substituted NH-pyrroles. The reaction was carried out under conditions that are not particularly harsh, the components that were used to initiate it are not difficult to obtain, and it could to tolerate a wide variety of functional groups (Scheme 55).64
 |
| | Scheme 55 Synthesis of pyrroles catalyzed by Cat. 19. | |
2.1.3. Carbazole. Carbazole is a tricyclic aromatic heterocycle with a core pyrrole ring joined by two benzene rings. The term “carbazole” encompasses tricyclic molecular frameworks and diverse fused carbazoles, including tetracyclic (comprising 5-, 6-, and 7-membered rings), pentacyclic, hexacyclic, and heptacyclic structures.78 This distinctive structure endows carbazole and its derivatives with considerable chemical stability, electrical characteristics, and biological activity, making them advantageous in several scientific and commercial applications. Carbazole derivatives are prevalent in both natural and manufactured substances, including alkaloids such as murrayanine and ellipticine, which have significant anticancer and antibacterial activities. Carbazoles have demonstrated a diverse range of biological activities including anticancer,79 antiepileptics,80 antibacterial,81 anti-inflammatories,82 antioxidative,83 analgesic,84 antidiarrheal,85 antihistaminic,86 neuroprotective,87 and pancreatic lipase inhibitory88 characteristics (Scheme 56).
 |
| | Scheme 56 Chemical structures of bioactive compounds containing a carbazole framework. | |
In the study by P. Zhang et al. (2020), they successfully synthesized clausine and glycozoline, alkaloids produced from carbazole, via a transition-metal and exogenous-oxidant-free C–H bond amination method (Scheme 57).89 This work presented a methodology for conducting environmentally sustainable electrochemical C–H bond dehydrogenative amination. This method has potential to be employed for the production of favored carbazole moieties, which are useful in various contexts. It was underlined that this approach has an eco-friendly and sustainable nature due to its scalability, relatively moderate conditions, and widespread applicability.
 |
| | Scheme 57 Synthesis of carbazole-derived alkaloids catalyzed by Cat. 21. | |
C. Gioia et al. (2008) published a protocol for the preparation of carbazole frames from 3-vinylindoles via a Diels–Alder reaction in the presence of an acid-base organo-catalyst (Scheme 58).90 The activation of 3-vinylindoles for catalytic asymmetric processes can potentially be facilitated by the interaction between a base and the N–H moiety. This was the case if the substrate was used correctly. Diels–Alder reactions use a wide variety of 1-amino-substituted dienes as the diene substrate.
 |
| | Scheme 58 Synthesis of carbazole frameworks catalyzed by Cat. 22. | |
In the study by J. Wang et al. (2015), they introduced a novel and comparatively less harmful approach for the production of carbazoles via tandem iodocyclization, involving 1,2-alkyl migration and subsequent aromatization. This methodology focused on the synthesis of carbazoles (Scheme 59),91 providing an approach for the synthesis of iodocarbazoles. This strategy involves performing tandem iodocyclization in conjunction with migration and aromatization. This technique of successive cascading may be carried out quickly at ambient temperature and in a very short period.
 |
| | Scheme 59 Synthesis of carbazoles catalyzed by ICl and K2CO3. | |
S. Yaragorla et al. (2018) first reported the production of 3-iodocarbazoles via the iodo-cycloisomerization of 1-(indol-3-yl)-1-arylbut-3-yn-2-ols (Scheme 60).92 The synthesis commenced with a cascade 5-endo-spirocyclization, and subsequently selective 1,2-alkyl migration as the second step in the synthesis process. The solvent employed in this study was AcOEt, and the researchers conducted their work with a focus on atom and step economy.
 |
| | Scheme 60 Synthesis of 3-iodocarbazoles catalyzed by I2. | |
The production of 1,3-dihydroxy-2-carboxycarbazole from the reaction between indole-3-acetic acid and Meldrum acid has undergone significant enhancements through the implementation of an innovative and very efficient methodology, which was introduced by K. Liu et al. (2015) (Scheme 61).93
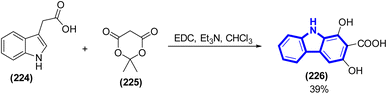 |
| | Scheme 61 Synthesis of 1,3-dihydroxy-2-carboxycarbazole. | |
Using the Cat. 23-catalyzed ring-opening and annulation of 2-amidodihydrofurans, a safe and efficient method for synthesizing carbazoles was established by J. Zhao et al. (2015) (Scheme 62).94 This method was based on the ring-opening of 2-amidodihydrofurans at r.t. Synthesis with strong chemical selectivity and regioselectivity may be successfully scaled up using this method.
 |
| | Scheme 62 Synthesis of trisubstituted carbazoles catalyzed by Cat. 24. | |
S. Biswas et al. (2015) presented a methodology for the synthesis of multi-substituted carbazoles by reacting α,β-substituted nitro olefins with 2-(3-formyl-1H-indol-2-yl)acetates in an aqueous environment, utilizing Cat. 19 as the catalyst (Scheme 63).95 This all-in-one, oxidant-free, mild, environmentally friendly technique may prove to be an effective alternative synthetic strategy for fast access to 3-alkyl-substituted carbazole frameworks that are structurally similar to physiologically active natural carbazoles.
 |
| | Scheme 63 Synthesis of substituted carbazoles catalyzed by Cat. 19. | |
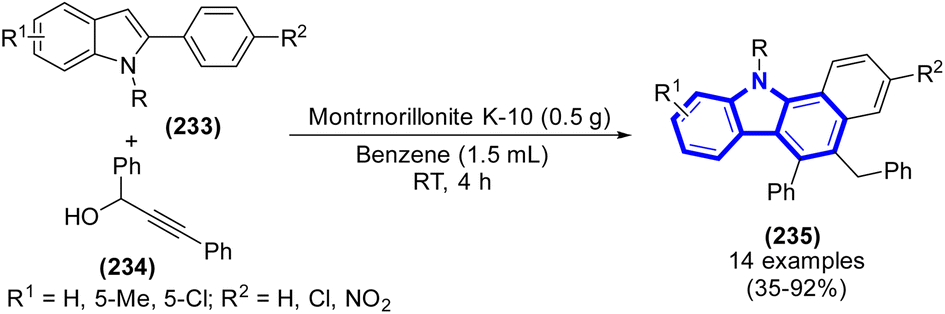
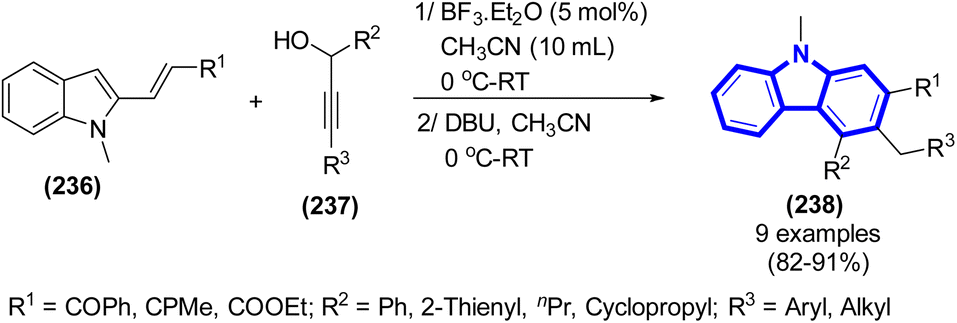
The development of an effective two-step synthesis method to benzo[a]carbazoles starting from 2-arylindoles was accomplished by J. W. Lim et al. (2015) (Scheme 64).96 Through a series of consecutive propargylations, propargyl-allenyl isomerization, and simultaneous 6π-electrocyclization procedure, it was possible to achieve the effective synthesis of benzo[a]carbazoles using 2-arylindoles as the starting material.
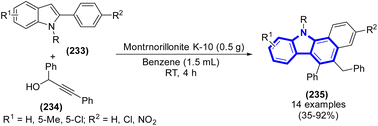 |
| | Scheme 64 Synthesis of benzo[a]carbazoles catalyzed by montmorillonite K-10. | |
This innovative one-pot [4 + 2]-benzannulation technique was employed to obtain substituted carbazoles, which was illustrated by C. Raji Reddy et al. (2016) (Scheme 65).97 The aforementioned technique has numerous advantages, including the utilization of moderate and metal-free reaction conditions, a wide range of applicable substrates, and the availability of easily accessible starting materials, ensuring its accessibility.
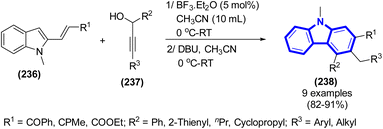 |
| | Scheme 65 Synthesis of substituted carbazoles. | |
An intermolecular annulation of acetyl indoles with alkynes catalyzed by Cat. 24 was created by H. Wang et al. (2017) (Scheme 66).98 Conditions that were mild and devoid of metals allowed the formation of carbazoles smoothly.
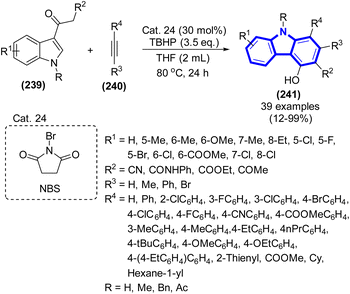 |
| | Scheme 66 Synthesis of carbazoles catalyzed by Cat. 24. | |
D. Cao et al. (2018) devised a broad methodology for the synthesis of 4-carbazolamine derivatives by employing a vinylogous Michael addition/cyclization/isomerization/elimination reaction between 3-nitroindoles and alkylidene malononitriles (Scheme 67).99 This method did not require the use of transition metals in any step of the process.
 |
| | Scheme 67 Synthesis of multi-substituted 4-carbazolamine derivatives. | |
Under metal-free conditions, an effective method for the conversion of indole to carbazole was created by S. Chen et al. (2016) (Scheme 68).100 In this method, indoles, ketones, and nitroalkenes are assembled into a single compound using a three-component technique. The manufacture of a varying range of functionalized carbazoles was demonstrated by the implementation of the indole-to-carbazole process, with yields ranging from satisfactory to outstanding. This method has several advantages, such as metal-free reaction condition, excellent regioselectivity, wide functional group tolerance, and readily available starting materials.
 |
| | Scheme 68 Synthesis of carbazoles catalyzed by NH4I. | |
An effective one-pot method for converting indole-to-carbazole that only requires two steps was discovered by S. Chen et al. (2017) (Scheme 69).101,102 The very efficient tricomponent synthesis of multi-substituted carbazoles commences with the use of indoles, ketones, and alkenes, with oxygen serving as the oxidizing agent in the reaction. Oxygen was the only oxidant that was used in this process, which did not include any transition metals. The current approach makes it possible to assemble a wide range of substituted carbazole products virtually. It also has a high degree of regioselectivity and can tolerate several functional groups.
 |
| | Scheme 69 Synthesis of carbazole frameworks. | |
Y. Li et al. (2016) presented the formal [4 + 2] cycloaddition of 3-nitroindoles, which resulted in the formation of chiral dihydrocarbazole frames (Scheme 70).103 Chiral dihydrocarbazole scaffolds could be produced in yields ranging from good to outstanding (up to 87%) and enantioselectivities ranging from modest to high (up to 97% ee) when 3-nitroindoles were used in conjunction with an organocatalyst. Under moderate reaction conditions, a [4 + 2] cycloaddition/elimination cascade occurred throughout the reaction.
 |
| | Scheme 70 Synthesis of chiral dihydrocarbazole frameworks catalyzed by Cat. 25. | |
The synthesis of carbazoles via carbene-catalyzed oxidative formal [4 + 2]-annulation of enals with 2-methyl-3-oxoacetate indoles has been established as an efficient method. This technique, introduced by D. Liu et al. (2018), provides a direct and swift pathway for the synthesis of carbazoles (Scheme 71).104 Also, this metal-free reaction has a wide substrate tolerance under moderate conditions.
 |
| | Scheme 71 Synthesis of carbazoles catalyzed by Cat 26. | |
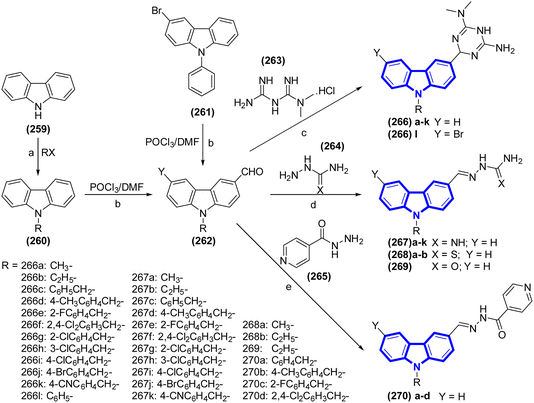
Hu-Ri Piao et al. (2021) proposed a method for the design, synthesis and evaluation of carbazole derivatives as potential antimicrobial agents (Scheme 72).105 Most of the compounds had significant inhibitory effects against several bacterial strains, including a multidrug-resistant clinical isolate and one fungal strain, with MICs ranging from 0.5 to 16 mg mL−1. Compounds 266f and 267d exhibited the highest inhibitory effects, with MICs ranging from 0.5 to 2 mg mL−1. Structure–activity relationship analysis and docking experiments indicated that the dihydro triazine moiety enhances the antibacterial efficacy and diminishes the toxicity of the carbazole derivatives. In vitro enzyme activity studies indicated that the binding of compound 266f to dihydrofolate reductase may explain the antibacterial action.
 |
| | Scheme 72 Synthesis and evaluation of carbazole derivatives. | |
Wu et al. (2017) described a novel transition metal-free one-pot approach for the synthesis of N-arylcarbazoles (Scheme 73). In the presence of atmospheric oxygen, anilines were reacted with cyclohexanones using KI as a promoter, CH3SO3H as an acid, and toluene as the solvent. Subsequently, iodine was added and the mixture heated at 160 °C to get N-arylcarbazoles. In this transformation, the nitrogen sources were aniline and cyclohexanone, with the aryl ring and the additional two aryl rings specified accordingly. A series of derivatives was synthesized, with yields in the range of 41% to 90%. A potential mechanism for this transformation was also suggested.106
 |
| | Scheme 73 Synthesis of N-arylcarbazoles using transition metal-free one-pot reaction. | |
Guo and colleagues (2012) successfully developed a metal-free and direct approach for the N-arylation of carbazoles using diaryliodonium salts. The reaction occurred between carbazoles and diaryliodonium salts to obtain the desired products. In this reaction, KOtBu was used as the base and toluene as the solvent at 50 °C. A series of derivatives was synthesized, with yields ranging from traces to 92% (Scheme 74).107
 |
| | Scheme 74 Synthesis of N-arylcarbazoles using diaryiodonium salts. | |
Chakrabarty et al. (2013) suggested a methodology for synthesizing carbazoles using aryne and nitrosoarenes. The major product was treated with nitrosoarenes to get N-arylcarbazoles. The procedure used Cat. 27 and dimethyl ether (DME) solvent, yielding the major products in yields of 51–76% (Scheme 75).108
 |
| | Scheme 75 Synthesis of N-arylcarbazoles catalyzed by Cat. 27. | |
In 2021, Feofanov and colleagues reported a technique for the synthesis of N-arylated carbazole that did not involve the use of transition metals. In the absence of transition metals, 2,2′-difluoro-1,1′-biphenyls were subjected to treatment with para-substituted anilines, generating the products with a yield in the range of 22–85%. NaH was used in dimethyl sulfoxide solvent at a temperature of 80 °C to carry out the reaction (Scheme 76).109
 |
| | Scheme 76 Synthesis of arylated carbazoles. | |
2.2. Five-membered heterocycles containing two nitrogen atoms
2.2.1. Imidazolidine. Imidazolidine is a saturated five-membered heterocyclic molecule including two nitrogen atoms located at positions 1 and 3. It is structurally similar to imidazoline and imidazole, although it diverges owing to its completely saturated ring structure, which affects its chemical reactivity and biological activity.110 Imidazolidine and its derivatives have attracted considerable attention in medicinal chemistry owing to their diverse pharmacological characteristics. Imidazolidines participate in a variety of crucial biological processes, including fungicidal,111 antibacterial,112 tyrosine phosphatase inhibitors,113 and antiviral110 activities, which together with N,N′-dibenzyl-2-arylimidazolidines, N,N′-bisaminoalkylimidazolidines, and N,N-dihydroxyphenylimidazolidines have attracted significant interest (Scheme 77).114,115
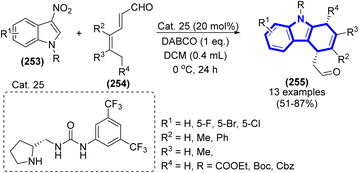
 |
| | Scheme 77 Chemical structures of bioactive compounds containing an imidazolidine framework. | |
A. N. Komogortsev et al. (2021) published a simple procedure for synthesizing new imidazole-2-thione derivatives incorporating allomaltol fragments via a one-pot three-step reaction among allomaltol, arylglyoxals, primary amines, and alkyl or aryl isothiocyanates (Scheme 78).116 In addition to moderate reaction conditions, atom economy, and rapid workup technique that does not need any purification, this procedure offers several other advantages. Moreover, XRD was used to identify the structures of an intermediate and one of the products.
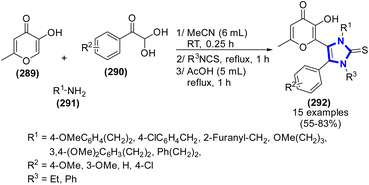 |
| | Scheme 78 Synthesis of imidazole-2-thione derivatives. | |
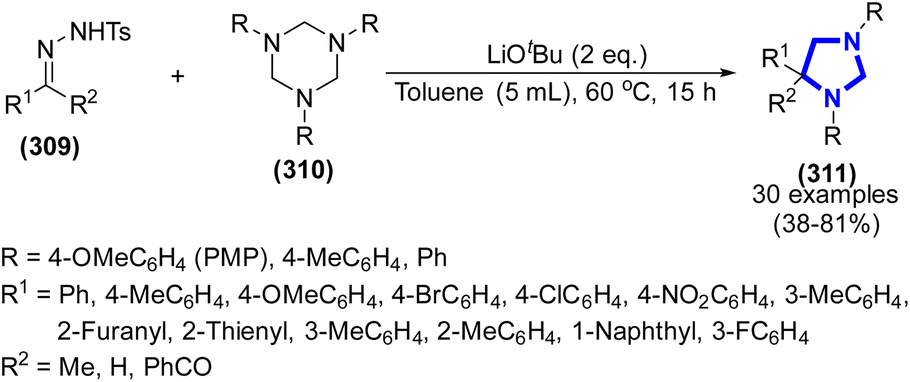
Simple modification of the reaction solvent for the preparation of a variety of imidazolidine frameworks under blue LED irradiation, leading too good to excellent yields, was researched by X. Cheng et al. (2021) (Scheme 79).117 The formation of the products was achieved through the cycloaddition reaction between α-diazo esters and hexahydro-1,3,5-triazines in dichloromethane (DCM) at ambient temperature for a duration of 12 h, without the use of any catalyst. More importantly, based on the findings of the control tests and density functional theory calculations, a viable reaction mechanism was presented.
 |
| | Scheme 79 Visible light-promoted synthesis of imidazolidine frameworks without a catalyst. | |
A. Casnati et al. (2019) employed phosphazene base Cat. 28 as an organo-catalyst for the synthesis of imidazolidin-2-ones and imidazole-2-ones from propargylic urea through an intramolecular hydroamidation (Scheme 80).118 When 5 mol% of Cat. 28 and 4 mL of MeCN were used and the reaction was conducted at r.t., the target product were obtained in moderate to excellent yields. Based on the findings of density functional theory (DFT), it was determined that the predominant pathway for the production of imidazole-2-ones involves a base-mediated isomerization process, leading to the formation of an allenamide intermediate.
 |
| | Scheme 80 Synthesis of imidazole-2-ones catalyzed by Cat. 28. | |
A. Angyal et al. (2019) documented a one-step three-component reaction involving α-amino acids, isatins, and 2H-azirines, resulting in the synthesis of a range of 1,3-diazaspiro[bicyclo[3.1.0]hexane]oxindole frameworks (Scheme 81).119 When DMSO was employed as the solvent at a temperature of 60 °C and the reaction time of 8 h, the products were obtained in moderate to high yields. This methodology showed several advantages such as endoselectivity, compatibility of a wide range of substrates, gentle reaction conditions, and high yields.
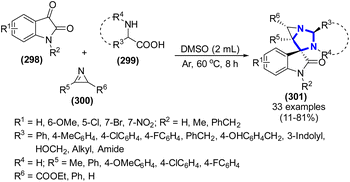 |
| | Scheme 81 Synthesis of 1,3-diazaspiro[bicyclo[3.1.0]hexane]oxindole frameworks. | |
In a study conducted by B. Zhao et al. (2018), a novel catalyst derived from 1-indanone-derived isothiocyanate was investigated for its application in the diastereo- and enantio-selective Mannich/cyclization reaction. The objective of this research was to synthesize a series of bispirocyclic indanone–thioimidazolidine–oxindoles, which possess two neighboring spiro-quaternary stereocenters (Scheme 82).120 With Cat. 29 (5 mol%), DCM as the solvent, and r.t., the desired product yields were good to outstanding after 24–48 h. Moreover, the effectiveness of the reaction was assessed through a large-scale reaction, giving 82% yield (>25:1 dr, >99% ee).
 |
| | Scheme 82 Synthesis of bispirocyclic indanone–thioimidazolidine–oxindoles catalyzed by Cat. 29. | |
Y.-L. Qian et al. (2017) conducted a 1,3-dipolar cycloaddition reaction using isatin, amino acid, and isatin-derived ketimine, resulting in the synthesis of dispirooxindole-imidazolidine frameworks. This reaction, which does not involve the use of additives, allows the formation of a diverse array of frameworks containing vicinal quaternary carbon centers (Scheme 83).121 This transformation possesses several advantages, including a wide range of applicable substrates, mild reaction conditions, and high product yields ranging from good to extraordinary. The synthesis was also performed on the gram scale, resulting in a product yield of 94% and a diastereomeric ratio of 85:15.
 |
| | Scheme 83 Synthesis of various dispirooxindole-imidazolidine frameworks. | |
P. Liu et al. (2017) reported a new technique to synthesize imidazolidines via the [2 + 1 + 2]-cycloaddition process between tosylhydrazones and hexahydro-1,3,5-triazines using LiOtBu as the catalyst and toluene as the solvent (Scheme 84).122 The advantageous features of this method are metal-free, modest to fine yields, and no need to separate the tosylhydrazones. Notably, the dual functions of LiOtBu were demonstrated to either liberate the diazo or boost the cycloaddition.
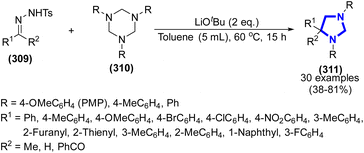 |
| | Scheme 84 Synthesis of imidazolidines catalyzed by LiOtBu. | |
A combination of thiazolium salt/triethylamine (Cat. 30) was found to be effective for initiating the domino process to prepare a variety of imidazolidine-2-thiones by G. Di Carmine et al. (2017) (Scheme 85).123 The reaction was performed via the intramolecular aza–acetalization reaction between aldehydes and benzylidenethioureas, leading to good to outstanding product yields. Moreover, this approach is considered diastereoselective (up to 99:1 dr) and effective because of the excellent yield of up to 95% in the gram-scale reaction.
 |
| | Scheme 85 Synthesis of imidazolidine-2-thiones catalyzed by Cat. 30. | |
P. V. Balaji et al. (2016) investigated the aminooxygenation between 1,2-diamine derivatives and styrene to form geminal diaminated imidazolidine utilizing Cat. 31 as the catalyst (Scheme 86).124 The target compounds were obtained in high yields after an 8 h reaction at r.t. in p-TSA and DCM as the solvent. This protocol is advantageous because of its high diastereoselectivity, good yields, and absence of metal.
 |
| | Scheme 86 Synthesis of geminal diaminated imidazolidine catalyzed by Cat. 31. | |
M. A. Tabarki et al. (2016) reported the application of a series of N-substituted isothiocyanates for the ring opening of N-alkylaziridine-2-carboxylates to create trans-imidazolidine-2-thiones without a catalyst (Scheme 87).125 This methodology showed many advantages such as regio- and stereo-selectivity, without additives, and high product yields. Furthermore, it was shown that the generation of the products was determined by the steric hindrance and electronic influence of the N-substituents in the substrates.
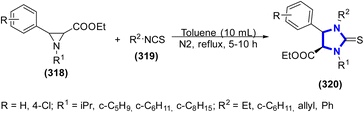 |
| | Scheme 87 Synthesis of trans-imidazolidine-2-thiones without a catalyst. | |
V. Satheesh et al. (2016) employed Cat. 32 as a catalyst and tBuOOH in water (T-Hydro) as an oxidizing agent for the preparation of imidazolidines (Scheme 88).126 Numerous N-aryl and 2-aryl substituted compounds were examined, resulting in the synthesis of products with favorable yields. The method in question exhibits several notable characteristics, including the use of water as the solvent, the absence of metals, regioselectivity, ease of product separation, and a wide range of applicable substrates.
 |
| | Scheme 88 Synthesis of imidazolidines catalyzed by Cat. 32. | |
Under the catalysis of Cat. 33, DCM as a solvent, and a temperature of 0 °C, the asymmetric 1,3-dipolar reaction between methanamines and methyl aryl ketimines to form N-sulfinylaldimines with modest to outstanding yields was reported by C. Izquierdo et al. (2016) (Scheme 89).127 Importantly, in the gram-scale of 5.3 mmol, the products were produced in high yields (83%) with high diastereoselectivity (>98:2).
 |
| | Scheme 89 Synthesis of N-sulfinylaldimines catalyzed by DPP. | |
Y.-H. Sun et al. (2015) reported the preparation of a wide range of dispirooxindole-imidazolidine frameworks via the self-1,3-dipolar cycloaddition of N-methylisatins and benzylamines in the presence of 3,5-dinitrobenzoic acid and 4 Å MS in THF as the solvent at r.t. (Scheme 90).128 It was also the first time that a range of new dispirooxindole-imidazolidines with two quaternary carbons and three stereogenic centers were synthesized in high yields and with exceptional stereoselectivities.
 |
| | Scheme 90 Synthesis of dispirooxindole-imidazolidines catalyzed by Cat. 34 in the presence of 4 Å MS. | |
M. Bai et al. (2015) employed commercial quinine as a catalyst for the domino Mannich-cyclization reactions of 3-isothiocyanato oxindoles and imines to synthesize spiro[imidazolidine-2-thione-4,30-oxindole] skeletons (Scheme 91).129 The intended products were obtained in high yields under ideal reaction conditions. This method exhibited notable characteristics, including remarkable diastereo- and enantio-selectivity (achieving ratios of up to 99:1 dr and 97% ee), the ease of obtaining the catalyst, and the low amount of catalyst required (1 mol%).
 |
| | Scheme 91 Synthesis of spiro[imidazolidine-2-thione-4,30-oxindole] skeletons catalyzed by Cat. 35. | |
A cyclo-condensation reaction between heteroarylamines and benzaldehyde derivatives to form imidazolidines with guanidinium chloride in glyoxal was reported by A. Olyaei et al. (2013) (Scheme 92).130 This methodology, which is now being used, has several advantages, including high efficiency and universality, distinct reaction profiles, and a one-pot two-step synthetic procedure. Additionally, this was the first time that heteroarylamines, arylaldehydes, and glyoxal were used in a reaction to obtain products that were needed.
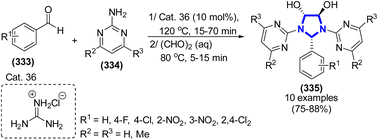 |
| | Scheme 92 Synthesis of imidazolidines catalyzed by Cat. 36. | |
A wide range of imidazoline-2-thiones derivatives created by aliphatic primary amines and vinyliminophosphorane, which was the product of the reaction between α-azidoketone with aromatic aldehydes, were reported by H. Xie et al. (2012) (Scheme 93).131 The advantages of this method are its simple procedure, moderate reaction conditions, excellent yields, and readily available starting material.
 |
| | Scheme 93 Synthesis of imidazoline-2-thione derivatives. | |
In a kinetically regulated method, the Dieckmann ring closure of hemiaminal ether and imidazolidines efficiently yields bicyclic substrates using a chemoselective ring closure. Additionally, hemiaminal and aminal heterocyclic systems containing –NCH (Ar)X– (X = O and NR) are accessible. According to the findings by Moloney et al. (2023), this procedure was not unsuccessful. The activity of these systems is at its highest in a particular well-defined region of chemical space (Scheme 94). These systems demonstrate significant antibacterial activity, at least against Gram-positive pathogens.132
 |
| | Scheme 94 Synthesis of imidazoline-2-thione derivatives. | |
Angelo Frongia and colleagues (2024) indicated that the [3 + 2]-annulation process involving thiourea, an ambident dinucleophile, and cyclobutanone, which features a new pull–pull alkene system, theoretically exhibits various chemo- and regio-selectivity profiles. Their report outlined the efficient synthesis of cyclobutanones and demonstrated their reaction with thioureas, which was mechanistically explained by the relevant steric and electronic effects. The [3 + 2]-annulation occurs under moderate, additive-free conditions, yielding previously unreported cyclobutane-fused imidazolidine-2-thione and thiazolidine-2-imines in satisfactory yields (Scheme 95).133
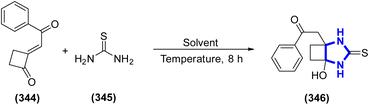 |
| | Scheme 95 Synthesis of 2-(phenacylethylidene)cyclobutanones. | |
Andrea Spallarossa and colleagues (2022) demonstrated that imidazolidine-2-thione interacted with atypical Vilsmeier adducts derived from dimethylacetamide and other acyl chlorides exhibiting distinct electronic and steric properties (Scheme 96). The type of acyl chloride reagent influences the synthesis of mono- or di-acylated thiourea derivatives. Semi-empirical computer simulations were used to identify the influencing component of the reaction. The reaction of benzoylimidazolidin-2-thiones with acyl chlorides was performed to evaluate the chemical versatility of mono-acylated derivatives, which have therapeutic significance. The derivatives of a restricted library of asymmetric di-acyl thioureas did not exhibit cytotoxic effects on SKOV-3 and MCF-7 cancer cell lines. Furthermore, in silico analyses demonstrated robust pharmacokinetics and drug-like characteristics for mono- and di-acylated thioureas. These features indicate that the synthesized compounds are attractive non-cytotoxic chemical frameworks for medicinal chemistry.134
 |
| | Scheme 96 Synthesis of mono- and di-acylated imidazolidine-2-thione derivatives. | |
2.2.2. Imidazole (imidazoline). Imidazole, also known as 1,3-diaza-2,4-cyclopentadiene, is a flat cyclic structure composed of five atoms, consisting of three carbon (C) atoms and two nitrogen (N) atoms, located at the 1 and 3 positions in the ring. Some representatives of this framework are histidine and histamine. Imidazoles have many applications because of their antifungal,135 anti-protozoan,136 and anti-hypotensive137 properties. Moreover, imidazole derivatives are also used in photography and electronics (Scheme 97).138
 |
| | Scheme 97 Chemical structures of bioactive compounds containing an imidazole framework. | |
A. A. Abdelhamid et al. (2020) employed IL pyrrolidinium hydrogen sulfate as a catalyst for the one-pot preparation of 1H-imidazole-1-carboxylates from a multi-component mixture of 1,2-diphenylethane-1,2-dione, NH4OAc, aromatic aldehydes, and ethyl glycinate hydrochloride (Scheme 98).139 After 20–50 min of reaction at 80 °C, the desired product yields were good to outstanding. Then, the intermediate was used to react with hydrazine hydrate to form 1H-(imidazole-1-yl)acetohydrazides at 25 °C in EtOH after 17–45 min, leading to good to excellent yields. This protocol is highly advantageous due to its short reaction time, ease of implementation, nonchromatographic approach, and high yields. Moreover, the in vivo experiment demonstrated the antioxidant action of the products.
 |
| | Scheme 98 Synthesis of ethyl-4,5-diphenyl-2-(substituted)-1H-imidazole-1-carboxylate catalyzed by Cat. 37. | |
Y. Tian et al. (2019) published a report on the acid-catalyzed synthesis of imidazole derivatives from amidines and sulfoxonium ylides in DCE at 110 °C after 12 h (Scheme 99).140 When CF3COOH was employed as the catalyst, the product yields were boosted. The advantages of this method are its large scope of aromatic functional groups, strong regioselectivity, cost-effectiveness, and ecologically benign property.
 |
| | Scheme 99 Synthesis of imidazole derivatives catalyzed by trifluoroacetic acid. | |
M. R. Albayati et al. (2019) described a method to use Cat. 38 as the catalyst for the preparation of a series of multi-substituted imidazole derivatives through the mixing of benzil, NH4OAc, phenethylamine, or C4H9NH2 with various aromatic aldehydes (Scheme 100).141 The main advantages of this approach are its relatively rapid reaction times, high yield, and simplicity of stabilization.
 |
| | Scheme 100 Synthesis of multi-substituted imidazole derivatives catalyzed by Cat. 38. | |
A [3 + 2] heterocycloaddition approach for the synthesis of cyanoaryl-imidazolines from cyanobenzene derivatives and azomethine ylides catalyzed by trifluoroacetic acid, followed by the in situ oxidation of the crude product to form 4-arylimidazole frameworks was demonstrated by M. Beuvin et al. (2018) (Scheme 101).142 Computational results indicated that the heterocycloaddition process was preferred from both a kinetic and thermodynamic standpoint.
 |
| | Scheme 101 Synthesis of 4-aryl imidazole frameworks catalyzed by trifluoroacetic acid. | |
S. Evjen et al. (2017) showed a report about the combination of alkyldicarbonyl compounds, as well as appropriate alkyl aldehydes and ammonia to create a series of polyalkylated N–H and N-alkyl imidazoles (Scheme 102).143 The main advantages of this approach are its readily available starting materials, high yield, and adaptability for mass production at a reasonable cost.
 |
| | Scheme 102 Synthesis of N-alkyl/aryl imidazoles. | |
In the study reported by C.-Y. Chen et al. (2013), they demonstrated a multicomponent reaction among internal alkynes, aldehydes, and anilines. The reaction was facilitated by pivalic acid and conducted in a combination of water and DMSO. The outcome of this reaction was the formation of imidazole moieties (Scheme 103).144 This approach is a feasible alternative to the classic transition metal-catalyzed procedures because of its ambient conditions, easy work-up technique, and excellent product yields. Furthermore, some of the products outperformed their analogs in terms of fluorescence emission and UV absorption.
 |
| | Scheme 103 Synthesis of imidazole moieties catalyzed by pivalic acid. | |
G. Mloston et al. (2011) introduced the synthesis of two imidazoline-frameworks via the reaction of ethyl 2-hydroxyimino-3-oxobutanoate, racemic amino alcohols, and formaldehyde in EtOH as the solvent (Scheme 104).145 The experimental procedure employed in this study was devoid of any metal catalysts. However, it was observed that the resulting product yields obtained were quite modest in magnitude.
 |
| | Scheme 104 Synthesis of imidazolone frameworks in ethanol. | |
Albrecht et al. (2011) reported the enantiocontrolled synthesis of a variety of optically active 2,5-disubstituted imidazoles with outstanding (92–97% ee) in modest to good yields (44–76%) via a two-step method. (Scheme 105).146 The experimental procedure involved the use of a single reaction sequence of two steps, namely the organocatalytic epoxidation or aziridination of α,β-unsaturated aldehydes, followed by [3 + 2]-annulation. The technique detailed in the study has several advantages, including the utilization of low catalyst loadings, commercially and readily available starting materials, and the implementation of moderate reaction conditions.
 |
| | Scheme 105 Synthesis of optically active 2,5-disubstituted imidazoles catalyzed by Cat. 39. | |
L. Preti et al. (2010) reported the microwave-assisted 1,5-electrocyclization of azavinyl azomethine ylides using 1,2-diaza-1,3-dienes, which led to the formation of a diverse range of 3H-imidazole-4-carboxylates (Scheme 106).147 It is worth noting that this procedure is simple and effective without isolating the intermediates and it can be conducted in a multicomponent manner.
 |
| | Scheme 106 Synthesis of 2-substituted 3H-imidazole-4-carboxylates under MW. | |
Owing to the efficacy and simplicity of the four-component reaction involving 1,2-diketones, aldehydes, primary amines, and ammonium acetate, it has been recognized as a viable approach for synthesizing 1,2,4,5-tetrasubstituted imidazoles (Scheme 107). This method had received significant attention from scientists around the world. Many research works have been published using various catalysts such as silica gel/NaHSO4 support,148 silica-supported perchloric acid,149 mercaptopropylsilica,150 poly(AMPS-co-AA),151 and zeolite BEA,152 and catalyst-free procedure.153
 |
| | Scheme 107 Synthesis of 1,2,4,5-polyimidazoles catalyzed by metal-free catalysts. | |
1-Substituted, 2-aryl, 4,5-phenylimidazole was synthesized by Bourissou et al. (2000) utilizing benzil, benzonitrile, and primary amines on the surface of silica gel under solvent-free conditions. The MW was used to produce modest efficiencies (Scheme 108).154
 |
| | Scheme 108 Synthesis of substituted, 2-aryl, 4,5-phenylimidazoles. | |
Benzyl, 3-methoxy-4-hydroxybenzaldehyde, and ammonium acetate were used in the synthesis of triaryl-imidazole by Nalage et al. (2010). The synthesis was carried out in the presence of polyethylene glycol and subjected to MW for a period of 5 min. These researchers achieved yields of up to 71% (Scheme 109).155
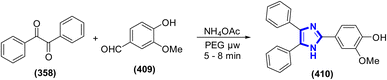 |
| | Scheme 109 Synthesis of triaryl-imidazole in the presence of polyethylene. | |
Acetic acid, aromatic aldehydes, phenylglyoxal, and ammonium acetate were utilized as components in the synthesis of disubstituted imidazole by Husain et al. (2009). Next, the 1,2,4-disubstituted imidazoles were combined with chlorobenzene in the presence of triethylamine and tetrahydrofuran (THF), which resulted in the production of a new product, which had a yield of 74% (Scheme 110).156
 |
| | Scheme 110 Synthesis of disubstituted imidazoles. | |
Sharma et al. (2024) documented the SAR-guided synthesis of powerful, selective, and artemisinin-synergistic imidazole derivatives with potential in vitro antiplasmodial and in vivo antimalarial efficacy. The findings indicated that the compound with 4,5-triphenyl and 2-naphthalene substitution on the imidazole ring had the most significant antiplasmodial action (0.5 μM), although Im2 demonstrated favorable resistance indices (1.06–1.72) and selectivity indices (>133.3). Despite Im22 demonstrating elevated resistance indices (4–4.6), it showed significant synergistic efficacy with ART against PfINDO. Im2 and Im5 demonstrated significant in vivo antimalarial efficacy, prolonging the lifespan of infected mice to 28.6 ± 4.2 and 27.4 ± 4 days, respectively, in contrast to the untreated control group, which survived 21.3 ± 3.5 days (Scheme 111).157
 |
| | Scheme 111 A novel series of 2,4,5-triphenyl-1H-imidazole derivatives. | |
2.2.3. Pyrazolidine. Formed from three carbon atoms and two neighboring nitrogen atoms, pyrazolines (sometimes called dihydropyrazole) are mono-unsaturated 5-membered heterocyclic compounds.158 Three tautomeric forms of pyrazoline exist, i.e., 1-, 2-, and 3-pyrazoline. Many pyrazolidine frameworks display substantial anti-inflammatory, antidepressant, anticancer, antibacterial, and antiviral properties such as oxyphenbutazone and acetylpyrazoline derivatives (Scheme 112).159–162
 |
| | Scheme 112 Chemical structures of bioactive compounds containing a pyrazolidine framework. | |
T. Gieshoff et al. (2017) discovered a new way to get 1,2-diarylpyrazolidin-3,5-diones from easily available dianilides without using harmful hydrazine building blocks (Scheme 113).163,164 This strategy is a viable alternative to the traditional methods because of sustainability, affordable starting materials, large substrate scope, and moderate to good yields.
 |
| | Scheme 113 Synthesis of 1,2-diarylpyrazolidin-3,5-diones via electrochemical method. | |
D. Wang et al. (2013) described the [3 + 2] annulation of azomethine imine with trifluoroethylidene malonate to create trifluoromethyl-containing pyrazolidine scaffolds (Scheme 114).165 Under the optimal conditions of DCM as the solvent, ambient temperature, and reaction time of 2 h, the target products were obtained in good to exceptional yields. This is a method with high diastereoselectivity (up to 99:1) and mild conditions. On a larger scale of 0.5 mmol, an excellent yield of 98% and >99:1 dr was achieved.
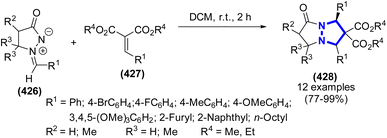 |
| | Scheme 114 Synthesis of trifluoromethyl-containing pyrazolidine scaffolds in DCM. | |
A domino aza-Michael/hemiacetal reaction between di-substituted hydrazine and α,β-unsaturated aldehydes to form pyrazolidine derivatives under the catalysis of pyrrolidine in CH2Cl2 was demonstrated by Z.-C. Geng et al. (2012) (Scheme 115).166 The desired products were acquired in substantial yields under the optimal reaction conditions. This process was regioselectively regulated, had a broad substrate scope, and was devoid of any additives.
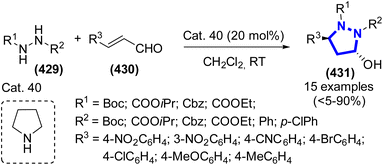 |
| | Scheme 115 Synthesis of pyrazolidine derivatives catalyzed by Cat. 40. | |
M. Fernandez et al. (2012) introduced an aza-Michael/hemiaminalization reaction between α,β-unsaturated aldehydes and N,N′-disubstituted hydrazides to form pyrazolidines with the catalysis of readily available silyl diarylprolinol ether in toluene solvent (Scheme 116).167 Although the product yields were quite low, this was a regioselective and metal-free protocol.
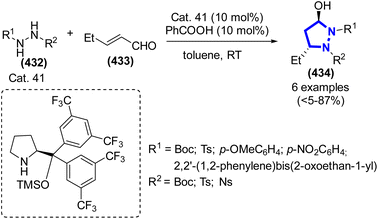 |
| | Scheme 116 Synthesis of pyrazolidines catalyzed by Cat. 41. | |
The 1,3-diamination between cinnamic aldehydes and di-1,2-N-tert-butoxycarbonyl (Boc)-protected hydrazine to form 3-hydroxypyrazolidines catalyzed by easily accessible chiral amines Cat. 42 was introduced by L. Deiana et al. (2012) (Scheme 117).168 This was a chemo- and enantio-selective-controlled (>99% ee) aza-Michael/hemiaminal domino sequence, leading too good to exceptional yields.
 |
| | Scheme 117 Synthesis of 3-hydroxypyrazolidines catalyzed by Cat. 42. | |
Remziye Azra Kartop and colleagues (2023) documented the design and synthesis of a series of new pyrazolidine derivatives from N-acyl hydrazones using N-triflylphosphoramide (NTPA)-catalyzed [3 + 2] cycloaddition (Scheme 118). The structural elucidation of the produced compounds was performed using several spectroscopic techniques, including IR, NMR, MS, and elemental analysis. The cytotoxic efficacy of the pyrazolidine derivatives was assessed against human lung cancer (A549), pancreatic cancer (PANC 1), prostate cancer (DU-145), breast cancer (MCF-7), and human dermal fibroblasts (HDF) using the MTS test. Compounds 440 and 441 (IC50 = 12.5–21.8 μM) exhibited superior antiproliferative activity compared to all other compounds against the A549 human lung cancer cell line and the PANC1 pancreatic cancer cell line, respectively (Scheme 119).169
 |
| | Scheme 118 Creation of novel pyrazolidines through [3 + 2] cycloaddition. | |
 |
| | Scheme 119 Chemical structures of bioactive compounds containing a pyrazole framework. | |
2.2.4. Pyrazole (pyrazoline). Pyrazoles are five-membered heterocyclic compounds with two contiguous nitrogen atoms at positions 1 and 2. This distinctive structure confers considerable chemical stability and biological activity to pyrazole derivatives, making them advantageous in medicinal chemistry, agrochemicals, and materials science.170 Many pyrazole moieties exhibit antitubercular, antimicrobial, anti-consultant, anticancer, and antiviral activities and act as selective COX-2 inhibitors such as celecoxib and epirizole.171–175K. D. Dwivedi et al. (2020) employed water extract from banana peels (WEB) as a catalyst for the preparation of pyrano[2,3-c]pyrazoles from the reaction between arylidene malononitriles and pyrazolone at r.t. after 30 min (Scheme 120).176 This was a green and broad synthesis technique that did not use any harmful organic solvents and ligands. Also, this methodology is versatile and may be effectively employed for a diverse array of substrates, resulting in yields ranging from high to exceptional. This approach offers several advantages, including quick reaction, environmentally favorable reaction conditions, and facile product separation without the need for column chromatography.
 |
| | Scheme 120 Synthesis of pyrano[2,3-c]pyrazoles catalyzed by WEB. | |
In the study conducted by H. F. Zhang et al. (2014), they presented a comprehensive analysis on the synthesis of a diverse array of functionalized fluorinated dihydropyrano[2,3-c]pyrazoles. The synthesis of pyrazoles from the (Z)-isomer was discussed. The reaction between 1H-pyrazol-5(4H)-ones and malononitrile was conducted in chloroform as the solvent at 0 °C (Scheme 121).177 The application of Cat. 44 as the catalyst resulted in the successful synthesis of the desired products through a domino Michael addition and Thorpe-Ziegler type cyclization. The reaction exhibited high yields, reaching up to 98%, and satisfactory enantioselectivity, with enantiomeric excess values of up to 90%.
 |
| | Scheme 121 Synthesis of functionalized dihydropyrano[2,3-c]pyrazoles catalyzed by Cat. 44. | |
With the catalytic activity of readily accessible primary amine produced from hydroquinine and 2-FC6H4CO2H, a series of tetrahydropyrano[2,3-c]pyrazoles was synthesized via the reaction between alkylidene pyrazolones and cyclic ketones/pentanal, which was reported by R. Maity et al. (2017) (Scheme 122).178 This enantioselective cascade Michael-hemiketalization reaction led to moderate to high yields of products containing three adjacent stereogenic centers.
 |
| | Scheme 122 Synthesis of tetrahydropyrano[2,3-c]pyrazoles catalyzed by Cat. 45. | |
Y. Wu et al. (2018) reported the utilization of quinine-derived squaramide as a catalyst for the successful synthesis of tetrahydropyrano[2,3-c]pyrazoles. This technique involved the reaction of 1,3-enynes, 1-aryl-3-substituted pyrazolones, and methyl vinyl ketone (Scheme 123).179 This protocol produced the desired products in good to exceptional yields from easily accessible starting materials without the isolation of the intermediate. The diastereomeric ratios were determined to be >20:1 dr by 1H-NMR analysis and the ee value by HPLC analysis was in the range of 82–95%.
 |
| | Scheme 123 Synthesis of tetrahydropyrano[2,3-c]pyrazoles catalyzed by Cat. 46. | |
A. Dandia et al. (2018) described the succession of oxidation, Michael addition, cyclization, and dehydration reaction between benzyl alcohol and 1H-pyrazol-5(4H)-one to prepare pyrano-2-one derivatives in water at r.t. (Scheme 124).180 It was also concluded that water was necessary for the production of C–C bonds and other phases of the reaction. The intrinsic benefits of this protocol include chemoselectivity, reduced reaction time, high yields, easy work-up approach, no need for chromatographic purification, and the use of green solvents, namely water.
 |
| | Scheme 124 Synthesis of pyrano-2-one derivatives in water. | |
V. Ramesh et al. (2016) reported a five-component method using various aromatic aldehydes, thiophenols, 3-oxobutanoates, phenylhydrazine, and malononitrile for the synthesis of thioether-containing dihydropyrano[2,3-c]pyrazoles after 20 min at 120 °C (Scheme 125).181 The aforementioned synthetic approach exhibited several advantages, such as its expeditious establishment, absence of purification protocols, swift reaction kinetics with favorable atom efficiency, and notable product yields ranging from 81% to 86%.
 |
| | Scheme 125 Synthesis of dihydropyrano[2,3-c]pyrazoles under solvent-free conditions. | |
This study encompasses a diverse array of pyrano[2,3-c]pyrazoles. The synthesis of 3′-carboxylate derivatives was elucidated by V. L. Gein et al. (2017) through a multi-component reaction involving diethyl oxalacetate sodium salt, isatin, malononitrile, and hydrazine hydrate. The reaction was conducted in the presence of acetic acid within ethanol as the solvent (Scheme 126).182 The yields of the required goods exhibited favorable to exceptional performance.
 |
| | Scheme 126 Synthesis of pyrano[2,3-c]pyrazole derivatives catalyzed by CH3COOH. | |
A. Alizadeh et al. (2014) devised a facile regioselective MCR comprised of ninhydrin, malononitrile, hydrazine derivatives, and β-keto esters or dimethyl acetylene dicarboxylate to synthesize spiro-pyranopyrazoles and oxa-aza[3.3.3]propellanes in the presence of the catalytically active piperidine (Scheme 127).183 The simplicity, regioselectivity, without metal catalyst or column chromatography purification, and high yields of this approach make it valuable.
 |
| | Scheme 127 Synthesis of spiro-pyranopyrazoles and oxa-aza-[3.3.3]propellanes catalyzed by Cat. 16 in ethanol. | |
A four-component cascade reaction involving N-phenyl hydrazine, various aromatic aldehydes, ethylacetoacetate, and nitroketene-N,S-acetal to produce pyranopyrazole frameworks catalyzed by piperidine at 120 °C under solvent-free conditions was introduced by K. Jayabal et al. (2014) (Scheme 128).184 Through a sequence of condensation-Knoevenagel–Michael-annulation reaction, the products were obtained in high yields, ranging from good to outstanding. This technology is notable owing to its rapid response time, great yield, low cost, simple operation, and huge structural variety.
 |
| | Scheme 128 Synthesis of pyranopyrazole frameworks catalyzed by Cat. 16. | |
In EtOH as the solvent and triethylamine as the catalyst, a cascade multicomponent reaction of hydrazine, dimethyl acetylene dicarboxylate, isatin, and malononitrile or ethyl cyanoacetate to form pyrano[2,3-c]pyrazole derivatives was accomplished by C. Wang et al. (2015) (Scheme 129).185 Additionally, this reaction has the benefits of utilizing an easily accessible starting material, applying mild reaction conditions, and exhibiting a straightforward operational procedure.
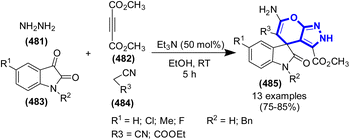 |
| | Scheme 129 Synthesis of pyrano[2,3-c]pyrazole derivatives catalyzed by Et3N. | |
Singh and colleague (2017) detailed an environmentally friendly and efficient method, i.e., the one-pot, multi-component synthesis of pyrano[2,3-c]pyrazoles facilitated by visible light, achieved without the use of catalysts or solvents (Scheme 130). This protocol is characterized by its cost-effectiveness, nontoxic nature, absence of catalysts and solvents, accessible reactants, and lack of requirement for specialized glassware and a photo-reactor system. The current methodology was further broadened by employing diethyl acetylene dicarboxylate as the reactant instead of ethylacetoacetate (Scheme 131).186
 |
| | Scheme 130 Synthesis of pyrano[2,3-c]pyrazoles catalyzed by metal-free catalysts. | |
 |
| | Scheme 131 Chemical structures of bioactive compounds containing a thiazolidine/isothiazolidine framework. | |
2.2.5. Thiazolidine/isothiazolidine. Thiazolidine is a cyclic molecule consisting of five members, wherein a thioether group is located at position 1 and an amine group is located at position 3. Thiazolidine and its derivatives have demonstrated antibacterial, anti-inflammatory, anticonvulsant, antimalarial, analgesic, anti-HIV, and anticancer activities.187–191A. Daghlavi et al. (2020) reported a Nef-isocyanide multicomponent reaction employing alkyl chloroglyoxalates, cyclohexyl isocyanide, and cyclic thiourea derivatives in the presence of graphene oxide-supported phosphoric acid to synthesize some hydroxy-thiazolidine-4-carboxylate derivatives (Scheme 132).192 The advantages of this method include the utilization of widely accessible chemicals, reusability of the catalyst, high yields, few environmental dangers, no requirement for stereoisomer separation, and as a result, a reduced number of total stages. The catalyst was employed for a series of five consecutive reactions without any decrease in the overall reaction yield observed.
 |
| | Scheme 132 Synthesis of hydroxy-thiazolidine-4-carboxylate derivatives catalyzed by phosphoric acid supported on graphene oxide. | |
G. Rainoldi et al. (2018) conducted a Ugi-type three-component reaction involving oxindole-based thiazolines, various isocyanides, and carboxylic acids in fluorinated solvents at ambient temperature. This reaction resulted in the formation of functionalized spirooxindole-fused thiazolidines (Scheme 133).193 The required compounds were obtained in satisfactory to favorable yields, exhibiting significant diastereoselectivity, efficient separation, and straightforward workup techniques.
 |
| | Scheme 133 Synthesis of spirooxindole-fused thiazolidine derivatives. | |
In the study by B. Kaboudin et al. (2018), they utilized Et3N as the catalyst and EtOH as the solvent to facilitate a multi-component condensation-cyclization process. This reaction involved the combination of hydrazine, allyl isothiocyanate, α-haloketone, and various aldehydes to produce thiazolidine-4-one and 3H-thiazole derivatives (Scheme 134).194 This approach is notable for its excellent yields, easy set-up, easily accessible starting ingredients, and simple reaction procedure without the isolation of the intermediate.
 |
| | Scheme 134 Synthesis of a wide range of thiazolidine-4-one and 3H-thiazoles catalyzed by Et3N. | |
H. Medini et al. (2015) introduced a synthetic route for a series of thiazoline-2-thione derivatives from secondary β-amino alcohols via electrolysis using carbon disulfide under the catalysis of an electrogenerated base (Scheme 135).195 The resulting compounds were obtained in yields of varying degrees, ranging from minor to significant.
 |
| | Scheme 135 Synthesis of thiazoline-2-thione derivatives in the presence of CS2. | |
A one-pot three-component reaction among 1,3-diketones, cyanates, and ethyl chloroacetate to synthesize a wide range of thiazolidine/oxazolidine derivatives in [Et3NH][HSO4] IL (20 mol%) was described by A. M. Malla et al. (2015) (Scheme 136).196 Moreover, the IL in this technique has both catalytic and medium engineering capabilities, which is a unique aspect of this approach. The advantages of this protocol include its outstanding yield, moderate reaction conditions, large substrate scope, decreased chemical waste, short reaction time, easy operating method, straightforward catalyst manufacture, and retained catalytic activity even after being recycled up to four times.
 |
| | Scheme 136 Synthesis of thiazolidine/oxazolidine derivatives catalyzed by [Et3NH][HSO4]. | |
A. Ziyaei-Halimehjani et al. (2012) reported the reaction among allyl amines, carbon disulfide, and iodine to synthesize thiazolidine-2-thiones through regiospecific iodocyclization in tetrahydrofuran at ambient temperature (Scheme 137).197 This protocol is easy, effective, and regiospecific, with good to high product yields.
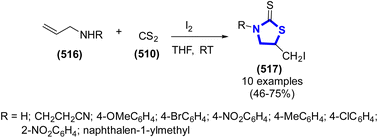 |
| | Scheme 137 Synthesis of thiazolidine-2-thiones catalyzed by I2. | |
B. R. P. Kumar et al. (2006) reported the microwave-induced reaction between thiazolidine-2,4-dione or thiazolidine-2-thioxo-thiazolidine-4-one and different aryl and heteroaryl aldehydes via Knoevenagel condensation in the presence of piperidine, activated silica gel, and acetic acid at the MW power of 900 W for 6–8 min to produce a wide range of thiazolidine-2,4-diones or 2-thioxothiazolidine-4-ones, respectively (Scheme 138).198 This protocol is advantageous because of its quick reaction speeds, high yields, no use of solvent, and no pollutants generated. Moreover, compared to conventional heating, microwave heating showed comparatively higher yields.
 |
| | Scheme 138 Synthesis of thiazolidine-2,4-dione or 2-thioxothiazolidine-4-one derivatives under MW. | |
Takumi Abe et al. showed that indoline hemiaminals yield 2,5-diaryl-4-hydroxythiazolines via a thioamidation/ring-switch sequence (2024). The effectiveness of this transformation hinges on the use of thioamide as a thiazoline precursor under transitory tautomeric regulation. This transformation involves gentle reaction conditions with favorable yields and extensive functional group tolerance (17 samples, up to 99% yield) (Scheme 139).199
 |
| | Scheme 139 Synthesis of thiazolo[4,5-b]indoles. | |
Xiantao Ma et al. disclosed an innovative approach for synthesizing 2-substituted-4,5-dihydrothiazol-4-ols via the cyclization of thioamides and 1,4-dithiane-2,5-diol in the presence of Et3N (2025). Stable hydroxy-substituted thiazole compounds were obtained from substituted thioamides. The reaction conditions are straightforward, the substrate scope is extensive, and the target compounds were acquired in yields of up to 82% (Scheme 140).200
 |
| | Scheme 140 Synthesis of 2-substituted-4,5-dihydrothiazol-4-ols. | |
2.3. Five-membered heterocycles containing at least three nitrogen atoms
2.3.1. Triazole. In the class of nitrogen-containing heterocyclic compounds, triazoles stand out as having particularly advantageous therapeutic uses. The structure of five-membered triazoles may be broken down into two distinct categories, i.e., 1,2,3-triazole and 1,2,4-triazole. Both 1,2,3- and 1,2,4-triazoles have structural features that allow them to support a wide variety of substituents around their core structures. This presents an opportunity for the advancement of several novel bioactive products. Triazole compounds have a broad range of pharmacological activities, including antibacterial,201,202 analgesic,203,204 anti-inflammatory, local anesthetic,205,206 antineoplastic,207,208 antiviral,209 antiproliferative,210 and anticancer211 activities (Scheme 141).
 |
| | Scheme 141 Chemical structures of bioactive compounds containing a triazole framework. | |
In a study conducted by Venigalla et al. (2020), an ultrasonic-assisted reaction was employed to synthesize a range of triazole derivatives using aldehydes and semicarbazide in EtOH at r.t. Notably, the reaction proceeded without the use of a catalyst (Scheme 142).212 This Knoevenagel-cyclic condensation has many advantages such as ecological friendliness, clean and simple operation, cost-effectiveness, fast reaction time, easy set-up, high output, and reduced waste generation.
 |
| | Scheme 142 Synthesis of triazole derivatives in the absence of a catalyst. | |
In the study conducted by A. Aksenov et al. (2020), the utilization of polyphosphoric acid (PPA) as a catalyst was investigated for the purpose of synthesizing 1,2,4-triazolo[4,3-a]quinolines. This synthesis involved the reaction between 2-hydrazinylquinolines and nitroalkanes, which was carried out at 130 °C (Scheme 143).213 This synthetic route resulted in good to outstanding product yields. In addition, several triazoloquinolines showed intriguing action in promoting the differentiation of neuroblastoma cancer cells.
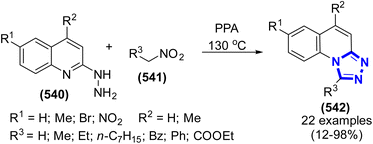 |
| | Scheme 143 Synthesis of 1,2,4-triazolo[4,3-a]quinoline derivatives catalyzed by PPA. | |
N. Guo et al. (2019) developed a method for the preparation of 1,2,3-triazole derivatives via the multicomponent reaction among primary amines, 1,3-dicarbonyls, and tosyl azide catalyzed by AcOH in DCM at 90 °C (Scheme 144).214 The present cycloaddition reaction, employing readily accessible starting materials, resulted in satisfactory to outstanding yields of the respective products.
 |
| | Scheme 144 Preparation of 1,2,3-triazole derivatives catalyzed by acetic acid. | |
A wide variety of 1,2,3-triazole-fused isoindoles produced by a one-pot two-step intramolecular azide–alkyne cycloaddition was published by N. Okamoto et al. (2018) (Scheme 145).215 The first step was the creation of a gem-diazide intermediate from o-alkynylbenzaldehydes and trimethylsilyl azide under the catalysis of trimethylsilyl trifluoromethanesulfonate in DCE at 0 °C to r.t. Following a reaction period of one hour, 4 Å molecular sieves and toluene were introduced, accompanied by an increase in temperature to 100 °C. This process resulted in the formation of the intended products, with yields ranging from good to outstanding.
 |
| | Scheme 145 Synthesis of 1,2,3-triazole-fused isoindoles catalyzed by TMSOTf. | |
A. V. Bogolyubsky et al. (2018) conducted a one-pot two-step conversion of various thioureas to 1,2,4-aminotriazoles derivatives (Scheme 146).216 The first step was the S-alkylation of thioureas using 1,3-propanesultone in NEt3, followed by consecutive ring closure with hydrazides to form 3-amino-1,2,4-triazoles. The advantages of this protocol include its cost- and time-effective manner, readily available starting ingredients, and no need for poisonous and volatile solvents but it has moderate yields.
 |
| | Scheme 146 Synthesis of 3-amino-1,2,4-triazole moieties catalyzed by NEt3. | |
In the study conducted by V. O. Filimonov et al. (2017), they synthesized of a range of 1,2,3-triazoles employing 2-cyanothioacetamides and sulfonyl azides using EtONa as the catalyst and EtOH as the solvent. The reaction took place at −10 °C (Scheme 147).217 The synthetic route used in this investigation yielded the intended compounds with results ranging from good to exceptional.
 |
| | Scheme 147 Synthesis of 1,2,3-triazoles catalyzed by EtONa. | |
W. Li et al. (2013) demonstrated the 1,3-dipolar cycloaddition of a wide range of α,β-unsaturated aldehydes and azides to form triazole-olefin derivatives in the presence of diethylamine and DBU in DMSO solvent at 60 °C and reaction time of 2 h (Scheme 148).218 The advantages of this approach include the use of a simple and low-cost catalyst, large substrate scope, easily accessible starting ingredients, simple operation, and high product yields.
 |
| | Scheme 148 Synthesis of triazole-olefin scaffolds catalyzed by amine. | |
2.3.2. Oxadiazole. Oxadiazoles are five-membered heterocyclic compounds including one oxygen atom and two nitrogen atoms in their ring structure, which exist in four isomeric forms including 1,2,3-oxadiazole, 1,2,4-oxadiazole, 1,3,4-oxadiazole, and 1,2,5-oxadiazole. Among them, 1,3,4-oxadiazole is the most extensively researched owing to its notable biological and pharmacological attributes.219 Oxadiazole derivatives have a wide variety of biological properties, including anti-cancer,220 anti-inflammatory,221 fungicidal,222 herbicidal,223 anti-HIV,224 antibacterial,225 and plant growth regulator actions (Scheme 149).226
 |
| | Scheme 149 Chemical structures of bioactive compounds containing an oxadiazole framework. | |
The intermolecular electrochemical cyclization of various α-keto acids and acylhydrazines was developed for the preparation of 1,3,4-oxadiazoles in nBu4NOAc and methanol by F. Lu et al. (2020) (Scheme 150).227 This protocol is advantageous because of the absence of transition metals or other oxidants, gentle reaction conditions, large substrate scope and great yields. The practicality of this methodology was examined using a 5-mmol-scale reaction, and the yield was shown to have no difference compared to the scale of 0.5 mmol.
 |
| | Scheme 150 Synthesis of 1,3,4-oxadiazole ring systems catalyzed by nBu4NOAc. | |
V. N. Telvekar et al. (2013) reported the rearrangement of 1,2-dioximes of α-diketones to Beckmann product 1,2,4-oxadiazoles using diphosphorus tetraiodide in DCM at r.t. (Scheme 151).228 This procedure using a new, gentle agent afforded the products in outstanding yields.
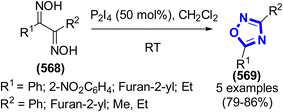 |
| | Scheme 151 Synthesis of 1,2,4-oxadiazole derivatives catalyzed by P2I4. | |
Under the oxidative effect of oxone and catalytic activity of NEt3, the oxidative desulfurization of various thiosemicarbazides to oxadiazole moieties using iodobenzene was examined by K. N. Patel et al. (2012) (Scheme 152).229 This synthetic route using easily accessible oxidants afforded matching compounds in high to exceptional yields.
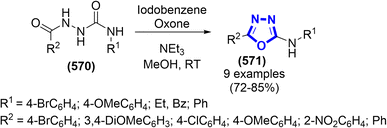 |
| | Scheme 152 . Synthesis of oxadiazole moieties catalyzed by NEt3. | |
The electrocyclization process involving the reaction of semicarbazone, formed from semicarbazide hydrochloride and various aldehydes, in the presence of sodium ethoxide, results in the formation of oxadiazoles. This reaction takes place in acetonitrile with lithium perchlorate as the electrolyte, with a platinum electrode serving as the catalyst. This methodology was first reported by L. K. Sharma et al. (2010) (Scheme 153).230 The implementation of this environmentally sustainable approach led to favorable outcomes in terms of product yields, ranging from satisfactory to exceptional.
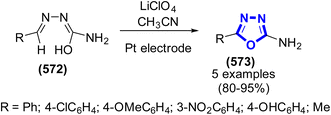 |
| | Scheme 153 Synthesis of oxadiazole frameworks catalyzed by LiClO4 via an electrolysis method. | |
V. V. Sureshbabu et al. (2008) presented a method for the synthesis of various 1,2,4-oxadiazole-linked orthogonally urethane-protected dipeptides via a two-step reaction between N-protected amino acyl fluoride and amino acid-derived amidoxime (Scheme 154).231 The first step was catalyzed by N-methylmorpholine in EtOH as the solvent at r.t. after 15 min and NaOAc was used as the catalyst in the second stage. This protocol afforded products with average to great yields.
 |
| | Scheme 154 Synthesis of 1,2,4-oxadiazole-linked orthogonally urethane-protected dipeptides catalyzed by Cat. 47. | |
The microwave-assisted coupling of amidoximes with carboxylic acids to prepare 1,2,4-oxadiazoles catalyzed by O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU)/1-hydroxybenzotriazole hydrate (HOBt) and excess N,N-diisopropylethylamine (DIPEA) in >80% yield was reported by M. D. Evans et al. (2003) (Scheme 155).232 Subsequently, employing a statistical design methodology, the optimal reaction conditions were predicted.
 |
| | Scheme 155 Synthesis of six 1,2,4-oxadiazole derivatives catalyzed by TBTU/HOBt and DIPEA. | |
2.3.3. Thiadiazole. Thiadiazole is a five-membered ring consisting of two nitrogen atoms, two carbon atoms, and a sulfur atom. Thiadiazole and its derivatives show numerous bioactivities such as antimicrobial,233,234 anti-inflammatory,235 anti-cancer,236,237 anticonvulsant,238 antidepressant,239 antioxidant,240 radioprotective,241 and anti-leishmanial.242,243 Also, several medications on the market contain a thiadiazole ring system, including acetazolamide, methazolamide, and sulfamethazole (Scheme 156).
 |
| | Scheme 156 Chemical structures of bioactive compounds containing a thiadiazole framework. | |
Y. Han et al. (2018) developed an approach for the synthesis of various 2-amino-1,3,4-thiadizole derivatives from thiosemicarbazide through intramolecular oxidative coupling in the presence of phenyl iodide and H2O2 with AcOH (Scheme 157).244 The current approach used readily available starting materials with moderate reaction conditions, adequate to exceptional product yields, and the exclusion of metal catalysts, hence improving the environmental sustainability of the process.
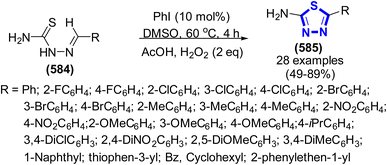 |
| | Scheme 157 Synthesis of 2-amino-1,3,4-thiadiazole catalyzed by PhI. | |
A synthetic route to various 4,5-functionalized 1,2,3-thiadiazoles from triazole catalyzed by a strong base in either protic (such as water, with or without acid) or aprotic (such as ethyl acetate) solvent at ambient temperature was described by V. O. Filimonov et al. (2017) (Scheme 158).217 This metal-free Cornforth-type rearrangement generated the products in great to outstanding yields. Based on DFT calculations, the process of rearrangement proceeds by initially generating a diazo molecule as an intermediate species. This rearrangement can be facilitated by acids, which catalyze the reaction through the protonation of the oxygen atom in the sulfonamide group.
 |
| | Scheme 158 Synthesis of 4,5-functionalized 1,2,3-thiadiazoles. | |
A. Feddouli et al. (2004) introduced the 1,3-dipolar cycloaddition of nitrilimines created by the in situ reaction between α-bromoglyoxalates and triethylamine with (1R)-thiocamphor to form various camphane-2-carboxylic acid ethyl esters in DCM at ambient temperature (Scheme 159).245 The structures of the pure 1,3,4-thiadiazoles diastereoisomers were thoroughly determined using spectroscopic methods, and based on the X-ray investigations, it was deduced that the absolute configuration of the C5 sporadic carbon is (R).
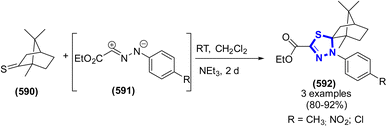 |
| | Scheme 159 Synthesis of 1,3,4-thiadiazole frameworks. | |
The reaction between ketothione and hydrazonoyl chlorides to synthesize dithiolo[1,4]thiazine derivatives in the presence of NEt3 was introduced by S. Barriga et al. (2001) (Scheme 160).246 This 1,3-dipolar cycloaddition approach gave the products in high yields.
 |
| | Scheme 160 Synthesis of dithiolo[1,4]thiazines catalyzed by NEt3. | |
Y. Song et al. (1999) demonstrated the alkylation of thiadiazolethiones with alkyl chloride or alkyl iodide in the presence of NaOH in THF to form 1,3,4-thiadiazoles (Scheme 161).247 This protocol gave the desired products in modest to high yields. Moreover, following mass screening and SAR tests, 1,3,4-thiadiazole was found to be a powerful, selective, and orally active COX-2 inhibitor.
 |
| | Scheme 161 Synthesis of 1,3,4-thiadiazole derivatives catalyzed by NaOH. | |
2.3.4. Tetrazole. Tetrazoles are five-membered heterocyclic compounds characterized by the presence of four nitrogen atoms in their ring structure. They have considerable chemical stability, acidity equivalent to carboxylic acids, and robust hydrogen bonding potential, making them advantageous in medicines, materials science, and coordination chemistry.248 Tetrazole derivatives are extensively used in medicinal chemistry as bioisosteres of carboxylic acids, enhancing metabolic stability and membrane permeability in drug design. Tetrazole moieties display many bioactivities such as antiviral,249,250 anticancer,251,252 antibacterial,253 antifungal,254 antioxidant,255 and inhibitors of metabolic processes.256D. P. Zarezin et al. (2018) reported the azido-Ugi multicomponent reaction of trimethylsilyl azide, cytisine, formaldehyde, and various isocyanides in the presence of MeOH solvent to synthesize tetrazole derivatives of cytisine (Scheme 163).257 This was a useful synthetic route because either aliphatic, aromatic, functionalized, or sterically hindered isocyanides could be employed, and the product yields were excellent.
Z. N. Tisseh et al. (2012) developed a synthetic route for catalyst-free cascade Knoevenagel condensation/1,3 dipolar cycloaddition, producing various 5-substituted-tetrazoles (Scheme 162).258 The experimental procedure involved the execution of a three-component reaction utilizing aromatic aldehydes, malononitrile, and NaN3 in an aqueous environment at 50 °C. This reaction yielded high quantities of products. Because there was no catalyst or additional agent, this innovative method allowed the use of water and avoided ecologically hazardous traditional organic solvents, as well as quick set-up and minimal waste generation.
 |
| | Scheme 162 Chemical structures of bioactive compounds containing a tetrazole framework. | |
 |
| | Scheme 163 Synthesis of tetrazole derivatives of cytisine. | |
3. 6-Membered rings
3.1. Six-membered heterocycles containing one nitrogen atom
3.1.1. Piperidine. Piperidine is a saturated heterocyclic molecule with six rings and one nitrogen atom. Its adaptable and non-aromatic configuration confers substantial chemical and biological attributes, establishing it as a crucial scaffold in pharmaceutical chemistry, organic synthesis, and materials research.259 Piperidine derivatives are prevalent in bioactive compounds and pharmacological agents in medical chemistry. They demonstrate a wide range of pharmacological actions, including analgesic, antihypertensive, depressive, antipsychotic, antiviral, and anticancer properties. Numerous prominent pharmaceuticals, including Risperidone (antipsychotic), Loperamide (antidiarrheal), and Paroxetine (antidepressant), contain a piperidine ring, underscoring its significance in drug design and optimization (Scheme 164 and 165).260,261
 |
| | Scheme 164 Synthesis of 5-substituted-tetrazole moieties without a catalyst. | |
 |
| | Scheme 165 Chemical structures of bioactive compounds containing a piperidine framework. | |
Hamid Reza Shaterian and Kobra Azizi (2013) reported a green procedure for the synthesis of highly functionalized piperidine derivatives (Scheme 166).262 The five-component reactions of aromatic aldehydes, aniline, and β-ketoester were conducted under the catalysis of an acidic IL. A range of acidic ILs was examined, including [Hmim]HSO4, [TMG]ClO4, and [TMG]TFA. The reaction mixture was stirred at 100 °C for 10 min in the absence of any solvent, resulting in the desired outcome. In particular, the IL has potential to be efficiently removed from the reaction media, and subsequently utilized in many cycles. The employed procedure demonstrated efficacy, diastereoselectivity, and convenience as a one-pot method.
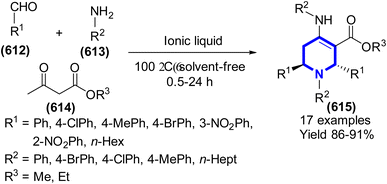 |
| | Scheme 166 Synthesis of functionalized piperidine catalyzed by an ionic liquid. | |
In the study conducted by Zhou et al. (2016), they documented the utilization of borane as a catalyst for the metal-free transfer hydrogenation of pyridines (Scheme 167).263 A diverse range of pyridines was produced through the utilization of piperidine derivatives, ammonia borane as the hydrogen source, and tris(perfluorophenyl)borane as the catalyst. The reactions were conducted using toluene as the solvent at 120 °C for 6 h, generating products in yields ranging from 44% to 88%, accompanied by cis-selectivity that varied from moderate to outstanding.
 |
| | Scheme 167 Synthesis of piperidine derivatives catalyzed by tris(perfluorophenyl)-borane. | |
Hongwei Zhang and Kilian Muniz (2017) described a way to selectively create piperidine via intramolecular catalytic Csp3–H amination (Scheme 168).264 In the experimental procedure, the reactions were performed in the presence of molecular iodine and NBS, utilizing a visible light source, over a duration of 4 h at ambient temperature. The reaction was investigated using sulfonimides, trimethyl-silylethylsulfonyl, and 2-bromophenyl sulfonyl under the optimized conditions. The corresponding piperidination products were obtained in medium to high yield. The experimental procedure showcased a technique characterized by gentle and consistent conditions, enabling the targeted synthesis of 2-arylsubstituted piperidines.
 |
| | Scheme 168 Synthesis of piperidines catalyzed by iodine and N-bromosuccinimide (NBS) (Cat. 26). | |
Audumbar Patil et al. (2018) developed a metal-free green procedure for the preparation of piperidine scaffolds from formaldehyde, aromatic aniline, and 1,3-dicarbonyl compounds (Scheme 169).265 2,2,2-Trifuoroethanol was used as the catalyst with a strong acidic property, which played a crucial function in increasing the rate of the reaction and initiating it.
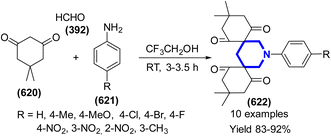 |
| | Scheme 169 Synthesis of piperidines catalyzed by 2,2,2-trifluoroethanol. | |
Rene Ebule et al. (2019) published an aza-Pummerer protocol for the synthesis of piperidines from homoallylic (Scheme 170).266 The present investigation demonstrated that the combination of hydrochloric acid (HCl) and N,N-dimethylpropyleneurea (DMPU) facilitates the generation of the (thiomethyl)methyl carbenium ion from dimethyl sulfoxide (DMSO) under mild conditions. High yields of 4-chloropiperidines were obtained through the cyclization of aromatic and aliphatic amines using the HCl/DMPU reagent. The aza-Pummerer method, facilitated by a combination of 5 + 1 protic acids, demonstrates a broad substrate scope. Under these conditions, the reaction exhibits high chemical yields, while effectively accommodating diverse functional groups. Furthermore, the scalability of this approach is noteworthy (Scheme 171).
 |
| | Scheme 170 Synthesis of piperidines catalyzed by HCl/DMU. | |
 |
| | Scheme 171 Chemical structures of bioactive compounds containing a pyridine framework. | |
3.1.2. Pyridine. Pyridine is a six-membered aromatic heterocycle with one nitrogen atom in its ring. The structural resemblance of pyridine to benzene, together with the electron-withdrawing characteristics of nitrogen, bestows it and its derivatives distinct chemical and electronic qualities, making them exceedingly desirable in many scientific and commercial applications.267 Pyridine serves as a crucial scaffold in medicinal chemistry, significantly improving bioavailability, metabolic stability, and target binding in several pharmaceutical medicines. Drugs containing pyridine have a diverse array of pharmacological effects. In contemporary medicine, pyridine derivatives are also becoming an increasingly important class of compounds with antimicrobial,268 antiviral,269 antioxidant,270 and anticancer271 activities.P. Baumgarten and A. Dornow (1939) reported a process for the synthesis of 2,3-disubstituted pyridine derivatives (Scheme 172).272 The reactions were performed via the condensation of alkyl or vinyl amines with 1,3-dicarbonyl derivatives.
 |
| | Scheme 172 Synthesis of pyridines under catalyst-free conditions. | |
Michael Robinson et al. (1992) prepared pyridines from enamino nitriles (Scheme 173).273 Under acidic conditions (using NH4OAc/HOAc), the target pyridine compounds were afforded in about 42% yield from the cyclization of chalcones and 1-amino-2-cyanocyclopentes.
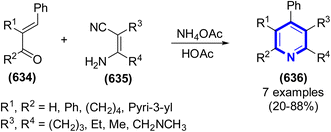 |
| | Scheme 173 Synthesis of pyridines catalyzed by AcOH. | |
Bagley et al. (2001) described a unique adaptation of the Bohlmann–Rahtz pyridine synthesis method (Scheme 174).274 In this method, the catalyst employed was either AcOH or Amberlyst-15. In a single synthetic step conducted at 50 °C, a diverse array of functionalized pyridines was derived through the cyclization reaction between enamino esters and alkynone substrates.
 |
| | Scheme 174 Synthesis of pyridines catalyzed by Amberlyst-15 ion exchange resin or HOAc. | |
In the study by Bagley et al. (2002), they developed a novel MW approach for the preparation of pyridines, employing the Bohlmann–Rahtz reaction (Scheme 175).275 Under microwave irradiation, the ethyl aminocrotonate reacted with various alkynones throughout a two-step procedure including Michael addition of enamine and alkynone and cyclodehydration to pyridine. The experiment was performed at 170 °C in toluene solvent, utilizing irradiation at either 150 or 160 W for a duration of 20 min.
 |
| | Scheme 175 Synthesis of pyridines under catalyst-free conditions. | |
In 2002, Kelly and Lebedev proposed a methodology for synthesizing a selection of unsymmetrical bridged terpyridines (Scheme 176).276 The synthesis of the pyridines was achieved through the condensation reaction between amine and carbonyl molecules. The present methodology employed intra- and inter-molecular Michael reactions as crucial steps.
 |
| | Scheme 176 Synthesis of unsymmetrical bridged terpyridines catalyzed by AcOH. | |
Xin Xiong et al. (2004) showed a novel mild condensation approach for the preparation of poly-substituted pyridines (Scheme 177).277 The reaction involving alkynones, 1,3-dicarbonyl compounds, and NH4OAc was conducted under reflux in alcoholic solvents for a duration of 24 h, in the absence of a catalyst. The present study showcased a novel heteroannulation reaction involving three components, which was successfully conducted using highly efficient and gentle reaction conditions. The pyridines that corresponded to the given conditions were obtained in yields ranging from modest to good and obtained as single regioisomers.
 |
| | Scheme 177 Synthesis of pyridines under catalyst-free conditions. | |
The mild aromatization of 1,4-dihydropyridines under metal-free conditions was reported by Xinqiang Fang et al. (2007) (Scheme 178).278 This method for the oxidation aromatization of 1,4-dihydropyridines to symmetrical and unsymmetrical pyridines was performed at r.t. in the presence of Cat. 48 as the organocatalyst. The reaction mixture was refluxed in anhydrous acetonitrile solvent under an oxygen atmosphere, while exposed to irradiation from a 150 W high-pressure mercury lamp. The light emitted from the lamp was filtered to exclude wavelengths below 310 nm. This study showcases the efficacy of 9-phenyl-10-methylacridinium as a very efficient photocatalyst. This organocatalyst has a significant level of recoverability and reusability.
 |
| | Scheme 178 Synthesis of pyridines catalyzed by Cat. 48. | |
In the study by Shujiang Tu et al. (2007), they documented the utilization of a Krohnke reaction as a means to synthesize terpyridines (Scheme 179).279 The experimental protocol involved a one-pot reaction using 2-acetylpyridines, aromatic aldehydes, and ammonium acetate, employing either MW or conventional heating. Various terpyridines were afforded in aqueous medium. This protocol is a convenient, economic, and environmentally friendly process.
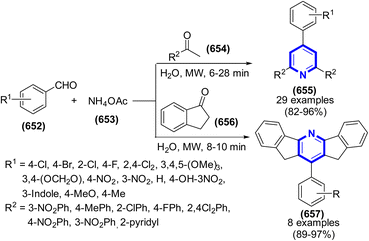 |
| | Scheme 179 Synthesis of terpyridines under catalyst-free conditions. | |
Kazuhiro Yoshida et al. (2009) reported the discovery of a novel synthesis pathway for the production of 3-hydroxypyridines (Scheme 180).280 The reactions were conducted by treating pyridin-3-ones with Cat. 12 in dimethylformamide at ambient temperature for a duration of 1 h. The corresponding products were generated in high yields. The primary advantages of this approach are its simplicity, flexibility, and ability to prevent the production of inseparable regioisomers.
 |
| | Scheme 180 Synthesis of 3-hydroxypyridines catalyzed by Cat. 12. | |
In the study by Boecker et al. (1986), they successfully synthesized derivatives of pyridines with 4-aryl and 4-alkyl substitutions via the enzymatic oxidation of 1,4-dihydropyridines (Scheme 181).281 Cytochrome P-450 enzyme was used as an efficient oxidase biocatalyst in this process.
 |
| | Scheme 181 Synthesis of 4-substituted pyridines catalyzed by cytochrome P-450 enzyme. | |
Jhillu S. Yadav et al. (2000) synthesized pyridines via the aromatization of Hantzsch 1,4-dihydropyridines (Scheme 182).282 The aromatization method was performed by refluxing Hantzsch 1,4-dihydropyridines with iodine in methanol. Various alkyl, benzyl, aryl, and heterocyclic 4-substituted pyridines were afforded in high yields. This protocol is highly advantageous over existing methods because of its simple and mild reaction conditions.
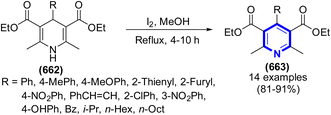 |
| | Scheme 182 Synthesis of pyridines catalyzed by molecular iodine. | |
Natsuki Nakamichi et al. (2004) developed a method for the activated carbon-promoted oxidative aromatization of Hantzsch 1,4-dihydropyridines (Scheme 183).283 In the presence of molecular oxygen, 1,4-dihydropyridines were treated in xylene or AcOH using activated carbon as the catalyst. The reaction mixture was placed in a three-necked flask and subjected to stirring at 120 °C for 30 min. The corresponding pyridine derivatives were obtained from a series of Hantzsch 1,4-dihydropyridines with substituents on their 4-position. This is a simple, environmentally friendly, economical, and operationally simple procedure without the use of metal oxides or organic oxidizing agents.
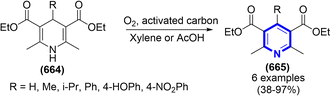 |
| | Scheme 183 Synthesis of pyridines catalyzed by activated carbon. | |
J. S. Yadav et al. (2006) aromatized 1,4-dihydropyridines using iodoxybenzoic acid as an efficient oxidant (Scheme 184).284 The experiment was conducted using DMSO as the solvent, at a temperature in the range of 80–85 °C. The reaction mixture was heated for a period of 2–4 h depending on the structure of the substrate. A wide variety of pyridine derivatives was produced in elevated yields. The advantages of this approach include the use of inexpensive oxidants, mild reaction conditions, and easy workup.
 |
| | Scheme 184 Synthesis of pyridines catalyzed by iodoxybenzoic acid. | |
Xiaojing Wei et al. (2014) proposed a novel metal-free-mediated oxidation aromatization technique for the conversion of 1,4-dihydropyridines to pyridines (Scheme 185).285 The process of aromatization of 1,4-dihydropyridines was facilitated through the use of Cat. 49 as a photosensitizer. Instead of using a photoredox catalyst and inorganic oxidant, the advantages of this protocol are the use of the organic dye eosin-Y and molecular oxygen.
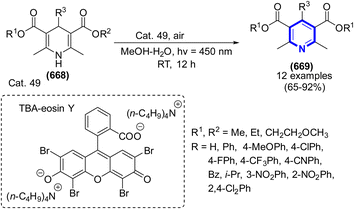 |
| | Scheme 185 Synthesis of pyridines catalyzed by Cat. 49. | |
In the study by Mohammad Movassaghi et al. (2007), they presented a technique enabling the synthesis of pyridine derivatives via a single-step and convergent approach (Scheme 186).286 The cyclization reactions were conducted under controlled conditions in dichloromethane at temperatures ranging from −78 °C to 0 °C. These reactions involved the direct condensation of N-vinyl/aryl amides with several π-nucleophile reagents, including ynamide, ynamine, alkyl vinyl ether, and silyl ether. The combinations were subjected to heating with trifluoromethanesulfonic anhydride (Tf2O) in the presence of 2-chloropyridine (2-ClPyr) as a base additive. A range of polysubstituted pyridines was synthesized in high yields.
 |
| | Scheme 186 Synthesis of pyridines catalyzed by 2-chloropyridine. | |
Brindaban C. Ranu et al. (2007) developed an improved process for the three-component synthesis of pyridines using task-specific ILs (Scheme 187).287 Stirring was used to execute the three-component condensations of aromatic aldehydes, malononitrile, and thiophenols, in the presence of 1-butyl-3-methylimidazolium hydroxide ion liquid ([Bmim]OH). A wide range of aromatics, as well as heteroaromatic pyridines, yielded good to exceptional results. The reactions were very clean, simple, and easy to work up. This is a green protocol because of the absence of hazardous organic solvents in the reaction and the reusability of ILs.
 |
| | Scheme 187 Synthesis of pyridines catalyzed by [Bmim]OH. | |
A general and efficient method for the 2-amination of pyridines was reported by Jingjun Yin et al. (2007) (Scheme 188).288 The pyridine N-oxide derivatives were treated with tert-butylamine in the presence of p-toluenesulfonic anhydride. This mixture was mixed in trifluorotoluene at 5–20 °C, followed by deprotection with trifluoroacetic acid (70 °C, 2–6 h). High yields of substituted pyridines were achieved under improved one-pot conditions.
 |
| | Scheme 188 Synthesis of pyridines catalyzed by p-toluenesulfonic anhydride. | |
Yaojia Jiang et al. (2014) researched a new strategy for the synthesis of pyridines via ring expansion of 2-allyl-2H-azirines (Scheme 189).289 It was claimed that the initial stage involved the formation of 1-azatrienes through the opening of 2-allyl-2H-azirines using Cat. 12 as a catalyst under metal-free conditions. Subsequently, the 1-azatrienes obtained were subjected to 6π-electrocyclization, resulting in the formation of pyridines. A diverse range of pyridines, bearing aryl, alkyl, and heterocyclic substituents, was synthesized with excellent efficiency, resulting in high to exceptional yields.
 |
| | Scheme 189 Synthesis of pyridines catalyzed by Cat. 12. | |
A new method for the synthesis of pyridines through a one-pot metal-free route was described by Hongbo Wei et al. (2015) (Scheme 190).290 The investigated technique is predicated on the utilization of cascade reactions involving aldehydes, phosphorus ylides, and propargyl azide. This approach occurred in three steps involved a Wittig reaction, Staudinger reaction, aza-Wittig reaction, 6π-3-azatriene electrocyclization, and 1,3-hydrogen shift. The present study described a novel and highly efficient metal-free one-pot synthetic protocol for the preparation of polysubstituted pyridines. This approach demonstrates several benefits, such as operational simplicity and the capacity to accept various functional groupings.
 |
| | Scheme 190 Synthesis of pyridines under catalyst-free conditions. | |
Huawen Huang et al. (2016) published a method for the transition-metal-catalyzed preparation of polysubstituted pyridines (Scheme 191).291 The reactions were conducted using O-acetylketoximes and acroleins in the presence of iodine and triethylamine. A diverse array of functional N-heterocycles was synthesized in moderate to excellent product yields with favorable chemo-selectivity. The investigation and proposal of the probable reaction mechanism for the metal-free synthesis of pyridines were conducted via mechanistic experiments. This comprehensive mechanism elucidated the function of iodine and triethylamine as activating agents for the N–O bond in the heterocyclization of oximes, thereby facilitating the process.
 |
| | Scheme 191 Synthesis of pyridines catalyzed by iodine. | |
Shen et al. (2015) developed a method for the synthesis of polysubstituted pyridines via the direct β-C(sp3)–H functionalization of enaminones, which offers a practical approach (Scheme 192).292 The two-step process in the one-pot annulation of 1-arylethylamines and ynones consists of an intermolecular Michael addition reaction followed by an intramolecular condensation. A wide range of pyridines was prepared under metal-free conditions in excellent yield and high regioselectivity. This technique exhibited compatibility with a wide range of substrates, demonstrated excellent atom efficiency, and showcased the significant synthetic potential of the resulting compounds.
 |
| | Scheme 192 Synthesis of pyridines catalyzed by an organic base. | |
Debashis Majee et al. (2016) proposed an innovative synthetic approach to produce 4,6-diarylpicolinates. This method involves a domino reaction between cyclic sulfamidate imines and Morita–Baylis–Hillman acetates derived from nitroolefins/nitrodienes (Scheme 193).293 The 5-membered cyclizations were performed using Cat. 19 as an organobase under metal-free conditions at 55 °C. A range of 4,6-diarypicolinate compounds was synthesized in high to outstanding yields.
 |
| | Scheme 193 Synthesis of pyridines catalyzed by Cat. 19. | |
Similarly, other metal-free and solvent-free protocols for the synthesis of diversely substituted picolinates were developed by Soumen Biswas et al. (2017) (Scheme 194).294 The formation of picolinate derivatives was achieved through a sequential reaction involving five- or six-membered cyclic sulfamidate imines and β,γ-unsaturated α-ketocarbonyls. This reaction was conducted under neat conditions and facilitated by MW. Under the optimized reaction conditions, the annulations were promoted by Cat. 19 as a solid organic base. The corresponding products were obtained including 4,6-disubstituted-picolinates, picolinates, and 4,6-diaryl-2-benzoyl/cinnamoyl-pyridines in good yields.
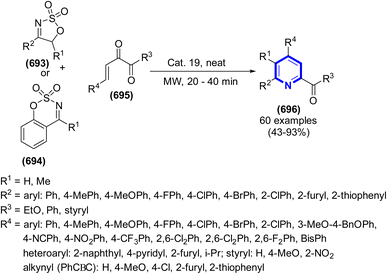 |
| | Scheme 194 Synthesis of pyridines catalyzed by Cat. 19. | |
In a study conducted by Yejun Gao et al. (2019), a method for synthesizing asymmetrical 2,6-diarylpyridines from α,β,γ,δ-unsaturated ketones was demonstrated. This process involved the Michael addition of ammonium formate, followed by annulation (Scheme 195).295 The cyclization process was performed in toluene solvent in ambient air at 110 °C for 16 h. A range of aliphatic and aromatic 2,4-dien-1-one substrates was employed to explore the annulation process. The corresponding products were obtained in high yields. This method offers numerous significant benefits, such as a simple and expedited work-up procedure, a wide range of substituents, lack of additives, outstanding yields, and a high level of regioselectivity.
 |
| | Scheme 195 Synthesis of 2,6-diarylpyridines under catalyst-free conditions. | |
Ghada Asskar et al. (2019) demonstrated the conversion of primary amines into pyridinium salts (Scheme 196).296 In this experimental procedure, glutaconaldehyde was employed as a substitute reagent for the zinc salt. In the present investigation, the degradation was effectively inhibited in the presence of amines. The conversion of primary amines to pyridinium salts was conducted using a Brønsted acid catalyst at 90 °C for 1 h. To synthesize a wide range of pyridines, a variation in the counterion and aniline was surveyed, resulting in excellent conversions.
 |
| | Scheme 196 Synthesis of pyridinium salts catalyzed by a Brønsted acid. | |
Inwon Kim et al. (2020) successfully devised a method for functionalizing the C4-pyridine scaffold using 1,4-dihydropyridines via photoinduced charge transfer (Scheme 197).297 The present study investigated the utilization of electron donor–acceptor complexes formed between N-amidopyridinium salts and 1,4-dihydropyridine derivatives, employing visible light irradiation as the primary stimulus. The resultant compounds were derived from different radicals of 1,4-dihydropyridines, including alkyl, acyl, and carbamoyl groups. This technique shows notable advantages, particularly in terms of superior regiocontrol, as well as the utilization of mild and metal-free settings. Notably, this strategy does not necessitate the use of a photocatalyst and may be effectively implemented under visible-light-promoted conditions.
 |
| | Scheme 197 Synthesis of pyridines under the catalyst-free conditions. | |
A new metal-free strategy for the synthesis of 2,3-disubstituted pyridines was reported by Peng Gao et al. (2020) (Scheme 198).53 The production of 2,3-disubstituted pyridines was achieved by means of a specific oxidative cyclization process involving N-hydroxyalkyl enamines and iodoxybenzoic acid. The process of synthesizing pyridines can be achieved using a two-step procedure, which entails the oxidation of alcohols and the subsequent condensation of aldehydes with the α-carbon of enamines. The reaction was conducted in tetrahydrofuran at 120 °C for 12 h. This is an efficient metal-free protocol for the synthesis of pyridines due to the use of environment-friendly substrates, wide reagent scope, mild conditions, and good efficiency.
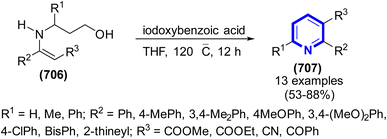 |
| | Scheme 198 Synthesis of pyridines catalyzed by iodoxybenzoic acid. | |
Jindian Duan et al. (2020) researched NH4I-triggered [4 + 2] cyclization for the synthesis of pyridines under metal-free conditions (Scheme 199).298 The strategy development in this study involved the annulation of α,β-unsaturated ketoxime acetates with N-acetyl enamides. The reaction mixture of ketoxime-enoate and N-acetyl enamide was heated in tetrahydrofuran with a certain amount of ammonium iodide and additive (Na2S2O4, NaHSO3, triethyl amine, or pyridine) at 120 °C for 8 h under a nitrogen atmosphere. A variety of polysubstituted pyridine derivatives was synthesized using an array of substrates, yielding adequate to outstanding results. A credible mechanism was suggested for this reaction based on the experimental results.
 |
| | Scheme 199 Synthesis of pyridines catalyzed by NH4I. | |
Jingjun Yin et al. (2007) synthesized 2-aminopyridines from pyridine N-oxides (Scheme 200).288 The products were obtained via a one-pot reaction using Ts2O and tBuNH2. The crude reaction mixture was treated with trifluoroacetic acid, which provided the tert-butyl group. This protocol is an efficient procedure for the 2-amination of 2-unsubstituted pyridine derivatives (Scheme 201).
 |
| | Scheme 200 Synthesis of 2-aminopyridines using Ts2O and t-BuNH2. | |
 |
| | Scheme 201 Chemical structures of bioactive compounds containing a quinoline/isoquinoline framework. | |
3.1.3. Quinoline and isoquinoline. Quinoline/isoquinoline are members of a class of organic compounds in the aromatic heterocyclic series. The compounds have a distinctive double-ring configuration including a benzene ring and a pyridine ring, which are joined at two neighboring carbon atoms. The ring that makes up benzene is composed of six carbon atoms, whereas the ring that makes up pyridine is composed of five carbon atoms and one nitrogen atom.299 These compounds are present in a wide variety of bioactive natural ingredients, medications that are now utilized in clinical practice, and pharmaceuticals, including anticancer,300 antibacterials,301 antitumor,302 and anti-inflammatory drugs.301Jingjun Yin et al. (2007) synthesized (iso)quinolones through a simple method (Scheme 202).288 In a single-step procedure, quinoline/isoquinoline N-oxide were transformed into 2-amino(iso)quinolines by employing Ts2O and tBuNH2. Subsequently, the unrefined reaction mixture was subjected to treatment with trifluoroacetic acid, resulting in the introduction of the tert-butyl group. This procedure enabled the highly effective one-pot 2-amination of 2-unsubstituted quinolones.
 |
| | Scheme 202 Synthesis of (iso)quinolones using Ts2O and t-BuNH2. | |
A simple and free-metal protocol for the preparation of 3-iodoquinolines was reported by Shaukat Ali et al. (2011) (Scheme 203).303 Their investigation involved the utilization of the 6-endo-dig iodocyclization methodology, employing mild reaction conditions. The reaction included the agitation of 2-tosylaminophenylprop-1-yn-3-ols and molecular iodine in methanol, followed by heating at 60 °C for an appropriate period. The target quinolones were achieved in modest to exceptional yields through this efficient and highly regioselective methods.
 |
| | Scheme 203 Synthesis of 3-iodoquinolines catalyzed by iodine. | |
Gang Shan et al. (2011) illustrated a novel one-step methodology for the preparation of 2-substituted quinolines via [3 + 3]-cyclization (Scheme 204).304 The experimental procedure involved the reaction between 3-ethoxycyclobutanones and aromatic amines, which took place in dichloromethane solvent under ambient conditions. To promote the reaction, BF3·OEt2 was added to the reaction mixture. A broad functional group tolerance was observed under the optimized reaction conditions.
 |
| | Scheme 204 Synthesis of quinolines catalyzed by BF3·OEt2. | |
In the study by Fei-Ping Ma et al. (2012), they devised a very effective and environmentally friendly approach to produce quinoline derivatives (Scheme 205).305 The Friedlander heteroannulation process was employed to carry out this approach, resulting in the synthesis of 2-aminoaryl ketones and α-methylene ketones in high yields. This process utilized a deep eutectic solvent that was non-toxic, making it both unique and very successful in an environmentally friendly manner.
 |
| | Scheme 205 Synthesis of quinoline catalyzed by L-(+)-tartaric-DMU. | |
A novel one-pot route for the synthesis of quinolones using a phosphine catalyst was described by San Khong et al. (2012) (Scheme 206).306 The process of annulating activated acetylenes with o-tosylamidobenzaldehydes or o-tosylamidophenones was carried out via Michael addition and aldol cyclization. The solution was agitated in acetonitrile at r.t. in triphenylphosphine. Then, the dihydroquinoline intermediates were treated with an HCl solution, leading to detosylated products. A variety of 3-substituted and 3,4-disubstituted quinolones was produced in high yield.
 |
| | Scheme 206 Synthesis of quinolones catalyzed by triphenylphosphine. | |
In the study conducted by Hassan Kefayati et al. (2012), they investigated the synthesis of acridin-1(2H)-ones in ILs (Scheme 207).307 The synthesis of a three-component condensation process involving 5,5-dimethyl-1,3-cyclohexadione, anilines, and isatin was conducted using 1-hexyl-3-methylimidazolium hydrogen sulfate ([HMIm]HSO4) as the solvent. The reaction was carried out under the influence of US for brief periods. The employed approach demonstrated a high level of efficiency and environmental friendliness in the synthesis of pyrroloacridine derivatives, resulting in significant yields and a straightforward process for isolating the desired products.
 |
| | Scheme 207 Synthesis of acridin-1(2H)-ones catalyzed by [HMIm]HSO4. | |
Qinghe Gao et al. (2014) reported in detail the utilization of a formal [3 + 2 + 1] annulation strategy for the synthesis of quinolines by a Povarov-type reaction (Scheme 208).308 The three-component reactions of methyl ketones, arylamines, and styrenes were performed in dimethyl sulfoxide at 100 °C for 4 h. In this study, molecular iodine played an important role as the catalyst. The corresponding products were afforded with various groups on the phenyl ring, such as electron-rich, bearing electron-neutral, and electron-deficient groups in the phenyl ring of methyl ketone and aniline.
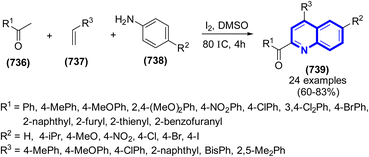 |
| | Scheme 208 Synthesis of quinolines catalyzed by iodine. | |
Ya-Nan Huang et al. (2016) illustrated a protocol for the synthesis of quinolinones and 2-(pseudo)haloquinolines using 4-dimethylaminopyridine as an organocatalyst (Scheme 209).309 2-Alkenyl-anilines were treated with di-tert-butyl dicarbonate in dichloromethane with 4-dimethylaminopyridine at ambient temperature. Under mild conditions, a variety of quinolines was afforded through 6π-electron cyclization.
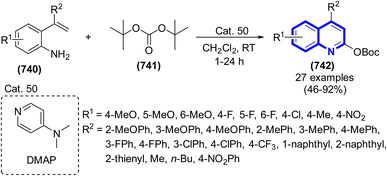 |
| | Scheme 209 Synthesis of quinolinones catalyzed by Cat. 50. | |
In the study by Xia Wu et al. (2017), they developed a new formal [4 + 2] pathway for the production of quinolones (Scheme 210).310 The reaction involving the formation of cyclic compounds from methyl ketones and arylamines was carried out using 1,4-dithane-2,5-diol as the ethylene source. This process was performed in dimethyl sulfoxide using a molecular iodine catalyst at 100 °C for 4 h. The target 2-acylquinolines were provided from various substrates in good yield. The authors presented a hypothesized mechanism that encompasses four distinct steps, namely iodination, Kornblum oxidation, Povarov reaction, and aromatization, based on the findings obtained from control trials.
 |
| | Scheme 210 Synthesis of quinolones catalyzed by iodine. | |
A novel method for the synthesis of quinolone derivatives through a three-component was described by Hao Wang et al. (2017) (Scheme 211).311 The process of cyclization involving aryl diazonium compounds, nitriles, and alkynes was conducted at a temperature of 60 °C in the absence of any additives and catalyst. This straightforward and quick approach yielded a collection of quinoline derivatives in high yield.
 |
| | Scheme 211 Synthesis of quinolines under catalyst-free conditions. | |
Jia-Chen Xiang et al. (2017) presented a detailed procedure for the synthesis of functionalized quinolines via a three-component reaction conducted in the absence of metal catalysts (Scheme 212).312 The annulation of aniline and two amino acids was performed in dimethyl sulfoxide with the assistance of HI. In addition, the mixture was subjected to the addition of molecular iodine, followed by agitation at 100 °C for 10 h. Under the optimum conditions, various mono- and di-substituted products were obtained in good yields.
 |
| | Scheme 212 Synthesis of quinolines catalyzed by I2/HI. | |
Tao-Shan Jiang et al. (2018) synthesized quinolines from anilines, acetophenones, and dimethyl sulfoxide in air (Scheme 213).313 The one-pot reaction for the synthesis of quinolone derivatives was facilitated by the utilization of methanesulfonic acid as a catalyst in this procedure. This process had an excellent substrate and functional group tolerance, generating the products in high yields.179
 |
| | Scheme 213 Synthesis of quinolines catalyzed by methanesulfonic acid. | |
In the same year, So Young Lee et al. (2018) prepared 2-substituted quinolines using a nucleophilic organic catalyst (Scheme 214).314 The cyclization of 2-aminochalcone occurred in the presence of a benzylamine catalyst in water as a green solvent. A wide substrate scope was studied, leading to the corresponding products in good yield. The advantages of the approach include its simple workup, easy product isolation, and available substrates.
 |
| | Scheme 214 Synthesis of quinolines catalyzed by benzylamine. | |
A simple and efficient route for the chemoselective synthesis of 3-aroylquinolines was developed by Pan Zhou et al. (2018) (Scheme 215).315 The process of cyclization involving 2-aminoaryl ketones and N,N-dimethylenaminones was seen to take place in a solvent mixture comprised of water and ethanol. The utilization of PTSA as a very effective catalyst was employed, with the reaction mixture being subjected to a temperature of 100 °C for 6 h. The benefits associated with employing this particular methodology include favorable reaction conditions, a wide applicability, abundant substrate availability, straightforward procedural steps, and significant product yields.
 |
| | Scheme 215 Synthesis of 3-aroylquinolines catalyzed by p-toluenesulfonic acid. | |
Jiang Nan et al. (2019) prepared 2-fluoroalkylated quinolines via [5 + 1] annulation under metal-free conditions (Scheme 216).316 2-Vinylanilines reacted with polyfluoroalkanoic acids at 140 °C. Additional types of fluoroalkylated quinolones were produced in good yield. This process was demonstrated to be an efficient synthetic route for fluoro-containing organic compounds under catalyst- and additive-free conditions.
 |
| | Scheme 216 Synthesis of 2-fluoroalkylated quinolines under catalyst-free conditions. | |
Next, Jiang Nan et al. (2020) provided a detailed account of a methodology for the synthesis of quinolone derivatives under metal-free conditions (Scheme 217).317 This approach was performed via domino condensation and aza-Prins cyclization. 2-Alkenylanilines reacted with β-dicarbonyl compounds in hexafluoroisopropanol at 100 °C for 2 h using p-toluenesulfonic acid as the catalyst. Various products were prepared from a wide substrate scope.
 |
| | Scheme 217 Synthesis of quinolones catalyzed by p-toluenesulfonic acid. | |
Dongping Cheng et al. (2020) reported an alternative method for synthesizing 2,4-diarylquinoline compounds (Scheme 218).318 The experimental procedure involved the utilization of oxidative cyclization, employing chloranil as the oxidizing agent. The reaction mixture was heated to a temperature of 80 °C using dichloroethane as the solvent. A diverse array of o-allylaniline derivatives was utilized in the synthesis of 2,4-diarylquinolines (Scheme 219).
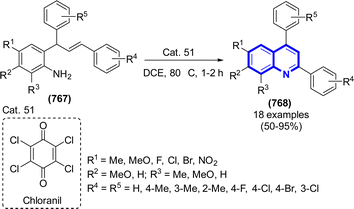 |
| | Scheme 218 Synthesis of 2,4-diarylquinolines catalyzed by Cat. 51. | |
 |
| | Scheme 219 Chemical structures of bioactive compounds containing a dihydropyridine framework. | |
3.1.4. Dihydropyridine. Dihydropyridines, also referred to as DHPs, are a class of extremely small chemical compounds that have a structure derived from pyridine. The pyridine structure can exist in a total of five isomeric forms, among which the 1,2-dihydro- and 1,4-dihydro-pyrdine structures are the most common ones.319 Because of their pharmacological and biological properties, such as antihypertensive,320 antianginal, and calcium channel blocker for cardiovascular disease,321 these precursors are significant.Polyhydroquinoline derivatives were developed by Majid M. Heravi et al. (2010). The synthesis of a Brønsted acid IL was achieved via a four-component Hantzsch reaction (Scheme 220).322 The solution containing aldehyde, ethylaceto-acetate, dimedone, and ammonium acetate was subjected to reflux in ethanol with the aid of Cat. 52. Various polyhydroquinoline derivatives, including aromatic, aliphatic, and heterocyclic compounds, were obtained using the aforementioned processes. The employed technique demonstrated a straightforward, effective, and environmentally benign approach.
 |
| | Scheme 220 Synthesis of polyhydroquinolines catalyzed by Cat. 52. | |
Jing-Yu He et al. (2015) synthesized 1,4-dihydropyridine-3,5-dicarboxylates under the catalysis of a Brønsted IL (Scheme 221).323 Under ultrasonic irradiation, the three-component reaction of aromatic aldehydes, methyl propionate, and ammonium carbonate was carried out in Cat. 53. Under the optimized reaction conditions, various 1,4-dihydropyridine derivatives were prepared at 60 °C for 2 to 10 min in good yield. This simple experimental procedure is considered a green approach with reusing and recycling abilities.
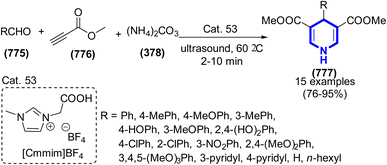 |
| | Scheme 221 Synthesis of 1,4-dihydropyridine catalyzed by Cat. 53. | |
In the study conducted by Jadhvar et al. (2015), a novel environmentally friendly approach was proposed for the synthesis of polyhydroquinoline frameworks. This method involved the utilization of a Brønsted acidic IL as a catalyst (Scheme 222).324 The polyhydroquinoline frameworks were synthesized via the condensation of substituted salicylaldehyde, dimedone, ethyl acetoacetate, and ammonium acetate. The reaction mixture was subjected to heating at a temperature of 80 °C with stirring, in the presence of [Msim]Cl. The corresponding polyhydroquinoline derivatives were produced from a wide range of substrates.
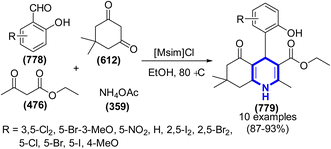 |
| | Scheme 222 Synthesis of polyhydroquinolines catalyzed by [Msim]Cl. | |
Manisha R. Bhosle et al. (2018) investigated a novel methodology for the synthesis of N-substituted decahydroacridine-1,8-dione derivatives (Scheme 223).325 A choline chloride: urea deep eutectic mixture was prepared and used as a recyclable organocatalyst and medium/solvent. The procedure was executed by a multicomponent reaction including a diverse array of aromatic aldehydes, dimedone, and aromatic amines under gentle conditions. The combination of choline chloride and urea has the potential to function as an environmentally friendly catalyst. The advantages of this protocol are catalytic activity, quick catalyst synthesis, straightforward workup protocol, and environmentally friendly conditions (Scheme 224).
 |
| | Scheme 223 Synthesis of decahydroacridine-1,8-diones catalyzed by a ChCl: urea mixture. | |
 |
| | Scheme 224 Chemical structures of bioactive compounds containing a diazinane framework. | |
3.2. Six-membered rings with two nitrogen-atom-containing heterocycles
3.2.1. Diazinane. Diazinanes feature a cyclohexane-like six-membered ring structure, with two of their carbons substituted by nitrogens. Diazinanes exist as three distinct isomers, which are characterized by the spatial arrangement of the nitrogen atoms in their respective molecular structures. The three isomers are denoted as 1,2-diazinane, 1,3-diazinane, and 1,4-diazinane. It has been shown that some diazinane derivatives exhibit anticonvulsant,326 anticancer,327 anti-inflammatory,328 and vasorelaxant properties.329Audumbar Patil et al. (2018) described an effective process for the preparation of 1,3-diazinane derivatives (Scheme 225).265 This protocol was conducted at ambient temperature in 2,2,2-trifluoroethanol. It has the advantages of gentle reaction conditions, no column chromatographic purification, and good yield.
 |
| | Scheme 225 Synthesis of 1,3-diazinane under catalyst-free conditions. | |
Sangram Gore et al. (2011) reported a study on a sustainable methodology for the synthesis of dihydropyrimidinone derivatives using a green approach, namely through the utilization of the Biginelli reaction (Scheme 226).330 The multicomponent condensation procedures were executed using a one-pot method, employing low melting combinations of L-(+)-tartaric acid and urea, all under moderate conditions. Additionally, several additional melts were examined, including citric acid–DMU, D-(−)-fructose–DMU, sorbitol–DMU–NH4Cl, and D-(+)-mannose–DMU. This work presented a process that demonstrates ecologically favorable qualities, therefore providing a way for synthesizing dihydropyrimidinone. The deep eutectic solvents played many roles in this approach, acting as the solvent, catalyst, and reactant.
 |
| | Scheme 226 Synthesis of dihydropyrimidinones catalyzed by a deep eutectic solvent. | |
In 2016, Farzaneh Mohamadpour et al. prepared 3,4-dihydro-pyrimidin-2-(1H)-one via a saccharin (Cat. 54)-catalyzed multiple component reaction (Scheme 227).331 The reaction involving a combination of aromatic aldehydes, alkyl acetoacetate, and urea was conducted by subjecting the mixture to heating at 80 °C for a certain duration, while utilizing saccharin as an effective organic catalyst. Following the optimization of the reaction conditions, a diverse array of 3,4-dihydropyrimidin-2-(1H)-one scaffolds was obtained in high yields within a short reaction period (Scheme 228).
 |
| | Scheme 227 Synthesis of 3,4-dihydropyrimidin-2-(1H)-ones catalyzed by Cat. 54. | |
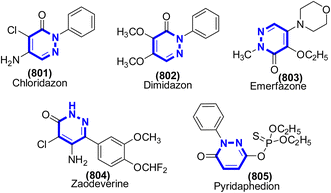 |
| | Scheme 228 Chemical structure of bioactive compounds containing a pyridazine framework. | |
3.2.2. Pyridazine (1,2-diazine) and cinnoline. Pyridazine is a six-membered aromatic heterocycle including two nitrogen atoms located at positions 1 and 2. This distinctive nitrogen configuration greatly affects the electrical characteristics, making pyridazine and its derivatives essential in pharmaceutical chemistry, agrochemicals, and materials research.332 Pyridazine-based compounds have been extensively investigated in medicinal chemistry for their pharmacological potential. They demonstrate diverse biological actions, including anticancer, antihypertensive, anti-inflammatory, antidiabetic, and antibacterial properties. These derivatives exhibit a broad spectrum of biological activities, including cardiovascular,333 antidiabetic,334 anti-AIDS,335 anticancer,336 and anticonvulsant.3373-Amino-5-arylpyridazine-4-carbonitriles were synthesized by J. Khalafy et al. (2013) (Scheme 229).338 The one-pot three-component reactions were performed between malononitrile and arylglyoxals in with N2H4·H2O at ambient temperature in H2O and EtOH for 1 h. This was a simple process for synthesizing various 3-amino-5-arylpyridazine-4-carbonitriles with biological and pharmaceutical applications.
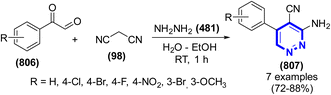 |
| | Scheme 229 Synthesis of arylpyridazine-4-carbonitriles under catalyst-free conditions. | |
Nader Ghaffari Khaligh et al. (2018) devised a highly efficient methodology for the synthesis of 4-amidocinnolines utilizing a metal and halogen-free approach (Scheme 230).339 The investigation involved the utilization of a one-pot reaction method, whereby o-alkynylaniline and nitrile were subjected to tbutyl nitrite, a metal-free diazotization reactant. Especially, saccharin was utilized as a green catalyst, playing a significant role in this reaction. The reaction mixture was refluxed for a duration of 6 h. The scope of this technique was investigated under the optimal reaction conditions, wherein a diverse set of 2-alkynylanilines and nitrile substrates was employed to obtain the corresponding 4-amidocinnoline derivatives. This method was demonstrated to be a systematic protocol with several advantages such as a metal-free conditions and environment-friendly workup.
 |
| | Scheme 230 Synthesis of 4-amidocinnolines catalyzed by Cat. 54. | |
A green regioselective method for the assembly of novel 7,8-dihydrocinnoline-5(6H)-ones was shown by Jabbar Khalafy et al. (2012) (Scheme 231).340 The experimental procedure involved the execution of a one-pot three-component reaction, whereby arylglyoxals, 1,3-cyclohexanedione, and dimedone were combined. The mixture was supplemented with hydrazine hydrate, serving as a catalyst. The reaction mixture was stirred in water, which served as a green solvent, at a temperature in the range of 5–8 °C (using an ice bath) for a duration of 20–40 min. A series of target cinnoline derivatives was obtained from various substrates after recrystallization from ethanol. The present study described a novel and environmentally friendly procedure for the synthesis of 7,8-dihydrocinnolin-5(6H)-one derivatives, characterized by their green and efficient protocol, which offers the advantage of a straightforward workup process.
 |
| | Scheme 231 Synthesis of 7,8-dihydrocinnolin-5(6H)-ones under catalyst-free conditions. | |
Guan-Hua Ma et al. (2014) prepared polyfunctionalized cinnoline-4-carboxamides under microwave irradiation (Scheme 232).341 The experiment involved the implementation of a regioselective ring-opening process on 6,7-dihydrobenzofuran-4(5H)-ones, followed by cyclization with hydrazine hydrate. The mixture was subjected to microwave irradiation at 60 °C for 25 min. In addition, the synthesis of benzofuran-4(5H)-one derivatives via a three-component domino reaction was conducted using cyclic-1,3-dione molecules, arylglyoxals, and alkyl isocyanides under identical reaction conditions. Thus, this is an eco-friendly two-step domino synthesis of cinnoline derivatives avoiding the use of transition metal catalysts.
 |
| | Scheme 232 Synthesis of cinnolines under catalyst-free conditions. | |
Tuyet-Anh Dang Thi et al. (2015) synthesized novel functionalized dihydro-benzo[h]cinnoline-5,6-diones via a one-pot multi-component reaction (Scheme 233).342 A mixture of hydrazines, aromatic aldehydes, and naphthoquinone was heated in tert-butanol for 30–60 min. A series of naphthoquinones was provided in this study. The detailed mechanism for the formation of naphthoquinones was proposed including Knoevenagel condensation, Michael addition, keto–enol tautomerization, and elimination. Besides, the cytotoxic activity of the target products was surveyed and described.
 |
| | Scheme 233 Synthesis of dihydrobenzo[h]cinnoline-5,6-diones under catalyst-free conditions. | |
Gopal Chandru Senadi et al. (2016) developed a protocol for the synthesis of 4-amido-cinnolines via a one-pot cascade reaction (Scheme 234).343 The construction of 4-amido-cinnoline derivatives was performed employing 2-alkynylanilines, tbutyl nitrite, and nitriles in the presence of BF3-etherate at ambient temperature. Based on the data obtained from control experiments, the reaction mechanism was proposed to explain the creation of two novel C–N bonds. This mechanism involves a sequential process of diazotization using tBuONO and the subsequent nucleophilic addition of the alkyne to the BF3-coordinated diazonium ion. Subsequently, the nitrile compound was introduced into the intermediate vinyl cation, resulting in the formation of the desired end product. A range of 4-amido-cinnoline derivatives was synthesized with satisfactory to excellent yields using a metal-free approach.
 |
| | Scheme 234 Synthesis of 4-amido-cinnolines catalyzed by BF3-etherate. | |
A simultaneous one-pot protocol for the preparation of 2-arylcinnolin-6-one moieties was described by Hamad M. Al-Matar et al. (2018) (Scheme 235).344 The experimental procedure involved the cyclization of 3-oxo-2-arylhydrazonopropanal derivatives with acetoacetanilide and triethylamine in an ethanol solvent. This study examined three distinct methodologies in the experimental procedure, namely refluxing for a duration of 2–4 h, microwave irradiation at 80 °C at a power output of 200 W for a period of 3–10 min, and US at 80 °C for a duration of 40–80 min. In this study, a variety of 2-arylcinnolin-6(2H)-one derivatives was prepared following the optimization of the method. Additionally, a comprehensive mechanistic route was proposed (Scheme 236).
 |
| | Scheme 235 Synthesis of 2-arylcinnolin-6-one catalyzed by triethylamine. | |
 |
| | Scheme 236 Chemical structures of bioactive compounds containing a phthalazine framework. | |
3.2.3. Phthalazine. Phthalazine is a fused bicyclic heterocyclic molecule with a pyridazine ring fused to a benzene ring. This structural framework confers distinctive electrical and chemical characteristics, making phthalazine derivatives significant in pharmaceutical chemistry, materials research, and industrial applications.345 Phthalazine-based compounds in medicinal chemistry exhibit a variety of pharmacological properties, such as anticancer, anti-inflammatory, antibacterial, antihypertensive, anticonvulsant, and enzyme inhibitory effects. Many medications displaying pharmacological effects, in particular antidiabetic,346 anticancer,347 antithrombotic,348 anti-inflammatory,349 and analgesic350 properties, possess the phthalazine scaffold as a pharmacophoric characteristic.Mohsen Shekouhy and Alireza Hasaninejad (2012) published a one-pot strategy for the preparation of 2H-indazolo[2,1-b]phthalazinetriones under catalyst-free conditions (Scheme 237).351 The four-component reactions among dimedone, benzaldehyde, hydrazinium hydroxide, and phthalic anhydride were carried out at r.t. under US. An IL, specifically 1-butyl-3-methylimidazolium bromide, was employed as an environmentally friendly solvent, which exhibited the advantageous characteristic of facile reusability. Many organic transformations for the synthesis of phthalazinetriones derivatives were studied, which were obtained in yields ranging from fair to outstanding.
 |
| | Scheme 237 Synthesis of phthalazinetriones catalyzed by 1-butyl-3-methylimidazolium bromide. | |
Similarly, Alireza Hasaninejed et al. (2012) conducted the synthesis of 2H-indazolo[2,1-b] phthalazinetrione derivatives using a one-pot four-component condensation approach, employing biodegradable polymeric catalysts (Scheme 238).352 A combination of aromatic aldehydes, phthalic anhydride, hydrazinium hydroxide, and 1,3-diketo compounds, namely 1,3-cyclohexanedione and 5,5-dimethyl 1,3-cyclohexanedione, was heated to 80 °C in the absence of a solvent. The current study used sulfuric acid-modified polyethylene glycol-6000 (PEG-OSO3H) as an effective and eco-friendly catalyst, demonstrating significant reusability. A series of target phthalazine compounds was obtained in high yields after recrystallization from hot ethanol.
 |
| | Scheme 238 Synthesis of phthalazinetriones catalyzed by Cat. 55. | |
In addition, phthalhydrazide was used as a reactant in an approach for the synthesis of pyrazolo[1,2-b]phthalazine-5,10-dione (Scheme 239).353 For example, R. Ghahremanzadeh et al. (2008) prepared 1H-pyrazolo[1,2-b]phthalazine-5,10-diones via a one-pot three-component reaction in IL as the solvent. The experimental procedure involved conducting multiple component reactions using phthalhydrazide, aromatic aldehydes, and malononitrile/ethyl cyanoacetate. A catalytic quantity of PTSA was employed as the catalyst, and the reactions were performed at 100 °C. In this study, the 1-butyl-3-methylimidazolium bromide IL ([Bmim]Br) was employed as an environmentally friendly solvent. Notably, this IL was efficiently recovered and reused in subsequent cycles. This cyclocondensation was demonstrated as an efficient synthetic approach to obtain phthalazine compounds.
 |
| | Scheme 239 Synthesis of phthalazine-5,10-dione catalyzed by PTSA. | |
In the same year, a one-pot synthetic protocol to obtain 2H-indazolo[2,1-b]phthalazine-triones was described by Maryam Sayyafi et al. (2008) (Scheme 240).354 The condensation reaction involving aromatic aldehydes, phthalhydrazide, and dimedone was conducted in the absence of a solvent. The reaction mixture was subjected to heating at 80 °C in the presence of a PTSA catalyst for a duration of several minutes. A variety of the matching products was produced in outstanding yield without any by-product.
 |
| | Scheme 240 Synthesis of phthalazine-triones catalyzed by p-toluenesulfonic acid. | |
An US one-pot methodology for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones was reported by Mohammad Reza Nabid et al. (2010) (Scheme 241).355 Under US, triethylamine was demonstrated as an effective catalyst in the annulation of aromatic aldehydes, phthalhydrazide, and malononitrile/ethyl cyanoacetate. The process was conducted in ethanol at 50 °C for 60 min, furnishing the respective compounds in excellent yields.
 |
| | Scheme 241 Synthesis of phthalazine-5,10-diones catalyzed by triethylamine. | |
Ramin Ghorbani-Vaghei et al. (2011) described a method for the one-pot preparation of 2H-indazolo[2,1-b]phthalazinetrione derivatives under solventless conditions (Scheme 242).356 Aldehydes, phthalhydrazide, and dimedone compounds were subjected to a three-component reaction under reflux conditions at a temperature in the range of 80–100 °C. This reaction was facilitated by the use of N-halosulfonamide catalysts, namely Cat. 56 and Cat. 57. A diverse range of aliphatic and aromatic 2H-indazolo[2,1-b]phthalazinetrione derivatives was synthesized with high efficiency, resulting in outstanding yields.
 |
| | Scheme 242 Synthesis of phthalazinetriones catalyzed by Cat. 56 or Cat. 57. | |
In the study conducted by Farzaneh Mohamadpour et al. (2016), they introduced an alternative and cost-effective method for synthesizing 3,4-dihydro-pyrimidin-2-(1H)-one derivatives (Scheme 243).331 The synthesis of phthalimide condensate was carried out with the application of heat at 90 °C for 2.5–4 h, using hydrazine monohydrate, benzaldehyde, and malononitrile as the reactants. This technique employed saccharin as an environmentally sustainable and ecologically friendly organocatalyst. A range of pyrazolo[1,2-b]phthalazine-5,10-dione derivatives was successfully synthesized in satisfactory yield using aldehyde derivatives possessing electron-donating and electron-withdrawing groups (Scheme 244).
 |
| | Scheme 243 Synthesis of pyrimidin-2-(1H)-ones catalyzed by Cat. 54. | |
 |
| | Scheme 244 Chemical structures of bioactive compounds containing a pyrimidine framework. | |
3.2.4. Pyrimidine (1,3-diazine) and quinazoline. Pyrimidine, also known 1,3-diazine, is formed by the substitution of two meta-oriented CH units in benzene with nitrogen atoms. The replacement nomenclature refers to quinazoline as 1,3-diazanaphthalene, whereas the fusion nomenclature refers to it as benzo[d]pyrimidine. The reported biological effects of pyrimidines include antibacterial,357 antitumor,358 and tyrosine kinase inhibitory effects.359 Quinazoline is a benzo-fused pyrimidine, which is classified by the fusion nomenclature as benzo[d]pyrimidine. Quinazoline derivatives have been identified to exhibit anticancer,360 anti-inflammation,361 and antibacterial362 therapeutic activities.Krishna Nand Singh et al. (2012) developed the protocol for the synthesis of benzopyranopyrimidines (Scheme 245). The experimental procedure involved the utilization of 1-butyl-3-methylimidazolium tetrafluoroborate IL ([Bmim]BF4) as a catalyst, conducted at ambient temperature in the absence of any solvent. The IL was successfully employed for up to five consecutive cycles without experiencing any substantial decrease in quantity or effectiveness.363
 |
| | Scheme 245 Synthesis of benzopyranopyrimidines catalyzed by [Bmim]BF4. | |
In 2006, Serena Ferrini et al. showed an efficient three-step synthetic approach to prepare 2,4-disubstituted quinazolines (Scheme 246).364 The Fries rearrangement of anilides occurred via a photochemical reaction, affording 2-aminoacylbenzene. Cyclization of 2,4-disubstituted quinazolines was achieved under MW conditions in ammonium formate. In this study, a range of 2,4-dialkyl/aryl quinazolines and benzoquinazolines was synthesized using the described methodology, resulting in significant yields.
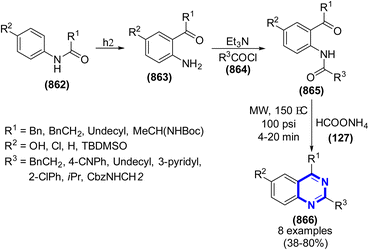 |
| | Scheme 246 Synthesis of 2,4-disubstituted quinazolines. | |
Minoo Dabiri et al. (2010) outlined a methodology for the synthesis of quinazolines via a one-pot reaction, employing a Brønsted acidic IL as the catalyst (Scheme 247).365 The condensation process was carried out using a three-component system consisting of 2-aminobenzophenones, formaldehyde or aromatic aldehydes, and ammonium acetate. The reaction mixture was subjected to thermal treatment at 80 °C for 2 h, while being exposed to 1-methylimidazolium trifluoroacetate ([Hmim]TFA). The catalyst composed of an IL demonstrated a high degree of ease in its separation and subsequent reuse, with the capability of being employed multiple times. The investigation encompassed a wide range of substrates, resulting in the successful synthesis of the desired compounds in high yields.
 |
| | Scheme 247 Synthesis of quinazoline catalyzed by [Hmim]TFA. | |
Jintang Zhang et al. (2011) reported a procedure that demonstrates high efficiency and novelty for the synthesis of 2-phenylquinazoline derivatives (Scheme 248).366 The process of combining 2-aminobenzophenones and benzylic amines was carried out using a sequential reaction that involved sp3C–H functionalization. The substrate mixture was supplemented with aqueous tbutyl hydroperoxide and iodide. The procedure involved subjecting the sample to heating at 90 °C for 12 h. A variety of 2-phenylquinazolines was produced from 2-aminobenzoketones with benzylic amines.
 |
| | Scheme 248 Synthesis of 2-phenylquinazolines catalyzed by I2/TBHP. | |
Zhan-Hui Zhang et al. (2012) synthesized quinazoline derivatives under catalyst-free conditions (Scheme 249).367 The experimental procedure involved conducting a one-pot three-component reaction using 2-aminoaryl ketones, aldehyde, and ammonium acetate as reactants. The reaction took place in a solvent composed of a low melting sugar-urea-salt mixture under aerobic oxidation conditions. A range of deep eutectic solvents (DESs) was examined, including citric acid–DMU, L-(+)-tartaric acid–choline chloride, mannose–DMU–NH4Cl, lactose–DMU–NH4Cl, maltose–DMU–NH4Cl, and D-(−)-fructose–DMU, L-(+)-tartaric acid–DMU. The advantage of the protocol include its simple work-up, eco-friendly method, and green reaction medium.
 |
| | Scheme 249 Synthesis of quinazolines catalyzed by a low melting point mixture. | |
Yizhe Yan et al. (2015) developed a innovate procedure for synthesizing quinazolines and quinazolinones (Scheme 250).368 The experimental procedure involved the utilization of iodine as a catalyst for the oxidative C(sp3)–H amination/C–N cleavage of tertiary amines. The domino ring annulation was conducted under an oxygen atmosphere. The reaction mixtures of alkyl amine and 2-aminobenzophenone or 2-aminobenzamide were heated in DMSO at 120 °C for 20 h. The advantage of this protocol is its simple workup, peroxide- and metal-free conditions, wide substrate scope, and high yield.
 |
| | Scheme 250 Synthesis of quinazolines and quinazolinones catalyzed by iodine. | |
A protocol for the synthesis of quinazolines and dihydroquinazolines was reported by Santanu Hati and Subhabrata Sen (2016) (Scheme 251).369 The reaction involved the utilization of o-aminobenzylamine and aliphatic, aromatic, and heteroaromatic aldehydes and conducted in the presence of Cat. 15 and acetonitrile as the solvent. The corresponding quinazolines and dihydroquinazolines were obtained with 2 equivalents or 1 equivalent of o-iodoxybenzoic acid, respectively. This is an efficient approach with several benefits including accessible starting ingredients, easy workup, high yield, and mild reaction conditions.
 |
| | Scheme 251 Synthesis of quinazolines and dihydroquinazolines catalyzed by o-iodoxybenzoic acid. | |
Dewal S. Deshmukh et al. (2018) synthesized 2-arylquinazolines through benzylic sp3 H–C bond amination under the catalysis of I2 (Scheme 252).370 The reaction occurred between benzylamines and 2-aminobenzaldehydes, 2-aminobenzophenones, or 2-aminobenzyl alcohols with O2 as a green oxidizing agent. Besides, molecular iodine was added to the mixture under solvent-free conditions. Various matching 2-arylquinazolines were prepared in fair to outstanding yields.
 |
| | Scheme 252 Synthesis of 2-arylquinazolines catalyzed by iodine. | |
D. S. Deshmukh and B. M. Bhanage (2020) prepared substituted quinazolines via a sulfur-mediated decarboxylative coupling method (Scheme 253).370 The reaction involving the condensation of 2-nitrobenzyl alcohols and arylacetic acids was carried out by refluxing in DMSO, with urea serving as the nitrogen source. In addition, a promoter consisting of Cat. 19 base and elemental sulfur was included in the mixture. This study successfully synthesized a range of quinazoline derivatives with excellent efficiency.
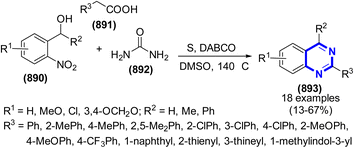 |
| | Scheme 253 Synthesis of quinazolines catalyzed by elemental sulfur. | |
Jiuxi Chen et al. (2007) presented a novel approach to the synthesis of 2,3-dihydroquinazolin-4(1H)-on derivatives. This process involved the utilization of ILs, specifically Cat. 58, or water-ion liquids, under catalyst-free conditions (Scheme 254).371 The aforementioned reactions were conducted by means of direct cyclocondensation of anthranilamides and aldehydes, as well as a one-pot three-component cyclocondensation involving isatoic anhydrides, ammonium acetate, and aldehydes. The green technique employed in this study involved the integration of straightforward experimental protocols, as well as the facilitation of efficient recovery and reusability of the newly developed reaction media. This approach was characterized by moderate reaction conditions and demonstrated favorable yields.
 |
| | Scheme 254 Synthesis of 2,3-dihydroquinazolin-4(1H)-on in ILs. | |
Ahmad Shaabani et al. (2008) conducted the synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives. This was achieved using a condensation reaction involving 2-aminobenzamide and a range of alkyl, aryl, and alicyclic aldehydes/ketones. The process was carried out at ambient temperature (Scheme 255).372 The experiment was conducted with the addition of a catalytic quantity of ammonium chloride in ethanol. The condensation reaction demonstrated excellent efficiency due to its ability to produce significant yields and achieve rapid reaction times.
 |
| | Scheme 255 Synthesis of 2,3-dihydroquinazolin-4(1H)-one catalyzed by ammonium chloride. | |
Lobo et al. (2012) employed an acidic eutectic combination to synthesize a diverse range of 2,3-dihydroquinazolin-4(1H)-one derivatives (Scheme 256).373 The synthesis involving the combination of isatoic anhydride, aldehydes, and aromatic amines was conducted in methanol solvent at 65 °C. High yields were obtained for the aromatic and heterocyclic substitution at the 2,3-positions of quinazolinone.
 |
| | Scheme 256 Synthesis of 2,3-dihydroquinazolin-4(1H)-one catalyzed by a deep eutectic solvent. | |
Moni Sharma et al. (2011) developed an efficient procedure for the synthesis of dihydroquinazolinones and spiroquinazolinone (Scheme 257).374 This protocol was performed via the cyclocondensation reaction of anthranilamide and aldehyde at r.t. with a cyanuric chloride catalyst to provide products in high yield. This procedure was demonstrated to be a highly succinct, effective, gentle, and straightforward technique (Scheme 258).
 |
| | Scheme 257 Synthesis of dihydroquinazolinones and spiroquinazolinone catalyzed by cyanuric chloride. | |
 |
| | Scheme 258 Chemical structures of bioactive compounds containing a benzo[4,5]imidazo[1,2-a]pyrimidine framework. | |
3.2.5. Benzo[4,5]imidazo[1,2-a]pyrimidine. Benzimidazole-fused pyrimidines belong to a class of N-heterocyclic compounds, and the N-fused hybrid structures of these compounds typically exhibit unique biological activities that are not present in the majority of homonuclear scaffolds. Benzimidazole-fused pyrimidines, also known as benzo[4,5]imidazo[1,2-a]pyrimidines, were discovered to exhibit distinct fluorescence characteristics as well as diverse biological activities. Their biological effects include antibacterial,375 antiviral,376 antiulcer,377 anti-HIV,378 anticancer,379 antiproliferative,380 and anti-inflammatory381 capabilities.C. S. Yao et al. (2008) illustrated a straightforward simple protocol for the preparation of benzo[4,5]imidazo[1,2-a]pyrimidine scaffolds (Scheme 259).382 The researchers conducted a three-component reaction involving aldehydes, a β-dicarbonyl compounds, and 2-aminobenzimidazole. This reaction was catalyzed by sulfamic acid and performed under solvent-free conditions. A range of benzo[4,5]imidazo[1,2-a]pyrimidine derivatives was successfully synthesized in favorable yields under the optimal reaction conditions and using different reagents.
 |
| | Scheme 259 Synthesis of benzo[4,5]imidazo[1,2-a]pyrimidines catalyzed by Cat. 60. | |
Next, C. Yao et al. (2009) synthesized 1H-pyrimido[1,2-a]benzimidazole frameworks from aryl aldehydes, 1,3-dicarbonyl compounds and 1H-benzo[d]imidazole-2-amine solvent at 90 °C (Scheme 260).383 The methodology employed in this study can be classified as environmentally friendly, given that it utilizes an IL known as [Bmim][BF4], which possesses green characteristics. This IL could be recycled and reused several times without considerable degradation in its catalytic efficacy. The developed process was distinguished by its cost-effectiveness, efficiency, simplicity, and eco-friendliness.
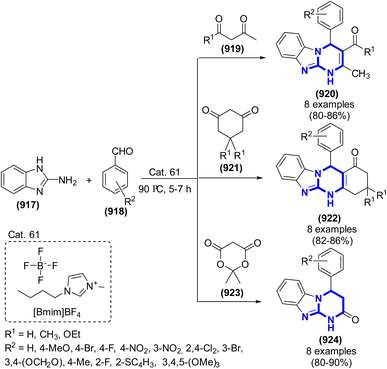 |
| | Scheme 260 Synthesis of 4-ayrl-3,4-dihydro-1H-pyrimido[1,2-a]benzimidazole catalyzed by Cat. 61. | |
Dharma Rao et al. (2011) reported a straightforward and effective protocol for the preparation of 3,4-dihydropyrimidin-2-(1H)one and benzo[4,5]imidazo/thioazo[1,2-a]pyrimidine moieties (Scheme 261).384 In their study, the researchers employed Cat. 62 as an innovative catalyst within the context of the Biginelli three-component coupling reaction. This reaction involved the simultaneous combination of an aldehyde, a 1,3-ketoester, and diamines in a single reaction vessel. The employed approach demonstrated a straightforward and effective nature, resulting in a significant yield.
 |
| | Scheme 261 Synthesis of 3,4-dihydropyrimidin-2-(1H)one catalyzed by Cat. 62. | |
Junhua Liu et al. (2012) developed a straightforward method for the preparation of benzo[4,5]imidazo[1,2-a]pyrimidine and benzo[1,2,4]triazolo[1,5-a]pyrimidine moieties (Scheme 262).385 The experimental procedure involved the synthesis of the desired compounds an aqueous medium, with thiamine hydrochloride serving as the catalyst. This methodology exhibited several advantages, including a straightforward workup procedure, mild reaction conditions, short reaction time, and environmentally friendly process.
 |
| | Scheme 262 Synthesis of benzo[4,5]imidazo[1,2-a]pyrimidine and benzo[1,2,4]triazolo[1,5-a]pyrimidine catalyzed by Cat. 63. | |
Many multifunctional 2-amino-5-cyano-4-[(2-aryl)-1H-indol-3-yl]-6 hydroxypyrimidines were prepared by Ragini Gupta et al. (2014) (Scheme 263).386 The multicomponent synthesis involving 3-formylindole, cyanoethylacetate, and guanidine hydrochloride was conducted using a milder base and under the optimized reaction conditions. Compared with refluxing, microwave irradiation and grindstone technology are simple procedures because of their short reaction time and higher yield (Scheme 264).
 |
| | Scheme 263 Synthesis of hydroxypyrimidines under MW. | |
 |
| | Scheme 264 Chemical structures of bioactive compounds containing a pyrazine and quinoxaline framework. | |
3.2.6. Pyrazine and quinoxaline. The quinoxaline family consists of a group of aromatic heterocycles. These compounds are formed through the fusion of two six-membered aromatic rings, with one of the rings containing two nitrogen atoms placed symmetrically at the 1 and 4 positions. Quinoxaline derivatives possess a diverse array of physicochemical and biological actions, including antibacterial,387 antiviral,388 and anticancer389 activity.Edward C. Taylor and Donald J. Dumas (1981) prepared pyrazine through the reaction of dicyanide with ammonia (Scheme 265).390 A mixture of (prop-1-en-2-yl)carbonimidoyl dicyanide and ammonia was refluxed in methanol. 2-Amino-3-cyano-5-(dimethoxymethyl)pyrazine was obtained in 85% yield.
 |
| | Scheme 265 Synthesis of pyrazine catalyzed under catalyst-free conditions. | |
The [4 + 2] cycloaddition of diiminosuccinonitrile with 1,2-dimethoxyethylene and ynamines for the synthesis of pyrazine was reported by Fukunaga et al. (1984) (Scheme 266).391 Tetrahydropyrazine was obtained in high yield. 1,2-Dimethoxyethylene, phenyl(diethylamino)acetylene, and diethyl-amino-1-propyne were the ynamines used in this protocol.
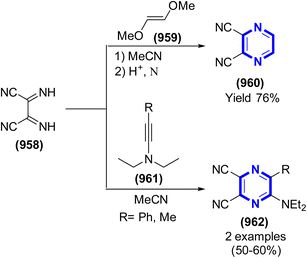 |
| | Scheme 266 Synthesis of pyrazine. | |
N. Sato et al. (1992) reported a protocol for the synthesis of 3-phenylthiopyrazinecarbonitrile by condensing 2,3-diamino-3-phenylthioacrylonitrile with glyoxal (Scheme 267).392 The reaction mixture was subjected to reflux in methanol for a duration of 5 h under PTSA. The resultant compounds were produced in high yield upon recrystallization using methanol as the solvent. This was an important step in the synthetic method for 3 alkoxy-and 3-aminopyrazinecarbonitriles.
 |
| | Scheme 267 Synthesis of 3-phenylthiopyrazinecarbonitrile catalyzed by PTSA. | |
Weijiang Zhang (2001) conducted a study on the regioselective synthesis of thieno-[2,3-b]pyrazine (Scheme 268).393 Phenylglyoxal was substituted under Sato's experimental conditions, resulting in the formation of a mixture of 5-phenyl-3-phenylthiopyrazinecarbonitrile and 6-phenyl-3-phenylthiopyrazinecarbonitrile, without exhibiting any regioselectivity. Thus, to solve this problem, the authors added excess trifluoroacetic acid (TFA) to a mixture of 2,2-diethoxyacetophenone and 2,3-diamino-3-phenylthioacrylonitrile instead of PTSA. The reaction mixture was refluxed in propan-2-ol at r.t. for a duration of 22–24 h. The result of the experiment showed that 6-phenyl-3-phenylthio-pyrazinecarbonitrile was obtained in good yield. Especially, this catalyst exhibited high selectivity, where the ratio of 6-phenyl and 5-phenyl derivatives was 15.5:1.
 |
| | Scheme 268 Synthesis of thieno[2,3-b]pyrazines catalyzed by trifluoroacetic acid. | |
A novel process for the synthesis of quinoxalines using Brønsted acid hydrotrope-combined catalyst was developed by Arjun Kumbhar et al. (2012) (Scheme 269).394 The combination of 1,2-diketones and 1,2-diamines was subjected to agitation with PTSA in an aqueous solution at r.t. The catalyst could be reused for the next cycles. Thus, this approach is considered an efficient and green protocol with a straightforward work-up, producing a good yield of quinoxaline compounds.
 |
| | Scheme 269 Synthesis of quinoxalines catalyzed by PTSA. | |
Another environmentally friendly procedure for the preparation of 2,3-diphenyl quinoxaline was reported Pranita Mahadik (2014) (Scheme 270).395 2,3-Diphenyl quinoxalines also were synthesized from o-phenylenediamine and benzil with PTSA within a short reaction time under mild conditions. Three different practical techniques were surveyed including ultrasound and stirring in ethanol or water. Compared with traditional methods, this approach is a more convenient such as avoiding the use of harmful solvents, simple workup, cleaner reactions, and being highly efficient.
 |
| | Scheme 270 Synthesis of 2,3-diphenyl quinoxaline catalyzed by PTSA. | |
Similarly, Zeyuan Zhang et al. (2016) also prepared quinoxaline derivatives using PTSA as a catalyst (Scheme 271).396 The one-pot reaction of benzene-1,2-diamines with 2-hydroxyethanone was performed using DMSO as the oxidant. This was a two-step process including oxidation and annulation to obtain the target compounds in high yield.
 |
| | Scheme 271 Synthesis of quinoxalines catalyzed by PTSA. | |
The one-pot preparation of quinoxaline scaffolds using a natural Brønsted acid was published by Radhakrishnan Mahesh et al. (2011) (Scheme 272).397 The reaction was based on the condensation between o-phenylenediamines and 1,2-dicarbonyl compounds with a certain amount of citric acid as a catalyst. The reaction mixture was agitated in ethanol for a duration ranging from 1 to 90 min. A variety of corresponding quinoxaline derivatives was obtained from many 1,2-dicarbonyl compounds such as benzil, diethyl ketomalonate, and pyruvic acid. This protocol is considered an efficient and mild reaction due to its simple workup, use of a natural catalyst, and short time.
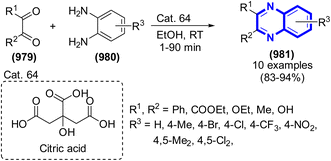 |
| | Scheme 272 Synthesis of quinoxalines catalyzed by Cat. 64. | |
In 2013, Jabbar Khalafy et al. reported a regioselective process for the synthesis of 3-arylpyrido[2,3-b]pyrazines (Scheme 273).398 The condensation reactions of arylglyoxals with 2,3-diaminopyridine were carried out in N,N-dimethylformamide and ethanol at 90 °C to obtain pyrido[2,3-b]pyrazine derivatives. The advantages of this protocol include its high yields, quick reaction times, and simple workup.
 |
| | Scheme 273 Synthesis of 3-arylpyrido[2,3-b]pyrazines. | |
Loghman Moradi et al. (2013) described an effective one-pot method for the preparation of pyrrolo[1,2-a]pyrazines (Scheme 274).399 The three-component reaction of ethylenediamine, acetylenic esters, and nitrostyrene was performed by refluxing in acetonitrile for 24 h. Sulfamic acid was used as a catalyst in this approach. Pyrrolo[1,2-a]pyrazine moieties were produced from a variety of acetylenic esters and nitrostyrene derivatives.
 |
| | Scheme 274 Synthesis of pyrrolo[1,2-a]pyrazines catalyzed by sulfamic acid. | |
In a study conducted by Shen et al. (2015), a very efficient method for synthesizing pyrrolobenzo-1,4-diazines with quaternary stereocenters was documented (Scheme 275).400 The procedure involved the utilization of the Pictet–Spengler reaction, specifically employing 2-(1H-pyrrol-1-yl)anilines and α-ketoamides as reactants. The reaction was conducted in toluene at 40 °C for 72 h. The chiral Brønsted acid catalyst employed in this process was spirocyclic phosphoric acid. The present study demonstrates that the chiral phosphoric acid catalyst utilizes unforeseen arene C–H⋯N hydrogen bonding to facilitate its activation and stereoinduction.
 |
| | Scheme 275 Synthesis of pyrrolobenzo-1,4-diazines catalyzed by Cat. 65. | |
K. Durga Rao Viswanadham et al. (2014) synthesized quinoxalines via iodine-mediated oxidative cyclization (Scheme 276).401 The corresponding products were afforded through the annulation of aryl acetylenes, arylethenes, or aromatic ketones with 1,2-diamines in high yield. This strategy is an efficient multi-pathway-coupled domino approach.
 |
| | Scheme 276 Synthesis of quinoxalines catalyzed by iodine. | |
Frédéric Lassagne et al. (2014) reported a protocol for the synthesis of quinoxalines and pyrido[2,3-b]pyrazines using saccharin as an organic catalyst (Scheme 277).402 The experiment involved conducting a cyclocondensation reaction between 1,2-arylenediamines and 1,2-dicarbonyl compounds in methanol at r.t., with relatively brief reaction durations. A series of target heterocycle products was obtained after completion of the reaction with high regioselectivity. Saccharin was demonstrated as an efficient organocatalyst for this strategy.
 |
| | Scheme 277 Synthesis of quinoxalines and pyrido[2,3-b]pyrazines catalyzed by saccharin. | |
Gordon W. Gribble and co-workers discovered method for the three-step synthesis of masked 2,3-diaminoindole from 2-iodo-3-nitro-1-(phenylsulfonyl)indole (2016). Treatment with trifluoroacetic acid produced the unstable 2,3-diamino-1-(phenylsulfonyl)indole, which could be captured by α-dicarbonyl compounds to provide 5H-pyrazino[2,3-b]indoles. This methodology enhances the established syntheses of 5H-pyrazino[2,3-b]indoles and analogous fused indoles by the condensation of isatins with phenylenediamines (Scheme 278).403
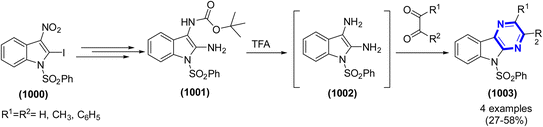 |
| | Scheme 278 Synthesis of 2,3-diaminoindole from 2-iodo-3-nitro-1-(phenylsulfonyl)indole. | |
Chhanda Mukhopadhyay and colleague reported the development of an exceptional oxidative coupling/cyclization technique for the synthesis of 5H-pyrazino[2,3-b]indole compounds (2023). This technique involves two sequential cross-coupling reactions, C–N coupling followed by C–C coupling, both catalyzed by molecular iodine under aerobic conditions. Electron-rich and electron-deficient benzylamines were used with various isatins to synthesize a range of 5H-pyrazino[2,3-b]indoles in good to exceptional yields (Scheme 279).404
 |
| | Scheme 279 Synthesis of 5H-pyrazino[2,3-b]indole. | |
Yongmin Ma and colleague described a metal-free method for the synthesis of dihydropyrazino[2,3-b]indoles in 2-MeTHF, using a green solvent at r.t (2024). A collection of functionalized dihydropyrazino[2,3-b]indoles (42 instances, 61–85%) was synthesized from widely accessible 2-aminoacetophenones, isocyanates, and 1,2-diamines in favorable yields, even on a gram scale, by straightforward filtering without the need for column chromatography purification. The current technology employs organocatalysts, an environmentally benign solvent, a domino approach, moderate conditions, easily obtainable substrates, large-scale synthesis, and wide substrate range (Scheme 280).405
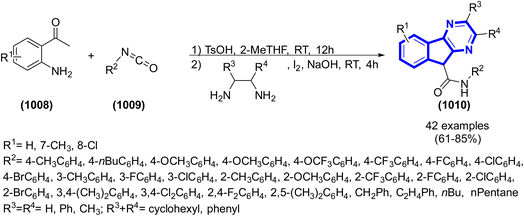 |
| | Scheme 280 Synthesis of dihydropyrazino[2,3-b]indoles in 2-MeTHF. | |
3.3. Six-membered heterocycles containing three nitrogen atoms
3.3.1. Triazinane. The synthesis of nitrogen-containing heterocyclic compounds has attracted significant attention due to their efficacy in interacting with diverse biological receptors, exhibiting high binding affinity.406 This article highlighted the triazine moiety, which has a wide range of biological properties and has evolved into a crucial heterocyclic scaffold, making it one of the most thoroughly researched heterocycles. The triazine structure is a heterocyclic ring analogous to the six-membered benzene ring, with three carbon atoms substituted by nitrogen atoms. The isomers of triazine are differentiated by the arrangement of their nitrogen atoms, designated as 1,2,3-triazine (994), 1,2,4-triazine (995), and 1,3,5-triazine (996) (Scheme 281).407
 |
| | Scheme 281 Various isomers of triazine. | |
The condensation of formaldehyde with nitriles was researched by M. A. Gradsten and M. W. Pollock in 1948 (Scheme 282).408 The reaction was performed under the catalysis of sulfuric acid. Similarly, M. A. Gradsten et al. (1950) synthesized hexahydro-1,3,5-tripropionyltriazine via the condensation of aldehyde and propionitrile. The reaction mixture was heated at a temperature in the range of 95 °C to 105 °C. The triazine compound was synthesized, resulting in a yield of 62%.409 Two years later, propionitrile, acrylonitrile, and benzonitrile were used as reagents in this condensation by Villiam Emmon et al., where solvent systems for this reaction were studied.410
 |
| | Scheme 282 Synthesis of triazine catalyzed by sulfuric acid. | |
A. T. Nielsen et al. (1973) researched this reaction via a condensation reaction (Scheme 283).411 Firstly, aldehyde ammonia hydrates were prepared via the nucleophile addition of aldehyde and ammonia. Next, amino-1-alkanols were converted to triazines after 2 h. A wide range of 2,4,6-trialkyl-1,3,5-hexahydrotriazine derivatives was prepared in a high yield.
 |
| | Scheme 283 Synthesis of 2,4,6-trialkyl-1,3,5-hexahydrotriazine. | |
Paraformaldehyde condensation with diamine was described by Jeannette M. García et al. (2014) (Scheme 284).412 The reaction was conducted in dimethyl sulfoxide at 50 °C or 185 °C, leading to the formation of 1,3,5-substituted hexahydro-1,3,5-triazines in 70% yield.
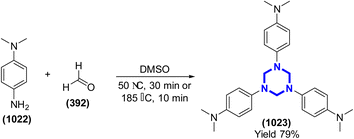 |
| | Scheme 284 Synthesis of hexahydro-1,3,5-triazines. | |
Tongtong Yang et al. (2019) reported a unique approach for the synthesis of 1,3,5-triazinane derivatives (Scheme 285).413 The condensation of hexafluoro-2,2-propanediamine and formaldehyde with tert-butylamine was carried out at 65 °C for 12 h. Specifically, 1-(tert-butyl)-4,4-bis(trifluoromethyl)-1,3,5-triazinane was obtained in high yield without further purification (Scheme 286).
 |
| | Scheme 285 Synthesis of 1,3,5-triazinane. | |
 |
| | Scheme 286 Chemical structures of bioactive compounds containing a 1,2,3-triazine-framework. | |
3.3.2. 1,2,3-Triazine. The 1,2,3-triazine heterocyclic system is composed of three carbon atoms and three nitrogen atoms arranged asymmetrically within a six-membered ring structure, one of which is composed of three nitrogen atoms arranged symmetrically at the 1, 2, and 3 positions. 1,2,3-Triazine is an important ingredient in many different types of medications, including antitumor,414 antibacterial,415 and antifungal drugs.416Terumitsu Kaihoh et al. (1987) synthesized halo-1,2,3-triazines (Scheme 287).417 1-Aminopyrazoles were treated with chlorine and bromine. Especially, triazines or halogenated 1-aminopy were not observed in this process. Various halo-1,2,3-triazines were obtained in high yields through this simple one-pot reaction.
 |
| | Scheme 287 Synthesis of halo-1,2,3-triazines. | |
Similarly, Akio Ohsawa et al. (1988) continuously researched the synthesis of 1,2,3-triazines using the same method (Scheme 288).418 The reactions were conducted between N-aminopyrazoles with various halogenating reagents such as Cl2, Br2, I2, Br–Cl, I–Cl, I–Br, N-chlorosuccinimide, and N-bromosuccinimide. Under the optimized conditions, many halogenated and unhalogenated 1,2,3-triazines were produced using this approach.
 |
| | Scheme 288 Synthesis of halogenated and unhalogenated 1,2,3-triazines. | |
The one-pot synthesis of fluorescent 2,5-dihydro-1,2,3-triazine frameworks was established by Richard N. Butler et al. (2006) (Scheme 289).419,420 The corresponding triazines were afforded via the reaction of 1,2,3-triazolium-1-aminides and propiolate esters involving Huisgen cycloaddition and rearrangements. The reaction mixture was refluxed in anhydrous acetone for a duration of 24 h. A diverse array of 1,2,3-triazine derivatives was synthesized in satisfactory yields (Scheme 290).
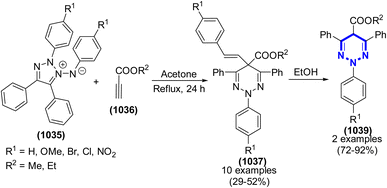 |
| | Scheme 289 Synthesis of 2,5-dihydro-1,2,3-triazine. | |
 |
| | Scheme 290 Chemical structures of bioactive compounds containing a 1,2,4-triazine framework. | |
3.3.3. 1,2,4-Triazine. The 1,2,4-triazine heterocyclic system is comprised of a six-membered ring structure consisting of three carbon atoms and three nitrogen atoms arranged asymmetrically, one of which is composed of three nitrogen atoms arranged symmetrically at the 1, 2, and 4 positions. 1,2,4-Triazines and their oxo derivatives show a wide variety of biological activities, including antibacterial,421 antifungal,422 anticancer,423 and anti-inflammatory424 properties.W. W. Paudler and J. M. Barton (1966) synthesized 1,2,4-triazine-3-carboxylate via a simple condensation reaction (Scheme 291).425 A mixture of glyoxal with ethyl oxalamidrazonate was stirred in ethanol at r.t. for 36 h, affording the target compounds.
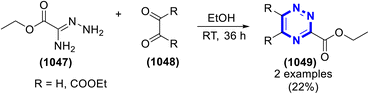 |
| | Scheme 291 Synthesis of 1,2,4-triazine-3-carboxylate. | |
Shahnaz Rostamizadeh and Kamran Sadeghi (2002) prepared 1,2,4-triazines through a one-pot three-component reaction (Scheme 292).426 A mixture of acid hydrazide, 1,2-diketones, and ammonium acetate was irradiated under a microwave source. The reaction was conducted on the surface of silica gel in the presence of a tertiary amine for the appropriate time. This protocol is efficient due to its rapid reaction rate, simple workup, and good yield.
 |
| | Scheme 292 Synthesis of 1,2,4-triazines catalyzed by silica gel. | |
A similar protocol for the preparation of 1,2,4-triazines from fatty acid hydrazides was studied by Abdul Rauf et al. (2007) (Scheme 293).427 The condensation of 1,2-diketones with MW was conducted on solid inorganic substrates under solvent-free conditions. The fatty-substituted triazine compounds were obtained in excellent yield and screened for antimicrobial activity.
 |
| | Scheme 293 Synthesis of 3,5,6-trisubstituted-1,2,4-triazines catalyzed by silica gel. | |
In 2007, a green one-pot synthesis protocol of 1,2,4-triazines in the Brønsted acidic IL was reported by T. M. Potewar et al. (Scheme 294).428 A mixture of hydrazide acids, 1,2-diketone compounds, and ammonium acetate was stirred at 100 °C for short reaction times. Cat. 66 ([Hbim]BF4) was used as the catalyst and solvent. Various 1,2,4-triazine compounds were afforded in excellent isolated yields.
 |
| | Scheme 294 Synthesis of 1,2,4-triazines catalyzed by Cat. 66. | |
Fabian Krauth et al. (2010) synthesized 1,2,4-triazine derivatives via cyclization (Scheme 295).429 Firstly, hydrazonoyl chlorides were reacted with amines such as dimethylamine, pyrrolidine, piperidine, and morpholine in dioxane at r.t. for 12 h. Next, a mixture of amidrazone intermediate compounds and formaldehyde was refluxed in ethanol using PTSA as a catalyst for 1–2 h. Finally, the target product was recrystallized and surveyed for antiproliferative and cytotoxic activity.
 |
| | Scheme 295 Synthesis of 1,2,4-triazine catalyzed by p-toluenesulfonic acid. | |
Riyaj S. Tamboli et al. (2014) prepared 5,6-diaryl-1,2,4-triazines through a one-pot reaction (Scheme 296).430 A mixture of diketones, thiosemicarbazides, and methyl iodide was stirred at 70 °C for 20 min. A mixture of DMSO and 1,3-dibutylimidazolium bromide (1:10 w/w) was used as the solvent in this process. A regioisomeric mixture of target 1,2,4-triazines was obtained from unsymmetrical diketone compounds. In particular, the IL exhibited a high degree of recoverability and was successfully reused multiple times.
 |
| | Scheme 296 Synthesis of 5,6-diaryl-1,2,4-triazines in an ionic liquid. | |
In the publication by Tang et al., they presented a concise and effective approach towards the synthesis of 1,2,4-triazine derivatives using domino cyclization (Scheme 297).431 The condensation was conducted between amidrazones and ketone or alkyne compounds in the presence of SeO2 or NIS-TsOH systems (N-iodosuccinimide and PTSA) as oxidants. The reaction mixture was subjected to thermal treatment at 110 °C for 4 h. A series of corresponding 1,2,4-triazines was prepared in moderate to high yield.
 |
| | Scheme 297 Synthesis of 1,2,4-triazine catalyzed by SeO2 or NIS-TsOH systems. | |
Yafeng Liu et al. (2017) presented an innovative approach for the production of 1,2,4-triazine compounds by utilizing of two interconnected domino reactions (Scheme 298).432 A mixture of N-aminobenzamidine and arylacetaldehydes or arylethyl alcohols was heated at 100 °C for 2 h in the presence of iodine and Cat. 15 additive, respectively. This study focused on investigating high functional group tolerance, which resulted in the production of the desired compounds in exceptional yields. The employed approach, devoid of transition metals, exhibited characteristics of simplicity, environmental friendliness, and high efficiency in the realm of heterocyclic synthesis.
 |
| | Scheme 298 Synthesis of 1,2,4-triazine catalyzed by o-iodoxybenzoic acid. | |
A one-pot protocol for the synthesis of 1,2,4-triazines was described by O. V. Shabunina et al. (2018) (Scheme 299).433 This process was conducted via the condensation of different hydrazones and aldehydes, with the addition of glacial AcOH to the reaction mixture. The annulation reaction occurred at ambient conditions, specifically at r.t., for the designated duration. The aforementioned methodology was deemed advantageous due to its practicality, straightforward experimental procedure, accessible substrates, and notable efficiency in terms of product yield.
 |
| | Scheme 299 Synthesis of 1,2,4-triazines catalyzed by glacial AcOH. | |
In the same year, Matthew S. Dowling et al. prepared 3,6-disubstituted-1,2,4-triazine scaffolds via an annulation reaction (Scheme 300).434 A mixture of β-keto-N-acylsulfonamide and hydrazine was mixed with Brønsted acid solution (HCl or TsOH) and heated at 45–60 °C for 18–24 h. Especially, two approaches were studied with different starting materials. Consequently, a diverse array of 3,6-disubstituted-1,2,4-triazines was synthesized by including various sp3-linked substituents at both the C-3 and C-6 positions.
 |
| | Scheme 300 Synthesis of 3,6-disubstituted-1,2,4-triazines catalyzed by Brønsted acid solution. | |
Lu Zhang et al. (2018) described a novel protocol for the synthesis of 2,5-dihydro-1,2,4-triazines and 1,2,4-triazines (Scheme 301).435 The initial step was the [3 + 3] cyclization reaction between ethyl diazoacetate and imine, which was carried out with the assistance of Cat. 12 as the catalyst. The combination was subjected to stirring at ambient temperature for a duration of 15 to 40 min in the presence of N,N-dimethylformamide. Then, tetrahydro-1,2,4-triazine was treated with 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ) as an oxidant and continuously stirred for the appropriate time and temperature. Dihydro-1,2,4-triazines and 1,2,4-triazines were synthesized under different reaction conditions. The former was obtained by conducting the reaction at 0 °C for 30 min, while the latter was created by carrying out the reaction at r.t. for a period of 8 h. The technique employed in this study demonstrated high efficiency, showcasing several advantageous features such as a streamlined one-pot process, a wide range of applicable substrates, excellent tolerance towards various functional groups, and mild reaction conditions (Scheme 302).
 |
| | Scheme 301 Synthesis of dihydro-1,2,4-triazines and 1,2,4-triazines. | |
 |
| | Scheme 302 Chemical structures of bioactive compounds containing a 1,3,5-triazine framework. | |
3.3.4. 1,3,5-Triazine. The 1,3,5-triazine heterocyclic system consists of a six-membered ring structure of three carbon atoms and three nitrogen atoms. The nitrogen atoms are positioned asymmetrically within the ring, with a symmetrical arrangement of three nitrogen atoms at 1, 3, and 5 positions. 1,3,5-Triazine and its derivatives have been shown to be efficient antibacterial,436 antimalarial,437 anticancer,438 antimicrobial,439 and antitubercular440 agents.Haruo Ogura et al. (1981) illustrated a strategy for the synthesis of triazinethione analogues (Scheme 303).441 The reaction of a carboximidamide with isothiocyanate was performed in acetonitrile at r.t. Then, triethyl orthoformate and xylene were added to the mixture and heated, and the desired triazinethiones were obtained via the cyclization of isothiobiuret.
 |
| | Scheme 303 Synthesis of triazinethione. | |
The one-pot preparation of (2-oxo-1,2-dihydropyridin-3-yl)-1,3,5-triazines was shown by Tomohiro Okawa et al. (1997) (Scheme 304).442 This is an approach with multiple steps including the intermolecular aza-Wittig reaction of methyl 2-(N-triphenylphosphoranylidene)amino-nicotinate with aryl isocyanates and heterocyclization by primary amines. Pyrido[1,2-a][1,3,5]triazines, the key intermediates, were isolated and characterized. Therefore, the transformation mechanism was proposed to elucidate the process by which the desired products are formed.
 |
| | Scheme 304 Synthesis of (2-oxo-1,2-dihydropyridin-3-yl)-1,3,5-triazines. | |
Shie and Fang (2006) employed the microwave-assisted technique to manufacture triazines through the cycloaddition of intermediate nitriles with dicyandiamide (Scheme 305).443 A mixture of aromatic aldehyde and iodine dissolved in ammonia water and tetrahydrofuran (THF) was introduced in a round-bottom flask and subjected to stirring for a duration of 1 h at ambient temperature. This reaction yielded the desired nitrile compound. Subsequently, the inclusion of dicyandiamide in the amalgamation ensued, followed by subjecting the amalgamation to irradiation in a microwave reactor operating at 80 °C for 15–30 min. Diamino-1,3,5-triazines were synthesized in a significant yield.
 |
| | Scheme 305 Synthesis of triazines. | |
M. Hemdan and M. Elshahawi (2007) synthesized 1,3,5-triazines from 3-oxo-5,6-diphenyl-2,3-dihydropyridazine-4-carbonyl isothiocyanate (Scheme 306).444 A mixture of isothiocyanate and guanidine HCl was refluxed, furnishing 1,3,5-triazine derivatives. This reaction was catalyzed by a certain amount of triethyl amine. The corresponding products were obtained in a yield of 71%.
 |
| | Scheme 306 Synthesis of 1,3,5-triazines catalyzed by triethyl amine. | |
A cyclocondensation process for the synthesis of pyridyl-substituted 1,3,5-triazines was developed by Laetitia Le Falher et al. (2014) (Scheme 307).445 The reaction between 4H-pyrido[1,3]oxazin-4-ones and amidines was carried out at ambient temperature or using MW. A diverse array of triazine compounds with varying properties was successfully synthesized from three distinct derivatives of 4H-pyrido[1,3]oxazin-4-one, yielding satisfactory to exceptional results. The hypothesized reaction mechanism for this change is considered to be feasible. This protocol is a rapid, efficient, and economic one-step synthetic preparation.
 |
| | Scheme 307 Synthesis of pyridyl-substituted 1,3,5-triazines under catalyst-free conditions. | |
Antonio Herrera et al. (2014) reported a one-pot approach for the synthesis of 1,3,5-triazine derivatives through the controlled cross-cyclotrimerization of nitriles (Scheme 308).446 The formation of 2,4-disubstituted-6-substituted 1,3,5-triazines was achieved through the reaction between a nitrile compound and either triflic anhydride or triflic acid. The one-pot reaction was conducted by subjecting the mixture to a temperature in the range of 65–110 °C for a duration of 24 h. A diverse range of triazine derivatives was synthesized from two distinct nitriles, resulting in yields varying from moderate to high.
 |
| | Scheme 308 Synthesis of 1,3,5-triazine. | |
Issa Yavari and Shabnam Mosaferi (2017) prepared 2H-pyrido[1,2-a][1,3,5]triazine-2-selenones via a one-pot reaction (Scheme 309).447 Initially, acyl chlorides were subjected to a reaction with potassium selenocyanate in acetone under ambient conditions for a duration of 10 min, resulting in the formation of acyl isoselenocyanates. Subsequently, pyridin-2-amine was introduced in the solution, which was then subjected to continuous stirring at ambient temperature for 8 h, resulting in the formation of pyrido[1,2-a][1,3,5]triazines. The operational simplicity, variety of reactants, easy workup, and potential diversity are advantages of this protocol.
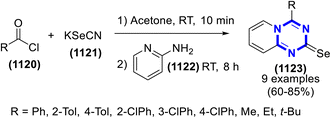 |
| | Scheme 309 Synthesis of 2H-pyrido[1,2-a][1,3,5]triazine-2-selenones. | |
A catalyst-free methodology for the preparation of 1,3,5-triazine-2,4-dithione scaffolds was developed by Gui-Feng Kang and Gang Zhang (2020) (Scheme 310).448 The implementation of this approach involved the utilization of three–component processes involving arylaldehydes, thiourea, and trialkyl orthoformates. The combination of substrates was subjected to thermal treatment in N,N-dimethylformamide at 80 °C for 1 h. A range of targeted substituted triazinethione compounds was synthesized in moderate to high yields via a one-pot process involving substituted aldehydes and thiourea. The investigation and proposal of a comprehensive plausible mechanism were based on control experiments. The method employed in this study is a direct and efficient strategy for synthesizing 4-aryl-6-(alkylthio)-3,4-dihydro-1,3,5-triazine-2(1H)-thione derivatives.
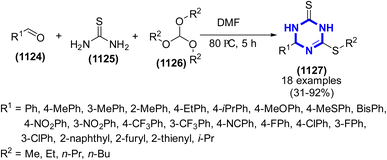 |
| | Scheme 310 Synthesis of 4-aryl-6-(alkylthio)-3,4-dihydro-1,3,5-triazine-2(1H)-thiones. | |
3.4. Six-membered rings with four nitrogen-atom-containing heterocycles
3.4.1. 1,2,3,4-Tetrazine. Tetrazine is a chemical compound characterized by a six-membered aromatic ring with four nitrogen atoms incorporated within its structure. In many cases, 1,2,3,4-tetrazines were obtained by fusing them into an aromatic ring system. Once this was done, the tetrazines were then stabilized as the dioxide derivatives, which were also referred to as v-tetrazines.449Aleksandr M. Churakov et al. prepared benzo-1,2,3,4-tetrazine-1,3-dioxides (Scheme 311).450 The annulation of 2-(tert-butyl-azoxy)aniline derivatives and oxodiazonium ion could be carried out via two different methods such as nitration of N-nitroamines or treatment of diazonium salts with peracids under basic conditions.
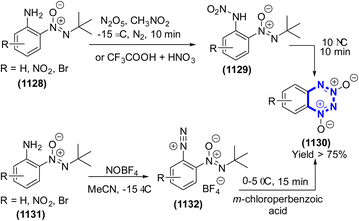 |
| | Scheme 311 Synthesis of benzo-1,2,3,4-tetrazine-1,3-dioxides. | |
Similarly, Thomas M. Klapotke et al. (2012) described a process for the synthesis of 5,7-dinitrobenzo-1,2,3,4-tetrazine-1,3-dioxide (Scheme 312).451 Initially, a combination of 2-tert-butyl-1-(2-anilino)-diazine-1-oxide and dinitrogen pentoxide was subjected to treatment in dichloromethane at −20 °C. Subsequently, the mixture was agitated for 15 min at 0 °C under a controlled nitrogen environment. The reaction mixture was subjected to heating at 80 °C for 2 h subsequent to the addition of nitric acid and oleum solution. The tetrazine-1,3-dioxide of interest was acquired in a yield of 45%.
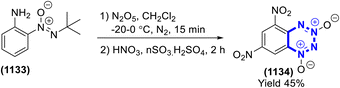 |
| | Scheme 312 Synthesis of 5,7-dinitrobenzo-1,2,3,4-tetrazine-1,3-dioxide catalyzed by nitric acid and oleum solution. | |
A novel protocol for the synthesis of 1,2,3,4-tetrazine 1,3-dioxides was described by Alexey A. Voronin et al. (2014) (Scheme 313).452 The annulation was carried out between 4-amino-5-(tert-butyl-NNO-azoxy)-2 R-2H-1,2,3-triazoles or their 1-oxides in a nitric acid–sulfuric acid–anhydride acetic system. The mixture was heated to ambient temperature and agitated for a duration of approximately one hour.
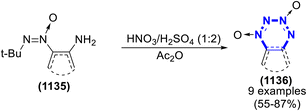 |
| | Scheme 313 Synthesis of 1,2,3,4-tetrazine-1,3-dioxides catalyzed by nitric acid–sulfuric acid–anhydride acetic system. | |
In addition, similar research was also performed by Alexey A. Voronin et al. (2017) (Scheme 314).453 A range of N-alkyl and N-aryl substituted derivatives of 1,2,3,4-tetrazine-1,3-dioxides was synthesized via an annulation process involving N-aryl-1,2,3-triazoles.
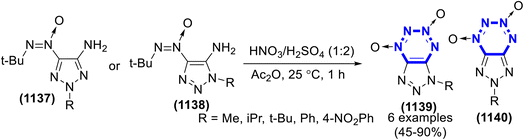 |
| | Scheme 314 Synthesis of N-alkyl and N-aryl substituted 1,2,3,4-tetrazine-1,3-dioxides catalyzed by nitric acid–sulfuric acid–anhydride acetic system. | |
In another four-step process, Michael S. Klenov et al. (2015) reported the synthesis of 6-(tert-butyl-NNO-azoxy)-5-methylthio-1,2,3,4-tetrazine-1,3-dioxide (Scheme 315).454 This reaction was performed via the treatment of azoxyalkene with excess BF3·Et2O at 5 °C. After 72 h, the 1,2,3,4-tetrazine-1,3-dioxide product was obtained in 20% yield with by-products (Scheme 316).
 |
| | Scheme 315 Synthesis of 6-(tert-butyl-NNO-azoxy)-5-methylthio-1,2,3,4-tetrazine-1,3-dioxide catalyzed by BF3·Et2O. | |
 |
| | Scheme 316 Chemical structure of a bioactive compound containing a 1,2,3,5-tetrazine framework. | |
3.4.2. 1,2,3,5-Tetrazine. Tetrazine is a chemical compound characterized by a six-membered aromatic ring with four nitrogen atoms incorporated within its structure. 1,2,3,5-Tetrazines, commonly referred to as tetrazines, have demonstrated notable biological activity as antiproliferative agents.455Paola Barraja et al. (2004) showed a strategy for the synthesis of [1,2,3,5]tetrazino-[5,4-a]indole-4-one (Scheme 317).455 A mixture of 2-diazoindoles and alkyl or aryl isocyanates was dissolved in anhydrous dichloromethane at 0 °C and agitated at ambient temperature for 1–2 h. A diverse array of target products was successfully synthesized in high yields following the purification process involving column chromatography and recrystallization.
 |
| | Scheme 317 Synthesis of [1,2,3,5]tetrazino[5,4-a]indole-4-one catalyzed under catalyst-free conditions. | |
A novel protocol for preparing 1,2,3,5-tetrazines was reported by Richard N. Butler et al. (1988 and 1990) (Scheme 318).456,457 This method is based on the cycloadditions of aryl N-sulphinylamines with substituted triazolium imides. The mixture reaction was refluxed in benzene for 48 h.
 |
| | Scheme 318 Synthesis of 1,2,3,5-tetrazine under catalyst-free conditions. | |
Then, Zhi-Chen Wu et al. (2019) also applied this reaction for the multi-gram scale preparation of N2,4,6,N5-tetrasubstituted 2,5-dihydro-1,2,3,5-tetrazines (Scheme 319).458
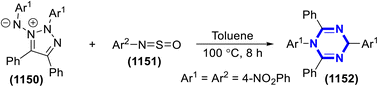 |
| | Scheme 319 Synthesis of N2,4,6,N5-tetrasubstituted 2,5-dihydro-1,2,3,5-tetrazines under catalyst-free conditions. | |
3.4.3. 1,2,4,5-Tetrazine. Tetrazine is a chemical compound with a six-membered aromatic ring with the inclusion of four nitrogen atoms. The 1,2,4,5-tetrazines have been extensively utilized in several fields and have gained significant recognition. The existence of a class of chemicals known as 3,6-disubstituted 1,2,4,5-tetrazines, alternatively referred to as s-tetrazines, has been documented. Studies on the particular biological activity of 1,2,4,5-tetrazines as an antitumor agent have been conducted (Scheme 320).459
 |
| | Scheme 320 Chemical structures of bioactive compounds containing a 1,2,4,5-tetrazine framework. | |
M. D. Coburn et al. (1991) prepared 1,2,4,5-tetrazine via the condensation of hydrazinecarbohydrazonhydrazide chloride and acetylacetone (Scheme 321).460 Similarly, Jochen Kerth et al. (2002) also synthesized 1,2,4,5-tetrazine via the same two-step process. Firstly, a dihydrotetrazine derivative was synthesized via the condensation of pentan-2,4-dione giving nitrate and the salt of hydrazinecarbohydrazonhydrazide. Then, it was oxidized by gaseous N2O4, affording 3,6-bis(3,5-dimethylpyrazol-1-yl)-1,2,4,5-tetrazine in 99% yield.461
 |
| | Scheme 321 Synthesis of 1,2,4,5-tetrazine under catalyst-free conditions. | |
Guo-Wu Rao et al. (2006) synthesized various 1,2,4,5-tetrazine derivatives (Scheme 322).462 In this study, two routes were used the preparation of 1,2,4,5-tetrazine. The first method was performed via the annulation of nitrile and hydrazine hydrate in the presence of elemental sulfur and ethanol. Besides, the azine compound was refluxed with hydrazine hydrate, leading to tetrazine. 3,6-Disubstituted-1,4-dihydro-1,2,4,5-tetrazines were treated via the oxidation of a mixture of sodium nitrite in an acetic acid solution.
 |
| | Scheme 322 Synthesis of various 1,2,4,5-tetrazines under catalyst-free conditions. | |
In the study by Karver et al. (2011), they presented a straightforward approach for the production of 1,2,4,5-tetrazines through [4 + 2] cycloaddition (Scheme 323).463 In the first step, carbonitrile, imidate ester, or amidine pair reacted in hydrazine at r.t. for the appropriate time. Then, the oxidation of dihydrotetrazine intermediates was performed by adding a mixture of sodium nitrite in an HCl solution.
 |
| | Scheme 323 Synthesis of 1,2,4,5-tetrazines under catalyst-free conditions. | |
A green protocol for the preparation of tetrazines was described by Zheng Fang et al. (2013) (Scheme 324).464 Briefly, gem-difluoroalkenes were treated with hydrazine in N,N-dimethylformamide. Subsequently, ethyl acetate and a base were added to the mixture. The experimental procedure involved studying the reaction under an oxygen atmosphere, at a temperature consistent with the ambient conditions. The symmetric and asymmetric 3,6-disubstituted 1,2,4,5-tetrazines were synthesized in excellent yields.
 |
| | Scheme 324 Synthesis of tetrazines under catalyst-free conditions. | |
In a study conducted by Chen Li et al. (2015), a novel methodology was developed for the synthesis of 3,6-unsymmetrically disubstituted-1,2,4,5-tetrazines through a one-pot process (Scheme 325).465 The cyclization of nitriles and hydrazine hydrate was performed by refluxing in ethanol at 78 °C. Especially, elemental sulfur was used as the catalyst. A series of target 3,6-unsymmetrically disubstituted-1,2,4,5-tetrazines was successfully produced in good yield.
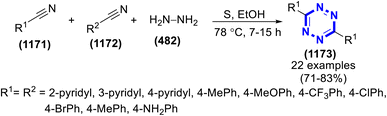 |
| | Scheme 325 Synthesis of 1,2,4,5-tetrazines catalyzed by elemental sulfur. | |
A novel sym-tetrazine was prepared by Tawfik A. Khattab (2018) through the condensation of nitrile with hydrazine (Scheme 326).466 The reaction was carried out between 4-hydroxybenzonitrile and hydrazine at 90 °C for 16 h, leading to 4,4′-(1,2,3,6-tetrahydro-1,2,4,5-tetrazine-3,6-diyl)diphenol. The intermediate underwent oxidation when exposed to a combination of sodium nitrite and acetic acid. This procedure successfully yielded 4,4′-(1,2,4,5-tetrazine-3,6-diyl)diphenol.
 |
| | Scheme 326 Synthesis of tetrazines under catalyst-free conditions. | |
Y.-Z. Ji et al. (2019) synthesized 1,2,4,5-tetrazine derivatives via the NCS-mediated chlorination of arylaldehyde-derived N-tosylhydrazones (Scheme 327).467 The arylaldehyde-derived arylsulfonyl-hydrazones underwent a reaction with N-chlorosuccinimide in the presence of a base. A diverse array of 1,2,4,5-tetrazine derivatives was synthesized in high yields using a wide range of hydrazone substrates, all in the absence of any metal catalysts.
 |
| | Scheme 327 Synthesis of 1,2,4,5-tetrazines. | |
4 Conclusions and outlook
This review summarized the current progress in the synthesis of N-heterocyclic frameworks, namely 5- and 6-membered rings, using one-pot methodologies devoid of transition metal catalysts. These techniques facilitate the synthesis of several valuable N-heterocyclic frameworks of significant interest using efficient, time-saving, and atom-economical procedures.
Despite the extensive applicability of N-heterocyclic frameworks, particularly 5- and 6-membered rings, several hurdles and untapped potential persist in the pursuit of developing more universal, efficient, sustainable, and practical synthetic methodologies. We provide a compilation of suggestions for further endeavors. The restricted substrate range can be broadened under moderate and environmentally friendly reaction conditions. Emphasis should be placed on the installation of potentially transformable groupings in effective medications and pharmaceuticals. Recent results on the electrochemical oxidative annulation of N-heterocycles indicate that this area still offers significant opportunities for synthetic advancements and noteworthy enhancements. The deliberate amalgamation of efficient chemical synthesis and enzymatic methods for the creation of various heterocyclic molecules signifies a promising future trajectory. Enzymatic methods, especially biocatalysis and enzyme-mediated cyclization, have distinct benefits including elevated regio- and stereo-selectivity, gentle reaction conditions, and little environmental effect. Recent progress in enzyme engineering and directed evolution has broadened the capabilities of enzymatic techniques for the synthesis of heterocycles, facilitating the development of new scaffolds with improved pharmacological attributes. Advancing further, in-depth investigations of enzyme-catalyzed processes, with chemical techniques, will be crucial in formulating sustainable and adaptable synthetic solutions. The ongoing advancement of N-heterocyclic chemistry will undoubtedly provide significant prospects for synthetic and medicinal chemists, fostering innovation in drug development and materials research. Undoubtedly, the advancement of enriched N-heterocyclic chemistry will persist, providing many prospects for both synthetic and medicinal chemists in the development of diverse heterocyclic frameworks of pharmacological significance.
Data availability statements
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Appendix
Abbreviation
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DFT | Density functional theory |
| dr | Diastereomeric ratio |
| ee | Enantiomeric excess |
| er | Enantiomeric ratio |
| HPLC | High-performance liquid chromatography |
| IL | Ionic liquid |
| MCR | Multi-component reaction |
| MIC | Minimum inhibitory concentration |
| MW | Microwave irradiation |
| NBS | N-Bromosuccinimide |
| PTSA | p-Toluenesulfonic acid |
| r.t. | Room temperature |
| SAR | Specific absorption rate |
| US | Ultrasonic irradiation |
Notes and references
- N. Kerru, S. V. Bhaskaruni, L. Gummidi, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, Synth. Commun., 2019, 49, 2437–2459 CrossRef CAS.
- N. Kerru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 142, 179–212 CAS.
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CAS.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 CAS.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 Search PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CAS.
- J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2019, 38, 36–46 CAS.
- X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CAS.
- C. M. Santos, M. Freitas and E. Fernandes, Eur. J. Med. Chem., 2018, 157, 1460–1479 CAS.
- P. N. Kalaria, S. C. Karad and D. K. Raval, Eur. J. Med. Chem., 2018, 158, 917–936 CAS.
- X. Li, J. Xu and Z.-G. Xu, Org. Chem. Front., 2024, 11, 4041–4053 CAS.
- P. A. Wieczorkiewicz, T. M. Krygowski and H. Szatylowicz, Phys. Chem. Chem. Phys., 2024, 26, 19398–19410 CAS.
- K. H. Nguyen, T. H. Nguyen, H. B. Phan, H. T. Nguyen and P. H. Tran, Next Mater., 2025, 8, 100536 Search PubMed.
- A. A. Bhat, N. Tandon, I. Singh and R. Tandon, J. Mol. Struct., 2023, 1283, 135175 CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- A. V. Smolobochkin, A. S. Gazizov, A. R. Burilov and M. A. Pudovik, Chem. Heterocycl. Compd., 2016, 52, 753–765 CrossRef CAS.
- A. S. Gazizov and A. V. Smolobochkin, Chem. Heterocycl. Compd., 2018, 54, 683–685 CrossRef CAS.
- J. Li, Y. Ye and Y. Zhang, Org. Chem. Front., 2018, 5, 864–892 RSC.
- F.-Y. Chen, L. Xiang, G. Zhan, H. Liu, B. Kang, S.-C. Zhang, C. Peng and B. Han, Tetrahedron Lett., 2020, 61, 151806 CrossRef CAS.
- X. Zhang, W. Qiu, J. Evans, M. Kaur, J. P. Jasinski and W. Zhang, Org. Lett., 2019, 21, 2176–2179 CrossRef CAS PubMed.
- Z. J. Wu, S. R. Li and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 130, 14266–14270 CrossRef.
- D. Kowalczyk, J. Wojciechowski and Ł. Albrecht, Synthesis, 2017, 49, 880–890 CAS.
- Z. Huang, Y. Bao, Y. Zhang, F. Yang, T. Lu and Q. Zhou, J. Org. Chem., 2017, 82, 12726–12734 CrossRef CAS PubMed.
- W. Shi, C.-M. Bai, K. Zhu, D.-M. Cui and C. Zhang, Tetrahedron, 2014, 70, 434–438 CAS.
- D. Chen, F. Liang, D. Feng, M. Xian, H. Zhang, H. Liu and F. Du, Chem. Eng. J., 2016, 300, 177–184 CAS.
- D. En, G.-F. Zou, Y. Guo and W.-W. Liao, J. Org. Chem., 2014, 79, 4456–4462 Search PubMed.
- G. Talavera, E. Reyes, J. L. Vicario, L. Carrillo and U. Uria, Adv. Synth. Catal., 2013, 355, 653–658 CAS.
- C. Guo, J. Song and L.-Z. Gong, Org. Lett., 2013, 15, 2676–2679 CAS.
- E. Gómez-Torres, D. A. Alonso, E. Gómez-Bengoa and C. Najera, Org. Lett., 2011, 13, 6106–6109 Search PubMed.
- F. Yu, X. Sun, Z. Jin, S. Wen, X. Liang and J. Ye, Chem. Commun., 2010, 46, 4589–4591 CAS.
- A. Chaudhary, J. M. Khurana, G. Khanna and M. Saroha, ChemistrySelect, 2018, 3, 6334–6337 CAS.
- L. D. Nguyen, K. H. Nguyen, P. N. Nguyen, K. N. Tran, D. D. Le, P. H. Tran and H. T. Nguyen, J. Chem. Technol. Biotechnol., 2025, 100, 121–137 CAS.
- M. K. Scott, G. E. Martin, D. L. DiStefano, C. L. Fedde, M. J. Kukla, D. L. Barrett, W. J. Baldy, R. J. Elgin, Jr., J. M. Kesslick and J. R. Mathiasen, et al., J. Med. Chem., 1992, 35, 552–558 CAS.
- E. Mateev, M. Georgieva and A. Zlatkov, J. Pharm. Pharm. Sci., 2022, 25, 24–40 CAS.
- M. S. El-Gaby, A. M. Gaber, A. A. Atalla and K. A. Abd Al-Wahab, Farmaco, 2002, 57, 613–617 CAS.
- P. S. Vieira, T. Souza, R. V. Honorato, L. M. Zanphorlin, K. U. Severiano, S. A. Rocco, A. H. C. de Oliveira, A. T. Cordeiro, P. S. L. Oliveira, P. O. de Giuseppe and M. T. Murakami, Biochem. Biophys. Res. Commun., 2017, 488, 461–465 CAS.
- M. J. Palmer, X. Deng, S. Watts, G. Krilov, A. Gerasyuto, S. Kokkonda, F. El Mazouni, J. White, K. L. White, J. Striepen, J. Bath, K. A. Schindler, T. Yeo, D. M. Shackleford, S. Mok, I. Deni, A. Lawong, A. Huang, G. Chen, W. Wang, J. Jayaseelan, K. Katneni, R. Patil, J. Saunders, S. P. Shahi, R. Chittimalla, I. Angulo-Barturen, M. B. Jiménez-Díaz, S. Wittlin, P. K. Tumwebaze, P. J. Rosenthal, R. A. Cooper, A. C. C. Aguiar, R. V. C. Guido, D. B. Pereira, N. Mittal, E. A. Winzeler, D. R. Tomchick, B. Laleu, J. N. Burrows, P. K. Rathod, D. A. Fidock, S. A. Charman and M. A. Phillips, J. Med. Chem., 2021, 64, 6085–6136 CAS.
- M. Xiong, X. Liang, Y. Zhou and Y. Pan, J. Org. Chem., 2021, 86, 4986–4993 CAS.
- M. Ghandi, M. Khodadadi and A. Abbasi, J. Heterocycl. Chem., 2021, 58, 478–487 CAS.
- D. Chang, J. Chen, Y. Liu, H. Huang, A. Qin and G.-J. Deng, J. Org. Chem., 2020, 86, 110–127 Search PubMed.
- Q.-X. Zi, C.-L. Yang, K. Li, Q. Luo, J. Lin and S.-J. Yan, J. Org. Chem., 2019, 85, 327–338 CrossRef PubMed.
- R. Zhang, Z. Zhang, K. Wang and J. Wang, J. Org. Chem., 2020, 85, 9791–9800 CrossRef CAS PubMed.
- J. Zhang, M. Liu, C. Li, Y.-J. Xu and L. Dong, Org. Chem. Front., 2020, 7, 420–424 RSC.
- L. Xu, L. Wu, T. Chen, S. Xu, C. Huang, Y. Wang, Q. You and J. Shen, ChemistrySelect, 2020, 5, 655–659 CAS.
- L. Wei, S.-M. Xu, Z. Jia, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2020, 56, 9691–9694 CAS.
- Z. Su, S. Wang, N. Luo and C. Wang, Synlett, 2020, 31, 1022–1026 CAS.
- R. Shrestha, H. D. Khanal, W. G. Yang, S. H. Kim, J. J. Shim and Y. R. Lee, Asian J. Org. Chem., 2020, 9, 1070–1075 CAS.
- N. A. Mir, P. Ramaraju, S. Vanaparthi, S. Choudhary, R. P. Singh, P. Sharma, R. Kant, R. Singh, M. Sankaranarayanan and I. Kumar, New J. Chem., 2020, 44, 16329–16339 CAS.
- V. A. Mamedov, E. A. Khafizova, N. E. Algaeva, S. K. Latypov and O. G. Sinyashin, J. Org. Chem., 2020, 85, 9887–9904 CAS.
- H. Jia, D. Min, T. Guo, M. Wu, X. Wang, J. Liu and S. Tang, J. Org. Chem., 2020, 85, 14847–14857 CAS.
- Q.-W. Gui, X. He, W. Wang, H. Zhou, Y. Dong, N. Wang, J.-X. Tang, Z. Cao and W.-M. He, Green Chem., 2020, 22, 118–122 CAS.
- N. E. Golantsov, A. S. Golubenkova, A. A. Festa, A. V. Varlamov and L. G. Voskressensky, Org. Lett., 2020, 22, 4726–4731 CAS.
- P. Gao, H.-J. Chen, Z.-J. Bai, M.-N. Zhao, D. Yang, J. Wang, N. Wang, L. Du and Z.-H. Guan, J. Org. Chem., 2020, 85, 7939–7951 CAS.
- I. V. Efimov, M. D. Matveeva, R. Luque, V. A. Bakulev and L. G. Voskressensky, Eur. J. Org Chem., 2020, 2020, 1108–1113 CAS.
- E. Dhanasekar, T. Kannan, R. Venkatesan, P. T. Perumal and J. Kamalraja, J. Org. Chem., 2020, 85, 9631–9649 CAS.
- S. Choudhary, J. Yadav, A. P. Pawar, S. Vanaparthi, N. A. Mir, E. Iype, R. Sharma, R. Kant and I. Kumar, Org. Biomol. Chem., 2020, 18, 1155–1164 CAS.
- T. Yang, K.-H. Wang, D. Huang, P. Li, Z. Deng, Y. Su and Y. Hu, Tetrahedron, 2019, 75, 2291–2297 CAS.
- V. Rajeshkumar, C. Neelamegam and S. Anandan, Synthesis, 2019, 51, 4023–4033 CAS.
- L. Meng, X. Chi, X. Sun, C. Cao, B. Ai, Q. Liu, P. Zhao, Z. Zhao, Y. Dong and H. Liu, Org. Biomol. Chem., 2019, 17, 7669–7673 CAS.
- J. A. Malone, C. E. Toussel, F. R. Fronczek and R. Kartika, Org. Lett., 2019, 21, 3610–3614 CrossRef CAS PubMed.
- V. Kumar, A. Awasthi, A. Metya and T. Khan, J. Org. Chem., 2019, 84, 11581–11595 CrossRef CAS PubMed.
- B. S. Karki, L. Devi, A. Pokhriyal, R. Kant and N. Rastogi, Chem. – Asian J., 2019, 14, 4793–4797 CrossRef CAS PubMed.
- X. Gao, P. Wang, Q. Wang, J. Chen and A. Lei, Green Chem., 2019, 21, 4941–4945 RSC.
- X. Chang, X. Yang, Z. Chen and W. Zhong, Synlett, 2019, 30, 1431–1436 CrossRef CAS.
- A. Balu Atar, E. Han, D. H. Sohn and J. Kang, Synth. Commun., 2019, 49, 1181–1192 CrossRef CAS.
- E. Ghabraie, S. Balalaie, M. Bararjanian, H. R. Bijanzadeh and F. Rominger, Tetrahedron, 2011, 67, 5415–5420 CrossRef CAS.
- A. Khammas, C. Yolacan and F. Aydogan, Russ. J. Gen. Chem., 2018, 88, 2680–2683 CAS.
- C. Zhou, H. Zheng, Y. Chen, G. Mao and G.-J. Deng, J. Org. Chem., 2023, 88, 1533–1544 CAS.
- K. Afratis, J. M. Bateman, B. F. Rahemtulla, O. Hughes, B. C. Milgram, T. A. Mulhern and E. P. A. Talbot, Org. Lett., 2023, 25, 461–465 CAS.
- T. Abbaspour, G. Firouzzadeh Pasha and M. Tajbakhsh, Appl. Organomet. Chem., 2023, 37, e6933 CAS.
- P. Chacko and K. Shivashankar, Tetrahedron, 2018, 74, 1520–1526 CAS.
- J. C. Borghs, L. M. Azofra, T. Biberger, O. Linnenberg, L. Cavallo, M. Rueping and O. El-Sepelgy, ChemSusChem, 2019, 12, 3083–3088 CAS.
- L. Kumar S, A. Servesh, S. J. Chundattu, S. Tabassum and S. Govindaraju, Mol. Diversity, 2024, 29, 1761–1787 Search PubMed.
- C.-X. Li, R.-J. Liu, K. Yin, L.-R. Wen and M. Li, Org. Biomol. Chem., 2017, 15, 5820–5823 CAS.
- T. Pavithra, D. B. Rajkumar, K. Gnanaoli, S. N. Sunil Gowda, N. Devipriya and C. U. Maheswari, ChemistrySelect, 2023, 8, e202204564 CAS.
- S. Pervaram, D. Ashok, C. V. R. Reddy, M. Sarasija and A. Ganesh, Chem. Data Collect., 2020, 29, 100508 CAS.
- T. Yu, F. Ji, D. Huang, Y. Gao, Z. Shi, X. Sha, J. Xu, S. You, M. Zhang and Q. Sha, Org. Chem. Front., 2021, 8, 5716–5721 CAS.
- H. T. Nguyen, P. N. Nguyen, T. Van Le, T. H. Nguyen, L. D. Nguyen and P. H. Tran, RSC Adv., 2023, 13, 28623–28631 RSC.
- G. Wang, S. Sun and H. Guo, Eur. J. Med. Chem., 2022, 229, 113999 CrossRef CAS PubMed.
- X. Zhu, Y. He, Z. Liu, Z. Zhu, Y. He, J. Qiu, D. Liu, M. Mo, P. Wang, X. Tian and P. Xu, Appl. Mater. Today, 2020, 19, 100559 CrossRef.
- Y. J. Xue, M. Y. Li, X. J. Jin, C. J. Zheng and H. R. Piao, J. Enzyme Inhib. Med. Chem., 2021, 36, 295–306 CAS.
- Y. Chen, N. Cao, H. Lv, K. Zeng, J. Yuan, X. Guo, M. Zhao, P. Tu and Y. Jiang, Phytochemistry, 2020, 170, 112186 CrossRef CAS PubMed.
- Y. Tachibana, H. Kikuzaki, N. H. Lajis and N. Nakatani, J. Agric. Food Chem., 2003, 51, 6461–6467 CAS.
- J. A. Scatina, D. R. Hicks, M. Kraml and M. N. Cayen, Xenobiotica, 1989, 19, 991–1002 CAS.
- S. Mandal, A. Nayak, M. Kar, S. K. Banerjee, A. Das, S. N. Upadhyay, R. K. Singh, A. Banerji and J. Banerji, Fitoterapia, 2010, 81, 72–74 CAS.
- J. H. Burckhalter, V. C. Stephens and L. A. Hall, J. Am. Pharm. Assoc., 1950, 39, 271–273 CAS.
- Y.-P. Liu, J.-M. Guo, Y.-Y. Liu, S. Hu, G. Yan, L. Qiang and Y.-H. Fu, J. Agric. Food Chem., 2019, 67, 5764–5771 CAS.
- R. Birari, S. K. Roy, A. Singh and K. K. Bhutani, Nat. Prod. Commun., 2009, 4, 1089–1092 CAS.
- P. Zhang, B. Li, L. Niu, L. Wang, G. Zhang, X. Jia, G. Zhang, S. Liu, L. Ma and W. Gao, Adv. Synth. Catal., 2020, 362, 2342–2347 CAS.
- C. Gioia, A. Hauville, L. Bernardi, F. Fini and A. Ricci, Angew. Chem., Int. Ed., 2008, 47, 9236–9239 CAS.
- J. Wang, H.-T. Zhu, Y.-F. Qiu, Y. Niu, S. Chen, Y.-X. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2015, 17, 3186–3189 CAS.
- S. Yaragorla, D. Bag, R. Dada and K. J. Jose, ACS Omega, 2018, 3, 15024–15034 CAS.
- K. Liu and S. Zhang, ACS Med. Chem. Lett., 2015, 6, 894–897 CAS.
- J. Zhao, P. Li, C. Xia and F. Li, Chem. - Eur. J., 2015, 21, 16383–16386 CAS.
- S. Biswas, A. Dagar, A. Srivastava and S. Samanta, Eur. J. Org Chem., 2015, 2015, 4493–4503 CAS.
- J. W. Lim, S. H. Kim, J. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2015, 36, 1351–1359 CAS.
- C. Raji Reddy, R. Rani Valleti and U. Dilipkumar, Chem. - Eur. J., 2016, 22, 2501–2506 CAS.
- H. Wang, Z. Wang, Y.-L. Wang, R.-R. Zhou, G.-C. Wu, S.-Y. Yin, X. Yan and B. Wang, Org. Lett., 2017, 19, 6140–6143 CrossRef CAS PubMed.
- D. Cao, A. Ying, H. Mo, D. Chen, G. Chen, Z. Wang and J. Yang, J. Org. Chem., 2018, 83, 12568–12574 CrossRef CAS PubMed.
- S. Chen, Y. Li, P. Ni, H. Huang and G.-J. Deng, Org. Lett., 2016, 18, 5384–5387 CrossRef CAS PubMed.
- S. Chen, Y. Li, P. Ni, B. Yang, H. Huang and G.-J. Deng, J. Org. Chem., 2017, 82, 2935–2942 CrossRef CAS PubMed.
- S. Chen, L. Wang, J. Zhang, Z. Hao, H. Huang and G.-J. Deng, J. Org. Chem., 2017, 82, 11182–11191 CrossRef CAS PubMed.
- Y. Li, F. Tur, R. P. Nielsen, H. Jiang, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 1020–1024 CrossRef CAS PubMed.
- D. Liu, Y. Gao, J. Huang, Z. Fu and W. Huang, J. Org. Chem., 2018, 83, 14210–14217 CrossRef CAS PubMed.
- Y.-J. Xue, M.-Y. Li, X.-J. Jin, C.-J. Zheng and H.-R. Piao, J. Enzyme Inhib. Med. Chem., 2021, 36, 296–307 CrossRef PubMed.
- J. Wu, X. Chen, Y. Xie, Y. Guo, Q. Zhang and G.-J. Deng, J. Org. Chem., 2017, 82, 5743–5750 CrossRef CAS PubMed.
- F. Guo, L. Wang, P. Wang, J. Yu and J. Han, Asian J. Org. Chem., 2012, 1, 218–221 CrossRef CAS.
- S. Chakrabarty, I. Chatterjee, L. Tebben and A. Studer, Angew. Chem., 2013, 52, 2968–2971 CrossRef CAS PubMed.
- M. Feofanov, V. Akhmetov, R. Takayama and K. Amsharov, Org. Biomol. Chem., 2021, 19, 7172–7175 RSC.
- S. P. Swain and S. Mohanty, ChemMedChem, 2019, 14, 291–302 CrossRef CAS PubMed.
- Y. A. Ammar, M. A. El-Sharief, M. M. Ghorab, Y. A. Mohamed, A. Ragab and S. Y. Abbas, Curr. Org. Synth., 2016, 13, 466–475 CrossRef CAS PubMed.
- G. S. de Carvalho, R. M. Dias, F. R. Pavan, C. Q. Leite, V. L. Silva, C. G. Diniz, D. T. de Paula, E. S. Coimbra, P. Retailleau and A. D. da Silva, Med. Chem., 2013, 9, 351–359 CrossRef CAS PubMed.
- X. Liang, H. Fu, P. Xiao, H. Fang and X. Hou, Bioorg. Chem., 2020, 103, 104124 CrossRef CAS PubMed.
- M. C. Caterina, I. A. Perillo, L. Boiani, H. Pezaroglo, H. Cerecetto, M. González and A. Salerno, Bioorg. Med. Chem., 2008, 16, 2226–2234 CrossRef CAS PubMed.
- V. Sharma and M. Khan, Eur. J. Med. Chem., 2001, 36, 651–658 CrossRef CAS PubMed.
- A. N. Komogortsev, V. G. Melekhina, B. V. Lichitsky and M. E. Minyaev, J. Heterocycl. Chem., 2021, 58, 864–872 CrossRef CAS.
- X. Cheng, B.-G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CAS.
- A. Casnati, A. Perrone, P. P. Mazzeo, A. Bacchi, R. Mancuso, B. Gabriele, R. Maggi, G. Maestri, E. Motti and A. s. Stirling, J. Org. Chem., 2019, 84, 3477–3490 CAS.
- A. Angyal, A. Demjén, V. Harmat, J. n. Wolfling, L. G. Puskás and I. Kanizsai, J. Org. Chem., 2019, 84, 4273–4281 CAS.
- B.-L. Zhao and D.-M. Du, Org. Lett., 2018, 20, 3797–3800 CAS.
- Y.-L. Qian, P.-J. Xia, J. Li, Q.-L. Zhao, J.-A. Xiao, H.-y. Xiang and H. Yang, Org. Biomol. Chem., 2017, 15, 8705–8708 CAS.
- P. Liu, G. Xu and J. Sun, Org. Lett., 2017, 19, 1858–1861 CAS.
- G. Di Carmine, D. Ragno, C. De Risi, O. Bortolini, P. P. Giovannini, G. Fantin and A. Massi, Org. Biomol. Chem., 2017, 15, 8788–8801 CAS.
- P. V. Balaji and S. Chandrasekaran, Tetrahedron, 2016, 72, 1095–1104 Search PubMed.
- M. A. Tabarki and R. Besbes, Tetrahedron Lett., 2016, 57, 3832–3836 CAS.
- V. Satheesh, M. Sengoden and T. Punniyamurthy, J. Org. Chem., 2016, 81, 9792–9801 CAS.
- C. Izquierdo, F. Esteban, J. L. G. Ruano, A. Fraile and J. Aleman, Org. Lett., 2016, 18, 92–95 CAS.
- Y.-H. Sun, Y. Xiong, C.-Q. Peng, W. Li, J.-A. Xiao and H. Yang, Org. Biomol. Chem., 2015, 13, 7907–7910 CAS.
- M. Bai, B.-D. Cui, J. Zuo, J.-Q. Zhao, Y. You, Y.-Z. Chen, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2015, 71, 949–955 CAS.
- A. Olyaei, M. K. Karimi and R. Razeghi, Tetrahedron Lett., 2013, 54, 5730–5733 CrossRef CAS.
- H. Xie, J.-C. Liu, L. Wu and M.-W. Ding, Tetrahedron, 2012, 68, 7984–7990 CrossRef CAS.
- L. Saney, T. Panduwawala, X. Li, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Org. Biomol. Chem., 2023, 21, 4801–4809 RSC.
- S. Barranco, A. Pagnanini, F. Cuccu, P. Caboni, R. Guillot, D. J. Aitken and A. Frongia, Org. Biomol. Chem., 2024, 22, 8048–8053 RSC.
- A. Scarsi, M. Ponassi, C. Brullo, C. Rosano and A. Spallarossa, Mol. Diversity, 2023, 27, 1285–1295 CrossRef CAS PubMed.
- S. H. Bae, J. H. Park, H. G. Choi, H. Kim and S. H. Kim, Biomol. Ther., 2018, 26, 494–502 CrossRef CAS PubMed.
- J. S. de Araújo, A. García-Rubia, V. Sebastián-Pérez, T. D. Kalejaiye, P. Bernardino da Silva, C. R. Fonseca-Berzal, L. Maes, H. P. De Koning, M. N. C. Soeiro and C. Gil, Antimicrob. Agents Chemother., 2019, 63, e02156 Search PubMed.
- H. Karppanen, P. Paakkari, A. L. Orma and I. Paakkari, Agents Actions, 1979, 9, 84–86 CrossRef CAS PubMed.
- A. Bhatnagar, P. Sharma and N. Kumar, Int. J. PharmTech Res., 2011, 3, 268–282 CAS.
- A. A. Abdelhamid, H. A. Salah and A. A. Marzouk, J. Heterocycl. Chem., 2020, 57, 676–685 CrossRef CAS.
- Y. Tian, M. Qin, X. Yang, X. Zhang, Y. Liu, X. Guo and B. Chen, Tetrahedron, 2019, 75, 2817–2823 CrossRef CAS.
- M. R. Albayati, A. A. Marzouk and A. A. Abdelhamid, J. Heterocycl. Chem., 2019, 56, 1514–1519 CrossRef CAS.
- M. Beuvin, M. Manneveau, S. Diab, B. Picard, M. Sanselme, S. R. Piettre, J. Legros and I. Chataigner, Tetrahedron Lett., 2018, 59, 4487–4491 CrossRef CAS.
- S. Evjen and A. Fiksdahl, Synth. Commun., 2017, 47, 1392–1399 CrossRef CAS.
- C.-Y. Chen, W.-P. Hu, P.-C. Yan, G. C. Senadi and J.-J. Wang, Org. Lett., 2013, 15, 6116–6119 CrossRef CAS PubMed.
- G. Mlostoń, E. Obijalska and H. Heimgartner, J. Fluorine Chem., 2011, 132, 951–955 CrossRef.
- Ł. Albrecht, L. K. Ransborg, A. Albrecht, L. Lykke and K. A. Jørgensen, Chem. - Eur. J., 2011, 17, 13240–13246 CrossRef PubMed.
- L. Preti, O. A. Attanasi, E. Caselli, G. Favi, C. Ori, P. Davoli, F. Felluga and F. Prati, Eur. J. Org Chem., 2010, 2010, 4312–4320 CrossRef PubMed.
- A. R. Karimi, Z. Alimohammadi, J. Azizian, A. A. Mohammadi and M. R. Mohammadizadeh, Catal. Commun., 2006, 7, 728–732 CrossRef CAS.
- S. Kantevari, S. V. N. Vuppalapati, D. O. Biradar and L. Nagarapu, J. Mol. Catal. A:Chem., 2007, 266, 109–113 CrossRef CAS.
- C. Mukhopadhyay, P. K. Tapaswi and M. G. B. Drew, Tetrahedron Lett., 2010, 51, 3944–3950 CrossRef CAS.
- A. Mohammadi, H. Keshvari, R. Sandaroos, B. Maleki, H. Rouhi, H. Moradi, Z. Sepehr and S. Damavandi, Appl. Catal., 2012, 429, 73–78 CrossRef.
- J. J. Gabla, S. R. Mistry and K. C. Maheria, Catal. Sci. Technol., 2017, 7, 5154–5167 RSC.
- A. Khodairy, A. M. Ali and M. T. El-Wassimy, J. Heterocycl. Chem., 2017, 54, 3342–3349 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- S. V. Nalage, M. B. Kalyankar, V. S. Patil, S. V. Bhosale, S. U. Deshmukh and R. P. Pawar, Open Catal. J., 2010, 3, 58–61 CrossRef CAS.
- A. Husain, S. Drabu and N. Kumar, Acta Pol. Pharm., 2009, 66, 243–248 CAS.
- M. Ratanlal, D. Mohankrishnan, V. Jayaram, D. Sahal and G. V. M. Sharma, ChemistrySelect, 2024, 9, e202302636 CrossRef CAS.
- A. A. Bhat, I. Singh, N. Tandon and R. Tandon, Eur. J. Med. Chem., 2023, 246, 114954 CrossRef CAS PubMed.
- B. Marshall and J. R. Warren, Lancet, 1984, 323, 1311–1315 CrossRef PubMed.
- A. Nomura, G. N. Stemmermann, P.-H. Chyou, G. I. Perez-Perez and M. J. Blaser, Ann. Intern. Med., 1994, 120, 977–981 CrossRef CAS PubMed.
- J. Labenz and G. Börsch, Gut, 1994, 35, 19–22 CrossRef CAS PubMed.
- J. Parsonnet, G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman, N. Orentreich and R. K. Sibley, N. Engl. J. Med., 1991, 325, 1127–1131 CrossRef CAS PubMed.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CAS.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CAS.
- D. Wang, H. P. Deng, Y. Wei, Q. Xu and M. Shi, Eur. J. Org Chem., 2013, 2013, 401–406 CAS.
- Z.-C. Geng, J. Chen, N. Li, X.-F. Huang, Y. Zhang, Y.-W. Zhang and X.-W. Wang, Beilstein J. Org. Chem., 2012, 8, 1710–1720 CAS.
- M. Fernandez, E. Reyes, J. L. Vicario, D. Badia and L. Carrillo, Adv. Synth. Catal., 2012, 354, 371–376 CAS.
- L. Deiana, G. L. Zhao, H. Leijonmarck, J. Sun, C. W. Lehmann and A. Córdova, ChemistryOpen, 2012, 1, 134–139 CAS.
- Y. Biliz, B. Hasdemir, H. B. Küçük, S. Yıldırım, F. Kocabaş and R. A. Kartop, J. Mol. Struct., 2024, 1295, 136813 CAS.
- H. T. Nguyen, T. V. Le and P. H. Tran, J. Environ. Chem. Eng., 2021, 9, 105228 CAS.
- N. Nayak, J. Ramprasad and U. Dalimba, Bioorg. Med. Chem. Lett., 2015, 25, 5540–5545 CrossRef CAS PubMed.
- N. C. Desai and D. V. Vaja, Int. Lett. Chem., Phys. Astron., 2018, 77, 35–52 Search PubMed.
- N. D. Amnerkar and K. P. Bhusari, Eur. J. Med. Chem., 2010, 45, 149–159 CrossRef CAS PubMed.
- A. Balbi, M. Anzaldi, C. Macciò, C. Aiello, M. Mazzei, R. Gangemi, P. Castagnola, M. Miele, C. Rosano and M. Viale, Eur. J. Med. Chem., 2011, 46, 5293–5309 CrossRef CAS PubMed.
- O. I. El-Sabbagh, M. M. Baraka, S. M. Ibrahim, C. Pannecouque, G. Andrei, R. Snoeck, J. Balzarini and A. A. Rashad, Eur. J. Med. Chem., 2009, 44, 3746–3753 CrossRef CAS PubMed.
- K. D. Dwivedi, B. Borah and L. R. Chowhan, Front. Chem., 2020, 7, 944 CrossRef PubMed.
- H.-F. Zhang, Z.-Q. Ye and G. Zhao, Chin. Chem. Lett., 2014, 25, 535–540 CrossRef CAS.
- R. Maity and S. C. Pan, Org. Biomol. Chem., 2017, 15, 8032–8036 RSC.
- C. Y. Wu and K. Chen, ChemistrySelect, 2018, 3, 3500–3504 CrossRef.
- A. Dandia, S. Bansal, R. Sharma and V. Parewa, ChemistrySelect, 2018, 3, 9785–9789 CrossRef CAS.
- V. Ramesh, S. Shanmugam and N. S. Devi, New J. Chem., 2016, 40, 9993–10001 RSC.
- V. L. Gein, T. M. Zamaraeva and P. A. Slepukhin, Tetrahedron Lett., 2017, 58, 134–136 CrossRef CAS.
- A. Alizadeh and F. Bayat, Helv. Chim. Acta, 2014, 97, 694–700 CrossRef CAS.
- K. Jayabal and T. P. Paramasivan, Tetrahedron Lett., 2014, 55, 2010–2014 CrossRef CAS.
- C. Wang, Y.-H. Jiang and C.-G. Yan, Chin. Chem. Lett., 2015, 26, 889–893 CrossRef CAS.
- B. P. Tripathi, A. Mishra, P. Rai, Y. K. Pandey, M. Srivastava, S. Yadav, J. Singh and J. Singh, New J. Chem., 2017, 41, 11148–11154 RSC.
- S. Pandeya, D. Sriram, G. Nath and E. DeClercq, Eur. J. Pharm. Sci., 1999, 9, 25–31 CrossRef CAS PubMed.
- V. Gududuru, E. Hurh, J. T. Dalton and D. D. Miller, Bioorg. Med. Chem. Lett., 2004, 14, 5289–5293 CrossRef CAS PubMed.
- S. Bhattacharya, B. Roy, S. Mana and S. Kumar, Pharma Res., 2009, 2, 79–83 Search PubMed.
- J. Balzarini, B. Orzeszko, J. K. Maurin and A. Orzeszko, Eur. J. Med. Chem., 2007, 42, 993–1003 CrossRef CAS PubMed.
- M.-H. Shih and F.-Y. Ke, Bioorg. Med. Chem., 2004, 12, 4633–4643 CrossRef CAS PubMed.
- A. Daghlavi, E. Kowsari, M. Abdouss, M. H. Ghasemi and E. Asadi, Res. Chem. Intermed., 2020, 46, 3593–3605 CrossRef CAS.
- G. Rainoldi, F. Begnini, M. De Munnik, L. Lo Presti, C. M. Vande Velde, R. Orru, G. Lesma, E. Ruijter and A. Silvani, ACS Comb. Sci., 2018, 20, 98–105 CrossRef CAS PubMed.
- B. Kaboudin and J. Abbasi Shiran, J. Sulfur Chem., 2018, 39, 633–645 CAS.
- H. Medini, N. H. Mekni and K. Boujlel, J. Sulfur Chem., 2015, 36, 653–659 CrossRef CAS.
- A. M. Malla, M. Parveen, F. Ahmad, S. Azaz and M. Alam, RSC Adv., 2015, 5, 19552–19569 CAS.
- A. Ziyaei-Halimehjani, K. Marjani and A. Ashouri, Tetrahedron Lett., 2012, 53, 3490–3492 CAS.
- B. P. Kumar, M. Nanjan, B. Suresh, M. Karvekar and L. Adhikary, J. Heterocycl. Chem., 2006, 43, 897–903 CAS.
- K. Yamada, T. Tsubogo, H. Kanazawa, S. Ishizuka, K. Ohyama, M. Kaida and T. Abe, Eur. J. Org Chem., 2024, 27, e202301130 CAS.
- M. Zhang, Y. Chen, J. Liu, X. Yan, K. Huang and X. Ma, Asian J. Org. Chem., 2025, 14, e202400558 CAS.
- M. Chen, S. Lu, G. Yuan, S. Yang and X. Du, Heterocycl. Commun., 2000, 6, 421–426 CAS.
- E. A. Sheremet, R. I. Tomanov, E. V. Trukhin and V. M. Berestovitskaya, Russ. J. Org. Chem., 2004, 40, 594–595 CAS.
- H. N. Hafez, H.-A. S. Abbas and A.-R. B. A. El-Gazzar, Acta Pharm., 2008, 58, 359–378 CAS.
- K. M. Banu, A. Dinakar and C. Ananthanarayanan, Indian J. Pharm. Sci., 1999, 61, 202 CAS.
- L. P. Guan, Q. H. Jin, G. R. Tian, K. Y. Chai and Z. S. Quan, J. Pharm. Pharm. Sci., 2007, 10, 254–262 CAS.
- A. Passannanti, P. Diana, P. Barraja, F. Mingoia, A. Lauria and G. Cirrincione, Heterocycles, 1998, 6, 1229–1235 Search PubMed.
- R. Gujjar, A. Marwaha, F. El Mazouni, J. White, K. L. White, S. Creason, D. M. Shackleford, J. Baldwin, W. N. Charman and F. S. Buckner, J. Med. Chem., 2009, 52, 1864–1872 CrossRef CAS PubMed.
- B. A. Johns, J. G. Weatherhead, S. H. Allen, J. B. Thompson, E. P. Garvey, S. A. Foster, J. L. Jeffrey and W. H. Miller, Bioorg. Med. Chem. Lett., 2009, 19, 1802–1806 CrossRef CAS PubMed.
- S. Manfredini, C. B. Vicentini, M. Manfrini, N. Bianchi, C. Rutigliano, C. Mischiati and R. Gambari, Bioorg. Med. Chem., 2000, 8, 2343–2346 CrossRef CAS PubMed.
- A. Duran, H. Dogan and S. Rollas, Il Farmaco, 2002, 57, 559–564 CrossRef CAS PubMed.
- Z. Xu, S. J. Zhao and Y. Liu, Eur. J. Med. Chem., 2019, 183, 111700 CrossRef CAS PubMed.
- L. S. Venigalla, S. Maddila and S. B. Jonnalagadda, J. Iran. Chem. Soc., 2020, 17, 1539–1544 CrossRef CAS.
- N. A. Aksenov, A. V. Aksenov, N. K. Kirilov, N. A. Arutiunov, D. A. Aksenov, V. Maslivetc, Z. Zhao, L. Du, M. Rubin and A. Kornienko, Org. Biomol. Chem., 2020, 18, 6651–6664 RSC.
- N. Guo, X. Liu, H. Xu, X. Zhou and H. Zhao, Org. Biomol. Chem., 2019, 17, 6148–6152 RSC.
- N. Okamoto, T. Sueda, H. Minami and R. Yanada, Tetrahedron Lett., 2018, 59, 1461–1464 CrossRef CAS.
- A. V. Bogolyubsky, O. Savych, A. V. Zhemera, S. E. Pipko, A. V. Grishchenko, A. I. Konovets, R. O. Doroshchuk, D. N. Khomenko, V. S. Brovarets and Y. S. Moroz, ACS Comb. Sci., 2018, 20, 461–466 CrossRef CAS PubMed.
- V. O. Filimonov, L. N. Dianova, K. A. Galata, T. V. Beryozkina, M. S. Novikov, V. S. Berseneva, O. S. Eltsov, A. T. Lebedev, P. A. Slepukhin and V. A. Bakulev, J. Org. Chem., 2017, 82, 4056–4071 CrossRef CAS PubMed.
- W. Li, Q. Jia, Z. Du and J. Wang, Chem. Commun., 2013, 49, 10187–10189 RSC.
- M. Taha, N. H. Ismail, S. Imran, M. Q. B. Rokei, S. M. Saad and K. M. Khan, Bioorg. Med. Chem., 2015, 23, 4155–4162 CrossRef CAS PubMed.
- T. Glomb, K. Szymankiewicz and P. Świątek, Molecules, 2018, 23, 3361 CrossRef PubMed.
- G. Chawla, B. Naaz and A. A. Siddiqui, Mini-Rev. Med. Chem., 2018, 18, 216–233 CAS.
- D. R. Faria, R. C. Melo, G. S. Arita, K. M. Sakita, F. A. V. Rodrigues-Vendramini, I. R. G. Capoci, T. C. A. Becker, P. S. Bonfim-Mendonça, M. S. S. Felipe, T. I. E. Svidzinski and E. S. Kioshima, Pathogens, 2021, 10, 314 CrossRef CAS PubMed.
- Y.-E. Wang, D. Yang, L. Dai, J. Huo, L. Chen, Z. Kang, J. Mao and J. Zhang, J. Agric. Food Chem., 2022, 70, 2510–2519 CrossRef CAS PubMed.
- W. A. El-Sayed, F. A. El-Essawy, O. M. Ali, B. S. Nasr, M. M. Abdalla, A. A. Abdel-Rahman and Z. Naturforsch C, J. Biosci., 2009, 64, 773–778 CAS.
- T. Glomb and P. Świątek, Int. J. Mol. Sci., 2021, 22, 6979 CrossRef CAS PubMed.
- M. Luczynski and A. Kudelko, Appl. Sci., 2022, 12, 3756 CrossRef CAS.
- F. Lu, F. Gong, L. Li, K. Zhang, Z. Li, X. Zhang, Y. Yin, Y. Wang, Z. Gao and H. Zhang, Eur. J. Org Chem., 2020, 2020, 3257–3260 CrossRef CAS.
- V. N. Telvekar and B. S. Takale, Synth. Commun., 2013, 43, 221–227 CrossRef CAS.
- K. N. Patel, N. C. Jadhav, P. B. Jagadhane and V. N. Telvekar, Synlett, 2012, 23, 1970–1972 CrossRef CAS.
- L. Sharma, S. Kumar, S. Singh and R. Singh, Russ. J. Electrochem., 2010, 46, 34–40 CrossRef CAS.
- V. V. Sureshbabu, H. P. Hemantha and S. A. Naik, Tetrahedron Lett., 2008, 49, 5133–5136 CrossRef CAS.
- M. D. Evans, J. Ring, A. Schoen, A. Bell, P. Edwards, D. Berthelot, R. Nicewonger and C. M. Baldino, Tetrahedron Lett., 2003, 44, 9337–9341 CrossRef CAS.
- S. Jazayeri, M. H. Moshafi, L. Firoozpour, S. Emami, S. Rajabalian, M. Haddad, F. Pahlavanzadeh, M. Esnaashari, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2009, 44, 1205–1209 CrossRef CAS PubMed.
- S. N. Swamy, B. Priya, B. Prabhuswamy, B. Doreswamy, J. S. Prasad and K. S. Rangappa, Eur. J. Med. Chem., 2006, 41, 531–538 CrossRef CAS PubMed.
- U. Salgın-Gökşen, N. Gökhan-Kelekçi, Ö. Göktaş, Y. Köysal, E. Kılıç, Ş. Işık, G. Aktay and M. Özalp, Bioorg. Med. Chem., 2007, 15, 5738–5751 CrossRef PubMed.
- A. T. Mavrova, D. Wesselinova, Y. A. Tsenov and P. Denkova, Eur. J. Med. Chem., 2009, 44, 63–69 CrossRef CAS PubMed.
- K. B. Zheng, J. He and J. Zhang, Chin. Chem. Lett., 2008, 19, 1281–1284 CrossRef CAS.
- A. Gupta, P. Mishra, S. Pandeya, S. K. Kashaw, V. Kashaw and J. P. Stables, Eur. J. Med. Chem., 2009, 44, 1100–1105 CAS.
- N. Ö. Can, Ö. D. Can, D. Osmaniye and Ü. Demir Özkay, Molecules, 2018, 23, 716 CrossRef PubMed.
- I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. Ul-Haq, A. Khan, S. Ali and M. I. Choudhary, Eur. J. Med. Chem., 2010, 45, 5200–5207 CAS.
- C. Kuş, G. Ayhan-Kılcıgil, S. Özbey, F. B. Kaynak, M. Kaya, T. Çoban and B. Can-Eke, Bioorg. Med. Chem., 2008, 16, 4294–4303 Search PubMed.
- M. Behrouzi-Fardmoghadam, F. Poorrajab, S. K. Ardestani, S. Emami, A. Shafiee and A. Foroumadi, Bioorg. Med. Chem., 2008, 16, 4509–4515 CAS.
- A. Foroumadi, S. Emami, S. Pournourmohammadi, A. Kharazmi and A. Shafiee, Eur. J. Med. Chem., 2005, 40, 1346–1350 CAS.
- Y. Han, Y. Sun, A. Abdukader, B. Liu and D. Wang, Catal. Lett., 2018, 148, 3486–3491 CAS.
- A. Feddouli, M. Y. A. Itto, A. Hasnaoui, D. Villemin, P. A. Jaffrès, J. S. D. O. Santos, A. Riahi, F. Huet and J. C. Daran, J. Heterocycl. Chem., 2004, 41, 731–735 CAS.
- S. Barriga, P. Fuertes, C. F. Marcos, D. Miguel, O. A. Rakitin, C. W. Rees and T. Torroba, J. Org. Chem., 2001, 66, 5766–5771 CAS.
- Y. Song, D. T. Connor, A. D. Sercel, R. J. Sorenson, R. Doubleday, P. C. Unangst, B. D. Roth, V. G. Beylin, R. B. Gilbertsen and K. Chan, J. Med. Chem., 1999, 42, 1161–1169 CAS.
- C. Gao, L. Chang, Z. Xu, X.-F. Yan, C. Ding, F. Zhao, X. Wu and L.-S. Feng, Eur. J. Med. Chem., 2019, 163, 404–412 CAS.
- D. C. Crosby, X. Lei, C. G. Gibbs, B. R. McDougall, W. E. Robinson Jr and M. G. Reinecke, J. Med. Chem., 2010, 53, 8161–8175 CAS.
- P. Zhan, H. Liu, X. Liu, Y. Wang, C. Pannecouque, M. Witvrouw and E. De Clercq, Med. Chem. Res., 2010, 19, 652–663 CAS.
- T. V. Serebryanskaya, T. Yung, A. A. Bogdanov, A. Shchebet, S. A. Johnsen, A. S. Lyakhov, L. S. Ivashkevich, Z. A. Ibrahimava, T. S. Garbuzenco and T. S. Kolesnikova, J. Inorg. Biochem., 2013, 120, 44–53 CAS.
- M. Arshad, A. R. Bhat, S. Pokharel, J.-E. Kim, E. J. Lee, F. Athar and I. Choi, Eur. J. Med. Chem., 2014, 71, 229–236 CrossRef CAS PubMed.
- M. Y. Wani, A. R. Bhat, A. Azam, D. H. Lee, I. Choi and F. Athar, Eur. J. Med. Chem., 2012, 54, 845–854 CrossRef CAS PubMed.
- A. B. Salake, A. S. Chothe, S. S. Nilewar, M. Khilare, R. S. Meshram, A. A. Pandey and M. Kathiravan, J. Chem. Biol., 2014, 7, 29–35 CrossRef PubMed.
- J. Figueiredo, M. Ismael, J. Pinheiro, A. Silva, J. Justino, F. Silva, M. Goulart, D. Mira, M. Araújo and R. Campoy, Carbohydr. Res., 2012, 347, 47–54 CrossRef CAS PubMed.
- K. Pegklidou, C. Koukoulitsa, I. Nicolaou and V. J. Demopoulos, Bioorg. Med. Chem., 2010, 18, 2107–2114 CrossRef CAS PubMed.
- D. P. Zarezin, A. M. Kabylda, V. I. Vinogradova, P. V. Dorovatovskii, V. N. Khrustalev and V. G. Nenajdenko, Tetrahedron, 2018, 74, 4315–4322 CrossRef CAS.
- Z. N. Tisseh, M. Dabiri, M. Nobahar, H. R. Khavasi and A. Bazgir, Tetrahedron, 2012, 68, 1769–1773 CrossRef CAS.
- R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234–12235 CrossRef CAS PubMed.
- M. De Angelis, C. Sappino, E. Mandic, M. D'Alessio, M. G. De Dominicis, S. Sannino, L. Primitivo, P. Mencarelli, A. Ricelli and G. Righi, Tetrahedron, 2021, 79, 131837 CrossRef CAS.
- S. R. Chowdhury, J. Gu, Y. Hu, J. Wang, S. Lei, M. S. Tavallaie, C. Lam, D. Lu, F. Jiang and L. Fu, Eur. J. Med. Chem., 2021, 222, 113541 CrossRef CAS PubMed.
- H. R. Shaterian and K. Azizi, J. Mol. Liq., 2013, 180, 187–191 CrossRef CAS.
- Q. Zhou, L. Zhang, W. Meng, X. Feng, J. Yang and H. Du, Org. Lett., 2016, 18, 5189–5191 CrossRef CAS PubMed.
- H. Zhang and K. Muñiz, ACS Catal., 2017, 7, 4122–4125 CrossRef CAS.
- A. Patil and R. Salunkhe, Res. Chem. Intermed., 2018, 44, 3337–3348 CrossRef CAS.
- R. Ebule, S. Mudshinge, M. H. Nantz, M. S. Mashuta, G. B. Hammond and B. Xu, J. Org. Chem., 2019, 84, 3249–3259 CrossRef CAS PubMed.
- P. N. Nguyen, L. H. T. Nguyen, T. L. H. Doan, P. H. Tran and H. T. Nguyen, RSC Adv., 2024, 14, 7006–7021 RSC.
- V. Chavan, S. Sonawane, M. Shingare and B. Karale, Chem. Heterocycl. Compd., 2006, 42, 625–630 CrossRef CAS.
- E. Abele, R. Abele and E. Lukevics, Chem. Heterocycl. Compd., 2003, 39, 825–865 CrossRef CAS.
- A. A. Fadda, M. A. Berghot, F. A. Amer, D. S. Badawy and N. M. Bayoumy, Arch. Pharm., 2012, 345, 378–385 CrossRef CAS PubMed.
- M. El-Naggar, H. Almahli, H. S. Ibrahim, W. M. Eldehna and H. A. Abdel-Aziz, Molecules, 2018, 23 Search PubMed.
- P. Baumgarten and A. Dornow, Ber. Dtsch. Chem. Ges., 1939, 72, 563–566 CrossRef.
- J. M. Robinson, L. W. Brent, C. Chau, K. A. Floyd, S. L. Gillham, T. L. McMahan, D. J. Magda, T. J. Motycka and M. J. Pack, J. Org. Chem., 1992, 57, 7352–7355 CrossRef CAS.
- M. C. Bagley, J. W. Dale and J. Bower, Synlett, 2001, 2001, 1149–1151 CrossRef.
- M. C. Bagley, R. Lunn and X. Xiong, Tetrahedron Lett., 2002, 43, 8331–8334 CAS.
- T. R. Kelly and R. L. Lebedev, J. Org. Chem., 2002, 67, 2197–2205 CAS.
- X. Xiong, M. C. Bagley and K. Chapaneri, Tetrahedron Lett., 2004, 45, 6121–6124 CAS.
- X. Fang, Y.-C. Liu and C. Li, J. Org. Chem., 2007, 72, 8608–8610 CAS.
- S. Tu, R. Jia, B. Jiang, J. Zhang, Y. Zhang, C. Yao and S. Ji, Tetrahedron, 2007, 63, 381–388 CAS.
- K. Yoshida, F. Kawagoe, K. Hayashi, S. Horiuchi, T. Imamoto and A. Yanagisawa, Org. Lett., 2009, 11, 515–518 CAS.
- R. H. Boecker and F. P. Guengerich, J. Med. Chem., 1986, 29, 1596–1603 CAS.
- J. S. Yadav, B. V. S. Reddy, G. Sabitha and G. S. K. K. Reddy, Synthesis, 2000, 2000, 1532–1534 Search PubMed.
- N. Nakamichi, Y. Kawashita and M. Hayashi, Synthesis, 2004, 2004, 1015–1020 Search PubMed.
- J. Yadav, B. Reddy, A. Basak, G. Baishya and A. V. Narsaiah, Synthesis, 2006, 2006, 451–454 Search PubMed.
- X. Wei, L. Wang, W. Jia, S. Du, L. Wu and Q. Liu, Chin. J. Chem., 2014, 32, 1245–1250 CAS.
- M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CAS.
- B. C. Ranu, R. Jana and S. Sowmiah, J. Org. Chem., 2007, 72, 3152–3154 CAS.
- J. Yin, B. Xiang, M. A. Huffman, C. E. Raab and I. W. Davies, J. Org. Chem., 2007, 72, 4554–4557 CAS.
- Y. Jiang, C.-M. Park and T.-P. Loh, Org. Lett., 2014, 16, 3432–3435 CrossRef CAS PubMed.
- H. Wei, Y. Li, K. Xiao, B. Cheng, H. Wang, L. Hu and H. Zhai, Org. Lett., 2015, 17, 5974–5977 CrossRef CAS PubMed.
- H. Huang, J. Cai, L. Tang, Z. Wang, F. Li and G.-J. Deng, J. Org. Chem., 2016, 81, 1499–1505 CrossRef CAS PubMed.
- J. Shen, D. Cai, C. Kuai, Y. Liu, M. e. Wei, G. Cheng and X. Cui, J. Org. Chem., 2015, 80, 6584–6589 CAS.
- D. Majee, S. Biswas, S. M. Mobin and S. Samanta, J. Org. Chem., 2016, 81, 4378–4385 CrossRef CAS PubMed.
- S. Biswas, D. Majee, S. Guin and S. Samanta, J. Org. Chem., 2017, 82, 10928–10938 CrossRef CAS PubMed.
- Y. Gao, R. Chen and Y. Ma, Synthesis, 2019, 51, 3875–3882 CrossRef CAS.
- G. Asskar, M. Rivard and T. Martens, J. Org. Chem., 2019, 85, 1232–1239 CrossRef PubMed.
- I. Kim, S. Park and S. Hong, Org. Lett., 2020, 22, 8730–8734 CrossRef CAS PubMed.
- J. Duan, L. Zhang, G. Xu, H. Chen, X. Ding, Y. Mao, B. Rong, N. Zhu and K. Guo, J. Org. Chem., 2020, 85, 8157–8165 CrossRef CAS PubMed.
- M. Hassani, W. Cai, K. H. Koelsch, D. C. Holley, A. S. Rose, F. Olang, J. P. Lineswala, W. G. Holloway, J. M. Gerdes and M. Behforouz, J. Med. Chem., 2008, 51, 3104–3115 CrossRef CAS PubMed.
- J. Iqbal, S. A. Ejaz, I. Khan, E. Ausekle, M. Miliutina and P. Langer, Daru, 2019, 27, 613–626 CrossRef CAS PubMed.
- S. S. Zhang, Q. W. Tan and L. P. Guan, Mini-Rev. Med. Chem., 2021, 21, 2261–2275 CrossRef CAS PubMed.
- Y.-T. Duan, Y.-F. Yao, D.-J. Tang, N. j. Thumar, S. B. Teraiya, J. A. Makawana, Y.-L. Sang, Z.-C. Wang, X.-X. Tao, A.-Q. Jiang and H.-L. Zhu, RSC Adv., 2014, 4, 20382–20392 RSC.
- S. Ali, H.-T. Zhu, X.-F. Xia, K.-G. Ji, Y.-F. Yang, X.-R. Song and Y.-M. Liang, Org. Lett., 2011, 13, 2598–2601 CrossRef CAS PubMed.
- G. Shan, X. Sun, Q. Xia and Y. Rao, Org. Lett., 2011, 13, 5770–5773 CAS.
- F.-P. Ma, G.-T. Cheng, Z.-G. He and Z.-H. Zhang, Aust. J. Chem., 2012, 65, 409–416 CrossRef CAS.
- S. Khong and O. Kwon, J. Org. Chem., 2012, 77, 8257–8267 CAS.
- H. Kefayati, F. Narchin and K. Rad-Moghadam, Tetrahedron Lett., 2012, 53, 4573–4575 CAS.
- Q. Gao, S. Liu, X. Wu and A. Wu, Org. Lett., 2014, 16, 4582–4585 CAS.
- Y.-N. Huang, Y.-L. Li, J. Li and J. Deng, J. Org. Chem., 2016, 81, 4645–4653 CrossRef CAS PubMed.
- X. Wu, X. Geng, P. Zhao, J. Zhang, X. Gong, Y.-d. Wu and A.-x. Wu, Org. Lett., 2017, 19, 1550–1553 CAS.
- H. Wang, Q. Xu, S. Shen and S. Yu, J. Org. Chem., 2017, 82, 770–775 CrossRef CAS PubMed.
- J.-C. Xiang, Z.-X. Wang, Y. Cheng, S.-Q. Xia, M. Wang, B.-C. Tang, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2017, 82, 9210–9216 CrossRef CAS PubMed.
- T.-S. Jiang, X. Wang and X. Zhang, Tetrahedron Lett., 2018, 59, 2979–2982 CrossRef CAS.
- S. Y. Lee and C.-H. Cheon, J. Org. Chem., 2018, 83, 13036–13044 CAS.
- P. Zhou, B. Hu, K. Rao, L. Li, J. Yang, C. Gao, F. Wang and F. Yu, Synlett, 2018, 29, 519–524 CAS.
- J. Nan, Y. Hu, P. Chen and Y. Ma, Org. Lett., 2019, 21, 1984–1988 Search PubMed.
- J. Nan, P. Chen, Y. Zhang, Y. Yin, B. Wang and Y. Ma, J. Org. Chem., 2020, 85, 14042–14054 CrossRef CAS PubMed.
- D. Cheng, X. Yan, J. Shen, Y. Pu, X. Xu and J. Yan, Synthesis, 2020, 52, 1833–1840 CAS.
- W. A. Carroll, K. A. Agrios, R. J. Altenbach, S. A. Buckner, Y. Chen, M. J. Coghlan, A. V. Daza, I. Drizin, M. Gopalakrishnan and R. F. Henry, J. Med. Chem., 2004, 47, 3180–3192 CAS.
- M. De Luca, G. Ioele and G. Ragno, Pharmaceutics, 2019, 11, 85 CAS.
- D. J. Triggle, Cell. Mol. Neurobiol., 2003, 23, 293–303 CAS.
- M. M. Heravi, M. Saeedi, N. Karimi, M. Zakeri, Y. S. Beheshtiha and A. Davoodnia, Synth. Commun., 2010, 40, 523–529 CrossRef CAS.
- J.-Y. He, H.-Z. Jia, Q.-G. Yao, S.-J. Liu, H.-K. Yue, H.-W. Yu and R.-S. Hu, Ultrason. Sonochem., 2015, 22, 144–148 Search PubMed.
- S. Jadhvar, H. Kasraliker, S. Goswami, A. Chakrawar and S. Bhusare, Res. Chem. Intermed., 2017, 43, 7211–7221 CrossRef CAS.
- M. R. Bhosle, D. Nipte, J. Gaikwad, M. A. Shaikh, G. M. Bondle and J. N. Sangshetti, Res. Chem. Intermed., 2018, 44, 7047–7064 CrossRef CAS.
- Z. Quan, X. Sun and L. Guan, Chem. Biol. Drug Des., 2009, 73, 313–319 CrossRef PubMed.
- M. C. Cardia, S. Distinto, E. Maccioni, A. Plumitallo, L. Sanna, M. L. Sanna and S. Vigo, J. Heterocycl. Chem., 2009, 46, 674–679 CrossRef CAS.
- M. I. Hegab, N. A. A. Taleb, S. M. Hasabelnaby and A. Goudah, Eur. J. Med. Chem., 2010, 45, 1267–1277 CrossRef PubMed.
- F. M. Awadallah, W. I. El-Eraky and D. O. Saleh, Eur. J. Med. Chem., 2012, 52, 14–21 CrossRef CAS PubMed.
- S. Gore, S. Baskaran and B. Koenig, Green Chem., 2011, 13, 1009–1013 CAS.
- F. Mohamadpour, M. T. Maghsoodlou, R. Heydari and M. Lashkari, J. Iran. Chem. Soc., 2016, 13, 1549–1560 CAS.
- H. T. Nguyen and P. H. Tran, RSC Adv., 2016, 6, 98365–98368 CAS.
- M. Szumilak and A. Stanczak, Molecules, 2019, 24, 2271 CAS.
- R. M. Butnariu and I. I. Mangalagiu, Bioorg. Med. Chem., 2009, 17, 2823–2829 CAS.
- E. M. Flefel, W. A. Tantawy, W. I. El-Sofany, M. El-Shahat, A. A. El-Sayed and D. N. Abd-Elshafy, Molecules, 2017, 22, 148 Search PubMed.
- D. Mantu, D. Maftei, D. Iurea, C. Ursu and V. Bejan, Med. Chem. Res., 2014, 23, 2909–2915 CAS.
- R. Sivakumar, N. Anbalagan, V. Gunasekaran and J. T. Leonard, Biol. Pharm. Bull., 2003, 26, 1407–1411 CAS.
- J. Khalafy, M. Rimaz, S. Farajzadeh and M. Ezzati, S. Afr. J. Chem., 2013, 66, 179–182 CAS.
- N. G. Khaligh, T. Mihankhah, M. R. Johan and J. J. Ching, Monatsh. Chem., 2018, 149, 1083–1087 CAS.
- J. Khalafy, M. Rimaz, M. Ezzati and R. H. Prager, Bull. Korean Chem. Soc., 2012, 33, 2890–2896 CAS.
- G.-H. Ma, X.-J. Tu, Y. Ning, B. Jiang and S.-J. Tu, ACS Comb. Sci., 2014, 16, 281–286 CAS.
- T. A. D. Thi, L. Decuyper, H. T. Phuong, D. V. Ngoc, H. T. Nguyen, T. T. Nguyen, T. Do Huy, H. H. Nguyen, M. D’hooghe and T. Van Nguyen, Tetrahedron Lett., 2015, 56, 5855–5858 Search PubMed.
- G. C. Senadi, B. S. Gore, W.-P. Hu and J.-J. Wang, Org. Lett., 2016, 18, 2890–2893 CrossRef CAS PubMed.
- H. M. Al-Matar, K. M. Dawood and W. M. Tohamy, RSC Adv., 2018, 8, 34459–34467 RSC.
- J. Sangshetti, S. K. Pathan, R. Patil, S. Akber Ansari, S. Chhajed, R. Arote and D. B. Shinde, Bioorg. Med. Chem., 2019, 27, 3979–3997 CrossRef CAS PubMed.
- B. L. Mylari, E. R. Larson, T. A. Beyer, W. J. Zembrowski, C. E. Aldinger, M. F. Dee, T. W. Siegel and D. H. Singleton, J. Med. Chem., 1991, 34, 108–122 CrossRef CAS PubMed.
- K. M. Amin, F. F. Barsoum, F. M. Awadallah and N. E. Mohamed, Eur. J. Med. Chem., 2016, 123, 191–201 Search PubMed.
- A. Sugimoto, H. Tanaka, Y. Eguchi, S. Ito, Y. Takashima and M. Ishikawa, J. Med. Chem., 1984, 27, 1300–1305 CrossRef CAS PubMed.
- A. Khalil, M. Berghot and M. Gouda, Eur. J. Med. Chem., 2009, 44, 4448–4454 Search PubMed.
- M. Van der Mey, H. Boss, D. Couwenberg, A. Hatzelmann, G. J. Sterk, K. Goubitz, H. Schenk and H. Timmerman, J. Med. Chem., 2002, 45, 2526–2533 Search PubMed.
- M. Shekouhy and A. Hasaninejad, Ultrason. Sonochem., 2012, 19, 307–313 Search PubMed.
- A. Hasaninejed, M. R. Kazerooni and A. Zare, Catal. Today, 2012, 196, 148–155 Search PubMed.
- R. Ghahremanzadeh, G. I. Shakibaei and A. Bazgir, Synlett, 2008, 2008, 1129–1132 Search PubMed.
- M. Sayyafi, M. Seyyedhamzeh, H. R. Khavasi and A. Bazgir, Tetrahedron, 2008, 64, 2375–2378 Search PubMed.
- M. R. Nabid, S. J. T. Rezaei, R. Ghahremanzadeh and A. Bazgir, Ultrason. Sonochem., 2010, 17, 159–161 CrossRef CAS PubMed.
- R. Ghorbani-Vaghei, R. Karimi-Nami, Z. Toghraei-Semiromi, M. Amiri and M. Ghavidel, Tetrahedron, 2011, 67, 1930–1937 CrossRef CAS.
- A. Bhat, R. Dongra and R. Selokar, Int. J. Pharma Bio Sci., 2014, 5, 422–430 CAS.
- E. M. Grivsky, S. Lee, C. W. Sigel, D. S. Duch and C. A. Nichol, J. Med. Chem., 1980, 23, 327–329 Search PubMed.
- A. M. Thompson, A. J. Bridges, D. W. Fry, A. J. Kraker and W. A. Denny, J. Med. Chem., 1995, 38, 3780–3788 CrossRef CAS PubMed.
- A. E. Wakeling, S. P. Guy, J. R. Woodburn, S. E. Ashton, B. J. Curry, A. J. Barker and K. H. Gibson, Cancer Res., 2002, 62, 5749–5754 CAS.
- A. Baba, N. Kawamura, H. Makino, Y. Ohta, S. Taketomi and T. Sohda, J. Med. Chem., 1996, 39, 5176–5182 CrossRef CAS PubMed.
- R. Rohini, P. M. Reddy, K. Shanker, A. Hu and V. Ravinder, Eur. J. Med. Chem., 2010, 45, 1200–1205 CrossRef CAS PubMed.
- A. K. Gupta, K. Kumari, N. Singh, D. S. Raghuvanshi and K. N. Singh, Tetrahedron Lett., 2012, 53, 650–653 CrossRef CAS.
- S. Ferrini, F. Ponticelli and M. Taddei, Org. Lett., 2007, 9, 69–72 CrossRef CAS PubMed.
- M. Dabiri, P. Salehi and M. Bahramnejad, Synth. Commun., 2010, 40, 3214–3225 CAS.
- J. Zhang, D. Zhu, C. Yu, C. Wan and Z. Wang, Org. Lett., 2010, 12, 2841–2843 Search PubMed.
- Z.-H. Zhang, X.-N. Zhang, L.-P. Mo, Y.-X. Li and F.-P. Ma, Green Chem., 2012, 14, 1502–1506 Search PubMed.
- Y. Yan, Y. Xu, B. Niu, H. Xie and Y. Liu, J. Org. Chem., 2015, 80, 5581–5587 Search PubMed.
- S. Hati and S. Sen, Synthesis, 2016, 48, 1389–1398 Search PubMed.
- D. S. Deshmukh and B. M. Bhanage, Synlett, 2018, 29, 979–985 Search PubMed.
- J. Chen, W. Su, H. Wu, M. Liu and C. Jin, Green Chem., 2007, 9, 972–975 CAS.
- A. Shaabani, A. Maleki and H. Mofakham, Synth. Commun., 2008, 38, 3751–3759 Search PubMed.
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Catal. Commun., 2012, 27, 179–183 CAS.
- M. Sharma, S. Pandey, K. Chauhan, D. Sharma, B. Kumar and P. M. Chauhan, J. Org. Chem., 2012, 77, 929–937 CAS.
- O. Algul, A. Meric, S. Polat, N. Didem Yuksek and M. S. Serin, Cent. Eur. J. Chem., 2009, 7, 337–342 Search PubMed.
- T. CHIBA, S. SHIGETA and Y. NUMAZAKI, Biol. Pharm. Bull., 1995, 18, 1081–1083 Search PubMed.
- K. TERASHIMA, O. MURAOKA and M. ONO, Chem. Pharm. Bull., 1995, 43, 1985–1991 CAS.
- S. M. Sondhi, R. P. Verma, V. K. Sharma, N. Singhal, J. L. Kraus, M. Camplo and J.-C. Chermann, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 122, 215–225 CAS.
- H. T. Abdel-Mohsen, F. A. Ragab, M. M. Ramla and H. I. El Diwani, Eur. J. Med. Chem., 2010, 45, 2336–2344 CAS.
- A. Nowicka, H. Liszkiewicz, W. P. Nawrocka, J. Wietrzyk, K. Kempińska and A. Dryś, Cent. Eur. J. Chem., 2014, 12, 1047–1055 Search PubMed.
- S. M. Sondhi, S. Rajvanshi, M. Johar, N. Bharti, A. Azam and A. K. Singh, Eur. J. Med. Chem., 2002, 37, 835–843 Search PubMed.
- C. S. Yao, S. Lei, C. H. Wang, C. X. Yu, Q. Q. Shao and S. J. Tu, Chin. J. Chem., 2008, 26, 2107–2111 Search PubMed.
- C. Yao, S. Lei, C. Wang, T. Li, C. Yu, X. Wang and S. Tu, J. Heterocycl. Chem., 2010, 47, 26–32 Search PubMed.
- G. D. Rao, B. Acharya, S. Verma and M. Kaushik, Tetrahedron Lett., 2011, 52, 809–812 CAS.
- J. Liu, M. Lei and L. Hu, Green Chem., 2012, 14, 840–846 Search PubMed.
- R. Gupta, A. Jain, Y. Madan and E. Menghani, J. Heterocycl. Chem., 2014, 51, 1395–1403 CAS.
- D. P. Singh, S. K. Deivedi, S. R. Hashim and R. G. Singhal, Pharmaceuticals, 2010, 3, 2416–2425 Search PubMed.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, Molecules, 2020, 25, 2784 Search PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, J. Mol. Struct., 2022, 1253, 132220 CAS.
- E. C. Taylor and D. J. Dumas, J. Org. Chem., 1981, 46, 1394–1402 CAS.
- T. Fukunaga and R. W. Begland, J. Org. Chem., 1984, 49, 813–821 CrossRef CAS.
- N. Sato, N. Matsui and J. Heterocyclic, Chem, 1992, 29, 1689–1692 CAS.
- W. Zhang, A. R. Haight, K. L. Ford and S. I. Parekh, Synth. Commun., 2001, 31, 725–730 CrossRef CAS.
- A. Kumbhar, S. Kamble, M. Barge, G. Rashinkar and R. Salunkhe, Tetrahedron Lett., 2012, 53, 2756–2760 CrossRef CAS.
- P. Mahadik, D. Jagwani and R. Joshi, Int. j. Innov. Res. Sci. Eng. Technol., 2014, 1, 482–490 Search PubMed.
- Z. Zhang, C. Xie, L. Feng and C. Ma, Synth. Commun., 2016, 46, 1507–1518 CrossRef CAS.
- R. Mahesh, A. K. Dhar, T. S. TVNV, S. Thirunavukkarasu and T. Devadoss, Chin. Chem. Lett., 2011, 22, 389–392 CrossRef CAS.
- J. Khalafy, A. Marjani and M. Haghipour, Curr. Chem. Lett., 2013, 2, 21–26 Search PubMed.
- L. Moradi, M. Piltan, H. Rostami and G. Abasi, Chin. Chem. Lett., 2013, 24, 740–742 CrossRef CAS.
- X. Shen, Y. Wang, T. Wu, Z. Mao and X. Lin, Chem. - Eur. J., 2015, 21, 9039–9043 CrossRef CAS PubMed.
- K. D. R. Viswanadham, M. P. Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC.
- F. Lassagne, F. Chevallier and F. Mongin, Synth. Commun., 2014, 44, 141–149 CAS.
- P. Z. Mannes, E. O. Onyango and G. W. Gribble, J. Org. Chem., 2016, 81, 12478–12481 CAS.
- D. Bera, R. Sarkar, P. Saha, P. Ghosh and C. Mukhopadhyay, Chem. Commun., 2023, 59, 7771–7774 CAS.
- J. Fang, J. Fang, Y. Rao, H. Qiu, Z. Pan and Y. Ma, Org. Biomol. Chem., 2024, 22, 2043–2048 CAS.
- J. Sharma and R. Kaushal, Russ. J. Gen. Chem., 2024, 94, 1794–1814 CAS.
- B. Groenendaal, D. J. Vugts, R. F. Schmitz, F. J. J. de Kanter, E. Ruijter, M. B. Groen and R. V. A. Orru, J. Org. Chem., 2008, 73, 719–722 CAS.
- M. Gradsten and M. Pollock, J. Am. Chem. Soc., 1948, 70, 3079–3081 CAS.
- W. O. Teeters and M. A. Gradsten, in Organic Syntheses, 2003, pp. 51 Search PubMed.
- W. D. Emmons, H. A. Rolewicz, W. N. Cannon and R. M. Ross, J. Am. Chem. Soc., 1952, 74, 5524–5525 CAS.
- A. T. Nielsen, R. L. Atkins, D. W. Moore, R. Scott, D. Mallory and J. M. LaBerge, J. Org. Chem., 1973, 38, 3288–3295 CAS.
- J. M. García, G. O. Jones, K. Virwani, B. D. McCloskey, D. J. Boday, G. M. ter Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb and A. M. Alabdulrahman, Science, 2014, 344, 732–735 Search PubMed.
- T. Yang, Z. Xu, Z. Meng and L. Zhai, ChemistrySelect, 2019, 4, 6338–6341 Search PubMed.
- S. Cascioferro, B. Parrino, V. Spanò, A. Carbone, A. Montalbano, P. Barraja, P. Diana and G. Cirrincione, Eur. J. Med. Chem., 2017, 142, 74–86 Search PubMed.
- K. Kobari, I. Takakura, M. Nakatomi, S. Sogame and C. Uylangco, Bull. W. H. O., 1970, 43, 365–371 Search PubMed.
- J. C. A. Hunt, E. Briggs, E. D. Clarke and W. G. Whittingham, Bioorg. Med. Chem. Lett., 2007, 17, 5222–5226 CAS.
- T. Kaihoh, T. Itoh, A. Ohsawa, M. Okada, C. Kawabata and H. Igeta, Chem. Pharm. Bull., 1987, 35, 3952–3954 Search PubMed.
- A. Ohsawa, T. Kaihoh, T. Itoh, M. Okada, C. Kawabata, K. Yamaguchi and H. Igeta, Chem. Pharm. Bull., 1988, 36, 3838–3848 CrossRef.
- R. N. Butler, A. M. Fahy, A. Fox, J. C. Stephens, P. McArdle, D. Cunningham and A. Ryder, J. Org. Chem., 2006, 71, 5679–5687 CrossRef CAS PubMed.
- R. N. Butler, A. M. Fahy, A. Fox, J. C. Stephens, P. McArdle, D. Cunningham and A. G. Ryder, Tetrahedron Lett., 2006, 47, 1721–1724 CAS.
- K. Sztanke, A. Sidor-Wójtowicz, J. Truchlińska, K. Pasternak and M. Sztanke, Ann. Univ. Mariae Curie-Sklodowska Sect. D Med., 2004, 59, 342–345 Search PubMed.
- L. X. Li, J. Jiao, X. B. Wang, M. Chen, X. C. Fu, W. J. Si and C. L. Yang, Molecules, 2018, 23, 746 Search PubMed.
- A. Gornowicz, A. Szymanowska, M. Mojzych, R. Czarnomysy, K. Bielawski and A. Bielawska, Molecules, 2021, 26, 2045 CrossRef CAS PubMed.
- P. Mullick, S. A. Khan, T. Begum, S. Verma, D. Kaushik and O. Alam, Acta Pol. Pharm., 2009, 66, 379–385 CAS.
- W. W. Paudler and J. M. Barton, J. Org. Chem., 1966, 31, 1720–1722 CrossRef CAS.
- S. Rostamizadeh and K. Sadeghi, Synth. Commun., 2002, 32, 1899–1902 CrossRef CAS.
- A. Rauf, S. Sharma and S. Gangal, Arkivoc, 2007, 16, 137–147 Search PubMed.
- T. Potewar, R. Lahoti, T. Daniel and K. Srinivasan, Synth. Commun., 2007, 37, 261–269 CrossRef CAS.
- F. Krauth, H.-M. Dahse, H.-H. Rüttinger and P. Frohberg, Bioorg. Med. Chem., 2010, 18, 1816–1821 CrossRef CAS PubMed.
- R. S. Tamboli, R. Giridhar, H. M. Mande, S. R. Shah and M. R. Yadav, Synth. Commun., 2014, 44, 2192–2204 CrossRef CAS.
- D. Tang, J. Wang, P. Wu, X. Guo, J.-H. Li, S. Yang and B.-H. Chen, RSC Adv., 2016, 6, 12514–12518 RSC.
- Y. Liu, X. Guo, D. Tang, J. Wang, P. Wu, J. Han and B. Chen, Chin. J. Chem., 2017, 35, 1222–1226 CrossRef CAS.
- O. Shabunina, A. Krinochkin, D. Kopchuk, G. Zyryanov, V. Rusinov and O. Chupakhin, Russ. J. Org. Chem., 2018, 54, 812–814 Search PubMed.
- M. S. Dowling, W. Jiao, J. Hou, Y. Jiang and S. Gong, J. Org. Chem., 2018, 83, 4229–4238 CrossRef CAS PubMed.
- L. Zhang, J. J. Chen, S. S. Liu, Y. X. Liang and Y. L. Zhao, Adv. Synth. Catal., 2018, 360, 2172–2177 Search PubMed.
- S. Singh, M. K. Mandal, A. Masih, A. Saha, S. K. Ghosh, H. R. Bhat and U. P. Singh, Arch. Pharm., 2021, 354, e2000363 Search PubMed.
- P. Gogoi, A. Shakya, S. K. Ghosh, N. Gogoi, P. Gahtori, N. Singh, D. R. Bhattacharyya, U. P. Singh and H. R. Bhat, J. Biochem. Mol. Toxicol., 2021, 35, e22682 CrossRef CAS PubMed.
- S. Cascioferro, B. Parrino, V. Spanò, A. Carbone, A. Montalbano, P. Barraja, P. Diana and G. Cirrincione, Eur. J. Med. Chem., 2017, 142, 523–549 CrossRef CAS PubMed.
- K. M. Al-Zaydi, H. H. Khalil, A. El-Faham and S. N. Khattab, Chem. Cent. J., 2017, 11, 39 CrossRef PubMed.
- N. Sunduru, L. Gupta, V. Chaturvedi, R. Dwivedi, S. Sinha and P. M. Chauhan, Eur. J. Med. Chem., 2010, 45, 3335–3345 CrossRef CAS PubMed.
- H. Ogura, H. Takahashi and O. Sato, Chem. Pharm. Bull., 1981, 29, 1838–1842 CrossRef CAS.
- T. Okawa, N. Osakada, S. Eguchi and A. Kakehi, Tetrahedron, 1997, 53, 16061–16082 CAS.
- J.-J. Shie and J.-M. Fang, J. Org. Chem., 2007, 72, 3141–3144 CAS.
- M. M. Hemdan and M. M. Elshahawi, J. Chem. Res., 2009, 2009, 75–77 Search PubMed.
- L. L. Falher, O. Ben Ayad, O. Ziyaret, A. Mamontov, C. Botuha, S. Thorimbert and F. Slowinski, J. Org. Chem., 2014, 79, 6579–6589 CAS.
- A. Herrera, A. Riano, R. Moreno, B. Caso, Z. D. Pardo, I. Fernandez, E. Saez, D. Molero, A. Sanchez-Vazquez and R. Martinez-Alvarez, J. Org. Chem., 2014, 79, 7012–7024 CAS.
- I. Yavari and S. Mosaferi, Monatsh. Chem., 2017, 148, 963–966 CAS.
- G.-F. Kang and G. Zhang, Beilstein J. Org. Chem., 2020, 16, 1447–1455 CAS.
- W.-X. Hu, G.-W. Rao and Y.-Q. Sun, Bioorg. Med. Chem. Lett., 2004, 14, 1177–1181 CAS.
- A. M. Churakov, O. Y. Smirnov, S. L. Ioffe, Y. A. Strelenko and V. A. Tartakovsky, Eur. J. Org Chem., 2002, 2002, 2342–2349 Search PubMed.
- T. M. Klapötke, D. G. Piercey, J. Stierstorfer and M. Weyrauther, Propellants, Explos. Pyrotech., 2012, 37, 527–535 Search PubMed.
- A. A. Voronin, V. P. Zelenov, A. M. Churakov, Y. A. Strelenko, I. V. Fedyanin and V. A. Tartakovsky, Tetrahedron, 2014, 70, 3018–3022 CAS.
- A. A. Voronin, A. M. Churakov, M. S. Klenov, Y. A. Strelenko, I. V. Fedyanin and V. A. Tartakovsky, Eur. J. Org Chem., 2017, 2017, 4963–4971 CAS.
- M. S. Klenov, O. V. Anikin, A. M. Churakov, Y. A. Strelenko, I. V. Fedyanin, I. V. Ananyev and V. A. Tartakovsky, Eur. J. Org Chem., 2015, 2015, 6170–6179 CAS.
- P. Barraja, P. Diana, A. Lauria, A. Montalbano, A. M. Almerico, G. Dattolo and G. Cirrincione, Bioorg. Med. Chem., 2005, 13, 295–300 CrossRef CAS PubMed.
- G. A. áO'Halloran and P. D. áO'Shea, J. Chem. Soc. Perkin Trans. I, 1990, 2527–2536 Search PubMed.
- R. N. Butler, D. Cunningham, P. McArdle and G. A. O'Halloran, J. Chem. Soc., Chem. Commun., 1988, 232–234 RSC.
- Z.-C. Wu and D. L. Boger, J. Am. Chem. Soc., 2019, 141, 16388–16397 CrossRef CAS PubMed.
- F. Xu, Z.-z. Yang, J.-r. Jiang, W.-g. Pan, X.-l. Yang, J.-y. Wu, Y. Zhu, J. Wang, Q.-Y. Shou and H.-g. Wu, Bioorg. Med. Chem. Lett., 2016, 26, 3042–3047 CrossRef CAS PubMed.
- M. Coburn, G. Buntain, B. Harris, M. Hiskey, K. Y. Lee and D. Ott, J. Heterocycl. Chem., 1991, 28, 2049–2050 CAS.
- J. Kerth and S. Löbbecke, Propellants, Explos. Pyrotech., 2002, 27, 111–118 CAS.
- G.-W. Rao and W.-X. Hu, Bioorg. Med. Chem. Lett., 2006, 16, 3702–3705 CAS.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjug. Chem., 2011, 22, 2263–2270 CAS.
- Z. Fang, W.-L. Hu, D.-Y. Liu, C.-Y. Yu and X.-G. Hu, Green Chem., 2017, 19, 1299–1302 CAS.
- C. Li, H. Ge, B. Yin, M. She, P. Liu, X. Li and J. Li, RSC Adv., 2015, 5, 12277–12286 RSC.
- T. A. Khattab, Helv. Chim. Acta, 2018, 101, e1800009 CrossRef.
- Y.-Z. Ji, H.-J. Li, Y. Liu and Y.-C. Wu, Synthesis, 2020, 52, 69–74 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
The Thai Nguyen
abc,
Vinh Thanh Chau Doan
abc,
Trinh Hao Nguyen
*ab,
The Thai Nguyen
abc,
Vinh Thanh Chau Doan
abc,
Trinh Hao Nguyen