
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential Michael addition, cross-coupling and [3 + 2] cycloaddition reactions within the coordination sphere of chiral Ni(II) Schiff base complexes derived from dehydroamino acids: pathways to the asymmetric synthesis of structurally diverse O-substituted serine and threonine analogs†
Emma A. Khachatryana,
Lusine Yu. Sahakyana,
Anna S. Tovmasyanb,
Gagik S. Melikyana,
Henrik A. Panosyanc,
Anna F. Mkrtchyan *ab,
Norio Shibatad,
Andrei V. Malkov*e and
Ashot S. Saghyan*ab
*ab,
Norio Shibatad,
Andrei V. Malkov*e and
Ashot S. Saghyan*ab
aInstitute of Pharmacy, Yerevan State University, 1 Alex Manoogian Str., 0025 Yerevan, Armenia. E-mail: anna_mkrtchyan@ysu.am; Fax: +374 60 710 410; Tel: +374 60 710 427
bScientific and Production Center “Armbiotechnology” of NAS RA, 14 Gyurjyan Str., 0056 Yerevan, Armenia
cScientific and Technological Center of Organic and Pharmaceutical Chemistry of NAS RA, 26 Azatutyan Ave., 0014 Yerevan, Armenia
dDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Showa-ku, Nagoya 466-8555, Japan
eDepartment of Chemistry, Loughborough University, University Road, Loughborough, LE11 3TU, UK
First published on 4th April 2025
Abstract
An approach to the synthesis of a series of novel, enantiomerically pure analogs of β-hydroxy-α-amino acids is reported. The method involves the introduction of the acetylene group into their side chain, followed by further elaboration of the terminal alkyne moiety. The asymmetric synthesis of alkyl- and aryl-substituted derivatives of (S)-O-propargylserine and (S)-allo-O-propargylthreonine (de >90%) was achieved through the nucleophilic Michael addition of the deprotonated congeners of propargyl alcohols to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the square-planar Ni(II) Schiff base complexes of dehydroamino acids (dehydroalanine and dehydroaminobutyric acid) with the chiral auxiliary (S)-BPB. Both (S)-O-propargylserine and (S)-allo-O-propargylthreonine were isolated with high enantiomeric purity (81–98% ee). The terminal alkyne group was further modified: Glaser reaction enabled formation of the dienyne products; Sonogashira cross-coupling gave rise to arylacetylene motifs, whereas [3 + 2]-cycloaddition reactions with 2-nirtophenylazide produced analogs of O-substituted (S)-serine and (S)-allo-threonine containing a 1,2,3-triazole group. All target amino acids were isolated with high enantiomeric purity (ee >98%). The developed approach provides an opportunity to synthesize new O-substituted analogs of β-hydroxy-α-amino acids with a diverse set of substituents in the side chain.
C bond of the square-planar Ni(II) Schiff base complexes of dehydroamino acids (dehydroalanine and dehydroaminobutyric acid) with the chiral auxiliary (S)-BPB. Both (S)-O-propargylserine and (S)-allo-O-propargylthreonine were isolated with high enantiomeric purity (81–98% ee). The terminal alkyne group was further modified: Glaser reaction enabled formation of the dienyne products; Sonogashira cross-coupling gave rise to arylacetylene motifs, whereas [3 + 2]-cycloaddition reactions with 2-nirtophenylazide produced analogs of O-substituted (S)-serine and (S)-allo-threonine containing a 1,2,3-triazole group. All target amino acids were isolated with high enantiomeric purity (ee >98%). The developed approach provides an opportunity to synthesize new O-substituted analogs of β-hydroxy-α-amino acids with a diverse set of substituents in the side chain.
Introduction
Optically active non-proteinogenic amino acids are crucial structural components of many pharmacologically active peptides, antibiotics, and other pharmaceuticals.1–4 Numerous non-proteinogenic amino acids and peptides containing aliphatic, aromatic, and heterocyclic substituents in their side chains have been synthesized and extensively studied for their biological activities.5–8 Among these, derivatives of serine and threonine occupy a prominent place due to their significant medical applications as therapeutic agents and integral components of complex drug formulations. For instance, the anticancer drug Taxol contains phenylisoserine,9 antibiotics such as azaserine and O-diazoacetyl-L-serine10 are used as antitumor agents, L-threo-α-amino-β-mescanbutanoic acid is the active aglycone of the antibiotic nisin 11, and L-threo-α-amino-β-methoxybutanoic acid12 is a key active ingredient in the therapeutic drug FR900359.13An important research direction involves the development of efficient methods to synthesize chiral, polyfunctional β-hydroxy-α-amino acid derivatives from simple starting materials. One promising approach employs O-alkylated derivatives of serine and threonine, such as O-methoxy-L-threonine and O-methoxy-methylene-L-threonine, which serve as innovative nitrogen sources and facilitate the incorporation of non-natural amino acids into jadomycin analogs produced by Streptomyces venezuelae ISP5230 cultures.14 Furthermore, (R)- and (S)-O-benzylserine and (R)- and (S)-O-(naphthalen-2-ylmethyl)serine have been utilized to develop subtype-selective agonists targeting the glycine site of NMDA receptors, focusing on the GluN1 subunit in combination with GluN2A-D subunits.15 Additionally, H-DL-β-(3,4-dihydroxyphenyl)-DL-Ser-OH demonstrated potent activity against S. aureus, including methicillin-resistant strains.16 O-Glycosylated amino acids, such as serine and threonine derivatives, have shown activity in inhibiting Trypanosoma cruzi trans-sialidase.17 On the other hand, alkynes are gaining attention in medicinal chemistry and the pharmaceutical industry due to their role in constructing complex chemical molecules.18 Alkynes are also key pharmacophore elements of acetylene antibiotics and several anticancer and antituberculosis drugs. Among the various alkyne-containing compounds with antitumor, antibacterial, antimicrobial, antifungal, and phototoxic properties, molecules with the acetylene bond adjacent to a hydroxyl group are particularly promising. Examples include steroid drugs like Ethynylestradiol, Gestodeno, Efavirenz, Mifepristone, and Calichoamicin,19,20 as well as falcarinol and falcarindiol, compounds isolated from Panax notoginseng that demonstrate anti-inflammatory, antibacterial, and anticancer activities.21 The inclusion of an alkyne group in the side chain significantly enhances the potential for molecular modification. For example, O-propargylserine derivatives incorporated into peptides can undergo oxidative α,ω-diyne coupling to produce novel peptidic macrocycles.22 Moreover, in situ click chemistry can be employed to obtain highly potent and selective inhibitors of cyclooxygenase-2 isozymes.23 The alkyne group also plays an important role in the total synthesis of natural products through reactions such as Sonogashira coupling.24
Therefore, β-hydroxy-α-amino acid analogs containing an alkyne group in the side chain, specifically when the hydroxyl group is separated from the alkyne by one carbon atom, represent a promising class of compounds with notable pharmacological activities. The presence of a terminal alkyne moiety facilitates the synthesis of more complex analogs through cross-coupling reactions, creating opportunities for the design of novel drug candidates.
Developing and optimizing methodologies for the synthesis of O-substituted derivatives of β-hydroxy-α-amino acids, with a particular emphasis on incorporating an alkyne moiety into their side chains, represents a highly relevant and promising approach in modern synthetic chemistry.
Among the well-known routes to O-substituted derivatives of β-hydroxy-α-amino acids, a photoinduced decarboxylative radical O-alkylation of serine and threonine proposed by Yoshimi25 can be noted (Scheme 1).
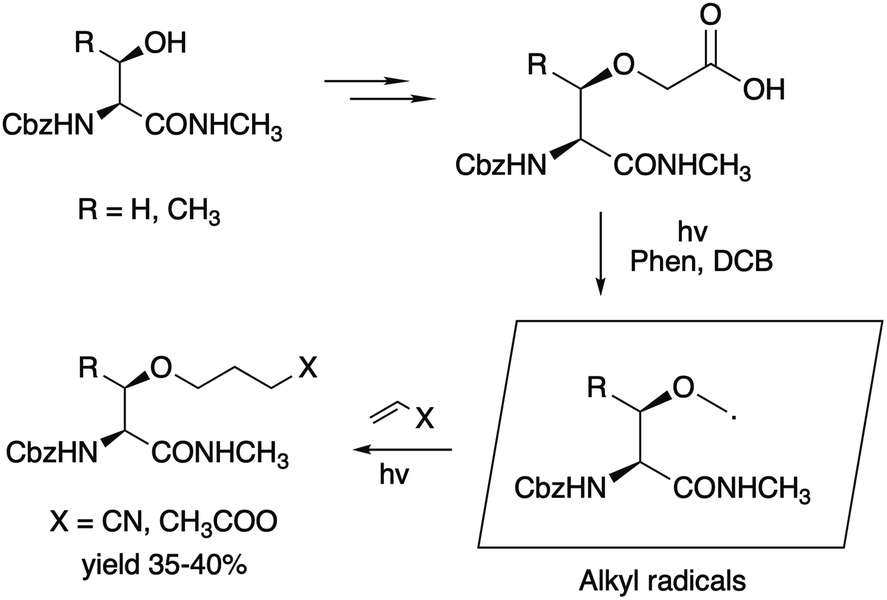 | ||
| Scheme 1 Photoinduced decarboxylative radical method for O-alkylation of serine. Phen = phenanthroline, DCB = 1,4-dicyanobenzene. | ||
An alternative approach to enantioenriched O-substituted derivatives of β-hydroxy-α-amino acids was introduced by the group of Belokon, which involved nucleophilic Michael addition to the CC double bond in chiral NiII Schiff base complexes of dehydroamino acids with BPB. However, the O-nucleophiles were limited to simple methanol and ethanol26 (Scheme 2).
 | ||
| Scheme 2 Approach to the synthesis of enantioenriched O-substituted derivatives of β-hydroxy-α-amino acids using nucleophilic Michael addition to the CC bond of chiral NiII complexes of Schiff base of dehydroamino acids with BPB. | ||
In the present work, the Michael addition approach is employed to introduce alkyne moiety into the side chain of β-hydroxy-α-amino acids, where the reactivity of terminal alkynes is exploited for further modification. The main focus was on synthesizing propargyl analogs where the β-hydroxyl and acetylene moiety are separated by one carbon atom, which is important in the context of the biological activity of the final products.
Results and discussion
Square-planar Ni(II) complexes of the Schiff bases of dehydroalanine (1) and (E)-dehydro-aminobutyric acid (1′) with the chiral auxiliary (S)-2-N-(N′-benzylprolyl)aminobenzophenone (S-BPB) were used as the substrates. They were obtained by the previously reported method.27 Propargyl alcohol 2a and its substituted aliphatic and aromatic derivatives 2b–f, were used as nucleophiles. The nucleophilic addition to the CC double bond in 1 and 1′ was carried out under conditions of base catalysis (Scheme 3).
 | ||
Scheme 3 The addition of the nucleophiles to the CC bond of complexes 1 and 1′. Conditions: (i) HCl; (ii) Dowex 8, H+; (iii) EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1). | ||
The progress of the nucleophilic addition was monitored by TLC by the disappearance of the starting complexes 1 and 1′ and the establishment of thermodynamic equilibrium between the diastereomers of the addition products. The process is thermodynamically controlled and provides a high diastereomeric excess of the main (S,S)- and (S,S,S)-diastereomers of the products containing substituted (S)-O-propargylserines and (S)-allo-propargylthreonine, respectively.
The structure and absolute configuration of the main diastereomeric complexes were rigorously established by physicochemical methods after the purification of the products by chromatography on silica.
The (S)-absolute configuration of the α-carbon atom of the amino acid moiety of the complexes was assigned using the positive sign of [α]D in analogy with previously reported similar Ni(II) complexes of other amino acids.2 Additional verification was obtained by the circular dichroism (CD) spectra: positive Cotton effects of the main diastereomeric complexes 3a–f and 4a in the 500–580 nm region indicated (S)-absolute configuration of the α-carbon atom of the amino acid moiety (see ESI†). For comparison, the CD spectra of (S,S)- and (S,R)-diastereomers of the similar alanine complexes with known absolute configuration unambiguously established by X-ray crystallography, are also given.
The absolute configuration of the α-carbon of the amino acid moiety in 4a was determined by 1H NMR based on the value of the chemical shift of its β-methyl protons. As reported earlier,27 the isopropyl group of the (S)-valine moiety in complexes with a similar structure was rigidly fixed in space with the pro-S methyl group located under the Ni2+ ion. This leads to the magnetic deshielding of the protons of this group due to the magnetic anisotropy of the Ni atom, resulting in a downfield shift of their signal (up to 1.8–1.9 ppm). In subsequent studies, this approach was used to assign the absolute configuration of the β-carbon of the threonine fragment in similar complexes.27
It was shown that the location of the methyl group under the metal ion was possible only in the case of (R)-configuration of the β-carbon atom, i.e. of the threo absolute configuration of the amino acid moiety of the complex, and it is disfavored for steric reasons for the allo-isomers. Signals of the same methyl protons in the spectra of aminobutyric acid complexes of the (2S,3S)-allo configuration appear in the range of 1.05–1.1 ppm.
This phenomenon was also observed in the 1H NMR spectra of the diastereoisomers of complex 4a. The upfield shift (1.07 ppm) of the methyl group signal of the O-propargylthreonine fragment of the main diastereomer of 4a favors the (S)-allo-configuration (Fig. 1, A), whereas the downfield shift of the same signals (1.75 ppm) for the minor diastereomer of 4a indicates the threo configuration of their amino acid moiety (Fig. 1, B). The (S,S)-allo-configuration of the amino acid moiety of this complex was confirmed by X-ray analysis (Fig. 2).
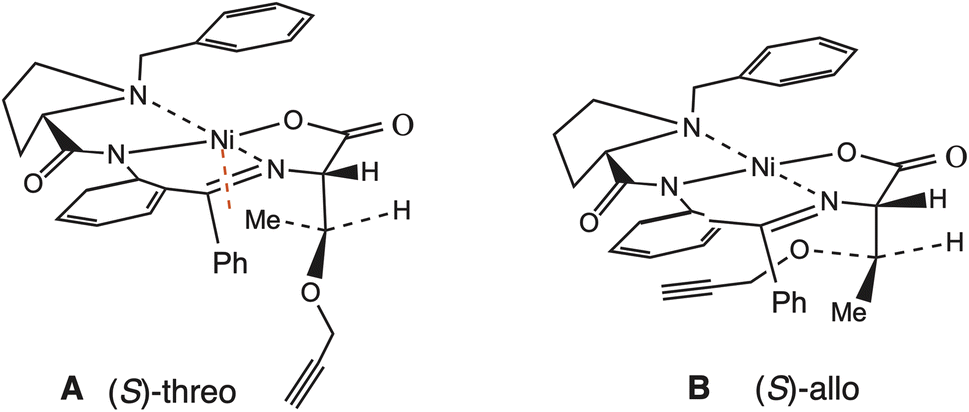 | ||
| Fig. 1 The spatial arrangement of the groups of the O-propargylthreonine fragment of complex 4a: (S)-threo configuration (A), (S)-allo-configuration (B). | ||
 | ||
| Fig. 2 Structure of complex 4a based on X-ray diffraction data. | ||
The ratio of the diastereomers in the addition products was determined from the 1H NMR spectra by integrating the methylene protons signals of the N-benzyl group; in the case of complexes 3a and 4a, this was also verified by chiral HPLC analysis of the corresponding amino acids (5a, 6a) obtained by acid hydrolysis of a mixture of diastereomeric complexes by ion-exchange demineralization, before chromatographic purification. (S,2S)-Diastereomers of 3a–f and (S,2S,3S)-diastereomer of 4a were formed with high diastereomeric excess. The results are shown in Table 1.
| Entry | Sub-strate | Nucleophile | Product 3, 4 | ||
|---|---|---|---|---|---|
| (1S,2S)/(1S,2R)a% (ee)b | Conv.c, % | ||||
| a Determined from the NMR spectra of the mixtures of diastereomeric complexes.b Determined by chiral HPLC analysis of the amino acid, isolated after acid hydrolysis of the reaction mixture, before chromatography.c Conversion at the stage of nucleophilic addition determined by NMR.d (S,S,S/S,S,R). | |||||
| 1 | 1 | 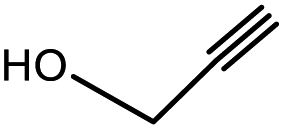 |
3a | 97.4/2.6 (93.8) | 90 |
| 2 | 1 |  |
3b | 95.8/4.2 | Trace |
| 3 | 1 | 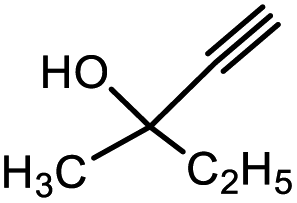 |
3c | 96.3/3.7 | 82 |
| 4 | 1 |  |
3d | 97.5/2.5 | 78 |
| 5 | 1 | 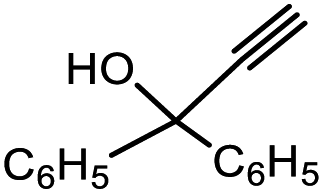 |
3e | 98/2 | 70 |
| 6 | 1 | 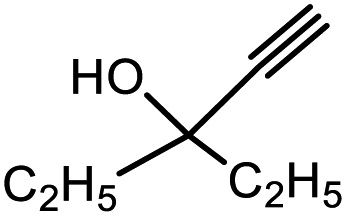 |
3f | 95.2/4.8 | Trace |
| 7 | 1′ | 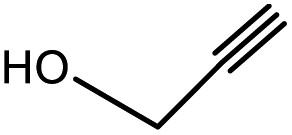 |
4a | 95.8/4.2d (90.6) | 90 |
In the nucleophilic addition of unsubstituted propargyl alcohol, the reaction proceeds efficiently, as observed with substrates 1 and 1′. However, the introduction of substituents generally leads to a decrease in reaction yield. Notably, the addition of substituted propargyl alcohols 2b–f to the dehydroaminobutyric acid complex (1′) was unsuccessful, likely due to steric hindrance at the reaction site, which may impede nucleophilic attack.
A closer analysis of the conversion data for complex 1 reveals that symmetrical dialkyl substituted nucleophiles 3b and 3f reacted sluggishly, whereas bulkier nucleophiles such as 3c, 3d, and 3e achieve significantly higher conversions. This observation suggests that steric hindrance might not be a single factor influencing the reactivity of tertiary alcohols. Without computational modeling of the transition states, it would be too premature to speculate on the reasons for this phenomenon.
The pure target amino acids were isolated using a standard protocol: a mixture of diastereomeric complexes 3a and 4a was subjected to acid hydrolysis, and ion-exchange demineralization, followed by recrystallization from aqueous ethanol.27 In this protocol, the initial chiral auxiliary (S)-BPB is regenerated with a nearly quantitative yield (>97%) without loss of enantiomeric purity, which allows it to be reused.
Using the outlined methodology, pure amino acids 5a and 6a were obtained in crystalline form, with enantiomeric purities exceeding 98% ee, as confirmed by chiral HPLC analysis. These results suggest that the procedure can be efficiently applied to obtain other amino acids with high enantiomeric purity.
The terminal alkyne groups in the side chain of the amino acid moiety of the synthesized complexes offer an opportunity for further synthetic modification. In this work, cross-coupling and [3 + 2]-cycloaddition reactions were investigated using complexes 3a and 4a featuring unsubstituted O-propargyl group (Scheme 4 and Table 2).
 | ||
| Scheme 4 Cross-coupling and [3 + 2]-cycloaddition reactions of the complexes 3a and 4a. Conditions: (i) 2M HCl, 50 °C; (ii) Dowex 8, H+; (iii) EtOH/H2O (1:1). | ||
| Initial complex | Reagent | Product complex | Amino acid | |||
|---|---|---|---|---|---|---|
| Conversionb (%) | Yieldc (%) | |||||
| a Diastereomeric purity of complexes 7–12 (according to NMR data) and enantiomeric purity (ee) of isolated amino acids 13–16 (according to HPLC data) exceed 98%.b Conversion determined by NMR.c Yield of the isolated product pure by NMR. | ||||||
| 1 | 3a | — | 7 | 58% | 13 | 47% |
| 2 | 4a | — | 8 | 56% | 14 | 43% |
| 3 | 3a | C6H5I | 9 | 66% | — | — |
| 4 | 4a | C6H5I | 10 | 90% | — | — |
| 5 | 3a | 2-NO2C6H4N3 | 11 | 83% | 15 | 44% |
| 6 | 4a | 2-NO2C6H4N3 | 12 | 80% | 16 | 45% |
The Sonogashira coupling enabled the introduction of the phenyl group to the terminal acetylene in complexes 3a and 4a (Scheme 4, route A). Complexes 7 and 8 were obtained with good yields, and then were converted to the target amino acids (S)-13 and (S,S)-14 using the standard method. During the Sonogashira reaction, a side product was formed, which was isolated and identified as a dimerization product. Following this, a Glaser homocoupling reaction was optimized (route B), to enable an efficient synthesis of dimeric complexes 9 and 10 with good chemical yields. The structures of these complexes were confirmed by NMR spectroscopy.
In a [3 + 2]-cycloaddition, 2-nitrophenyl azide was reacted with the terminal alkyne of 3a and 4a to furnish complexes 11 and 12 in good yields, which were converted to the target amino acids 15 and 16 containing a 1,2,3-triazole group in the side chain (route C). Investigation of other propargyl-containing complexes (3b–f) in cross-coupling reactions is currently underway in our laboratories.
In the reactions shown in Scheme 4, the starting complexes 3a and 4a were used as pure (S,S)- and (S,S,S)-diastereoisomers. It is worth noting that the chiral α-C of these amino acid fragments does not directly participate in the formation of a new carbon–carbon bond in the cross-coupling reactions. However, weakly basis conditions of the reaction may cause partial epimerization of this stereogenic center. Therefore, the chiroptical data of the intermediate complexes and the isolated final amino acids were examined. The analysis of the diastereomeric purity of intermediate complexes 7–12 by NMR spectroscopy and chiral HPLC analysis of amino acids 13–16 revealed that in all the reactions, the stereochemical integrity remained intact. This was additionally confirmed by the circular dichroism (CD) method (see ESI†). The positive Cotton effects observed in the region of 500–580 nm in the CD spectra of complexes 7–12 indicate the (S)-absolute configuration of the α-C atom of the amino acid moieties. The target amino acids were isolated from diastereomerically pure complexes 9–12 using standard procedures.27 The structure and enantiomeric purity (81–98% ee) of recrystallized amino acids 13–16 were confirmed by the appropriate spectral and analytical techniques.
Conclusions
A highly selective asymmetric synthesis of a series of O-propargyl-substituted β-hydroxy-α-amino acids has been developed involving nucleophilic Michael addition of substituted propargyl alcohols to the double bond of square-planar NiII Schiff base complexes of dehydroamino acids (dehydroalanine and dehydroaminobutyric acid) and the chiral auxiliary (S)-BPB. Enantiomerically pure (S)-O-propargylserine and (S)-allo-O-propargylthreonine were obtained. All nucleophilic addition reactions proceed with high diastereomeric excess (de >90%). The terminal alkyne group of the synthesized analogs of O-propargyl-substituted serine and threonine was further modified through Sonogashira cross-coupling with iodobenzene, Glaser dimerization and the [3 + 2]-cycloaddition with 2-nitrophenyl azide. The latter reaction produced derivatives of O-substituted (S)-serine and (S)-allo-threonine containing a 1,2,3-triazole group in the side chain. The coupling and cycloaddition reactions proceeded with the retention of the stereochemical integrity. The target amino acids were obtained with high enantiomeric purity (81–98% ee). The developed approach creates an opportunity for the synthesis of new O-substituted β-hydroxy-α-amino acids with a diverse set of substituents in the side chain.Experimental
Materials
All starting reagents were obtained from commercial sources and used without further purification. The initial complexes 1 and 1′ were prepared following literature protocols.26,27 TLC analyses were performed on glass plates coated with silica gel 60 F254. Column chromatography was performed on silica (60 × 120 mesh) using glass columns. Melting points (mp) were determined by ≪Electrothermal≫. 1H and 13C NMR spectra (≪Mercury-300 Varian≫» 300 MHz respectively) were recorded using TMS as an internal standard (0 ppm). Elemental analyses were done by elemental analyzer EURO EA 3000.The enantiomeric purity of the amino acids was determined by HPLC (≪Waters Alliance 2695 HPLC System≫) on the chiral phase Nautilus-E column, 5 μm, 4.6 mm × 250 mm (BioChemMak ST, Moscow, Russia), and a mixture of 20% MeOH and 80% 88 mM aqueous solution KH2PO4 was used as the eluent. The optical rotation was measured on a Perkin Elmer-341 polarimeter. LCMS analysis was performed on Shimadzu LCMS 2020 with Prominence-I LC-2030C 3D. The CD analyses were carried out with Chirascan™ V100.
General procedure for nucleophilic Michaele addition to complex 1
Complex 1 (2.0 g, 3.9 mmol) was dissolved in 3.5 mL of CH3CN in a round bottom flask. Separately, NaOH (0.156 g, 3.9 mmol) was dissolved in 3.5 mL of propargyl alcohol and then added to the solution of complex 1. The reaction mixture was stirred at room temperature; the reaction progress was monitored by thin-layer chromatography (TLC) on silica plates using a 3:1 ethyl acetate/chloroform mixture as a mobile phase.
Upon completion of the reaction, the mixture was extracted with ethyl acetate and distilled water. The organic layer was dried over anhydrous MgSO4 to remove residual water, followed by filtration through filter paper. The filtrate was concentrated to dryness under reduced pressure and the resulting residue was recrystallized from acetone to obtain the purified product.
General procedure of nucleophile Michaele addition reaction of complex 1′
Complex 1′ (10.0 g, 0.0191 mol) was dissolved in 100 mL of a 0.3 M solution of sodium salt of propargyl alcohol in propargyl alcohol. The reaction mixture was stirred at 50–55 °C under a nitrogen atmosphere. The reaction progress was monitored by TLC on silica using a 3:1 ethyl acetate/chloroform mixture as a mobile phase. After completion, the reaction mixture was neutralized with concentrated acetic acid and evaporated to dryness under reduced pressure. The resulting residue was recrystallized from acetone to obtain the purified product.
General procedure for Sonogashira reaction
To a mixture of iodobenzene (0.78 mL, 7.0 mmol) and 5 mL of triethylamine, PdCl2(PPh3)2 (0.12 g, 0.1 mmol) and copper iodide (0.067 g, 0.3 mmol) were added in a pressure tube under an argon atmosphere. Subsequently, a mixture of 1,4-dioxane (5 mL) and complex 3a or 4a (3.5 mmol) was added to the reaction vessel. The reaction mixture was heated at 60 °C with stirring. The progress of the reaction was monitored by TLC on silica using a 3:1 ethyl acetate/chloroform mixture as a mobile phase. Upon completion, the reaction mixture was extracted with ethyl acetate and distilled water. The organic phase was dried over anhydrous MgSO4 to remove residual moisture and filtered through filter paper. The filtrate was concentrated to dryness under reduced pressure, and the residue was recrystallized from acetone to yield the purified product.
General procedure for Glaser reaction
To a mixture of 1,4-dioxane (5 mL) and triethylamine (5 mL), copper iodide (0.67 g, 3.5 mmol) and complex 3a or 4a (3.5 mmol) were added. The reaction mixture was stirred at room temperature, and the progress of the reaction was monitored by TLC on silica using a 3:1 chloroform/acetone mixture as a mobile phase. After the reaction was complete, the mixture was extracted with ethyl acetate and distilled water. The organic phase was dried over anhydrous MgSO4 to remove residual moisture and filtered through filter paper. The filtrate was concentrated to dryness under reduced pressure, and the residue was recrystallized from acetone to obtain the purified product.
General procedure for [3 + 2]-cycloaddition reactions
1-Azido-2-nitrobenzene (1.16 g, 7.0 mmol) was dissolved in 5 mL of 1,4-dioxane in a round-bottom flask under an argon atmosphere at room temperature. Copper iodide (0.067 g, 0.3 mmol) was added to the solution, and the mixture was stirred for 5 minutes. Triethylamine (0.738 mL, 5.2 mmol) was then added, and stirring was continued for 30 minutes. Subsequently, complex 3a or 4a (3.5 mmol) was added, and the reaction mixture was stirred at 60 °C. The progress of the reaction was monitored by TLC on silica using a 3:1 ethyl-acetate/chloroform mixture as a mobile phase. After completion, the reaction mixture was extracted with ethyl acetate and distilled water. The organic layer was dried over anhydrous MgSO4 to remove any residual water, and then filtered through filter paper. The filtrate was concentrated to dryness under reduced pressure, and the residue was recrystallized from acetone to yield the purified product.
General procedure for isolation of amino acids
Isolation of α-amino acids from the complexes was carried out following the literature protocol.26 The suspension of the complex in CH3OH was slowly added to a vigorously stirred 2 M aqueous HCI at 50–60 °C. After the complexes were decomposed (10–20 min), the free amino acids were purified using a cation-exchange column (Dowex-50 (H+ form)).Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 4a has been deposited at the CCDC under 2395037 and can be obtained from https://www.ccdc.cam.ac.uk/structures/ (free of charge).Author contributions
Emma A. Khachatryan: investigation, formal analysis, methodology. Lusine Yu. Sahakyan: methodology, investigation. Anna S. Tovmasyan: methodology, investigation. Gagik S. Melikyan: methodology, investigation. Henrik A. Panosyan: data analysis. Anna F. Mkrtchyan: conceptualization, funding acquisition, supervision, project administration, writing – review & editing. Norio Shibata: supervision, funding acquisition. Andrei V. Malkov: conceptualization, supervision, writing – review & editing. Ashot S. Saghyan: resources, conceptualization, funding acquisition, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support was provided by joint research projects SCS 21AG-1D013, 23AA-1D021 and ISTC AM-2705.References
- M. A. T. Blaskovich, Unusual Amino Acids in Medicinal Chemistry, J. Med. Chem., 2016, 59(24), 10807–10836, DOI:10.1021/acs.jmedchem.6b00319.
- A. S. Saghyan and P. Langer, Asymmetric Synthesis of Optically Pure Non-proteinogenic Amino Acids, Wiley-Vch Verlag, 2016 Search PubMed.
- C. Cativiela, M. D. Diaz-de-Villegas and J. A. Galvez, Asymmetric Synthesis of .beta.-Lactams. Highly Diastereoselective Alkylation of Chiral 2-Cyano Esters, J. Org. Chem., 1994, 59(9), 2497–2505, DOI:10.1021/jo00088a034.
- N. Wang, H. Mei, G. Dhawan, W. Zhang, J. Han and V. A. Soloshonok, New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine, Molecules, 2023, 28(9), 3651, DOI:10.3390/molecules28093651.
- M. A. Fik-Jaskółka, A. F. Mkrtchyan and A. S. Saghyan, et al., Spectroscopic and SEM evidences for G4-DNA binding by a synthetic alkyne-containing amino acid with anticancer activity, Spectrochim. Acta, Part A, 2020, 229, 117884, DOI:10.1016/j.saa.2019.117884.
- A. S. Saghyan, G. M. Mkrtchyan and A. S. Dadayan, et al., Asymmetric synthesis of enantiomerically enriched (S)-α-aminopropionic acids containing heterocyclic side chains, Tetrahedron: Asymmetry, 2013, 24(4), 229–232, DOI:10.1016/j.tetasy.2013.01.014.
- A. S. Saghyan, H. M. Simonyan and S. G. Petrosyan, et al., Thiophenyl-substituted triazolyl-thione l-alanine: asymmetric synthesis, aggregation and biological properties, Amino Acids, 2014, 46(10), 2325–2332, DOI:10.1007/s00726-014-1782-3.
- Z. T. Gugkaeva, M. P. Stukalova, A. F. Smol’yakov, A. T. Tsaloev, V. I. Maleev and V. A. Larionov, Asymmetric Metal-Templated Approach to Amino Acids with a CF3-Containing 3,2’-Pyrrolidinyl Spirooxindole Core via a Michael/Mannich [3 + 2]-Cycloaddition Reaction, Adv. Synth. Catal., 2023, 366(5), 1205–1211, DOI:10.1002/adsc.202301214.
- D. G. I. Kingston, The shape of things to come: Structural and synthetic studies of taxol and related compounds, Phytochemistry, 2007, 68(14), 1844–1854, DOI:10.1016/j.phytochem.2006.11.009.
- R. F. Pittillo and D. E. Hunt, Azaserine and 6-Diazo-5 -Oxo-L-Norleucine (DON), in Antibiotics, ed. D. Gottlieb and P. D. Shaw, Springer, Berlin, Heidelberg, 1967, DOI:10.1007/978-3-662-38439-8_39.
- M. Musiejuk and P. Kafarski, Engineering of Nisin as a Means for Improvement of Its Pharmacological Properties: A Review, Pharmaceuticals, 2023, 16(8), 1058, DOI:10.3390/ph16081058.
- Y. Qian, C. Jing, S. Liu and W. Hu, A highly enantioselective four-component reaction for the efficient construction of chiral β-hydroxy-α-amino acid derivatives, Chem. Commun., 2013, 49(26), 2700, 10.1039/c3cc40546j.
- M. C. T. Hartman, K. Josephson, C. W. Lin and J. W. Szostak, An Expanded Set of Amino Acid Analogs for the Ribosomal Translation of Unnatural Peptides, PLoS One, 2007, 2(10), e972, DOI:10.1371/journal.pone.0000972.
- D. L. Jakeman, S. Farrell, W. Young, R. J. Doucet and S. C. Timmons, Novel jadomycins: incorporation of non-natural and natural amino acids, Bioorg. Med. Chem. Lett., 2005, 15(5), 1447–1449, DOI:10.1016/j.bmcl.2004.12.082.
- A. R. Maolanon, R. Risgaard and S. Y. Wang, et al., Subtype-Specific Agonists for NMDA Receptor Glycine Binding Sites, ACS Chem. Neurosci., 2017, 8(8), 1681–1687, DOI:10.1021/acschemneuro.7b00117.
- K. Ikeda, T. Shoda, Y. Demizu and G. Tsuji, Discovery of non-proteinogenic amino acids inhibiting biofilm formation by S. aureus and methicillin-resistant S. aureus, Bioorg. Med. Chem. Lett., 2021, 48, 128259, DOI:10.1016/j.bmcl.2021.128259.
- M. B. Martins-Teixeira, V. L. Campo and M. Biondo, et al., α-Selective glycosylation affords mucin-related GalNAc amino acids and diketopiperazines active on Trypanosoma cruzi, Bioorg. Med. Chem., 2013, 21(7), 1978–1987, DOI:10.1016/j.bmc.2013.01.027.
- T. T. Talele, Acetylene Group, Friend or Foe in Medicinal Chemistry, J. Med. Chem., 2020, 63(11), 5625–5663, DOI:10.1021/acs.jmedchem.9b01617.
- A. Z. Aris, A. S. Shamsuddin and S. M. Praveena, Occurrence of 17α-ethynylestradiol (EE2) in the environment and effect on exposed biota: a review, Environ. Int., 2014, 69, 104–119, DOI:10.1016/j.envint.2014.04.011.
- B. Costa and N. Vale, Efavirenz: History, Development and Future, Biomolecules, 2022, 13(1), 88, DOI:10.3390/biom13010088.
- M. Lakshmi Haritha Bharathi, D. Ramesh and B. G. Vijayakumar, et al., Acetylene containing 2-(2-hydrazinyl)thiazole derivatives: design, synthesis, andin vitroandin silicoevaluation of antimycobacterial activity against Mycobacterium tuberculosis, RSC Adv., 2022, 12(14), 8771–8782, 10.1039/d2ra00928e.
- S. Verlinden, N. Geudens, J. C. Martins, D. Tourwé, S. Ballet and G. Verniest, Oxidative α,ω-diyne coupling as an approach towards novel peptidic macrocycles, Org. Biomol. Chem., 2015, 13(36), 9398–9404, 10.1039/c5ob01153a.
- A. Bhardwaj, J. Kaur, M. Wuest and F. Wuest, In situ click chemistry generation of cyclooxygenase-2 inhibitors, Nat. Commun., 2017, 8, 1, DOI:10.1038/s41467-016-0009-6.
- D. Wang and S. Gao, Sonogashira coupling in natural product synthesis, Org. Chem. Front., 2014, 1(5), 556–566, 10.1039/c3qo00086a.
- T. Yamamoto, T. Iwasaki, T. Morita and Y. Yoshimi, Strategy for O-Alkylation of Serine and Threonine from Serinyl and Threoninyl Acetic Acids by Photoinduced Decarboxylative Radical Reactions: Connection between Serine/Threonine and Carbohydrates/Amino Acids at the Side Chain, J. Org. Chem., 2018, 83(7), 3702–3709, DOI:10.1021/acs.joc.8b00061.
- Y. N. Belokon, A. S. Sagyan and S. A. Djamgaryan, et al., General method for the asymmetric synthesis of anti-diastereoisomers of β-substitutedL-2-aminobutanoic acids via chiral nickel(II) Schiff's base complexes of dehydroaminobutanoic acid. X-Ray crystal and molecular structure of the nickel(II) complex of the Schiff's base from [(benzylprolyl)amino]benzophenone and dehydroaminobutanoic acid, J. Chem. Soc., Perkin Trans. 1, 1990,(8), 2301–2310, 10.1039/p19900002301.
- Y. N. Belokon, A. S. Sagyan, S. M. Djamgaryan, V. I. Bakhmutov and V. M. Belikov, Asymmetric synthesis of β-substituted α-amino acids via a chiral NiII complex of dehydroalanine, Tetrahedron, 1988, 44(17), 5507–5514, DOI:10.1016/s0040-4020(01)86056-7.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2395037. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra00910c |
| This journal is © The Royal Society of Chemistry 2025 |