
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid thiazolyl–benzylidene–phenol metal complexes as novel chemotherapeutic agents with anti-topoisomerase I activity in human breast carcinoma: synthesis, in vitro and in silico studies†
Ahmed M. Alharbia,
Mona Katary bc,
Khulud M. Alshehrid,
Basim H. Asghare,
Mohmed M. Omranf,
Reda F. M. Elshaarawygh,
Amira Milii,
Hani S. Hafez*j and
Rozan Zakryak
bc,
Khulud M. Alshehrid,
Basim H. Asghare,
Mohmed M. Omranf,
Reda F. M. Elshaarawygh,
Amira Milii,
Hani S. Hafez*j and
Rozan Zakryak
aDepartment of Chemistry, Faculty of Sciences, Umm Al-Qura University, Makkah, Saudi Arabia. E-mail: amaharbi@uqu.edu.sa
bChemistry Department, Faculty of Science, Port-Said University, Port-Said, Egypt. E-mail: monakatary@gmail.com
cDepartment of Basic Sciences, Horus University, Egypt. E-mail: mkatary@horus.edu.eg
dDepartment of Biology, Al-Baha University, Saudi Arabia. E-mail: kalshehri@bu.edu.sa
eDepartment of Chemistry, Faculty of Sciences, Umm Al-Qura University, Makkah, Saudi Arabia. E-mail: bhasghar@uqu.edu.sa
fChemistry Department, Faculty of Science, Helwan University, Helwan, Egypt. E-mail: Mohmed.Omran@gmail.com
gDepartment of Chemistry, Faculty of Science, Suez University, 43533 Suez, Egypt. E-mail: reda.elshaarawy@suezuniv.edu.eg
hInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine Universität Düsseldorf, Düsseldorf, Germany
iDepartment of Biology, Al-Baha University, Al Aqiq, Saudi Arabia. E-mail: ahajsalah@bu.edu.sa
jDepartment of Zoology, Faculty of Science, Suez University, 43533 Suez, Egypt. E-mail: hani.hafez@suezuniv.edu.eg
kChemistry Department, Faculty of Science, Port-Said University, Port-Said, Egypt. E-mail: rzakrya@yahoo.com
First published on 17th June 2025
Abstract
A new ligand, benzylidene–phenol–thiazole (HBHTP), and its M(II) complexes (M = Co, Ni, Cu, or Zn) were synthesized using a hybrid pharmacophore approach. The structures were optimized using density functional theory (DFT) calculations. MTT cytotoxicity assay showed that CuBHTP was the most effective and least toxic to normal cells, with the highest toxicity against MCF-7 cells. CuBHTP was more selective than staurosporine, with a selectivity index (SI) of 4.2 for cancer MCF-7 cells compared to 2.5 for healthy MCF10a cells. Compared with novobiocin, it exhibited significant inhibitory effects on aromatase cytochrome 19A and reduced Hsp90 expression. The treatment also revealed significant upregulation of the apoptotic marker P53 and inhibitory effects on tubulin β, SULF1,2, and bFGF gene expression levels compared to the untreated MCF7 carcinoma. Furthermore, CuBHTP significantly inhibited topoisomerase I and cell proliferation by inducing cell cycle arrest in G1 and S phases. The CuBHTP complex is a highly effective anticancer agent, and molecular docking studies have confirmed its binding to grooves and topoisomerase I. Therefore, ligand/copper may shed new light on the inhibitory mechanisms of cancer cell proliferation through its ability to form DNA adducts.
1 Introduction
The incidence of breast cancer (BC), the most common cancer among women, has markedly increased over the past decade.1 The heterogeneity of BC, with up to ten molecular subtypes, challenges the development of effective treatments. Conventional chemotherapy remains the primary treatment for patients with BC, but there are limited molecular therapeutic options. Clinical management is challenging because of the restricted therapies and clinicopathological heterogeneity. Apoptosis is critical for growth and homeostasis of multicellular organisms. Surviving cells with DNA damage contribute to cancer progression. Cancer cells exhibit morphological features such as membrane blebbing, chromatin condensation, and apoptotic bodies.2Topoisomerase I (TOPI), a type IB enzyme, controls the DNA structure during transcription, recombination, replication, and repair.3 It breaks one strand, causing single-strand breaks in DNA. Cancer cells require a high TOPI activity for rapid division, and their inhibition can cause double-strand breaks and cell death. Topoisomerase I inhibitors, including camptothecin, exhibit dose-limiting adverse effects and poor water solubility, which lead to bone marrow toxicity and gastrointestinal disorders. Developing novel topoisomerase I inhibitors with reduced side effects is crucial, as these agents effectively impair cancer cell genetic stability4 and induce DNA strand breaks.5,6 TOPI–DNA cleavage complexes (TOPIcc) are formed by inhibition of re-ligation by carnitine palmitoyltransferase (CPT) in the TOPI catalytic cycle. TOPIcc exhibits cytotoxic activity during the S-phase because its interaction with the replication machinery leads to DNA breakdown and cell death.7
Molecular hybridization is a promising strategy for drug design, particularly for anticancer therapy. By combining multiple pharmacophores into a single entity, hybrid drug design offers the potential to overcome limitations associated with conventional anticancer drugs. The ability to address pharmacokinetic limitations and enhance therapeutic efficacy is crucial for the development of innovative treatments.8 As advancements in this field continue, molecular hybridization holds great promise in the development of novel and effective anticancer therapies.
Thiazoles are important in medicinal chemistry because of their wide range of biological activities, which make them valuable for drug discovery. Synthesis of thiazole-based therapeutic agents is central to the development of effective treatments for various diseases. These compounds can significantly affect drug discovery and therapeutic development. Tiazofurin, dasatinib,9 and thiazole hybrids have shown promising results in clinical trials for cancer therapy.10 Benzylidene derivatives have gained attention in pharmaceutical research because of their diverse biological activities, demonstrating potential in anticancer,11 antimicrobial, antiviral,12,13 anti-inflammatory, and antioxidant applications. Phenol derivatives are promising candidates owing to their antimicrobial, antioxidant, and anti-inflammatory properties.14 Combretastatin A4, which combines benzylidene and phenol pharmacophores, has emerged as a promising anticancer agent that targets the tumor vasculature, making it valuable for clinical applications.15
When designing novel metal-based anticancer agents, the selection of appropriate metal ions is crucial, because their properties significantly affect the stability, reactivity, and pharmacological activity of the resulting complexes. We selected cobalt (Co), nickel (Ni), copper (Cu), and zinc (Zn) for complexation with the novel ligand benzylidene–phenol–thiazole (HBHTP), based on their coordination chemistry, redox behavior, and biological significance. Cobalt(II) was selected for its ability to form stable octahedral complexes and redox activity, which can induce oxidative stress in cancer cells.16 Cobalt complexes have shown promise for interacting with DNA and inhibiting topoisomerase.17 Nickel(II) was chosen because of its capacity to form square-planar or octahedral complexes that facilitate DNA intercalation, potentially disrupting cancer cell proliferation. Nickel complexes have been shown to be cytotoxic to cancer cell lines.18 Copper(II) was prioritized because of its biological relevance and redox properties, which enable the generation of reactive oxygen species (ROS) that target cancer cells.19 Copper complexes exhibit anticancer potential through DNA-binding, topoisomerase inhibition, and modulation of signalling pathways.19 Zinc(II) was selected because of its role as an enzyme cofactor and its ability to form tetrahedral complexes. Zinc complexes have the potential to stabilize DNA-binding motifs and inhibit tumor-associated enzymes.20 These metals were chosen for their ability to form complexes with nitrogen- and oxygen-containing ligands such as HBHTP.21 By studying these metal complexes, we aimed to understand how metal ion properties influence their anticancer activity and mechanisms of action, following the hybrid pharmacophore strategy to enhance their therapeutic efficacy.
The present study was constructed into two pathways with the following two aims: (1) to use molecular hybridization to construct a new thiazole hybrid (HBHTP) (Fig. 1) and its metal(II) complexes (M = Co, Ni, Cu, Zn) to investigate their biological role in minimizing and diminishing cellular proliferation and induction of apoptotic mechanisms through cell cycle arrest, DNA content, P53 expression, and triggering apoptosis; and (2) to investigate the role of the synthesized thiazole hybrid and its metal complexes in inhibiting the gene expression of aromatase, sulfatase (Sulf1,2), and basic fibroblast growth factor (bFGF), which are the main factors that affect tumorigenesis, recurrence, and treatment resistance, and their ability to inhibit topoisomerase and form CT adducts through the metals.
 | ||
| Fig. 1 Design of thiazole hybrid by combining multiple pharmacophores. | ||
2 Materials and methods
The ESI† contains the specifications of the reagents and their commercial suppliers. Analytical grade organic solvents were supplied by ADWIC Chemical Reagent Co. Ltd (Cairo, Egypt). 2-(4-Chlorobenzylidene)hydrazine-1-carbothioamide (1) was obtained from a previous study. The experimental method used to prepare 2-hydroxyphenacyl bromide (2) is described in the ESI.† Silica gel plates were used for thin-layer chromatography (TLC) to monitor reactions. The ESI† provides a complete description of the devices used for the characterization of novel compounds.2.1. Synthesis of 2-(2-(2-(4-chlorobenzylidene)hydrazineyl)thiazol-4-yl)phenol (HBHTP, 3)
The ligand was prepared following a protocol adapted from Saydam and Yilmaz,22 however, with slight modifications. Briefly, thiosemicarbazone (1) (0.42 g, 2 mmol, 2.0 equiv.) anhydrous sodium acetate (0.66 g, 8 mmol, 8.0 equiv.) and ethanol (40 mL) were added to a 100 mL round-bottom flask (RBF). After stirring the content for 10 min at room temperature, 2-hydroxyphenacyl bromide (2) (0.43 g, 2 mmol, 2.0 equivalent) was added, and the reaction mixture was allowed to reflux for 6–8 h. The progress and completion of the reaction were monitored by TLC, using a mixture of chloroform/ethyl acetate (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the mobile phase. After cooling to ambient temperature, the precipitate was collected by filtration, washed with ethanol (3 × 5 mL), and dried under vacuum to yield HBHTP. The product was obtained as a faint yellow solid in an 81% yield. mp 221–223 °C. The details of the structural characterization of the ligand are provided in the ESI.†
:1) as the mobile phase. After cooling to ambient temperature, the precipitate was collected by filtration, washed with ethanol (3 × 5 mL), and dried under vacuum to yield HBHTP. The product was obtained as a faint yellow solid in an 81% yield. mp 221–223 °C. The details of the structural characterization of the ligand are provided in the ESI.†
2.2. General procedure for the preparation of metal complexes (4a–d)
The ligand was prepared following a protocol adapted from Saydam and Yilmaz,22 however, with slight modifications. Briefly, an ethanolic solution of the ligand (1.0 mmole HBHTP/10 mL EtOH) and metal(II) chlorides (1.0 mmole salt/10 mL EtOH) was refluxed for 5 h in the presence of three drops of an aqueous ammonia solution to produce the corresponding M(II) complexes (M = Co, Ni, Cu, Zn). After the allotted reflux time, the reaction mixture was allowed to cool to room temperature and the solid products were filtered out. The products were collected, washed three times with 3 mL ethanol, and dried overnight under vacuum at room temperature.2.3. In silico studies
Molecular docking studies were conducted following geometry optimization of the benzylidene–phenyl–thiazole (HBHTP) ligand and its copper complex (CuBHTP). The optimized compounds were subjected to an active-site molecular docking simulation after converting their file formats into PDBQT using the Open Babel software (version 2.3.2). The protein molecular target (topoisomerase I ID: 1T8I) was downloaded from the Protein Data Bank PDB (https://www.rcsb.org). The targeted protein was prepared by removing all heteroatoms and water molecules using the Biovia Discovery Studio software. SWISS-PDBVIEWER (version 4.1.0) (SPDBV) was used to minimize energy consumption and check for any missing parts of the selected receptor. AutoDock 4.2 software23 was used to implement the active-site molecular docking process. The process began with the incorporation of polar hydrogens into the targeted protein, followed by the inclusion of Kollman charges and the assignment of Gasteiger charges. The grid box was configured to cover the entire active site of the protein, spanning (60, 60, 60) points along the attributes (X = 23.255, Y = −5.866, Z = 30.659) with a spacing of 0.375. The docking parameters were managed using genetic algorithms, 50 conformations/poses were detected, and the best conformer was selected considering the lowest free energy of binding, which is represented by ligand-receptor non-covalent interactions (NCIs). LigPlus (LigPlot+ v.2.2.8) was used to study the hydrophobic interactions between docked drug-like molecules and the target protein. The best pose and the 3D and 2D images of the best conformers were visualized using Biovia Discovery Studio.2.4. Cytotoxicity and cell viability assay
The human breast cancer cell line MCF-7 and human breast epithelial cell line MCF10A were obtained from the VACSERA Tissue Culture Unit in Egypt. The MTT test was used to evaluate the in vitro cytotoxicity of the newly synthesized HBHTP ligand (3) and its complexes (4a–d) against human breast carcinoma (MCF-7) to induce endothelial cell death, in comparison with the clinical anticancer medication staurosporine (STP) as a positive control. MCF-7 and MCF10A cells were seeded in triplicate in 96-well Corning plates and treated with 50 μM of each compound. The MTT reagent was added 72 h post-treatment, followed by a 4 hour incubation period.24 The formazan crystals were then dissolved in 100 μL of dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm and 630 nm using a ROBONIK P2000 ELIZA multi-detection microplate reader. The effects of chemicals on cell viability were determined using the following equation:A: absorbance of the tested compound; A0: absorbance of the blank (no cells, positive control); ANC: absorbance of the negative control (only cells, no treatment).

2.5. Aromatase (CYP19A) activity assay kit (fluorometric)
The CuBHTP was tested using an in vitro aromatase inhibition assay. Aromatase inhibition was quantified by measuring the fluorescence intensity of fluorescein standards using an aromatase (CYP 19A) Activity Assay Kit (Biovision).25 Briefly, CuBHTP was preincubated with a reaction mixture containing the enzyme and substrate for 60 min at 37 °C. Subsequently, the fluorescence of the mixture was measured using a TECAN fluorescence spectrophotometer (1038708 Männedorf, Switzerland) at excitation and emission wavelengths of 488 nm and 527 nm, respectively.2.6. Enzyme-linked immunosorbent assay quantitative estimation of Hsp90 and P53
The effect of CuBHTP on HSP90α expression in MCF-7 cells was compared with that of the control group treated with novobiocin, a potent inhibitor of HSP90α that triggers DNA-triggered apoptosis in MCF-7 breast cancer cells, using a screening assay kit (BPS Bioscience catalog # 50317) and cell homogenates specifically designed for HSP90α (C-terminal) inhibition.Tumor suppressor protein P53 was assessed using an assay kit (NB 200-103). Homogenized cells were stored at −80 °C and centrifuged. The optical density was measured using a microplate reader at 450 nm. Average absorbance values were calculated for the standards, controls, and samples. A standard curve was plotted using the mean absorbance against concentration. The data were normalized to β-actin levels and expressed in ng mL−1 using a calibration curve from the Hsp90 dilutions. This was repeated twice using a ROBONIK P2000 ELISA READER (MIDC Industrial Area, Mahape, Navi Mumbai – 400710, India).
2.7. Tubulin β enzyme assay
In vitro assessment of β-tubulin in the tissue homogenate extracts was performed using a Simple-Step ELISA kit.26 The cells were grown in DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 mg per mL insulin (Sigma), and 1% penicillin–streptomycin. The cells were plated at a density of 1.2–1.8 × 10000 cells per well in 100 μL of complete growth medium supplemented with 100 μL of the compound per well in a 96-well plate and incubated for 24 h. Tubulin was estimated using Tecan-spark READER (1038708 Männedorf, Switzerland) at an excitation wavelength of 360 nm and an emission wavelength of 450 nm.
2.8. Annexin V-FITC apoptosis detection
MCF-7 cells were seeded at a density of 1.0 × 105 cells per well in 6-well microtiter plates. After 24 h, the HCPT and lycorine solutions were used at concentrations of 3, 6, and 12 μmol L−1. An equal volume of RPMI-1640 medium was added to the control group. The cells were cultured for 48 h at 37 °C in a CO2 incubator. The supernatant was discarded and the cells were resuspended in a solution containing 100000 cells and phosphate-buffered saline (PBS). The cells were then mixed with propidium iodide (PI) and Annexin V-FITC and analyzed by flow cytometry (BD Biosciences, 2350 Qume Drive, San Jose, California 95131, United States) in an ice bath. The cell suspension was filtered through a 300-mesh nylon mesh before detection at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.27
2.9. Propidium iodide flow cytometry kit for cell cycle analysis
The cell cycle phases of MCF-7 cells were examined using a propidium iodide (PI) flow cytometry kit (ab139418).28 The cells were washed with phosphate-buffered saline, fixed with 70% ethanol for 30 min, centrifuged, washed twice with PBS, and stained with propidium iodide (PI). Cells were treated with ribonuclease A and 400 μL PI solution per million cells and incubated for 5–10 minutes at room temperature. The labeled cells were assessed by flow cytometry in a solution containing PI and RNase A using a BD FACS Caliber instrument (2350 Qume Drive, San Jose, California 95131, United States).2.10. Quantitative real-time (qPCR) estimation
Reverse transcriptase (RT) was used to produce complementary DNA (cDNA) by quantitative real-time PCR. Total mRNA was extracted from MCF-7 cells using the SV Total RNA Isolation System. SYBR Green I was used for real-time PCR (Applied Biosystems Real-Time PCR Instruments) amplification using Applied Biosystems software version 3.1. ATG gene expression, internal control (GAPDH gene expression), and non-template control (water) cDNA samples were amplified in duplicate by qPCR. The master mix in the PCR tubes was thoroughly mixed, without bubbling.29 The sulfatase 1 SULF1: F5′-AGACAGCCACTCACCTCTTCAG-3, R-5′TTCTGCCAGTGCCTCTTTGCTG-3′; SULF2: F5′-CTAGCTAGCAAAAAAGAAGATGGGCCCCC-3′, R-5′-CGGGATCCTTAACCTTCCCAGCCTTCCC-3′; bFGF: 5′-TCTTCCTGCGCATCCACC-3′ and reverse 5′-TCAGCTCTTAGCAGACATTGGAAGA-3′; TOPI 5′-GAACAAGCAGCCCGAGGATGAT-3′ and 5′-TGCTGTAGCGTGATGGAGGCAT-3′; TOPII: 5′-GGTCAGTTTGGAACTCGGCTTC-3′ and 5′-AGGAGGTTGTCATCCACAGCAG-3′ and housekeeping genes (GAPDH) were analyzed, and the Ct values were noted on the PCR data sheet for each of these genes. A correlation between the expression of the internal control and target genes was observed, as was the evaluation of gene expression in the control sample.2.11. Statistical analyses
Measurements were performed in quadruplicate for each experiment. One-way and two-way ANOVA were used with a significance threshold of P < 0.05, to determine the significance of the differences between the experimental groups. Data are presented as mean ± standard deviation (SD). IC50 values were determined using GraphPad Prism (v9.0) and standard errors (SE) were obtained from repeated measurements. Furthermore, 95% confidence intervals (CI) were calculated to evaluate the accuracy of IC50 estimates.3 Results and discussion
3.1. Chemistry
A step-by-step protocol used to prepare the thiazolylphenol ligand (HBHTP, 3) and its metal complexes is shown in Scheme 1. 4-Chlorobenzaldehyde was initially employed as a precursor to generate 4-chlorobenzylidene thiosemicarbazone (2) via a Schiff base condensation reaction with thiosemicarbazide in ethanol under reflux. α-Bromination of 2-hydroxyacetophenone was performed by reacting it with a mixture of Br2 and CH3COOH under stirring at room temperature to obtain phenacyl bromide (2). The desired ligand (HBHTP, 3) was obtained via intermolecular cyclocondensation by refluxing the ethanolic mixture of thiosemicarbazone (1) and phenacyl bromide (2) in the presence of a catalytic amount of anhydrous AcONa. To prepare metal(II) BHTP complexes, HBHTP was refluxed with M(II)Cl2·xH2O in an ethanolic solution with the addition of ammonia as a deprotonating agent (Scheme 1).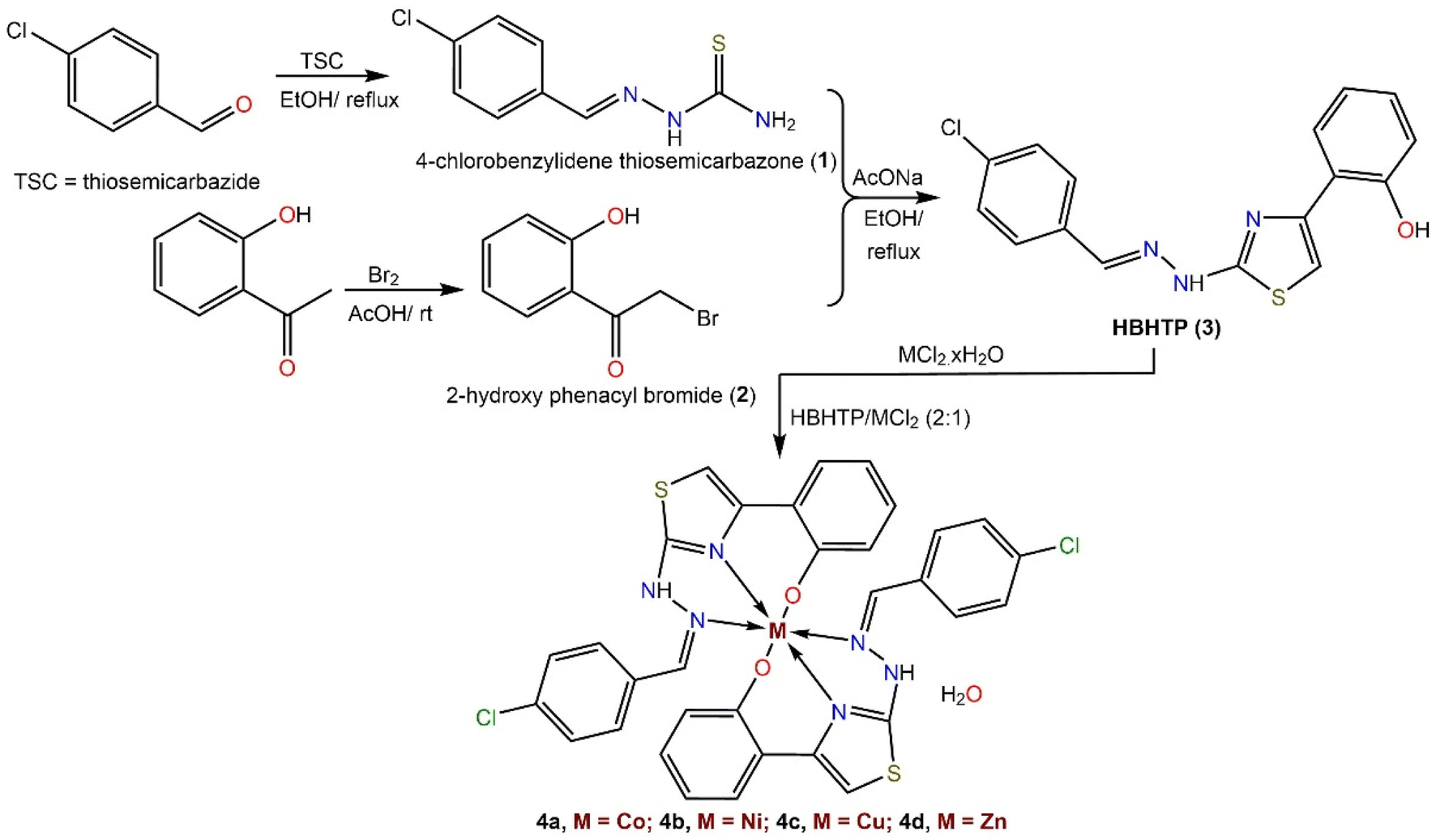 | ||
| Scheme 1 The step-by-step protocol used for the preparation of thiazolyl–phenol ligand (HBHTP, 3) and its complexes. | ||
3.2. Structural characterization
A new thiazolylphenol ligand (HBHTP, 3) and its M(II) complexes were obtained in reasonable yield. Consequently, the structures of the complexes were determined through elemental and spectral analyses, including FTIR, UV-vis, and magnetic measurements. The structures were compared with those of previously reported metal complex analogs (Table S1, ESI†). The elemental analysis results (CHNS) not only verified that the new compounds were formed successfully but also supported their proposed molecular structures.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N) and ν(N–N) stretching bands of the hydrazineyl group in the free ligand shifted to higher or lower frequencies than their free-state values of 1634 and 1525 cm−1, respectively, indicating that the HBHTP ligand was coordinated to the M(II) ions via the N atom.22 The IR spectrum of the ligand showed a medium band at 1601 cm−1 corresponding to its ν(CN) thiazole ring; this band was shifted to a lower frequency by approximately 13–25 cm−1 following complexation, indicating that it was modified upon coordination with metal ions. The band of the free ligand at 753 cm, assigned to ν(C–S–C), remained almost unchanged upon complexation, indicating that the sulfur atom was not involved in the coordination with the metal ions.32 Notably, the IR spectra of the complexes showed FTIR bands in the range – 441–491 cm−1, which corresponded to the stretching vibrations of ν(M–N). Other bands were observed between 669 and 696 cm−1 and were attributed to the stretching vibrations of ν(M–O).31 Overall, the IR spectral analysis indicated that the thiazole hybrid ligand (HBHTP, 3) functioned as a monoanionic (NNO) tridentate ligand.
N–N) and ν(N–N) stretching bands of the hydrazineyl group in the free ligand shifted to higher or lower frequencies than their free-state values of 1634 and 1525 cm−1, respectively, indicating that the HBHTP ligand was coordinated to the M(II) ions via the N atom.22 The IR spectrum of the ligand showed a medium band at 1601 cm−1 corresponding to its ν(CN) thiazole ring; this band was shifted to a lower frequency by approximately 13–25 cm−1 following complexation, indicating that it was modified upon coordination with metal ions. The band of the free ligand at 753 cm, assigned to ν(C–S–C), remained almost unchanged upon complexation, indicating that the sulfur atom was not involved in the coordination with the metal ions.32 Notably, the IR spectra of the complexes showed FTIR bands in the range – 441–491 cm−1, which corresponded to the stretching vibrations of ν(M–N). Other bands were observed between 669 and 696 cm−1 and were attributed to the stretching vibrations of ν(M–O).31 Overall, the IR spectral analysis indicated that the thiazole hybrid ligand (HBHTP, 3) functioned as a monoanionic (NNO) tridentate ligand.
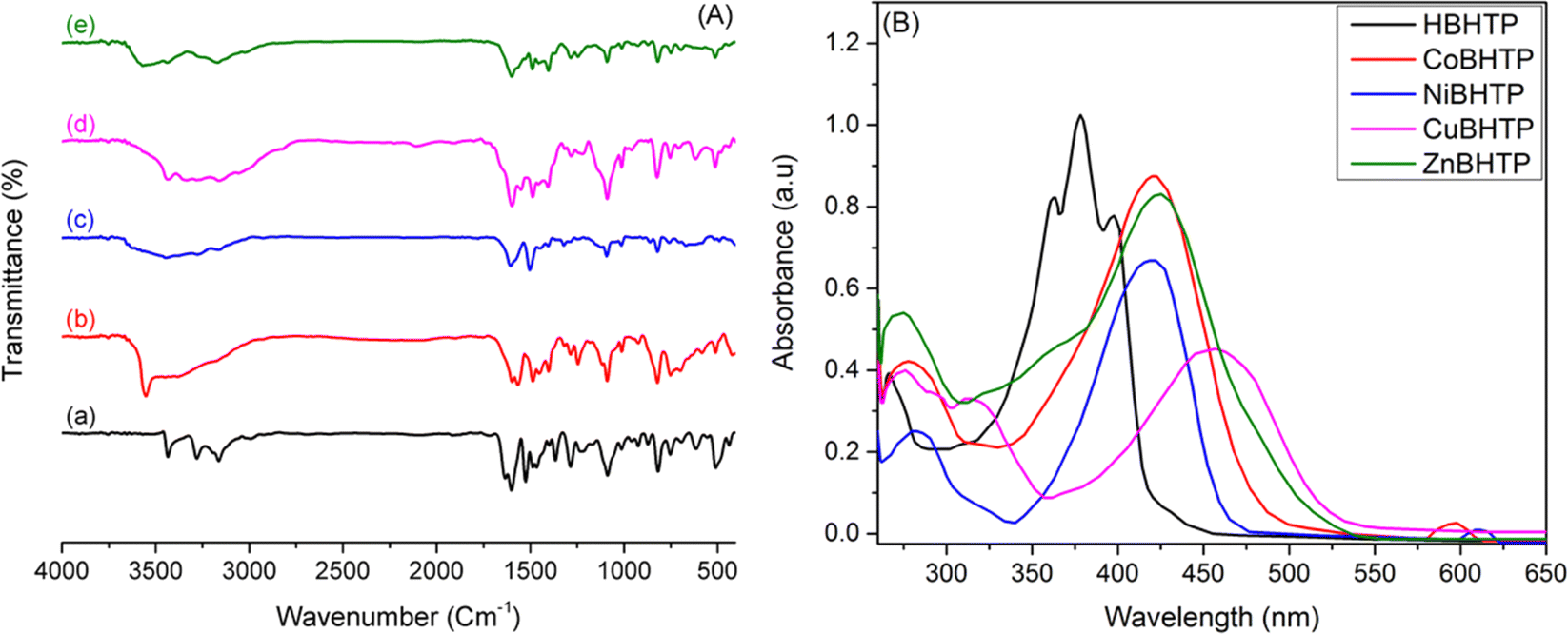 | ||
| Fig. 2 (A) FTIR spectra of (a) HBHTP, (b) CoBHTP, (c) NiBHTP, (d) CuBHTP, and (e) ZnBHTP; (B) UV-vis spectra of the HBHTP ligand and its complexes in DMSO solution. | ||
N chromophores of the thiazole ring and azomethine linkage, respectively.33 Bathochromic shifts were observed in these bands upon complexation, which is evidence of coordination between the thiazole ring and the azomethine group. The electronic spectrum of the CoBHTP complex exhibited a strong peak at 427 nm and a weak band at 599 nm, which can be attributed to the 4T1g(F) → 4A2g(P) and 4T1g(F) → 4T1g(P) transitions, respectively. It was hypothesized that the octahedral geometry of the CoBHTP complex is based on its electronic transition pattern, and the experimentally measured magnetic moment of 4.8 BM. The spectral bands at 612 and 417 nm in the NiBHTP complex are attributed to the 3A2g → 3T2g (F) and 3A2g → 3T1g(F) transitions, respectively. Furthermore, the hexa-coordinated form of the Ni complex is supported by a magnetic moment value of 2.9 BM, which correlates with the observed transitions.33 In the UV-vis spectrum of the CuBHTP complex, a significant band was detected at 455 nm, corresponding to the spin-allowed metal-to-ligand charge transfer (MLCT) d → π * transition, in addition to the characteristic ligand peaks. The observed blue shift aligns with previously reported studies.34,35 This transition is commonly observed in octahedral copper complexes and is accompanied by tailing towards higher wavelengths. Our hypothesis is supported by the fact that the experimentally measured magnetic moment of 1.5 BM was recorded, which also suggests the presence of a molecular interaction that may be created by a coordinating ligand.36,37UV-vis spectral monitoring (for 72 h) under physiological conditions was used to validate the physiological stability of the novel complexes. As depicted in Fig. S5 (ESI†), the UV-vis spectra of the CuBHTP complex did not exhibit any significant changes over the storage duration. This observation indicated the remarkable stability of this complex under physiological conditions. The absence of significant alterations in the absorption spectrum throughout the testing period demonstrated that the CuBHTP complex maintained its structural integrity and remained unaffected by time.
N moieties of the hydrazine fragments. Additionally, the singlet peak observed at δ 7.05 ppm could be attributed to the resonance of the phenolic proton. Finally, the set of signals observed in the chemical shift range of 8.26–6.86 ppm was assigned to the protons of the thiazolyl ring and the chlorobenzylidene moiety.3.3. In silico studies
 | ||
| Fig. 3 (A and B) Optimized geometrical structures of (A) the benzylidene–phenol–thiazole (HBHTP) ligand and (B) the copper complex (CuBHTP). (C and D) Molecular electrostatic potentials (MEP) of (C) HBHTP and (D) CuBHTP). | ||
Table S4 (ESI†) summarizes the outcomes of the natural bond orbital (NBOs) analysis for the free ligand and its complex to gain insight into the interaction strength. The results showed that the most negatively charged atoms in HBHTP were O22 = −0.683, N8 = −0.513, N12 = −0.409, and N13 = −0.214. These charges suggest the presence of electron-rich regions in which complexation occurs. The upper-center atom forms an octahedral sphere with N8, N13, and O22 of the two ligand molecules. Cu interacts with the N8 and N13 atoms via the P lone-pair orbitals. O22, which has a higher negative charge, participates in complexation and validates electron donation toward the N–Cu coordinate form. The coordination process did not involve N12, a secondary amine group, which may have contributed to intermolecular hydrogen bonding. The electron-rich sites of the complex were O1 = −0.343, O23 = −0.314, N21 = −0.277, N43 = −0.265, and N17 = −0.228.
Molecular electrostatic potential (MEP) was used to demonstrate the surface reactivity of HBHTP and CuBHTP. Fig. 3C and D show the significant reactive sections of the parent ligand (HBHTP) and its complex (CuBHTP), with yellow, orange, red, green, and blue indicating electronic potential values. Red denotes electron-rich (most negative) sites, whereas blue represents electron-poor (most positive) sites.38 The red curve in HBHTP comprises electron donors N8, N12, and N13, as confirmed by NBO analysis. Nucleophilic attack by N8, N13, and the orange-colored hydroxylic oxygen atom initiated a complex reaction. The oxygen atoms in the electron-rich CuBHTP complex were analyzed using NBOs analysis.
| Parameter | HBHTP | CuBHTP |
|---|---|---|
| a ΔE = ELUMO − EHOMO; ionization potential (I) = −EHOMO; electron affinity (A) = −ELUMO; chemical hardness (η) = (I − A)/2, chemical potential (μ) = −(I + A)/2, electronegativity (χ) = −(EHOMO + ELUMO)/2, electrophilicity index (ω) = μ2/2η, global softness (S) = 1/2η. | ||
| Total energy (a.u) | −1715.035 | −3625.335 |
| Dipole moment (debye) | 3.194 | 8.631 |
| EHOMO (eV) | −5.547 | −4.769 |
| ELUMO (eV) | −1.882 | −2.582 |
| ΔE (eV) | 3.665 | 2.187 |
| I (eV) | 5.547 | 4.769 |
| A (eV) | 1.882 | 2.582 |
| η (eV) | 1.832 | 1.094 |
| μ (eV) | −3.689 | −3.675 |
| S (eV) | 0.273 | 0.457 |
| χ (eV) | 3.714 | 3.675 |
| ω (eV) | 3.714 | 6.176 |
HBHTP exhibited a high ΔE value of 3.665 eV, suggesting excellent stability and the formation of a less reactive complex. In contrast, CuBHTP displayed a lower ΔE value of 2.187 eV, indicating a higher reactivity and lower stability. The values of I and A are related to EHOMO and ELUMO, respectively, indicating the ability of the compounds to donate or accept electrons. A comparison of CuBHTP and HBHTP revealed that CuBHTP exhibited a lower value (4.769 eV) and higher A value (2.582 eV) than HBHTP. This suggests that the CuBHTP has the potential to function as an electron donor. The global reactivity descriptors, namely, chemical hardness and global softness, depend on the parameters I and A, respectively. The compound CuBHTP exhibited a higher S value of 0.457 eV and a lower η value of 1.094 eV than HBHTP, indicating a higher reactivity.38 The electrophilicity index (ω) quantifies the change in energy of a system saturated with electrons. Fig. 4A and B show the energy values of the designed compounds. Fig. 4C and D show the HOMO and LUMO electronic orbital densities of the HBHTP and CuBHTP, respectively.
 | ||
| Fig. 4 (A and B) Description of the molecular orbitals with the calculated electronic transitions for (A) HBHTP and (B) CuBHTP. (C and D) Electronic distribution of FMOs for the optimized compounds (C) HBHTP (3), (D) CuBHTP (4c). | ||
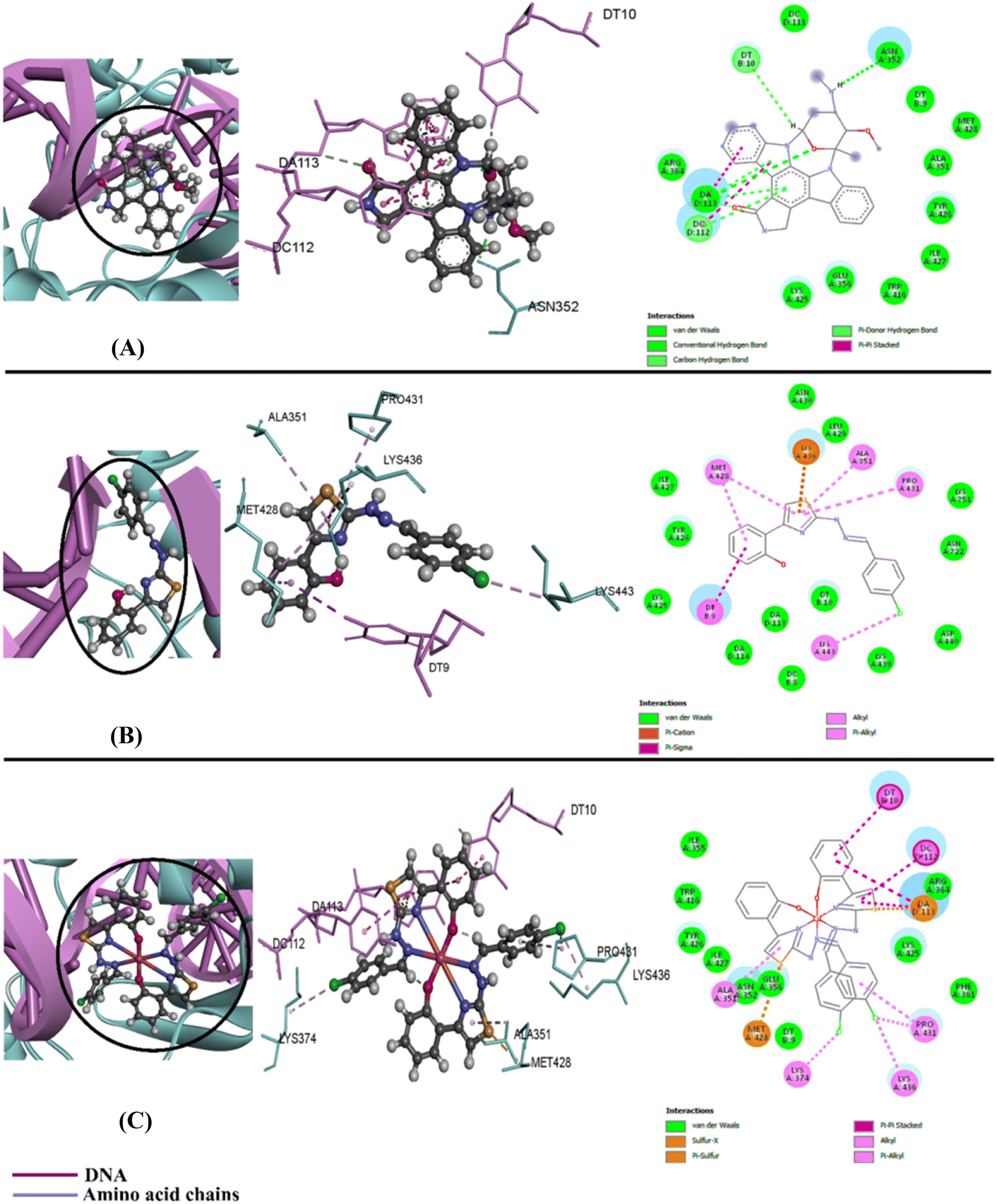 | ||
| Fig. 5 3D and 2D images of non-covalent interactions (NCIs) resulting from molecular docking of (A) staurosporine, (B) HBHTP, and (C) CuBHTP against the 1T8I receptor. | ||
The free HBHTP ligand predominantly interacts through van der Waals forces with hydrophobic amino acids, such as Ala351, Leu429, and Tyr426, which significantly contributes to the binding affinity by enhancing the molecular fit within the receptor pocket. Conversely, in addition to hydrophobic contacts with Ala35and Ile427, CuBHTP exhibited more robust hydrophobic interactions because of its coordination with the metal center, which induced additional π–π stacking with aromatic residues, such as Tyr426 and Trp416, thus increasing its overall stability when bound to TOPI. Interestingly, CuBHTP demonstrated an optimized design for optimal receptor occupancy and efficacy compared to the reference medication, which only showed hydrophobic interactions with Ala351 and Tyr426.
3.4. Cytotoxicity
The results revealed that the free ligand and its metal complexes exhibited varying degrees of cytotoxicities. Although the free ligand demonstrated moderate cytotoxicity, the metal complexes showed elevated levels of cytotoxicity, with the copper complex (CuBHTP, 4c) being the most cytotoxic against MCF-7 using IC50 (Fig. 6A–C). These findings suggest that the presence of metal ions (particularly Cu(II) ions) in the ligand structure enhances the cytotoxicity. The measured IC50 values, which represent the concentration of a compound required to inhibit cell growth by 50%, provided quantitative evidence of the cytotoxic effects of the ligands and their metal complexes. Therefore, CuBHTP exhibited the most significant cytotoxic effects on MCF7 cells compared to other metal ligands (Fig. 7A). | ||
| Fig. 6 (A and B) Dose-cytotoxicity correlation in terms of (A) optical density (O.D) and (B) MCF-7 viability; (C) the IC50 values (μg mL−1) of the new compounds against MCF-7 cell lines, as compared to a clinical drug (STP). | ||
 | ||
| Fig. 7 The impacts of the CuBHTP and staurosporine treatment on MCF7 and MCF10a cell lines. (A) The IC50, (B) the % of viable cells, mean ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
Interestingly, by comparing the IC50 values of the copper complex (CuBHTP) with those of its analogs (Table S6, ESI†), it was evident that the new complex exhibited a much stronger inhibitory effect on MCF-7 cell growth than the other complexes did. Therefore, a new Cu complex with enhanced cancer cell interactions may improve the anticancer activity in cancer research.
The efficacy of two treatments, CUBHTP and staurosporine (STP), were evaluated on MCF-10A (a normal epithelial breast cell line) and MCF-7 breast cancer cell lines, with potency measured as IC50 in μM, where lower values denote higher potency. CUBHTP exhibited an IC50 of approximately 40 μM in MCF-10A cells, indicating moderate activity, whereas its IC50 in MCF-7 cells was significantly lower, at approximately 10 μM, suggesting a higher potency with a statistically significant difference (P < 0.001). Conversely, STP demonstrated greater potency than CUBHTP in both cell lines, with an IC50 of approximately 10 μM in MCF-10A cells and an even lower IC50 of approximately 5 μM in MCF-7 cells, with a significant difference (P < 0.001) (Fig. 7A). Furthermore, the activity of STP in MCF-10A and MCF-7 cells showed no significant difference in activity between the two cell lines, lacking selectivity and revealing toxicity in both normal and cancer cell lines. Both treatments exhibited higher efficacy against MCF-7 cells, with statistically significant results compared with MCF-10A cells, underscoring their potential selectivity for MCF-7 cells. However, CUBHTP exhibited significant selectivity towards cancer cells compared to normal cells.
Additionally, treatment of MCF-10A cells with CUBHTP resulted in cell viability of approximately 70%, suggesting that this treatment effectively maintained a high proportion of viable cells, indicating lower toxicity or a protective effect of the treatment. In contrast, MCF-7 cells treated with CUBHTP exhibited a significantly reduced viability of approximately 40%, with a statistically significant difference (P < 0.001) compared to MCF-10A cells, highlighting CUBHTP's selective toxicity of CUBHTP toward MCF-7 cells while sparing MCF-10A cells. Conversely, STP treatment resulted in lower overall cell viability, with MCF-10A cells exhibiting approximately 40% and MCF-7 cells at approximately 30% viability, indicating that STP is more toxic to both cell lines. The difference in viability between MCF-10A and MCF-7 cells upon STP treatment was not significant (“ns”), indicating that STP did not exhibit selective toxicity in the two cell lines (Fig. 7B). Therefore, CUBHTP demonstrated a greater ability to protect MCF-10A cells (70% viability) than MCF-7 cells (40% viability), with a significant difference, whereas STP showed less protective capacity, with viabilities of 40% and 30% for MCF-10A and MCF-7 cells, respectively, with no significant selectivity. Thus, CUBHTP was more effective at maintaining MCF-10A cell viability when targeting MCF-7 cells. These findings are consistent with those of previous studies, indicating that the synthesized compound may serve as a targeted chemotherapeutic agent for tumor cells with chromosomal aberrations, developmental traits, and cancer-related expression characteristics.41 CuBHTP showed potential as a therapeutic agent against breast cancer MCF-7 cells while sparing non-tumorigenic MCF-10A cells, consistent with studies showing that compounds can selectively induce cytotoxicity in cancer cells through metabolic differences.42 STP exhibited non-selective toxicity, affecting both MCF-10A and MCF-7 cells, without significant differences in viability. This lack of selectivity may limit STP's therapeutic use of STP as it can damage healthy cells during chemotherapy.43 The differential response to CUBHTP may be attributed to the overexpression of receptors in MCF-7 cells targeted by the compound.44 Further studies are needed to elucidate CUBHTP's molecular targets of CUBHTP for clinical application.
Copper(II) complexes have emerged as promising anticancer agents owing to their redox activity, which generates reactive oxygen species (ROS) that target cancer cells.45 A copper(II) hydrazone complex showed an IC50 of 7.14 ± 0.05 μM against MCF-7 cells.46 Another copper(II) Schiff base complex had an IC50 of 10 μM with an SI of 2.19 The CuBHTP complex (IC50 5.2 μM, SI 4.2) shows enhanced potency through DNA binding and topoisomerase I inhibition by thiazole and phenol moieties. Cobalt(II), nickel(II), and zinc(II) complexes showed varied efficacies, with a cobalt(II) hydrazone complex showing an IC50 of 15 μM against MCF-7 cells.47 A nickel(II) salphen complex showed an IC50 of approximately 20 μM against MCF-7 cells, but demonstrated high selectivity for quadruplex DNA (>50-fold).48 Zinc(II) hydrazone complexes have IC50 values ranging from approximately 8 to 10 μM in MCF-7 cells, although they often lack selectivity.49
Cisplatin, a conventional platinum-based chemotherapeutic agent, shows an IC50 of 5.9–20 μM against MCF-7 cells. However, its clinical use is limited by its toxicity to normal cells (SI ∼1–2) and resistance development.19 CuBHTP exhibited a comparable or lower IC50 and higher SI values than cisplatin, indicating its advantage over cisplatin in terms of toxicity. Its multi-targeted mechanisms, including DNA adduct formation, topoisomerase I inhibition, and modulation of aromatase, SULF1,2, and bFGF gene expression, offer a broader scope than the DNA cross-linking mechanism of cisplatin. Compared to staurosporine, CuBHTP showed higher selectivity (SI: 4.2 vs. 2.5) and inhibited Hsp90 and tubulin β. CuBHTP demonstrated high potency (IC50 = 5.2 μM), selectivity (SI = 4.2), and multi-targeted activity, addressing toxicity and resistance. UV-vis spectroscopy confirmed the stability of CuBHTP in the assay medium, ensuring the activity of the intact complex.45 Notably, certain iridium(III) complexes with N-heterocyclic carbene ligands have achieved sub-micromolar IC50 values (0.3–4.8 μM) against K562 cells, surpassing the efficacy of cisplatin.50 Therefore, CuBHTP presents significant advantages over cisplatin, staurosporine, and other metal complexes because of its potency, selectivity, and multi-targeting mechanisms. These findings highlight the potential of the HBHTP platform in the development of novel anticancer agents.
3.5. Inhibitory effects of CuBHTP on aromatase (CYP19A)
Human aromatase (HA), which is key to estrogen synthesis, is a potential candidate for endocrine therapy of breast cancer. Aromatase inhibitors (AIs) block HA activity and reduce blood estrogen levels. AIs includes steroidal and nonsteroidal blockers, with steroidal blockers as natural HA substrates. Hormone therapy, which suppresses estrogen levels, is crucial in breast cancer treatment.51The results showed that CuBHTP substantially inhibited cytochrome 19A (CYP 19A) in MCF-7 cancer cells (P < 0.001), whereas letrozole significantly suppressed aromatase expression (P < 0.0001) (Fig. 8A). Although the activity of CuBHTP was lower than that of letrozole, it effectively suppressed aromatase with fewer adverse effects, meeting the clinical therapy needs. Understanding the structure of human aromatase could help in the development of innovative treatment candidates.52
 | ||
| Fig. 8 (A) Inhibitory activity of CuBHTP and letrozole on CYP19A; (B) inhibitory role of CuBHTP on Hsp90 in comparison to novobiocin; (C) upregulating effect on P53 expression in comparison to cisplatin, mean ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.6. Effect of CuBHTP on the quantitative expression of Hsp90 and P53
Moreover, MCF-7 cells showed high expression of Hsp90, and CuBHTP significantly reduced this expression (P < 0.001) compared to novobiocin, which reduced the expression (P < 0.01) (Fig. 8B). Condelli et al.53 revealed that the expression of heat shock proteins (HSPs), also known as molecular chaperones, increases in cells exposed to stressors, such as heat shock, chemical agents, and pathological abnormalities. According to Whitesell and Lindquist,54 the potential application of Hsp90 chaperones in cancer treatment has been emphasized, with the proposal that HSP90 inhibitors could be utilized as standalone therapies or in conjunction with other treatments. Furthermore, Condelli et al.53 found that HSP90α is crucial in cellular processes such as energy consumption, survival, and signalling, and its presence facilitates the invasion and migration of various cancer cells in both laboratory and living organisms.31,55CuBHTP elevated P53 expression in MCF7 cancer cells beyond the effects of cisplatin, leading to G1 phase arrest and reduced G2/M phase, apoptosis, and DNA repair (Fig. 8C). P53 mutations affect DNA binding and stability, thereby reducing the function associated with tumor development. Addressing these changes is crucial for effective tumor management.56 CuBHTP, a promising chemotherapeutic agent, restricts HSP90 expression in breast cancer cells, elevates thermoregulation, and augments P53 for disease eradication. Its role in apoptosis via P53 elevation and G1 phase arrest may involve Cu(II) and its active ligands. Alvarez et al.57 identified ligands with bi- and tri-thiazole groups in Cu complexes, showing C–H⋯Cl interactions that enabled the deformed copper cores to form binuclear copper complexes during crystallization. Reduced Hsp90 and increased p53 levels are associated with decreased RNA synthesis in cancer cells.58 Studies have shown that major chaperones, such as Hsp proteins, stabilize p53 by reducing its exposure to proteolytic enzymes in the proteasome.59
These chaperones are important targets for the anticancer activity of chemotherapeutic drugs and for cancer treatment. However, Hsps have been implicated in cancer progression and resistance to anticancer therapies.60 Under stress, Hsp synthesis increases, facilitating protein folding and suppressing hydrophobic structures associated with significant protein damage.61
3.7. Effect of CuBHTP on tubulin β enzyme activity expression
Analysis of the effects of MCF-7, CUBHTP, and colchicine on tubulin β levels (μg mL−1) (Fig. 9) showed that MCF-7 cells exhibited the highest expression of tubulin β at approximately 5 μg mL−1, whereas CUBHTP and colchicine showed lower levels (2 μg mL−1). Statistical analysis revealed significant differences between MCF-7 cells treated with both CUBHTP and colchicine (P < 0.001), whereas CUBHTP and colchicine showed no significant difference in their impact on tubulin β levels. The elevated expression of tubulin β in MCF-7 cells can be effectively suppressed by CUBHTP, as well as by colchicine. | ||
| Fig. 9 Role of CuBHTP on tubulin β enzyme levels in MCF7 cells, mean ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
This suggested that its activity may be attributed to its interaction with the tubulin/microtubule system and its ability to impede cell division. The antimitotic mechanism may involve binding to α/β-tubulin heterodimers or polymer.62 Colchicine binds to β-tubulin at the colchicine-binding site of the α–β-tubulin dimer interface, inhibits tubulin polymerization, and prevents microtubule assembly, thereby leading to mitotic arrest. Stanton et al.63 showed colchicine binds to β-tubulin with high affinity (Kd ≈ 1 μM), disrupting microtubule dynamics by stabilizing the tubulin dimer. This mechanism was confirmed by Ravelli et al.,64 who reported that CuBHTP inhibited tubulin β better than colchicine, which has toxicity limitations. Colchicine disrupts cellular processes in normal and cancerous cells by binding to β-tubulin, causing side effects, such as gastrointestinal toxicity, bone marrow suppression, and neurotoxicity. Finkelstein et al.65 The narrow therapeutic index of colchicine causes dose-limiting toxicity, with gastrointestinal effects in 80% of patients. Extended exposure can cause multiorgan failure, as noted by Nuki,66 in patients with renal or hepatic impairment.
Microtubules play a critical role in cellular processes such as cell shape, transportation, and chromosome segregation during mitosis and meiosis, forming mitotic spindles that are crucial for proper cellular function.67 Leandro-García et al.68 reported a high incidence of TUBB3 overexpression in brain cancer, other solid tumor types,69 and ovarian cancer.70,71 Consequently, chemotherapeutic agents that inhibit tumor cell microtubule synthesis and impede their function by blocking mitotic spindle fibers culminate in programmed cell death in both solid tumors and hematological cancers.46 Narvi et al.72 suggested that increased microtubule dynamics associated with TUBB3 could potentially result in resistance to drugs that target the microtubules.
3.8. Effect of CuBHTP on cell content and cell cycle phases
The effectiveness of the synthesized compounds in inhibiting MCF-7 cell growth depends on their ability to decrease DNA content and assess cell cycle progression through cell cycle arrest and cell growth inhibition in MCF-7 cells.73 MCF-7 cells were exposed to CuBHTP at a concentration equivalent to its IC50 value for 24 h. As shown in Fig. 10, CuBHTP treatment significantly reduced the number of MCF-7 cells, leading to a decrease in the proportion of cells in the G0/G1 phase from 61.43% to 55.12%, and an increase in the proportion of cells in the G2/M phase from 24.76% to 33.41%. Additionally, the proportion of cells in the S phase decreased from 13.803% to 11.47% compared with that of the untreated cells. | ||
| Fig. 10 Effect of CuBHTP on the cell cycle and apoptosis as determined by flow cytometry analysis. (I (A–F)) Cell cycle and (II (A)) DNA content and (B) Annexin V-FITC for apoptosis. | ||
The DNA content across different cell cycle phases (MCF7, CuBHTP/MCF7, NQO1, and STP/M) was used to assess the activity of CuBHTP and STP. In MCF7 cells, the DNA content distribution was 61.43% in G0/G1, 24.76% in S, and 13.81% in G2/M, indicating a high proportion of cells in G0–G1 phase, suggesting active cell division. For CuBHTP/MCF7 treated, the distribution shifted to 55.12% in G0/G1, 33.41% in S, and 11.47% in G2/M, showing a slight increase in G0/G1 and decrease in G2/M, which may indicate that CuBHTP induces partial arrest in G0/G1, slowing progression to G2/M (Fig. 10II(A)). Önem et al.74 revealed that purpurin combined with Cu(II) can cause DNA damage by generating ROS and forming DNA adducts through the Cu(I)/Cu(II) redox cycle, which may involve both mechanisms of carcinogenic effects. The effects of different treatments on MCF-7 cells were evaluated by measuring necrosis and early and late apoptosis using Annexin V-FITC staining. In the cancer control group, the distribution was 4.76% necrosis, 8.22% early apoptosis, and 15.97% late apoptosis, indicating minimal necrosis. In the CuBHTP/MCF7 treatment, necrosis decreased to 1.96%, early apoptosis 0.27%, and late apoptosis remained low at 0.14%, suggesting that CuBHTP induced a moderate increase in early apoptosis, with a slight increase in necrosis. For the STP/MCF7 treatment, necrosis was 2.2%, early apoptosis was 7.7%, and late apoptosis was 7.5%, showing a slight increase in necrosis and early apoptosis compared to the control, but with no significant late apoptosis (Fig. 10IIB). Overall, CuBHTP appeared to be effective in promoting early apoptosis and necrosis in MCF-7 cells, whereas both treatments had a limited effect on late apoptosis.
3.9. Quantitative real-time (qRT-PCR) estimation of gene expression
The effects of CuBHTP and staurosporine (STP) on MCF-7 cells were investigated for the expression of SULF1, SULF2, bFGF, TOPI, and TOPII. The treatment with CuBHTP resulted in a 1.2-fold increase. SULF1 and SULF2 showed fold changes of 0.7 and 0.5, with SULF2 reduction being significant (P < 0.001) and SULF1 less significant (P < 0.05). bFGF expression was decreased by 0.3-fold (P < 0.001). TOPI and TOPII showed 0.6-fold changes, with TOPI showing a significant reduction (P < 0.001), and TOPII showing moderate significance (P < 0.01) (Fig. 11A). CuBHTP significantly downregulated SULF2, bFGF, and TOPI expression, and had moderate effects on TOPII and SULF1 in MCF-7 cells. The downregulation of SULF2 and bFGF (P < 0.001) suggests interference with sulfatase activity and growth factor signalling and inhibiting proliferation and metastasis.75 The reduction in TOPI expression (P < 0.001) and TOPII (P < 0.01) implies that CuBHTP may inhibit DNA replication and transcription, akin to the action of topoisomerase inhibitors, such as camptothecin.76 These findings suggest the potential of CuBHTP as a multi-target cancer therapeutic agent. CuBHTP inhibited topoisomerase I (TOPI) and II (TOPII) in MCF-7 cells, as indicated by the reduced gene expression. This aligns with research where 4-benzoylthiosemicarbazides demonstrated TOPI and TOPII inhibition, with IC50 values of approximately 50 μM in MCF-7 cells.77 The mechanism of action of CuBHTP likely involves stabilization of the TOP–DNA complex, preventing DNA re-ligation, and inducing apoptosis. Treatment with STP of MCF-7 cells reduced SULF1 and SULF2 expression, with fold changes of 0.7 (P < 0.01) and 0.6 (P < 0.001), respectively. bFGF expression decreased by 0.4 (P < 0.001), whereas TOPI and TOPII showed fold changes of 0.6, with TOPI showing a significant reduction (P < 0.001) and TOPII showing moderate significance (P < 0.01) compared to MCF-7 cells (Fig. 11B). STP significantly downregulated SULF2, bFGF, and TOPI; moderately affected SULF1 and TOPII; and slightly upregulated MCF-7 cells. Staurosporine, a protein kinase inhibitor, exhibits broad-spectrum inhibitory effects. Downregulation of SULF2 and bFGF may disrupt sulfatase activity and growth factor signalling, potentially inhibiting cancer cell growth.78 The reduction in TOPI and TOPII expression is consistent with previous studies, indicating that staurosporine interferes with DNA replication and transcription.79 These results underscore STP's potential of STP as an anticancer agent and indicate that its non-selective nature could affect normal cells. Comparative analysis of the effects of CuBHTP and staurosporine (STP) on MCF-7 gene expression revealed their distinct activities. For SULF1, CuBHTP reduced its expression to approximately 0.7, whereas STP decreased it to approximately 0.6. Both compounds downregulated SULF2, with CuBHTP reducing it to ∼0.5, and STP reducing it to ∼0.6. bFGF was suppressed by both agents, with CuBHTP exhibiting a greater effect (∼0.3) than STP (∼0.4). For TOPI and TOPII, both compounds reduced the expression to ∼0.6. CuBHTP demonstrated strong or comparable inhibitory effects on SULF2, TOPIFGF, TOPI, and TOPII, whereas STP was more effective in suppressing SULF1. These findings suggested that CuBHTP may have a more targeted inhibitory profile than broader-acting STP. | ||
| Fig. 11 (A and B) Effect of CuBHTP and staurosporine treatment on SULF1, 2, and bFGF, TopI, II gene expression in MCF7 breast cancer cells, mean ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
Xu et al.80 stated that Sulf-1 plays a crucial role in regulating cell growth and death, making it a target for hepatocellular carcinoma therapy. Cancer cells initiate survival pathways. Inhibition of SULF1 and 2 suppresses growth factor signalling, contributing to chemotherapeutic activity.81,82 High SULF1 and SULF2 expression levels are correlated with poor prognosis with SULF1 priority dysregulated in HNSCC tissues and SULF2 promotes tumor growth by increasing fibroblast growth factor 7, which activates the PI3K/AKT and MAPK/ERK pathways to induce cell cycle progression.83,84
Topoisomerase I inhibits cell proliferation and causes cell cycle arrest in the G1 and G2 phases of the cell cycle. Oxocrebanine catalytically inhibits DNA degradation. The levels of TOPI and II are modulated by CuBHTP, and targeting these enzymes has anticancer potential. According to Wang and Tse-Dinh,2 TOP I and II inhibitors have limitations, including resistance and adverse effects. The authors have proposed dual inhibitors that target both enzymes. An additional aromatic ring enhances DNA intercalator affinity and increases cytotoxicity. Qian et al.85 found aminothiazonaphthalimide forms a DNA complex with anti-tumor activity in lung cancer cells. Bharti et al.86 reported copper(II) complexes cleave DNA via increased hydroxyl radical production, implying that DNA is largely free from RNA and proteins.
Benzylidene–phenol–thiazole (HBHTP) metal complexes, specifically CuBHTP, target multiple pathways in breast cancer via topoisomerase I (TOPI) inhibition. Topoisomerase II (TOPII) aids breast cancer progression via DNA replication,4 while TOPI resolves DNA supercoiling during transcription.87 TOPI inhibitors, such as camptothecin derivatives, are effective against metastatic breast cancer by stabilizing the TOPI–DNA cleavage complex and inducing DNA breaks and apoptosis.88 Molecular docking studies have shown that CuBHTP binds strongly to TOPI through thiazole and phenol moieties,77 complementing its DNA adduct (CT DNA adducts covalent modifications of DNA that occur when carcinogens or reactive chemicals bind to cytosine (C) and thymine (T) bases in DNA. These adducts can disrupt normal DNA replication and transcription, potentially leading to mutations, cancer or other genetic damage) formation, p53 upregulation, and Hsp90 and tubulin β inhibition. Targeting TOPI offers advantages over TOPII inhibitors, such as anthracyclines, which face resistance due to TOPII downregulation.89 CuBHTP significantly inhibited TOPI, coupled with G1 and S phase arrest and the upregulation of apoptotic markers, supporting its efficacy. Future research should explore the activity of CuBHTP against TOPII for its dual-targeting potential.4
The MCF-7 cell line (ER+/PR+/HER2−) was selected to evaluate the anticancer activity of M(II)–HBHTP complexes because of its widespread use in screening metal-based anticancer agents, enabling comparison with literature IC50 values.19 CuBHTP exhibited potent cytotoxicity against MCF-7 cells (IC50 = 5.2 ± 0.3 μM) with high selectivity (SI = 4.2 vs. MCF10A), indicating sensitivity to its mechanisms, including topoisomerase I (TOPI) inhibition. Gene expression analyses and molecular docking confirmed TOPI inhibition and downstream effects (p53 upregulation), validating the utility of MCF-7 cells despite their moderate TOPI expression.88 However, MCF-7 cells express lower levels of TOPI and TOPII than aggressive cell lines, such as MDA-MB-231, which may limit their relevance for TOP inhibitor studies.89 Therefore, targeting TOPI with CuBHTP is justified by its role in breast cancer, the docking-confirmed binding affinity of CuBHTP, and its potential to overcome TOPII inhibitor resistance in breast cancer cells. The MCF-7 cell line, despite moderate TOPI expression, was appropriate for the initial screening, highlighting the potential of CuBHTP as a TOPI-targeted anticancer agent and warranting further mechanistic and preclinical studies.
4 Conclusion
Chemotherapeutic research has shifted towards non-platinum-based compounds as anticancer medications with reduced toxicity. Copper compounds are of interest because of their endogenous nature and their DNA-damaging potential in humans. A novel ligand, the benzylidene–phenol–thiazole hybrid (HBHTP), was synthesized using a hybrid pharmacophore strategy. M(II) complexes were created and validated using FT-IR, UV-vis, and NMR spectroscopies. CuBHTP was the most active compound, exhibiting the lowest toxicity in MCF-10A cells. The copper complex exhibited superior cytotoxicity against MCF-7 breast cancer cells (IC50 = 5.2 μM) with high selectivity (SI = 4.2) compared to MCF10A cells, outperforming that of staurosporine (SI = 2.5). CuBHTP inhibited aromatase and reduced Hsp90 expression more effectively than novobiocin while upregulating P53 beyond the effects of cisplatin. It suppressed tubulin β, SULF1, SULF2, and bFGF gene expression, with significant reductions in SULF2, bFGF, and topoisomerase I (TOPI) expression (P < 0.001). CuBHTP induced cell cycle arrest in the G1 and S phases, reduced the DNA content, and promoted apoptosis (early: 8.22%; late: 15.97%). Molecular docking revealed that CuBHTP strongly binds to TOPI (−9.64 kcal mol−1), stabilizing TOPI–DNA complexes. Structural characterization validated the stability of the complexes, while in silico analyses supported CuBHTP reactivity. Compared to cisplatin and staurosporine, CuBHTP exhibited enhanced potency, selectivity, and multi-targeted activity. This study demonstrates the potential of CuBHTP as TOPIOP1-targeted chemotherapeutic agent, although it is limited by its in vitro scope and evaluation in MCF-7 and MCF10A cells. However, further in vivo studies are required to confirm this hypothesis.5 Study limitations
Although a study on benzylidene–phenol–thiazole (HBHTP) and its Cu(II) complex (CuBHTP) showed promising anticancer activity against MCF-7 cells, several limitations were observed. This study focused on in vitro cytotoxicity and molecular docking and did not include in vivo validation in animal models. Assessment of topoisomerase I expression is limited to molecular docking and gene expression data, without enzymatic activity assays. This study evaluated only the MCF-7 and MCF10A cell lines, limiting insights across cancer types. The mechanisms underlying the regulation of P53, Hsp90, tubulin β, SULF1,2, and bFGF have not yet been fully elucidated. Moreover, long-term effects on cell cycle arrest and resistance mechanisms have not been investigated. Future studies should address these gaps through in vivo experiments, using a broader range of cell lines, and mechanistic analysis. Future modifications to HBHTP, such as the incorporation of electron-donating groups, may further reduce the IC50 of CuBHTP, as suggested by the structure–activity relationship studies.Data availability
The data supporting the findings of this study are available upon request from the corresponding author. The data were not publicly available due to privacy or ethical restrictions.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors extend their appreciation to Umm Al-Qura University, Saudi Arabia for funding this research work through grant number: 25UQU4331270GSSR01.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- W. Wang and Y.-C. Tse-Dinh, Curr. Top. Med. Chem., 2019, 19, 730–740 CrossRef CAS PubMed.
- M. Will, J. Liang, C. Metcalfe and S. Chandarlapaty, Nat. Rev. Cancer, 2023, 23, 673–685 CrossRef CAS.
- Y. Pommier, ACS Chem. Biol., 2013, 8, 82–95 CrossRef CAS PubMed.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS PubMed.
- Y. Seol, H. Zhang, Y. Pommier and K. C. Neuman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16125–16130 CrossRef CAS PubMed.
- B. Zhao, P. Liu, T. Fukumoto, T. Nacarelli, N. Fatkhutdinov, S. Wu, J. Lin, K. M. Aird, H.-Y. Tang, Q. Liu, D. W. Speicher and R. Zhang, Nat. Commun., 2020, 11, 908 CrossRef CAS PubMed.
- I. Mancini, J. Vigna, D. Sighel and A. Defant, Molecules, 2022, 27, 4948 CrossRef CAS PubMed.
- H. Kantarjian, E. Jabbour, J. Grimley and P. Kirkpatrick, Nat. Rev. Drug Discov., 2006, 5, 717–718 CrossRef CAS PubMed.
- P.-C. Lv, K.-R. Wang, Y. Yang, W.-J. Mao, J. Chen, J. Xiong and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2009, 19, 6750–6754 CrossRef CAS PubMed.
- M. Anjalin, N. Kanagathara and A. R. B. Suganthi, Mater. Today: Proc., 2020, 33, 4751–4755 Search PubMed.
- S. K. Bharti, S. K. Patel, G. Nath, R. Tilak and S. K. Singh, Transition Met. Chem., 2010, 35, 917–925 CrossRef CAS.
- A. Rauf, M. K. Kashif, B. A. Saeed, N. A. Al-Masoudi and S. Hameed, J. Mol. Struct., 2019, 1198, 126866 CrossRef CAS.
- K. A. Scott, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2022, 65, 7044–7072 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- V. Thamilarasan, N. Sengottuvelan, A. Sudha, P. Srinivasan and G. Chakkaravarthi, J. Photochem. Photobiol., B, 2016, 162, 558–569 CrossRef CAS PubMed.
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- K. S. Kasprzak, F. W. Sunderman Jr and K. Salnikow, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2003, 533, 67–97 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- C. Hoppe, S. Kutschan, J. Dörfler, J. Büntzel, J. Büntzel and J. Huebner, Clin. Exp. Med., 2021, 21, 297–313 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, 1999 Search PubMed.
- S. Saydam and E. Yilmaz, Spectrochim. Acta, Part A, 2006, 63, 506–510 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- M. Y. Alfaifi, M. A.-E. Zein, A. A. Shati, M. A. Alshehri, S. E. I. Elbehairi, H. S. Hafez and R. F. M. Elshaarawy, J. Photochem. Photobiol., A, 2019, 385, 112083 CrossRef CAS.
- E. İnce Ergüç, S. Özcan Sezer and H. Gürer Orhan, Turk. J. Pharm. Sci., 2022, 19, 626–629 CrossRef PubMed.
- T. Zhu, S.-H. Wang, D. Li, S.-Y. Wang, X. Liu, J. Song, Y.-T. Wang and S.-Y. Zhang, Bioorg. Med. Chem. Lett., 2021, 37, 127698 CrossRef CAS PubMed.
- T. Xiaoling, M. A. Ashraf and Z. Yanyan, Pak. J. Pharm. Sci., 2016, 29, 2169–2172 Search PubMed.
- M. Roederer, Z. Darzynkiewicz and D. R. Parks, Methods Cell Biol., 2004, 75, 241–256 CrossRef PubMed.
- S. A. Bustin, AZ of Quantitative PCR, International University Line, La Jolla, CA, 2004, vol. 5 Search PubMed.
- M. Y. Alfaifi, S. E. I. Elbehairi, H. S. Hafez and R. F. M. Elshaarawy, J. Mol. Struct., 2019, 1191, 118–128 CrossRef CAS.
- S. E. I. Elbehairi, M. Y. Alfaifi, A. A. Shati, M. A. Alshehri, R. F. M. Elshaarawy and H. S. Hafez, Chem. Biol. Drug Des., 2020, 96, 1148–1161 CrossRef CAS PubMed.
- R. F. M. Elshaarawy, T. B. Mostafa, A. A. Refaee and E. A. El-Sawi, RSC Adv., 2015, 5, 68260–68269 RSC.
- S. Abubakar, G. A. Shallangwa and I. Abdulkadir, J. Umm. Al-qura Univ. Appl. Sci., 2024, 10, 613–623 CrossRef.
- A. A. Alzharani, J. Umm. Al-qura Univ. Appl. Sci., 2023, 9, 455–470 CrossRef.
- E. Chiyindiko, E. H. G. Langner and J. Conradie, Molecules, 2022, 27 DOI:10.3390/molecules27186033.
- A. A. Awaji, H. W. Alhamdi, K. M. Alshehri, M. Y. Alfaifi, A. A. Shati, S. E. I. Elbehairi, N. A.-F. Radwan, H. S. Hafez, R. F. M. Elshaarawy and M. Welson, J. Inorg. Biochem., 2025, 262, 112720 CrossRef CAS PubMed.
- R. F. M. Elshaarawy and C. Janiak, Arabian J. Chem., 2016, 9, 825–834 CrossRef CAS.
- H. A. Elbadawy, A. E. Ali, A. A. Elkashef, S. Foro and D. S. El-Sayed, Appl. Organomet. Chem., 2022, 36, e6793 CrossRef CAS.
- G. T. Tigineh, M. Abiye, A. Melese and A. Abebe, Chem. Biol. Drug Des., 2023, 101, 479–488 CrossRef CAS PubMed.
- A. Lauria, M. Ippolito and A. M. Almerico, J. Mol. Model., 2007, 13, 393–400 CrossRef CAS PubMed.
- X. Zheng, X. Gai, S. Han, C. D. Moser, C. Hu, A. M. Shire, R. A. Floyd and L. R. Roberts, Genes, Chromosomes Cancer, 2013, 52, 225–236 CrossRef CAS PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef CAS PubMed.
- A. Burguin, C. Diorio and F. Durocher, J. Pers. Med., 2021, 11, 808 CrossRef PubMed.
- B. Kinnel, S. K. Singh, G. Oprea-Ilies and R. Singh, Cancers, 2023, 15, 1320 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS.
- P. T. Lim, B. H. Goh and W.-L. Lee, 3 - Taxol: Mechanisms of action against cancer, an update with current research, Paclitaxel, ed. M. K. Swamy, T. Pullaiah and Z.-S. Chen, Academic Press, 2022, pp. 47–71, ISBN 9780323909518, DOI:10.1016/B978-0-323-90951-8.00007-2.
- P. Kumari, B. Ghosh and S. Biswas, J. Drug Targeting, 2016, 24, 179–191 CrossRef CAS PubMed.
- R. Paprocka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Coord. Chem. Rev., 2022, 452, 214307 CrossRef CAS.
- S.-R. Li, Y.-M. Tan, L. Zhang and C.-H. Zhou, Pharmaceutics, 2023, 15, 1348 CrossRef CAS PubMed.
- P. Štarha, Inorg. Chem. Front., 2025, 12, 897–954 RSC.
- G. Zucchini, E. Geuna, A. Milani, C. Aversa, R. Martinello and F. Montemurro, Int. J. Women's Health, 2015, 7, 551–563 Search PubMed.
- W. L. Miller, Trends Endocrinol. Metab., 2017, 28, 771–793 CrossRef CAS PubMed.
- V. Condelli, F. Crispo, M. Pietrafesa, G. Lettini, D. S. Matassa, F. Esposito, M. Landriscina and F. Maddalena, Cells, 2019, 8, 532 CrossRef PubMed.
- L. Whitesell and S. L. Lindquist, Nat. Rev. Cancer, 2005, 5, 761–772 CrossRef CAS PubMed.
- R. Bershawy, H. S. Hafez, S. S. El-Sakka, A. Hammad and M. H. Soliman, J. Biomol. Struct. Dyn., 2023, 42(23), 12885–12899 CrossRef.
- X. Wang, X. Song, W. Zhuo, Y. Fu, H. Shi, Y. Liang, M. Tong, G. Chang and Y. Luo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21288–21293 CrossRef CAS PubMed.
- N. Alvarez, F. Velluti, F. Guidali, G. Serra, M. G. Kramer, J. Ellena, G. Facchin, L. Scarone and M. H. Torre, Inorg. Chim. Acta, 2020, 508, 119622 CrossRef CAS.
- S. Zhang, C. Wang, B. Ma, M. Xu, S. Xu, J. Liu, Y. Tian, Y. Fu and Y. Luo, Cell Rep., 2020, 32(1), 107879 CrossRef CAS PubMed.
- L. Dubrez, S. Causse, N. Borges Bonan, B. Dumétier and C. Garrido, Oncogene, 2020, 39, 516–529 CrossRef CAS PubMed.
- S. Yang, H. Xiao and L. Cao, Biomed. Pharmacother., 2021, 142, 112074 CrossRef CAS PubMed.
- B. J. Lang, M. E. Guerrero, T. L. Prince, Y. Okusha, C. Bonorino and S. K. Calderwood, Arch. Toxicol., 2021, 95, 1943–1970 CrossRef CAS PubMed.
- B. C. Parker, M. Engels, M. Annala and W. Zhang, J. Pathol., 2014, 232, 4–15 CrossRef CAS PubMed.
- E. T. Rosolowsky, J. Skupien, A. M. Smiles, M. Niewczas, B. Roshan, R. Stanton, J. H. Eckfeldt, J. H. Warram and A. S. Krolewski, J. Am. Soc. Nephrol., 2011, 22, 545–553 CrossRef CAS PubMed.
- R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS PubMed.
- Y. Finkelstein, S. E. Aks, J. R. Hutson, D. N. Juurlink, P. Nguyen, G. Dubnov-Raz, U. Pollak, G. Koren and Y. Bentur, Clin. Toxicol., 2010, 48, 407–414 CrossRef CAS PubMed.
- G. Nuki, Curr. Rheumatol. Rep., 2008, 10, 218–227 CrossRef CAS PubMed.
- E. R. Smith, J.-Q. Wang, D.-H. Yang and X.-X. Xu, Drug Resistance Updates, 2022, 65, 100881 CrossRef PubMed.
- L. J. Leandro-García, S. Leskelä, I. Landa, C. Montero-Conde, E. López-Jiménez, R. Letón, A. Cascón, M. Robledo and C. Rodríguez-Antona, Cytoskeleton, 2010, 67, 214–223 CrossRef PubMed.
- M. C. Tsourlakis, P. Weigand, K. Grupp, M. Kluth, S. Steurer, T. Schlomm, M. Graefen, H. Huland, G. Salomon, T. Steuber, W. Wilczak, H. Sirma, R. Simon, G. Sauter, S. Minner and A. Quaas, Am. J. Pathol., 2014, 184, 609–617 CrossRef CAS PubMed.
- G. Ferrandina, G. F. Zannoni, E. Martinelli, A. Paglia, V. Gallotta, S. Mozzetti, G. Scambia and C. Ferlini, Clin. Cancer Res., 2006, 12, 2774–2779 CrossRef CAS PubMed.
- Y. Koh, B. Jang, S.-W. Han, T.-M. Kim, D.-Y. Oh, S.-H. Lee, C. H. Kang, D.-W. Kim, S.-A. Im, D. H. Chung, Y. T. Kim, T.-Y. Kim, Y.-W. Kim, J. H. Kim, D. S. Heo and Y.-J. Bang, J. Thorac. Oncol., 2010, 5, 320–325 CrossRef PubMed.
- E. Narvi, K. Jaakkola, S. Winsel, C. Oetken-Lindholm, P. Halonen, L. Kallio and M. J. Kallio, Br. J. Cancer, 2013, 108, 82–90 CrossRef CAS PubMed.
- D. Kaminskyy, A. Kryshchyshyn and R. Lesyk, Eur. J. Med. Chem., 2017, 140, 542–594 CrossRef CAS PubMed.
- A. N. Önem, K. Sözgen Başkan and R. Apak, ACS Omega, 2023, 8, 5103–5115 CrossRef PubMed.
- C. Zhu, L. He, X. Zhou, X. Nie and Y. Gu, Oncol. Rep., 2016, 35, 1318–1328 CrossRef CAS PubMed.
- S. Mastrangelo, G. Attina, S. Triarico, A. Romano, P. Maurizi and A. Ruggiero, Biomed. Pharmacol. J., 2022, 15, 553–562 CAS.
- J. L. Delgado, C.-M. Hsieh, N.-L. Chan and H. Hiasa, Biochem. J., 2018, 475, 373–398 CrossRef CAS.
- M. Tenhagen, P. J. Van Diest, I. A. Ivanova, E. Van Der Wall and P. Van Der Groep, Endocr.-Relat. Cancer, 2012, 19, 115–129 Search PubMed.
- M. W. Karaman, S. Herrgard, D. K. Treiber, P. Gallant, C. E. Atteridge, B. T. Campbell, K. W. Chan, P. Ciceri, M. I. Davis and P. T. Edeen, Nat. Biotechnol., 2008, 26, 127–132 CrossRef CAS PubMed.
- G. Xu, W. Ji, Y. Su, Y. Xu, Y. Yan, S. Shen, X. Li, B. Sun, H. Qian, L. Chen, X. Fu, M. Wu and C. Su, Oncotarget, 2014, 5, 5029–5039 CrossRef PubMed.
- W. Ji, J. Yang, D. Wang, L. Cao, W. Tan, H. Qian, B. Sun, Q. Qian, Z. Yin, M. Wu and C. Su, PLoS One, 2011, 6, e23274 CrossRef CAS PubMed.
- H. Liu, X. Fu, W. Ji, K. Liu, L. Bao, Y. Yan, M. Wu, J. Yang and C. Su, Hepatol. Res., 2013, 43, 516–525 CrossRef CAS PubMed.
- D.-Y. Liu, X.-M. Shen, F.-F. Yuan, O.-Y. Guo, Y. Zhong, J.-G. Chen, L.-Q. Zhu and J. Wu, Mol. Neurobiol., 2015, 52, 1467–1476 CrossRef CAS PubMed.
- T. Jian, Z. H. Chen, Z. Chen and D. Tan, Braz. J. Med. Biol. Res., 2020, 53(2), 8901 CrossRef.
- X. Qian, Z. Li and Q. Yang, Bioorg. Med. Chem., 2007, 15, 6846–6851 CrossRef CAS PubMed.
- S. K. Bharti and S. K. Singh, Med. Chem. Res., 2014, 23, 1004–1015 CrossRef CAS.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section (materials, spectral data, and computational analysis), tables (Tables S1–S6), and figures (Fig. S1–S4). See DOI: https://doi.org/10.1039/d5ra00889a |
| This journal is © The Royal Society of Chemistry 2025 |