
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Europium(III)/terbium(III) mixed metal–organic frameworks and their application as ratiometric thermometers with tuneable sensitivity in organic dispersion†
Madhura Joshi‡
a,
Maurizio Riesner‡ b,
Zhuang Wang‡c,
Sahba Mireskandaria,
Raju Nandaa,
Rebecca Elfriede Rebera,
Christoph Hubera,
Marcus Fischera,
Rachel Fainblatb,
Karl Mandelde,
Dorothea Wissera,
Doris Segets*cf,
Gerd Bacher*bf,
Florian M. Wisser*ag and
Martin Hartmann*ag
b,
Zhuang Wang‡c,
Sahba Mireskandaria,
Raju Nandaa,
Rebecca Elfriede Rebera,
Christoph Hubera,
Marcus Fischera,
Rachel Fainblatb,
Karl Mandelde,
Dorothea Wissera,
Doris Segets*cf,
Gerd Bacher*bf,
Florian M. Wisser*ag and
Martin Hartmann*ag
aErlangen Center for Interface Research and Catalysis (ECRC), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: florian.wisser@fau.de; martin.hartmann@fau.de
bWerkstoffe der Elektrotechnik, University of Duisburg-Essen (UDE), Carl-Benz-Straße 199, 47057 Duisburg, Germany. E-mail: gerd.bacher@uni-due.de
cInstitute for Energy and Materials Processes – Particle Science and Technology (EMPI-PST), University of Duisburg-Essen (UDE), Carl-Benz-Straße 199, 47057 Duisburg, Germany. E-mail: doris.segets@uni-due.de
dDepartment of Chemistry and Pharmacy, Inorganic Chemistry, Friedrich-Alexander Universität Erlangen-Nürnberg (FAU), Egerlandstrasse 1, 91058 Erlangen, Germany
eFraunhofer-Institute for Silicate Research ISC, Neunerplatz 2, 97082 Würzburg, Germany
fCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen (UDE), Germany
gErlangen Center for Functional Particle Systems (FPS), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Haberstr. 9a, 91058 Erlangen, Germany
First published on 9th April 2025
Abstract
The ability to engineer on a molecular level luminescent metal–organic frameworks (MOFs) enables the design of well-performing ratiometric, i.e., self-referencing, temperature sensors. Lanthanide-based MOFs stand out as luminescent temperature sensors due to their high luminescence intensity and the sharp emission lines of the lanthanides. The use of two different lanthanide cations, incorporated into the same MOF structure, is supposed to enable ratiometric temperature sensing. Herein, we present a series of mixed-metal EuxTb(1−x)BTC, in which the metal ions are homogeneously dispersed, as demonstrated by 1H solid state NMR spectroscopy. The EuxTb(1−x)BTC series shows controllable luminescent properties, which depend on the solvation of the lanthanide. The two MOFs in the series with the lowest Eu contents, namely Eu0.05Tb0.95BTC and Eu0.02Tb0.98BTC, are suitable candidates for ratiometric temperature sensing, achieving sensitivities of up to 2.0% K−1. As the fluorescence is affected by the presence of solvents, simultaneous ratiometric temperature and solvent sensing is possible with remarkable high thermal sensitivities of ca. 0.1% K−1 and ca. 0.2% K−1 for dispersions of Eu0.02Tb0.98BTC in acetonitrile and ethanol, respectively.
Introduction
In the field of chemical sensing, metal–organic frameworks (MOFs) have attracted much attention as they combine the advantages of high surface area and pore volume as well as crystallinity with tuneable structures and functionalities.1,2 Thus, MOFs have found applications in easy-to-read optical sensors,3–5 as well as in electrochemical sensing6,7 and fluorescent sensing.2,8–12 MOF-based fluorescence sensors stand out compared to other materials due to their modular synthesis, allowing adjustment of the MOF structure and chemical composition and, thus, tailoring of its optical properties. This has in the past allowed the design of fluorescence sensors with high target selectivity as well as high sensitivities and low limits of detection.12 MOF-based fluorescence sensors have found applications in various fields, including toxic metal ion detections in wastewater, gas and volatile organic compounds detection, but also for determination of physical properties such as pH, humidity and temperature.2,8–17To enable ratiometric sensing, i.e. self-calibrating sensing without the use of an external reference, at least two different luminescent centres are required.10 This can include two different metal complexes, e.g. two different lanthanides, two different types of quantum dots (QDs), or a combination of a QD and a metal complex. Lanthanide-based (Ln) MOFs are attracting increasing attention due to their good luminescence intensity and the sharp emission lines of the lanthanides.8,18 Thus, from two or more transitions, originating from the different emissive centres within the same material, the sensor signal is determined optically as the ratio between the probe-dependent emission intensities. In case of temperature sensing, the nature of the MOF, i.e., the combination of metal and organic linker, mainly determines the temperature range which can be accurately measured. MOFs as ratiometric temperature sensors might offer large potential as an in situ probe to detect local temperature changes during a given application, e.g. in biological applications and nanomedicine, in catalytic reactions or in adsorption processes.16 However, precise understanding of the temperature sensing under different conditions is required. Though it is known that the luminescence in lanthanide 1,3,5-benzene tricarboxylate (LnBTC) MOFs strongly depends on the experimental conditions, e.g. the presence of a solvent,19–21 systematic studies that allow understanding the interplay between the MOF material and its functional environment are yet rare.21
With regard to particle size and shape, most of the known pure Ln-containing MOFs show a preferred crystallization along one crystallographic direction leading typically to micrometer-sized, needle-shaped crystals.22–24 While this is very attractive for crystal structure determination, it may hamper their application in areas where size becomes important, such as in contrasting agents in bio-imaging, or in thin films. A prominent technique to control the size and shape of MOF crystals is the use of modulators. In their seminal work, Guo et al. reported the use of sodium acetate (NaOAc) as a suitable capping agent to reduce the particle size without affecting the crystal structure and properties of LnBTC MOFs.25 While the original protocol allowed for the synthesis of rod-like LnBTC of several tens of μm in length,8,25 the protocol reported by Guo et al.25 lead to the synthesis of almost spherical particles with diameters below 100 nm. Those LnBTC (nano)particles have found widespread application in luminescent sensing, including small molecule detection,20,26 and ratiometric temperature sensing,27 or in luminescent thin films.25,28–30 However, detailed understanding of Ln incorporation in mixed LnBTCs, and the impact of solvation on ratiometric temperature sensing remain yet unsolved.
Here we report the investigation of the ratiometric temperature sensing in a series of mixed europium and terbium BTC MOFs, labelled as EuxTb(1−x)BTC. The homogeneous incorporation of both cations was demonstrated by 1H MAS NMR spectroscopy, as the 1H chemical shift of the linker is sensitive to the presence of both lanthanides. Using the NaOAc modulator approach, the EuxTb(1−x)BTC series was obtained as nanoparticulate systems (nanorods), enabling their use not only in the dry solid state, but also dispersed in various solvents in the colloidal state. Temperature sensing was established on dry powders of Eu0.05Tb0.95BTC and Eu0.02Tb0.98BTC. Finally, we demonstrate the potential of dual emissive Eu/Tb MOFs towards simultaneous ratiometric temperature and solvent sensing. To this end, we have studied the temperature-dependent luminescence of Eu0.02Tb0.98BTC dispersed in ethanol or acetonitrile as model organic solvents. The emission intensities and the ratio between the Eu and Tb emission depend on both the temperature and the solvation of the MOF, thus allowing simultaneous temperature and solvent determination.
Results and discussion
Mixed-metal Eu/Tb BTC MOFs
The PXRD patterns of the as-synthesised EuxTb(1−x)BTC MOFs (x = 0.0 to 1.0) are very similar and show the characteristic peaks of the parent EuBTC19 and [TbBTC(H2O)]·(DMF)(H2O0.5) (Fig. 1a and S1†).31 The high similarity of the patterns confirms that a series of isostructural MOFs was obtained. As the materials were obtained as fine powders (Fig. S2†), they exhibit rather broad peaks. Thus, small changes in lattice parameters cannot be observed, which might be expected when gradually replacing Eu with Tb. In the series the estimated crystallite sizes, as obtained using the Scherrer equation, vary between 28 (TbBTC) and 46 nm (Eu0.02Tb0.98BTC, see Table S1†). All materials in the EuxTb(1−x)BTC series were obtained as needle-shaped nanorods (Fig. S3†). For the two most interesting materials, Eu0.05Tb0.95BTC and Eu0.02Tb0.98BTC (see below), the characteristic length of the particles is below 500 nm, with average lengths of 125 and 225 nm, respectively (Fig. S2 and S3†). | ||
| Fig. 1 (a) PXRD patterns of as-synthesised EuxTb(1−x)BTC MOFs. Bragg marker for [EuBTC (H2O)]·(H2O)1.5 (red, CCDC 617492 (ref. 19)) and [TbBTC(H2O)]·(DMF)(H2O)0.5 (green31). (b) TGA recorded under synthetic air (2 K min−1 heating rate), (c) IR spectra of EuxTb(1−x)BTC MOFs (same colour code as in (a)) together with the IR spectrum of H3BTC (dark grey) and NaOAc (light grey) and (d) N2 physisorption isotherms recorded at 77 K (same colour code as in (a)). Closed symbols denote the adsorption, open symbols indicate the desorption branch. | ||
The thermal stability and decomposition behaviour of all materials is very similar, as observed by thermogravimetric analysis (TGA) in air. Between room temperature and 275 °C, desorption of remaining ethanol, water and/or DMF occur, while network decomposition only starts above 350 °C. In line with the expected values for the two pure phases, the relative mass loss above 350 °C changes from approx. 49% in case of EuBTC to approx. 47% in case of TbBTC. At 600 °C Eu2O3 and Tb4O7 are obtained,32 at ∼1000 °C, Tb4O7 is deoxygenised yielding Tb2O3 (Fig. S4†).32,33 The results from TGA, combined with results from inductively coupled plasma optical emission spectroscopy (ICP OES), are thus indicative for an almost perfect 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between lanthanides and BTC. Indeed, using ICP OES, only traces of sodium (<1%, Table 1) are detected.
:1 stoichiometry between lanthanides and BTC. Indeed, using ICP OES, only traces of sodium (<1%, Table 1) are detected.
| Material | ωEua/wt% | ωTba/wt% | ωNaa/wt% | SBETb/m2 g−1 | Vpc/cm3 g−1 |
|---|---|---|---|---|---|
| a Determined by ICP OES analysis.b Determined from N2 physisorption experiments at 77 K, at 0.05 < p/p0.c Determined from N2 physisorption experiments at 77 K, at 0.99 p/p0. | |||||
| EuBTC | 31.88 ± 0.15 | — | 0.49 ± 0.01 | 410 | 0.24 |
| Eu0.75Tb0.25BTC | 23.09 ± 0.61 | 8.16 ± 0.05 | 0.60 ± 0.02 | 450 | 0.32 |
| Eu0.51Tb0.49BTC | 15.84 ± 0.91 | 15.81 ± 0.11 | 0.39 ± 0.11 | 630 | 0.31 |
| Eu0.25Tb0.75BTC | 7.79 ± 0.30 | 23.87 ± 0.19 | 0.22 ± 0.04 | 510 | 0.32 |
| Eu0.05Tb0.95BTC | 1.47 ± 0.08 | 33.41 ± 2.85 | 0.46 ± 0.07 | 500 | 0.28 |
| Eu0.02Tb0.98BTC | 0.62 ± 0.04 | 34.41 ± 0.66 | 0.38 ± 0.06 | 510 | 0.22 |
| TbBTC | — | 31.77 ± 0.24 | 0.18 ± 0.03 | 510 | 0.20 |
The almost perfect stoichiometry is further evidenced by IR spectroscopy. The vanishing of the characteristic υas(COO) vibration of H3BTC around 1720 cm−1 in all MOFs of the series highlights that hardly any missing cluster defect is present (Fig. 1c). Such a defect would give rise to only partially deprotonated H3BTC, which would in turn be characterised by its vibration around 1720 cm−1. Likewise, only traces of acetate might be present in this series of samples, as the characteristic CH vibrations around 2990 and 2930 cm−1 are barely visible. The apparent surface area of the series was evaluated using nitrogen as probe molecule. After activation at 250 °C in vacuum for at least 4 hours, all MOFs show type I isotherms. The BET areas vary between 410 and 630 m2 g−1 for EuBTC and Eu0.48Tb0.52BTC, respectively (Table 1). All MOFs show a pore volume of approx. 0.2 to 0.3 cm3 g−1 (Table 1). The BET areas of the MOFs are in line with literature reports on Tb and Eu MOFs,26,31 but are slightly lower as compared to their Ho, Tm, Lu and Dy counterparts.34
1H solid state NMR spectroscopy
Recently, Blahut et al. demonstrated the use of magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy as a powerful toolbox to obtain insight into fine details of the structure of paramagnetic MOFs.35–37 Here, we recorded 1H MAS NMR spectra at fast MAS of 30 kHz on the single lanthanide MOFs, EuBTC and TbBTC, and the mixed-metal phases EuxTb(1−x)BTC (Fig. 2a). Both single-metal MOFs are paramagnetic, however, Eu3+ in the ground state of the [Xe] 4f6 configuration often exhibits an angular momentum of 0, leading to no paramagnetic shift anisotropy, contact shift or pseudo-contact shift, whereas Tb3+ exhibits sizable values for these paramagnet-induced interactions.38 This is also reflected by different aspects of the 1H MAS NMR spectra: the 1H MAS NMR spectrum of pure EuBTC shows three resonances and well-defined spinning sidebands (Fig. 2a). We tentatively assign the resonance at approx. 1.6 ppm to the protons of the water molecule coordinated to the lanthanides in the MOF structure and the resonances at ca. 8 and −4 ppm to the two different proton positions of the aromatic protons of BTC present in the asymmetric unit of both EuBTC and TbBTC (Fig. 2b and c).19,31 For comparison, in the 1H NMR spectrum of paramagnetic Cu3BTC2, only one resonance for aromatic protons has been observed,39,40 since the asymmetric unit of Cu3BTC2 only contains one proton position.41 | ||
| Fig. 2 (a) 1H MAS NMR spectra of EuxTb(1−x)BTC recorded at 500 MHz. Asterisks denote spinning sidebands. To all spectra a line broadening of 300 Hz was applied. (b and c) View of the crystal structure of [TbBTC(H2O)]·(DMF)(H2O)0.5 (ref. 31) along the crystallographic a direction (b) and along the crystallographic c direction (c). Solvent molecules in the pore channel are omitted for clarity. Colour code: green: Tb, grey: C, white: H (crystallographic H1 position), blue: H (crystallographic H2 position), red: O (from BTC) and pink: O (from water, disordered between two positions). | ||
The 1H MAS NMR spectrum of pure TbBTC contains three resonances. The resonance of the water molecules is shifted to 4.7 ppm. The resonances of the aromatic protons are shifted to ca. 70 ppm and −62 ppm. These extreme values of 1H chemical shifts are caused by significant paramagnetic contributions, including shift anisotropy, contact shifts or pseudo-contact shifts induced by Tb3+. Interestingly, in the 1H MAS NMR spectra of the series of mixed metal EuxTb(1−x)BTC MOFs, with increasing Tb3+ content, we observe an almost continuous evolution of the chemical shifts of the two aromatic proton positions from one pure phase to the other. Thus each proton in the mixed metal EuxTb(1−x)BTC MOFs is influenced by paramagnetic effects of both metal ions. This confirms experimentally that the two cations are homogeneously distributed within the MOFs, leading to short Tb3+-to-Eu3+ interatomic distances (dTb–Eu ∼4.7 Å) and excluding the formation of large Eu- or Tb-rich domains or even co-crystallization of single metal MOFs. This unprecedented insight into the cation distribution is crucial for the understanding of other material properties including their photophysics (vide infra).
Room temperature luminescent properties
The photoluminescence (PL) spectra of the samples were measured in the solid state and dispersed in ethanol and acetonitrile. In the solid state, upon excitation at λex = 254 nm, the pure TbBTC shows the characteristic emissions centred at approx. 489 nm (5D4 → 7F6), 545 nm (5D4 → 7F5), 586 nm (5D4 → 7F4) and 622 nm (5D4 → 7F3), respectively (Fig. 3a). Likewise, the PL spectrum of EuBTC shows the lines characteristic of Eu-centred emissions at 595 nm (5D0 → 7F1), 616 nm (5D0 → 7F2), 653 nm (5D0 → 7F3) and 695 nm (5D0 → 7F4, Fig. 3b).44 The corresponding excitation spectra of both pure MOFs demonstrate an efficient sensitisation by the linker,18 as the excitation mainly occurs on ligand based π–π* transitions and Ln3+–O2−-charge transfer (CT) bands.45 We suppose that similarly to literature reports, ligand-to-metal charge transfer occurs from linker-based triplet states to 5D4 (Tb3+) and 5D2 (Eu3+) levels (Fig. 3d).42 In case of EuBTC, relaxation by internal conversion (IC) from the 5D2 into 5D0 energy level occurs. From the lowest exited levels (5D0 (Eu3+) or 5D4 (Tb3+)), transition of the electron into 7FJ levels is accompanied by emission of a photon.42,46 | ||
| Fig. 3 (a) PL (solid line, λex: 254 nm) and PL excitation (dotted line, λem: 543 nm) spectra of TbBTC, (b) PL (solid line, λex: 254 nm) and PL excitation (dotted line, λem: 615 nm) spectra of EuBTC, (c) PL spectra of EuxTb(1−x)BTC MOFs [x = 0.0 to 1.0] in the solid state (λex: 254 nm). Colour code as in Fig. 2a. Green labels mark Tb3+-based emission, red labels Eu3+-based emission. (d) Simplified Jablonski diagram showing basic photophysical processes according to ref. 42 and 43 LMCT: ligand-to-metal charge transfer, IC: internal conversion. Comparison of the solid-state PL spectrum with the PL spectra obtained for the MOF dispersions in ethanol and acetonitrile of (e) TbBTC and (f) EuBTC (λex: 300 nm). | ||
In the PL spectra of the EuxTb(1−x)BTC series, the intensities of the Tb3+-based emission lines decrease and vanish almost completely from an Eu content of 25% onwards (Fig. 3c and S5†). Such a decrease of the Tb3+ emission intensity upon doping the framework with Eu has also been observed in, e.g., series of [Tb2xEu2−2x(bdc)3(H2O)4]∞ or EuxTb1−xDMBDC MOFs.47–49 Recent work by Carneiro Neto et al.50 and Trannoy et al.14 suggest that the energy transfer occurs by a quadrupole–quadrupole mechanism, as the acceptor Eu3+ ions are homogeneously dispersed in the Tb3+ donor matrix (see above), resulting in a minimal distance between neighbouring Tb3+ and Eu3+ ions of 4.75 Å.31 A basic requirement for the energy transfer process among two species is the spectral overlap between donor emission (in our case Tb3+) and acceptor absorption (in our case Eu3+). This requirement is fulfilled at around 490 nm, 540 nm and 580 nm with the overlap between the 5D4 → 7F6, 5D4 → 7F5 and 5D4 → 7F4 emission of Tb3+ and the 7F0,1 → 5D2, the 7F0,1 → 5D1 and 7F0,1 → 5D0 absorption of Eu3+ (Fig. S6†).43,50 It is noteworthy that 7F1 → 5D0 exhibits larger oscillator strength than 7F0 → 5D0.51 The energy transfer becomes more efficient upon heating due to the increased number of energy transfer channels given by the thermal population of acceptor (Eu3+) 7F1 and 7F2 levels (Fig. 3d).43
The intensity of the Eu3+ emission lines at 595 nm (5D0 → 7F1), 616 nm (5D0 → 7F2), 653 nm (5D0 → 7F3) and 695 nm (5D0 → 7F4) increases for low Eu ratios and decreases for higher Eu ratios (Fig. 3c). The decrease in Eu3+ emission with increasing Eu content might be attributed to concentration quenching in form of energy migration from Eu3+ to a non-radiative recombination site or by cross relaxation with other (excited) Eu sites.48
The emission spectra of dispersions of the pure TbBTC in ethanol or in acetonitrile are very similar to the PL spectra in the solid state (Fig. 3e). The absence of any significant solvatochromic behaviour in TbBTC is corroborated by reports on molecular Tb3+ complexes.52–54 In case of EuBTC, significant solvatochromic effects are observed, in particular on the 5D0 → 7F2 (ΔJ = 2 manifold) and 5D0 → 7F4 (ΔJ = 4 manifold) transition (Fig. 3f). When dispersed in ethanol, the emission spectrum of EuBTC is characterised by a slight shift of the emission wavelength and a more pronounced Stark splitting of the ΔJ = 2 manifold: instead of one broad band, three well resolved bands can be distinguished. When replacing ethanol with acetonitrile, the Stark splitting of the 7F2 manifold is less pronounced, with an almost unaffected position of the highest energy transition. These changes in Stark splitting of the manifold have been correlated to the change of the solvent polarity. With increasing solvent polarity, the difference between lower energy transitions decreases, causing a less pronounced ligand field splitting of the 7FJ levels.55 Likewise, also more pronounced Stark splittings are observed for the ΔJ = 1 and ΔJ = 2 manifolds. For the ΔJ = 4 manifold, the Stark splitting is almost invisible when EuBTC is dispersed in acetonitrile, resulting in an increase in the relative intensities of the transition as compared to the PL spectra recorded in ethanol and in the dry solid state. Overall, the solvatochromic behaviour of the Eu3+ luminescent centres follows the same trends observed for organometallic Eu complexes.55 The observed changes are thus most likely caused by a change in the solvent polarity,55 but contributions from solvent coordination to open metal sites and/or H-bonding cannot be ruled out. The different solvatochromic behaviours of Eu3+ and Tb3+ luminescent centres in EuBTC and TbBTC are also observed in the EuxTb(1−x)BTC series (Fig. S7†), opening the possibility for dual ratiometric temperature and solvent sensing (vide infra).
Temperature-dependent solid-state luminescence properties
Temperature-dependent luminescence spectra were recorded for Eu0.05Tb0.95BTC and Eu0.02Tb0.98BTC in the temperature range from 200 to 330 K and from 173 to 423 K in the solid-state (Fig. 4a, b and S8†). The terbium-based emission, e.g. at 489 nm and 543 nm, decreases with increasing temperature. For the Eu3+-based emission, the temperature dependency is more complex. Between 200 and 280 K, the emission also decreases with increasing temperature, while above 280 K it increases again (Fig. 4c and S9†). This change in temperature dependency further confirms an energy transfer from donor Tb3+ levels to acceptor Eu3+ levels, which becomes more efficient in the EuxTb(1−x)BTC series above approx. 280 K. The thermal energy is then sufficient to allow for the thermal population of acceptor (Eu3+) 7F1 and 7F2 levels (Fig. 3d).43 As mentioned above, energy transfer becomes thus more efficient due to the increased number of energy transfer channels (see below). Moreover, this change in the temperature dependency of the Eu emission is not correlated to a phase transition of the MOFs or a phase separation into single metal LnBTCs, evidenced by the absence of any signal in dynamic scanning calorimetry experiments (Fig. S10†).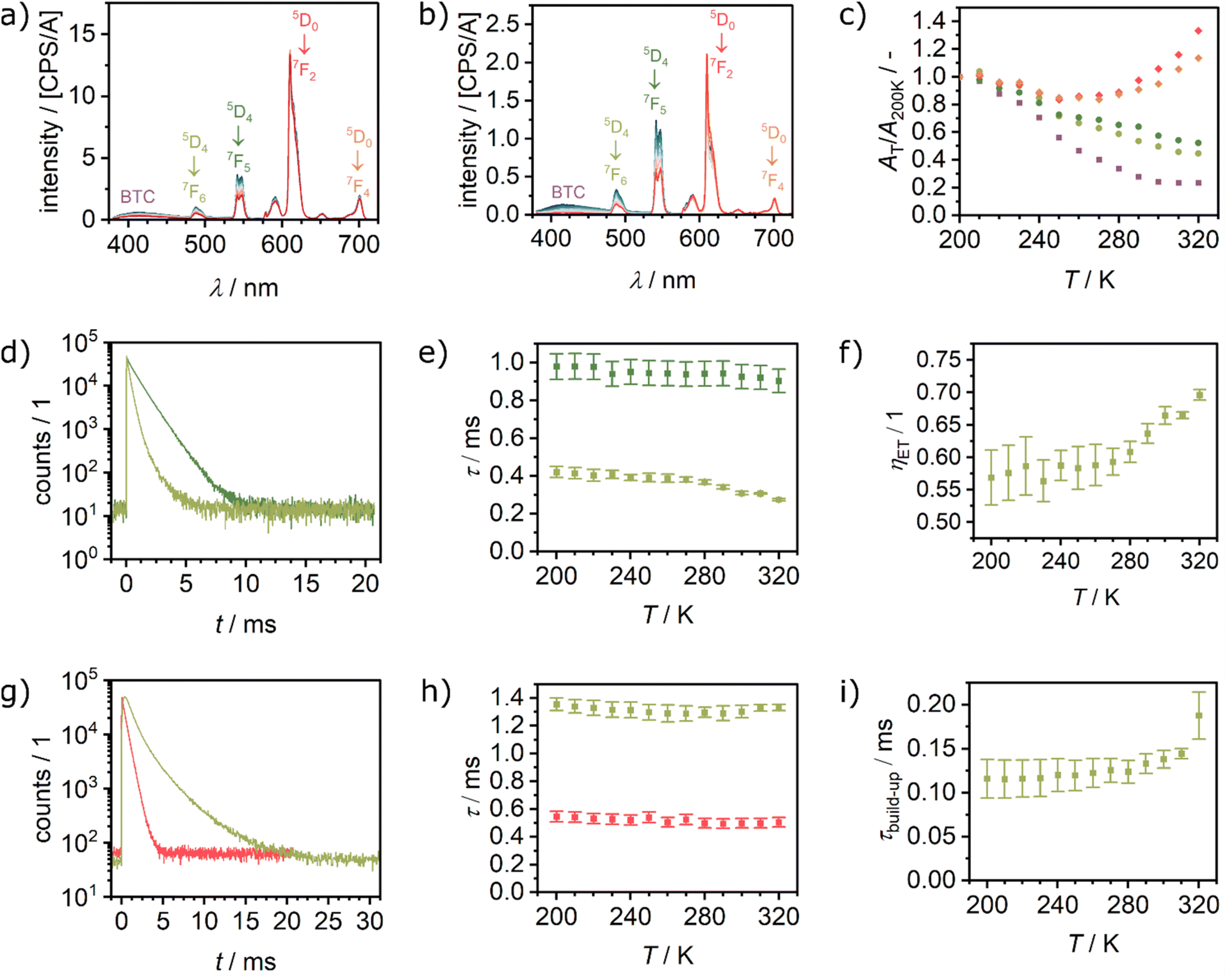 | ||
| Fig. 4 Temperature dependent luminescence spectra recorded between 200 and 330 K after excitation at 370 nm of (a) Eu0.05Tb0.95BTC and (b) Eu0.02Tb0.98BTC. (c) Corresponding evolution of the luminescence with temperature, expressed as area at a given temperature divided by the area at 200 K, for Eu0.02Tb0.98BTC. (d) Time correlated single photon counting (TCSPC) spectra of TbBTC (dark green) and Eu0.02Tb0.98BTC (light green) recorded at λem = 542 nm (Tb emission) and at 320 K. For all TCSPC spectra between 200 and 320 K see Fig. S12a and d.† (e) Evolution of Tb emission lifetime as a function of temperature in TbBTC (dark green) and Eu0.02Tb0.98BTC (light green). (f) Corresponding energy transfer efficiency from Tb to Eu, estimated from the Tb emission lifetimes shown in (b) using eqn (4). (g) TCSPC spectra of EuBTC (red) and Eu0.02Tb0.98BTC (light green) recorded at λem = 610 nm (Eu emission) and at 320 K. For all TCSPC spectra between 200 and 320 K see Fig. S12e and f.† (h) Evolution of Eu emission lifetime as a function of temperature in EuBTC (red) and Eu0.02Tb0.98BTC (light green). (i) Evolution of Eu emission build-up as a function of temperature in Eu0.02Tb0.98BTC (light green). | ||
For both materials, in the range from 200 to 280 K, the decrease in emission intensity is stronger for the Tb3+ than for the Eu3+-based emission. For single lanthanide Ln2(Hpcpa)3(H2O)5·H2O (H3pcpa = N-(4-carboxyphenyl)oxamic acid) a decrease of the emission of either Tb3+ or Eu3+ with increasing temperature (15 to 300 K) has been reported.44 For EuBTC doped with CsPbBr3 quantum dots,27 as well as Eu0.37Tb0.63BTC,18 an increase in Eu3+ emission with increasing temperature above 293 K has been reported. In contrast, for a series of EuxTb(1−x)-DMBDC (x = 0.0011, 0.0046, 0.0069; DMBDC = 2,5-dimethoxy-1,4-benzenedicarboxylate) a different behaviour is observed.56 While the Tb3+ emission still decreases with increasing temperature, the Eu3+ emission increases with increasing temperature in the range from 10 to 300 K. Interestingly, for pure Eu-DMBDC and pure Tb-DMBDC, a decrease of the emission with increasing temperature has been reported, the same temperature dependency behaviour as observed for pure TbBTC and EuBTC (Fig. S11†). This inversion of the temperature dependency of the Eu3+ emission when doped into the Tb-DMBDC matrix has been explained by an efficient energy transfer from Tb3+ to Eu3+.56
To confirm the energy transfer in the EuxTb1−xBTC series, we analysed the emission lifetimes of the pure lanthanide as well as mixed lanthanide materials as a function of the temperature (Fig. 4d, g and S12†). The Tb emission lifetime in TbBTC decreases from approx. 980 μs at 200 K to ∼900 μs at 320 K, while in Eu0.02Tb0.98BTC it decreases from approx. 420 to 270 μs (Fig. 4e and Table S2†). From those measurements, the efficiency of the energy transfer (ηET) from Tb to Eu can be estimated by comparing the Tb lifetime in TbBTC (τ0) with that in EuxTb1−xBTC materials (τmix) according to18,56
| ηET = 1 − τ0/τmix. | (1) |
The efficiency of the energy transfer increases from approx. 55% (200 K) to more than 70% (320 K, Fig. 4f). The energy transfer efficiency is comparable to the best efficiencies reported for Eu–Tb MOFs with comparable composition and Tb3+-to-Eu3+ interatomic distances (dTb–Eu < 4.5 Å, ηET ∼80%).14,57 It is much higher as compared to MOFs with larger Tb3+-to-Eu3+ interatomic distances (dTb–Eu > 7 Å), which cause significantly lower energy transfer efficiencies (20–40%).58
Interestingly, the build-up time constant of the Eu emission in Eu0.02Tb0.98BTC increases significantly compared to the pure EuBTC (Fig. 4i). The build-up also increases with increasing temperature from ∼120 μs (200 K) to ∼190 μs (320 K). Such an increase in the Eu build-up time constant has recently been attributed to a delayed Tb3+-to-Eu3+ energy transfer.59 Consequently, the lifetime of the Eu emission in the mixed Eu0.02Tb0.98BTC remains almost constant (∼1.2–1.3 ms), a significant increase compared to lifetimes between 550 μs (200 K) and 500 μs (320 K) in pure Eu BTC (Fig. 4h and Table S2†). Overall, these results, the shortening of the Tb lifetime, the increase in the Eu lifetime and the increase in the energy transfer efficiency, are in excellent agreement with experimental and computational models of the Tb-to-Eu energy transfer in MOFs.14,15
Ratiometric temperature sensing in the solid-state
For the application as ratiometric temperature sensor, we used the ratio
 | (2) |
 | (3) |
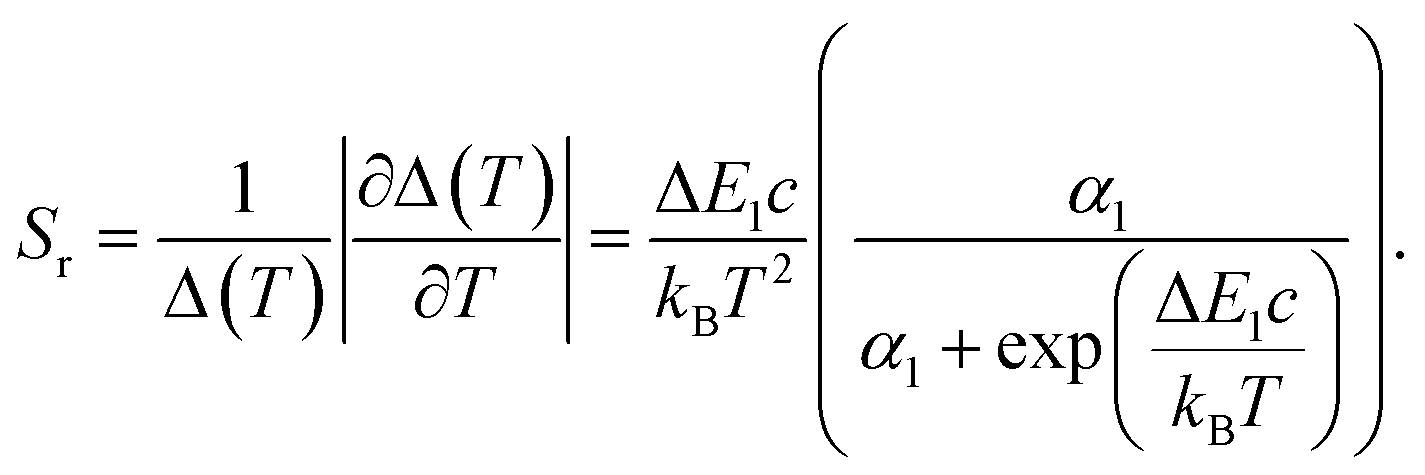 | (4) |
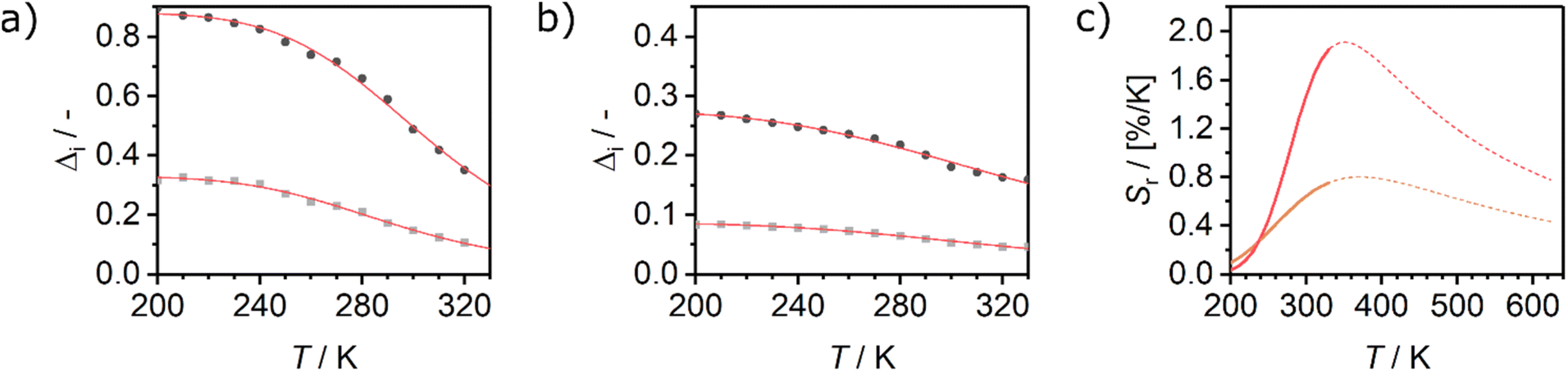 | ||
| Fig. 5 Evolution of the luminescence ratios Δi as a function of temperature, for (a) Eu0.02Tb0.98BTC and (b) Eu0.05Tb0.95BTC. Data for Δi based on Tb 5D4 → 7F5 transition are shown in dark grey, based on Tb 5D4 → 7F6 transition in light grey. (c) Evolution of relative sensitivities in the analysed temperature regime for Tb 5D4 → 7F5 transition. Sr of Eu0.02Tb0.98BTC (red), Sr of Eu0.05Tb0.95BTC (orange). Solid lines correspond to Sr curves in the calibrated temperature regime, dotted lines correspond to the extrapolated Sr curves limited by the thermal decomposition of the MOFs around 623 K (Fig. 1b). | ||
| Material | T/K | ΔE/cm−1 | Sm/[% K−1] | Tm/K | Ref. |
|---|---|---|---|---|---|
| a Average of two independent experiments. 1,3-bdc = 1,3-benzendicarboxylate; bdc = 1,4-benzenedicarboxylate; BPDA = biphenyl-3,5-dicarboxylate; ad = adipate; phth = phthalate; PDC = pyridine-3,5-dicarboxylate; N-BDC = 2-amino-1,4-benzenedicarboxylate. | |||||
| Eu0.05Tb0.95BTC | 200–330 | 1300 ± 100 | 0.75 | 330 | |
| 298–473 | 6100 | 1.19 | 347 | ||
| Eu0.02Tb0.98BTCa | 200–330 | 2600 ± 500 | 1.61 ± 0.24 | 330 | |
| 173–473 | 5500 ± 400 | 1.66 ± 0.39 | 335 ± 5 | ||
| Eu0.37Tb0.63BTC | 313–473 | 0.68 | 313 | 18 | |
| Tb0.85Eu0.15(OAc)(1,3-bdc)(H2O) | 150–350 | 500 | 0.44 | 236 | 14 |
| Tb0.99Eu0.01(bdc)1.5(H2O)2 | 290–320 | — | 0.14 | 318 | 61 |
| Tb0.8Eu0.2BPDA | 293–328 | — | 1.19 | 313 | 62 |
| Eu0.33Tb0.66(ad)0.5(phth)(H2O)2 | 303–423 | — | 1.21 | 303 | 63 |
| Eu0.05Tb1.95-PDC | 298–333 | — | 1.37 | 333 | 64 |
| [Tb1.9Eu0.1(N-BDC)3(DMF)4] | 100–293 | 918 | 2.6 | 190 | 15 |
| Tb5.94Eu0.06-UiO-66 | 255–295 | — | 4.9 | 295 | 65 |
| Fe3O4@ZrOBDC:Eu3+,Tb3+ | 153–573 | — | 0.7 | 543 | 66 |
When extrapolating the sensitivity Sr range up to the thermal stability limit of the MOFs around 623 K, we estimate the overall sensitivities to increase to ca. 2.1% K−1 (∼370 K) and ca. 0.8% K−1 (∼370 K) for Eu0.02Tb0.98BTC and Eu0.05Tb0.95BTC, respectively. Indeed, in the calibrated high temperature range between 298 and 473 K, maximum sensitivities Sm of up to 2.0% K−1 (335 K) and 1.19% K−1 (347 K) were achieved for Eu0.02Tb0.98BTC and Eu0.05Tb0.95BTC, respectively (Table 2). Note that the average Sm and Tm values obtained for Eu0.02Tb0.98BTC using two different setups and on independent samples are statistically the same. Moreover, the materials show no change in structural properties (Fig. S13 and S14a†) and only slight changes in photophysical properties (Fig. S14b and c†).
Interestingly for macroscopic Eu0.37Tb0.63BTC needles, preferred oriented along the crystallographic c-direction,18 the Sr value decreases above 313 K with increasing temperature. To shed light on the origin of the observed different luminescent properties of macroscopic needles (Eu0.37Tb0.63BTC) reported by Wang et al.18 and our materials, we analysed the emission lifetimes of the pure lanthanide as well as mixed lanthanide materials as a function of the temperature (Fig. 4d, g and S12†). The emission lifetime of both Eu and Tb behave quite differently in the pure LnBTC materials of different morphology: While we observe lifetimes of around 900 μs and 500 μs for Tb and Eu respectively at 320 K (Fig. 4e and h), Wang et al. observed much longer lifetimes of approx. 1300 μs for Tb (313–330 K).18 In EuxTb1−xBTC materials, the Tb emission lifetime is always shorter than in the pure TbBTC, with however shorter lifetimes in the case of the smaller particles (∼300 μs vs. ∼550 μs at 310 K).
Consequently, the more pronounced relative reduction of the Tb emission lifetime in EuxTb1−xBTC compared to TbBTC for the nanoscopic needles results in a higher energy transfer efficiency of approx. 70% at 320 K, compared to ∼58% at 313 K for macroscopic needles.
While the lifetime of the Eu emission in Eu0.02Tb0.98BTC shows hardly any temperature dependency (∼1.2–1.3 ms, Fig. 4h), Wang et al. observed an increase in lifetime for Eu in the mixed material as well. A possible reason for these differences in behaviour might be a smaller crystallite size in the case of our materials, different preferred orientation, different amount and nature of defects present in the materials (e.g. NaOAc vs. HCl used as modulators), or the different excitation wavelength used (385/370 nm vs. 296 nm). The excitation wavelength affects the ratios of Tb and Eu emission (Fig. S15†) and thus possibly the energy transfer. The use of different modulators might cause different coordination polyhedra and site symmetry in the materials, parameters which are known to impact the luminescence lifetime of e.g. Eu.67
Like in the Tb1−xEux(OAc)(1,3-bdc)(H2O) series reported by Trannoy et al.,14 we see a slightly higher maximum temperature for lower europium content. However, in the case of BTC frameworks, the highest sensitivity is also observed for the lowest europium content. A possible explanation might be that the temperature dependencies of the Tb centred transitions are less pronounced in Eu0.02Tb0.98BTC than in Eu0.05Tb0.95BTC (Fig. 4c and S9†). This is also seen by the increase in activation energy for the non-radiative decay with decreasing Eu content (Table 2).
Ratiometric temperature sensing in organic solvents
Eu0.02Tb0.98BTC was also investigated as temperature probe dispersed in ethanol (EtOH) or acetonitrile (ACN, Fig. 6) in the temperature range from 250 to 330 K. As for the pure MOFs (Fig. 3e and f), solvation with either ACN or EtOH tremendously affects the emission profile also for Eu0.02Tb0.98BTC. When dispersed in ACN, the Eu3+ 5D0 → 7F2 emission is stronger than the most intense Tb3+ emission (5D4 → 7F5), which might be explained by a better energy transfer from terbium to europium as compared to the dispersion in ethanol. The Tb3+ transitions are hardly affected by the nature of the solvent, with however a slightly lower intensity in ACN (Fig. 6c). Thus from Δi (eqn (1)) we can discern these solvents, as Δi(EtOH) is approx. twice as high as Δi(ACN), over the whole temperature range investigated (Fig. 6d and S16†). In contrast to the case in the solid state, where the Tb3+ transitions show a stronger thermal dependency than the Eu3+ transitions, in the dispersions both lanthanides are similarly affected. Therefore, we used a simplified linear expression to evaluate the temperature dependency of Δi. The maximal sensitivity drops to 0.06% K−1 (5D4 → 7F5) and 0.22% K−1 (5D4 → 7F5) when dispersed in acetonitrile and ethanol, respectively. Interestingly, the only other report on the solvent effect on temperature sensing in MOFs by Cadiau et al. reported an inverse behaviour: when Tb0.99Eu0.01(bdc)1.5(H2O)2 was dispersed in water, the sensitivity increased from 0.14% K−1 (dry MOF) to 0.31% K−1.61 | ||
| Fig. 6 Temperature dependent luminescence spectra recorded between 253 and 333 K after excitation at 254 nm of Eu0.02Tb0.98BTC dispersed in (a) ethanol (EtOH) and (b) acetonitrile (ACN). # marks the 2nd order laser peak. (c) Comparison of PL intensities of Tb3+ and Eu3+ transitions expressed as the ratio of the respective areas measured in EtOH and ACN. (d) Corresponding evolution of the luminescence ratios Δi as a function of temperature, for Δi based on Tb3+ 5D4 → 7F5 transition for dispersion in EtOH (purple) and ACN (blue). (e and f) Sr calculated using data for (e) Tb3+ 5D4 → 7F5 transition and (f) Tb3+ 5D4 → 7F6 transition. Sr of Eu0.02Tb0.98BTC dispersed in EtOH (purple) and ACN (blue). Solid lines correspond to Sr curves in the calibrated temperature regime, dotted lines correspond to the extrapolated Sr curves limited by the melting and boiling temperature of ACN and EtOH, respectively. | ||
Here we note that the sensitivity is not affected by slight agglomeration and/or sedimentation which occur in the suspensions (Fig. S17 and S18†). The intensities of the different transitions of both lanthanides are similarly affected (Fig. S17a†) due to their homogeneous incorporation within a particle (see above). Thus, the luminescence ratios Δi as a descriptor for the temperature sensing are not affected over time (Figure S17b†).
In summary, from Δi the solvents can readily be discerned, while temperature determination is possible using the temperature dependency of Δ and of the sensitivity Sr.
Conclusions
Using the acetate mediated synthesis protocol of LnBTCs, we obtained a series of EuxTb(1−x)BTC MOFs, in which the ratio of the two lanthanides is controlled by the synthesis gel composition. Using 1H MAS NMR spectroscopy, we demonstrated the homogeneous incorporation of both cations in the series of mixed metal EuxTb(1−x)BTC MOFs. The 1H NMR chemical shifts of the two crystallographically different protons change continuously in the series from one pure LnBTC MOF to the other, indicating the formation of a single crystalline phase with homogeneous distribution of the two different metals. The precise control of lanthanide stoichiometry and distribution allows to control the luminescent properties of the final material. While for Eu loadings above 25 mol%, the Tb3+ emission is completely quenched, in Eu0.05Tb0.95BTC still 20% of the Tb3+ emission intensity of the parent TbBTC remains, sufficient to be used as temperature sensor. Eu0.05Tb0.95BTC and Eu0.02Tb0.98BTC allow for ratiometric temperature sensing in the range between 200 and 330 K with unprecedented sensitivities for pure Tb–Eu MOFs. Interestingly, the emission profiles of Tb centres are hardly affected when the material is dispersed in different solvents, while for Eu centres the emission wavelengths of the transitions as well as the intensity ratios for the different transitions change in the presence of ethanol and acetonitrile. Therefore, the solvation of the MOFs should be considered when using them as ratiometric temperature sensors in the presence of solvents or vapours. This solvation dependency of the lanthanide emission can be rationalised and taken into account in such sensors, to determine temperature and discern between different solvents present as demonstrated here for acetonitrile and ethanol as model compounds.Experimental
Materials and methods
Eu(NO3)3·6H2O (Thermo Scientific, 99.9%), Tb(NO3)3·6H2O (Thermo Scientific, 99.99%), 1,3,5-benzene tricarboxylic acid (H3BTC, Alfa Aesar, 98%), N,N-dimethylformamide (DMF, Merck, 99.5%), NaOAc (Merck, 99%) and ethanol (Merck, >95%). All chemicals were used as supplied without further purification.Powder X-ray diffraction (PXRD) patterns were collected on a PANalytical Emryrean series 2 in Bragg–Brentano geometry using CuKα radiation at 2θ angles between 2 and 50°. Thermogravimetric analysis (TGA) of powder samples was measured on a Hi-Res TGA 2950 Thermogravimetric Analyzer (TA Instruments) in the temperature range from room temperature to 700 °C with a heating rate of 2 K min−1 in synthetic air and on a SDT 2960 Simultaneous DSC-TGA (TA Instruments) in the temperature range from room temperature to 1100 °C with a heating rate of 2 K min−1 in synthetic air. Nitrogen physisorption experiments were carried out using an ASAP 2000 (Micromeritics). Prior to measurements the samples were degassed at 250 °C under vacuum overnight. Inductively coupled plasma optical emission spectroscopy analysis (ICP OES) was done using a SPECTRO Ciros-CCD. Prior to ICP OES analysis, ∼100 mg of MOF powder were digested in aqua regia (9 ml HCl, 3 ml HNO3) inside a Teflon lined autoclave using a Berghof-Microwave Digester-Speedwave 4 (θmax = 200 °C, pmax = 50 bar, 37 min). IR spectra were recorded using a JASCO FT/IR-4100 in attenuated total reflectance geometry (ATR, resolution 2 cm−1, 64 scans).
Photoluminescence (PL) spectra of the suspensions were recorded using either a fluorescence spectrophotometer (Horiba Fluorolog) in the temperature range of −253 to 323 K or using a Jasco Spectrofluorometer FP-8500 at room temperature. PL spectra at room temperature of the solid samples were recorded using a Jasco Spectrofluorometer FP-8500 (λex = 256 nm).
Steady-state PL spectra in the low temperature regime (200–300 K) were recorded with a fluorescence spectrophotometer Fluorolog-3 (FL3-22) by Horiba Scientific with a xenon lamp operating at 370 nm as excitation source. Time resolved PL decays were recorded with a phosphorimeter (FL-1040A) integrated into the Fluorolog-3 setup with a pulsed xenon lamp operating at 370 nm as excitation source. Temperature adjustment between 200 and 330 K was achieved by using a JANIS ST-500 cryostat. Sample preparation for the temperature dependent PL spectroscopy (steady-state and time resolved) of the solid samples was done by dispersing the MOF powder in ethanol and drop casting 70 μl onto a silicon substrate.
Time correlated single photon counting (TCSPC) data were recorded using a phosphorimeter (FL-1040A) integrated into the Fluorolog-3 setup with a pulsed xenon lamp operating at 370 nm as excitation source. Temperature adjustment between 200 and 320 K was achieved by using a modified ST-300-MS-cryostat (Janis Research). The values reported in Table S2† are the amplitude average lifetime from deconvolution of the TCSPC data using:
 | (5) |
In case of the Eu emission in Eu0.02Tb0.98BTC materials, also the build-up time constant was integrated into the formula used for deconvolution:
 | (6) |
Deconvolution was done using Origin 2023.
Steady-state PL spectra in the high temperature regime (298–473 K) as well as the full temperature regime (173–473 K) were recorded with a home-build fibre optical fluorescence spectrophotometer consisting of an AvaSpec-HSC1024x58TEC-EVO spectrometer (Avantes) and a 385 nm LED (Thorlabs) as excitation source. Spectra were recorded in reflectance. Temperature adjustment between 173 K and 473 K was achieved by using a FTIRSP600 stage (Linkam Scientific Instruments). Solid samples were deposited as powders on quartz lids on top of the silver heating block inside the FTIRSP600 stage.
Scanning electron microscopy (SEM) images were obtained on a Carl Zeiss Gemini Ultra 55. Dynamic scanning calorimetry was recorded using a NETZSCH DSC 204F1 Phoenix equipped with a liquid nitrogen dewar. Samples were filled into aluminum pans, closed with an aluminum lid and analysed between −30 and 100 °C with a heating/cooling rate of 2 K min−1.
1H solid state NMR spectra were collected on a Varian 500 NMR spectrometer (1H resonance frequency at 499.86 MHz) equipped with a 1.6 mm probe at a spinning frequency of 30 kHz. Prior to NMR measurements, all samples were activated overnight at 250 °C under vacuum and packed in an Argon filled glovebox. The DEPTH sequence was used for recording all spectra. The π/2 and π pulse widths for proton were 2.5 μs and 5.0 μs, respectively. The recycle delay was 50 ms (T1 ≈ 9 ms) with an acquisition time of 3 ms. 1024 scans were accumulated. All 1H NMR spectra were referenced to 0.0 ppm to the central line of sodium trimethylsilylpropanesulfonate.
Synthetic procedures
General procedure for synthesis of nanorod EuxTb(1−x)BTC MOFs. The MOFs were synthesised following a modified literature procedure.25 In a typical synthesis of mixed LnBTC (Eu0.5Tb0.5BTC), 373.4 mg (4.5 mmol) NaOAc were dissolved in 57 ml DMF and 3 ml deionised H2O in a 100 ml round bottom flask equipped with a reflux condenser. Next, 339.8 mg (750 μmol) Tb(NO3)3·6H2O, 334.6 mg (750 μmol) Eu(NO3)3·6H2O and 321.7 mg (1.5 mmol) H3BTC were added and the synthesis mixture was heated to 110 °C and stirred at this temperature for 4 h (300 rpm). After cooling to room temperature, the white solid was isolated by centrifugation, washed with ca. 30 ml DMF and 30 ml absolute ethanol (three times). The white slurry was dried in an oven at 80 °C. Isolated yield: 471.3 mg (1.02 mmol, 68%).All EuxTb(1−x)BTC samples were prepared following the procedure described above and are denoted accordingly to the experimentally determined molar ratios as: EuxTb(1−x)BTC MOFs [x = 0 to 1].
Data availability
Data for this article, including information on the data sets used in Fig. 1–6 (raw data and data converted to Ascii or JCAMP files) and information on data processing, are available at https://doi.org/10.5281/zenodo.14773089.Author contributions
M. J.: investigation (MOF synthesis), writing – review & editing. M. R.: investigation (TD-PL solid-state, TCSPC), writing – review & editing. Z. W.: investigation (TD-PL dispersion), writing – review & editing. S. M.: investigation (MOF synthesis, TD-PL solid-state), formal analysis, writing – review & editing. R. N., C. H.: investigation (solid-state NMR), formal analysis, writing – review & editing. R. E. R.: investigation (additional characterisation), writing – review & editing. M. F.: investigation (additional characterisation), supervision, writing – review & editing. R. F., F. M. W.: conceptualization, supervision, formal analysis, writing – original draft, review & editing. K. M.: writing – review & editing. D. W.: conceptualization, supervision, writing – original draft, review & editing, funding acquisition. D. S., G. B., M. H.: conceptualization, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project-ID 416229255–CRC 1411, project BA1422/16-1 and Project-ID 539724755 – FPS Core Facility is gratefully acknowledged. The authors are grateful to Susanne Pachaly for ICP OES analysis and to Lilong Wu for help with material synthesis and data evaluation.Notes and references
- S. L. Griffin and N. R. Champness, A periodic table of metal-organic frameworks, Coord. Chem. Rev., 2020, 414, 213295 Search PubMed.
- J.-X. Wang, J. Yin, O. Shekhah, O. M. Bakr, M. Eddaoudi and O. F. Mohammed, Energy Transfer in Metal-Organic Frameworks for Fluorescence Sensing, ACS Appl. Mater. Interfaces, 2022, 14, 9970–9986 CrossRef CAS PubMed.
- P. Müller, F. M. Wisser, P. Freund, V. Bon, I. Senkovska and S. Kaskel, Optical Sensors Using Solvatochromic Metal-Organic Frameworks, Inorg. Chem., 2017, 56, 14164–14169 CrossRef PubMed.
- J. Wang, J. Wang, Y. Li, M. Jiang, L. Zhang and P. Wu, A europium(iii)-based metal–organic framework as a naked-eye and fast response luminescence sensor for acetone and ferric iron, New J. Chem., 2016, 40, 8600–8606 RSC.
- P. Müller, F. M. Wisser, V. Bon, R. Grünker, I. Senkovska and S. Kaskel, Postsynthetic Paddle-Wheel Cross-Linking and Functionalization of 1,3-Phenylenebis(azanetriyl)tetrabenzoate-Based MOFs, Chem. Mater., 2015, 27, 2460–2467 CrossRef.
- P. Freund, I. Senkovska and S. Kaskel, Switchable Conductive MOF-Nanocarbon Composite Coatings as Threshold Sensing Architectures, ACS Appl. Mater. Interfaces, 2017, 9, 43782–43789 CrossRef CAS PubMed.
- P. Freund, L. Mielewczyk, M. Rauche, I. Senkovska, S. Ehrling, E. Brunner and S. Kaskel, MIL-53(Al)/Carbon Films for CO 2 -Sensing at High Pressure, ACS Sustainable Chem. Eng., 2019, 7, 4012–4018 Search PubMed.
- Y. Xie, G. Sun, G. A. Mandl, S. L. Maurizio, J. Chen, J. A. Capobianco and L. Sun, Upconversion Luminescence through Cooperative and Energy-Transfer Mechanisms in Yb3+ -Metal-Organic Frameworks, Angew. Chem., Int. Ed., 2023, 62, e202216269 Search PubMed.
- M. A. M. Lucena, M. F. L. Oliveira, A. M. Arouca, M. Talhavini, E. A. Ferreira, S. Alves, F. H. Veiga-Souza and I. T. Weber, Application of the Metal-Organic Framework Eu(BTC) as a Luminescent Marker for Gunshot Residues: A Synthesis, Characterization, and Toxicity Study, ACS Appl. Mater. Interfaces, 2017, 9, 4684–4691 Search PubMed.
- H. Xu, C.-S. Cao, X.-M. Kang and B. Zhao, Lanthanide-based metal-organic frameworks as luminescent probes, Dalton Trans., 2016, 45, 18003–18017 Search PubMed.
- S. Roy, A. Chakraborty and T. K. Maji, Lanthanide–organic frameworks for gas storage and as magneto-luminescent materials, Coord. Chem. Rev., 2014, 273–274, 139–164 Search PubMed.
- Y. Shu, Q. Ye, T. Dai, Q. Xu and X. Hu, Encapsulation of Luminescent Guests to Construct Luminescent Metal-Organic Frameworks for Chemical Sensing, ACS Sens., 2021, 6, 641–658 Search PubMed.
- M.-L. Han, G.-X. Wen, W.-W. Dong, Z.-H. Zhou, Y.-P. Wu, J. Zhao, D.-S. Li, L.-F. Ma and X. Bu, A heterometallic sodium–europium-cluster-based metal–organic framework as a versatile and water-stable chemosensor for antibiotics and explosives, J. Mater. Chem. C, 2017, 5, 8469–8474 Search PubMed.
- V. Trannoy, A. N. Carneiro Neto, C. D. S. Brites, L. D. Carlos and H. Serier-Brault, Engineering of Mixed Eu 3+/Tb 3+ Metal-Organic Frameworks Luminescent Thermometers with Tunable Sensitivity, Adv. Opt. Mater., 2021, 9, 2001938 Search PubMed.
- A. Kourtellaris, W. Lafargue-Dit-Hauret, F. Massuyeau, C. Latouche, A. J. Tasiopoulos and H. Serier-Brault, Tuning of Thermometric Performances of Mixed Eu–Tb Metal–Organic Frameworks through Single-Crystal Coordinating Solvent Exchange Reactions, Adv. Opt. Mater., 2022, 10, 2200484 CrossRef CAS.
- J. Rocha, C. D. S. Brites and L. D. Carlos, Lanthanide Organic Framework Luminescent Thermometers, Chemistry, 2016, 22, 14782–14795 CrossRef CAS PubMed.
- Z. Zhou, X. Li, Y. Tang, C. C. Zhang, H. Fu, N. Wu, L. Ma, J. Gao and Q. Wang, Oxidative deoximation reaction induced recognition of hypochlorite based on a new fluorescent lanthanide-organic framework, Chem. Eng. J., 2018, 351, 364–370 Search PubMed.
- H. Wang, D. Zhao, Y. Cui, Y. Yang and G. Qian, A Eu/Tb-mixed MOF for luminescent high-temperature sensing, J. Solid State Chem., 2017, 246, 341–345 Search PubMed.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Luminescent Open Metal Sites within a Metal–Organic Framework for Sensing Small Molecules, Adv. Mater., 2007, 19, 1693–1696 Search PubMed.
- W. Yang, J. Feng, S. Song and H. Zhang, Microwave-assisted modular fabrication of nanoscale luminescent metal-organic framework for molecular sensing, ChemPhysChem, 2012, 13, 2734–2738 CrossRef CAS PubMed.
- Y. Xiao, L. Wang, Y. Cui, B. Chen, F. Zapata and G. Qian, Molecular sensing with lanthanide luminescence in a 3D porous metal-organic framework, J. Alloys Compd., 2009, 484, 601–604 CrossRef CAS.
- Q. Yao, A. Bermejo Gómez, J. Su, V. Pascanu, Y. Yun, H. Zheng, H. Chen, L. Liu, H. N. Abdelhamid, B. Martín-Matute and X. Zou, Series of Highly Stable Isoreticular Lanthanide Metal–Organic Frameworks with Expanding Pore Size and Tunable Luminescent Properties, Chem. Mater., 2015, 27, 5332–5339 CrossRef CAS.
- S. Pal, A. Bhunia, P. P. Jana, S. Dey, J. Möllmer, C. Janiak and H. P. Nayek, Microporous La-metal-organic framework (MOF) with large surface area, Chemistry, 2015, 21, 2789–2792 CrossRef CAS PubMed.
- H.-L. Jiang, N. Tsumori and Q. Xu, A series of (6,6)-connected porous lanthanide−organic framework enantiomers with high thermostability and exposed metal sites: scalable syntheses, structures, and sorption properties, Inorg. Chem., 2010, 49, 10001–10006 CrossRef CAS PubMed.
- H. Guo, Y. Zhu, S. Qiu, J. A. Lercher and H. Zhang, Coordination Modulation Induced Synthesis of Nanoscale Eu1-xTbx-Metal-Organic Frameworks for Luminescent Thin Films, Adv. Mater., 2010, 22, 4190–4192 Search PubMed.
- X. Lian and B. Yan, A lanthanide metal–organic framework (MOF-76) for adsorbing dyes and fluorescence detecting aromatic pollutants, RSC Adv., 2016, 6, 11570–11576 Search PubMed.
- J. Liu, Y. Zhao, X. Li, J. Wu, Y. Han, X. Zhang and Y. Xu, Dual-Emissive CsPbBr 3 @Eu-BTC Composite for Self-Calibrating Temperature Sensing Application, Cryst. Growth Des., 2020, 20, 454–459 Search PubMed.
- H. Brunckova, E. Mudra, L. Rocha, E. Nassar, W. Nascimento, H. Kolev, A. Kovalcikova, Z. Molcanova, M. Podobova and L. Medvecky, Preparation and characterization of isostructural lanthanide Eu/Gd/Tb metal-organic framework thin films for luminescent applications, Appl. Surf. Sci., 2021, 542, 148731 Search PubMed.
- T.-W. Duan and B. Yan, Hybrids based on lanthanide ions activated yttrium metal–organic frameworks: functional assembly, polymer film preparation and luminescence tuning, J. Mater. Chem. C, 2014, 2, 5098–5104 Search PubMed.
- D.-H. Chen, R. Haldar, B. L. Neumeier, Z.-H. Fu, C. Feldmann, C. Wöll and E. Redel, Tunable Emission in Heteroepitaxial Ln-SURMOFs, Adv. Funct. Mater., 2019, 29, 1903086 Search PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, Rod packings and metal-organic frameworks constructed from rod-shaped secondary building units, J. Am. Chem. Soc., 2005, 127, 1504–1518 Search PubMed.
- Z. Rzączyńska, A. Ostasz and S. Pikus, Thermal properties of rare earth elements complexes with 1,3,5-benzenetricarboxylic acid, J. Therm. Anal. Calorim., 2005, 82, 347–351 CrossRef.
- J. Zhang, H. Chen, J. Wang, D. Wang, D. Han, J. Zhang and S. Wang, Phase transformation process of Tb2O3 at elevated temperature, Scr. Mater., 2019, 171, 108–111 CrossRef CAS.
- M. Almáši, V. Zeleňák, J. Kuchár, S. Bourrelly and P. L. Llewellyn, New members of MOF-76 family containing Ho(III) and Tm(III) ions: Characterization, stability and gas adsorption properties, Colloids Surf., A, 2016, 496, 114–124 CrossRef.
- J. Blahut, A. L. Lejeune, S. Ehrling, I. Senkovska, S. Kaskel, F. M. Wisser and G. Pintacuda, Monitoring Dynamics, Structure, and Magnetism of Switchable Metal-Organic Frameworks via 1 H-Detected MAS NMR, Angew. Chem., Int. Ed., 2021, 60, 21778–21783 Search PubMed.
- F. Uhlig, M. B. Stammler, F. Meurer, I. G. Shenderovich, J. Blahut and F. M. Wisser, Monitoring structure and coordination chemistry of Co4O4-based oxygen evolution catalysts by nitrogen-14/-15 and cobalt-59 NMR spectroscopy, Dalton Trans., 2024, 53, 8541–8545 RSC.
- D. Šorm, J. Blahut, B. Bashta, I. Císařová, E. Vrbková, E. Vyskočilová and J. Sedláček, Complex isomerism influencing the textural properties of organometallic Cu(salen) porous polymers: paramagnetic solid-state NMR characterization and heterogeneous catalysis, Dalton Trans., 2024, 53, 12162–12175 Search PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Paramagnetic NMR in solution and the solid state, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- F. Gul-E-Noor, B. Jee, A. Pöppl, M. Hartmann, D. Himsl and M. Bertmer, Effects of varying water adsorption on a Cu3(BTC)2 metal-organic framework (MOF) as studied by 1H and 13C solid-state NMR spectroscopy, Phys. Chem. Chem. Phys., 2011, 13, 7783–7788 RSC.
- F. Gul-E-Noor, B. Jee, M. Mendt, D. Himsl, A. Pöppl, M. Hartmann, J. Haase, H. Krautscheid and M. Bertmer, Formation of Mixed Metal Cu 3– x Zn x (btc) 2 Frameworks with Different Zinc Contents: Incorporation of Zn 2+ into the Metal–Organic Framework Structure as Studied by Solid-State NMR, J. Phys. Chem. C, 2012, 116, 20866–20873 CrossRef CAS.
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, A chemically functionalizable nanoporous material, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- L. V. Meyer, F. Schönfeld and K. Müller-Buschbaum, Lanthanide based tuning of luminescence in MOFs and dense frameworks--from mono- and multimetal systems to sensors and films, Chem. Commun., 2014, 50, 8093–8108 RSC.
- M. O. Rodrigues, J. D. L. Dutra, L. A. O. Nunes, G. F. de Sá, W. M. de Azevedo, P. Silva, F. A. A. Paz, R. O. Freire and S. A. Júnior, Tb 3+ →Eu 3+ Energy Transfer in Mixed-Lanthanide-Organic Frameworks, J. Phys. Chem. C, 2012, 116, 19951–19957 CrossRef CAS.
- J. W. de Oliveira Maciel, M. A. Lemes, A. K. Valdo, R. Rabelo, F. T. Martins, L. J. Queiroz Maia, R. C. de Santana, F. Lloret, M. Julve and D. Cangussu, Europium(III), Terbium(III), and Gadolinium(III) Oxamato-Based Coordination Polymers: Visible Luminescence and Slow Magnetic Relaxation, Inorg. Chem., 2021, 60, 6176–6190 Search PubMed.
- K. Liu, H. You, G. Jia, Y. Zheng, Y. Song, M. Yang, Y. Huang and H. Zhang, Coordination-Induced Formation of One-Dimensional Nanostructures of Europium Benzene-1,3,5-tricarboxylate and Its Solid-State Thermal Transformation, Cryst. Growth Des., 2009, 9, 3519–3524 Search PubMed.
- V. G. Nosov, A. S. Kupryakov, I. E. Kolesnikov, A. A. Vidyakina, I. I. Tumkin, S. S. Kolesnik, M. N. Ryazantsev, N. A. Bogachev, M. Y. Skripkin and A. S. Mereshchenko, Heterometallic Europium(III)-Lutetium(III) Terephthalates as Bright Luminescent Antenna MOFs, Molecules, 2022, 27(18), 5763 Search PubMed.
- C. J. Liang, T. C. Wong, L. S. Hung, S. T. Lee, Z. R. Hong and W. L. Li, Self-quenching of excited europium ions in Eu(DBM) 3 bath-based organic electroluminescent devices, J. Phys. D: Appl. Phys., 2001, 34, L61–L64 Search PubMed.
- P.-A. Hansen, C. S. Granerød, Ø. Prytz and O. Nilsen, Controlling luminescence and quenching mechanisms in subnanometer multilayer structure of europium titanium oxide thin films, J. Lumin., 2019, 215, 116618 Search PubMed.
- V. Haquin, M. Etienne, C. Daiguebonne, S. Freslon, G. Calvez, K. Bernot, L. Le Pollès, S. E. Ashbrook, M. R. Mitchell, J.-C. Bünzli, S. V. Eliseeva and O. Guillou, Color and Brightness Tuning in Heteronuclear Lanthanide Terephthalate Coordination Polymers, Eur. J. Inorg. Chem., 2013, 2013, 3464–3476 CrossRef CAS.
- A. N. Carneiro Neto, R. T. Moura, A. Shyichuk, V. Paterlini, F. Piccinelli, M. Bettinelli and O. L. Malta, Theoretical and Experimental Investigation of the Tb 3+ → Eu 3+ Energy Transfer Mechanisms in Cubic A 3 Tb 0.90 Eu 0.10 (PO 4 ) 3 (A = Sr, Ba) Materials, J. Phys. Chem. C, 2020, 124, 10105–10116 CrossRef CAS.
- P. A. Tanner, Some misconceptions concerning the electronic spectra of tri-positive europium and cerium, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- W. Qin, Y. Zhang, W. Liu and M. Tan, Synthesis and infrared and fluorescence spectral properties of luminescent terbium and europium complexes with open-chain carboxylate crown ethers, Spectrochim. Acta, Part A, 2003, 59, 3085–3092 CrossRef PubMed.
- F. Dang, Y. Li and W. Liu, The new fluorescence enhancement system Tb-N-(2-pyridinyl) ketoacetamide-Et(3)N-Zn and its application, Spectrochim. Acta, Part A, 2007, 66, 676–680 CrossRef PubMed.
- S. M. Z. Al-Kindy, Z. Al-Harasi, F. E. O. Suliman, A. Al-Hamadi and A. Pillay, Terbium sensitized luminescence for the determination of ketoprofen in pharmaceutical formulations, J. Fluoresc., 2009, 19, 249–255 CrossRef CAS PubMed.
- J. D. Fradgley, A. T. Frawley, R. Pal and D. Parker, Striking solvent dependence of total emission and circularly polarised luminescence in coordinatively saturated chiral europium complexes: solvation significantly perturbs the ligand field, Phys. Chem. Chem. Phys., 2021, 23, 11479–11487 RSC.
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, A luminescent mixed-lanthanide metal-organic framework thermometer, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed.
- T. Amiaud, V. Jubera and H. Serier-Brault, A new highly sensitive cryogenic luminescent MOF thermometer built with pyromellitic acid, J. Mater. Chem. C, 2023, 11, 10951–10956 Search PubMed.
- J. Perez-Obando, J. Manzur, P. Fuentealba, J. Morales, A. Vega, R. Costa de Santana, A. N. Carneiro Neto and E. Spodine, Trichromatic color tuning strategy for emission of heterometallic EuIII/TbIII coordination polymers with triazolyl-substituted 4-methyl-phenoxo ligand, J. Rare Earths, 2024 DOI:10.1016/j.jre.2024.06.036.
- A. N. Carneiro Neto, E. Mamontova, A. M. P. Botas, C. D. S. Brites, R. A. S. Ferreira, J. Rouquette, Y. Guari, J. Larionova, J. Long and L. D. Carlos, Rationalizing the Thermal Response of Dual-Center Molecular Thermometers: The Example of an Eu/Tb Coordination Complex, Adv. Opt. Mater., 2022, 10, 2101870 Search PubMed.
- C. Brites, A. Millán and L. D. Carlos, Lanthanides in Luminescent Thermometry, vol. 49, pp. , pp. 339–427 Search PubMed.
- A. Cadiau, C. D. S. Brites, P. M. F. J. Costa, R. A. S. Ferreira, J. Rocha and L. D. Carlos, Ratiometric nanothermometer based on an emissive Ln3+-organic framework, ACS Nano, 2013, 7, 7213–7218 CrossRef CAS PubMed.
- D. Zhao, X. Rao, J. Yu, Y. Cui, Y. Yang and G. Qian, Design and Synthesis of an MOF Thermometer with High Sensitivity in the Physiological Temperature Range, Inorg. Chem., 2015, 54, 11193–11199 CrossRef CAS PubMed.
- T. Chuasaard, A. Ngamjarurojana, S. Surinwong, T. Konno, S. Bureekaew and A. Rujiwatra, Lanthanide Coordination Polymers of Mixed Phthalate/Adipate for Ratiometric Temperature Sensing in the Upper-Intermediate Temperature Range, Inorg. Chem., 2018, 57, 2620–2630 CrossRef CAS PubMed.
- X. Zhou, H. Wang, S. Jiang, G. Xiang, X. Tang, X. Luo, L. Li and X. Zhou, Multifunctional Luminescent Material Eu(III) and Tb(III) Complexes with Pyridine-3,5-Dicarboxylic Acid Linker: Crystal Structures, Tunable Emission, Energy Transfer, and Temperature Sensing, Inorg. Chem., 2019, 58, 3780–3788 CrossRef CAS PubMed.
- E. Djanffar, H. A. Bicalho, Z. Ajoyan, A. J. Howarth and H. Serier-Brault, Rare-earth UiO-66 for temperature sensing near room temperature, J. Mater. Chem. C, 2024, 12, 8024–8029 RSC.
- L. Thi Kieu Giang, W. M. Piotrowski, N. Thanh Huong, H. Thi Khuyen, P. Thi Lien, D. Manh Tien, N. Vu, N. Hai Yen, P. Thanh Binh, V. Duc Chinh and Ł. Marciniak, Temperature sensing and magnetic properties of the Fe3O4@ZrOBDC:Eu3+,Tb3+ MMOF, Opt. Mater., 2024, 157, 116050 Search PubMed.
- S. S. Mortensen, V. R. M. Nielsen and T. J. Sørensen, Contrasting impact of coordination polyhedra and site symmetry on the electronic energy levels in nine-coordinated Eu(III) and Sm(III) crystals structures determined from single crystal luminescence spectra, Dalton Trans., 2024, 53, 10079–10092 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization. See DOI: https://doi.org/10.1039/d5ra00822k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |