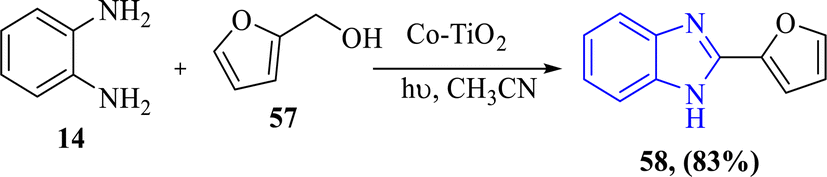DOI:
10.1039/D5RA00819K
(Review Article)
RSC Adv., 2025,
15, 22097-22127
Synthesis and SARs of benzimidazoles: insights into antimicrobial innovation (2018–2024)
Received
3rd February 2025
, Accepted 6th June 2025
First published on 30th June 2025
Abstract
Benzimidazole derivatives have garnered significant attention in medicinal chemistry owing to their versatile pharmacological properties, particularly their potent antimicrobial activity. This review comprehensively explores the advancements in the synthesis of benzimidazoles and their antimicrobial property evaluation from 2018 to 2024. Recent synthetic methodologies emphasize green chemistry approaches including solvent-free and catalyst-driven reactions, offering improved yields, selectivity, and environmental sustainability. Structural modifications, such as functionalization at positions 2 and 5/6 of the benzimidazole ring, were extensively investigated to enhance the antimicrobial efficacy against a broad spectrum of pathogens including multidrug-resistant bacterial and fungal strains. Furthermore, we elucidate the structure–activity relationships (SARs) of benzimidazole derivatives, enabling the rational design of highly potent antimicrobial agents. The mentioned period also witnessed the integration of hybrid molecules, wherein benzimidazoles were conjugated with other bioactive scaffolds to achieve synergistic antimicrobial effects.
1 Introduction
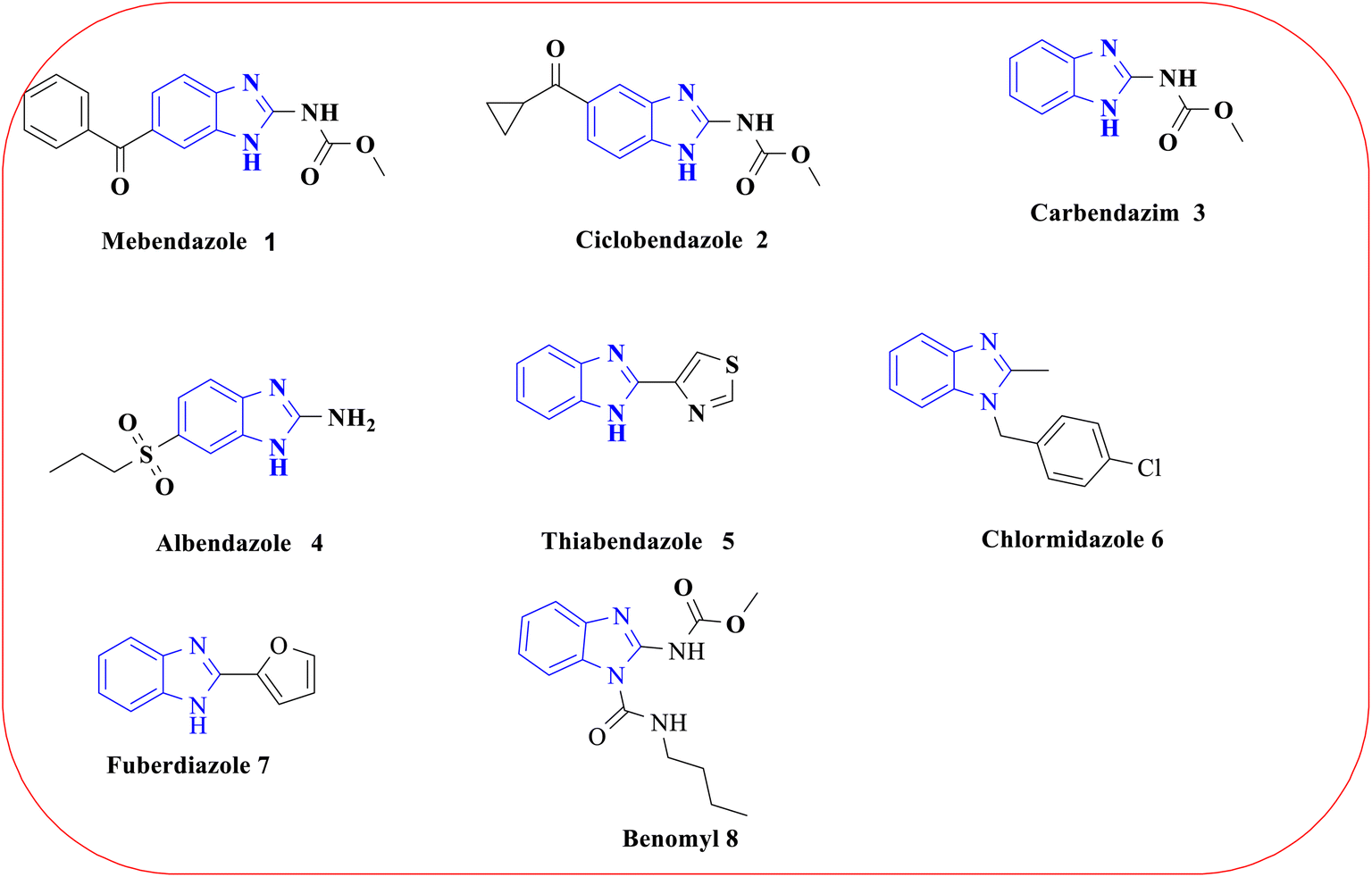
The history of heterocyclic chemistry first began in the 1800s alongside the progress of organic chemistry.1 Since 2020, nearly 65% of the literature in organic chemistry is based on heterocyclic chemistry.2 Heterocycles have important applications in the fields of chemistry, industry, and biology, as they play vital roles in the metabolic processes of all living cells.3 They typically consist of five- or six-membered rings and have at least one heteroatom, such as a nitrogen (N), oxygen (O), or sulfur (S) atom.4 Nitrogenous heterocyclic compounds have a significant influence in the process of discovering and developing drugs, primarily because they are commonly found in natural products and bioactive molecules.5 These compounds frequently exhibit a wide range of unique pharmacological effects, which make them interesting for researchers.6 Particularly, the benzimidazole ring has been extensively investigated in the field of medicinal chemistry since 1944,7 following the discovery of 5,6-dimethylbenzimidazole as a byproduct of vitamin B12 breakdown,8 owing to its structure similarity with DNA-purine nitrogen bases (adenine and guanine), and it consists of a bicyclic organic structure having an imidazole ring that contains two nitrogen atoms attached to one benzene ring.9,10 Multiple studies reported in the literature have presented an in-depth pharmacological framework of benzimidazoles and their derivatives,11 exhibiting potential biological activities such as antibacterial,12–14 antifungal,15,16 antiviral,17,18 antileishmanial,19,20 antimalarial21,22 and antiprotozoal23,24 functions. Currently, several benzimidazole-based drugs are available and commercially accessible, including mebendazole 1 (ref. 25), ciclobendazole 2 (ref. 26), carbendazim 3 (ref. 27), albendazole 4 (ref. 28), thiabendazole 5 (ref. 29), chlormidazole 6 (ref. 30), fuberidazole 7 (ref. 31) and benomyl 8 (ref. 27)(Fig. 1).32 Nevertheless, this review focused on the pharmacological properties of benzimidazole derivatives that have been evaluated between 2018 and 2024.
 |
| | Fig. 1 Some commercially available benzimidazole-based drugs. | |
1.1 Chemistry
The systematic nomenclature of the 1H-benzimidazole ring system is illustrated in structure 9. Although benzimidazole in 9 has been shown to have a proton at N1, there is, in fact, a quick exchange between the –NH– and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-nitrogen atoms, allowing for the appearance of two tautomers of the benzimidazole molecule. Tautomerism develops via an intermolecular mechanism involving two benzimidazole molecules 9 and 9a or upon interactions with a protic solvent like water33 (Fig. 2).
N-nitrogen atoms, allowing for the appearance of two tautomers of the benzimidazole molecule. Tautomerism develops via an intermolecular mechanism involving two benzimidazole molecules 9 and 9a or upon interactions with a protic solvent like water33 (Fig. 2).
 |
| | Fig. 2 Tautomerism of the benzimidazole nucleus. | |
1.2 Synthetic pathways
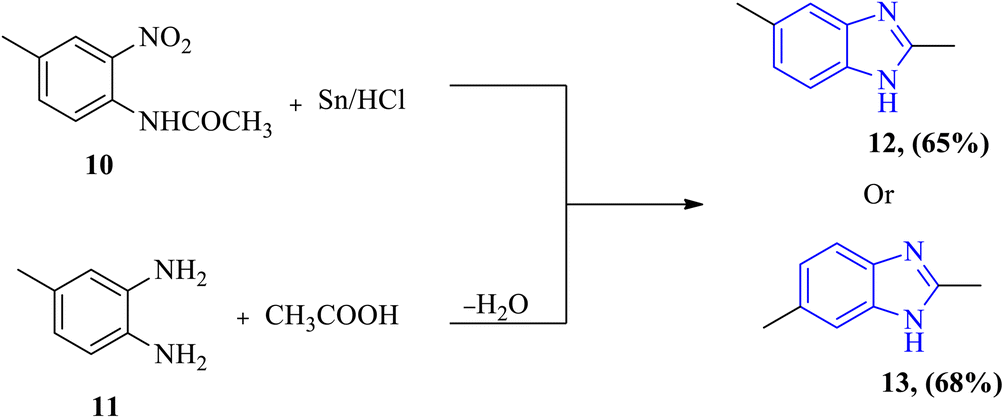
Due to the significant synthetic relevance and diverse bioactivities of benzimidazoles and their derivatives, efforts have been consistently made to create libraries of these molecules. Numerous synthetic processes have been designed and modified to achieve products with high yield and purity. An initial review revealed that the first benzimidazole 12 or 13 was produced in 1872 by Hoebrecker via the reduction of 4-methyl-2-nitroacetanilide 10.34 After several years, Ladenburg synthesized compounds 12 or 13 with a moderate yield by refluxing 3,4-diaminotoluene 11 with acetic acid; the term ‘Anhydrobase’ originated in the early literature to describe the loss of water during the synthesis of this type of chemical reaction (Fig. 3).35–37
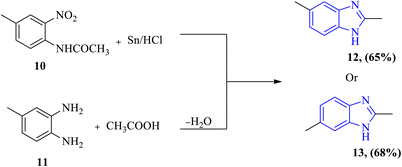 |
| | Fig. 3 The first schemes used for benzimidazole synthesis. | |
1.2.1 From carboxylic acids, esters or aldehydes. The primary synthesis methods for benzimidazole candidates via Phillip's method involve the condensation of O-phenylenediamine 14 with carboxylic acids 15, esters 17 or aldehydes 19 by cyclization under high acidic conditions such as hydrochloric acid, hot glacial acetic acid or boric acid. This method is widely utilized for the synthesis of various benzimidazoles (Scheme 1).38–40
 |
| | Scheme 1 Phillips method: synthesis of benzimidazole derivatives via the condensation of O-phenylenediamine 14 with carboxylic acids 15 under acidic conditions. | |
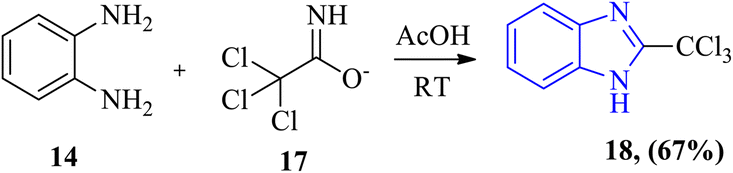
The proper imidate ester (trichloroacetimidate) 17 was employed as the starting material for the synthesis of benzimidazole derivatives by Alaqeel via reaction with O-phenylenediamine 14 or its salt to yield 2-trichloromethyl benzimidazole 18 at room temperature, which serves as a significant precursor for 2-carboxylic benzimidazoles with a good yield (Scheme 2).41
 |
| | Scheme 2 Synthesis of 2-trichloromethyl benzimidazole 18 from O-phenylenediamine 14 and trichloroacetimidate ester 17 at room temperature. | |
Benzimidazole derivative 20 can be synthesized from aldehydes, as reported by Heravi et al., via straightforward one-step synthesis from O-phenylenediamine 14 and 2-methylbenzaldehyde 19 in the presence of boric acid as an efficient catalyst under mild aqueous conditions (Scheme 3).42
 |
| | Scheme 3 One-step synthesis of benzimidazole derivative 20 from O-phenylenediamine 14 and 2-methylbenzaldehyde 19 using boric acid under mild aqueous conditions. | |
1.2.2 Metal-catalyzed reactions. Metal-catalyzed reactions have emerged as a cornerstone in the synthesis of benzimidazole derivatives, providing efficient, selective, and versatile routes for constructing these biologically significant heterocycles. Transition metal catalysts such as palladium (Pd) and copper (Cu) play a pivotal role in facilitating key transformations including cyclization and functionalization of benzimidazole scaffolds.43–45A metal-catalyzed reaction was demonstrated by Srimani et al. for the synthesis of benzimidazole through coupling aromatic diamines 14 with primary alcohols 21, as illustrated in Scheme 4. This reaction employs a phosphine-free tridentate NNS–manganese(I) complex as a catalyst, utilizing cost-effective bases such as KOH, t-BuOK, and K2CO3, resulting in 22 and 23 compounds with excellent yields (Scheme 4).46
 |
| | Scheme 4 Manganese(I)-catalyzed synthesis of benzimidazoles 22 and 23 from aromatic diamines 14 and primary alcohols 21 using a phosphine-free NNS–Mn complex. | |
A sulfur-based catalyst was shown to efficiently produce benzimidazole derivatives according to Nguyen et al. The reaction initiates with 1,2-dibenzyldisulfane 24 and O-phenylenediamine derivative 25 in N-methyl-2-pyrrolidone (NMP) and DMSO at 100 °C to obtain benzimidazole derivative 26.47 Moreover, in the same year, Nguyen reported a multi-component one-pot synthesis using maleic anhydride 28, benzyl amine 27, and O-phenylenediamine 14, which undergo oxidation, decarboxylation, and cyclization to form a benzimidazole product 29. In this reaction, sulfur functions as a catalyst, while DMSO serves as an oxidizing agent, resulting in a substantial yield of the benzimidazole scaffold (Scheme 5).48
 |
| | Scheme 5 Sulfur-catalyzed synthesis of benzimidazole 26 from 1,2-dibenzyldisulfane 24 and O-phenylenediamine derivative 25 in NMP/DMSO at 100 °C. One-pot, multi-component synthesis of benzimidazole 29 from benzylamine 27, maleic anhydride 28, and O-phenylenediamine 14 using a sulfur catalyst and DMSO as an oxidant. | |
Using a copper–palladium catalyst significantly improved the efficacy of the reaction between benzyl alcohol 30 and O-phenylenediamine 14 in the synthesis of benzimidazole 31, as illustrated by Mokhtari, which underwent a dehydrogenative coupling reaction as a novel catalyst under solvent-free conditions. The catalyst employed in this reaction is reusable (Scheme 6).49
 |
| | Scheme 6 Copper-palladium-catalyzed solvent-free synthesis of benzimidazole 31 from benzyl alcohol 30 and O-phenylenediamine 14 via dehydrogenative coupling. | |
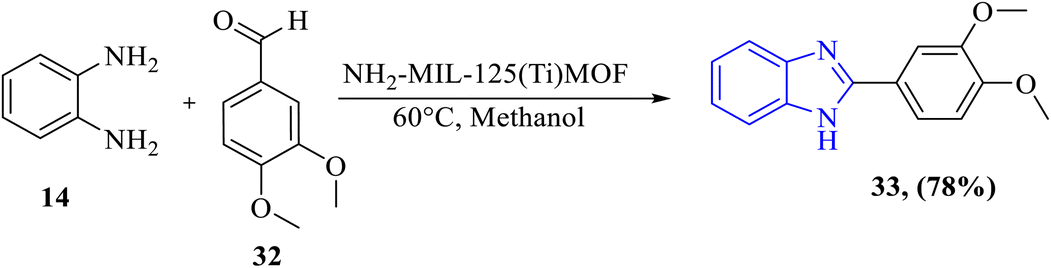
The research conducted by Sankar and his team in 2020 reported expeditious synthesis of benzimidazole 33 by the reaction of O-phenylenediamine 14 via oxidative cyclization using a metal–organic framework (MFO) as an efficient catalyst, as it increases the electrophilicity of 3,4-dimethoxybenzaldehyde 32 and enhances the rate of the reaction resulting in high yields with short time of the reaction (Scheme 7).50
 |
| | Scheme 7 MOF-catalyzed oxidative cyclization of O-phenylenediamine 14 with 3,4-dimethoxybenzaldehyde 32 for the rapid synthesis of benzimidazole 33. | |
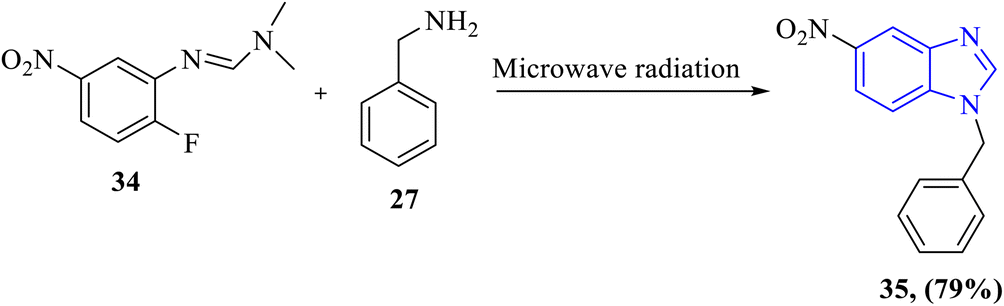
1.2.3 Metal-free benzimidazole synthesis. Metal-free synthesis of benzimidazoles has emerged as a sustainable and environmentally friendly alternative to traditional metal-catalyzed methods. These approaches align with the principles of green chemistry, avoiding the use of expensive, toxic, or scarce metal catalysts, and often operate under mild conditions with high yields.26,51,52 In 2019, Liu et al. presented a metal-free method for the synthesis of N-substituted benzimidazole 35 with microwave radiation. The reaction between fluoro-aryl formamidines 34 and derivatives of primary amines 27 gives high yields (Scheme 8).53
 |
| | Scheme 8 Metal-free microwave-assisted synthesis of N-substituted benzimidazole 35 from a fluoro-aryl formamidine 34 and a primary amine derivative 27. | |
A metal-free reaction including a simple reagent was performed by Mostafavi et al. for the synthesis of 37. In this reaction, O-phenylenediamine 14 and DMF 36 converted into benzimidazole using hexamethyldisilazane (HMDS) as a reagent. This reaction is free of any acid, transition metal, or solvent, yet it produces a great yield (Scheme 9).54
 |
| | Scheme 9 Metal-free synthesis of benzimidazole (37) from O-phenylenediamine 14 and DMF 36 using hexamethyldisilazane (HMDS). | |
The one-step reaction approach for the synthesis of benzimidazole was demonstrated by Feizpour et al., utilizing a cobalt ascorbic acid complex coated on TiO2 nanoparticles to synthesize benzimidazoles 42 that, upon exposure to intermediate 41, underwent aerobic photooxidative cyclization reaction to offer the target compound 42. The catalyst in this reaction is reusable, and the process is conducted by photocatalysis-based organic synthesis (Scheme 10).55
 |
| | Scheme 10 Photocatalytic one-step synthesis of benzimidazoles 42 via aerobic photooxidative cyclization of intermediate 41 using a cobalt ascorbate-TiO2 nanoparticle catalyst. | |
A sustainable approach for synthesizing benzimidazole 44 was developed by Gan et al. using the O-phenylenediamine derivative 43, in which it was condensed with dimethylformamide and its derivatives. In this approach, an imidazolium hydrochloride salt serves as the chemical initiator and activates formamide (Scheme 11).56
 |
| | Scheme 11 Sustainable synthesis of benzimidazole 44 via the condensation of O-phenylenediamine derivative 43 with dimethylformamide using imidazolium hydrochloride salt as an initiator. | |
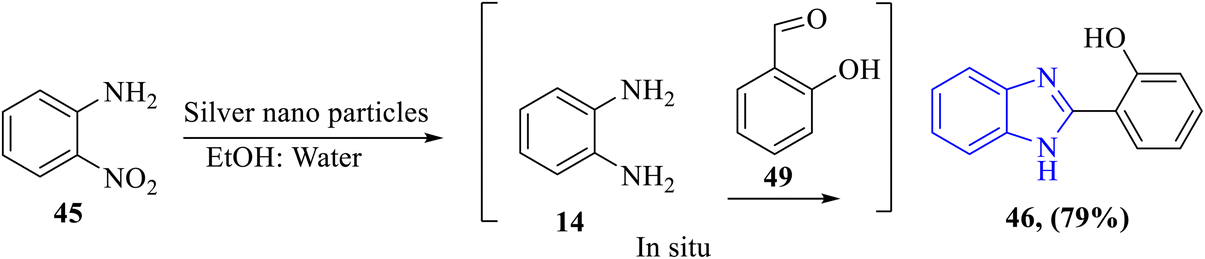
1.2.4 Green synthesis of benzimidazole. The green synthesis of benzimidazoles has emerged as a focal point in medicinal chemistry, aligning with the principles of sustainability, environmental responsibility, and energy efficiency. Recent advancements in this area emphasize the use of eco-friendly reagents, renewable resources, and energy-saving methodologies, reducing the environmental footprint while maintaining high synthetic efficiency.57–60A novel, environmentally friendly method for the synthesis of benzimidazole 46 was reported by Arya et al. via one-pot route synthesis of 2-nitroaniline 45 using novel silver nanoparticles from green algae as a catalytic agent, resulting in its reduced form 14 that reacts with appropriate aldehyde 49. Interestingly, it produced a high yield product (Scheme 12).61
 |
| | Scheme 12 Green one-pot synthesis of benzimidazole 46 via silver nanoparticle-catalyzed reduction of 2-nitroaniline 45 to O-phenylenediamine 14, followed by reaction with aldehyde 49. | |
The work by Di Gioia et al. in 2019 presented a method for the synthesis of benzimidazole scaffolds 50 and 51 via the reaction of O-phenylenediamine 14 with appropriate aldehydes 48 and 49 using Deep Eutectic Solvents (DESs) such as choline chloride 47, as it serves as a medium and reagent at the same time. It provides advantages concerning the yield regarding compound 50. Furthermore, the reaction with two molar amounts of aldehydes containing electron-withdrawing groups (as 2,4-dichlorobenzaldehyde) might occur owing to the electron-deficient aldehydes having a lower density of negative charge on the oxygen atom of the carbonyl group. Hence, they do not simply coordinate with the DES components, leading to the hydrogen bonding interaction, and therefore, the formation of the disubstituted product 51 is not favored (Scheme 13).62
 |
| | Scheme 13 Synthesis of benzimidazole 50 from O-phenylenediamine 14 and aldehyde 48 using deep eutectic solvents (choline chloride 47 serving as both the medium and the reagent). | |
In their research published in 2021, Thakore and his team reported the synthesis of polymer vesicles loaded with copper nanoparticles (CuNPs@vesicles) functioning as nanoreactors. These nanoreactors were utilized for the production of 2-substituted benzimidazole 53 in one-pot synthesis. The CuNPs@vesicles converted 2-nitroaniline into O-phenylenediamine 14, which subsequently serves as a precursor for the production of benzimidazole. The authors also investigated the impact of substituents on aldehydes. Thus, aldehydes substituted with electron-withdrawing groups (EWG) as 52 need shorter reaction times than those substituted with electron-releasing groups (ERG). The complete series of reactions occurs in an aqueous medium under ambient circumstances (Scheme 14).63
 |
| | Scheme 14 One-pot synthesis of 2-substituted benzimidazole 53 using CuNPs@vesicle nanoreactors, converting 2-nitroaniline into O-phenylenediamine 14, followed by reaction with substituted aldehydes 52 in an aqueous medium. | |
1.2.5 Photo-catalyzed reaction. Photo-catalyzed reactions have gained significant attention as innovative and sustainable approaches for synthesizing benzimidazole derivatives. Leveraging light energy to drive chemical transformations, these methodologies align with green chemistry principles, offering high efficiency, mild reaction conditions, and reduced environmental impact. This section highlights the recent advancements in photo-catalyzed strategies for benzimidazole synthesis.64–66In a study published in 2021, Montini et al. reported the synthesis of benzimidazole via a one-pot process through irradiation of dinitrobenzene 54 in 96% ethanol using Pt/TiO2–B, N as a photocatalyst, where the molar level of dinitrobenzene gradually reduced with total conversion achieved after approximately 17 hours. Throughout that time, the molar proportion of nitroaniline 45 increased rapidly within the first 2 hours, exceeding 30–40%, and remained within this range until the complete consumption of dinitrobenzene; subsequently, the quantity of nitroaniline decreased as it was reduced and transformed into O-phenylenediamine 14 that reacted with appropriate aldehyde 55 resulting in benzimidazole product 56 (Scheme 15).67
 |
| | Scheme 15 One-pot synthesis of benzimidazole 56 via photocatalytic reduction of dinitrobenzene (54) to O-phenylenediamine 14 followed by condensation with aldehyde 55 using a Pt/TiO2–B,N catalyst in ethanol. | |
The photocatalytic properties of cobalt-loaded titanium dioxide (Co–TiO2) under solar light exposure, as shown by Kumaraswamy et al. in 2022, reported an eco-friendly approach for the synthesis of 2-aryl benzimidazoles 58, where the Co–TiO2 photocatalysts exhibited superior catalytic performance relative to pure TiO2 attributed to greater charge separation and visible-light activity. The synthesis procedure performed with renewable solar energy produced good yields up to 85% with optimized 2% Co–TiO2 catalysts via a reaction with O-phenylenediamine 14 with primary alcohol furan-2-ylmethanol 57. This method provides the concepts of green chemistry (Scheme 16).68
 |
| | Scheme 16 Solar-light-driven synthesis of 2-aryl benzimidazole 58 from O-phenylenediamine 14 and furan-2-ylmethanol 57 using a cobalt-loaded TiO2 (Co–TiO2) photocatalyst. | |
The 2023 research by Abdelhamid et al. developed the synthesis of benzimidazole 60 by the reaction of O-phenylenediamine 14 with suitable aldehyde 59 utilizing a solid-state acid composed of zirconium oxosulfate embedded into carbon (ZrOSO4@C) in a single vessel as a catalyst allowing condensation and cyclization under light radiation at room temperature which resulted in high yields and purity (Scheme 17).69
 |
| | Scheme 17 Room-temperature light-assisted synthesis of benzimidazole 60 from O-phenylenediamine 14 and aldehyde 59 catalyzed using solid-state ZrOSO4@C acid. | |
1.2.6 Efficient synthetic routes for benzimidazole-based compounds. The synthesis of benzimidazole-based scaffolds has developed through different efficient strategies depending on the desired product and reaction conditions. Traditional methods including cyclocondensation of O-phenylenediamines with carboxylic acids, esters, or aldehydes remain highly efficient and widely used due to their simplicity and good yields.39,44 Furthermore, metal-catalyzed reactions such as copper- or palladium-mediated oxidative cyclization are particularly significant for synthesizing benzimidazole derivatives.46 Moreover, metal-free approaches are gaining attention for their lower toxicity especially in pharmaceutical applications where metal residues are undesirable.56 Green synthesis protocols, employing water and Deep Eutectic Solvents (DESs), often under solvent-free or microwave-assisted conditions, offer reduced reaction times and cleaner profiles.62 Finally, emerging photo-catalyzed methods using visible light activation have opened new avenues for environmentally sustainable benzimidazole synthesis under mild conditions without harsh reagents.67 In parallel, green synthetic strategies are increasingly recognized for their efficiency and compatibility with green chemistry principles. Taken together, these two approaches are distinguished as the most efficient and practical for the synthesis of benzimidazole scaffolds, offering a balance of accessibility and sustainability that aligns well with the current demands of medicinal and organic chemistry research.
1.2.7 Toxicological considerations and structural optimization in benzimidazole-based compounds. Evaluating the toxicity of benzimidazole-based compounds is essential during the early stages of their drug development, particularly due to their structural similarity to purine bases like adenine and guanine.70 Several benzimidazole derivatives reviewed here, such as those containing triazole, azomethine, or metal-coordinating groups, exhibit promising antimicrobial activities with favorable safety profiles, as evidenced by in vitro cytotoxicity data and brine shrimp assays. Planar aromaticity, the presence of reactive functional groups such as aldehydes, and the ability of a molecule to chelate metal ions are known as structural features that may contribute to toxicity through off-target binding.71,72 To control toxicological liabilities, the rational design that includes minimizing lipophilicity may reduce nonspecific binding and ensure metabolic stability to prevent bioactivation into toxic intermediates. Furthermore, compounds such as Ni(II) complexes suggest that controlled coordination chemistry can enhance safety as mentioned before for compound 61a.73 Hence, future benzimidazole derivatives should be optimized not only for their potency but also for their ADMET properties. This can be achieved through in silico prediction tools and preliminary evaluations that aimed to minimize mutagenicity, carcinogenicity, and organ-specific toxicity.
1.2.8 Evaluation of nitrosamine risk in benzimidazole-based scaffolds. While benzimidazole-based scaffolds are generally not classified as high-risk frameworks for nitrosamine formation, it is important to assess the structural features of synthesized derivatives for potential risk. In the present review, the majority of the synthesized compounds including Schiff bases, triazole hybrids, and metal complexes do not possess secondary or tertiary amines directly attached to the benzimidazole core that were regularly linked in nitrosamine formation. However, a few compounds such as those featuring N-alkylated benzimidazoles may contain nitrosamine precursors, particularly if exposed to nitrosating agents under acidic conditions during synthesis. Furthermore, synthetic routes that include formamidine intermediates, DMF, or dimethylamine derivatives may theoretically introduce trace nitrosamine risk.74 To date, nitrosamine risk assessments are not reported for benzimidazole derivatives at the discovery stage, but considering their increasing regulatory importance, especially FDA, such evaluations should become a routine component of early-stage drug development. Rational design strategies aimed to minimize the risk of nitrosatable amines, substituting with safer reagents and monitoring residual solvents. Collectively, future studies on benzimidazole-based compounds should integrate predictive nitrosamine risk models.
2 Pharmacological activities
2.1 Antibacterial activities
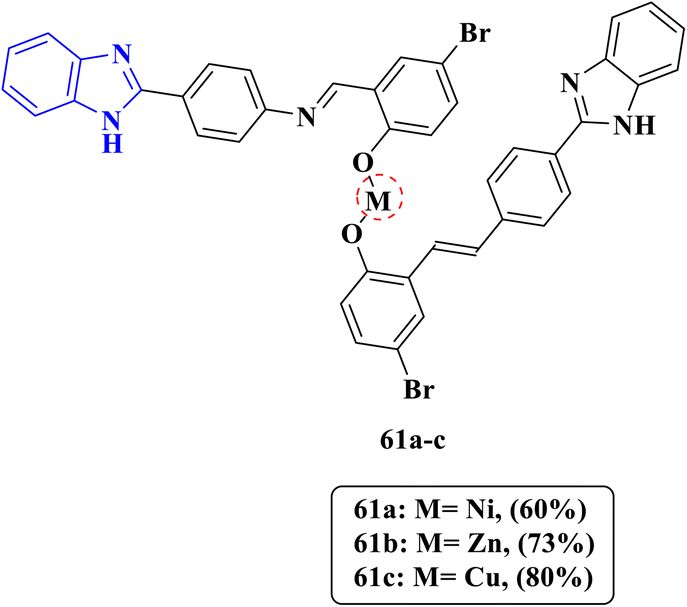
Due to the isosteric nature of benzimidazole and its derivatives with purine molecules, a competitive interaction occurs, leading to the inhibition of nucleic acid synthesis, which results in cell damage and cell death. Additionally, it has been noted that some benzimidazole compounds have the ability to interfere with the process of folate biosynthesis in microbial cells, thus blocking the formation of folates resulting in the inhibition of bacterial growth. Mahmood et al. designed and synthesized benzimidazole derivatives containing metals incorporated with an azomethine group, leading to the formation of complexes, which seem to be responsible for various biological activities with high safety margin. Their mode of action depends on binding to DNA. thus preventing the replication process of genetic materials. Compounds 61a–c were investigated against one Gram-positive bacterial strain Micrococcus luteus and one Gram-negative strain Escherichia coli (Fig. 4). Compound 61a displayed outstanding antibacterial activities with effective inhibition zone diameters (DIZ = 16.8 mm and 18.8 mm) comparable to the inhibition zone diameter values of the reference drug kanamycin (DIZ = 24.6 mm and 21.2 mm), against M. luteus and E. coli, respectively. Furthermore, compounds 61b and 61c containing other metals, such as Zn and Cu, exhibited activity only against E. coli (DIZ = 18.9 mm and 11.8 mm, respectively) (Table 1). The results were confirmed through the inhibition of DNA gyrase via binding activity by a thermal denaturation test, and as the temperature of the double-stranded DNA solution increases, the double helix undergoes denaturation due to the dissociation of hydrogen bonds between the base pairs. Therefore, it can be concluded that the Ni complex is a strong DNA binder due to its high lipophilicity increasing penetration through lipid membranes, facilitating the interaction with DNA by intercalation; then, the Cu complex is the weakest DNA binder, representing an advantage in terms of safety, as the Ni complex displayed no toxicity in the brine shrimp assay, with the exception of the Cu complex which showed slight toxicity.73
 |
| | Fig. 4 Chemical structures of metal-complexed benzimidazole derivatives 61a–c evaluated for antibacterial activity. | |
Table 1 Antibacterial activity diameter inhibition zones of compounds 61a–c compared to kanamycin against Micrococcus luteus and Escherichia coli
| Compound |
Diameter inhibition zone (mm) |
| M. luteus |
E. coli |
| 61a |
16.8 |
18.8 |
| 61b |
11 |
18.9 |
| 61c |
14.9 |
11.8 |
| Kanamycin |
6.8 |
8.4 |
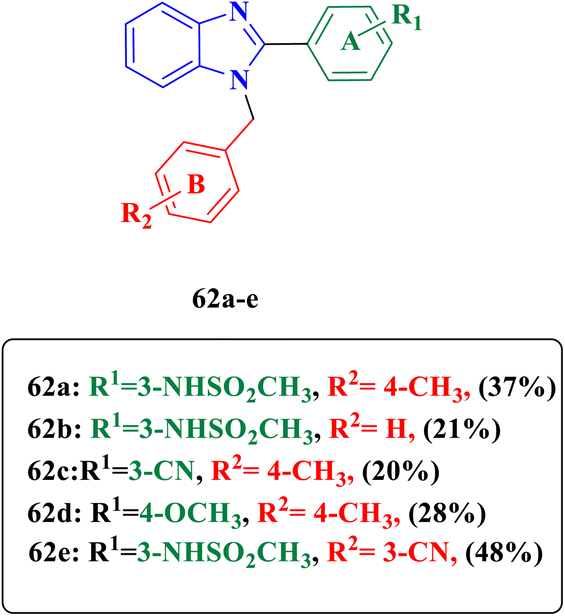
Dokla and collaborators designed and synthesized N-alkyl-2-substituted-1H-benzimidazole derivatives 62a–d and evaluated their activity against one bacterial strain E. coli (TolC mutant strain) (Fig. 5), as well as the presence of an outer membrane in most Gram-negative bacteria imparts an additional barrier against the penetration of many antibiotics. Efflux pumps might also be a reason for the lack of antimicrobial activity; hence, designated compounds were co-administrated with colistin (membrane disrupting antibiotic) to detect if the outer membrane impedes the antibacterial activity of the synthesized compounds. Compound 62a was the derivative with the best activity against E. coli (MIC = 2 μg mL−1) compared to the reference linezolid (MIC = 8 μg mL−1). Moreover, compounds 62b and 62c displayed good activity (MIC = 16 μg mL−1). However, compounds 62d and 62e exhibited weak activity against E. coli (MIC values > 128 μg mL−1) (Table 2). Moreover, compound 62a represents an excellent safety profile against human colorectal (Caco-2) and monkey kidney epithelial cells. Structure activity relationship (SAR) revealed that according to ring A introduction substituents at meta position give excellent antimicrobial activity 62a–c also 3-NHSO2CH3 group is optimal for activity while substitution at para positions diminished antibacterial activity as compound 62d. Furthermore, ring B substitution at para position gives outstanding antimicrobial activity to 62c compared to meta substituents 62e.75
 |
| | Fig. 5 N-alkyl-2-substituted benzimidazoles 62a–e evaluated with colistin for enhanced Gram-negative activity. | |
Table 2 Minimum inhibitory concentrations (MIC) of compounds 62a–e against E. coli (TolC mutant strain) in the presence of colistin, with comparison to linezolid
| Compound |
Minimum inhibition concentration (μg mL−1) |
| E. coli |
| 62a |
2 |
| 62b |
16 |
| 62c |
16 |
| 62d |
>128 |
| 62e |
>128 |
| Linezolid |
8 |
In 2018, Bistrović and colleagues designed novel scaffolds of benzimidazole derivatives by variation in positioning 2,5 with phenyl substituents on the triazole ring, and the amidino moieties allowed the development of compounds with comparable activity as an antibacterial agent. Compounds 63a–e were investigated against two Gram-positive bacterial strains, namely, Methicillin-resistant Staphylococcus aureus (MRSA) and E. faecalis, and three Gram-negative bacteria, namely, E. coli, K. pneumoniae and P. aeruginosa (Fig. 6). Compounds 63a displayed broad-spectrum antibacterial activities against MRSA and E. faecalis (MIC = 16 μg mL−1 and 32 μg mL−1 respectively), comparable to the MIC value of the reference drug ampicillin (4 μg mL−1 and 1 μg mL−1, respectively) and excellent activity against E. coli and K. pneumoniae with MIC values of 4 μg mL−1 and 8 μg mL−1, respectively, in comparison with the MIC value of the reference drug ceftazidime (8 μg mL−1 and >128 μg mL−1) with high affinity to DNA presenting high potency against extended-spectrum β-lactamase (ESBL)-producing E. coli and K. pneumoniae. Compound 63c exhibited greater antibacterial activity against only Gram-positive bacterial strains than compound 63a with MIC values of 8 μg mL−1 and 32 μg mL−1 with respect to the reference drug ampicillin. Furthermore, compounds 63d and 63e demonstrated less antibacterial activity only against Gram-positive bacteria with MIC values ranging from 128 μg mL−1 to 256 μg mL−1, while compound 63b showed no activity against all bacterial strains (Table 3). Furthermore, the active compounds still had less antibacterial activities than ampicillin, which might be due to irreversible binding in cell wall biosynthesis. In contrast, the designated compounds have alternative modes of action, including DNA interaction and potential membrane disruption, which are inherently less potent in the initial stages of development. These findings support the need for future studies focused on optimizing triazole substitution patterns or combining these compounds with ampicillin to enhance antibacterial efficacy through dual mechanisms. According to SAR evaluations, it was observed that the type of amidino moiety had positive impact one the activity in case of no substitution gave wide spectrum compound 63a on both Gram-positive and Gram-negative bacteria while the lipophilic character of isopropyl and chloro substitutions increased Gram-positive selectivity, especially against MRSA strain 63c while introduction of bulky methoxy group at para position revealed diminished or no antibacterial against all tested strains 63b and 63e.76
 |
| | Fig. 6 Structures of benzimidazole–triazole hybrids with amidino moieties 63a–e, synthesized for evaluation against Gram-positive and Gram-negative bacteria. | |
Table 3 MIC values of benzimidazole–triazole derivatives 63a–e against Gram-positive and Gram-negative bacterial strains, against ampicillin and ceftazidime
| Compound |
Minimum inhibition concentration (μg mL−1) |
| MRSA |
E. faecium |
E. coli |
K. pneumoniae |
| 63a |
16 |
32 |
4 |
8 |
| 63b |
— |
256 |
— |
— |
| 63c |
8 |
32 |
— |
— |
| 63d |
128 |
128 |
— |
— |
| 63e |
256 |
256 |
— |
— |
| Ampicillin |
4 |
1 |
— |
— |
| Ceftazidime |
— |
— |
8 |
>128 |
In 2021, Rashdan et al., reported that benzimidazole derivatives 64a–c (Fig. 7) induced highly expected antimicrobial activity through inhibition DNA gyrase subunit B leading to disruption of DNA synthesis, subsequently, cell death. DNA gyrase is considered as an essential bacterial enzyme that is included in the control of topological transitions of DNA, therefore the enzyme has been selected as a therapeutic target for many antimicrobial agents. Compounds were screened against one Gram-positive bacteria, S. aureus, and two Gram-negative bacteria, E. coli and P. aeruginosa, to evaluate their antibacterial efficacy. Compounds 64a and 64b exhibited excellent antibacterial activity against S. aureus, E. coli and P. aeruginosa with an inhibition zone diameter ranging from 17 mm to 29 mm compared to that of the reference drug ciprofloxacin (20 mm, 23 mm and 21 mm, respectively). Furthermore, compound 64c showed good antibacterial activity against S. aureus with an inhibition zone diameter of 23 mm and moderate activity against P. aeruginosa with an inhibition zone diameter of 13 mm with respect to the inhibition zone diameter of the control drug ciprofloxacin of 20 mm and 21 mm, respectively (Table 4). Moreover, it did not prove any activity against E. coli. Moreover, molecular docking studies for designated compounds 64a and 64b displayed similar binding modes towards the targeted enzyme DNA gyrase B as the co-crystallized ligand ciprofloxacin with Thr165. The SAR findings suggested that the presence of chlorine atom in the phenyl ring at position-4 in derivative 64b was favorable, also existence of thiadiazole ring 64a and 64b essential for activity and give antibacterial action against all tested strains thus could be illustrated in compound 64c.77
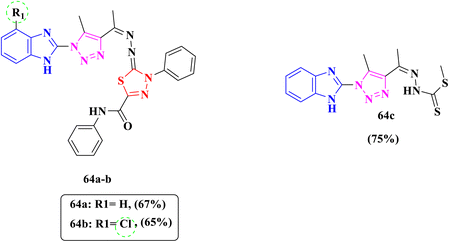 |
| | Fig. 7 Benzimidazole-based compounds 64a–c developed as antibacterial agents targeting the DNA gyrase B subunit. | |
Table 4 Diameter inhibition zone of compounds 64a–c against S. aureus, E. coli, and P. aeruginosa, compared to ciprofloxacin
| Compound |
Diameter inhibition zone (mm) |
| S. aureus |
E. coli |
P. aeruginosa |
| 64a |
24 |
25 |
17 |
| 64b |
29 |
21 |
19 |
| 64c |
23 |
— |
13 |
| Ciprofloxacin |
20 |
23 |
21 |
Deswal and his team designed a novel series of benzimidazole-1,2,3-triazole-indoline derivatives exemplified by 65a–b using a click reaction (Fig. 8). Compounds were screened against two bacterial strains to evaluate their activity: one Gram-negative bacteria, E. coli, and one Gram positive bacteria S. aureus. They displayed significant antibacterial activity. Compound 65a showed excellent activity against E. coli and S. aureus with MIC values of 0.026 μg mL−1 and 0.031 μg mL−1, respectively, compared to the control drug norfloxacin with MIC values of 0.039 and 0.020 μg mL−1, whereas Compound 65b exhibited excellent activity against E. coli and good activity against S. aureus with MIC values of 0.030 μg mL−1 and 0.060 μg mL−1, respectively, comparable to the MIC reference drug norfloxacin (Table 5). The notable broad-spectrum antibacterial activity of the compounds particularly 65a proved to be associated with the presence of a hydrophilic moiety as the pyridine ring at position 2 of the benzimidazole nucleus and typically electron-withdrawing group, as (NO2) on the indole moiety was optimal for activity. However, the presence of a lipophilic moiety as the methyl group proved to be selective against E. coli 65b.78
 |
| | Fig. 8 Benzimidazole–triazole–indoline hybrids 65a–b designed as antibacterial agents against E. coli and S. aureus. | |
Table 5 MIC values of 65a–b against E. coli and S. aureus compared to norfloxacin
| Compound |
Minimum inhibition concentration (μg mL−1) |
| E. coli |
S. aureus |
| 65a |
0.026 |
0.031 |
| 65b |
0.030 |
0.060 |
| Norfloxacin |
0.039 |
0.020 |
The research by Saber et al. proved that a 1,2,3-triazole moiety could serve as a linker connected to the benzimidazole via a cycloaddition reaction. This parameter plays an essential role in membrane permeation into the microbial cell, and the resulting bio isosteric effects on peptide linkage through hydrogen bond formation, dipole–dipole interaction and π stacking interactions led to high-affinity binding with biological targets increasing membrane permeation into the microbial cell. Moreover, the intermolecular interaction of the designated compounds was determined by Hirshfeld surface analyses; in addition, the Monte Carlo method was used to investigate the interfacial interaction of these derivatives with iron, copper and aluminum surfaces and provide better anti-corrosion properties for iron than copper and aluminum. Moreover, substitution on N1 on the triazole moiety with aliphatic ester chain, compound 66a, gave outstanding broad-spectrum antibacterial activities compared to substitution with the benzyl ring, compound 66b, that had selectivity only against Gram-positive bacteria, while increasing the length of aliphatic chain abolished the antimicrobial activity of 66c. Compounds 66a–c were investigated for their antimicrobial activity against one Gram-positive bacteria, S. aureus, and two Gram-negative bacterial strains, E. coli and P. aeruginosa (Fig. 9). Compound 66a exhibited greater antibacterial activity against S. aureus and E. coli with an equipotent MIC value of 3.12 μg mL−1 compared with the MIC value of the control drug chloramphenicol, 12.50 μg mL−1 and 6.25 μg mL−1, respectively, while compound 66b showed excellent activity particularly on S. aureus with an MIC value of 3.12 μg mL−1 and mild activity against E. coli with an MIC value of 25 μg mL−1, comparable to the MIC value of the reference drug chloramphenicol. However, compound 66c displayed weak antibacterial activity with MIC values of 25 μg mL−1, 12.50 μg mL−1 and 25 μg mL−1 against S. aureus, E. coli and P. aeruginosa, respectively. Compounds 66a and 66b had less antibacterial action on P. aeruginosa with MIC values of 12.50 μg mL−1 and 25 μg mL−1, respectively, compared to the MIC value of 6.25 μg mL−1 of the reference drug chloramphenicol (Table 6).79
 |
| | Fig. 9 Benzimidazole–triazole hybrids 66a–c tested against S. aureus, E. coli, and P. aeruginosa. | |
Table 6 Minimum inhibitory concentrations (MIC, μg mL−1) of compounds 66a–c against S. aureus, E. coli, and P. aeruginosa, compared with chloramphenicol
| Compound |
Minimum inhibition concentration (μg mL−1) |
| S. aureus |
E. coli |
P. aeruginosa |
| 66a |
3.125 |
3.125 |
12.5 |
| 66b |
3.125 |
25 |
12.5 |
| 66c |
25 |
12.5 |
25 |
| Chloramphenicol |
12.5 |
6.25 |
6.25 |
Al-blewi and his team proved that antibacterial activity is affected by substitutions on the benzimidazole ring, while the antibacterial potency of the mono-substituted benzimidazole 67a was found to be less than that of the di-substituted benzimidazole 67b–c, which may be attributed to the synergistic effect between the benzimidazole and sulfonamoyl nucleus and the enhanced lipophilicity of the bis-substituted derivatives that positively affects the activity against Gram-negative tested strains. Compounds 67a–c were investigated for their antimicrobial activity against two Gram-positive bacteria, Bacillus cereus and S. aureus, and two Gram-negative bacterial strains, E. coli and P. aeruginosa (Fig. 10). Compound 67b showed outstanding antibacterial activity against all the tested bacterial strains with an equipotent MIC value of 32 μg mL−1 against B. cereus and S. aureus comparable to the MIC value of the reference drug ciprofloxacin 8 μg mL−1 and 4 μg mL−1, respectively, and an equipotent MIC value of 64 μg mL−1 against E. coli and P. aeruginosa in comparison with the MIC value of ciprofloxacin 8 μg mL−1 and 4 μg mL−1, respectively. Moreover, derivative 67c displayed good antibacterial activity against all tested strains with an equipotent MIC value of 64 μg mL−1. Furthermore, compound 67a exhibited less antibacterial properties against B. cereus and S. aureus strains with an equal MIC value of 64 μg mL−1 comparable to the control drug ciprofloxacin and against E. coli and P. aeruginosa with MIC values of 256 μg mL−1 and 128 μg mL−1, respectively, with respect to the MIC value of the control drug ciprofloxacin (Table 7). Toxicity results for both compounds displayed good safety margin with neither carcinogenicity nor mutagenicity. In addition, in silico ADMET evaluation of the designated compound 67b meets the criteria of drug-likeness and obeys Lipinski's rule of five, presenting a good candidate of further research and development.80
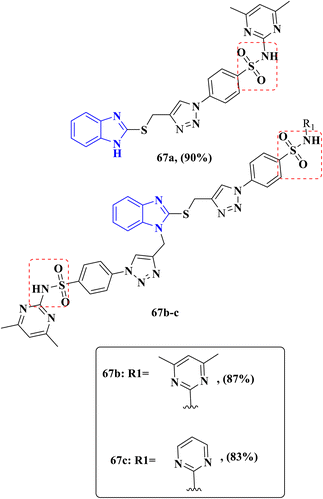 |
| | Fig. 10 Antibacterial benzimidazole derivatives 67a–c evaluated against Gram-positive and Gram-negative strains. | |
Table 7 MIC values of compounds 67a–c against four bacterial strains, benchmarked with ciprofloxacin
| Compound |
Minimum inhibition concentration (μg mL−1) |
| B. cerus |
S. aureus |
E. coli |
P. aeruginosa |
| 67a |
64 |
64 |
128 |
256 |
| 67b |
32 |
32 |
64 |
64 |
| 67c |
64 |
64 |
64 |
64 |
| Ciprofloxacin |
8 |
4 |
4 |
8 |
The study by Aparna and collaborators utilized similar techniques to obtain novel 4-methoxybenzimidazole derivatives 68a–d (Fig. 11). The SAR study revealed that the synthesized compounds exhibited good antibacterial activity with MIC values of 58.78 μg mL−1 to 130.28 μg mL−1 in comparison with the reference drug ciprofloxacin 10 μg mL−1. All the derivatives showed higher selectivity against Gram-positive strains than against Gram-negative strains except in derivative 68b that had selectivity toward E. coli and the activity was provided by shifting in positioning on the phenyl ring attributed to the triazole moiety, through its incorporation with an atom of large radius as the chlorine group at position-4. Moreover, compound 68a exhibited the highest potency against S. aureus and E. coli, attributed to hydrophilic substituents, as the carboxylic acid group at position 3 and the methyl substitution at the benzoic ring affected the potency negatively. Compound 68a showed significant antibacterial activity against Gram-positive bacteria S. aureus with an MIC value of 58.78 μg mL−1 and mild activity against Gram-negative bacteria E. coli with an MIC value of 68.74 μg mL−1, comparable to the MIC value of 10 μg mL−1 of the reference drug ciprofloxacin. However, compounds 68b and 68c showed comparable activities against S. aureus and E. coli bacteria, where compound 68b showed MIC values of 106.83 μg mL−1 and 91.45 μg mL−1 compared to the MIC value of the reference drug ciprofloxacin, respectively. Nevertheless, compound 68c exhibited MIC values of 83.74 μg mL−1 and 118.46 μg mL−1, when compared to the MIC value of the reference drug ciprofloxacin. Compound 68d also demonstrated less antibacterial activity against both strains S. aureus and E. coli with MIC values of 130.28 μg mL−1 and 140.26 μg mL−1, respectively, when compared with the MIC value of the control drug ciprofloxacin (Table 8). Furthermore, molecular modelling studies for the designated compound 68a revealed a moderate inhibitory activity of enol acyl carrier protein reductase (FabI) interactions with amino acid residues Phe96, Met99, Tyr147, Tyr157 and Met160.81
 |
| | Fig. 11 4-Methoxybenzimidazole derivatives 68a–d synthesized for bacterial evaluation against S. aureus and E. coli. | |
Table 8 MIC data for 68a–d against S. aureus and E. coli, using ciprofloxacin as a reference drug
| Compound |
Minimum inhibition concentration (μmol mL−1) |
| S. aureus |
E. coli |
| 68a |
58.78 |
68.74 |
| 68b |
106.86 |
91.45 |
| 68c |
83.47 |
118.46 |
| 68d |
130.28 |
140.26 |
| Ciprofloxacin |
10 |
10 |
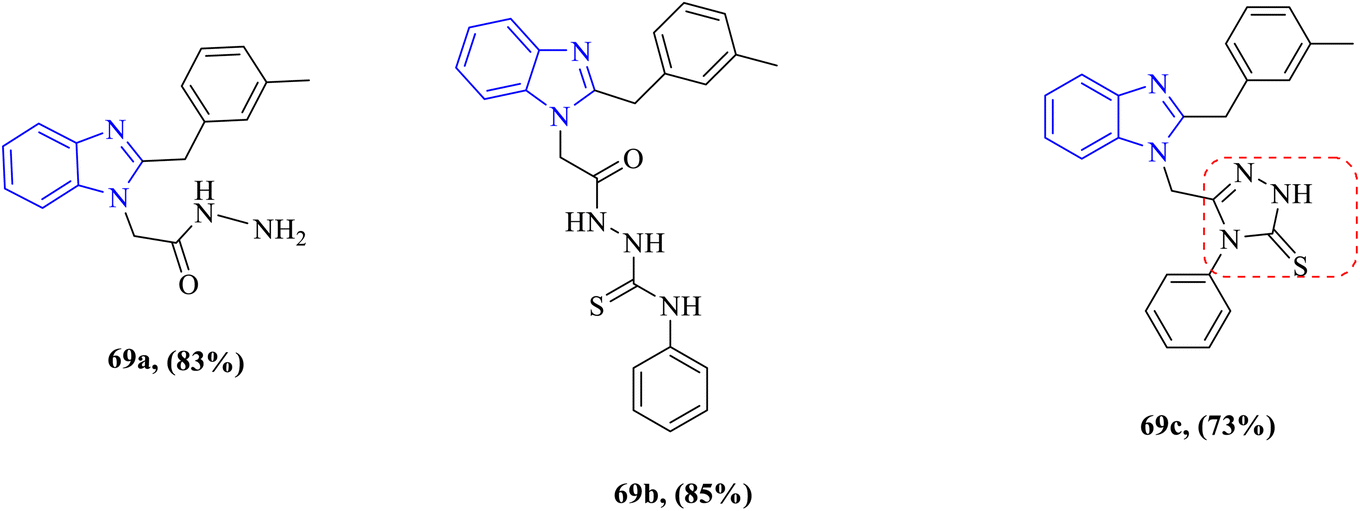
The antibacterial properties of some benzimidazole derivatives 69a–c were investigated by Kantar et al. against one Gram-positive bacteria B. subtilis and one Gram-negative bacteria Yersinia pseudotuberculosis (Fig. 12). The antibacterial activity ranged from good to moderate. Compound 69a exhibited broad-spectrum activities against both tested strains with equipotent MIC values of 62.5 μg mL−1 against the MIC value of the control drug ampicillin 100 μg mL−1, while compound 69b had high selective activity against B. subtilis with an MIC value of 62.5 μg mL−1 and intense activity against Y. pseudotuberculosis with an MIC value of 250 μg mL−1 with respect to the reference drug ampicillin; however, compound 69c exhibited the highest potency (MIC = 31.25 μg mL−1 and 62.5 μg mL−1) against B. subtilis and Y. pseudotuberculosis, respectively, compared to the drug ampicillin (Table 9), and this high potency is probably due to the combination of the benzimidazole nucleus and the 1,2,4-triazole ring.82
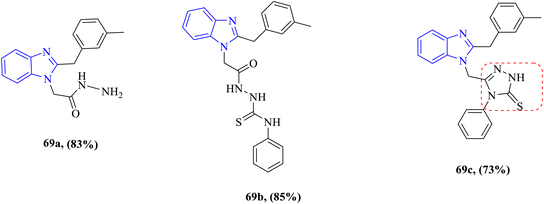 |
| | Fig. 12 Benzimidazole derivatives 69a–c tested against B. subtilis and Y. pseudotuberculosis. | |
Table 9 MIC activity values of compounds 69a–c against B. subtilis and Y. pseudotuberculosis against ampicillin
| Compound |
Minimum inhibition concentration (μg mL−1) |
| B. subtilis |
Y. pseudotuberculosis |
| 69a |
62.5 |
62.5 |
| 69b |
62.5 |
250 |
| 69c |
31.25 |
62.5 |
| Ampicillin |
100 |
100 |
Nandwana et al. proved that polycyclic aromatic compounds 70a–d offered highly active antibacterial properties (Fig. 13), where compound 70a exhibited the highest potency that may be explained by hybridization between the benzimidazole nucleus, quinazoline and triazole ring than the other azole derivatives 70b–d. Those compounds were investigated against two Gram-positive bacteria, S. aureus and B. subtilis, and three Gram-negative bacteria, E. coli, Salmonella typhi and Pseudomonas putida, where compound 70a showed significant activity against S. aureus and B. subtilis with an equipotent MIC value of 8 μg mL−1 compared to the MIC value of 6.25 μg mL−1 of the reference drug ciprofloxacin and displayed a greater equal MIC activity value of 4 μg mL−1 against P. putida and E. coli compared to the MIC value of 6.25 μg mL−1 of the reference drug ciprofloxacin. Compound 70b showed good antibacterial activity against S. aureus and B. subtilis with MIC values of >8 μg mL−1 and 8 μg mL−1, respectively, and significant activity against E. coli and P. putida with MIC values of >8 μg mL−1 and 8 μg mL−1 when compared to the control drug ciprofloxacin. Furthermore, compound 70c also showed moderate activity with an equipotency MIC value of 16 μg mL−1 against both Gram-positive strains and Gram-negative bacteria E. coli and S. typhi comparable to the MIC value of the reference drug ciprofloxacin 6.25 μg mL−1. Compound 70d showed less antibacterial activity against Gram-positive strains with equal MIC values of 64 μg mL−1 and > 2 μg mL−1 for both Gram-negative strains E. coli and P. putida, respectively, in comparison to ciprofloxacin (Table 10). Furthermore, compound 70a modulated the biofilm formation through biocidal mechanism either by degradation of the extracellular matrix or weakened the biofilm, which is an extracellular substance providing high resistance to microbes against phagocytosis and other components of the body's defense system. In addition, the bactericidal assay of propidium iodide and live-dead bacterial cell screening by using a mixture of acridine orange/ethidium bromide cause cell death, which might be due to considerable changes in the bacterial cell membrane. The synthesized compounds were also assessed for hemolytic activity, which indicated an acceptable standard towards human blood cells.83
 |
| | Fig. 13 Structures of polycyclic benzimidazole hybrids 70a–d incorporating triazole and quinazoline moieties for antibacterial evaluation. | |
Table 10 MIC values of compounds 70a–d against S. aureus, B. subtilis, E. coli, P. putida, and S. typhi, with ciprofloxacin as reference
| Compound |
Minimum inhibition concentration (μg mL−1) |
| S. aureus |
B. subtilis |
P. putida |
E. coli |
| 70a |
8 |
8 |
4 |
4 |
| 70b |
>8 |
8 |
8 |
>8 |
| 70c |
16 |
16 |
16 |
16 |
| 70d |
64 |
64 |
>32 |
>32 |
| Ciprofloxacin |
6.25 |
6.25 |
6.25 |
6.25 |
The incorporation of 2-mercaptobenzimidazole with a 1,2,4-triazole moiety by Al-Majidi and colleagues yielded promising antibacterial compounds 71a–c (Fig. 14). Compounds were evaluated for their antibacterial activity against Gram-positive bacteria S. aureus and Gram-negative bacteria P. aeruginosa. Compounds 71a and 71b displayed powerful activity against S. aureus with inhibition zone diameters of 18 mm and 19 mm when compared to the 33 mm of control drug amoxicillin, respectively, and moderate activity against P. aeruginosa with inhibition zone diameters of 14 mm and 11 mm when compared to the 32 mm of drug amoxicillin. Furthermore compound 71c presented good activity against S. aureus with an inhibition zone diameter of 17 mm compared to the control drug amoxicillin; however, the activity against P. aeruginosa is a little effective, as indicated by an inhibition zone diameter of 15 mm compared to the antibiotic amoxicillin (Table 11).84
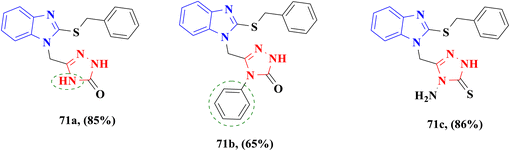 |
| | Fig. 14 2-Mercaptobenzimidazole–triazole derivatives 71a–c tested against S. aureus and P. aeruginosa. | |
Table 11 Inhibition zone diameters of compounds 71a–c against S. aureus and P. aeruginosa, compared with amoxicillin
| Compound |
Diameter inhibition zone (mm) |
| S. aureus |
P. aeruginosa |
| 71a |
18 |
14 |
| 71b |
19 |
11 |
| 71c |
17 |
11 |
| Amoxicillin |
33 |
32 |
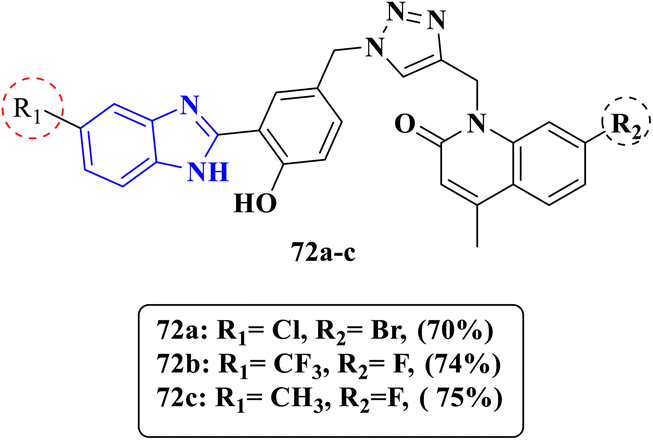
Research findings of Elgadil and his team proved that synthesized scaffolds of benzimidazole derivatives incorporated with a triazole ring induce antibacterial activity by inhibiting the activity of topoisomerase II (DNA gyrase), which is essential for bacterial cell survival. Compounds 72a–c were investigated for their activity against one Gram-positive bacteria S. aureus and one Gram-negative bacteria E. coli (Fig. 15). Compound 72a displayed greater activity against S. aureus than the reference drug ampicillin with an MIC value of 0.07 μm mL−1 comparable to 0.27 μm mL−1 and excellent activity against E. coli with an MIC value of 0.14 μm mL−1 when compared to the MIC value of the reference drug ampicillin 0.26 μm mL−1. Moreover, compound 72b exhibited outstanding antibacterial activities against both bacterial strains with an equipotent MIC value of 0.14 μm mL−1 with respect to the MIC value of the reference drug ampicillin. Compound 72c showed less antibacterial activity against S. aureus with MIC values of 10.11 μm mL−1 and 2.53 μm mL−1 against E. coli, respectively, than ampicillin (Table 12). In addition, molecular docking studies were conducted to detect the binding modes comparable to the co-crystalized ligand ampicillin, and the designated compound 72a displayed an extra binding mode compared to ampicillin through H-bond interaction with ARG458, DG9 and DG8 and interaction residues with DC13 and DG9. SAR analysis revealed that the introduction of electron-withdrawing groups as Cl and –CF3 at position 5 gave highly potent antibacterial compounds 72a and 72b with a wide spectrum against both Gram-positive and Gram-negative bacteria. Their potency was nearly two folds more than that of ampicillin. However, derivatives bearing an electron-donating methyl substituent 72c exhibited the lowest potency.85
 |
| | Fig. 15 Benzimidazole–triazole hybrids 72a–c designed to inhibit bacterial DNA gyrase tested against S. aureus and E. coli. | |
Table 12 MIC values of compounds 72a–c against S. aureus and E. coli, compared to ampicillin
| Compound |
Minimum inhibition concentration (μg mL−1) |
| S. aureus |
E. coli |
| 72a |
0.07 |
0.14 |
| 72b |
0.14 |
0.14 |
| 72c |
10.11 |
2.53 |
| Ampicillin |
0.27 |
0.26 |
Benzimidazole derivatives fused to a quinazoline moiety have been reported by Korrapati et al. and evaluated for their antibacterial activity (Fig. 16). The synthesized compounds were tested against three Gram-positive bacteria, S. aureus, B. subtilis and M. luteus, and one Gram-negative bacteria, K. planticola. Compounds 73a–d displayed excellent antibacterial activity with MIC values ranging from 3.9 μg mL−1 to >125 μg mL−1 through inhibition of DNA gyrase, leading to the inhibition of replication, transcription and recombination, which resulted in the death of the microbial organism. Compound 73c exhibited outstanding activity against S. aureus, B. subtilis and M. luteus with an MIC value of 3.9 μg mL−1 comparable to the MIC value of the reference drug ciprofloxacin 0.9 μg mL−1 (Table 13). The good activity of compounds 73a and 73b attributed to substitution with a fluorine atom generally against tested bacterial strains, particularly S. aureus, while replacing the fluorine atom with the nitro group, led to improvement in the antibacterial activity, particularly against S. aureus, B. subtilis and M. luteus. Furthermore, molecular docking studies of the designated compound 73c displayed binding interactions similar to the co-crystalized ligand ciprofloxacin with amino acid residues Ile90 and Asn46. Overall, the synthesized derivatives exhibited better selectivity against Gram-positive bacteria than Gram-negative bacteria, electron-donating group substitution, methoxy substitution in compound 73d, diminished the activity.86
 |
| | Fig. 16 Benzimidazole–quinazoline hybrids 73a–d targeting DNA gyrase for antibacterial activity. | |
Table 13 MIC values of compounds 73a–d against Gram-positive and Gram-negative bacterial strains
| Compound |
Minimum inhibition concentration (μg mL−1) |
| S. aureus |
B. subtilis |
M. luteus |
k. planticola |
| 73a |
7.8 |
7.8 |
7.8 |
>125 |
| 73b |
7.8 |
7.8 |
7.8 |
>125 |
| 73c |
7.8 |
3.9 |
3.9 |
>125 |
| 73d |
>125 |
>125 |
>125 |
>125 |
| Ciprofloxacin |
0.9 |
0.9 |
0.9 |
0.9 |
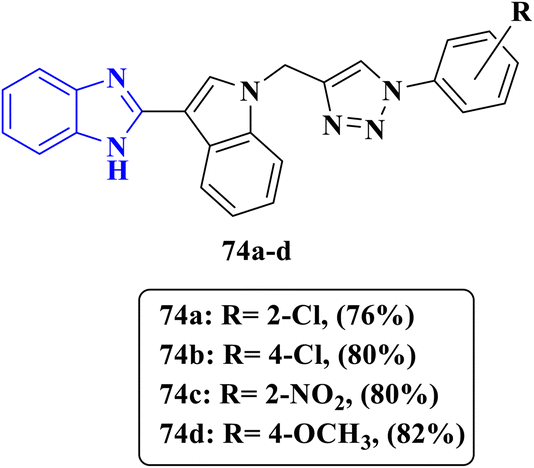
2.1.1 Antifungal activity. Ashok and his team synthesized novel benzimidazoles linked to indole and 1,2,3 triazole moieties via a microwave-assisted click reaction. Compounds 74a–d were designed and tested for their potential antifungal activity against Candida albicans (Fig. 17). Compounds 74a and 74c showed excellent antifungal activity with an equal MIC value of 6.25 μg mL−1 comparable to the MIC value of 1.56 μg mL−1 of the reference drug fluconazole by inhibiting ergosterol synthesis, a vital component of fungal cell membranes, while compound 74d exhibited weak antifungal activity with an MIC value of 100 μg mL−1 (Table 14). In addition, physicochemical parameters such as the drug score were calculated, and the results showed that the designed compounds 74a and 74c displayed favorable drug score values. The structure–activity relationship indicated that substitution with electron-withdrawing groups either at position 2 or at position 4 had equal positive effects on the antifungal activity; however, introducing substitutions with other electron-donating groups as the methoxy group decreases the antifungal activity.87
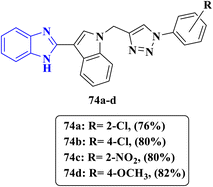 |
| | Fig. 17 Benzimidazole–triazole derivatives 74a–d designed for antifungal evaluation against C. albicans. | |
Table 14 MIC values of compounds 74a–d against C. albicans
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
| 74a |
6.25 |
| 74b |
6.25 |
| 74c |
6.25 |
| 74d |
100 |
| Fluconazole |
1.56 |
In 2023, Mallikanti et al. reported that incorporation of benzimidazole and triazole pharmacophores significantly improved antifungal efficacy. The planar structure of these compounds 75a–d likely facilitates the π–π stacking and T-shaped interactions within the cavity of target receptors. Compounds 75a–d were screened against two fungal strains C. albicans and A. niger (Fig. 18). Compounds 75a and 75b showed excellent equipotency antifungal activities through interfering with fungal cell division with an inhibition zone diameter value of 24 μg mL−1, whereas compound 75c displayed a high inhibition zone diameter of 23 μg mL−1 comparable to the inhibition zone value of 18 μg mL−1 of the reference drug griseofulvin against C. albicans, while they revealed outstanding activity against A. niger with inhibition zone diameters of 23, 24 and 25 μg mL−1, respectively in comparison to griseofulvin drug. However, compound 75d presented an inhibition zone diameter of 9 μg mL−1 against C. albicans and 10 μg mL−1 against A. niger comparable to the control drug griseofulvin (Table 15). Moreover, molecular docking studies were conducted against secreted aspartic proteinase (Sap)1 specifically as it plays a crucial role in superficial candida infections. The designated compound 75c presented extra binding interactions as the co-crystalized ligand griseofulvin with H-bond interactions with Asp32, Asp86, Ser88, Thr222, and Tyr225 and hydrophobic interactions with Val12, Ile30, Tyr84, Gly85, Ser88, Ile119, Ala303, and Ile305 of Sap. The results clearly indicated that introduction of electron withdrawing groups such as CN and F at positions 3 and 4 on the phenyl ring increase antifungal activity while substitution with electron donating groups as methyl reducing antifungal activity. Moreover 2,4 difluro-moiety at position 5 optimal for activity.88
 |
| | Fig. 18 Benzimidazole–triazole conjugates 75a–d optimized for antifungal activity. | |
Table 15 Inhibition zone diameters of compounds 75a–d against C. albicans and A. niger
| Compound |
Diameter inhibition zone (μg mL−1) |
| C. albicans |
A. niger |
| 75a |
24 |
23 |
| 75b |
24 |
24 |
| 75c |
23 |
25 |
| 77d |
9 |
10 |
| Griseofulvin |
18 |
18 |
Benzimidazole derivatives containing 1,2,4 triazole moieties were synthesized by Kankite and his team to obtain hybrids 76a–c (Fig. 19). Compounds were tested against one fungal strain C. albicans in vitro with an additional benefit of in vivo screening by the kidney burden test. Compound 76a exhibited excellent antifungal activity at a concentration of 0.0075 μM mL−1 (Table 16), which is equipotent to fluconazole activity by inhibiting ergosterol biosynthesis through binding to 14-α-demethylase (CYP51) causes membrane dysfunction resulted in death of the fungi. While compounds 76b and 76c showed less antifungal activity at equal values, 0.015 μM mL−1. Furthermore, a molecular modeling study was conducted to detect appropriate binding with the targeted enzyme CYP51, the designated compound 76a is positioned perpendicular to the porphyrin plane, and a heme iron is highly coordinated with a ring nitrogen N4 as the co-crystalized ligand fluconazole, presenting the same binding. The structure–activity relationship illustrated that the antifungal activity increased through the introduction of phenyl ring at position 1, while the antifungal activity reduced with the increase in the alkyl length of N1 of the benzimidazole nucleus (methyl to ethyl). Compounds displayed high safety margin supported by kidney burden test so inducing highly safe antifungal candidates.89
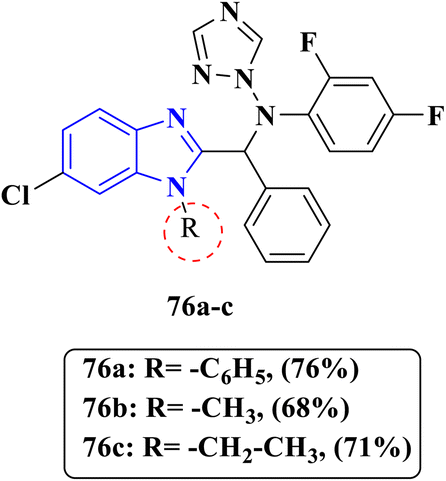
 |
| | Fig. 19 Benzimidazole–triazole hybrids 76a–c acting on CYP51 for antifungal activity. | |
Table 16 MIC values of compounds 76a–c against C. albicans
| Compound |
Minimum inhibition concentration (μM mL−1) |
| C. albicans |
| 76a |
0.0075 |
| 76b |
0.015 |
| 76c |
0.015 |
| Fluconazole |
0.0075 |
In the in vitro studies by Evren et al., new benzimidazole derivatives including triazole moieties were synthesized to evaluate their activity. Compounds 77a–c were screened for antifungal activity against C. albicans fungal strains (Fig. 20). Compounds 77a exhibited outstanding activity against the fungal strain with an MIC value of 3.9 μg mL−1 rather than the MIC value of the reference drug ketoconazole 7.8 μg mL−1 acting by the inhibition of ergosterol biosynthesis, which resulted in fungicidal action while compound 77b showed equipotent MIC as the control drug ketoconazole. However, compound 77c implied weak antifungal activity with an MIC value >1 mg mL−1 (Table 17). Furthermore, molecular docking analysis against 14-α-demethylase (CYP51) revealed that the designated compound 77a displayed similar binding modes as the co-crystalized ligand through pi–pi stacking interactions with Hie377. Moreover, extra binding modes with hydrogen bond interactions with Met508 and hydrophobic interactions with Leu376 and Tyr64 are observed. In addition, ADMET studies were performed for the synthesized compounds and presented compounds have good oral bioavailability with high safety margin. Evidently, direct attachment between triazole and phenyl rings resulting in compounds with planar conformation as compounds 77a and 77b increased the antifungal potency.90
 |
| | Fig. 20 Benzimidazole–triazole compounds 77a–c evaluated for antifungal potency against C. albicans. | |
Table 17 MIC values of compounds 77a–c against C. albicans
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
| 77a |
3.9 |
| 77b |
7.8 |
| 77c |
>1 mg mL−1 |
| Ketoconazole |
7.8 |
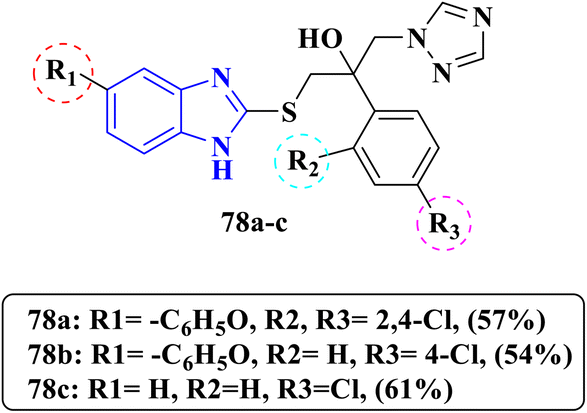
In recent investigations by Ghobadi et al., a series of benzimidazole-bearing triazole moieties were designed as a hybrid of mebendazole 78a–c against two fungal strains C. albicans and C. neoformans (Fig. 21). Compounds 78a and 78b exhibited excellent antifungal activity against C. albicans with an equipotent MIC value <0.063 μg mL−1, their MIC values were 2- to 8-fold higher than that of the reference drug fluconazole (0.5 μg mL−1). Compound 78a also showed good antifungal activity against C. neoformans with an MIC value of 1 μg mL−1 and compound 78b exhibited outstanding activity against C. neoformans with an MIC value of 0.125 μg mL−1, when compared to the MIC value of the reference drug fluconazole 0.5 μg mL−1. However, compound 78c displayed weak antifungal activity against both fungal strains with MIC values of 4 μg mL−1 and 16 μg mL−1, respectively, comparable to the MIC value of the reference drug fluconazole (Table 18). According to SAR analysis, substitution at position 5 in the benzimidazole with a bulky benzoyl group positively affected the antifungal activity. However, a simplification strategy by removing a benzoyl moiety gave weak antifungal agents, while variation in halogens substituents in the phenyl ring gave equipotent compounds, but it mainly depends on the type of the side chain. Docking studies as mentioned displayed high binding in the active site of lanosterol 14α-demethylase (CYP51) through a coordination bond between the N4 in the triazole ring and heme iron with a distance 2.7 Å as well as, the designated synthesized compounds78a and 78b displayed desirable ADMET properties and drug-likeness more than the reference drug fluconazole. In addition, they showed high profile safety margin with no carcinogenic probability.91
 |
| | Fig. 21 Benzimidazole–triazole analogs 78a–c screened against Candida and Cryptococcus species. | |
Table 18 MIC values of compounds 78a–c against C. albicans and C. neoformans
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
C. neoformans |
| 78a |
<0.063 |
1 |
| 78b |
<0.063 |
0.125 |
| 78c |
4 |
16 |
| Fluconazole |
0.5 |
0.5 |
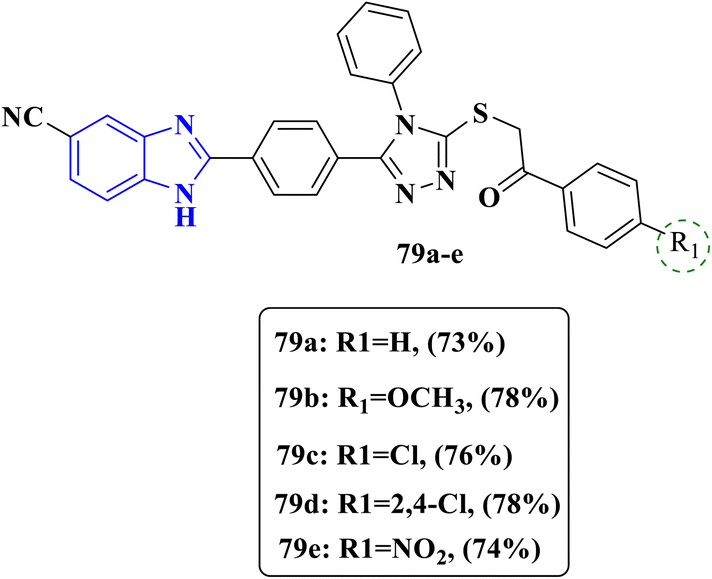
A novel series of benzimidazole hybridized with 1,2,4 triazole moiety was designed by Güzel and colleagues, compounds were investigated to evaluate their antifungal activity against four fungal strains of Candida, C. albicans, C. glabrata, C. krusei and C. parapsilopsis (Fig. 22). Compounds 79a–d exhibited outstanding activity against only one fungal strain C. glabrata, compounds 79b–79c showed an equipotent MIC value of 0.97 μg mL−1 greater than the MIC value of 1.95 μg mL−1 of the reference drug voriconazole, while compound 79d displayed antifungal activity with an MIC value equal to that of the control drug voriconazole (Table 19). Furthermore, a molecular docking study was conducted on the most active compounds 79b–79c and displayed binding interactions similar to voriconazole with Tyr118, His377 and Hem601 residues, and the interactions with HEM present in 14 α demethylase (CYP51) were seen as π–π stacking and π-cation interactions, presenting high antifungal activity found in the designated compounds. In addition, Scanning Electron Microscopy with Energy-Dispersive X-ray analysis (SEM-EDX) was used to observe the surface morphology of C. glabrata before and after the test compound treatment, the SEM results displayed cell wall deformations and loss of membrane integrity as the reference drug voriconazole. The SAR analysis proved that all compounds substituted at position 4 either with bulky groups as a methoxy group or with large radius groups as a chlorine atom fruitfully affect the antifungal activity. Moreover, the phenyl ring was substituted with different groups; from the screening results, we can conclude that potent compounds 79a–79c bear chloro and methoxy groups. Substitution mainly in the phenyl ring at position C-4 was crucial for the anticandidal activity.92
 |
| | Fig. 22 Benzimidazole–triazole hybrids 79a–e evaluated for anticandidal activity. | |
Table 19 MIC values of compounds 79a–e against C. glabrata
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. glabrata |
| 79a |
1.95 |
| 79b |
0.97 |
| 79c |
0.97 |
| 79d |
1.95 |
| 79e |
1.95 |
| Voriconazole |
1.95 |
The work by Aaghaz and his team reported novel benzimidazole scaffolds bearing thiazole and morphiline moieties inducing significant antifungal activities, and compounds 80a–c were investigated against three fungal strains C. neoformans, C. albicans and C. parapsilosis (Fig. 23). Compound 80a displayed little antifungal activity against the tested fungal strains with an equivalent MIC value of 81.6 μg mL−1 comparable to the MIC value of the control drug amphotericin 1 μg mL−1, while compound 80b exhibited excellent antifungal activity against the mentioned strains with MIC values of 2.4 μg mL−1, 4.9 μg mL−1 and 19.6 μg mL−1, respectively, comparable to amphotericin drug. Moreover, compound 80c exhibited good activity against both C. neoformans and C. albicans with MIC values of 9.7 μg mL−1 and 38.7 μg mL−1, respectively, and minor antifungal activities against C. parapsilosis with an MIC value of 155.9 μg mL−1 with respect to the MIC value of amphotericin (Table 20). Moreover, propidium iodide (PI) uptake study by confocal laser scanning microscopy test was utilized to detect permeability to cell membrane through a red fluorescent dye found in PI, and a promising result was observed regarding compound 80b followed by staining with PI, leading to the permeabilization of this compound inside the fungal cells, and this has been confirmed by the presence of red fluorescence in the treated cells. In addition, the Time Kill Assay test was performed to detect time intervals required to completely inhibit the growth of fungal cells, and it was observed that compound 80b significantly kills C. neoformans after 8 h when compared to amphotericin B that inhibits the growth of Cryptococcal cells after 4 h, presenting an advantage of sustained activity over time. Moreover, the designated compounds displayed direct coordination with nitrogen of the azole ring to the HEM ferric ion in CYP51 belonging to cytochrome P450 that is required for the biosynthesis of ergosterol in fungi. On top of that, in vitro mammalian cell cytotoxicity of 80b was performed against the human cancer cell line (HeLa) as well as normal human cell line (HEK-293) at different concentrations. The designated compound 80b did not exhibit any significant toxicity. SAR studies demonstrated that thiazole and morpholine moieties are optimal for activity. Furthermore, substitution with a methoxy group at the para position diminished antifungal activity towards all fungi strains, while substitution at the meta position enhanced antifungal activity against all fungal strains, and introducing an electron-donating group as the methyl group at position 6 in the benzimidazole core offered promising antifungal activities particularly against C. neoformans and C. albicans 80b; however, substitution with an electron-withdrawing group as the fluorine atom produced significant antifungal activities only towards C. neoformans.93
 |
| | Fig. 23 Benzimidazole–thiazole–morpholine hybrids 80a–c with promising antifungal profiles. | |
Table 20 MIC values of compounds 80a–c against three Candida species
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. neoformans |
C. albicans |
C. parapsilosis |
| 80a |
81.6 |
81.6 |
81.6 |
| 80b |
2.4 |
4.9 |
19.6 |
| 80c |
9.7 |
38.7 |
155.9 |
| Amphotericin B |
1 |
1 |
1 |
Through molecular hybridization, Cevik and collaborators synthesized bioactive derivatives 81a–c in a single molecule as benzimidazole hybrids with oxadiazole moieties, providing anticandida properties through the inhibition of 14α-demethylase (Fig. 24). Compounds were screened against different species of candida fungal strains C. albicans, C. glabrata and C. kruesi using ketoconazole as the reference drug. Compound 81a reported an excellent MIC value of 0.78 μg mL−1 against C. albicans and good antifungal activity with an MIC value of 3.12 μg mL−1 against both C. glabrata and C. kruesi, when compared to the MIC value of the reference drug ketoconazole 1.56 μg mL−1. Compound 81b displayed excellent antifungal activity against C. albicans with an MIC value of 0.78 μg mL−1, mild antifungal activity against C. glabrata with an MIC value of 3.12 μg mL−1 and outstanding antifungal activity against C. kruesi with an MIC value of 1.56 μg mL−1 comparable to the ketoconazole drug. Compound 81c exhibited significant antifungal activity against the tested fungal strains with MIC values of 1.56 μg mL−1 for both C. albicans and C. glabrata and 0.78 μg mL−1 against C. kruesi with respect to the reference drug ketoconazole 1.56 μg mL−1 (Table 21). The results were supported with molecular docking studies, displaying binding modes directly with an HEM ferrous ion in the active site of CYP51 as ketoconazole. Furthermore, molecular dynamics were performed at 100 ns simulations, and the designated compounds displayed high stability with low RMSD aligned with strong binding to the HEM. Compound 81b showed the most stable binding followed by 81c and 81a. On top of that, all tested compounds showed high therapeutic safety margin by these studies and the targeted compounds offer further support to the biological results. Structure–activity relationship showed that either the absence of substitution or the introduction of an electron-withdrawing chlorine atom at position 5 enhanced antifungal activity against Candida albicans, as observed in compounds 81a and 81b, while substitution with electron donating group as methyl group showed equipotent activity to the reference drug ketoconazole 81c against both fungi strains C. albicans, C. glabrata. They also displayed outstanding potent antifungal activity against C. kruesi compared with ketoconazole.94
 |
| | Fig. 24 Benzimidazole–oxadiazole conjugates 81a–c targeting Candida species. | |
Table 21 MIC values of compounds 81a–c against C. albicans, C. glabrata, and C. krusei
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
C. glabrata |
C. krusei |
| 81a |
0.78 |
3.12 |
3.12 |
| 81b |
0.78 |
3.12 |
1.56 |
| 81c |
1.56 |
1.56 |
0.78 |
| Ketoconazole |
1.56 |
1.56 |
1.56 |
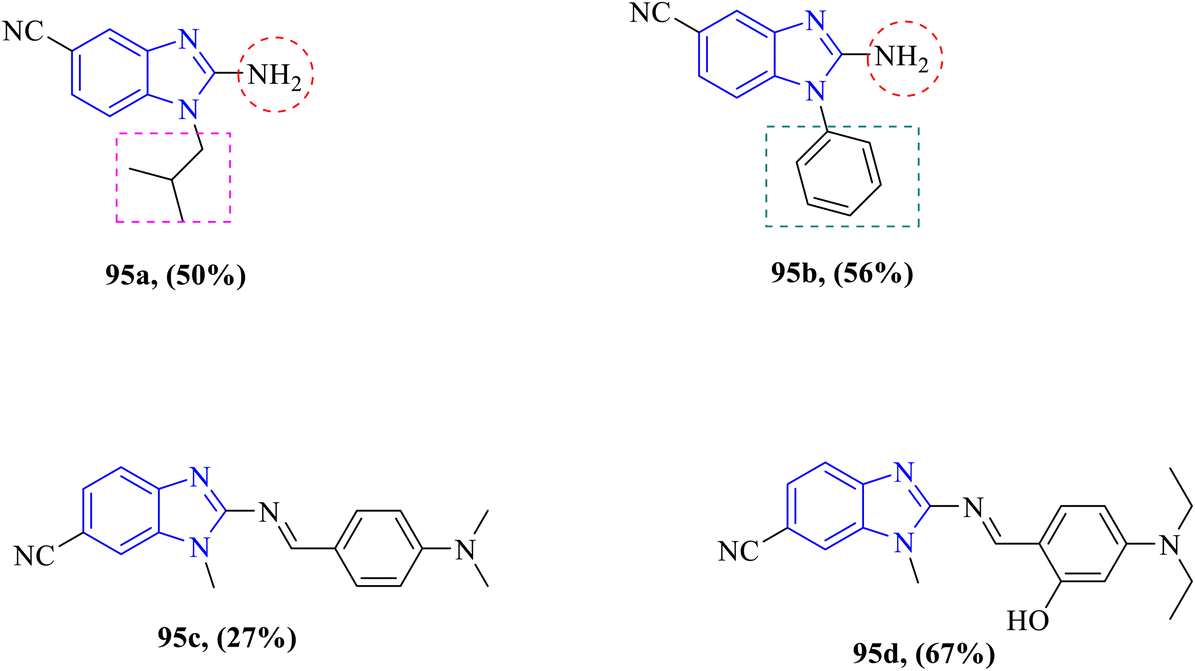
In their 2023 study, Pham et al. designed and synthesized novel benzimidazole scaffolds 82a–82c via the alkylation of N1 of benzimidazole inducing antifungal activity (Fig. 25). Compounds were screened against two fungal strains of C. albicans and A. niger. Compound 82b displayed excellent antifungal activity with MIC values of 16 μg mL−1 against C. albicans when compared to the reference drug fluconazole 4 μg mL−1 and outstanding activity against A. niger with an MIC value of 32 μg mL−1 compared to the MIC of the fluconazole drug 128 μg mL−1. Compound 82a showed less antifungal activity against both fungal strains with an equal MIC value of 512 μg mL−1 against C. albicans and A. niger with respect to the fluconazole drug. Moreover, compound 82c exhibited minor antifungal activity against C. albicans with an MIC value of 128 μg mL−1 looking at the MIC value of fluconazole drug and good antifungal activity against A. niger with equipotent MIC value of fluconazole drug 128 μg mL−1 compared to fluconazole (Table 22). Moreover, molecular docking studies revealed that compound 82b established one strong hydrogen bond with Tyr225 resembling the co-crystallized ligand fluconazole with N-myristoyltransferase (NMT), which is crucial for fungal viability. Moreover, it is involved in key processes such as cell wall synthesis, morphogenesis, and signal transduction. Additionally, compound 82b exhibited an excellent ADMET profile with a high safety margin, and hence, it is recommended to be a lead-like compound for further drug development. SAR analysis revealed that the benzyl moiety was optimal for activity when replaced with an allyl moiety, where the antifungal activity was reduced in 82a. Moreover, substitution on the phenyl ring at position 4 with large radius groups such as chlorine, as in compound 82b, was associated with enhanced antifungal activity.in compound 82b, was associated with enhanced antifungal activity. Moreover, the introduction of one more chlorine atom at position 3 diminished activity as in derivative 82c.95
 |
| | Fig. 25 N1-substituted benzimidazoles 82a–c screened for antifungal activity. | |
Table 22 MIC values of compounds 82a–c against C. albicans and A. niger
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
A. niger |
| 82a |
512 |
512 |
| 82b |
16 |
32 |
| 82c |
128 |
128 |
| Fluconazole |
128 |
128 |
Morcoss and collaborators designed novel benzimidazole scaffolds incorporated with hydrazone moieties 83a–e and then investigated their antimicrobial activity against two fungal strains C. albicans and C. neoformans (Fig. 26). Compounds 83a–e exhibited significant antifungal activity against C. albicans and C. neoformans with MIC values ranging from 4 μg mL−1 to >32 μg mL−1, comparable to the reference drug fluconazole with values 0.125 μg mL−1 and 8 μg mL−1, respectively. Compound 83a showed excellent activity comparable to other compounds with MIC values of 4 μg mL−1 and 16 μg mL−1 against the two fungal strain, respectively, comparable to the MIC value of the control drug fluconazole. Compound 83c revealed good antifungal activity against C. albicans and C. neoformans with an MIC value of 16 μg mL−1 when compared to the reference drug fluconazole, while compound 83d displayed good activity against C. albicans with an MIC value of 8 μg mL−1 and an MIC value >32 μg mL−1 against C. neoformans. However, both compounds 83b and 83e displayed MIC more than 32 μg mL−1 with less antifungal activity (Table 23). Additionally, Sterol Quantitation Method (SQM) used a spectrophotometric assay to measure the effect of antifungal drugs that inhibit sterol biosynthesis, and compound 83a efficiently inhibited the ergosterol biosynthesis in the fungal cell membrane by binding to sterol 14α-demethylase like the reference drug ketoconazole. Furthermore, the designated compound 83a exhibited high safety margin against red blood cells and human embryonic kidney cells at a concentration up to 32 μg mL−1. Moreover, compound 83a exhibited excellent pharmacokinetic properties, characterized by high oral bioavailability. The results were supported with molecular docking studies and revealed that compound 83a showed binding modes interaction with HEM as the co-crystallized ligand fluconazole. SAR analysis could be interpreted that an unsubstituted phenyl ring and the introduction of a chlorine atom at position 4 increased the activity against both C. albicans and C. neoformans while substitution with chlorine atom at positions 2,4 increased antifungal activity against only C. albicans; however, substitution with either electron-donating groups or small halogen groups as fluorine atoms abolished antifungal activity.96
 |
| | Fig. 26 Benzimidazole–hydrazone derivatives 83a–e with antifungal efficacy. | |
Table 23 MIC values of compounds 83a–e against C. albicans and C. neoformans
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
C. neoformans |
| 83a |
4 |
16 |
| 83b |
>32 |
>32 |
| 83c |
16 |
16 |
| 83d |
8 |
>32 |
| 83e |
>32 |
>32 |
| Fluconazole |
0.125 |
0.125 |
In 2021, Chaurasi and his team designed and synthesized benzimidazole nucleoside analogues containing a glycoside linkage, as variable chemical scaffolds yielded targeted derivatives 84a–84c (Fig. 27), and compounds were investigated against four fungal strains C. albicans, A. niger, Aspergillus flavus and Fusarium oxysporum. Notably synergistic effects of these moieties offered excellent activity against three fungal strains C. albicans, A. flavus and F. oxysporum. Compounds 84a and 84c exhibited significant antifungal activity against C. albicans, A. flavus and F. oxysporum, with MIC values ranging from 0.78 μg mL−1 to 12.5 μg mL−1 compared with the MIC values of 3.12, 1.56 and 12.5 μg mL−1 of the reference drug ketoconazole, respectively. Furthermore, compound 84b showed lesser antifungal activity against tested strains, with MIC values of 50, 25 and 100 μg mL−1 respectively, than the control drug ketoconazole (Table 24). Density functional theory (DFT) calculations were performed to study the electronic properties of compounds 84a and 84c and to support the biological results, and demonstrated that compounds with lower HOMO–LUMO energy gaps, particularly compound 84a, exhibited excellent antifungal activity against C. albicans as fluconazole. The reduced bandgap energy (ΔE) facilitated efficient intramolecular charge transfer and stronger interactions with fungal biomolecules. SAR analysis proved that position 4 in the phenyl ring is important for antifungal activity and it must be free of substitutions for the most potent antifungal agent 84a or incorporated with hydrophilic group as OH group 84c; however, increasing lipophilicity through substitution with electron-withdrawing groups as bromine diminished the activity 84b. Moreover, in silico ADMET study supported that the compounds could be developed to be good antifungal candidates.97
 |
| | Fig. 27 Benzimidazole–isoxazole hybrids 84a–c against fungal strains. | |
Table 24 MIC values of compounds 84a–c against fungal strains
| Compound |
Minimum inhibition concentration (μg mL−1) |
| C. albicans |
A. flavus |
F. oxysporum |
| 84a |
1.56 |
1.56 |
6.25 |
| 84b |
50 |
25 |
100 |
| 84c |
3.12 |
0.78 |
12.5 |
| Ketoconazole |
3.12 |
1.56 |
12.5 |
2.1.2 Dual (antibacterial and antifungal) activities. A novel series of benzimidazole scaffolds bearing quinolone hybrids 85a–85d, 86a–86b and 87a–87e were synthesized and investigated by Wang et al. for their antimicrobial activities (Fig. 28). All compounds were screened against three strains of Gram-positive bacteria (MRSA, E. faecalis and S. aureus), four strains of Gram-negative bacteria (K. pneumoniae, E. coli, P. aeruginosa and A. baumanii) and four fungal strains (C. albicans, C. tropicalis, A. fumigatus and C. parapsilosis). Compounds 85a–85d exhibited excellent antibacterial activity against MRSA with MIC values ranging from 8 μg mL−1 to 128 μg mL−1 compared to the reference drugs norfloxacin and clinafloxacin with MIC values >512 μg mL−1 and significant activity against K. pneumoniae with MIC values ranging from 8 μg mL−1 to 256 μg mL−1 compared to the reference drugs norfloxacin and clinafloxacin with MIC values 32 μg mL−1 and >512 μg mL−1, respectively, while compounds 86a–86b displayed good activity against MRSA with MIC values 8 μg mL−1 and 128 μg mL−1 and gentle activity against P. aeruginosa with equal MIC values 32 μg mL−1 comparable to the MIC values of the reference drugs of 32 μg mL−1 and 4 μg mL−1 respectively. Furthermore, series of compounds 87a–87e showed the most outstanding activity against both MRSA with MIC values ranging from 8 μg mL−1 to 256 μg mL−1 and P. aeruginosa with MIC values ranging from 1 μg mL−1 to 16 μg mL−1, particularly compound 87b presented excellent antibacterial activity against P. aeruginosa with an MIC value of 1 μg mL−1, for which the potency was 4- and 32-fold greater than that of the reference drugs norfloxacin and clinafloxacin (Table 25). Furthermore, molecular docking study revealed that compound 87b has strong inhibitory activity to topoisomerase IV altering DNA topology by generating a double-stranded break in the genetic material presenting similar binding modes to the Gly419 residue and base D16 of DNA through hydrogen bonding at distances of 2.4 and 2.1 A° with the co-crystallized ligand moxifloxacin. Moreover, the designated compound 87b displayed excellent binding modes as the co-crystallized ligand with fluconazole with HEM. Additionally, the derivative 87b displayed high safety profile against human laryngeal carcinoma epithelial cells. The SAR insights for compounds 85a–d revealed that the length of the aliphatic chain bearing in the benzimidazole nucleus affected by length of aliphatic chain, since short ethyl chain demonstrated greater efficacy as antibacterial agents than those with hexadecyl chain. In addition, it was shown that the unsaturated aliphatic chains have a less inhibitory effect, thus presenting poor antibacterial activity in compounds 86a–b, and the presence of a fluorine atom increases the antibacterial activity and inducing a wide spectrum against Gram-positive and Gram-negative bacteria. Mentioned compounds exhibited outstanding antifungal activity against C. tropicalis with MIC values ranging from 1 μg mL−1 to 256 μg mL−1 with respect to the MIC value of the reference drug fluconazole 256 μg mL−1. Specifically, compound 87b showed better antifungal activity with 256-fold more than the control drug fluconazole. In addition, alkyl-unsaturated allyl substituted compounds 85a–85d and 86a–b had moderate antifungal activity. Moreover, it became clear that fluorobenzyl compounds 87b–c exhibited similar or even more potent antifungal properties compared with chlorobenzyl compounds 87d–e. This suggests that the presence of a fluorine atom on the phenyl groups had a positive effect on inhibiting microbial growth.98
 |
| | Fig. 28 Benzimidazole derivatives 85a–d, 86a–b and 87a–e designed for dual antimicrobial activity. | |
Table 25 MIC values of compounds 85a–d, 86a–b and 87a–e against bacterial and fungal strains
| Compound |
Minimum inhibition concentration (μg mL−1) |
| MRSA |
K. pneumoniae |
P. aeruginosa |
C. tropicalis |
| 85a |
16 |
8 |
64 |
16 |
| 85b |
8 |
8 |
512 |
16 |
| 85c |
16 |
8 |
256 |
128 |
| 85d |
128 |
256 |
256 |
256 |
| 86a |
8 |
64 |
32 |
128 |
| 86b |
128 |
128 |
32 |
512 |
| 87a |
64 |
4 |
4 |
4 |
| 87b |
8 |
16 |
1 |
1 |
| 87c |
128 |
4 |
16 |
64 |
| 87d |
64 |
64 |
8 |
256 |
| 87e |
256 |
256 |
32 |
256 |
| Norfloxacin |
>512 |
32 |
32 |
— |
| Clinafloxacin |
>512 |
>512 |
4 |
— |
| Fluconazole |
— |
— |
— |
256 |
The research by Yadv and colleagues designed novel benzimidazole derivatives 88a–88f incorporated with benzoyl and different substituted acetamides for inducing the antimicrobial activity (Fig. 29). The synthesized compounds were investigated for their activities against five bacterial strains (S. aureus, B. cereus, B. subtilis, S. typhi and E. coli), and two fungal strains (C. albicans and A. niger). Compounds 88a–d displayed excellent antibacterial activity against S. aureus with an equipotency MIC value of 0.027 μM mL−1 comparable to the MIC value of the reference drug cefadroxil of 0.37 μM mL−1, while compounds 88e–f had good antibacterial activity with MIC values 0.031 μM mL−1 and 0.030 μM mL−1. Furthermore, compounds 88a–d exhibited significance antifungal activity against A. niger, with an equipotency MIC value of 0.027 μM mL−1 comparable to the MIC value of the reference drug fluconazole 0.47 μM mL−1, while compounds 88e–f showed good antifungal activity with MIC values 0.031 μM mL−1 and 0.030 μM mL−1 when compared to the control drug fluconazole (Table 26). From the obtained results, we can conclude that the antimicrobial activity were not positively affected by substitutions on the phenyl ring, either with electron-withdrawing groups as Cl, Br and NO2 or with electron-donating group as CH3 and OCH3 against all bacterial and fungal tested strains.99
 |
| | Fig. 29 Benzimidazole derivatives 88a–f evaluated for dual antimicrobial activity. | |
Table 26 MIC values of compounds 88a–f against S. aureus and A. niger
| Compound |
Minimum inhibition concentration (μM mL−1) |
| S. aureus |
A. niger |
| 88a |
0.027 |
0.027 |
| 88b |
0.027 |
0.027 |
| 88c |
0.027 |
0.027 |
| 88d |
0.027 |
0.027 |
| 88e |
0.031 |
0.031 |
| 88f |
0.030 |
0.030 |
| Cefadroxil |
0.037 |
— |
| Fluconazole |
— |
0.047 |
Carvacrol is a phenolic monoterpene derivative produced naturally, where hybrid compounds for medical use were developed. Bhoi et al. designed benzimidazole derivatives 89a–f developed from 2-formyl carvacrol screened and evaluated against one Gram-positive bacteria S. aureus and two Gram-negative bacteria P. aeruginosa, E. coli, and two fungal strains (C. albicans and A. niger) (Fig. 30). Compounds 89a–d showed exceptional activity against S. aureus with MIC values ranging from 12.5 μg mL−1 to 250 μg mL−1 comparable to the MIC value of the reference drug ciprofloxacin 50 μg mL−1 and excellent activity against P. aeruginosa and E. coli with MIC values ranging from 12.5 μg mL−1 to 250 μg mL−1 when compared to the MIC value of the control drug chloramphenicol 50 μg mL−1. Compound 89b displayed outstanding antibacterial activity against E. coli while compound 89c displayed the lowest MIC value leading to greater antibacterial activity against S. aureus strains. Compounds 89e–f displayed equipotence activity against C. albicans with an MIC value of 250 μg mL−1 comparable to the reference drug griseofulvin with an MIC value of 500 μg mL−1 (Table 27). The ADME predictions displayed that compounds 89e and 89f have reliable pharmacokinetic properties. Structure–activity relationship can be illustrated by increasing the antibacterial activity either through bioisosteric replacement by introduction pyridine moiety instead of phenyl ring towards Gram-positive bacteria 89c or by introducing electron-donating group as the methyl group increases the activity against Gram-negative bacteria 89b; however, substitution at position 6 with halogens increases antifungal activity against C. albicans 89e–f.100
 |
| | Fig. 30 Carvacrol-derived benzimidazole hybrids 89a–f screened for antibacterial and antifungal activity. | |
Table 27 MIC values of compounds 89a–f against S. aureus, P. aeruginosa, E. coli, and C. albicans
| Compound |
Minimum inhibition concentration (μg mL−1) |
| S. aureus |
P. aeruginosa |
E. coli |
C. albicans |
| 89a |
50 |
100 |
62.5 |
500 |
| 89b |
62.5 |
50 |
12.5 |
500 |
| 89c |
12.5 |
50 |
25 |
500 |
| 89d |
250 |
250 |
50 |
500 |
| 89e |
125 |
125 |
100 |
250 |
| 89f |
500 |
250 |
100 |
250 |
| Ciprofloxacin |
50 |
— |
— |
— |
| Chloramphenicol |
— |
50 |
50 |
— |
| Griseofulvin |
— |
— |
— |
500 |
Recently, Liu and his team designed novel benzimidazole derivatives inducing the antimicrobial activity, where they were incorporated with chalcone 90a–c, N-alkyl benzimidazolyl chalcones 91a–c and pyrimidines moieties 92a–c and 93a–99c (Fig. 31). The compounds were investigated against four Gram-positive bacteria (Staphylococcus aureus, Methicillin-Resistant Staphylococcus aureus, Bacillus subtilis and Micrococcus luteus) and six Gram-negative bacteria (Bacillus proteus, Escherichia coli, Pseudomonas aeruginosa, Bacillus typhi, Escherichia coli and Shigella dysenteriae) and five fungi (Candida albicans and Candida mycoderma, Candida utilis, Saccharomyces cerevisiae and Aspergillus flavus). Compounds 90a–c displayed good antibacterial activity against B. subtilis and B. proteus with MIC values ranging from 4 μg mL−1 to 512 μg mL−1 comparable to the MIC value of the reference drug chloromycin 32 μg mL−1. Compound 90a exhibited an equipotence MIC activity value of 8 μg mL−1 against B. subtilis with chloromycin and excellent MIC activity value 4 μg mL−1 against B. proteus than the control drug chloromycin; moreover, they revealed great activity against C. utilis with MIC values ranging from 16 μg mL−1 to 512 μg mL−1. Compound 90a demonstrated an equipotence MIC value of 16 μg mL−1 compared to the reference drug fluconazole. Compounds 91a–c proved perfect activity against S. aureus and P. aeruginosa with MIC values ranging from 0.5 μg mL−1 to 512 μg mL−1 compared to the MIC values of the reference drug norfloxacin of 8 μg mL−1 and 1 μg mL−1, respectively. Compound 91a exhibited higher activity against S. aureus and P. aeruginosa than norfloxacin with MIC values of 4 μg mL−1 and 0.5 μg mL−1. Furthermore, they expressed outstanding activity against C. utilis ranging from 16 μg mL−1 to 512 μg mL−1. Compound 91a exhibited equipotence high activity against C. utilis with an MIC value of 16 μg mL−1 comparable to the reference drug fluconazole. Compounds 92a–c outlined excellent antibacterial activity against S. aureus and P. aeruginosa with MIC values ranging from 8 μg mL−1 to 512 μg mL−1 comparable to the MIC values of the reference drug chloromycin of 8 μg mL−1 and 16 μg mL−1, respectively. Compound 92c displayed outstanding antibacterial activity with an equipotence MIC value of 8 μg mL−1 compared to the reference drug chloromycin against S. aureus and great activity against P. aeruginosa more than the control drug chloromycin with MIC values of 8 μg mL−1 and 16 μg mL−1 against chloromycin drug. In addition, they revealed excellent antifungal activity against C. utilis ranging from 2 μg mL−1 to 8 μg mL−1 when compared with the MIC value of the reference drug fluconazole 16 μg mL−1. Compound 92b proved antifungal activity that is 8-fold more potent than the fluconazole drug. Compounds 93a–c showed excellent antibacterial activity against S. aureus and P. aeruginosa with MIC values ranging from 2 μg mL−1 to 128 μg mL−1 comparable to the MIC value of the reference drug chloromycin. Compounds 93a–93c displayed good antibacterial activity with MIC values of 2 μg mL−1 and 8 μg mL−1 against S. aureus comparable to the MIC value of 8 μg mL−1 of the reference drug chloromycin and great activity against P. aeruginosa with MIC values of 16 μg mL−1 and 4 μg mL−1, respectively. In addition, they exhibited exceptional antifungal activity against A. flavus with an MIC value ranging from 1 μg mL−1 to 256 μg mL−1 with respect to the MIC value of the reference drug fluconazole 250 μg mL−1 (Table 28). Compound 90d showed excellent activity that is 256-fold more than that of fluconazole drug. In addition, DNA binding studies were performed to detect interaction with DNA by intercalation, groove binding, or electrostatic interactions. The results indicated that the designated compound 93c highly binds to DNA and blocks replication, explaining its antimicrobial effects. Moreover, molecular docking studies displayed that compound 93c strongly binds to DNA gyrase, resulting in the inhibition of DNA replication via forming a hydrogen bond with Asp1083 through NH and NH2 groups, and additional interactions were detected with Arg1122, Met1121, Ala1120, and Tyr1087. Furthermore, Molecular Electrostatic Potential (MEP) analysis was used to visualize how a drug or ligand would fit and bind to its target; as a result, compound 93c showed strong positive regions around NH2 and NH, favorable for target binding supporting of molecular docking study. On top of that, compound 93c exhibited low toxicity against human hepatocyte (LO2) cells, suggesting a good safety profile. The SAR results indicated that chalcone and pyrimidine moieties are optimal for antimicrobial activity, in case of chalcone derivatives as in derivatives 90a–c and 91a–c, introduction of electron donating groups as methyl group increase the activity towards both bacterial and fungal strains while introducing electron withdrawing groups as chlorine or fluorine atoms resulted in minor antimicrobial activities. While alkylation of NH in the benzimidazole ring with different small and long aliphatic chains, studies revealed that propyl chain has more potent antimicrobial activity > pentyl > heptyl. However, pyrimidine derivatives 91a–c and 92a–c offered promising antimicrobial activities and also the chain length of alkylated NH is essential for activity as chalcone derivative substitution in the para position with an electron-withdrawing group as chlorine atom 92b increased activity towards bacterial strains, while substitution with an electron-donating group as methyl group 92c increased antifungal activity towards C. utilis. Additionally, NH-free benzimidazole analogs, such as in the designated compounds 93a–c, exhibited potent antimicrobial activity, introduction of electron donating group as methyl group at position 4 increase antibacterial activity against Gram-negative bacteria P. aeruginosa and outstanding antifungal activity against A, flavus, while incorporation with electron withdrawing group as fluorine atom increase activity towards Gram-positive bacteria S. aureus.101
 |
| | Fig. 31 Benzimidazole hybrids 90a–c, 91a–c, 92a–c and 93a–c designed for dual antimicrobial evaluation. | |
Table 28 Antimicrobial MIC data for benzimidazole derivatives 90a–c, 91a–c, 92a–c and 93a–c
| Compound |
Minimum inhibition concentration (μg mL−1) |
| B. subtilis |
B. proteus |
S. aureus |
P. aeruginosa |
C. utilis |
A. flavus |
| 90a |
16 |
4 |
8 |
4 |
16 |
16 |
| 90b |
64 |
32 |
16 |
128 |
>512 |
512 |
| 90c |
64 |
512 |
128 |
512 |
64 |
512 |
| 91a |
4 |
512 |
4 |
0.5 |
16 |
128 |
| 91b |
256 |
64 |
512 |
512 |
512 |
16 |
| 91c |
512 |
512 |
512 |
512 |
>512 |
512 |
| 92a |
512 |
128 |
512 |
128 |
512 |
512 |
| 92b |
64 |
4 |
16 |
64 |
256 |
256 |
| 92c |
16 |
16 |
8 |
8 |
64 |
64 |
| 93a |
2 |
128 |
2 |
16 |
256 |
256 |
| 93b |
64 |
8 |
64 |
128 |
4 |
4 |
| 93c |
8 |
1 |
8 |
4 |
1 |
1 |
| Chloromycin |
32 |
32 |
8 |
16 |
— |
— |
| Norfloxacin |
2 |
4 |
8 |
1 |
— |
— |
| Fluconazole |
— |
— |
— |
— |
16 |
256 |
2.1.3 Antiviral activity. Francesconi et al. investigated two series of benzimidazole derivatives (thio), semicarbazones and hydrazones, synthesized from 5-acetyl benzimidazoles to evaluate their antiviral efficacy (Fig. 32). Compounds 94a and 94b displayed a dual inhibitory activity against Influenza A and Human corona virus as the first benzimidazole derivatives reported activity against Corona virus. Compound 94a exhibited good activity against both Influenza A and HCV with EC50 = 38 μM and 56 μM, respectively, comparable to the EC50 of the reference drug ribavirin 7.5 μM; moreover, compound 94b showed outstanding activity against both viruses with EC50 = 25 μM and 38 μM, respectively, when compared to the reference drug ribavirin. Compounds 94c and 94d exhibited potent and selective activity against respiratory syncytial virus (RSV) by inhibiting viral RNA synthesis. Their EC₅₀ values were (7 μM and 2.4 μM, respectively) comparable to the reference drug ribavirin (6.7 μM) (Table 29). Molecular modelling studies for the designated compounds 94c and 94d properly bind to the exposed surface of the RSV F protein, and they bound within the hydrophobic pocket through π–π stacking and cation-π interactions with F137, F140, and F488. Furthermore, compounds 94c and 94d showed a favorable profile in terms of lipophilicity, with a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value below 5 (Lipinski rules) and also demonstrated complete absorption at the human intestinal membrane compared to the reference compound, exhibiting greater oral bioavailability and lower binding to plasma proteins. SAR analysis revealed that regarding Influenza A and Corona virus, the activity is attributed to thiosemicarbazone especially when compared with the benzyl ring; the nature of the substituent also does not have a significant effect on antiviral activity as the unsubstituted derivative (H) 94b had high potency comparable with electron-withdrawing (Cl) 94a. Furthermore, compounds featuring 2-benzotriazol in addition to thiosemicarbazone and hydrazone moieties 94c and 94d showed selective potency against Respiratory syncytial virus (RSV), comparable to the approved antiviral drug ribavirin.102
P value below 5 (Lipinski rules) and also demonstrated complete absorption at the human intestinal membrane compared to the reference compound, exhibiting greater oral bioavailability and lower binding to plasma proteins. SAR analysis revealed that regarding Influenza A and Corona virus, the activity is attributed to thiosemicarbazone especially when compared with the benzyl ring; the nature of the substituent also does not have a significant effect on antiviral activity as the unsubstituted derivative (H) 94b had high potency comparable with electron-withdrawing (Cl) 94a. Furthermore, compounds featuring 2-benzotriazol in addition to thiosemicarbazone and hydrazone moieties 94c and 94d showed selective potency against Respiratory syncytial virus (RSV), comparable to the approved antiviral drug ribavirin.102
 |
| | Fig. 32 Benzimidazole derivatives 94a–d evaluated for antiviral activity against RSV, Influenza, and Corona virus. | |
Table 29 EC50 values of synthesized benzimidazole derivatives 94a–d against RSV, Influenza, and Corona virus
| Compound |
Effective concentration 50 (μM) |
| Influenza virus |
Corona virus |
Respiratory syncytial virus |
| 94a |
38 |
56 |
— |
| 94b |
25 |
38 |
— |
| 94c |
— |
— |
7 |
| 94d |
— |
— |
2.4 |
| Ribavirin |
7.5 |
7.5 |
6.7 |
Novel substituted benzimidazole derivatives were designed by Beč et al. and evaluated against different viruses as Human corona virus, Influenza virus, Yellow fever virus, Zika virus and Sindbis virus (Fig. 33). Moreover, they studied the impact of the synthesized compounds on the biological activity of substituents positioned at N atom of the benzimidazole nuclei and type of substituents attached to the phenyl ring. Compounds 95a and 95b displayed high selectivity against Zika virus through inhibition of its replication through inhibition of RNA-dependent RNA polymerase with EC50 = 43.1 μM and 46.4 μM, respectively, if compared with the EC50 of the reference drug ribavirin >250 μM, while compounds 95c and 95d showed poor activity against all tested viral strains with EC50 > 100 μM (Table 30). The SAR studies revealed that novel benzimidazole derivatives bearing free NH2 group at position 2 retain or increase antiviral activity. Also cyano group is optimal for activity at position 5 and shifting in positioning at 6-position badly affect potency while variation in substituents at position 1 with aliphatic chains afford derivatives 95a with high potent antiviral activity than substitution with phenyl ring 95b. While N-substitution with Schiff base derivatives such as bearing 4-N,N-dimethyl amino- or 4-N,N diethylamino-2-hydroxyohenyl ring 95c and 95d proved to be lacking any antiviral activity.103
 |
| | Fig. 33 Benzimidazole analogs 95a–d developed for antiviral testing against Zika virus. | |
Table 30 Antiviral activity (EC50) of benzimidazole derivatives 95a–d against Zika virus
| Compound |
Effective concentration 50 (μM) |
| Zika virus |
| 95a |
43.1 |
| 95b |
46.4 |
| 95c |
>100 |
| 95d |
>100 |
| Ribavirin |
>250 |
In 2023, Srivastava et al. designed and synthesized novel pyrimidine hybrids and screened them for their antiviral activity against different viruses such as Human immunodeficiency virus (HIV), Para-influenza-3 virus, Reovirus, Sindbis virus, Coxsackie virus and Yellow fever virus (Fig. 34). Compound 96a displayed high selectivity and potency against Coxsackie virus acting by inhibition of its replication through inhibition of RNA-dependent RNA polymerase leading to mutagenesis and suppression of viral replication with outstanding EC50 = 0.026 μM comparable to the EC50 > 250 μM of the reference drug ribavirin. Furthermore, compounds 96d and 96e exhibited excellent activity against HIV with IC50 values of 6.65 μg mL−1 and 15.82 μg mL−1, respectively, which indicated them as promising anti-HIV agents. While compounds 96b and 96c showed poor antiviral activity against all tested viruses with IC50 > 100 μg mL−1 (Table 31). Notably, molecular modelling studies revealed that compounds 96d and 96e exhibited π–π interactions with Tyr318 at a distance of 5.0 A° and π–π and π–cation interactions with Tyr318 and Lys103 amino acid residues, respectively, in the hydrophobic pocket of RT protein. Moreover, Molecular Dynamics (MD) Simulation was simulated over 50 ns to support docking studies, revealing that compound 96d have higher stability than HIV-RT:nevirapine. In addition, the physicochemical properties and ADMET prediction for the designated compound 96d displayed good oral bioavailability with high profile safety margin. SAR analysis revealed that antiviral activity against Coxsackie virus based on the phenyl ring at position 2 must be free of any substitutions and pyrimidine at the N1 of the benzimidazole core 96a. Additionally, for anti-HIV activity, the introduction of halogens such as chlorine atom at para position of phenyl ring substituted from benzimidazole scafold at position 2, along with the incorporation of a five-membered ring at position 2 were found to be favorable. Furthermore, methyl substitution at position 5 of the N-1 pyrimidine ring enhanced the activity, as observed in compounds 96d and 96e.104
 |
| | Fig. 34 Benzimidazole hybrids 96a–e evaluated for antiviral activity against Coxsackie virus and HIV. | |
Table 31 EC50 values of designated benzimidazole 96a–e against Coxsackie virus and HIV
| Compound |
Effective concentration 50 (μg mL−1 & μM−1) |
| Coxsackie virus |
HIV |
| 96a |
0.026 |
— |
| 96b |
— |
>100 |
| 96c |
— |
>100 |
| 96d |
— |
6.65 |
| 96e |
— |
15.82 |
| Ribavirin |
>250 |
>250 |
3 General structure–activity relationship (SAR) study
The structure–activity relationship (SAR) describes the relation between a drug molecule's chemical or three-dimensional structure and its biological activity. This relationship is explored by altering the compound's structure to observe changes in its biological activity or potency. Specifically, the introduction of new functional groups and variations in their positions on the parent nucleus of a biologically active compound are evaluated to assess their impact on biological effects.
Antibacterial activity of benzimidazoles can be affected by:
• Metal complexation of benzimidazoles with metals such as nickel (Ni), particularly when substituted at position 2 as in compound 61a, significantly enhances DNA intercalation and antibacterial activity.
• Meta-substituents on the 2-phenyl ring of benzimidazole like 3-NHSO2CH3 in compound 62a enhance the antibacterial potency, whereas para-substituents in compound 62d diminish activity.
• Unsubstituted amidino groups as in compound 63a in 5-position contribute to broad-spectrum antibacterial activity. Moreover, the incorporation of lipophilic groups like isopropyl in compound 63c increases selectivity towards Gram-positive bacteria, especially MRSA.
• Introduction of electron-withdrawing groups as Cl and CF3 in position-5 in compounds 72a and 72b significantly boosts antibacterial potency, while electron-donating groups like methyl in compound 72c reduce activity.
• Molecular hybridization with other heterocycles (e.g., quinazoline and triazole in compound 70a) at position-2 enhances the antibacterial activity.
• Aliphatic ester chains on the triazole ring at position-2 in compound 66a had broad-spectrum activity, while bulkier or aromatic groups in compound 66b tend to narrow the spectrum towards Gram-positive bacteria.
• The incorporation of hydrophilic groups such as carboxylic acid in compound 68a improves activity compared to other lipophilic substitutions.
• Bulky groups such as methoxy group at position 1 on the heterocycle as in compound 73d negatively impact antibacterial activity.
Antifungal activity of benzimidazoles can be affected by:
• Substitution at position 5 with bulky benzoyl groups in compounds 78a and 78b significantly enhances antifungal potency. In contrast, applying a simplification strategy on the structure as in compound 78c reduces the activity.
• Introduction of hydrophilic groups on the 2-phenyl ring of benzimidazole core as hydroxyl groups in compound 84a or free of substitution in 84c derivative improve activity, while lipophilic groups such as bromine in 84b diminish efficacy.
• Electron-withdrawing groups such as chlorine in compounds 79a–c and fluorine in compounds 75a and 75b at position 2 of the phenyl ring were associated with enhanced antifungal activity. Conversely, substitution with electron-donating groups like methoxy or methyl, as in 74d and 82a, reduced antifungal potency.
• Introducing triazole and oxadiazole substituents at position 2 of the benzimidazole scaffold was found to be essential for antifungal activity, enhancing binding affinity to HEM and increasing efficacy against the tested fungal strains, as observed in compounds 76a, 78a, and 81a–c.
• Molecular hybridization with thiazole and morpholine at position-2 in compounds 80a–c improves activity, especially against C. neoformans as morpholine increases membrane penetration. Moreover, introduction of a hydrazone moiety at position-5 in 83a–c derivatives enhances selectivity and potency, particularly with chlorine substitution at position 4 of the phenyl ring.
• Planar conformations (e.g. in compounds 77a and 77b) enhance π–π interactions with fungal enzymes, thus enhancing the antifungal activity.
Dual antibacterial and antifungal activities of benzimidazoles can be affected by:
• Short aliphatic chains (ethyl) at position-1 on the benzimidazole core enhance antibacterial activity as in compound 85a and 91a whereas longer chains reduce it in compound 85d, 91b and 91c.
• Benzimidazole incorporated with benzoyl moieties affected by substitution properties as electron withdrawing groups as bromine and nitro on the phenyl ring in derivatives 88a–d significantly enhance antimicrobial activity, while electron donating groups like methyl and methoxy in compounds 88e–f reduce it.
• In carvacrol-based benzimidazole, bioisosteric replacement of the phenyl ring with a pyridine ring in the 89c derivative enhances the activity against S. aureus, while the introduction of an electron-donating group like methyl group in compound 89b positively affected the activity against E. coli, furthermore introduction of halogens at position 6 enhance the activity against C. albicans as in compounds 89e–f.
• The design of benzimidazole–chalcone hybrids at position 2 generally resulted in enhanced antimicrobial activity if incorporated with electron donating groups as CH3 group 90a on the other hand electron withdrawing groups like F, Cl diminished activity 90b and 90c or with pyrimidine moieties incorporated with electron with drawing group Cl improve antibacterial activity 92b while introduction electron donating group CH3 enhance antifungal activity 92c.
Antiviral activity of benzimidazoles can be affected by:
• Molecular hybridization with a thiosemicarbazone moiety at position-5 and the presence of an unsubstituted phenyl ring at position-2 are essential for antiviral activities against Influenza A and Corona virus 94b than 94a.
• The introduction of a benzotriazole moiety at position-2 enhances the selectivity for RSV (compounds 94c and 94d).
• For anti-Zika virus activity, a free –NH2 group at position 2 is essential for optimal efficacy as compounds 95a and 95b than other substituted compounds 95c and 95d, also presence of cyano group at position-5 is optimal for antiviral activity, moreover, N1 substitution with aliphatic chains 95a has favored antiviral activity over phenyl rings 95b.
• Introducing a pyrimidine moiety at position 1 of the benzimidazole ring, along with an unsubstituted phenyl ring at position 2, as in compound 96a, was found to be crucial for activity against Coxsackie virus. In the case of HIV, substitution at the para-position of the phenyl ring with a large radius atom such as chlorine, as in compound 96d, enhanced antiviral activity.
4 Conclusion and future prospects
The period from 2018 to 2024 has witnessed substantial advancements in the synthesis and antimicrobial evaluation of benzimidazole derivatives, reaffirming their significance in medicinal chemistry. Novel synthetic methodologies, particularly those aligned with green chemistry principles, have facilitated efficient and sustainable production of these compounds. Structural modifications and the development of hybrid molecules have demonstrated remarkable antimicrobial activity, even against multidrug-resistant pathogens. SAR studies have further refined our understanding of structure–activity relationships, paving the way for the rational design of highly potent derivatives. Despite these achievements, challenges remain, such as the need for improved selectivity, reduced toxicity, and enhanced pharmacokinetic profiles for clinical applications. Future research should focus on the exploration of benzimidazole-based nanomaterials and drug delivery systems hold promise for enhancing the therapeutic efficacy and overcoming resistance mechanisms. The continued emphasis on interdisciplinary approaches and collaborative efforts will be crucial in unlocking the full potential of benzimidazoles as next-generation antimicrobial agents, addressing the global crisis of antimicrobial resistance.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
References
- Z. Hosseinzadeh, A. Ramazani and N. Razzaghi-Asl, Anti-cancer nitrogen-containing heterocyclic compounds, Curr. Org. Chem., 2018, 22(23), 2256–2279 CrossRef CAS.
- T. Qadir, et al., A review on medicinally important heterocyclic compounds, Open Med. Chem. J., 2022, 16(1), e187410452202280 CrossRef CAS.
- M. Hossain and A. K. Nanda, A review on heterocyclic: Synthesis and their application in medicinal chemistry of imidazole moiety, Science, 2018, 6(5), 83–94 CAS.
- V. J. Ram, et al., The Chemistry of Heterocycles: Nomenclature and Chemistry of Three to Five Membered Heterocycles, Elsevier, 2019 Search PubMed.
- K. Singh, et al., Insights into the structure activity relationship of nitrogen-containing heterocyclics for the development of antidepressant compounds: An updated review, J. Mol. Struct., 2021, 1237, 130369 CrossRef CAS.
- C. Verma, et al., Coordination bonding and corrosion inhibition potential of nitrogen-rich heterocycles: Azoles and triazines as specific examples, Coord. Chem. Rev., 2023, 488, 215177 CrossRef CAS.
- S. Banerjee, et al., A critical review of benzimidazole: Sky-high objectives towards the lead molecule to predict the future in medicinal chemistry, Results Chem., 2023, 6, 101013 CrossRef CAS.
- Y. N. A. Soh, et al., Composition and biotransformational changes in soluble microbial products (SMPs) along an anaerobic baffled reactor (ABR), Chemosphere, 2020, 254, 126775 CrossRef CAS PubMed.
- H. Hernández-López, C. J. Tejada-Rodríguez and S. Leyva-Ramos, A panoramic review of benzimidazole derivatives and their potential biological activity, Mini-Rev. Med. Chem., 2022, 22(9), 1268–1280 CrossRef PubMed.
- S. K. Manna, T. Das and S. Samanta, Polycyclic benzimidazole: Synthesis and photophysical properties, ChemistrySelect, 2019, 4(30), 8781–8790 CrossRef CAS.
- P. K. Singh and O. Silakari, Benzimidazole: journey from single targeting to multitargeting molecule, in Key Heterocycle Cores for Designing Multitargeting Molecules, Elsevier, 2018, pp. 31–52 Search PubMed.
- X. Yang, et al., A New Discovery towards Novel Skeleton of Benzimidazole-Conjugated Pyrimidinones as Unique Effective Antibacterial Agents, Chin. J. Chem., 2022, 40(22), 2642–2654 CrossRef CAS.
- H. A. AlDifar, et al., Synthesis of Benzimidazole and Phthaloylamino Acid derivatives and Antibacterial Activity, J. Med. Chem. Sci., 2023, 6, 1975–1984 CAS.
- V. Kamat, et al., Synthesis, molecular docking, antibacterial, and anti-inflammatory activities of benzimidazole-containing tricyclic systems, J. Chin. Chem. Soc., 2021, 68(6), 1055–1066 CrossRef CAS.
- K. Mahmood, et al., Synthesis, characterization and biological evaluation of novel benzimidazole derivatives, J. Biomol. Struct. Dyn., 2020, 38(6), 1670–1682 CAS.
- E. Mulugeta and Y. Samuel, Synthesis of Benzimidazole-Sulfonyl Derivatives and Their Biological Activities, Biochem. Res. Int., 2022, 2022(1), 7255299 Search PubMed.
- B. T. B. Hue, et al., Benzimidazole derivatives as novel zika virus inhibitors, ChemMedChem, 2020, 15(15), 1453–1463 CrossRef CAS PubMed.
- X. Huo, et al., Design, synthesis, in vitro and in vivo anti-respiratory syncytial virus (RSV) activity of novel oxizine fused benzimidazole derivatives, Eur. J. Med. Chem., 2021, 224, 113684 CrossRef CAS PubMed.
- J. C. Sánchez-Salgado, et al., Systematic search for benzimidazole compounds and derivatives with antileishmanial effects, Mol. Diversity, 2018, 22, 779–790 CrossRef PubMed.
- N. Razzaghi-Asl, et al., Insights into the current status of privileged N-heterocycles as antileishmanial agents, Mol. Diversity, 2020, 24, 525–569 CrossRef CAS PubMed.
- G. A. Dziwornu, et al., Antimalarial benzimidazole derivatives incorporating phenolic mannich base side chains inhibit microtubule and hemozoin formation: structure–activity relationship and in vivo oral efficacy studies, J. Med. Chem., 2021, 64(8), 5198–5215 CrossRef CAS PubMed.
- M. Leshabane, et al., Benzimidazole derivatives are potent against multiple life cycle stages of Plasmodium falciparum malaria parasites, ACS Infect. Dis., 2021, 7(7), 1945–1955 CrossRef CAS PubMed.
- V. M. Patel, et al., N-Mannich bases of benzimidazole as a potent antitubercular and antiprotozoal agents: Their synthesis and computational studies, Synth. Commun., 2020, 50(6), 858–878 CrossRef CAS.
- A. Moreno-Herrera, et al., Recent advances in the development of broad-spectrum antiprotozoal agents, Curr. Med. Chem., 2021, 28(3), 583–606 CAS.
- J.-Y. Chai, B.-K. Jung and S.-J. Hong, Albendazole and mebendazole as anti-parasitic and anti-cancer agents: an update, Korean J. Parasitol., 2021, 59(3), 189 CrossRef CAS PubMed.
- N. T. Chung, V. C. Dung and D. X. Duc, Recent achievements in the synthesis of benzimidazole derivatives, RSC Adv., 2023, 13(46), 32734–32771 RSC.
- S. Bai, et al., Research Progress on Benzimidazole Fungicides: A Review, Molecules, 2024, 29(6), 1218 CrossRef CAS PubMed.
- J. Jacob, et al., Clinical efficacy and safety of albendazole and other benzimidazole anthelmintics for rat lungworm disease (neuroangiostrongyliasis): a systematic analysis of clinical reports and animal studies, Clin. Infect. Dis., 2022, 74(7), 1293–1302 CrossRef CAS PubMed.
- M. Budetić, et al., Review of Characteristics and Analytical Methods for Determination of Thiabendazole, Molecules, 2023, 28(9), 3926 CrossRef PubMed.
- I. Celik, et al., Synthesis, Molecular Docking, Dynamics, Quantum-Chemical Computation, and Antimicrobial Activity Studies of Some New Benzimidazole–Thiadiazole Hybrids, ACS Omega, 2022, 7(50), 47015–47030 CrossRef CAS PubMed.
- S. B. F. Fawzi, et al., Benzimidazole and Its Derivatives: Exploring Their Crucial Role in Medicine and Agriculture: A Short Review, Al-Kitab J. for Pure Sci., 2024, 8(02), 125–137 CrossRef.
- B. Pathare and T. Bansode, biological active benzimidazole derivatives, Results Chem., 2021, 3, 100200 Search PubMed.
- R. S. Keri, et al., Comprehensive review in current developments of benzimidazole-based medicinal chemistry, Chem. Biol. Drug Des., 2015, 86(1), 19–65 Search PubMed.
- M. Faheem, et al., A review on the modern synthetic approach of benzimidazole candidate, ChemistrySelect, 2020, 5(13), 3981–3994 Search PubMed.
- Y. Bansal and O. Silakari, The therapeutic journey of benzimidazoles: A review, Bioorg. Med. Chem., 2012, 20(21), 6208–6236 CrossRef CAS PubMed.
- N. D. Mahurkar, et al., Benzimidazole: A Versatile Scaffold for Drug Discovery and Beyond-A Comprehensive Review of Synthetic Approaches and Recent Advancements in Medicinal Chemistry, Results Chem., 2023, 101139 Search PubMed.
- M. S. Vasava, et al., Benzimidazole: A milestone in the field of medicinal chemistry, Mini-Rev. Med. Chem., 2020, 20(7), 532–565 Search PubMed.
- V. A. S. Pardeshi, et al., A review on synthetic approaches of benzimidazoles, Synth. Commun., 2021, 51(4), 485–513 Search PubMed.
- S. Thapa, S. L. Nargund and M. S. Biradar, A systematic review on diverse synthetic route and pharmacological activities of benzimidazole as optimized lead, J. Ultra Chem., 2022, 18(2), 24–36 CrossRef CAS.
- M. Patel, et al., A Review of Approaches to the Metallic and Non-Metallic Synthesis of Benzimidazole (BnZ) and Their Derivatives for Biological Efficacy, Molecules, 2023, 28(14), 5490 CrossRef CAS PubMed.
- S. I. Alaqeel, Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review, J. Saudi Chem. Soc., 2017, 21(2), 229–237 Search PubMed.
- M. R. Poor Heravi and M. Ashori, Boric acid catalyzed convenient synthesis of benzimidazoles in aqueous media, J. Chem., 2013, 2013(1), 496413 CrossRef.
- A. Mehra and R. Sangwan, Synthesis and Pharmacological Properties of the Benzimidazole Scaffold: A Patent Review, ChemistrySelect, 2023, 8(45), e202300537 CrossRef CAS.
- P. P. Nair, A. Jayaraj and C. A. Swamy P, Recent advances in benzimidazole based NHC-metal complex catalysed cross-coupling reactions, ChemistrySelect, 2022, 7(4), e202103517 CrossRef CAS.
- B. Zou, Q. Yuan and D. Ma, Synthesis of 1, 2-disubstituted benzimidazoles by a Cu-catalyzed cascade aryl amination/condensation process, Angew. Chem., Int. Ed., 2007, 46(15), 2598–2601 CrossRef CAS PubMed.
- K. Das, A. Mondal and D. Srimani, Selective synthesis of 2-substituted and 1, 2-disubstituted benzimidazoles directly from aromatic diamines and alcohols catalyzed by molecularly defined nonphosphine manganese (I) complex, J. Org. Chem., 2018, 83(16), 9553–9560 CrossRef CAS PubMed.
- T. B. Nguyen, L. P. A. Nguyen and T. T. T. Nguyen, Sulfur-Catalyzed Oxidative Coupling of Dibenzyl Disulfides with Amines: Access to Thioamides and Aza Heterocycles, Adv. Synth. Catal., 2019, 361(8), 1787–1791 CrossRef CAS.
- L. A. Nguyen, P. Retailleau and T. B. Nguyen, Elemental Sulfur/DMSO-Promoted Multicomponent One-pot Synthesis of Malonic Acid Derivatives from Maleic Anhydride and Amines, Adv. Synth. Catal., 2019, 361(12), 2864–2869 CrossRef CAS.
- J. Mokhtari and A. H. Bozcheloei, One-pot synthesis of benzoazoles via dehydrogenative coupling of aromatic 1, 2-diamines/2-aminothiophenol and alcohols using Pd/Cu-MOF as a recyclable heterogeneous catalyst, Inorg. Chim. Acta, 2018, 482, 726–731 CrossRef CAS.
- V. Sankar, et al., Metal–organic framework mediated expeditious synthesis of benzimidazole and benzothiazole derivatives through an oxidative cyclization pathway, New J. Chem., 2020, 44(3), 1021–1027 RSC.
- D. M. Dissanayake and A. K. Vannucci, Transition-Metal-Free and Base-Free Electrosynthesis of 1 H-Substituted Benzimidazoles, ACS Sustain. Chem. Eng., 2018, 6(1), 690–695 CrossRef CAS.
- K. Kant, et al., Recent Advancements in Strategies for the Synthesis of Imidazoles, Thiazoles, Oxazoles, and Benzimidazoles, ChemistrySelect, 2023, 8(47), e202303988 CrossRef CAS.
- X. Liu, et al., CN bond formation and cyclization: A straightforward and metal-free synthesis of N-1-alkyl-2-unsubstituted benzimidazoles, Tetrahedron Lett., 2019, 60(15), 1057–1059 CrossRef CAS.
- H. Mostafavi, et al., Synthesis of 1 H-1, 3-benzimidazoles, benzothiazoles and 3 H-imidazo [4, 5-c] pyridine using DMF in the presence of HMDS as a reagent under the transition-metal-free condition, Chem. Pap., 2018, 72, 2973–2978 CrossRef CAS.
- F. Feizpour, M. Jafarpour and A. Rezaeifard, A tandem aerobic photocatalytic synthesis of benzimidazoles by cobalt ascorbic acid complex coated on TiO2 nanoparticles under visible light, Catal. Lett., 2018, 148, 30–40 CrossRef CAS.
- Z. Gan, et al., Imidazolium chloride-catalyzed synthesis of benzimidazoles and 2-substituted benzimidazoles from o-phenylenediamines and DMF derivatives, Tetrahedron, 2018, 74(52), 7450–7456 CrossRef CAS.
- S. K. Bagaria, N. Jangir and D. K. Jangid, Green and eco-compatible iron nanocatalysed synthesis of benzimidazole: A review, Sustainable Chem. Pharm., 2023, 31, 100932 CrossRef CAS.
- S. Singhal, et al., Recent Trends in the Synthesis of Benzimidazoles From o-Phenylenediamine via Nanoparticles and Green Strategies Using Transition Metal Catalysts, J. Heterocycl. Chem., 2019, 56(10), 2702–2729 CrossRef CAS.
- N. Kaur, et al., Metallovesicles as smart nanoreactors for green catalytic synthesis of benzimidazole derivatives in water, J. Mater. Chem. A, 2019, 7(29), 17306–17314 RSC.
- M. Nardi, et al., A review on the green synthesis of benzimidazole derivatives and their pharmacological activities, Catalysts, 2023, 13(2), 392 CrossRef CAS.
- A. Arya, V. Mishra and T. S. Chundawat, Green synthesis of silver nanoparticles from green algae (Botryococcus braunii) and its catalytic behavior for the synthesis of benzimidazoles, Chem. Data Collect., 2019, 20, 100190 CrossRef CAS.
- M. L. Di Gioia, et al., Green synthesis of privileged benzimidazole scaffolds using active deep eutectic solvent, Molecules, 2019, 24(16), 2885 CrossRef PubMed.
- F. Shukla, M. Das and S. Thakore, Copper nanoparticles loaded polymer vesicles as environmentally amicable nanoreactors: a sustainable approach for cascading synthesis of benzimidazole, J. Mol. Liq., 2021, 336, 116217 CrossRef CAS.
- L. Ravindar, et al., Cross-Coupling of C−H and N−H Bonds: A Hydrogen Evolution Strategy for the Construction of C−N Bonds, Eur. J. Org Chem., 2022, 2022(31), e202200596 CrossRef CAS.
- D. Kaldhi, et al., Organocatalytic oxidative synthesis of C2-functionalized benzoxazoles, naphthoxazoles, benzothiazoles and benzimidazoles, Tetrahedron Lett., 2019, 60(3), 223–229 CrossRef CAS.
- T. T. Bui and H. K. Kim, Recent Advances in Photo-mediated Radiofluorination, Chem.–Asian J., 2021, 16(16), 2155–2167 CrossRef CAS PubMed.
- T. Montini, et al., Sustainable photocatalytic synthesis of benzimidazoles, Inorg. Chim. Acta, 2021, 520, 120289 CrossRef CAS.
- G. Kumaraswamy, et al., An efficient photocatalytic synthesis of benzimidazole over cobalt-loaded TiO2 catalysts under solar light irradiation, J. Photochem. Photobiol., A, 2022, 429, 113888 CrossRef CAS.
- H. N. Abdelhamid, I. M. Mekhemer and A.-A. M. Gaber, Metal-organic frameworks (MOFs)-derived ZrOSO4@ C for photocatalytic synthesis of benzimidazole derivatives, Mol. Catal., 2023, 548, 113418 CrossRef CAS.
- M. Gaba and C. Mohan, Development of drugs based on imidazole and benzimidazole bioactive heterocycles: recent advances and future directions, Med. Chem. Res., 2016, 25, 173–210 CrossRef CAS.
- R. M. LoPachin and T. Gavin, Molecular mechanisms of aldehyde toxicity: a chemical perspective, Chem. Res. Toxicol., 2014, 27(7), 1081–1091 Search PubMed.
- D. Hernández-Romero, et al., First-row transition metal compounds containing benzimidazole ligands: An overview of their anticancer and antitumor activity, Coord. Chem. Rev., 2021, 439, 213930 Search PubMed.
- K. Mahmood, et al., Synthesis, DNA binding and antibacterial activity of metal (II) complexes of a benzimidazole Schiff base, Polyhedron, 2019, 157, 326–334 CrossRef CAS.
- M. A. Argirova, et al., New 1 H-benzimidazole-2-yl hydrazones with combined antiparasitic and antioxidant activity, RSC Adv., 2021, 11(63), 39848–39868 RSC.
- E. M. Dokla, et al., Development of benzimidazole-based derivatives as antimicrobial agents and their synergistic effect with colistin against gram-negative bacteria, Eur. J. Med. Chem., 2020, 186, 111850 CrossRef CAS PubMed.
- A. Bistrović, et al., Synthesis, anti-bacterial and anti-protozoal activities of amidinobenzimidazole derivatives and their interactions with DNA and RNA, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 1323–1334 CrossRef PubMed.
- H. R. Rashdan, et al., Synthesis, identification, computer-aided docking studies, and ADMET prediction of novel benzimidazo-1, 2, 3-triazole based molecules as potential antimicrobial agents, Molecules, 2021, 26(23), 7119 CrossRef CAS PubMed.
- L. Deswal, et al., Synthesis, antimicrobial and α-glucosidase inhibition of new benzimidazole-1, 2, 3-triazole-indoline derivatives: a combined experimental and computational venture, Chem. Pap., 2022, 76(12), 7607–7622 CrossRef CAS.
- A. Saber, et al., New 1, 2, 3-triazole containing benzimidazolone derivatives: Syntheses, crystal structures, spectroscopic characterizations, Hirshfeld surface analyses, DFT calculations, anti-corrosion property anticipation, and antibacterial activities, J. Mol. Struct., 2021, 1242, 130719 CrossRef CAS.
- F. F. Al-Blewi, et al., Design, synthesis, ADME prediction and pharmacological evaluation of novel benzimidazole-1, 2, 3-triazole-sulfonamide hybrids as antimicrobial and antiproliferative agents, Chem. Cent. J., 2018, 12, 1–14 CrossRef PubMed.
- Y. Aparna, et al., Synthesis and Antimicrobial Activity of Novel Bis-1, 2, 3-triazol-1 H-4-yl-substituted Aryl Benzimidazole-2-thiol Derivatives, Russ. J. Gen. Chem., 2020, 90, 1501–1506 CrossRef CAS.
- G. K. Kantar, et al., Synthesis and antimicrobial activity of some new triazole bridged benzimidazole substituted phthalonitrile and phthalocyanines, Rev. Roum. Chim., 2018, 63(1), 59–65 Search PubMed.
- N. K. Nandwana, et al., Design and synthesis of imidazo/benzimidazo [1, 2-c] quinazoline derivatives and evaluation of their antimicrobial activity, ACS Omega, 2018, 3(11), 16338–16346 CrossRef CAS PubMed.
- M. Al-Majidi, et al., Synthesis and Identification of Some New Derivatives of ([Benzyl Thio) Benzimidazole-N-(Methylene-5-Yl)]-4, 5-Di Substituted 1, 2, 4-Triazole and Evaluation of Their Activity as Antimicrobial and Anti-Inflammatory Agents, Iraqi J. Sci., 2021, 62(4), 1054–1065 CrossRef.
- K. El Gadali, et al., Synthesis, structural characterization and antibacterial activity evaluation of novel quinolone-1, 2, 3-triazole-benzimidazole hybrids, J. Mol. Struct., 2023, 1282, 135179 CrossRef.
- S. B. Korrapati, et al., In-silico driven design and development of spirobenzimidazo-quinazolines as potential DNA gyrase inhibitors, Biomed. Pharmacother., 2021, 134, 111132 Search PubMed.
- D. Ashok, et al., Conventional and microwave-assisted synthesis of new indole-tethered benzimidazole-based 1, 2, 3-triazoles and evaluation of their antimycobacterial, antioxidant and antimicrobial activities, Mol. Diversity, 2018, 22, 769–778 CrossRef CAS PubMed.
- V. Mallikanti, et al., Synthesis, antimicrobial activity and molecular docking of novel benzimidazole conjugated 1, 2, 3-triazole analogues, Chem. Data Collect., 2023, 45, 101034 CrossRef CAS.
- R. S. Kankate, P. S. Gide and D. P. Belsare, Design, synthesis and antifungal evaluation of novel benzimidazole tertiary amine type of fluconazole analogues, Arabian J. Chem., 2019, 12(8), 2224–2235 CrossRef CAS.
- A. E. Evren, İ. Çelik and U. A. Çevik, Synthesis, molecular docking, in silico ADME and antimicrobial activity studies of some new benzimidazole-triazole derivatives, Cumhur. Sci. J., 2021, 42(4), 795–805 Search PubMed.
- E. Ghobadi, et al., Design, synthesis and biological activity of hybrid antifungals derived from fluconazole and mebendazole, Eur. J. Med. Chem., 2023, 249, 115146 CrossRef CAS PubMed.
- E. Güzel, et al., Synthesis of benzimidazole-1, 2, 4-triazole derivatives as potential antifungal agents targeting 14α-demethylase, ACS Omega, 2023, 8(4), 4369–4384 CrossRef PubMed.
- S. Aaghaz, et al., Synthesis, biological evaluation and mechanistic studies of 4-(1, 3-thiazol-2-yl) morpholine-benzimidazole hybrids as a new structural class of antimicrobials, Bioorg. Chem., 2023, 136, 106538 CrossRef CAS PubMed.
- U. A. Çevik, et al., Synthesis, molecular modeling, quantum mechanical calculations and ADME estimation studies of benzimidazole-oxadiazole derivatives as potent antifungal agents, J. Mol. Struct., 2022, 1252, 132095 CrossRef.
- E. C. Pham, et al., N, 2, 6-Trisubstituted 1 H-benzimidazole derivatives as a new scaffold of antimicrobial and anticancer agents: design, synthesis, in vitro evaluation, and in silico studies, RSC Adv., 2023, 13(1), 399–420 RSC.
- M. M. Morcoss, et al., Design, synthesis, mechanistic studies and in silico ADME predictions of benzimidazole derivatives as novel antifungal agents, Bioorg. Chem., 2020, 101, 103956 Search PubMed.
- H. Chaurasia, et al., Molecular modelling, synthesis and antimicrobial evaluation of benzimidazole nucleoside mimetics, Bioorg. Chem., 2021, 115, 105227 CrossRef CAS PubMed.
- Y. N. Wang, et al., Discovery of Benzimidazole–Quinolone Hybrids as New Cleaving Agents toward Drug-Resistant Pseudomonas aeruginosa DNA, ChemMedChem, 2018, 13(10), 1004–1017 CrossRef CAS PubMed.
- S. Yadav, et al., Synthesis and evaluation of antimicrobial, antitubercular and anticancer activities of 2-(1-benzoyl-1 H-benzo [d] imidazol-2-ylthio)-N-substituted acetamides, Chem. Cent. J., 2018, 12, 1–14 CrossRef PubMed.
- R. T. Bhoi, J. D. Rajput and R. S. Bendre, An efficient synthesis of rearranged new biologically active benzimidazoles derived from 2-formyl carvacrol, Res. Chem. Intermed., 2022, 1–22 Search PubMed.
- H.-B. Liu, et al., Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation, Eur. J. Med. Chem., 2018, 143, 66–84 CrossRef CAS PubMed.
- V. Francesconi, et al., Synthesis and biological evaluation of novel (thio) semicarbazone-based benzimidazoles as antiviral agents against human respiratory viruses, Molecules, 2020, 25(7), 1487 CrossRef CAS PubMed.
- A. Beč, et al., Novel biologically active N-substituted benzimidazole derived Schiff bases: Design, synthesis, and biological evaluation, Molecules, 2023, 28(9), 3720 CrossRef PubMed.
- R. Srivastava, et al., Exploring antiviral potency of N-1 substituted pyrimidines against HIV-1 and other DNA/RNA viruses: Design, synthesis, characterization, ADMET analysis, docking, molecular dynamics and biological activity, Comput. Biol. Chem., 2023, 106, 107910 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Asmaa M. AboulMagd
b,
Asmaa M. AboulMagd