
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structural insights and bio-evaluation of N-phenoxyethylisatin hydrazones as potent α-glucosidase inhibitors†
Saba Mehreenah,
Muhammad Imran Ali a,
Sidra tul Munthab,
Mehwash Ziac,
Aman Ullah*d,
Saeed Ullahe,
Ajmal Khanef,
Javid Hussaing,
Muhammad U. Anware,
Ahmed Al-Harrasi*e and
Muhammad Moazzam Naseer*a
a,
Sidra tul Munthab,
Mehwash Ziac,
Aman Ullah*d,
Saeed Ullahe,
Ajmal Khanef,
Javid Hussaing,
Muhammad U. Anware,
Ahmed Al-Harrasi*e and
Muhammad Moazzam Naseer*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan. E-mail: moazzam@qau.edu.pk
bPeking University Institute of Advanced Agriculture Sciences, Weifang, Shandong, China
cDepartment of Chemistry, Allama Iqbal Open University, Islamabad 44000, Pakistan
dDepartment of Agricultural, Food, and Nutritional Science, 4-10 Agriculture/Forestry Centre, University of Alberta, Edmonton, AB T6G 2P5, Canada. E-mail: ullah2@ualberta.ca
eNatural and Medical Sciences Research Centre, University of Nizwa, Birkat Almouz 616, Oman. E-mail: aharrasi@unizwa.edu.om
fDepartment of Chemical and Biological Engineering, College of Engineering, Korea University, Seoul 02841, Republic of Korea
gDepartment of Biological Sciences & Chemistry, College of Arts and Sciences, University of Nizwa, Nizwa, Oman
hDepartment of Chemistry, Faculty of Sciences, The University of Haripur, KP, 22620, Pakistan
First published on 7th May 2025
Abstract
Effective α-glucosidase inhibitors are vital for managing type 2 diabetes, emphasizing the need for novel and potent compounds. A series of novel N-phenoxyethylisatin hydrazones 1(a–l) have been synthesized and characterized by their spectral data, and in the case of 1l by its single crystal X-ray analysis. All the synthesized compounds were in vitro evaluated for their inhibition potential against the α-glucosidase enzyme. Interestingly, most of these compounds exhibited significant inhibitory activity against the α-glucosidase enzyme, with IC50 values ranging from 3.64 ± 0.13 to 94.89 ± 0.64 μM compared to the standard drug, acarbose (IC50 = 873.34 ± 1.67 μM). The compound 1e was found to be the most active compound of the series having an IC50 value of 3.64 ± 0.13 μM. Molecular docking studies revealed a binding score of −9.7 kcal mol−1 for 1e, slightly surpassing that of acarbose (−9.4 kcal mol−1). Unlike acarbose, which primarily relies on hydrogen bonding, the binding interactions of 1e are dominated by π-interactions. ADMET profiling confirmed favourable pharmacokinetics for these compounds, including good oral bioavailability, balanced hydrophilicity, and minimal predicted toxicity. These findings highlight the potential of these compounds as promising candidates for the development of more effective treatments for hyperglycemia.
1. Introduction
As obesity rates continue to rise, diabetes mellitus (DM) has emerged as a major health challenge of the 21st century, ranking among the leading causes of global mortality.1–6 Classified into two main types, type I DM (insulin-dependent) and type II DM (non-insulin-dependent), the latter, often associated with postprandial hyperglycemia, accounts for approximately 90–95% of diabetic cases.1–6 Elevated postprandial blood glucose levels in diabetic individuals trigger the generation of reactive oxygen species (ROS), leading to severe health complications including cardiovascular diseases,7 nephropathy,8 retinopathy,9 synaptic plasticity,10 thrombosis,11 and Alzheimer's disease.12 Consequently, controlling postprandial hyperglycemia becomes a critical concern for managing diabetes.13 One approach to address this hyperglycemia is by inhibiting the digestion of dietary carbohydrates, a process mediated by the enzyme α-glucosidase in the small intestine.14 Inhibition of α-glucosidase activity reduces carbohydrate digestion, offering a therapeutic strategy to lower blood glucose levels.14 Although commercially available α-glucosidase inhibitors such as acarbose, voglibose, and miglitol are in use, they are associated with various side effects and efficacy issues.15,16 For example, acarbose can cause abdominal distention, diarrhea, liver dysfunction, and hepatotoxicity.17 Voglibose has been linked to dizziness, nausea, and vomiting upon initial use.18 Miglitol, while generally well-tolerated, may cause gastrointestinal issues like bloating, diarrhea, and flatulence, and in rare cases, can lead to elevated liver enzymes and hypoglycemia when combined with other antidiabetic medications.19 Hence, there is a need to explore new, more effective, and readily accessible α-glucosidase inhibitors.Isatin derivatives particularly hydrazones, hold a prominent position in medicinal chemistry owing to their structural versatility and diverse pharmacological activities.20,21 Incorporating hydrazide functionality into the isatin scaffold significantly enhances its pharmacological potential, facilitating the development of compounds with superior bioactivity and selectivity.20,21 Isatin hydrazones have been widely studied for their ability to inhibit key enzymes such as tyrosine kinase,22 α-glucosidase,23 and cholinesterases,24 highlighting their potential as therapeutic candidates for managing diabetes, cancer, neurodegenerative diseases, and other health conditions. Furthermore, the synthetic accessibility and structural diversity of these conjugates enable the creation of extensive chemical libraries, advancing drug discovery efforts.20,21 Thus, the study of isatin derivatives particularly hydrazones, represents a fertile area of research with significant potential to deliver novel therapeutic agents targeting a broad spectrum of diseases and disorders.
Building upon our ongoing research on isatin derivatives,24–26 herein we report a series of novel hydrazone derivatives of N-phenoxyethylisatin (1a–1l). The structure of compound 1l was unambiguously confirmed via X-ray single-crystal analysis. The synthesized compounds were evaluated in vitro for their α-glucosidase inhibitory activity, revealing that most exhibited significant potency. Additionally, in silico analyses were conducted to assess their ADME properties, toxicity profiles, and molecular docking interactions, offering detailed insights into the binding mechanisms of these inhibitors within the enzyme's active site.
2. Results and discussion
2.1 Chemistry
Synthesis of N-phenoxyethyl isatin hydrazones 1(a–l) was initiated with the preparation of N-phenoxyethylisatin C and a series of hydrazides F intermediates (Scheme 1). Both C (ref. 27) and F intermediates were obtained in good to excellent yields starting from simple and commercially available substituted phenols and aryl carboxylic acids, respectively each in two steps. The intermediates C and F were finally condensed in ethanol solvent containing a catalytic amount of glacial acetic acid. The reaction provided good to excellent yields (70–87%) of the desired products in 8 hours (Scheme 1). The structure of all the synthesized hydrazone derivatives 1(a–l) was established by collecting their spectral data. Additionally, the structure of 1l was unambiguously determined by means of its single crystal diffraction analysis.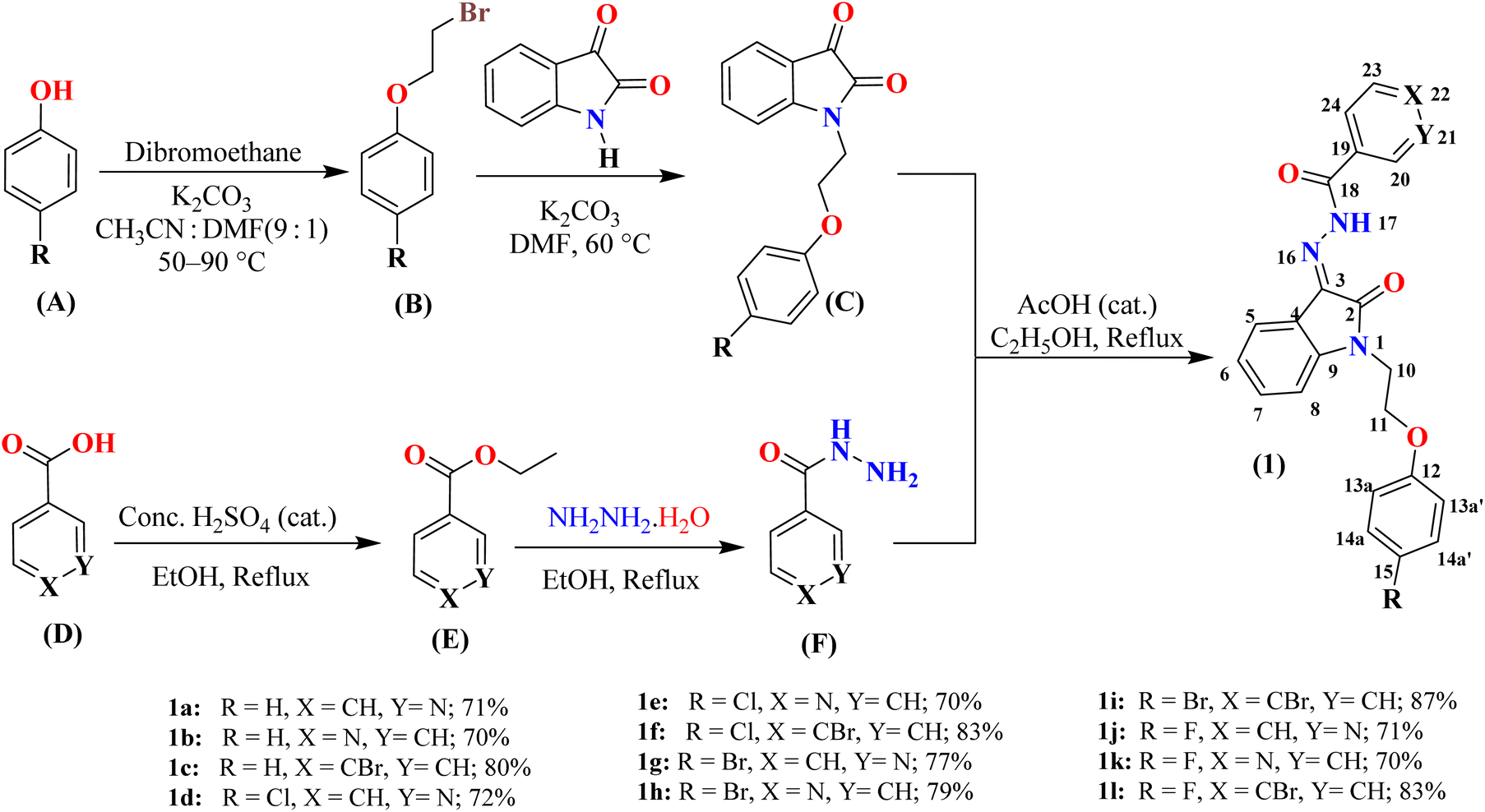 | ||
| Scheme 1 Synthesis of N-phenoxyethyl isatin hydrazones 1(a–l). The atoms are numbered solely for the purpose of assigning signals in the 1H and 13C NMR spectra (see experimental section for details). | ||
In IR spectroscopy, the synthesis of N-phenoxyethyl isatin hydrazones 1(a–l) was indicated by the appearance of new bands corresponding to N–H stretching vibrations in the range of 3194–3250 cm−1 and imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations at 1652–1660 cm−1. The absence of the keto carbonyl (CO) stretch of N-phenoxyethyl isatin intermediate, alongside the presence of characteristic stretching vibrations for lactam (CO) at 1698 cm−1 and amide (CO) at 1676–1683 cm−1, further supported the formation of the products. Additionally, the presence of absorption bands at 1608–1616 cm−1, 1466–1488 cm−1 for aromatic (CC) stretches, at 1242–1269 cm−1 for Csp2–O ether stretches, and at 1152–1166 cm−1 for Csp3–O ether stretches provided further evidence of product formation.
N) stretching vibrations at 1652–1660 cm−1. The absence of the keto carbonyl (CO) stretch of N-phenoxyethyl isatin intermediate, alongside the presence of characteristic stretching vibrations for lactam (CO) at 1698 cm−1 and amide (CO) at 1676–1683 cm−1, further supported the formation of the products. Additionally, the presence of absorption bands at 1608–1616 cm−1, 1466–1488 cm−1 for aromatic (CC) stretches, at 1242–1269 cm−1 for Csp2–O ether stretches, and at 1152–1166 cm−1 for Csp3–O ether stretches provided further evidence of product formation.
The successful formation of N-phenoxyethyl isatin hydrazones 1(a–l) was also established by their 1H NMR data. A characteristic downfield signal for the N–H proton (H-17) was observed in the range of 13.47–14.19 ppm, confirming the formation of hydrazones. Additionally, a multiplet at 4.10–4.31 ppm corresponding to four methylene protons (H-10 and H-11) supported the structure of the desired hydrazones. The singlet for one proton appeared at 9.06–9.26 ppm for H-20 for nicotinic acid hydrazones whereas a broad signal or multiplet of these protons along with H-21, 22, 23, and 24 was observed at 7.42–8.91 ppm in other cases. The total integration of signals in the aliphatic and aromatic regions provided additional evidence supporting product formation.
In the 13C NMR spectra, the appearance of a characteristic signal at 131.6–132.3 ppm, corresponding to the imine carbon (C-3), and the absence of the keto carbonyl signal at 182.38–183.81 ppm (from intermediate C) confirmed the formation of N-phenoxyethyl isatin hydrazones 1(a–l). Signals observed at 164.0–164.2 ppm and 161.7–162.0 ppm were attributed to two carbonyl groups: the amide carbonyl of the isatin ring and the hydrazone carbon (C-18), respectively. Additional evidence was provided by the carbon signals in the aromatic (109.9–158.0 ppm) and aliphatic (39.6–65.5 ppm) regions, consistent with the expected structure of the products. Furthermore, HRMS data unequivocally confirmed the formation of desired compounds (see experimental section for details).
2.2 X-ray single crystal analysis
Single crystals of the N-phenoxyethyl isatin hydrazone 1l, suitable for X-ray diffraction analysis, were successfully grown from a concentrated DMSO solution over one week at ambient temperature (Table 1). Compound 1l crystallized in orthorhombic crystal system with a centrosymmetric Pbca space group. Its molecular structure (ORTEP diagram), along with crystallographic atom labeling is illustrated in Fig. 1.| Crystal data | 1l |
|---|---|
| CCDC | 2355357 |
| Chemical formula | C23H17BrFN3O3 |
| Mr | 482.30 |
| Crystal system, space group | Orthorhombic, Pbca |
| Temperature (K) | 296 |
| a, b, c (Å) | 11.9327 (14), 11.8361 (13), 29.118 (3) |
| V (Å3) | 4112.5 (8) |
| Z | 8 |
| Radiation type | Mo Kα |
| μ (mm−1) | 2.04 |
| Crystal size (mm) | 0.80 × 0.31 × 0.05 |
| Data collection | |
| Diffractometer | Bruker APEX-II CCD |
| Absorption correction | Multi-scan SADABS |
| Tmin, Tmax | 0.448, 0.745 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 208, 3537, 2632 208, 3537, 2632 |
| Rint | 0.080 |
| (sin θ/λ)max (Å−1) | 0.591 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.042, 0.110, 1.10 |
| No. of reflections | 3537 |
| No. of parameters | 283 |
| No. of restraints | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δ〉max, Δ〉min (e Å−3) | 0.21, −0.58 |
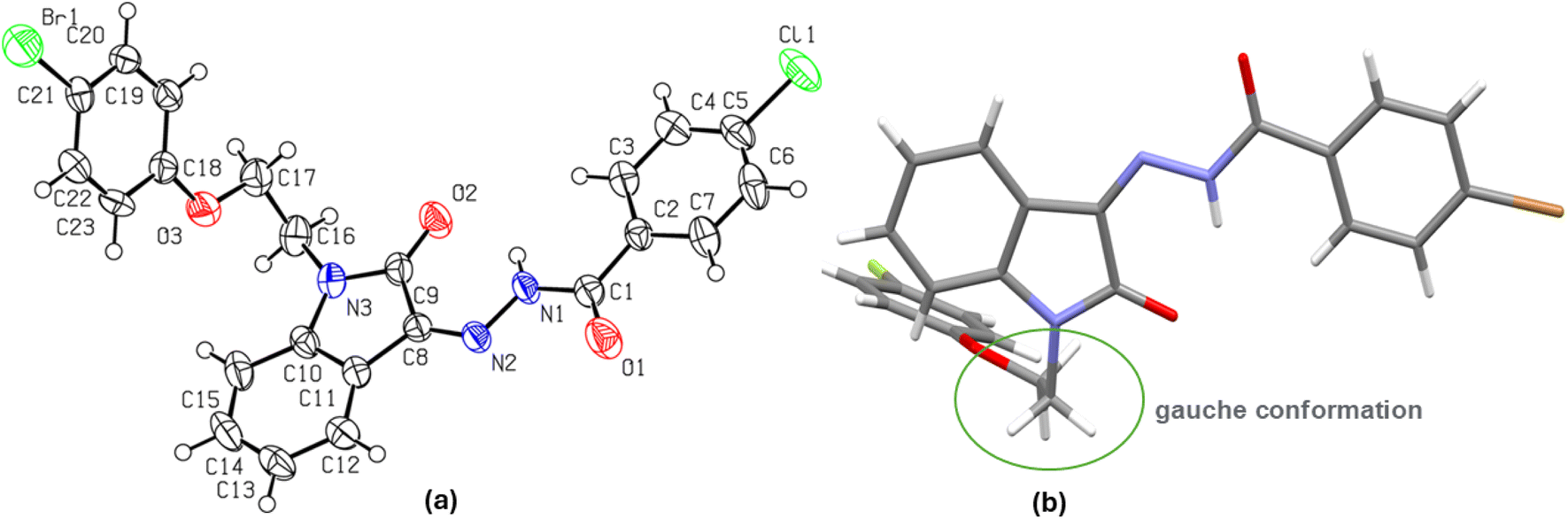 | ||
| Fig. 1 (a) The molecular structure (ORTEP diagram) of N-phenoxyethyl isatin hydrazone 1l. Displacement ellipsoids are drawn at the 50% probability level, (b) showing gauche conformation of ethylene linker. | ||
The isatin nucleus demonstrates partial antiaromaticity due to its 8π-electron Huckel antiaromatic system.28 However, conjugation at position 3 via the hydrazide moiety alters this character, introducing a resonance form consistent with a 10π-electron Huckel aromatic system Fig. 2. In the solid-state structure of compound 1l, a strong intramolecular hydrogen bond between the 2-carbonyl oxygen atom of the isatin ring and the hydrazide amidic NH group [N(1)–H(1)⋯O(2) 2.021 Å] enforces cis geometry around the imine bond, stabilizing the aromatic resonance form through charge separation. The isatin and p-bromophenyl rings, connected via a carboxylic acid hydrazide moiety, exhibit near planarity with dihedral angles of N(1)–N(2)–C(8)–C(9) −1.23° and O(1)–C(7)–C(4)–C(5) −175.72° around the central N-imino amide fragment N(2)–N(1)–C(7)–O(1) −3.19°. The planarity of p-bromophenyl ring with N-imino amide fragment can be attributed to lone pair-π repulsive and weak [C(3)–H(3)⋯O(1) 2.468 Å] attractive interactions.29 This extended planarity (vide infra) enhances the potential for π-π stacking interactions of isatin hydrazide conjugate fragment. This planar isatin hydrazide conjugate (vide supra) however adopts a nearly perpendicular orientation regarding the p-fluorophenoxy pendant linked through an ethylene bridge. The ethylene bridge itself is present in a staggered gauche conformation, with both aryl fragments (isatin conjugated with hydrazide and p-fluorophenoxy) forming a dihedral angle of N(3)–C(16)–C(17)–O(3) −68.89°. Importantly, the solid-state packing of compound 1l is dominated mainly by two types of non-covalent interactions i.e., antiparallel π⋯π stacking (3.840 Å distance between the centres of stacked isatin phenyl ring and p-bromophenyl ring of hydrazide fragment) and a self-complimentary CH⋯O [C(17)–H(17B)⋯O(2) 2.625 Å & C(23)–H(23)⋯O(2) 2.425 Å ] contacts.
 | ||
| Fig. 2 (a) Canonical forms of hydrazide conjugates of isatin showing antiaromatic and aromatic systems, (b) antiparallel π⋯π stacking interactions, (c) a self-complimentary CH⋯O contacts in the solid-state packing of compound 1l. | ||
2.3 Bio-evaluation of N-phenoxyethyl isatin hydrazones 1(a–l)
To discover the anti-diabetic N-phenoxyethyl isatin hydrazones 1(a–l), all twelve compounds were subjected to an in vitro α-glucosidase inhibition assay. Out of all tested hydrazide conjugates, ten compounds displayed highly potent anti-diabetic potential (IC50 ranging from 3.64 ± 0.13 to 94.89 ± 0.64 μM) when compared to the α-glucosidase inhibitor (marketed drug), acarbose (IC50 = 873.34 ± 1.67 μM) (Table 2). For comparison, the reported IC50 value of acarbose in the literature is 750.0 ± 1.5.30 To elucidate the structure–activity relationship (SAR), the compound series was categorized into three distinct groups (A, B, and C), each consisting of four analogs sharing identical X and Y substituents while differing in the nature of the R-group (H, Cl, Br, F) on the phenoxy ring.| Compd | R | X | Y | Percent inhibition (0.5 mM) | IC50 ± μM (SEM) |
|---|---|---|---|---|---|
| 1a | H | CH | N | 75.63 | 94.89 ± 0.64 |
| 1b | H | N | CH | 94.13 | 4.29 ± 0.13 |
| 1c | H | CBr | CH | 90.60 | 17.28 ± 0.30 |
| 1d | Cl | CH | N | 92.94 | 5.73 ± 0.24 |
| 1e | Cl | N | CH | 94.65 | 3.64 ± 0.13 |
| 1f | Cl | CBr | CH | 94.46 | 4.97 ± 0.21 |
| 1g | Br | CH | N | 92.09 | 12.05 ± 0.24 |
| 1h | Br | N | CH | 89.92 | 42.64 ± 0.38 |
| 1i | Br | CBr | CH | 94.91 | 4.58 ± 0.11 |
| 1j | F | CH | N | 36.00 | N/A |
| 1k | F | N | CH | 39.30 | N/A |
| 1l | F | CBr | CH | 93.13 | 8.24 ± 0.29 |
| Acarbose | — | — | — | 59.37 (1 mM) | 873.34 ± 1.67 |
Group A (X = CH, Y = N), comprising compounds 1a (R = H), 1d (R = Cl), 1g (R = Br), and 1j (R = F), demonstrated a pronounced influence of halogen substitution on the phenoxy ring, with a more significant impact on α-glucosidase inhibitory activity compared to the corresponding analogs in Groups B and C. Notably, compound 1a (R = H), which lacks a halogen substituent, exhibited the weakest inhibitory activity, with an IC50 value of 94.89 ± 0.64 μM. This renders it the least potent α-glucosidase inhibitor not only within Group A but across the entire series. In contrast, the introduction of a chlorine atom in compound 1d (R = Cl) significantly enhanced inhibitory activity, yielding an IC50 value of 5.73 ± 0.24 μM. This compound emerged as the most active member within Group A and the fourth most potent inhibitor among all derivatives studied. Substitution with a bromo group in compound 1g (R = Br) led to a moderate decrease in potency relative to 1d, resulting in an IC50 of 12.05 ± 0.24 μM; however, it remained substantially more active than the unsubstituted analog 1a. Interestingly, the incorporation of a fluoro substituent in compound 1j (R = F) led to a complete loss of inhibitory activity, and as a result, its IC50 value could not be determined.
Group B (X = N, Y = CH), containing compounds 1b (R = H), 1e (R = Cl), 1h (R = Br), and 1k (R = F), exhibited the highest overall α-glucosidase inhibitory activity among the three groups and includes the two most potent compounds in the series. Within this group, compound 1b (R = H) demonstrated notable inhibition with an IC50 value of 4.29 ± 0.13 μM. Substitution of the hydrogen atom with a chlorine atom in compound 1e (R = Cl) led to a further enhancement in inhibitory activity, rendering it the most potent α-glucosidase inhibitor of the entire series, with an IC50 value of 3.64 ± 0.13 μM. In contrast, incorporation of a bromine substituent in compound 1h (R = Br) significantly reduced inhibitory efficacy, resulting in an IC50 value of 42.64 ± 0.38 μM. Similar to compound 1j in Group A, the introduction of a fluorine atom in compound 1k (R = F) led to a loss of inhibitory activity; hence, its IC50 value was also not determined.
Group C (X = CBr, Y = CH), consisting of compounds 1c (R = H), 1f (R = Cl), 1i (R = Br), and 1l (R = F), exhibited α-glucosidase inhibitory activity that was intermediate relative to Groups A and B. Within this group, compound 1c (R = H) displayed relatively low activity, with an IC50 value of 17.28 ± 0.30 μM, ranking as the fifth least potent inhibitor in the series. Substitution with a chlorine atom in compound 1f (R = Cl) significantly enhanced activity, resulting in an IC50 value of 4.97 ± 0.21 μM. Notably, compound 1i (R = Br) exhibited the highest potency within Group C, with an IC50 value of 4.58 ± 0.11 μM, positioning it as the third most active inhibitor in the entire series. Interestingly, in contrast to the pronounced activity loss observed with the fluorine-substituted analogs in Groups A (1j) and B (1k), compound 1l (R = F) retained moderate inhibitory activity, yielding an IC50 value of 8.24 ± 0.29 μM. These findings suggest that the CBr substitution at the X-position may mitigate the deactivating effect of fluorine on α-glucosidase inhibition.
A careful analysis of the α-glucosidase inhibitory trend observed across all three groups, based on variation of the R substituent at the para-position of the phenoxy ring, reveals the following order of activity: Cl > Br > H > F. This trend suggests that the size and electronegativity of the substituents, play a dominant role in modulating inhibitory potency. Although hydrogen and fluorine are comparable in size, the marked difference in their electronegativities appears to adversely affect enzyme inhibition. Chlorine, by contrast, provided an optimal combination of size and electronegativity, emerging as the most effective substituent for enhancing inhibitory activity. Replacement of chlorine with the bulkier bromine atom led to a decline in activity, likely due to larger size that may disrupt optimal interactions within the enzyme's active site. Similarly, the observed trend in inhibitory activity among the acid hydrazide scaffolds (X = N > X = CBr > Y = N) may be attributed to the enhanced ability of substituents at the 4-position to establish favorable interactions within the α-glucosidase active site. These observations underscore the importance of both steric and electronic parameters in dictating α-glucosidase inhibitory efficacy in this series.
2.4 Molecular docking studies
The ability of ligands to superimpose with receptor sites has long been considered an essential factor in drug design and development.31 Although molecular docking has some limitations such as solvent effects, flexibility of protein, and dependence on scoring function it is still being used for initial screening.31,32 Additionally, docking analysis combined with experimental data provides valuable insight to investigate the potential of drug candidates.33 To investigate the binding affinities and interactions of the most active compound 1e, and the standard drug acarbose, molecular docking studies were performed against α-glucosidase, and the results were compared (Fig. 3 and 4). In rational drug design and development, the ability of a drug candidate to effectively align and interact with the receptor site through molecular docking is a critical feature. When combined with experimental data, docking studies provide valuable insights into the potential of drug candidates.34,35 The enzyme α-glucosidase (EC 3.2.1.20) (PDB ID: 2zq0) is a hydrolase released by the intestine that catalyzes the hydrolysis of 1 → 4 glycosidic linkages in oligosaccharides and polysaccharides, yielding glucose.36 This enzyme plays a pivotal role in elevating blood glucose levels.36 α-Glucosidase inhibitors, such as acarbose, are utilized to mitigate hyperglycemia by controlling glucose levels, thereby preventing diabetes and related complications.37,38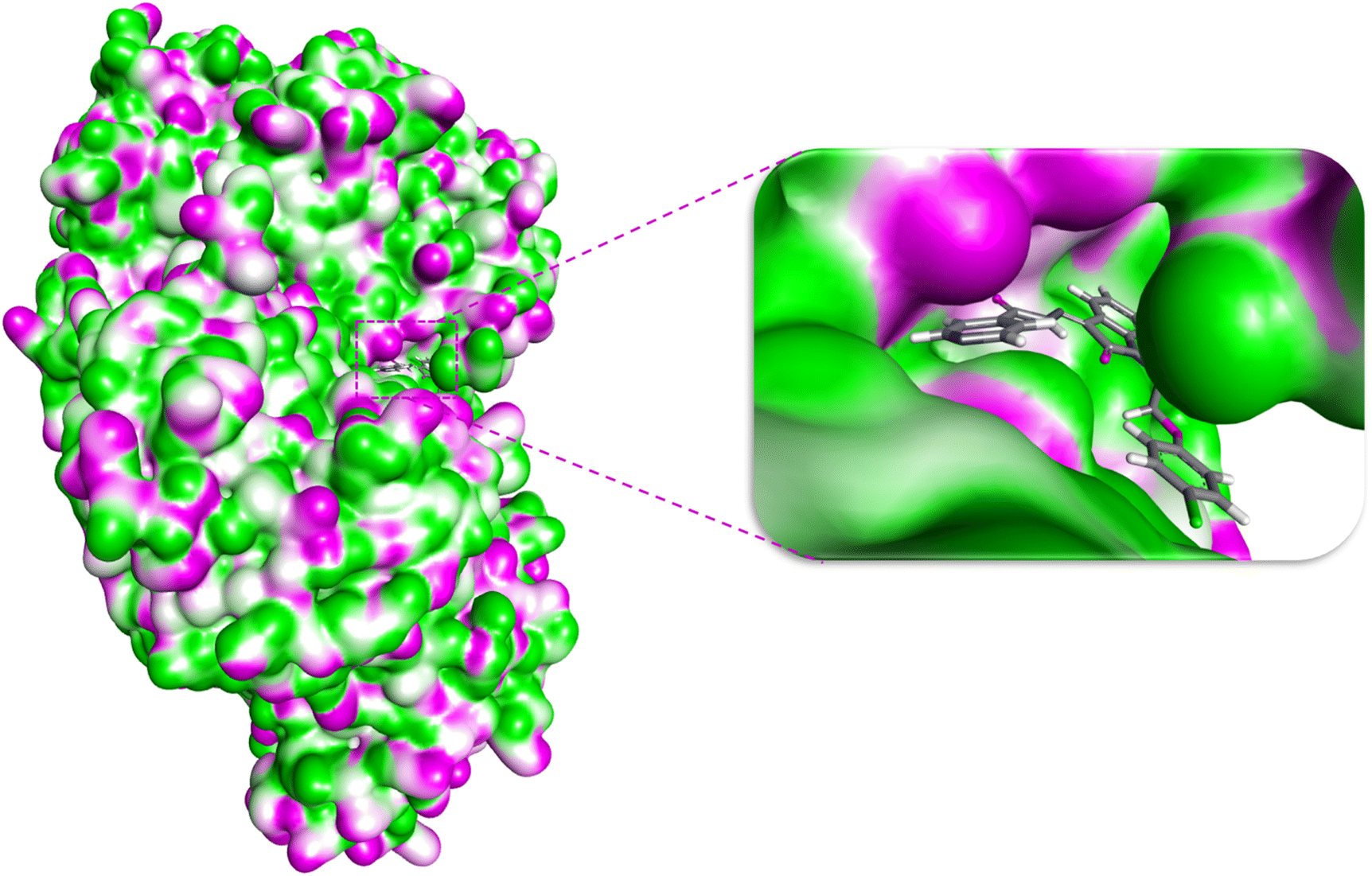 | ||
| Fig. 3 The docking pose of 1e within the binding pocket of α-glucosidase. | ||
 | ||
| Fig. 4 (a) 3D and (b) 2D interactions of compound 1e with α-glucosidase. (c) 3D and (d) 2D interactions of acarbose with α-glucosidase. | ||
Interestingly, the docking studies revealed a binding score of −9.7 kcal mol−1 for compound 1e against α-glucosidase, compared to −9.4 kcal mol−1 for acarbose (Table 3). These results suggest that compound 1e exhibits comparable or slightly superior binding affinity to α-glucosidase, reinforcing its potential as a promising α-glucosidase inhibitor.
| Compd | Docking score (kcal mol−1) | Receptor amino acid | Part of ligand | Interactions | Distance (Å) |
|---|---|---|---|---|---|
| 1e | −9.7 | LYS (A:274) | Pyridine ring | π-cation, H-bonding | 4.36, 3.92 |
| LYS (A:66) | CO (hydrazide) |
H-bonding | 3.76 | ||
| VAL (A:167) | Pyridine | π–σ | 3.83 | ||
| SER (B:404) | CO isatin |
Dipole dipole | 2.85 | ||
| ASN (B:403) | Ph-O-CH2 | Dipole dipole | 2.28, 2.38 | ||
| TYR (A:279) | Phenyl ring | Amide-π stacking | 3.67 | ||
| MET (A:280) | Pyridine | π–anion | 3.96 | ||
| MET (A:280) | O–H | H-bonding | 4.10 | ||
| Acarbose | −9.4 | VAL (A:249) | O–H | H-bonding | 3.14 |
| GLN (A:281) | C–O–C | H-bonding | 4.03 | ||
| ASN (A:488) | O–H | Donor–donor | 3.98 | ||
| TYR (A:407) | –CH3 | π–alkyl | 5.49 |
Fig. 4 illustrates the interactions of compound 1e with various amino acids in the binding pocket of α-glucosidase. The pyridine ring of isonicotinohydrazide forms three major interactions: one involving hydrogen bonding with LYS (A:274), with a bond distance of 3.92 Å, and the others involving π–cation, π–anion, and π–σ interactions with amino acids LYS (A:274), VAL (A:167), and MET (A:280), respectively. The carbonyl group (CO) of the hydrazide moiety also forms a strong hydrogen bond with LYS (A:66), with a bond distance of 3.76 Å. Additionally, the carbonyl (CO) of isatin and the ether linkage engage in dipole–dipole interactions with amino acids SER (B:404) and ASN (B:403), with bond distances of 2.85 Å and 2.28 Å, respectively. Another significant interaction occurs between the amino acid TYR (A:279) and the phenyl ring of the phenoxy group, involving amide-π stacking, with a bond distance of 3.67 Å. In contrast, due to the presence of abundant hydroxyl groups, acarbose depicted conventional H-bonding as prominent interactions. π-Alkyl interaction is also observed between the methyl group of acarbose and TYR (A:407) with a bond distance of 5.49 Å. In addition, there is also an unfavorable interaction present between the amino group of ASN (A:488) and the –OH group of acarbose (Fig. 4). Interaction details are summarized in Table 3. It is important to note that π interactions are dominant in compound 1e, while hydrogen bonding predominates in acarbose.
Additionally, the binding scores of selected N-phenoxyethyl isatin hydrazones (1b, 1c, 1d, 1f, 1g, 1i and 1l) were calculated and compared with their experimental IC50 values to explore the correlation between the observed biological activity and docking scores. As shown in Fig. 5, this correlation supports the alignment between the in silico and in vitro analyses.
 | ||
| Fig. 5 Correlation between IC50 and binding scores of N-phenoxyethyl isatin hydrazones. | ||
2.5 Drug likeness and molecular properties of conjugates 1(a–l)
To cross the blood–brain barrier and increase bioavailability, certain molecular properties are possessed by the most clinically used drugs.39 To check the drug-likeness of compounds 1(a–l), molecular properties like total polar surface area (TPSA), molecular volume, molar refractivity, number of rotatable bonds, lipophilicity, and hydrogen bond donors/acceptors were calculated and their alignment with Lipinski's rule of 5 (ref. 39) was checked. It is obvious from Table 4 that all the synthesized compounds 1(a–l) follow the Lipinski Rule of Five.| Compd | HBD | HBA | minlog P | nRotB | TPSA (Å2) | Molar refractivity | Molecular weight |
|---|---|---|---|---|---|---|---|
| 1a | 1 | 5 | 2.17 | 7 | 83.89 | 111.5 | 386.4 |
| 1b | 1 | 5 | 2.28 | 7 | 83.39 | 111.5 | 386.4 |
| 1c | 1 | 4 | 2.28 | 7 | 71.00 | 121.4 | 464.31 |
| 1d | 1 | 5 | 2.52 | 7 | 83.39 | 116.5 | 420.8 |
| 1e | 1 | 5 | 2.39 | 7 | 83.39 | 116.5 | 420.8 |
| 1f | 1 | 4 | 3.40 | 7 | 71.00 | 126.4 | 498.7 |
| 1g | 1 | 5 | 2.53 | 7 | 83.39 | 119.1 | 465.3 |
| 1h | 1 | 5 | 2.20 | 7 | 83.39 | 119.1 | 465.3 |
| 1i | 1 | 4 | 3.21 | 7 | 71.00 | 129.0 | 543.2 |
| 1j | 1 | 5 | 2.64 | 7 | 83.39 | 111.4 | 404.4 |
| 1k | 1 | 5 | 1.92 | 7 | 83.39 | 111.4 | 404.4 |
| 1l | 1 | 4 | 2.95 | 7 | 71.00 | 121.3 | 482.3 |
| Acarbose | 14 | 19 | −8.56 | 9 | 321.17 | 136.7 | 645.60 |
| Standard values | 5 | 10 | 5 | 10 | 140 | 130 | 500 |
These molecular properties of hydrazones 1(a–l) were compared with acarbose and it was revealed that acarbose showed marked deviation from every standard value except for the number of rotatable bonds. The bioavailability of compounds 1(a–l) and acarbose was also compared using a boiled egg diagram40 and bioavailability radar chart. It was observed that being a highly polar compound, acarbose is not present in yellow, white, or even in the gray region of the panel. While nicotinic or iso-nicotinic hydrazones are present in the white region indicating they have high intestinal absorption. While hydrazones with 4-Br substituent are present in the yellow region. This indicates they can easily cross the blood–brain barrier (BBB). In addition, all the hydrazide conjugates are shown by red dots i.e., they are PGP−. Furthermore, in the bioavailability radar chart, the acarbose showed remarkable deviations from every parameter except flexibility. In contrast, the highest scorer conjugate 1e shows only one deviation of unsaturation (Fig. 6).
 | ||
| Fig. 6 (a) Boiled-egg diagram of hydrazones 1(a–l); (b) bioavailability radar chart for acarbose; (c) bioavailability radar chart for compound 1e. | ||
Computational-based protocols such as, CADMA-Chem,41 Swiss ADME,40 or PkCSM42 are expected as threshold points to expedite further the discovery of orally available drugs that have the potential to stop or slow down human disorders. The physiological properties for instance, absorption, distribution, metabolism, excretion, and toxicity (ADMET) are studied using these protocols to check the efficacy and bioavailability of a drug. The physiological properties of 1(a–l) are shown in Tables 5, and S1.†
| ADMET properties | 1e | 1f | Acarbose | |
|---|---|---|---|---|
| Absorption | WS (log mol L−1) | −4.785 | −6.278 | −1.931 |
| IS (%abs) | 93.815 | 90.027 | 0 | |
| SP (log Kp) | −2.77 | −2.695 | −2.73 | |
| Distribution | BBBP (log BB) | −0.882 | −0.732 | −2.962 |
| CNSP (log PS) | −2.387 | −1.946 | −7.116 | |
| VDss (log L kg−1) | −0.423 | −0.238 | −0.781 | |
| Metabolism | CYP3A4 inhibitor | Yes | Yes | No |
| CYP1A2 inhibitor | No | No | No | |
| CYP2C19 inhibitor | No | No | No | |
| CYP2C9 inhibitor | Yes | Yes | No | |
| Excretion | TC (log mL min−1 kg−1) | −0.103 | −0.236 | 0.451 |
| Toxicity | AMES toxicity | No | No | No |
| Max. TD. (log mg kg−1 per day) | 0.059 | 0.234 | 0.58 | |
| ORAT (LD50) (mol kg−1) | 2.285 | 2.272 | 2.93 | |
| HT | No | No | No | |
| SS | No | No | No | |
| T. pyriformis toxicity (log μg L−1) | 0.539 | 0.516 | 0.285 | |
The absorption parameters include water solubility, intestinal solubility, and skin permeability (log Kp). The water solubility of 1e and 1f are −4.785 and −6.278 log mol−1 L−1 indicating that they are less soluble in water while acarbose showed −1.931 log mol−1 L−1 since it is more polar than hydrazones and can form a greater number of hydrogen bonds. The intestinal solubility of 1(e–f) is much higher than acarbose 93.815%, and 90.027%, respectively. Whereas acarbose showed only 0% intestinal absorption. The skin permeability for the three of them is quite matchable43 (Table 5). The central nervous system (CNS) and blood–brain barrier (BBB) absorbency and VDss values of all the novel conjugates were approximately equal to the normal values. For example, for 1e these values are −0.882 log BB, −2.387 log PS, and −0.423 log L kg−1 respectively. While acarbose exhibited very low distribution with the values of −2.962 log BB, −7.116 log PS, and −0.781 log L kg−1.44 Acarbose did not inhibit any of cytochrome P40 enzymes while 1(a–l) are found to show inhibitory action against CYP3A4, and CYP3A9 i.e., they can be co-administered with other drugs to increase their concentration in plasma.45 The excretion value for hydrazones 1(e–f) (−0.103 and −0.236 log mL min−1 kg−1) are lower than that of acarbose (0.451 log mL min−1 kg−1) indicating that acarbose can be eliminated more easily than hydrazide conjugates.46 The anticipated toxicity values are also relevant to the drug-likeness behavior of these hydrazones with high ORAT (LD50) and no hepatotoxicity and skin sensitization.47 The ADMET assessment underscores that these N-phenoxyethyl isatin hydrazones 1(a–l) have acceptable drug-like potency and very low toxicity.
3. Conclusions
In conclusion, we have synthesized and evaluated N-phenoxyethyl isatin hydrazones 1(a–l) for their α-glucosidase inhibitory potential. The in vitro studies demonstrated significant inhibitory activity, with IC50 values ranging from 3.64 ± 0.13 to 94.89 ± 0.64 μM, markedly more potent than the standard drug acarbose (IC50 = 873.34 ± 1.67 μM). The structure–activity relationship (SAR) analysis revealed that bromo substituents at the 4th position of the aryl ring in the hydrazide moiety, as well as a chloro substituent at the 4th position of the phenoxy ring, is beneficial for the biological activity. Molecular docking studies of compound 1e with α-glucosidase indicated a strong binding affinity (−9.7 kcal mol−1) with the enzyme's receptor site. Additionally, physicochemical property analysis confirmed favorable pharmacokinetics, including good oral bioavailability, balanced hydrophilicity, and minimal predicted toxicity. These findings underscore the potential of N-phenoxyethyl isatin hydrazones as promising alternatives to existing α-glucosidase inhibitors and highlight their suitability for further investigation as candidates for managing postprandial hyperglycemia.4. Experimental
The synthesis of substituted ethyl benzoates E and substituted benzohydrazides F are detailed in the ESI.†4.1 General procedure for the synthesis of N-phenoxyethylisatin hydrazones 1(a–l)
In a 100 mL round bottom flask, the N-phenoxyethylisatins C (ref. 24) (1.0 mmol) were dissolved in dry ethanol (20 mL) and refluxed with respective benzohydrazides F (1.2 mmol) in the presence of a catalytic amount of acetic acid for 8 hours. Lime-yellow to orange-colored products were precipitated out in the reaction mixture. Upon completion of the reaction, as indicated by TLC, precipitates were hot filtered and washed with cold ethanol, and dried to get pure N-phenoxyethylisatin hydrazones 1(a–l).![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) (cm−1); 3200 (N–H stretch), 3063 (Csp2–H stretch), 2921, 2885 (Csp3–H asymmetric and symmetric stretch), 1683 (CO stretch, lactam), 1676 (CO stretch, amide), 1658 (CN stretch, imine) 1614, 1484 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1166 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.11 (1H, s, H-17), 8.99 (1H, s, H-20), 8.61 (1H, d, 3J = 3.5 Hz, H-8), 7.81–7.98 (1H, m, H-22), 7.47 (1H, Brs, H-7), 6.92–7.33 (5H, m, H-14a, 14a′, 15, 23, 24), 6.61–6.81(2H, m, H-5, 6), 6.33–6.53 (2H, m, H-13a, 13a′), 4.22–3.99 (4H, m, H-10, 11). HRMS (ESI) m/z [M + H]+ calculated for C22H18N4O3: 387.4188, found: 387.4112. (cm−1) 3196 (N–H stretch), 3069 (Csp2–H stretch), 2924, 2881 (Csp3–H asymmetric and symmetric stretch), 1698 (CO stretch, lactam), 1683 (CO stretch, amide), 1658 (CN stretch, imine) 1615, 1484 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1161 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.19 (1H, s, H-17), 8.91 (2H, Brs, H-20, 24), 7.83–7.87 (3H, m, H-21, 23, 8), 7.46 (1H, t, 3J = 7.8 Hz, H-7), 7.24–7.27 (1H, m, H-5), 7.15–7.21 (3H, m, H-14a, 14a′, 15), 6.94–6.99 (1H, m, H-6), 6.83–6.86 (2H, m, H-13a, 13a′), 4.16–4.31 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H18N4O3: 387.4188, found: 387.4112. (cm−1); 3196 (N–H stretch), 3061 (Csp2–H stretch), 2924, 2881 (Csp3–H asymmetric and symmetric stretch), 1683 (CO stretch, lactam), 1678 (CO stretch, amide), 1659 (CN stretch, imine) 1614, 1482 (CC stretch, aromatic), 1254 (Csp2–O stretch, ether), 1151 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.07 (1H, s, H-17), 7.89–7.91 (3H, m, H-8, 20, 24), 7.67–7.73 (2H, m, H-21, 23), 7.45 (1H, t, 3J = 7.8 Hz, H-7), 7.25–7.30 (2H, m, H-14a, 14a′), 7.14–7.21 (2H, m, H-5, 15), 6.93–6.99 (1H, m, H-6), 6.84–6.86 (2H, m, H-13a, 13a′), 4.20–4.31 (4H, m, H-10, 11) 13C NMR (75 MHz, CDCl3) δ (ppm); 39.8 (C-10), 65.2 (C-11), 110.1 (C-8), 114.3 (C-13a, 13a′), 119.5 (C-4), 121.4 (C-15), 122.1 (C-6), 123.7 (C-21, 23), 129.5 (C-14a, 14a′, 20, 24), 129.5 (C-19), 130.9 (C-5), 131.6 (C-7), 132.2 (C-3), 141.8 (C-22), 143.2 (C-9), 158.0 (C-12), 158.7 (C-2), 162.0 (C-2), 164.0 (C-18); HRMS (ESI) m/z [M + H]+ calculated for C23H18BrN3O3: 465.3260, found: 465.3192. (cm−1); 3250 (N–H stretch), 3053 (Csp2–H stretch), 2972, 2921 (Csp3–H asymmetric and symmetric stretch), 1690 (CO stretch, lactam), 1680 (CO stretch, amide), 1660 (CN stretch, imine) 1613, 1488 (CC stretch, aromatic), 1260 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm) 14.10 (1H, s, H-17), 9.25 (1H, s, H-20), 8.84 (1H, d, 3J = 3.9 Hz, H-8), 8.28–8.32 (1H, m, H-22), 7.87 (1H, Brs, H-7), 7.42–7.51 (2H, m, H-23, 24), 7.32–7.37 (2H, m, H-14a, 14a′), 7.18 (1H, d, 3J = 7.8 Hz, H-5), 7.12 (1H, t, 3J = 7.5 Hz, H-6), 6.70–6.75 (2H, m, H-13a, 13a′), 4.18–4.28 (4H, m, H-10, 11); 13C NMR (75 MHz, CDCl3) δ (ppm); 39.6 (C-10), 65.5 (C-11), 109.9 (C-8), 113.7 (C-4), 116.2 (C-13a, 13a′), 119.4 (C-6), 122.1 (C-15), 123.8 (C-23), 128.1 (C-5), 131.7 (C-19), 132.3 (C-7, 14a, 14a′), 135.1 (C-3), 138.0 (C-24), 143.2 (C-9), 148.9 (C-22), 153.3 (C-20), 157.1 (C-12), 161.7 (C-2), 164.2 (C-18); HRMS (ESI) m/z [M + H]+ calculated for C22H17ClN4O3: 421.8608, found: 421.8534. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2976, 2921 (Csp3–H asymmetric and symmetric stretch), 1698 (CO stretch, lactam), 1684 (CO stretch, amide), 1659 (CN stretch, imine) 1616, 1484 (CC stretch, aromatic), 1269 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.86 (1H, s, H-17), 8.86–8.88 (2H, t, 3J = 5.7 Hz, H-21, 23), 7.79–7.80 (2H, m, H-20, 24), 7.64 (1H, d, 3J = 5.1 Hz, H-8), 7.51 (1H, t, 3J = 7.8 Hz, H-7), 7.40–7.42 (2H, m, H-14a, 14a′), 7.35 (1H, d, 3J = 8.1 Hz, H-5), 7.19 (1H, t, 3J = 7.8 Hz, H-6), 6.84–6.87 (2H, m, H-13a, 13a′), 4.20 (4H, Brs, H-10, 11) HRMS (ESI) m/z [M + H]+ calculated for C22H17ClN4O3: 421.8608, found: 421.8534. (cm−1); 3250 (N–H stretch), 3068 (Csp2–H stretch), 2985, 2896 (Csp3–H asymmetric and symmetric stretch), 1682 (CO stretch, lactam), 1678 (CO stretch, amide), 1656 (CN stretch, imine) 1615, 1480 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1153 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.80 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.5 Hz, H-8), 7.50 (1H, t, 3J = 7.5 Hz, H-7), 7.41 (2H, d, 3J = 8.7 Hz, H-14a, 14a′), 7.36 (1H, t, 3J = 8.1 Hz, H-5), 7.18 (1H, t, 3J = 7.5 Hz, H-6), 6.86 (2H, d, 3J = 8.7 Hz, H-13a, 13a′), 4.18–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17BrClN3O3: 499.7688, found: 499.7613. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2965, 2896 (Csp3–H asymmetric and symmetric stretch), 1696 (CO stretch, lactam), 1682 (CO stretch, amide), 1656 (CN stretch, imine) 1614, 1487 (CC stretch, aromatic), 1261 (Csp2–O stretch, ether), 1155 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.11 (1H, s, H-17), 9.26 (1H, s, H-20), 8.85 (1H, B, H-22), 8.31 (H, d, 3J = 7.5 Hz, H-24), 7.88 (1H, Brs, H-8), 7.43–7.48 (1H, m, H-7), 7.34–7.37 (3H, m, H-14a, 14a′, 23), 7.11–7.19 (2H, m, H-5, 6), 6.71–6.74 (2H, m, H-13a, 13a′), 4.22–4.24 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17BrN4O3: 466.3148, found: 466.3072. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2966, 2895 (Csp3–H asymmetric and symmetric stretch), 1696 (CO stretch, lactam), 1683 (CO stretch, amide), 1658 (CN stretch, imine) 1614, 1488 (CC stretch, aromatic), 1242 (Csp2–O stretch, ether), 1162 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm) 14.15 (1H, s, H-17), 8.87 (2H, B, H-20, 24), 7.86 (3H, Brs, H-8, 21, 23), 7.36–7.45 (1H, m, H-7), 7.33–7.36 (2H, m, H-14a, 14a′), 7.11–7.19 (2H, m, H-5, 6), 6.70–6.73 (2H, m, H-13a, 13a′), 4.21–4.24 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17BrN4O3: 466.3148, found: 466.3072. (cm−1); 3196 (N–H stretch), 3064 (Csp2–H stretch), 2974, 2921 (Csp3–H asymmetric and symmetric stretch), 1682 (CO stretch, lactam), 1676 (CO stretch, amide), 1654 (CN stretch, imine) 1614, 1480 (CC stretch, aromatic), 1243 (Csp2–O stretch, ether), 1152 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.81 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.5 Hz, H-8), 7.50 (1H, t, 3J = 7.8 Hz, H-7), 7.39–7.42 (2H, m, H-14a, 14a′), 7.33–7.36 (1H, m, H-5), 7.18 (1H, t, 3J = 7.5 Hz, H-6), 6.84–6.87 (2H, m, H-13a, 13a′), 4.18–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17Br2N3O3: 544.2228, found: 544.2152. (cm−1); 3194 (N–H stretch), 3053 (Csp2–H stretch), 2924, 2874 (Csp3–H asymmetric and symmetric stretch), 1694 (CO stretch, lactam), 1676 (CO stretch, amide), 1656 (CN stretch, imine) 1614, 1469 (CC stretch, aromatic), 1260 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm) 13.75 (1H, s, H-17), 9.06 (1H, s, H-20), 8.84 (1H, d, 3J = 4.5 Hz, H-22), 8.25 (1H, d, 3J = 8.1 Hz, H-24), 7.59–7.66 (2H, m, H-8, 23), 7.48 (1H, t, 3J = 7.8 Hz, H-7), 7.33 (1H, d, 3J = 7.8 Hz, H-5), 7.16 (1H, t, 3J = 7.5 Hz, H-6), 7.04–7.10 (2H, m, H-14a, 14a′), 6.86–6.91 (2H, m, H-13a, 13a′), 4.15–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17FN4O3: 405.4092, found: 405.4018. (cm−1); 3194 (N–H stretch), 3064 (Csp2–H stretch), 2940, 2898 (Csp3–H asymmetric and symmetric stretch), 1687 (CO stretch, lactam), 1678 (CO stretch, amide), 1658 (CN stretch, imine) 1616, 1466 (CC stretch, aromatic), 1261 (Csp2–O stretch, ether), 1159 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm) 13.47 (1H, s, H-17), 8.87 (2H, d, 3J = 6 Hz, H-20, 24), 7.79–7.81 (2H, m, H-21, 23), 7.64 (1H, d, 3J = 6.9 Hz, H-8), 7.51 (1H, t, 3J = 7.8 Hz, H-7), 7.35 (1H, d, 3J = 7.8 Hz, H-5), 7.18 (1H, t, 3J = 7.8 Hz, H-6), 7.05–7.11 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.16–4.26 (4H, m, H-10, 11). HRMS (ESI) m/z [M + H]+ calculated for C22H17FN4O3: 405.4092, found: 405.4018. (cm−1); 3194 (N–H stretch), 3061 (Csp2–H stretch), 2921, 2876 (Csp3–H asymmetric and symmetric stretch), 1684 (CO stretch, lactam), 1676 (CO stretch, amide), 1652 (CN stretch, imine) 1608, 1488 (CC stretch, aromatic), 1243 (Csp2–O stretch, ether), 1152 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.81 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.2 Hz, H-8), 7.50 (1H, t, 3J = 7.8 Hz, H-7), 7.35 (1H, d, 3J = 7.8 Hz, H-5), 7.19 (1H, t, 3J = 7.5 Hz, H-6), 7.05–7.11 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.17–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17BrFN3O3: 483.3172, found: 483.3097.
(cm−1); 3200 (N–H stretch), 3063 (Csp2–H stretch), 2921, 2885 (Csp3–H asymmetric and symmetric stretch), 1683 (CO stretch, lactam), 1676 (CO stretch, amide), 1658 (CN stretch, imine) 1614, 1484 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1166 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.11 (1H, s, H-17), 8.99 (1H, s, H-20), 8.61 (1H, d, 3J = 3.5 Hz, H-8), 7.81–7.98 (1H, m, H-22), 7.47 (1H, Brs, H-7), 6.92–7.33 (5H, m, H-14a, 14a′, 15, 23, 24), 6.61–6.81(2H, m, H-5, 6), 6.33–6.53 (2H, m, H-13a, 13a′), 4.22–3.99 (4H, m, H-10, 11). HRMS (ESI) m/z [M + H]+ calculated for C22H18N4O3: 387.4188, found: 387.4112. (cm−1) 3196 (N–H stretch), 3069 (Csp2–H stretch), 2924, 2881 (Csp3–H asymmetric and symmetric stretch), 1698 (CO stretch, lactam), 1683 (CO stretch, amide), 1658 (CN stretch, imine) 1615, 1484 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1161 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.19 (1H, s, H-17), 8.91 (2H, Brs, H-20, 24), 7.83–7.87 (3H, m, H-21, 23, 8), 7.46 (1H, t, 3J = 7.8 Hz, H-7), 7.24–7.27 (1H, m, H-5), 7.15–7.21 (3H, m, H-14a, 14a′, 15), 6.94–6.99 (1H, m, H-6), 6.83–6.86 (2H, m, H-13a, 13a′), 4.16–4.31 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H18N4O3: 387.4188, found: 387.4112. (cm−1); 3196 (N–H stretch), 3061 (Csp2–H stretch), 2924, 2881 (Csp3–H asymmetric and symmetric stretch), 1683 (CO stretch, lactam), 1678 (CO stretch, amide), 1659 (CN stretch, imine) 1614, 1482 (CC stretch, aromatic), 1254 (Csp2–O stretch, ether), 1151 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.07 (1H, s, H-17), 7.89–7.91 (3H, m, H-8, 20, 24), 7.67–7.73 (2H, m, H-21, 23), 7.45 (1H, t, 3J = 7.8 Hz, H-7), 7.25–7.30 (2H, m, H-14a, 14a′), 7.14–7.21 (2H, m, H-5, 15), 6.93–6.99 (1H, m, H-6), 6.84–6.86 (2H, m, H-13a, 13a′), 4.20–4.31 (4H, m, H-10, 11) 13C NMR (75 MHz, CDCl3) δ (ppm); 39.8 (C-10), 65.2 (C-11), 110.1 (C-8), 114.3 (C-13a, 13a′), 119.5 (C-4), 121.4 (C-15), 122.1 (C-6), 123.7 (C-21, 23), 129.5 (C-14a, 14a′, 20, 24), 129.5 (C-19), 130.9 (C-5), 131.6 (C-7), 132.2 (C-3), 141.8 (C-22), 143.2 (C-9), 158.0 (C-12), 158.7 (C-2), 162.0 (C-2), 164.0 (C-18); HRMS (ESI) m/z [M + H]+ calculated for C23H18BrN3O3: 465.3260, found: 465.3192. (cm−1); 3250 (N–H stretch), 3053 (Csp2–H stretch), 2972, 2921 (Csp3–H asymmetric and symmetric stretch), 1690 (CO stretch, lactam), 1680 (CO stretch, amide), 1660 (CN stretch, imine) 1613, 1488 (CC stretch, aromatic), 1260 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm) 14.10 (1H, s, H-17), 9.25 (1H, s, H-20), 8.84 (1H, d, 3J = 3.9 Hz, H-8), 8.28–8.32 (1H, m, H-22), 7.87 (1H, Brs, H-7), 7.42–7.51 (2H, m, H-23, 24), 7.32–7.37 (2H, m, H-14a, 14a′), 7.18 (1H, d, 3J = 7.8 Hz, H-5), 7.12 (1H, t, 3J = 7.5 Hz, H-6), 6.70–6.75 (2H, m, H-13a, 13a′), 4.18–4.28 (4H, m, H-10, 11); 13C NMR (75 MHz, CDCl3) δ (ppm); 39.6 (C-10), 65.5 (C-11), 109.9 (C-8), 113.7 (C-4), 116.2 (C-13a, 13a′), 119.4 (C-6), 122.1 (C-15), 123.8 (C-23), 128.1 (C-5), 131.7 (C-19), 132.3 (C-7, 14a, 14a′), 135.1 (C-3), 138.0 (C-24), 143.2 (C-9), 148.9 (C-22), 153.3 (C-20), 157.1 (C-12), 161.7 (C-2), 164.2 (C-18); HRMS (ESI) m/z [M + H]+ calculated for C22H17ClN4O3: 421.8608, found: 421.8534. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2976, 2921 (Csp3–H asymmetric and symmetric stretch), 1698 (CO stretch, lactam), 1684 (CO stretch, amide), 1659 (CN stretch, imine) 1616, 1484 (CC stretch, aromatic), 1269 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.86 (1H, s, H-17), 8.86–8.88 (2H, t, 3J = 5.7 Hz, H-21, 23), 7.79–7.80 (2H, m, H-20, 24), 7.64 (1H, d, 3J = 5.1 Hz, H-8), 7.51 (1H, t, 3J = 7.8 Hz, H-7), 7.40–7.42 (2H, m, H-14a, 14a′), 7.35 (1H, d, 3J = 8.1 Hz, H-5), 7.19 (1H, t, 3J = 7.8 Hz, H-6), 6.84–6.87 (2H, m, H-13a, 13a′), 4.20 (4H, Brs, H-10, 11) HRMS (ESI) m/z [M + H]+ calculated for C22H17ClN4O3: 421.8608, found: 421.8534. (cm−1); 3250 (N–H stretch), 3068 (Csp2–H stretch), 2985, 2896 (Csp3–H asymmetric and symmetric stretch), 1682 (CO stretch, lactam), 1678 (CO stretch, amide), 1656 (CN stretch, imine) 1615, 1480 (CC stretch, aromatic), 1258 (Csp2–O stretch, ether), 1153 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.80 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.5 Hz, H-8), 7.50 (1H, t, 3J = 7.5 Hz, H-7), 7.41 (2H, d, 3J = 8.7 Hz, H-14a, 14a′), 7.36 (1H, t, 3J = 8.1 Hz, H-5), 7.18 (1H, t, 3J = 7.5 Hz, H-6), 6.86 (2H, d, 3J = 8.7 Hz, H-13a, 13a′), 4.18–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17BrClN3O3: 499.7688, found: 499.7613. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2965, 2896 (Csp3–H asymmetric and symmetric stretch), 1696 (CO stretch, lactam), 1682 (CO stretch, amide), 1656 (CN stretch, imine) 1614, 1487 (CC stretch, aromatic), 1261 (Csp2–O stretch, ether), 1155 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm); 14.11 (1H, s, H-17), 9.26 (1H, s, H-20), 8.85 (1H, B, H-22), 8.31 (H, d, 3J = 7.5 Hz, H-24), 7.88 (1H, Brs, H-8), 7.43–7.48 (1H, m, H-7), 7.34–7.37 (3H, m, H-14a, 14a′, 23), 7.11–7.19 (2H, m, H-5, 6), 6.71–6.74 (2H, m, H-13a, 13a′), 4.22–4.24 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17BrN4O3: 466.3148, found: 466.3072. (cm−1); 3250 (N–H stretch), 3056 (Csp2–H stretch), 2966, 2895 (Csp3–H asymmetric and symmetric stretch), 1696 (CO stretch, lactam), 1683 (CO stretch, amide), 1658 (CN stretch, imine) 1614, 1488 (CC stretch, aromatic), 1242 (Csp2–O stretch, ether), 1162 (Csp3–O stretch, ether); 1H NMR (300 MHz, CDCl3) δ (ppm) 14.15 (1H, s, H-17), 8.87 (2H, B, H-20, 24), 7.86 (3H, Brs, H-8, 21, 23), 7.36–7.45 (1H, m, H-7), 7.33–7.36 (2H, m, H-14a, 14a′), 7.11–7.19 (2H, m, H-5, 6), 6.70–6.73 (2H, m, H-13a, 13a′), 4.21–4.24 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17BrN4O3: 466.3148, found: 466.3072. (cm−1); 3196 (N–H stretch), 3064 (Csp2–H stretch), 2974, 2921 (Csp3–H asymmetric and symmetric stretch), 1682 (CO stretch, lactam), 1676 (CO stretch, amide), 1654 (CN stretch, imine) 1614, 1480 (CC stretch, aromatic), 1243 (Csp2–O stretch, ether), 1152 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.81 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.5 Hz, H-8), 7.50 (1H, t, 3J = 7.8 Hz, H-7), 7.39–7.42 (2H, m, H-14a, 14a′), 7.33–7.36 (1H, m, H-5), 7.18 (1H, t, 3J = 7.5 Hz, H-6), 6.84–6.87 (2H, m, H-13a, 13a′), 4.18–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17Br2N3O3: 544.2228, found: 544.2152. (cm−1); 3194 (N–H stretch), 3053 (Csp2–H stretch), 2924, 2874 (Csp3–H asymmetric and symmetric stretch), 1694 (CO stretch, lactam), 1676 (CO stretch, amide), 1656 (CN stretch, imine) 1614, 1469 (CC stretch, aromatic), 1260 (Csp2–O stretch, ether), 1154 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm) 13.75 (1H, s, H-17), 9.06 (1H, s, H-20), 8.84 (1H, d, 3J = 4.5 Hz, H-22), 8.25 (1H, d, 3J = 8.1 Hz, H-24), 7.59–7.66 (2H, m, H-8, 23), 7.48 (1H, t, 3J = 7.8 Hz, H-7), 7.33 (1H, d, 3J = 7.8 Hz, H-5), 7.16 (1H, t, 3J = 7.5 Hz, H-6), 7.04–7.10 (2H, m, H-14a, 14a′), 6.86–6.91 (2H, m, H-13a, 13a′), 4.15–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C22H17FN4O3: 405.4092, found: 405.4018. (cm−1); 3194 (N–H stretch), 3064 (Csp2–H stretch), 2940, 2898 (Csp3–H asymmetric and symmetric stretch), 1687 (CO stretch, lactam), 1678 (CO stretch, amide), 1658 (CN stretch, imine) 1616, 1466 (CC stretch, aromatic), 1261 (Csp2–O stretch, ether), 1159 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm) 13.47 (1H, s, H-17), 8.87 (2H, d, 3J = 6 Hz, H-20, 24), 7.79–7.81 (2H, m, H-21, 23), 7.64 (1H, d, 3J = 6.9 Hz, H-8), 7.51 (1H, t, 3J = 7.8 Hz, H-7), 7.35 (1H, d, 3J = 7.8 Hz, H-5), 7.18 (1H, t, 3J = 7.8 Hz, H-6), 7.05–7.11 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.16–4.26 (4H, m, H-10, 11). HRMS (ESI) m/z [M + H]+ calculated for C22H17FN4O3: 405.4092, found: 405.4018. (cm−1); 3194 (N–H stretch), 3061 (Csp2–H stretch), 2921, 2876 (Csp3–H asymmetric and symmetric stretch), 1684 (CO stretch, lactam), 1676 (CO stretch, amide), 1652 (CN stretch, imine) 1608, 1488 (CC stretch, aromatic), 1243 (Csp2–O stretch, ether), 1152 (Csp3–O stretch, ether); 1H NMR (300 MHz, DMSO-d6) δ (ppm); 13.81 (1H, s, H-17), 7.84 (4H, Brs, H-20, 21, 23, 24), 7.64 (1H, d, 3J = 7.2 Hz, H-8), 7.50 (1H, t, 3J = 7.8 Hz, H-7), 7.35 (1H, d, 3J = 7.8 Hz, H-5), 7.19 (1H, t, 3J = 7.5 Hz, H-6), 7.05–7.11 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.17–4.26 (4H, m, H-10, 11); HRMS (ESI) m/z [M + H]+ calculated for C23H17BrFN3O3: 483.3172, found: 483.3097.4.2 α-Glucosidase inhibition assay
The inhibition of α-glucosidase (E.C.3.2.1.20) enzyme was performed by using assay 0.05 M phosphate buffer (pH 6.8) at 37 °C.48 At 37 °C for 15 minutes, the enzyme (2 units/2 mL) was incubated in phosphate-buffer with various concentrations of the tested substances dissolved in DMSO. After that, the substrate (0.7 mM, p-nitrophenyl-α-D-glucopyranoside) was added, and the variation in absorbance at 400 nm was measured using a spectrophotometer (xMarkTM Microplate Spectrophotometer, BIO-RAD) for 30 minutes. In the control, the tested compounds were replaced with DMSO-d6 (7.5 percent final). As a standard inhibitor, acarbose was utilized.4.3 Statistical analysis
The SoftMax Pro suite and Excel were used to analyse the obtained results for biological activity.Percent inhibition was calculated using the formula below.
 | (1) |
EZ-FIT (Perrella Scientific, Inc., USA) was used for IC50 calculations of all tested samples. To overcome the expected errors all experiments were performed in triplicate, and variations in the results are reported in Standard Error of Mean values (SEM).
 | (2) |
4.4 Molecular docking and ADMET assessment of N-phenoxyethylisatin hydrazones 1(a–l)
The docking protocol was followed by adopting previously available literature with a small modification.49 The 2D structures of ligands were drawn using ChemBioDraw50 which were converted into 3D using OpenBabel.51 The energy of ligands was minimized using the MMFF94 force field. After the removal of all heteroatoms and the ligand, AutoDock Vina52 was used to carry out docking simulations employing the 3D structure of bacterial α-glucosidase (PDB ID: 2zq0). The structure of α-glucosidase was refined, water molecules were removed, hydrogen atoms were added, and side chains were fixed. The grid box of 40 × 40 × 40 Å was created at the center of 7.156, 18.137, and −10.678 in each dimension with a grid spacing of 0.5 Å to determine the ligand–receptor interactions. The Lamarkian Genetic algorithm was used as a scoring function. The same protocol and cavity were used for acarbose and their binding modes and energies were compared. BIOVIA Discovery Studio (ver. 4.5)53 was utilized to visualize the interactions. Hydrazide conjugates 1(a–l) were compared against Lipinski's Rule of Five (Ro5), and their biochemical applications were evaluated using the online computational tool SwissADME.40 Moreover, the pharmacokinetic properties, such as the absorption, distribution, metabolism, excretion, and toxicity (ADMET), of the synthesized hydrazide conjugates were evaluated through an online server, pkCSM.42Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
MMN is grateful to the Quaid-i-Azam University and The World Academy of Sciences (TWAS) for financial support through University Research Fund (URF) and Project No. 13-419 RG/PHA/AS_CUNESCO FR: 3240279216, respectively.References
- K. L. Ong, L. K. Stafford, S. A. McLaughlin, E. J. Boyko, S. E. Vollset, A. E. Smith, B. E. Dalton, J. Duprey, J. A. Cruz and H. Hagins, Lancet, 2023, 402, 203–234, DOI:10.1016/S0140-6736(23)01301-6.
- S. A. Antar, N. A. Ashour, M. Sharaky, M. Khattab, N. A. Ashour, R. T. Zaid, E. J. Roh, A. Elkamhawy and A. A. Al-Karmalawy, Biomed. Pharmacother., 2023, 168, 115734, DOI:10.1016/j.biopha.2023.115734.
- A. Mushtaq, U. Azam, S. Mehreen and M. M. Naseer, Eur. J. Med. Chem., 2023, 249, 115119, DOI:10.1016/j.ejmech.2023.115119.
- P. T. O’gara, F. G. Kushner, D. D. Ascheim, D. E. Casey Jr, M. K. Chung, J. A. De Lemos, S. M. Ettinger, J. C. Fang, F. M. Fesmire and B. A. Franklin, Circulation, 2013, 127, 529–555, DOI:10.1161/CIR.0b013e3182742.
- M. I. Ali and M. M. Naseer, RSC Adv., 2023, 13, 30462–30490, 10.1039/D3RA05953G.
- R. I. Holt, C. S. Cockram, R. C. Ma and A. O. Luk, Diabetologia, 2024, 67, 1168–1180, DOI:10.1007/s00125-024-06102-x.
- X. Zhao, Y. Wang, R. Chen, J. Li, J. Zhou, C. Liu, P. Zhou, Z. Sheng, Y. Chen and L. Song, Cardiovasc. Diabetol., 2021, 20, 1–18, DOI:10.1186/s12933-021-01321-7.
- C. Mora-Fernández, V. Domínguez-Pimentel, M. M. de Fuentes, J. L. Górriz, A. Martínez-Castelao and J. F. Navarro-González, J. Physiol., 2014, 592, 3997–4012, DOI:10.1113/jphysiol.2014.272328.
- M. Miller-Meeks, Contemp. Intern. Med., 1994, 6, 13–15 Search PubMed.
- W. H. Gispen and G.-J. Biessels, Trends Neurosci., 2000, 23, 542–549, DOI:10.1016/S0166-2236(00)01656-8.
- N. Vazzana, P. Ranalli, C. Cuccurullo and G. Davì, Thromb. Res., 2012, 129, 371–377, DOI:10.1016/j.thromres.2011.11.052.
- Y. Kong, F. Wang, J. Wang, C. Liu, Y. Zhou, Z. Xu, C. Zhang, B. Sun and Y. Guan, Front. Aging Neurosci., 2020, 12, 217, DOI:10.3389/fnagi.2020.00217.
- S. R. Joshi, E. Standl, N. Tong, P. Shah, S. Kalra and R. Rathod, Expert Opin. Pharmacother., 2015, 16, 1959–1981, DOI:10.1517/14656566.2015.1070827.
- R. Tundis, M. R. Loizzo and F. Menichini, Mini-Rev. Med. Chem., 2010, 10, 315–331, DOI:10.2174/138955710791331007.
- L. Gong, D. Feng, T. Wang, Y. Ren, Y. Liu and J. Wang, Food Sci. Nutr., 2020, 8, 6320–6337, DOI:10.1002/fsn3.1987.
- R. D. Alharthy, S. B. Zahra, N. Fatima, A. Tabassum, S. Ullah, S. A. Halim, A. Khan, J. Hussain, A. Al-Harrasi and Z. Shafiq, J. Mol. Struct., 2023, 1288, 135783, DOI:10.1016/j.molstruc.2023.135783.
- Y.-Y. Zhang, L.-Q. Hou and T.-Y. Zhao, Exp. Clin. Endocrinol. Diabetes, 2014, 122, 373–378, DOI:10.1055/s-0034-1375676.
- Y. Bando, Y. Ushiogi, D. Toya, N. Tanaka and M. Fujisawa, Intern. Med., 1998, 37, 753–756, DOI:10.2169/internalmedicine.37.753.
- L. J. Scott and C. M. Spencer, Drugs, 2000, 59, 521–549, DOI:10.2165/00003495-200059030-00012.
- E. p. Başaran, S. Köprü, S. Akkoç and B. i. Türkmenoğlu, ACS Omega, 2024, 24, 26503–26518, DOI:10.1021/acsomega.4c03014.
- S. Kumar, J. M. Oh, P. Prabhakaran, A. Awasti, H. Kim and B. Mathew, Sci. Rep., 2024, 14, 1264, DOI:10.1038/s41598-024-51728-x.
- H. S. Al-Salem, M. Arifuzzaman, I. S. Issa and A. M. Rahman, Appl. Sci., 2021, 11, 3746, DOI:10.3390/app11093746.
- I. Abbasi, H. Nadeem, A. Saeed, H. A. A. Kharl, M. N. Tahir and M. M. Naseer, Bioorg. Chem., 2021, 116, 105385, DOI:10.1016/j.bioorg.2021.105385.
- S. Mehreen, A. Ullah, H. Nadeem, N. Dege and M. M. Naseer, RSC Adv., 2022, 12, 1788–1796, 10.1039/D1RA08286H.
- A. Mushtaq, R. Asif, W. A. Humayun and M. M. Naseer, RSC Adv., 2024, 14, 14051–14067, 10.1039/D4RA01937G.
- M. I. Ali, J. Hussain, M. U. Anwar, A. Al-Harrasi and M. M. Naseer, RSC Adv., 2025, 15, 1152–1162, 10.1039/D4RA08086F.
- S. Mehreen, M. Zia, A. Khan, J. Hussain, S. Ullah, M. U. Anwar, A. Al-Harrasi and M. M. Naseer, RSC Adv., 2022, 12, 20919–20928, 10.1039/D2RA03307K.
- M. A. R. Matos, M. S. Miranda, V. M. Morais and J. F. Liebman, Org. Biomol. Chem., 2003, 1, 2566–2571, 10.1039/B304405J.
- M. Zia, S. Hameed, A. Frontera, E. Irran and M. M. Naseer, CrystEngComm, 2021, 23, 3144–3151, 10.1039/D1CE00354B.
- F. Peytam, M. Adib, R. Shourgeshty, M. Mohammadi-Khanaposhtani, M. Jahani, S. Imanparast, M. A. Faramarzi, A. A. Moghadamnia, B. Larijani and M. Mahdavi, J. Mol. Struct., 2021, 1224, 129290, DOI:10.1016/j.molstruc.2020.129290.
- P. Agu, C. Afiukwa, O. Orji, E. Ezeh, I. Ofoke, C. Ogbu, E. Ugwuja and P. Aja, Sci. Rep., 2023, 13, 13398, DOI:10.1038/s41598-023-40160-2.
- L. David, P. A. Nielsen, M. Hedstrom and B. Norden, Curr. Comput.-Aided Drug Des., 2005, 1, 275–306, DOI:10.2174/1573409054367682.
- F. Stanzione, I. Giangreco and J. C. Cole, Prog. Med. Chem., 2021, 60, 273–343, DOI:10.1016/bs.pmch.2021.01.004.
- H. Alonso, A. A. Bliznyuk and J. E. Gready, Med. Res. Rev., 2006, 26, 531–568, DOI:10.1002/med.20067.
- M. I. Ali, H. Nazir, D. Mross, K. Jurkschat and M. M. Naseer, Chem. Biodiversity, 2025, e202403440, DOI:10.1002/cbdv.202403440.
- H. B. B. AG, Eur. J. Clin. Invest., 1994, 24, 3–10, DOI:10.1111/j.1365-2362.1994.tb02249.x.
- F. A. Van de Laar, Vasc. Health Risk Manage., 2008, 4, 1189–1195, DOI:10.2147/vhrm.s3119.
- N. Asano, Glycobiology, 2003, 13, 93–104, DOI:10.1093/glycob/cwg090.
- M. P. Pollastri, Curr. Protoc. Pharmacol., 2010, 49, 9–12, DOI:10.1002/0471141755.ph0912s49.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- E. G. Guzman-Lopez, M. Reina, A. Perez-Gonzalez, M. Francisco-Marquez, L. F. Hernandez-Ayala, R. Castañeda-Arriaga and A. Galano, Int. J. Mol. Sci., 2022, 23, 13246, DOI:10.3390/ijms232113246.
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072, DOI:10.1021/acs.jmedchem.5b00104.
- A. Beig, R. Agbaria and A. Dahan, PLoS One, 2013, 8, e68237, DOI:10.1371/journal.pone.0068237.
- M. S. Alavijeh, M. Chishty, M. Z. Qaiser and A. M. Palmer, NeuroRx, 2005, 2, 554–571, DOI:10.1602/neurorx.2.4.554.
- K. Venkatakrishnan, L. L. von Moltke, R. S. Obach and D. J. Greenblatt, Curr. Drug Metab., 2003, 4, 423–459, DOI:10.2174/1389200033489361.
- R. Masereeuw and F. G. Russel, Drug Metab. Rev., 2001, 33, 299–351, DOI:10.1081/DMR-120000654.
- E. A. Blomme and Y. Will, Chem. Res. Toxicol., 2016, 29, 473–504, DOI:10.1021/acs.chemrestox.5b00407.
- S. Ullah, M. Waqas, S. A. Halim, I. Khan, A. Khalid, A. N. Abdalla, H. A. Makeen, A. Ibrar, A. Khan and A. Al-Harrasi, Int. J. Biol. Macromol., 2023, 250, 126227, DOI:10.1016/j.ijbiomac.2023.126227.
- F. Peytam, M. Adib, R. Shourgeshty, L. Firoozpour, M. Rahmanian-Jazi, M. Jahani, S. Moghimi, K. Divsalar, M. A. Faramarzi and S. Mojtabavi, Sci. Rep., 2020, 10, 2595, DOI:10.1038/s41598-020-59079-z.
- G. W. Milne, Software Review of ChemBioDraw 12.0, ACS Publications, 2010 Search PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 1–14, DOI:10.1186/1758-2946-3-33.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461, DOI:10.1002/jcc.21334.
- D. S. Biovia, Discovery Studio, Dassault Syst'emes BIOVIA, San Diego, CA, USA, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2355357. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra00770d |
| This journal is © The Royal Society of Chemistry 2025 |