
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of tempering-induced microstructural evolution on the corrosion behaviour of high chromium cast iron
Jun-Seob Lee *ab,
Ochegereal Basanjavb,
Jun-Hyeong Leebc,
Jun-Seok Ohd and
Je-Hyun Leeab
*ab,
Ochegereal Basanjavb,
Jun-Hyeong Leebc,
Jun-Seok Ohd and
Je-Hyun Leeab
aSchool of Materials Science and Engineering, Changwon National University, Changwondaehak-ro, Changwon, 51140, South Korea. E-mail: junseoblee@changwon.ac.kr; Fax: +82-55-261-7017; Tel: +82-55-213-3691
bDepartment of Materials Convergence and System Engineering, Changwon National University, Changwondaehak-ro, Changwon, 51140, South Korea
cCOTEC, Ungnam-ro, Changwon, 51553, South Korea
di-Casting Tech Ltd, Gomo-ro, Gimhae, 50875, South Korea
First published on 7th May 2025
Abstract
This study investigated the effect of tempering heat treatment on the corrosion behaviour of high chromium cast iron (HCCI) in a solution of 0.5 mol dm−3 H2SO4 and 0.01 mol dm−3 HCl. After tempering, microstructural analyses revealed that the primary γ phase transformed into the α phase, accompanied by the precipitation of fine carbides and changes in the composition of the eutectic carbides. Electrochemical and surface analyses demonstrated that the galvanic effect, driven by the area ratio and distance between the carbides and the α phase, accelerated selective corrosion in heat-treated specimens compared to as-casted samples. The preferential dissolution was initiated in either the γ or α phase and propagated towards the eutectic γ phase or inter-eutectic carbides (α) for as-casted and heat-treated HCCI, respectively. Consequently, heat treatment accelerated the corrosion rate by intensifying galvanic interactions.
1. Introduction
High chromium cast iron (HCCI), characterised by a carbon (C) content of 2.0 to 3.0 wt% and a chromium (Cr) content of 14.0 to 28.0 wt%, is distinguished by its resistance to wear and corrosion in demanding environments, particularly in acidic chloride conditions. These properties render HCCI a suitable material for applications in rolls, moulds, and slurry pumps within flue gas desulphurisation (FGD) facilities.1–5Among the various types of HCCI used in slurry pumps, those containing 27 wt% Cr are particularly suggested for their corrosion resistance.2,6–12 However, in FGD systems, these alloys are susceptible to various forms of corrosion, including localised corrosion, stress corrosion cracking, and general corrosion when exposed to aggressive environments. To replicate the corrosive conditions typically encountered in FGD facilities, researchers prepare experimental solutions using a combination of sulfuric and hydrochloric acids.13–15
The HCCI is typically subjected to heat treatment (tempering at approximately 500 °C) to optimise its mechanical properties and corrosion resistance. The microstructure prior to tempering generally consists of primary (dendritic) austenite (γ) phases and eutectic γ+carbide phases. Following heat treatment, the primary γ phase transforms into the ferrite (α) phase, accompanied by the precipitation of fine carbides (M23C6: M = Fe and Cr) ranging from several nanometres to micrometres in size. Additionally, the eutectic γ phase that exists between the eutectic carbides transforms into the α phase, referred to as the inter-eutectic carbide (α) phase, after heat treatment.9,11,16–19 It has been reported that, although mechanical strength and hardness values increase following this heat treatment, there is a corresponding decrease in corrosion resistance.11 Wiengmoon et al. reported that Mo addition and subsequent heat treatments in 28 wt% Cr of HCCI increase hardness values through solid solution strengthening, martensitic transformation, and carbide precipitation, while simultaneously reducing corrosion resistance due to chromium depletion in the substrate and changes in phase distribution.18 However, the specific reasons for the changes in corrosion resistance and the corresponding microstructural explanations have not yet been fully elucidated. In particular, detailed information on the electrochemical energy differences between microstructural phases in HCCI is lacking, despite its importance for understanding the galvanic interactions that govern corrosion before and after the heat treatment.
The authors report20–23 indicated that HCCI 27Cr-x (2.1 wt% < x < 2.8 wt%) undergoes general corrosion before heat treatment, primarily due to galvanic effects between the carbide and γ phases. As the proportion of carbide phases (acting as inert cathodes) increases, the galvanic effect intensifies, accelerating the selective dissolution of the γ phase (acting as the local anode). This suggests that the corrosion behaviour of HCCI in FGD system slurry pumps is strongly influenced by galvanic interactions within its microstructure. Specifically, the galvanic corrosion of the γ phase in HCCI is influenced by the geometrical relation between the γ phase and adjacent carbides.
In practice, the HCCI utilized in various products typically undergoes tempering heat treatment at approximately 500 °C.3,4,16 This process modifies the microstructure to optimise mechanical properties, although it may adversely affect corrosion resistance. Therefore, understanding how heat treatment affects the corrosion behaviour of HCCI is important for optimal alloy design. Unlike our previous work,22 which focused on pre-heat treatment corrosion behaviour in Fe–27Cr–xC HCCI, the present study was designed to investigate the post-tempering corrosion mechanism, incorporating microstructural evolution and time-dependent behaviour through surface analyses.
This study explores the corrosion behaviour of HCCI in relation to microstructural changes before and after heat treatment. Specifically, the underlying reasons for corrosion behaviour are analysed based on electrochemical and surface characterisation, including the progression of corrosion over time in an acidic environment.
2. Experimental procedures
The HCCI specimen was fabricated with a chemical composition of 2.1 wt% C, 27 wt% Cr, 0.89 wt% Ni, and 0.86 wt% Mo, with the remainder composed of Fe (as-casted specimen). The heat-treatment specimen was prepared by performing a solution heat treatment at 1065 °C for 4 h, followed by air quenching. Tempering was then carried out at 500 °C for 4 h, after which the specimen was water quenched. Those of the HCCI specimens with a diameter of 26 mm were cut into 5 mm-thick and then ground using SiC paper to a finish of 3000 grit, followed by mirror polishing with a 1 μm diamond suspension.After surface preparation, the specimens were etched by immersion in Murakami's reagent (4 g K3[Fe(CN)6] + 8 g KOH + 100 ml H2O), and the microstructure was observed using an optical microscope. The surfaces of the non-etched specimens after mirror polishing were analysed using a field emission electron probe micro analyzer (EPMA; JEOL, JXA-8530F) in back-scattered electron (BSE) mode, and elemental mapping analysis of Fe, Cr, and C was conducted. Point analysis was performed based on the elemental mapping data obtained from EPMA to quantify the local composition.
The identification of crystallographic phases of HCCIs was performed using electron back-scattered diffraction (EBSD) patterns obtained from a scanning electron microscope (SEM: JSM-6510, JEOL). A database of crystallographic models was employed to approximate the diffraction patterns. Furthermore, phase fractions were quantitatively obtained from the EBSD phase maps using image analysis software (ImageJ), based on pixel area distribution for each identified phase. At least three different regions were analysed per sample to ensure reliability, and the mean values with standard deviations (mean ± SD) are presented in Table 1.
| FCC | BCC | M7C3 (hexagonal) | M23C6 (hexagonal) | |
|---|---|---|---|---|
| As-casted | 49.1 ± 5.6 | 13.7 ± 2.4 | 36.8 ± 3.5 | 0.4 ± 0.07 |
| Heat-treated | — | 64.9 ± 8.3 | 24.8 ± 3.3 | 10.3 ± 5.2 |
The three-electrode electrochemical system with a working electrode (HCCI, surface area of 0.25 cm2), a reference electrode (silver/silver chloride electrode in saturated KCl, SSE), and a counter electrode (amorphous carbon, surface area of 18 cm2), was utilised. The HCCI specimen was connected to copper wires, and the assembly was embedded in epoxy resin to form the working electrode. The surface of the working electrode was polished with SiC #800, and then the electrode potential was measured for 600 s before performing potentiodynamic polarisation at a rate of 1 mV s−1 in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl at room temperature. The experimental solution was selected as a simulated acidic solution for desulphurisation facilities, as it was suitable for assessing the corrosion behaviour of HCCI.13–15 Furthermore, after mirror polishing the surface of the working electrode, potentiostatic polarisation at −0.35 or 0.00 VSSE was conducted for 600 s in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl at room temperature, and the corrosion behaviour on the electrode surface was observed using an optical microscope. To ensure reproducibility, all electrochemical measurements were performed in triplicate, and the results were reported as mean values with corresponding standard deviations (mean ± SD).
To measure the corrosion rate over time, a 5 × 5 × 5 mm specimen was ground with SiC #100 and immersed in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl at room temperature for 24 h. After a specified duration of immersion, the specimen was taken out of the solution, and the weight loss was measured to calculate the corrosion rate using the following equation:
 | (1) |
Scanning Kelvin probe force microscopy (SKPFM) was conducted using a commercial atomic force microscope (XE-100, Park Systems), operated in electrostatic force microscope mode in the air at room temperature. A conductive cantilever (ElectricMulti 75G, Park Systems) with a chromium/platinum coated tip (radius <25 nm, a spring constant of 1–7 N m−1) was employed. The scans covered an area of 30 × 30 μm2 at a scan rate of 0.1 Hz. All measurements were independently repeated three times to confirm measurement consistency. The obtained surface potential data were analysed, and representative results are expressed as mean values with standard deviations (mean ± SD).
3. Results and discussions
3.1 Microstructural analysis before and after heat treatment
Fig. 1 shows the optical microscopic images of HCCI surfaces of (a) as-casted and (c) heat-treated. Generally, the microstructure of HCCI consists of an austenitic phase (with C content below 3 wt% in alloys containing 27 wt% Cr),4,6,9–11 which is composed of a primary γ phase, an eutectic γ phase, and an eutectic carbide phases.4–7 In Fig. 1 (a), the dendritic and remaining eutectic phases are presented. Based on previous research,20–23 it is estimated that the dendritic phase corresponds to the primary γ phase, while the eutectic phase consists of γ + carbide phases. When HCCI is heat-treated, several micrometres of precipitated phases are observed within the dendritic phase, while none are observed in the eutectic regions. Previous research16,17,20,22 indicates that after heat treatment, the face-centred cubic (FCC) crystal structure of the primary γ phase transforms into a body-centred cubic (BCC) crystal structure of the α phase and hexagonal crystal structure of the carbide phases at 790 °C. Since the heat-treatment temperature is 500 °C, it is reasonable to estimate that the microstructure of the heat-treated HCCI consists of α phases along with carbide phases.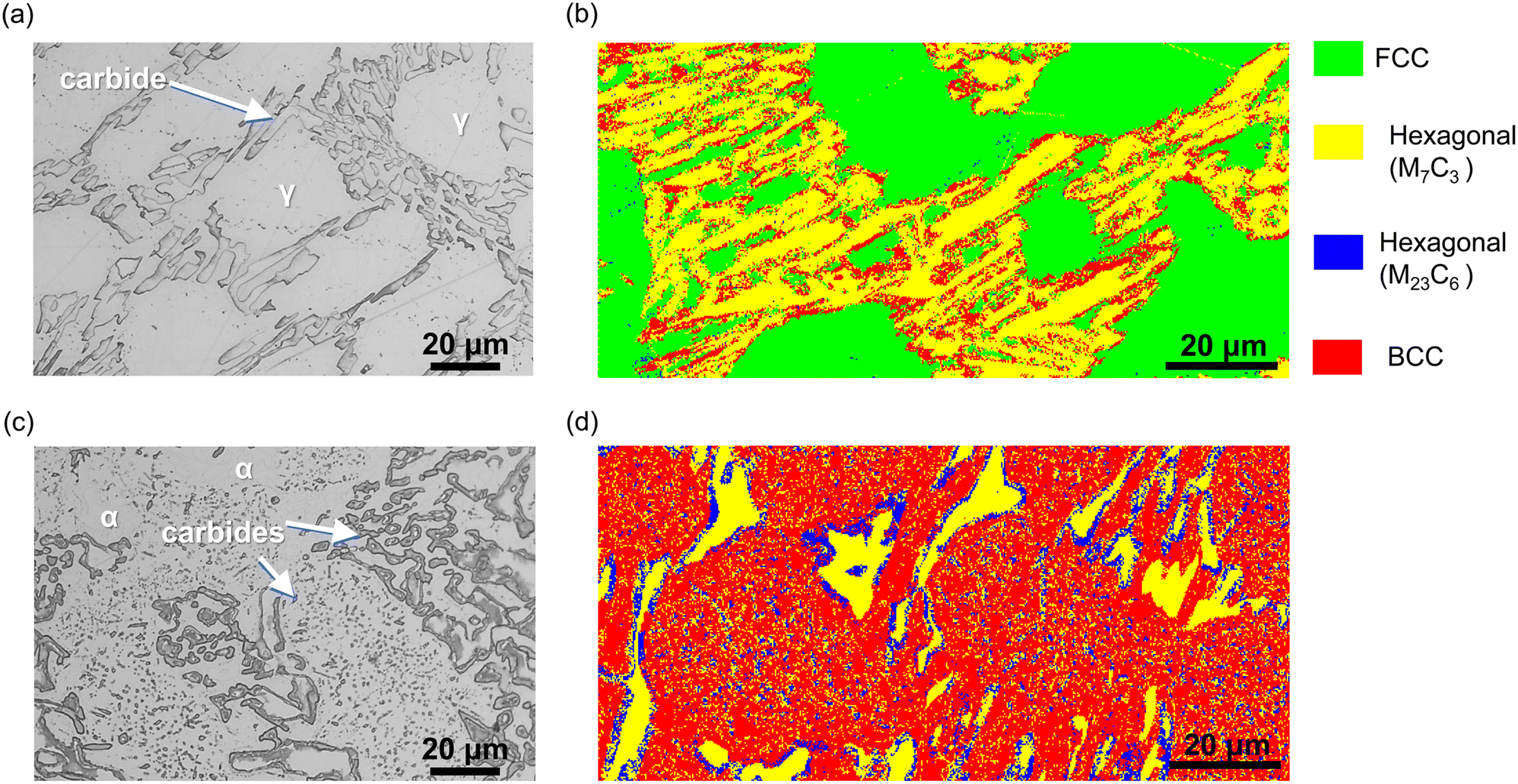 | ||
| Fig. 1 Optical microscopic and EBSD phase maps images of the HCCIs: (a and b) as-casted and (c and d) heat-teated. | ||
Fig. 1 also presents EBSD phase map images of the HCCIs: (b) as-casted and (d) heat-treated. The bright green, yellow, blue, and red colours correspond to the crystal structures of face-centred cubic (FCC), hexagonal (M7C3; M = Fe and Cr), hexagonal (M23C6; M = Fe and Cr), and body-centred cubic (BCC), respectively. For the as-casted HCCI, the FCC, BCC/BCT and hexagonal (M7C3) phases are identified. The dendritic region is characterised by FCC structure with BCC edges, while the eutectic region comprises hexagonal (M7C3 and M23C6) and FCC phases. The as-casted specimen shows phase fractions of 49.1 ± 5.6% FCC and 36.8 ± 3.5% M7C3 carbides, along with 13.7 ± 2.4% BCC and 0.4 ± 0.07% M23C6.
The BCC or body-centred tetragonal (BCT) phases are reported to transform at temperatures below 800 °C,3,16,17 suggesting that rapid cooling during the solidification of the HCCI promotes the formation of BCC/BCT phases at the edges of the FCC phases. In the heat-treated HCCI, BCC and hexagonal phases (M7C3 and M23C6) are identified. The FCC phase is completely transformed into BCC (64.9 ± 8.3%) after heat treatment, while the M7C3 carbide fraction decreases to 24.8 ± 3.3%, and the M23C6 fraction significantly increases to 10.3 ± 5.2%. The dendritic regions contain several micrometres of hexagonal phases (M7C3 and M23C6) embedded within the BCC phase. In contrast, the inter-eutectic carbide regions are characterised by BCC accompanied by hexagonal phases (M7C3 and M23C6), with M23C6 phases located at the edges of the M7C3 phases.
Fig. 2 presents back-scattered and elemental mapping images of the HCCI surfaces (a) as-casted and (b) heat-treated characterised using an electron probe micro analyser. Regardless of the heat treatment, the relatively dark regions (indicating higher Cr and C content in the BSE) and elemental mapping images correspond to carbides, while the brighter regions (indicating higher Fe content) represent the γ and α phases in the as-casted and heat-treated HCCIs, respectively. In the heat-treated specimens shown in Fig. 2(b), several micrometres of carbides precipitated in the α phase exhibit high Cr and C contents. Additionally, distinct concentration differences of Cr, Fe, and C are identified between the centre and edge regions of the eutectic M7C3 carbides. The edges of the carbides display relatively higher Fe content and lower Cr and C contents compared to the centres. After quantifying the stoichiometric values of the carbides (M7C3; M = Cr and Fe), the centre and edge are identified as (Cr39Fe13)C48 and (Cr36Fe24)C40; at%, respectively. The eutectic carbides (M7C3) are analysed in the as-casted condition as (Cr50Fe19)C31. This compositional difference between the centre and edge of the carbides indicates the diffusion of Cr, Fe, and C during the heat treatment process. The decrease in Cr and C concentration leads to the formation of the eutectic carbides (M23C6) at the edges of the eutectic carbides (M7C3).
 | ||
| Fig. 2 Back-scatted electron images and EPMA elemental mapping images of the HCCIs: (a) as-casted and (b) heat-treated. | ||
3.2 Potentiodynamic polarisation characteristics of HCCI
Fig. 3(a) shows the potentiodynamic polarisation curves of the as-casted and hea-treated HCCIs in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. All curves display active-to-passive transition behaviour from the corrosion potential to approximately 0.00 VSSE, indicating that the surfaces of HCCI experience active dissolution, regardless of the heat-treatment procedure. In those active regions of HCCIs, there are two distinct active regions: (i) from approximately −0.40 VSSE to approximately −0.30 VSSE and (ii) from approximately −0.30 VSSE to approximately 0.00 VSSE. This suggests that the active dissolution behaviour on the HCCI surface may not be identical across these regions. The critical current density (ic) in each region was determined by identifying the peak anodic current value within the respective active region of each polarisation curve. The ic in region (i) is higher for the as-casted HCCI, measuring 3.3 × 10−3 A cm−2, compared to the heat-treated HCCI, which measures 1.9 × 10−3 A cm−2. In contrast, the ic in region (ii) is lower for the as-casted HCCI, at 9.6 × 10−3 A cm−2, while the heat-treated HCCI shows a higher value of 15.8 × 10−3 A cm−2. This indicates that the active dissolution rates in regions (i) and (ii) differ between the as-casted and heat-treated HCCI surfaces. In this study, ic was selected instead of the conventional corrosion current density, as it offers a more meaningful representation of electrochemical behaviour in multi-phase systems with complex anodic transitions.15 | ||
| Fig. 3 (a) Potentiodynamic polarisation curves of the as-casted and heat-treated HCCIs immersed in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl, and optical microscopic images after the potentiostatic polarisation at (a) as-casted and (b) heat-treated at −0.35 VSSE or 0.0 VSSE in 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. The applied potentials correspond to the active dissolution regions identified in the polarisation curves, and the current remained relatively stable throughout the test duration. | ||
As the potential value increases from approximately 0.00 to 0.40 VSSE, the anodic current decreases, indicating that the HCCI surfaces are in a passive state. In the passive region, from approximately 0.40 VSSE to 1.00 VSSE, the passive current measures 1.9 × 10−4 A cm−2 for the as-casted HCCI surfaces and 7.7 × 10−4 A cm−2 for the heat-treated HCCI surfaces. This demonstrates that the passive surface formed on the as-casted HCCI is relatively less electrochemically conductive than that formed on the heat-treated HCCI. At potential values >1.00 VSSE, the anodic current increases significantly with polarisation, indicating the transpassive behaviour of the HCCIs, generally associated with oxygen evolution and further anodic reaction involving Cr species.
Fig. 3 also shows the optical microscopic images of (b) as-casted and (c) heat-treated HCCI surfaces before and after the potentiostatic polarisation at −0.35 VSSE or 0.00 VSSE, respectively, for 600 s in 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. The applied potentials of −0.35 VSSE and 0.00 VSSE correspond to the anodic dissolution peaks in the active regions (i) and (ii) identified in Fig. 3(a). For the as-casted HCCI surface, anodic dissolution predominates in the primary γ phase at −0.35 VSSE and in the eutectic γ phase at 0.00 VSSE. In the heat-treated HCCI surface, dissolution occurs in the α phase adjacent to precipitated carbides at −0.35 VSSE, and in the α phase located between the eutectic carbides at 0.00 VSSE. This indicates that preferential dissolution occurs at lower overpotentials in the primary γ or α phase compared to the eutectic γ or inter-eutectic carbide (α) phase in the active regions.
3.3 Corrosion progression and morphological changes during immersion
Fig. 4(a) illustrates the transient of electrode potentials during the open circuit condition of the as-casted and the heat-treated HCCI, monitored over 24 h in 0.5 mol dm−3 H2SO4 and 0.01 mol dm−3 HCl. In all the HCCIs, three stages of the potential transient are identified. During the initial immersion period of about 100 s (stage I), the potential values remain approximately −0.335 VSSE. Subsequently, in stage II, the values sharply increase to around −0.280 VSSE until approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. Finally, in stage III, the potential values increase to about −0.260 VSSE until 24 h of immersion. The potential transient and its values are similar during stage I, while the linear slope for stage II differs between the as-casted and heat-treated samples, estimated at 61 and 32 mV log(s)−1, respectively. This indicates that the potential shift for the as-casted sample is faster than that of the heat-treated sample during stage II. The potential transient time from stage I to II for as-casted and heat-treated HCCI are estimated as approximately 1000 and 400 s, respectively. In stage III, a similar slope value is observed.
000 s. Finally, in stage III, the potential values increase to about −0.260 VSSE until 24 h of immersion. The potential transient and its values are similar during stage I, while the linear slope for stage II differs between the as-casted and heat-treated samples, estimated at 61 and 32 mV log(s)−1, respectively. This indicates that the potential shift for the as-casted sample is faster than that of the heat-treated sample during stage II. The potential transient time from stage I to II for as-casted and heat-treated HCCI are estimated as approximately 1000 and 400 s, respectively. In stage III, a similar slope value is observed.
 | ||
| Fig. 4 (a) Transient electrode potential and (b) corrosion rates of as-casted and heat-treated HCCIs in 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl for 24 h. | ||
The significant change of the potential transient slope during immersion time in stages I and II can be discussed based on the active regions in Fig. 3(a). Where the active regions (i) and (ii) for as-casted and heat-treated (Fig. 3(a)) are in the electrode potential ranges of (i) from ca. −0.40 VSSE to −0.30 VSSE and (ii) from ca. −0.30 VSSE to 0.00 VSSE. The potential transition time and value from stage I to stage II are approximately 1000 s and −0.310 VSSE, respectively, for as-casted HCCI. In contrast, for heat-treated HCCI, the transition time and value are approximately 400 s and −0.320 VSSE, respectively. This indicates that the dissolution sequence during the transition from stage I to stage II differs depending on the heat treatment, proceeding from the primary γ phase to the eutectic γ phase in the as-casted HCCI, and from the α phase to the inter-eutectic carbide (α) phase in the heat-treated HCCI.
Fig. 4(b) shows the corrosion rates over time, calculated from the weight loss of the as-casted and heat-treated HCCI specimens that were removed from 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl at various time points chosen during a 24 h immersion period. The corrosion rates are shown for time intervals ranging from 150 s to 86400 s (24 h), reflecting the entire immersion period. The corrosion rate increases exponentially with immersion time. The corrosion rate sharply rises after approximately 600 s for heat-treated specimens and after about 1000 s for as-casted specimens. After 10000 s of immersion, the corrosion rate for the heat-treated specimen exceeds that of the as-casted specimen. Following approximately 20000 s, the corrosion rate continues to increase, but the rate of acceleration declines for all HCCI specimens, regardless of heat treatment. At the end of the 24-hour immersion, the corrosion rates are 248 mm year−1 for heat-treated specimens and 192 mm year−1 for as-casted specimens.
Fig. 5(a) shows optical microscopic images of (a) as-casted and (b) heat-treated HCCI surfaces during the immersion in 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. When the as-casted HCCI (Fig. 5(a)) is immersed in the solution, the interface around the carbides and γ phases is slightly corroded. By 300 s, the dissolution of the γ phase becomes evident, particularly in the primary γ phase. By 1200 s, the eutectic γ phases also exhibit evidence of dissolution, as shown in Fig. 4(b), indicating the transition from stage I to stage II in the dissolution sequence. As immersion reaches 2400 s, dissolution progresses further, affecting both primary and eutectic γ phases. Even after 21600 s (6 h), dissolution proceeds across both γ phases without selectivity, and in some regions, the corrosion depth exceeds several millimetres.
 | ||
| Fig. 5 Optical microscopic images of the HCCIs: (a) as-casted and (b) heat-treated, obtained after immersion for 0, 150, 300, 600, 1200, 2400, 7200 and 21600 s in 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. | ||
For heat-treated HCCI in Fig. 5(b), the interface around precipitated carbides and the α phase is predominantly corroded within the first 300 s of immersion. After 600 s of immersion, a significant change in the dissolution behaviour is observed, propagating into the inter-eutectic carbide (α) phase. This change signifies the transition from stage I to stage II of the corrosion process (Fig. 4(a)). During stage I, which lasts approximately 600 s, the dissolution primarily occurs in the α phase, where the previous dendritic structure existed before heat treatment, while the precipitated carbides remain relatively intact. However, once stage II commences, the corrosion rate of the inter-eutectic carbide (α) phase accelerates. As immersion continues to 21600 s (6 h), the dissolution of all α phases (dendritic and inter-eutectic carbides) is clearly observed. Although the carbide phase initially remains intact, prolonged dissolution eventually causes partial loss of the carbide phase itself.
3.4 Kelvin probe analysis
Fig. 6 presents the topographic and contact potential difference (CPD) images of the as-casted (a and b) and heat-treated (c and d) HCCI surfaces using scanning Kelvin probe force microscopy (SKPFM). The CPD value indicates the relative work function difference between the cantilever tip surface and the specimen surface. The relatively bright regions in Fig. 6(a) and (c) correspond to carbides, while the comparatively dark regions represent the γ or α phase. The average height of the areas identified as carbides is approximately 0.8 to 0.07 μm greater than the average height of the γ or α phase region. This disparity is attributed to the hardness difference between the γ or α phase and the carbides. It has been reported that the hardness value for eutectic carbide in hypoeutectic HCCI is approximately 9 GPa, while the hardness value of γ or α phase is around 6 to 7 GPa.24 Given that the carbides possess higher hardness values than the γ or α phase, a difference in surface protrusion persists even after the grinding and polishing for the SKPFM preparing process. | ||
| Fig. 6 Scanning Kelvin probe force microscopic (SKPFM) images of HCCI specimens: (a and c) topography and (b and d) contact potential difference (CPD) images for (a and b) as-casted and (c and d) heat-treated specimens. (e) Relative contact potential differences (ΔCPD) with respect to the primary γ phase (as-casted) and α phase (heat-treated). | ||
In Fig. 6(b) and (d), the mapping images of the CPD values reveal that the carbide phases—specifically, the carbides (M7C3) in the as-casted specimens and the carbides (M7C3 and M23C6) in the heat-treated specimens—display relatively higher CPD values than the γ or α phases. The CPD values for the carbides are approximately 40 mV and 20 mV higher than those of the γ or α phases in the as-casted and heat-treated specimens, respectively. For the heat-treated specimen, the CPD values in the eutectic carbides (specifically the M7C3 phase shown in Fig. 1(d)) are approximately 10 mV higher in the centre of the carbide phase than at the edge.
According to the EPMA point analysis results, after the heat treatment specimen, the Cr/Fe ratios of the eutectic carbide (M7C3) at the edge and the centre are 1.61 ± 0.45 and 2.89 ± 0.90, respectively. Additionally, the concentration of C decreases at the edge of the carbides. Generally, the C–Cr bond is less ionic (more covalent) than the C–Fe atomic bond in the carbide phases of HCCI.24,25 The decrease in the Cr/Fe ratio at the edge of the carbides results in a weakening of both covalent and metallic C–M bonds in these phases. Since the electron work function (EWF) is strongly associated with bonding strength, particularly in metallic bonds, higher EWF values are related to increased electron densities between metal atoms.25–27
Generally, when a particular area (or phase) on the surface exhibits a CPD value that is relatively higher than that of other regions, it indicates a greater work function difference between the cantilever tip and the substrate. An increase in the CPD value implies a more noble electrochemical tendency, allowing regions with higher CPD values to function relatively as cathodes. Since the carbide phases exhibit higher CPD values compared to the substrate γ or α phases, the γ or α phases surrounding the carbides act as local anodes, while the carbides themselves act as local cathodes.
To understand the galvanic behaviour between these anodes and cathodes—the carbides and other phases, respectively, in HCCI—it is useful to present these CPD values relative to the primary γ or α phases as ΔCPD in Fig. 6(e). Given that the CPD values of the primary γ and α phases are the lowest in each specimen, they act as anodes. For as-casted HCCI, the ΔCPD for the eutectic carbide (M7C3) and eutectic γ phase relative to the primary γ phase is 37 mV and 11 mV, respectively. For heat-treated HCCI, the ΔCPD for the eutectic (M7C3: at the centre and the edge), precipitated carbide (M23C6), and inter-eutectic carbide (α) phases relative to the α phase is 21 mV, 14 mV, 16 mV, and 9 mV, respectively. The ΔCPD values for those phases indicate that the γ and α phases are preferentially dissolved when exposed to acidic solutions. In the case of as-casted HCCI, the ΔCPD of the eutectic γ phase is more positive than that of the primary γ phases, suggesting that the dissolution of the primary γ phases will occur prior to that of the eutectic γ phases (Fig. 5(a)). For heat-treated HCCI, the α phase surrounded by the precipitated carbides (M23C6; ΔCPD relative to the α phase of 16 mV) preferentially dissolves compared to the α phases surrounded by the eutectic carbides (M7C3; ΔCPD relative to the inter-eutectic carbide phase (α) of 4 mV). This suggests that dissolution initiates from the α phase and progresses to the inter-eutectic carbide (α) phases over the immersion period.
The ΔCPD value for the eutectic carbide relative to the γ phase in the as-casted HCCI (38 mV) is higher than the ΔCPD values for the carbides (eutectic and precipitated) relative to the α phase in heat-treated HCCI (9 mV and 16 mV, respectively). This also suggests that the driving force for galvanic corrosion involving the γ phase and carbides is greater than that for the α phase. In general, galvanic corrosion rates are influenced by the area ratio and the distance between local anodes and cathodes. A larger cathode and a smaller anode, when those are positioned closer together, will lead to an accelerated rate of galvanic corrosion. In the HCCI specimens, local anodes (either γ or α phases) are surrounded by local cathodes (carbides), and their galvanic corrosion rates are strongly affected by the geometric arrangement between the anodes and cathodes. Given that heat treatment can increase the cathode/anode area ratio and reduce the distance between them—due to the presence of precipitated carbides—it can be suggested that the corrosion rate of the α phase following heat treatment is greater than that of the γ phase. The microstructural geometry—specifically the area ratio and distance between cathodic and anodic phases—plays a key role in galvanic corrosion, as supported by ΔCPD analysis (Fig. 6(e)).
As immersion progresses, the galvanic effect becomes increasingly important in accelerating corrosion, as the dissolution of matrix phases enhances cathode-to-anode interactions. During immersion, the surface area of the electrochemically inert carbide increases in three dimensions due to the preferential dissolution of the γ and α phases. As a result, the galvanic effect becomes increasingly significant, leading to an accelerated corrosion rate in HCCI. This suggestion is also supported by Fig. 5. When the immersion time exceeds approximately 1000 s—during the period characterised by accelerated corrosion rates for both as-casted and heat-treated HCCI—the galvanic effect in heat-treated specimens becomes more pronounced. This is attributed to the increasing distance and area effects between the carbides and the α phase, resulting in a substantial rise in the corrosion rate.
3.5 Implications and future perspectives
This study reports the corrosion behaviour of HCCI before and after tempering heat treatment. The corrosion rate becomes faster and more accelerated after heat treatment, attributed to the galvanic effect arising from the area ratio and distance between the carbides and the α phase. While previous research has mentioned an increase in corrosion rates following the heat treatment of HCCI,11 the specific reasons for this phenomenon have not been discussed in detail. One possible reason for this lack of information is the difficulty in analysing microstructural changes before and after heat treatment, as well as characterising the differences in electrochemical behaviour between phases.To the authors' knowledge, this study is the first to analyse the differences in the various microstructures present in HCCI before and after heat treatment, along with the impact of inter-electrode distance and area ratio on corrosion rates. This information is expected to provide crucial insights into the corrosion behaviour between those phases in HCCI, which is essential for the development of HCCIs requiring a balance between wear resistance and corrosion resistance for use in slurry pumps in FGD systems with acidic environments. Based on the information obtained regarding the galvanic effect from this study, future research will be proposed to optimise the corrosion rate of HCCI through design by adjusting the (Cr + Fe)/C ratio and refining the heat treatment process.
4. Conclusions
This study demonstrated that tempering heat treatment significantly influences the preferential dissolution behaviour of high chromium cast iron (HCCI) in a solution of 0.5 mol dm−3 H2SO4 + 0.01 mol dm−3 HCl. The phase transformation of the primary γ phase into the α phase and the precipitation of fine carbides during heat treatment played a important role in this corrosion process. Electrochemical analysis of the potentiodynamic polarisation curves and time-dependent changes in electrode potential indicated that corrosion initially occurred in the primary γ phase before heat treatment, and in the α phase after heat treatment, subsequently propagating towards the eutectic γ phase and inter-eutectic carbide (α) phase. The contact potential difference (CPD) values indicated that the carbide phases had higher CPD values than the γ and α phases, suggesting their role as local cathodes. This behaviour resulted in the surrounding γ or α phases functioning as local anodes, thereby further accelerating the dissolution process. As the corrosion progressed, this led to a strengthening of the galvanic interactions between local anodes and cathodes, which in turn contributed to the higher corrosion rates obtained in heat-treated HCCI compared to the as-casted condition.This study provided new insights into the relation between the galvanic effect and microstructural changes in HCCI before and after tempering. Inter-electrode distance and area ratio between carbides and the α phase were identified as key factors contributing to the accelerated corrosion. These findings provide guidance for designing HCCI with improved corrosion and wear resistance in acidic environments.
Data availability
All data used in this study were presented in the manuscript.Author contributions
Jun-Seob Lee: conceptualization; funding acquisition; investigation; methodology; project administration; validation; writing – original draft; writing – review & editing, Ochegereal Basanjav: formal analysis; Jun-Hyeong Lee: formal analysis; Jun-Seok Oh: resources, Je-Hyun Lee: funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (RS-2021-KP002514, Development of R&D engineers for combined cycle power plant technologies).References
- C. H. Pitt and Y. M. Chang, Corrosion, 1986, 42, 312 Search PubMed.
- A. Neville, F. Reza, S. Chiovelli and T. Revega, Metall. Mater. Trans. A, 2006, 37, 2339 Search PubMed.
- U. P. Nayak, M. A. Guitar and F. A. Mücklich, Metals, 2020, 10, 30 Search PubMed.
- J. R. Davis, ASM Specialty Handbook- Cast Irons, ASM International, 1996, pp. 107–122 Search PubMed.
- R. J. Chung, X. Tang, D. Y. Li, B. Hinckley and K. Dolman, Wear, 2009, 267, 356 Search PubMed.
- C. P. Tabrett, I. R. Sare and M. R. Ghomashchi, Int. Mater. Rev., 1996, 41, 59 Search PubMed.
- I. Chakrabarty and A. Basak, J. Mater. Sci. Lett., 1987, 6, 1399 Search PubMed.
- N. Fu, X. Tang, D. Y. Li, L. Parent and H. Tian, J. Solid State Electrochem., 2015, 19, 337 Search PubMed.
- A. Wiengmoon, J. T. H. Pearce and T. Chairuangsri, Mater. Chem. Phys., 2011, 125, 739 Search PubMed.
- A. Wiengmoon, T. Chairuangsri, N. Poolthong and J. T. H. Pearce, Mater. Sci. Eng., A, 2008, 480, 331 Search PubMed.
- H. D. T. Hong, H. N. Hong, M. N. Ngoc and Q. H. T. Ngoc, ISIJ Int., 2021, 61, 1660 Search PubMed.
- A. ElJersifi, K. Aouadi, M. B. Ali, A. Chbihi, N. Semlal, H. Bouaouine and S. Naamane, J. Mater. Eng. Perform., 2023, 32, 8262 Search PubMed.
- D. P. Le, W. S. Ji, J. G. Kim, K. J. Jeong and S. H. Lee, Corros. Sci., 2008, 50, 1195 Search PubMed.
- Z. Wang, D. Mei, W. Zhang, E. Han and Y. Wang, Engineering, 2011, 3, 653 Search PubMed.
- J.-S. Lee, K. Fushimi, T. Nakanishi, Y. Hasegawa and Y. S. Park, Corros. Sci., 2014, 89, 111 Search PubMed.
- J.-S. Oh, Y.-G. Song, B.-G. Choi, C. Bhamornsut, R. Nakkuntod, C.-Y. Jo and J.-H. Lee, Metals, 2021, 11, 1579 Search PubMed.
- D. Li, L. Liu, Y. Zhang, C. Ye, X. Ren, Y. Yang and Q. Yang, Mater. Des., 2009, 30, 340 Search PubMed.
- A. Wiengmoon1, N. Tareelap, S. Imurai, T. Chairuangsri and J. T. H. Pearce, Solid State Phenom, 2018, 283, 95 Search PubMed.
- V. Marimuthu and K. Kannoorpatti, J. Bio Tribo. Corros., 2016, 2, 29 Search PubMed.
- J.-S. Lee, J.-H. Lee, J.-S. Oh and J.-H. Lee, Corros. Sci. Technol., 2021, 20, 367 Search PubMed.
- J.-S. Lee, J.-H. Lee, J.-S. Oh, J. G. Kim and J.-H. Lee, Corros. Sci. Technol., 2022, 21, 418 Search PubMed.
- J.-S. Lee, J.-H. Lee, S.-Y. Rho, Y.-H. Lee, Y.-J. Lee, J.-S. Oh and J.-H. Lee, RSC Adv., 2023, 13, 19220 Search PubMed.
- B. Ochegerel, J.-S. Lee, J.-H. Lee, J.-S. Oh and J.-H. Lee, Corros. Sci. Technol., 2022, 22, 252 Search PubMed.
- J. Cui, L. Guo, H. Lu and D. Y. Li, Wear, 2017, 376–377, 587 Search PubMed.
- L. Guo, Y. Tang, J. Cui, J. Li, J. R. Yang and D. Y. Li, Scr. Mater., 2021, 190, 168 Search PubMed.
- M. Yoshitake, Y. Aparna and K. Yoshihara, Appl. Surf. Sci., 2001, 169, 666 Search PubMed.
- M. Kadowai, A. Saengdeejing, I. Muto, Y. Chen, T. Doi, K. Kawano, Y. Sugawara and N. Hara, J. Electrochem. Soc., 2021, 168, 11503 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |