DOI:
10.1039/D5RA00619H
(Review Article)
RSC Adv., 2025,
15, 8121-8155
Corey–Fuchs reaction enabled synthesis of natural products: a review
Received
26th January 2025
, Accepted 6th March 2025
First published on 17th March 2025
Abstract
Natural products can be derived from a vast array of animals, plants and microorganisms and are generally characterized by a wide spectrum of bioactive properties such as anti-viral, anti-cancer, anti-inflammatory and anti-bacterial properties. Synthesis of natural products is of paramount importance in various fields including medicine, biotechnology and agriculture. The Corey–Fuchs reaction, also known as the Ramirez Corey–Fuchs reaction, is a pivotal organic transformation and plays a significant role in the synthesis of intricate natural products and their analogues. This review article highlights the development of the Corey–Fuchs reaction in recent years towards the synthesis of complex natural products including polyketides, alkaloids, terpenoids and peptides.
1. Introduction
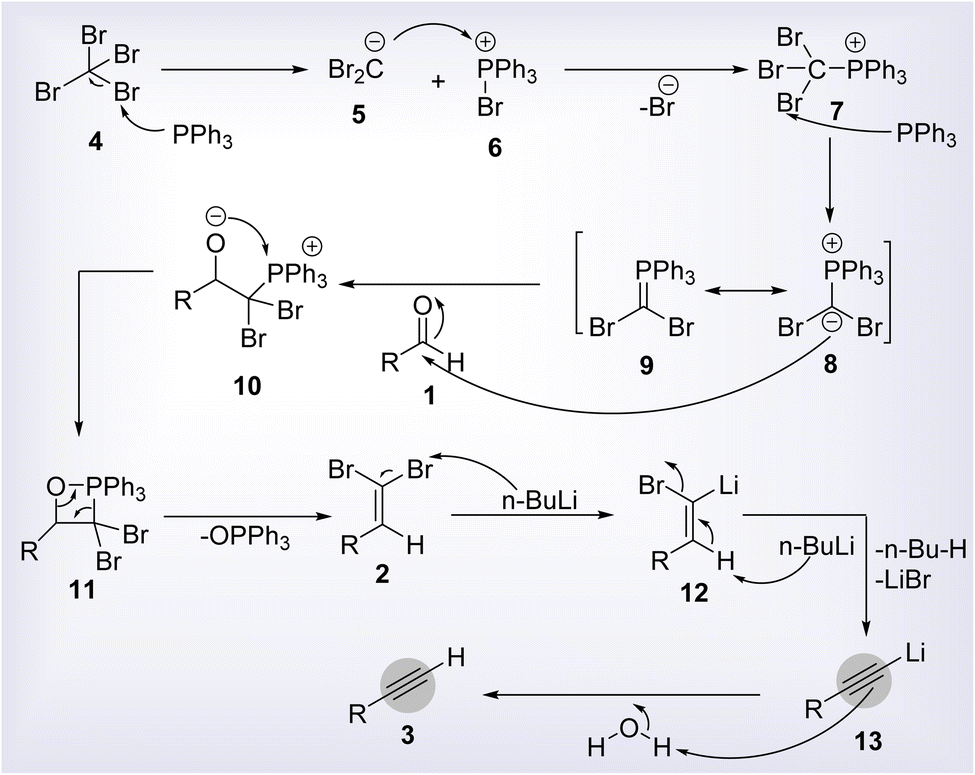
The Corey–Fuchs reaction was established by Nobel laureate E. J. Corey and his colleague P. L. Fuchs in 1972.1 It is a pioneering transformation in organic synthesis involving the construction of a terminal alkyne via one carbon homologation of aldehyde. The reaction protocol includes a two steps methodology entailing the formation of α,α-dibromoalkene from an aldehyde followed by its conversion to a terminal alkyne under the assistance of a strong base (Scheme 1). The general reaction mechanism involves the formation of phosphorus ylide 8 from the reaction of triphenylphosphine and carbon tetrabromide, succeeded by its treatment with aldehyde 1 to furnish dibromoolefin 2. In the next step, the lithium halogen exchange process and elimination lead to the synthesis of desired alkyne 3 (Scheme 2).2
 |
| | Scheme 1 General reaction of Corey–Fuchs protocol. | |
 |
| | Scheme 2 General mechanism for Corey–Fuchs reaction involving the synthesis of terminal alkyne. | |
Terminal alkynes constitute a primary class of organic substances that have gained considerable acclaim since the turn of the century due to their diverse utilization and applications in multiple fields. Over 400 naturally occurring substances possessing terminal alkyne as a building block, have been characterized by a range of biological active properties including anti-viral, anti-cancer, anti-microbial, anti-malarial and anti-inflammatory3 properties. Moreover, alkynes synthesized through Corey–Fuchs reaction, act as key intermediates in the assembly of various other natural products, such as macrolides,4 δ-lactone,5 alkaloids,6 unsaturated fatty acids,7 polyketides8 and terpenoids9 etc.
A well-utilized strategy for the construction of alkynes is the one-carbon homologation of carbonyl compounds. Building on Corey's pioneering studies in this field,10 numerous methodologies including Ramirez Corey–Fuchs reaction, Colvin rearrangement,11 Ohira–Bestmann reaction12 and Seyferth–Gilbert homologation11 have been come forth in the literature. Corey–Fuchs reaction is often found as an optimal method for the alkyne synthesis owing to mild reaction conditions, wide substrate scope and high yields over other phosphorous-catalyst based reactions.13 Ohira–Bestman reagent (dimethyl 1-diazo-2-oxopropylphosphonate) (termed as Seyferth–Gilbert homologation) was widely employed for the synthesis of terminal alkynes, however, these types of reagents are relatively expensive, precarious and also have limited applications for the preparation of acetylenic group starting from aldehydes.14 Moreover, Corey–Fuchs procedure allows for the preparation of enantiomerically pure products in contrast to Ohira–Bestmann reaction protocol as it is incompatible due to the racemization of the product.15
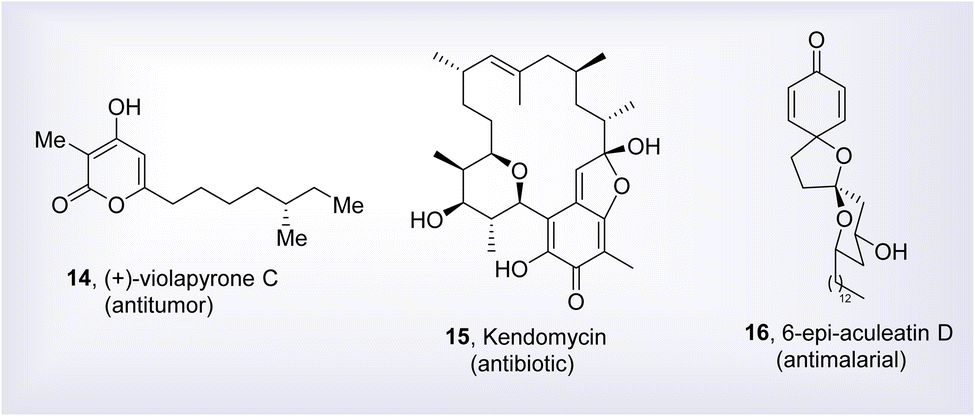
Various organic reactions are employed to carry out the total synthesis of naturally occurring compounds and other intricate biologically active heterocyclic functionalities.16–19 Surplus literature regarding the construction of several sophisticated natural products and other intricate organic compounds involving the Corey–Fuchs reaction as a main step, has been reported yet.20–23 Natural products have been the backbone of various biological active and potential lead compounds and their total synthesis has witnessed a pronounced advancement in recent years.24 In this perspective, Lee et al., in 2014, demonstrated the total synthesis of (+)-Violapyrone C employing Corey–Fuchs reaction. (+)-Violapyrone C 14 is featured with high cytotoxic activity against six human tumor cell lines.25 Similarly, in the same year, Sengoku and coworkers described the total synthesis of Kendomycin 15 utilizing the Corey–Fuchs reaction conditions.26 This natural product not only exhibit strong anti-tumor activity against human call lines but also show excellent anti-bacterial properties against Gram positive and negative bacteria. In 2015, Huang et al. devised the total synthesis of 6-epi-Aculeatin D 16 via Corey–Fuchs alkyne synthesis, which showcases strong anti-malarial activity with IC50 value of 0.18–3 μM against P. falciparum (Fig. 1).27
 |
| | Fig. 1 Structure of some biological important natural products having Corey–Fuchs reaction as main step in total synthesis. | |
The Corey–Fuchs reaction has endured notable breakthrough in the recent past and the need for a thorough and state-of-the-art review on this topic has gained great prominence. The most recent comprehensive review on this topic dates back to 2015, emphasizing the need for a contemporary assessment of its scope and applications.28 This manuscript intends to provide a latest (2016 to date) update on the cutting-edge development, showcasing the versatility and potential of this invaluable reaction in natural product synthesis.
2. Review of literature
2.1. Synthesis of polyketide-based natural products
2.1.1. Chatenaytrienin-4 synthesis. Acetogenin membranacin is a naturally occurring polyketide which belongs to a large group of fatty acid type compounds called annonaceous acetogenins. These natural products are characterized by a number of biological active properties such as pesticidal, cytotoxic, anti-malarial, anti-feedant, immunosuppressive and most likely, anti-tumor properties.29 Adrian and Stark30 in 2016, described modified strategy for the efficient stereodivergent construction of straight chain 1,5,9, n-polyenes and employed this for the first total synthesis of chatenaytrienin-4 24, which is the hypothetical biosynthetic forerunner of membranacin 25. The total synthesis commenced from the unexpensive 1-dodecyne 17 which was subjected to subsequent allylation to yield 18 followed by hydroboration to form bis-homopropargylic alcohol 19 in 70% yield. Next, the alcohol 19 was subjected to PCC/silica mediated oxidation in DCM to furnish aldehyde compound 20 (86%), followed by Corey–Fuchs alkyne synthesis reaction under the presence of CBr3 and PPh3 to furnish a dibromo compound 21 in conjugation with its lithiation and allylation in the presence of TBAI to furnish enediyne 22 in 80% yield. Further, enediyne 22 was subjected to chemoselective hydroboration in the presence of 9-BBN-dimer to deliver alcohol 23 in 63% yield. Carboxylic compound 24 was synthesized from 23 in a few steps followed by its treatment with hydroxy butanolide 25 in the presence of DMAP, ensued by Fries rearrangement and successive selective hydrogenation to deliver compound 26 (90%). Further, the hydroxyl group was protected under the presence of Tf2O followed by Lindlar's catalyst mediated reduction and then Pd-mediated reduction to finally achieve chatenaytrienin-4 27 (51% over 2 steps) in overall 6% yield in 15 linear steps (Scheme 3).
 |
| | Scheme 3 Total synthesis of chatenaytrienin-4 27 by Adrian and Stark. | |
2.1.2. Tetrangulol synthesis. Tetrangulol 35 is a naturally occurring quinone, which belongs to the group of angucyclines. This group of compounds exhibit a wide range of biological properties such as anti-cancer and anti-bacterial activities.31 Tetrangulol 35 was synthesized for the first time by Brown & Thomson in 1976.32 Since then, a number of synthetic methods have been reported on the synthesis of biological active tetrangulol.33 Owing to high medicinal properties, Ngwira34 et al., in 2016 reported the synthesis of tetrangulol by employing Suzuki–Miyaura coupling reaction & Corey–Fuchs alkyne synthesis as key synthetic steps. The total synthesis initiated via generation of substituted benzaldehyde 30 which is a key precursor, from commercially available 1,5-dihydroxynaphthalene 29 over few steps. Next, the compound 31 was subjected to reductive methylation followed by its reaction with n-BuLi and triisopropylborate to furnish boronic acid 32 (96%). The synthesized acid 32 underwent Suzuki–Miyaura coupling reaction in the presence of Pd(PPh3)4 to access 3-methoxy-5-methyl-2-(1,4,5-trimethoxynaphthalen-2-yl)benzaldehyde 33 in 80% yield. Compound 33 was then subjected to Corey–Fuchs reaction under the presence of CBr4 and PPh3 to furnish dibromoalkene 34 in conjugation with n-BuLi to form desired alkyne compound 35 in 88% yield. In the next step, formation of a mixture of tetracyclic product 36 (61%) and 37 (23%) took place when compound 35 was reacted in the presence of PtCl2 and PhMe. Both the products were separated via chromatography and compound 36 were further subjected to CAN oxidation followed by its treatment with boron tribromide to afford desired tetrangulol 38 in 97% yield (Scheme 4).
 |
| | Scheme 4 Total synthesis of tetrangulol 38 by Ngwira and coworkers. | |
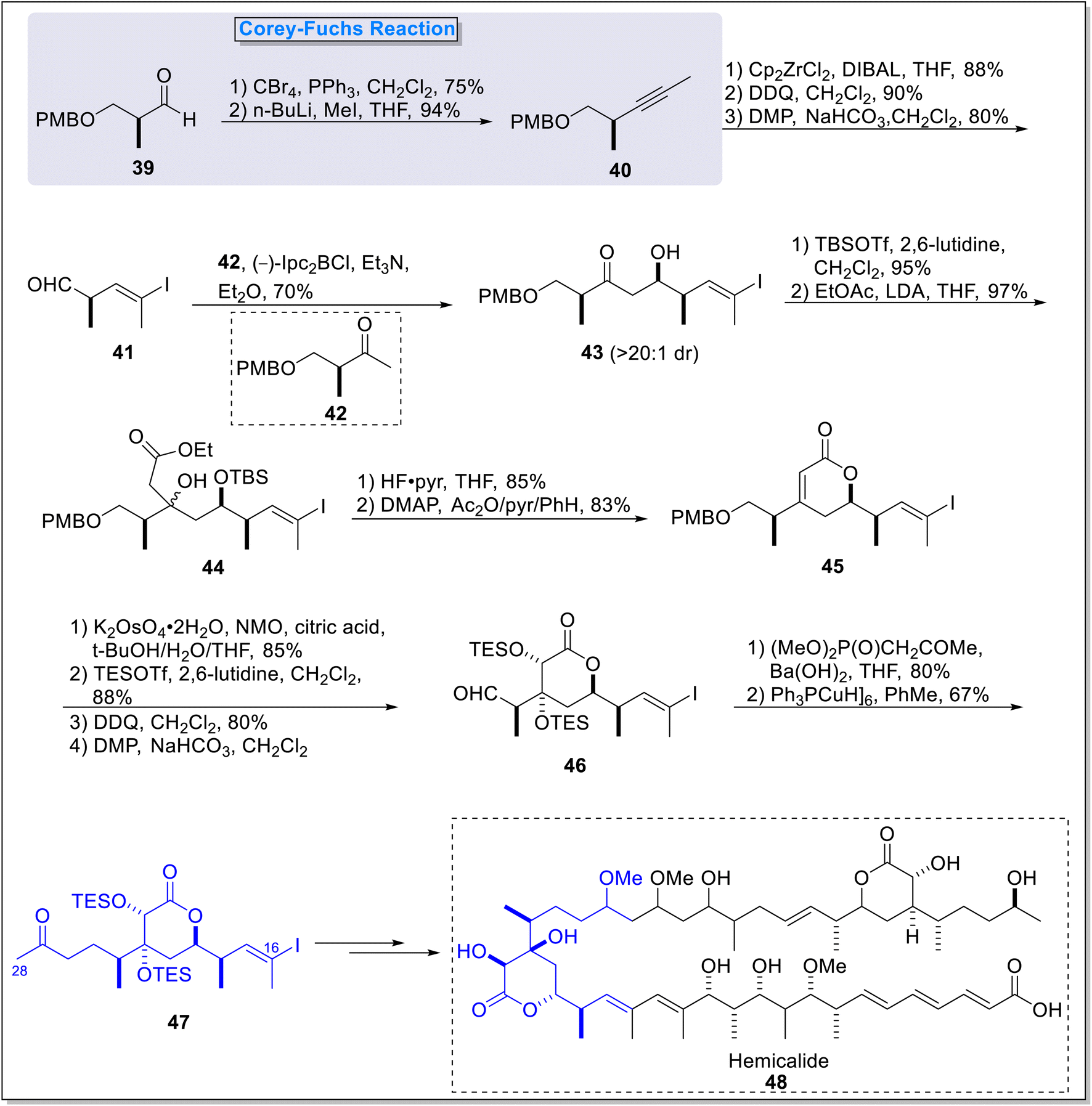
2.1.3. Synthesis of C16–C28 fragment of hemicalide. Hemicalide is a naturally occurring compound, fits into the class of polyketides.35 It is extracted from marine sponge Hemimycale sp. in the south pacific and featured with potent cytotoxic properties. Hemicalide exhibit excellent growth inhibitory potential against human cancer cell lines with IC50 values in picomolar, processing via tubulin targeting anti-mitotic mechanism.36,37 In this regard, MacGregor36 et al., in 2016 synthesized the advanced C16–C28 fragment 47 of the hemicalide 48 with restructured 18,19-syn geometry. The synthesis commenced with the formation of alkyne compound 40 (94%) under the Corey–Fuchs reaction followed by hydrozirconation and iodination in conjugation with subsequent 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) induced ether cleavage and oxidation to furnish (Z)-vinyl iodide 41 (80%). In the next step, vinyl iodide 41 were subjected to aldol addition reaction with PMB ether 42 under the conditions of ((–)-Ipc2BCl, Et3N) to generate a syn adduct 43 in 70% yield with exclusive diastereoselectivity. Next, subsequent formation of TBS ether and enolate addition in the presence of EtOAc and LDA led to the formation of ester 44 (97%), which was further subjected to TBS ether cleavage and induced lactonization in conjugation with elimination to furnish α-lactone 45 in 83% yield. Next, aldehyde compound 46 was achieved in 4 steps including syn-dihydroxylation and TES ether generation in the presence of TESOTf and 2,6-lutidine followed by PMB ether cleavage in DDQ and Dess–Martin periodinane (DMP) mediated oxidation. In the next step, Horner–Wadsworth Emmons olefination in the presence (MeO)2P(O)CH2COMe & Ba(OH)2 and Stryker reduction under the conditions of [Ph3PCuH]6 and PhMe subsequently led towards the synthesis of completed C16–C28 fragments 47 in 67% yield (Scheme 5).
 |
| | Scheme 5 Total synthesis of C16–C28 fragment of hemicalide 48 by MacGregor and coworkers. | |
2.1.4. Actinoallolide synthesis. Actinoallolides belong to the class of 12 or 14-membered macrolides, derived from culture medium of Actinoallomurus fulvus MK10-036.38 These compounds showcase a number of activities, such as in vitro anti trypanosomal potential against Trypanosoma brucei brucei GUTat 3.1 strain which is a bioweapon of Nagana disease in animals, Trypanosoma brucei rhodesiense which is a bioweapon of African Trypanosomiasis and Trypanosoma cruzi Tulahun strain which cause Chagas disease.38 Actinoallolide A 61 possessed most of the potent activities such as anti-trypanosomal activity against T. b. rhodesiense STIB900 strain and also exhibit cytotoxic properties against MRC-5 cell lines and MIC; >10 μg per paper disk against Gram-negative and Gram-positive bacteria.38 Since macrolides depict a variety of biological activities, actinoallolides are first naturally occurring macrolides possessing anti-trypanosomal properties. In 2016, Ohista39 et al., reported the construction of the key intermediate (+)-60 towards the convergent total synthesis of 61. The synthesis commenced from the synthesis of right part (–)-54 of the compound (+)-60. For this purpose, the compound 49 was transformed into asymmetric alcohol 50 (78%) via protection of hydroxyl group in the presence of MsCl, DMAP and pyridine followed by its treatment with cesium acetate under the presence of 18-crown-6, in conjugation with DIBAL-H mediated reduction. Next, alcohol 50 was subjected to acetalization followed by ozonolysis to furnish aldehyde compound 51 (79%) which was further reacted under the Corey–Fuchs reaction conditions to furnish alkyne 52. Next, PMP acetal ring opening furnished ketone 53 in 4 steps. The hydrozirconation of compound 53 followed by TIPS protection of hydroxyl group finally furnished first precursor (−)-54 (97%) for the synthesis of intermediate (+)-60. Next, the other coupling partners i.e., E-vinyl stannane (+)-56 and alkyne compound (+)-58 were also synthesized from commercially available mono-methyl hydroquinone 55 and methyl-3-hydroxy methyl propanoate 57 respectively over a few steps. Next, the construction of main carbon skeleton of the key intermediate (+)-60 was obtained from above synthesized precursors i.e., (−)-54 and (+)-58. As cross coupling reactions facilitate the robust synthesis of C–C and C–heteroatom bonds, accelerating the synthesis of coupled products.40,41 For this purpose, compound (+)-58 was subjected to Negishi cross coupling with (–)-54 to generate coupling product 59 (59%). In the next step, the compound 59 underwent hydrozirconation followed by Stille coupling with (+)-56 to finally furnish (+)-60 in 28% yield (Scheme 6).
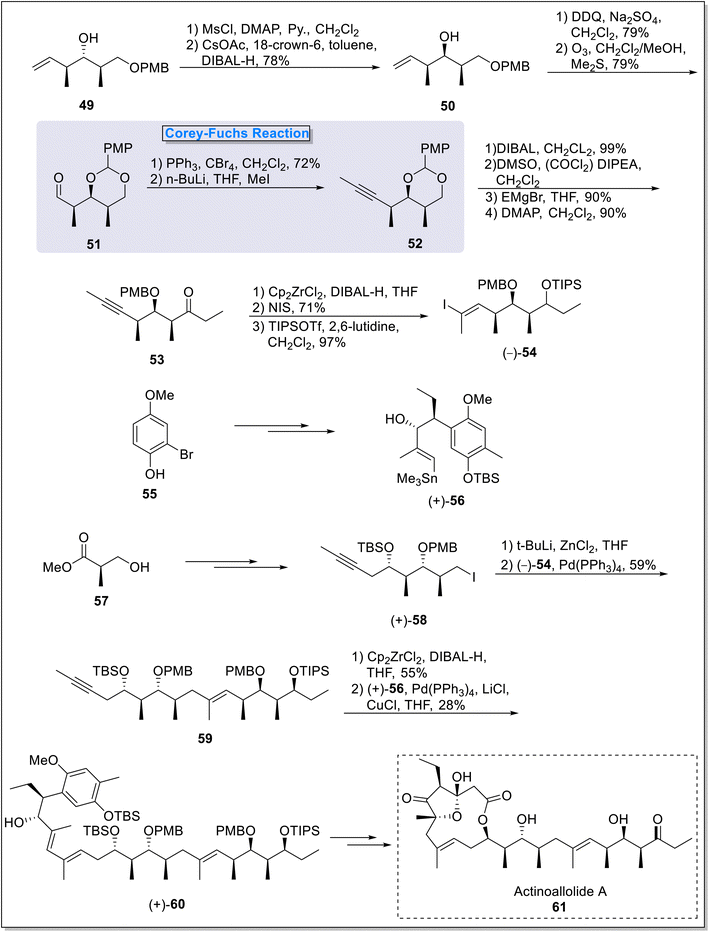 |
| | Scheme 6 Total synthesis of key intermediate of the actinoallolide A (+)-60 by Ohista and coworkers. | |
2.1.5. Orevactaene and epipyrone synthesis. Orevactaene 74 and epipyrone A 76 are significant naturally occurring scaffolds, comprised of 4-hydroxy-2-pyrone core structure.42 Both compounds were extracted from Epicoccum purpurascens strains and featured with a number of biological actives properties such as orevactaene depicts HIV-1 inhibitory and telomerase inhibitory properties.42 Whereas epipyrone A showcase potential against the influenza A virus and also act as a strong fungicide.43 Owing to the unique structural and biological properties, Preindl44 et al., in 2017 devised total synthesis of these intriguing moieties via a number of key steps such as Corey–Fuchs reaction, two alkyne cycloisomerization reaction and Stille coupling. The total synthesis began with the generation of first precursor 63 from commercially available D-arabinose 62 over a few steps. In the next step, alcohol 64 was transformed into propargyl alcohol 65 (51% over 3 steps) via subsequent oxidation and Corey–Fuchs reaction in conjugation with the treatment of resulting alkyne with paraformaldehyde. Compound 65 was then reacted with ethyl trifluoropyruvate 66 followed by hydroboration in the presence of the oxyboran succeeded by subsequent oxidation in the presence of trimethylamine N-oxide and Suzuki-type cross-coupling with 1-iodopropyne under the conditions of Pd catalysis furnished compound 67 (57% over 3 steps). Further, compound 67 was reacted with PhMe2SiLi to form silyl compound 68 (90%) which was converted to compound 69 over 4 steps yielding 89%. In the next step, commercially available epichlorohydrin 70 was subjected to alkylation under the presence of sodium acetylide in basic conditions followed by stannylcupration in conjugation with oxidation to yield 71. Next, Wittig olefination, reduction, oxidation and bora-Wittig reaction subsequently delivered heterodimetalated tetraene 73 (84%). Next, compound 73 was subjected to Stille coupling with compound 69 in the presence of Pd catalysis followed by Suzuki coupling reaction with compound 63 that resulted in formation of core structure of orevactaene. Next, deprotection using TSAF ultimately furnished desired 74. Apipyrone A 76 was also synthesized in the same manner from galactopyranosyl fluoride 75 over a few steps (Scheme 7).
 |
| | Scheme 7 Total synthesis of putative orevactaene 74 & epipyron A 76 by Preindl and coworkers. | |
2.1.6. (−)-Dactylolide synthesis. (−)-Dactylolide 85 is a naturally occurring compound fits into the class of cytotoxic macrolides, derived from Dactylospongia sp. of Vanuatu sponge.45 In 2018, Tanaka46 et al., published the convergent total synthesis of (−)-dectylolide 85, centered around the inter- and intramolecular allylation strategies. The synthesis commenced with the generation of key reactant 80 from known alcohol 77 via protection in the presence of MOMCl/iPr2NEt/DMAP succeeded by the DIBAL-H mediated reduction. Next, treated it with CBr4 and PPh3 to furnish dibromoalkene 78 via Corey–Fuchs reaction in conjugation with its reaction in the presence of n-BuLi and then ethyl chloroformate to generate ester compound 79 (88%). Precursor 80 was afforded from compound 79 over a few steps. Next, the other reacting partner 82 was also generated over a few steps from a known alcohol 81. Both the reacting partners 80 & 82 in hand, the total synthesis of (−)-dactylolide 85 was performed under various reaction conditions. First, both the reactants i.e., 80 & 82 were reacted via Mukaiyama aldol condensation in the presence of CMPI followed by its reduction with DIBAL-H in conjugation with treatment with (CH2ClCO)2O, pyridine and DMAP to afford α-acetoxy ether 83. Next, compound 83 was subjected to react with ZnBr2.OEt2 and MS5A succeeded by its reaction with TMSBr/TBAI in conjugation with MnO2 via oxidation (73% over 2 steps) and then Pinnick oxidation to access seco-acid 84 (99%). The compound 84 was subjected to react with MNBA and DMAP succeeded by its deprotection using TBAF in conjugation with Dess–Martin oxidation to synthesize (−)-dactylolide 85 in 88% yield (Scheme 8).
 |
| | Scheme 8 Total synthesis of (−)-dactylolide 85 by Tanaka and coworkers. | |
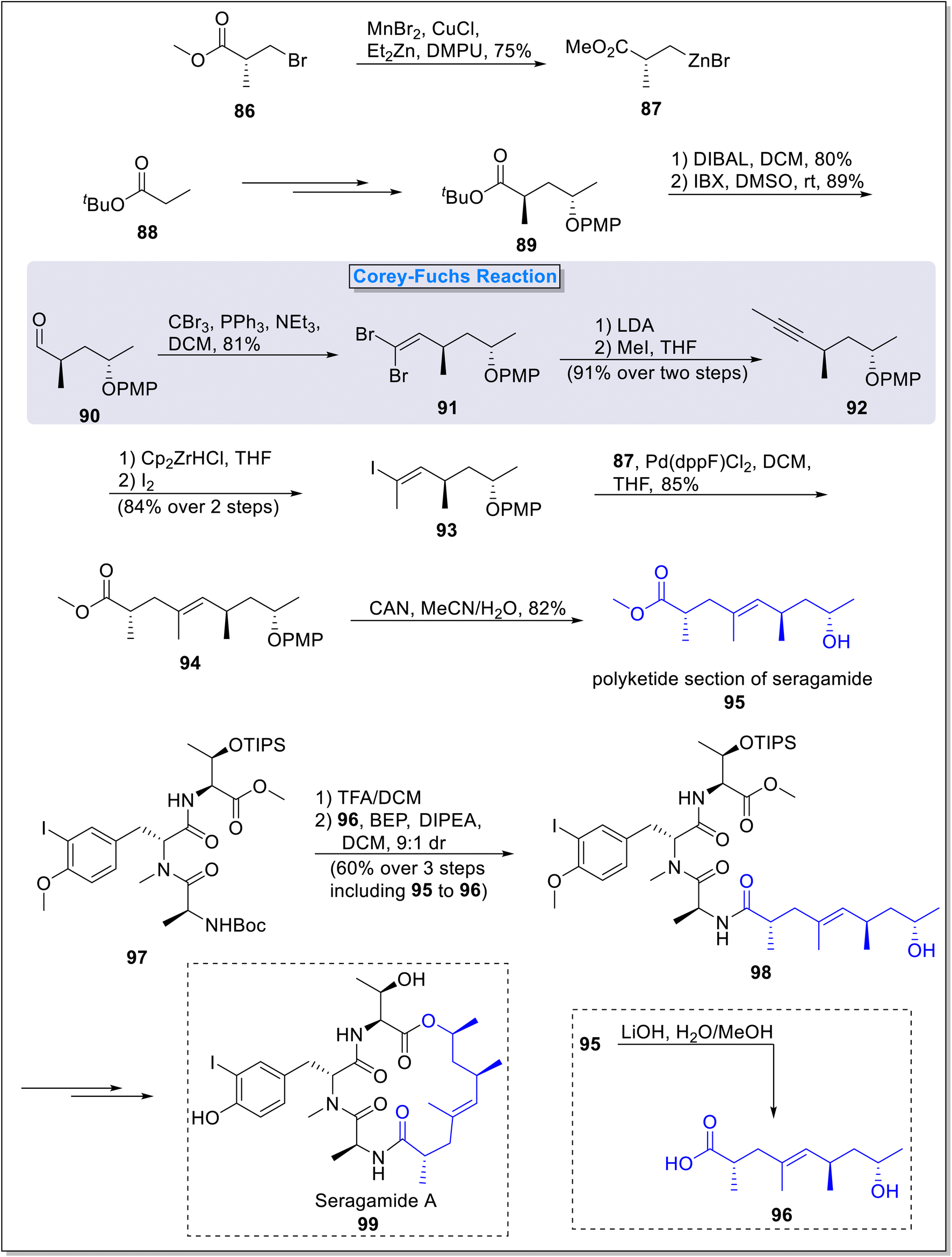
2.1.7. Synthesis of polyketide fragment of seragamide. Seragamides are naturally occurring compounds featured with excellent cytotoxic properties.47 These compounds were isolated from marine sponge Suberites japonicus.48 Lang and Lindel49 in 2019, described the total synthesis of the polyketide fragment of the Seragamide employing Corey–Fuchs reaction as a main step. The synthesis involved nine overall steps with 21% yield. The total synthesis commenced from the preparation of organozinc homoenolate 87 which was utilized as Negishi coupling partner, from the treatment of β-bromopropionic acid 86 in the presence of manganese bromide, copper chloride and di ethyl zinc. Next, tert-butyl ester 89 was synthesized from tert-butyl propionate 88 over a few steps. Compound 89 was then reduced in the presence of DIBAL followed by DMAP-catalyzed oxidation to obtain aldehyde 90. In the next step, aldehyde 90 was subjected to Corey–Fuchs reaction under the presence of carbon tetrabromide and triphenyl phosphine to form dibromoalkene 91 succeeded by its reaction with LDA and then methyl iodide to form internal alkyne 92 in 91% yield (over 2 steps). Next, compound 92 underwent subsequent hydrozirconation and iodination under the presence of Schwartz reagent and iodine to form (E)-olefin 93 in 84% yield (over 2 steps) with exclusive regio- and stereoselectivity. Compound 93 underwent Negishi coupling with homoenolate 87 under the presence of palladium catalysis in DCM to furnish protected nonenoic acid 94 in 85% yield. In the final step, PMP deprotection of 94 in the presence of CAN afforded fragment 95 in 82% yield. Next, tripeptide 97 was subjected to BEP-catalyzed reaction with compound 96 (synthesized from polyketide section 95 via saponification), to form peptide-polyketide open chain compound 98 in 60% yield (over 2 steps) with 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of diastereoselectivity. This open chain peptide-polyketide compound 98 was further transformed into natural product seragamide A 99 over few steps (Scheme 9).
:1 ratio of diastereoselectivity. This open chain peptide-polyketide compound 98 was further transformed into natural product seragamide A 99 over few steps (Scheme 9).
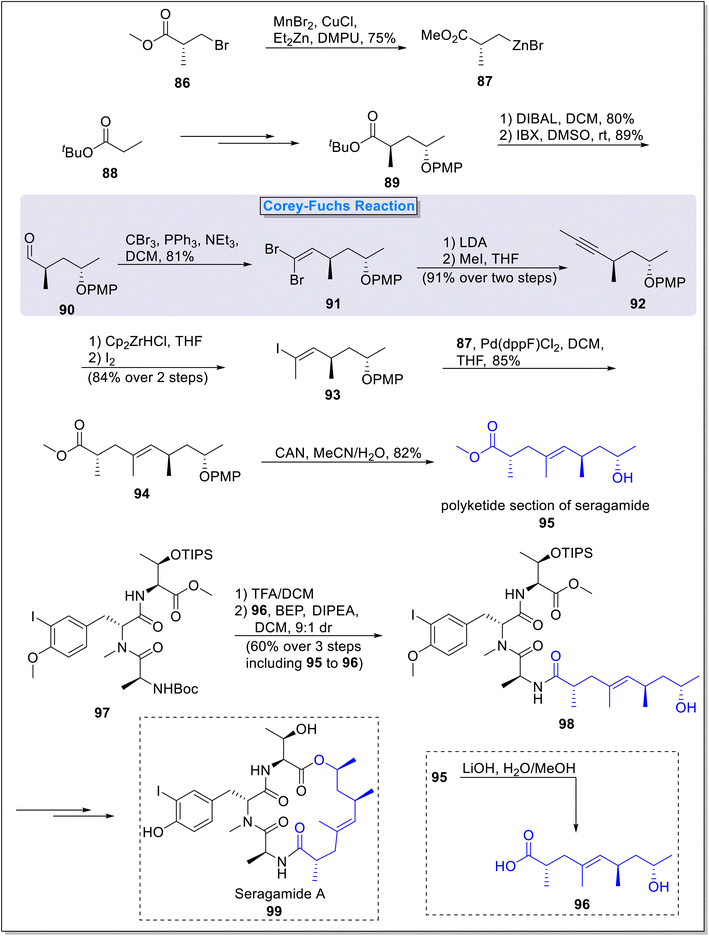 |
| | Scheme 9 Total synthesis of polyketide fragment of the natural product seragamide A 99 by Lang and Lindel. | |
2.1.8. (−)-Enigmazole A and (−)-15-O-methylenigmazole synthesis. (−)-Enigmazole A 110 and its natural chemical analog (−)-15-O-methylenigmazole 114 fits into the family of phosphomacrolides derived from marine sponge Cinachyrella enigmatica of New Guinean Papua.50 These naturally occurring compounds are featured by 18-membered macro-lactone possessing biological active properties. (−)-Enigmazole A 110 exhibit cytotoxic activity against the NCl 60 cell line, with a GI50 value of 1.7 μM.51 In 2020, Sakurai52 et al., reported the total synthesis of (−)-enigmazole A 110 and (−)-15-O-methylenigmazole 114 by employing a number of key reaction procedures and Corey–Fuchs alkyne synthesis reaction is one of them. The total synthesis commenced with protection of homoallylic alcohol 100 to generate TBS protected ether 101 (97%), which on oxidative cleavage, furnished aldehyde compound 102. The aldehyde 102 underwent two steps Corey–Fuchs reaction to access alkyne 104 in 94% yield in the presence of standard Corey–Fuchs conditions. Next, alkyne 104 was deprotected in the presence of n-BuLi followed by acetylide trapping with acetaldehyde succeeded by benzoyl chloride/pyridine mediated acylation and then TBAF catalyzed deprotection generated compound 105 (92% over 2 steps). In the next step, compound 105 was made to react with the synthesized carboxylic acid 106 in the presence of 3,4,6-Cl3C6H2COCl, Et3N and DMAP (Yamaguchi conditions) to furnish ester compound 107. Finally, (−)-enigmazole A 108 was synthesized from ester 107 over a few steps. The total synthesis of (−)-15-O-methylenigmazole 109 was also accessible from ester compound 107, which was transformed into natural product 109 over a few synthetic steps (Scheme 10).
 |
| | Scheme 10 Total synthesis of (−)-enigmazole A 108 & (−)-15-O-methylenigmazole 109 by Sakurai and coworkers. | |
2.1.9. Metacridamide B synthesis. Metacridamide B 120 fits into the class of macrolides, extracted for the first time from conidia of the entomopathogenic fungus M. acridum By Krasnoff et al., in 2012.53 It is a 17 membered macrocyclic secondary metabolite featuring biological active property i.e., anti-cytotoxicity with IC50 of 18.2 mM against HepG2/C3A.53 Due to its unique structural architecture with remarkable biological potential, researchers explore various synthetic pathways towards its total synthesis.54 In this regard, Sharma and coworkers55 in 2021 documented the total synthesis of metacridamide B in overall 27 steps. The key methodological steps involved Yamaguchi macrolactonization, Corey–Fuchs alkyne synthesis, Ru-catalyzed alkyne functionalization, Stille coupling and Micalizio coupling. The total synthesis was proceeded with the synthesis of a key intermediate 111. The alkyne moiety 111 (79%) was accomplished from primary alcohol 110 in two reaction steps including DMP oxidation and Corey–Fuchs alkyne synthesis reaction in the presence of TPP, CBr4 in conjugation with the use of n-BuLi. The synthesis of the second reactant aldehyde compound 114 was accomplished over few steps starting from an alkyne 112 and aldehyde compound 113. Next, both reacting entities in hands, coupling of 111 & 114 took place to yield enantiopure secondary alcohol 115 (80%) in three steps involving reaction with n-BuLi, DMP oxidation and asymmetric reduction in the presence of (S)-CBS catalysis. Secondary alcohol 115 was subjected to regioselective functionalization under the presence of Bu3SnH and ruthenium catalyst followed by its treatment with NIS in conjugation with Stille coupling in the presence of CuTC, Pd[PPh3]4 and [Ph2PO2][NBu4] to afford compound 116 in 84% yield. Further, protection of free hydroxyl with TBS group took place followed by selective deprotection in the presence of CSA (catalyst). Next, DMP oxidation was followed by Pinnick oxidation to furnish intermediate 117 in 77% yield over 2 steps. Intermediate 117 was further treated with L-phenylalanine methyl ester 118 under the presence of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) followed by PMB deprotection with DDQ to form alcohol compound 119 in 94% yield. In the final step, compound 119 was subjected to ester hydrolysis in the presence of Me3SnOH succeeded by Yamaguchi macrolactonization to generate TBS protected metacridamide B which on HF-pyridine catalyzed silyl deprotection gave metacridamide B 120 in 50% yield over 2 steps (Scheme 11).
 |
| | Scheme 11 Total synthesis of metacridamide B 120 by Sharma and coworkers. | |
2.1.10. Veramycin A, NFAT-133 & TM-123 synthesis. Veramycins along with their chemical analogues NFAT-133 and TM-123 are well known polyketides, were derived from culture broth of Streptomyces sp. from Sanofi microbial strain. These natural products showcased an enhanced baseline deoxy glucose uptake in a tailored L6 rat skeletal muscle cell line.56 In 2022, Dardić57 et al., reported a detailed total synthesis of α-pyrone featured veramycin A 135 and its congeners NFAT-133 126 and TM-123 132. The synthesis was commenced with the formation of Weinreb amide intermediate 123 (95%) from (S)-aldehyde 121 in two steps involving the aldol addition with benzyl-3-propionyloxazolidinone 122 in the presence of Bu2BOTf and NEt3 followed by oxazolidinone cleavage. Next, compound 123 was coupled with pinacol boronate 124 via Suzuki coupling in the presence of XPhos Pd G4 (as catalyst) to afford 125 in 96% yield. Compound 125 was efficiently converted to NFAT-133 126 in two steps entailing the formation of methyl ketone in the presence of Grignard reagent in conjugation with deprotection of TBS group in 25% overall yield. Next, to accomplish the synthesis of veramycin A 135 and TM-123 132, the intermediate 125 was converted into aldehyde 127 (97% yield) via TBS protection of hydroxyl group, succeeded by reduction in the presence of DIBAL-H. The prepared aldehyde 127 underwent two step Corey–Fuchs reaction in the presence of CBr4, PPh3, and NEt3 to yield dibromoalkene 128 followed by its treatment with butyl lithium and methyl chloroformate to furnish propiolic acid methyl ester 129 in 76% yield. Next, the ester 129 was reacted with t-butyl ester 88 & TMSE protected ester 132 to furnish cyclized intermediates 130 (84% yield) and 133 (94% yield) correspondingly under the Claisen reaction conditions. These cyclized precursors were then transformed into their corresponding pyrones i.e., veramycin A 134 and TM-123 131 in the presence of (SPhos)AuNTf2 (gold catalyst) and MeNO2/AcOH followed by deprotection of alcohols (in 16% and 12% overall yield respectively) (Scheme 12).
 |
| | Scheme 12 Total synthesis of veramycin A 134, NFAT-133 126 & TM-123 131 by Dardić and coworkers. | |
2.1.11. (±)-Parvistilbine B & (±)-stemenone B synthesis. (±)-Parvistilbine B 149 and (±)-stemenone B 148 are naturally occurring compounds that fit into the class of polyketides and extracted from the Stemona parviflora and Stemona tuberosa respectively, exhibiting profound historical roots in Chinese phytomedicines.58 Parvistilbine B was extracted from the same room which exhibit excellent nematocidal activity while (±)-stemenone B has anti-inflammatory potential with IC50 value of 2.97 μM.59 In 2022, Kozlowski60 et al., first proposed a new dearomative oxidation strategy for the construction of para-quinol moiety which is the core structure of both abovementioned natural compounds, then performed the total synthesis of these compounds. The main steps involving the total synthesis were Corey–Fuchs alkyne synthesis and Sonogashira-hydrogenation. The total synthesis began with the synthesis of alkyne precursors 137 and 140 from commercially available o-anisaldehyde 135 and p-hydroxy-2-anisaldehyde 136 respectively via Corey–Fuchs reaction. Next, the second coupling partner 144 was synthesized from dimethyl resorcinol 141 which was subjected to debromination to synthesize 142 (99%) succeeded by subsequent methylation and debromination to achieve 143 in 99% yield. Next, the compound 143 was subjected to Li–halogen exchange reaction and selective deprotection to furnish compound 144 with 55% yield. All the coupling partners 137, 140 & 144 in hand, Sonogashira coupling of 144 with 137 and 140 led to the formation of compounds 145 (51%) and 146 (45%) respectively, followed by hydrogenation–debenzylation and photo-catalytic dearomatization strategy to construct (±)-stemenone B 147 (86% over 2 steps) and (±)-parvistilbine B 148 (90% over 2 steps) respectively (Scheme 13).
 |
| | Scheme 13 Total synthesis of (±)-parvistilbine B 148 & (±)-stemenone B 147 by Kozlowski and coworkers. | |
2.1.12. Aigialomycin D synthesis. Aigialomycin D 160 is a fungal polyketide, derived from the marine mangrove fungus Aigialus parvus BCC 5311. This is a 14 membered resorcyclic acid lactone, featuring a number of bioactive properties including anti-malarial activity (IC50: 6.6 μg mL−1 for P. falciparum) and cytotoxicity in human cell lines (IC50: 3.0 & 18 μg mL−1 in KB and BC-1 cells respectively).61 Total synthesis of aigialomycin D was disclosed for the first time in 2004 by Danishefsky and coworkers in 18 steps and with 8% overall yield.62 In continuation of exploring the new ways towards the total synthesis of bioactive natural products, Sudhakar63 et al., in 2024 published the total synthesis of aigialomycin D 160. The key reactions in synthetic approach entailed Corey–Fuchs alkyne synthesis, Yamaguchi esterification and ring closing metathesis. The synthesis was initiated with epoxide 149, which was subjected to ring opening reaction with homoallyl magnesium bromides under the conditions of dry ether to generate an alcohol.64 The synthesized alcohol on reaction with BnBr and NaH furnished benzyl ether 150 in 86% yield. Next, benzyl ether 150 was transformed into diol under the acidic medium (70% aq. acetic acid) which on reaction with BzCl and Et3N got converted to monobezoate 151 in 81% yield. On treatment with TBSCl and imidazole, a TBS protected product was furnished which subjected debenzoylation in the presence of K2CO3 followed by Swern oxidation to form respective aldehyde 152. Aldehyde 152 underwent Corey–Fuchs reaction in the presence of CBr4 and PPh3 to access corresponding alkyne 153 in 77% yield. On treatment with n-BuLi, resulting acetylenic anion reacted with (S)-propylene oxide followed by its reaction with red-Al to give intermediate 154 in 74% yield. In the next step, the synthesis of another key precursor 157 took place from 2,4-dimethoxybenzoic acid 155, which was converted into aldehyde compound 156 in 2 steps including the synthesis of amide in the presence of thionyl chloride and its conversion to aldehyde 156 in the presence of n-BuLi and DMF. Next, Wittig olefination of aldehyde 156 consequently led to the formation of styrene derivative which on hydrolysis gave 157 in 82% yield. The prepared intermediate 157 subjected to Yamaguchi reaction to form an anhydride which on further condensation with above synthesized intermediate 154 in the presence of DMAP delivered ester 159 in 72% yield. Finally, the ester 159 transformed into aigialomycin D 160 (74% & 2.67% overall yield) in two steps entailing the reaction of 159 with Grubb's 2nd generation catalyst in conjugation with deprotection step in the presence of AlI3 in benzene (Scheme 14).
 |
| | Scheme 14 Total synthesis of aigialomycin D 160 by Sudhakar and coworkers. | |
2.2. Synthesis of alkaloid-based natural products
2.2.1. Synthesis of C22–C40 fragment of azaspiracids. Alkaloids are a diverse class of naturally products, comprise of millions of biologically active compounds.65 Azaspiracids are naturally occurring compounds fits into the category of lipophilic algal toxins (originally alkaloids), extracted from the dinoflagellates A. spinosum and A. poporum.66 These azaspiracids are responsible for various biological responses in higher animals as its consumption by human through shellfish may cause diarrhetic shellfish poisoning.67 Having intricate structural features along with bioactive properties, synthesis of these compounds has gained much interest. In this regard, Zhang68 et al. in 2016 demonstrated the total synthesis of C22–C40 region 173 of the azaspiracids utilizing Corey–Fuchs reaction, Nozaki–Hiyama–Kishi coupling and intramolecular-hetero-Michael addition reaction as main steps. The total synthesis commenced from the synthesis of TMS alkyne fragment 166 involving the anti-aldol reaction of (S)-lactate ketone 161 with easily available aldehyde 162 in the presence of c-Hex2BCl to access β-hydroxy ketone 163. Next, compound 163 was first protected and then reduction of keto-ester took place with the assistance of lithium borohydride to furnish 164 (78%) followed by sodium periodate mediated oxidative cleavage to furnish aldehyde compound 165. Next, the synthesis of TMS protected alkyne 166 (81%) was took place utilizing the Corey–Fuchs reaction followed by the silyl protection. Next, deprotection of the TBS group led the synthesis of compound 167 (90%), followed by its conversion to epimeric acetal 168 in the presence of DDQ in DCM. Epimeric acetal 168 underwent DIBAL-H mediated reduction followed by the Parikh–Doering oxidation to achieve compound 169 in 95% yield. Next, compound 170 was treated in the presence of NIS and AgOTf in DMF to prepare iodo-alkyne followed by Nozaki–Hiyama–Kishi coupling with 169 to furnish propargylic alcohol 171 in 95% yield. Next, acyclic intermediate 172 was generated over a few steps. The acyclic intermediate 172 underwent ring closing reaction over a few steps to finally generate C22–C40 fragment 173 of the naturally occurring azaspiracids (Scheme 15).
 |
| | Scheme 15 Total synthesis of C22–C40 fragment 173 of azaspiracids by Zhang and coworkers. | |
2.2.2. Tetradecapentaenoic acid synthesis. Tetradecapentaenoic acids, a class of poly-unsaturated fatty acid amides present in many tropical plant species such as Z. bungeanum, Z. piperitum and Z. ailanthoi.69 Until now, more than 50 compounds of poly-unsaturated fatty acids have been extracted from Zanthoxylum plant species displaying remarkable bioactivities. Amides of these fatty acids depict anti-helmintic, anti-oxidant, apoptotic and anti-proliferative potential. Tetradecapentaenoic acid amide derivative 178 also termed as sanshool derivative, extracted from Zanthoxylum bungeanum.70 Sanshool derivatives exhibit promising biological active properties as they act as power activator for cell apoptosis and also depict anti-cancer properties71 as cancer continues to pose as a causative agent for death worldwide.72 In 2019, Kolodiazhna & Kolodiazhnyi73 demonstrated the total synthesis of sanshool derivative 178 via employing Corey–Fuchs reaction as the key step. The total synthesis commenced with the reaction of known unsaturated aldehyde 174 in the presence of dibromomethylene phosphorane (generated via reaction of CBr4 and PPh3) to furnish dibromoalkene 175. Next, dibromoalkene 175 was treated with n-BuLi to form terminal alkyne 176 followed by its reaction with oxalyl dichloride in conjugation with triphenyl phosphine to form unsaturated ester compound 177. In the final step, desired tetradecapentaenoic acid amide 178 was obtained, upon treatment of ester compound 177 with n-BuLi (Scheme 16).
 |
| | Scheme 16 Total synthesis of tetradecapentaenoic acid amide 178 by Kolodiazhna & Kolodiazhnyi. | |
Same group of scientists74 also represented the stereoselective synthesis of tetradecapentaenoic acid derivatives i.e., 178 & 186. The key steps in the synthesis involved Wittig reaction, Ramirez–Corey–Fuchs reaction and Trost–Kazmaier rearrangement. The synthesis began with the synthesis of sorbaldehyde 180 from hexadienol 179 via Parikh–Doering oxidation in the presence of pyridine sulfur trioxide complex. Next step involved the synthesis of second reactant i.e., phosphonium salt 182 which was generated from aldehyde compound 181 in two steps. The above synthesized compound 180 underwent Wittig olefination reaction with phosphonium compound followed by lithium aluminum hydride mediated reduction to generate alcohol 184. Next, Parikh–Doering oxidation of alcohol 184 generated aldehyde compound 174 which was subjected to two steps Corey–Fuchs reaction in the presence of CBr4, PPh3 to form dibromoalkene 175 and then n-BuLi in conjugation with methyl chloroformate to furnish alkyne compound 185. Next, amides 178 and 186 were synthesized from compound 185 over a few steps (Scheme 17).
 |
| | Scheme 17 Total synthesis of tetradecapentaenoic acid derivatives 178 & 186 by Kolodiazhna & Kolodiazhnyi. | |
2.2.3. (−)-Chelonin A synthesis. (−)-Chelonin A 196 having 2,6-disubstituted morpholine core structure, was first extracted from the marine sponge Chelonaplysilla sp. in 1991.75 This compound showcased good anti-microbial potential against B. subtilis and also depicted 60% inhibition of phorbol myristate acetate triggered inflammation in the host animals.75 Hitherto, four total syntheses of chelonin A and an asymmetric synthesis of (+)-chelonin A (unnatural enantiomer) have been reported to date.76–79 Thus, Gunawana80 et al., in 2023 described the asymmetric total synthesis of (−)-chelonin A by employing various reaction processes i.e., Corey–Fuchs, 1,3-addition of rhodium carbenoid, and annulation reaction. The first step for the total synthesis of (−)-chelonin included the synthesis of two starting substrates i.e., bromohydrin (−)-188 and triazole precursor 194. In this regard, α-bromoketone 187 was converted to enantioenriched bromohydrin (−)-188 in the presence of Noyori catalyst 189 and HCO2H/HCO2K to furnish target product (−)-188 in 87% yield with exceptional enantiomeric excess (>80% ee). In the next step, indole-3-carbaldehyde 190 was subjected to N-tosylation, followed by Corey–Fuchs reaction to synthesize terminal alkyne 193 in 62% yield. For this purpose, the protected aldehyde 191 (synthesized via N-tosylation) was treated under the conditions of CBr4 and PPh3 to generate gem-dibromo alkene 193 followed by its reaction with n-BuLi to form alkyne 193. In the next step, alkyne 193 was then transformed into triazole 195 (75%) when treated with TsN3 under the catalytic conditions of Cu thiophene carboxylate (CuTc). The synthesized substrates i.e., triazole 194 and (−)-bromohydrin 188 were both subjected to the Rh2OcT4 catalyzed reaction followed by cyclization reaction in the presence of CH3CN & Cs2CO3 succeeded by its treatment with a mixture of trifluoroacetic acid and triethylsilane to generate the diastereoselective syn-compound 195 in 77% yield. In the last step, deprotection of protected morpholine 195 took place in the presence of Na naphthalenide to finally deliver enantioselective (−)-chelonin A 196 in 52% yield (Scheme 18).
 |
| | Scheme 18 Total synthesis of (−)-chelonin A 196 by Gunawana and coworkers. | |
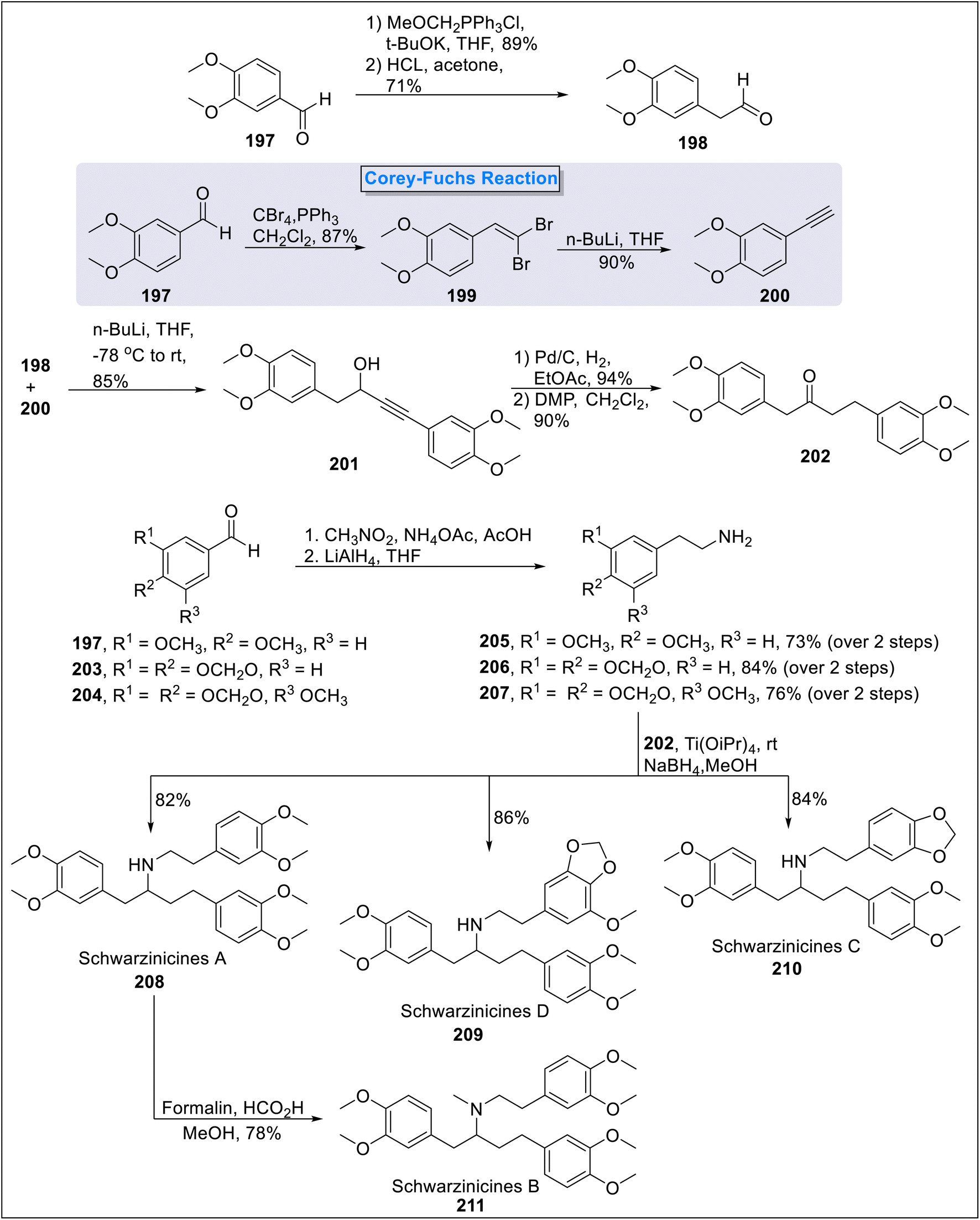
2.2.4. Schwarzinicines A–D synthesis. Schwarzinicines A–D, chemically referred as 1,4-diarylbutanoidphenethylamine derivatives are naturally occurring alkaloids, isolated from Ficus schwarzii.81 These natural products are attributed to vasorelaxant, anti-diarrheal, anti-ulcer and anti-inflammatory properties.82 Annapurna83 et al., in 2024 accomplished the total synthesis of these Schwarzinicines A–D compounds by employing Corey–Fuchs, Henry and Eschweiler–Clarke reactions. The synthesis began with the formation of 2-arylacetaldehyde 198 from veratraldehyde 197 in two steps via Wittig reaction and ether cleavage protocol in the presence of MeOCH2PPh3Cl, tBuOK and HCl. Alongside, phenyl acetylene 200 (90%) was synthesized via Corey–Fuchs reaction from veratraldehyde 197 in two steps under the conditions of CBr4, PPh3, n-BuLi. Next, phenyl acetylene 200 and 2-arylacetaldehyde 198 underwent coupling reaction in the presence of n-BuLi to furnish alkynol 201 (85%) followed by the reduction of triple bond and subsequent oxidation in the presence of Dess–Martin periodinane to access diaryl ketone 202 in 90% yield. In the next step, 2-arylethanamines 205, 206, 207 were synthesized from veratraldehyde 197, piperonal 203 and 3-methoxy-4,5-methylenedioxybenzaldehyde 204 respectively in the presence of CH3NO2, NH4OAc, AcOH and LiAlH4 in two steps. The synthesized 2-arylethanamines i.e., 205, 206 & 207 were reacted with diaryl ketone 202 in the presence of titanium(IV)isopropoxide and NaBH4 to furnish schwarzinicines A 208 (82%), schwarzinicines C 210 (84%) and Schwarzinicines D 209 (86%) respectively. Finally, schwarzinicines A 208 was converted into schwarzinicines B 211 via Eschweiler–Clarke methylation providing 78% yield (Scheme 19).
 |
| | Scheme 19 Total synthesis of schwarzinicines A–D 208–211 by Annapurna and coworkers. | |
2.3. Synthesis of polyunsaturated lipid-based natural products
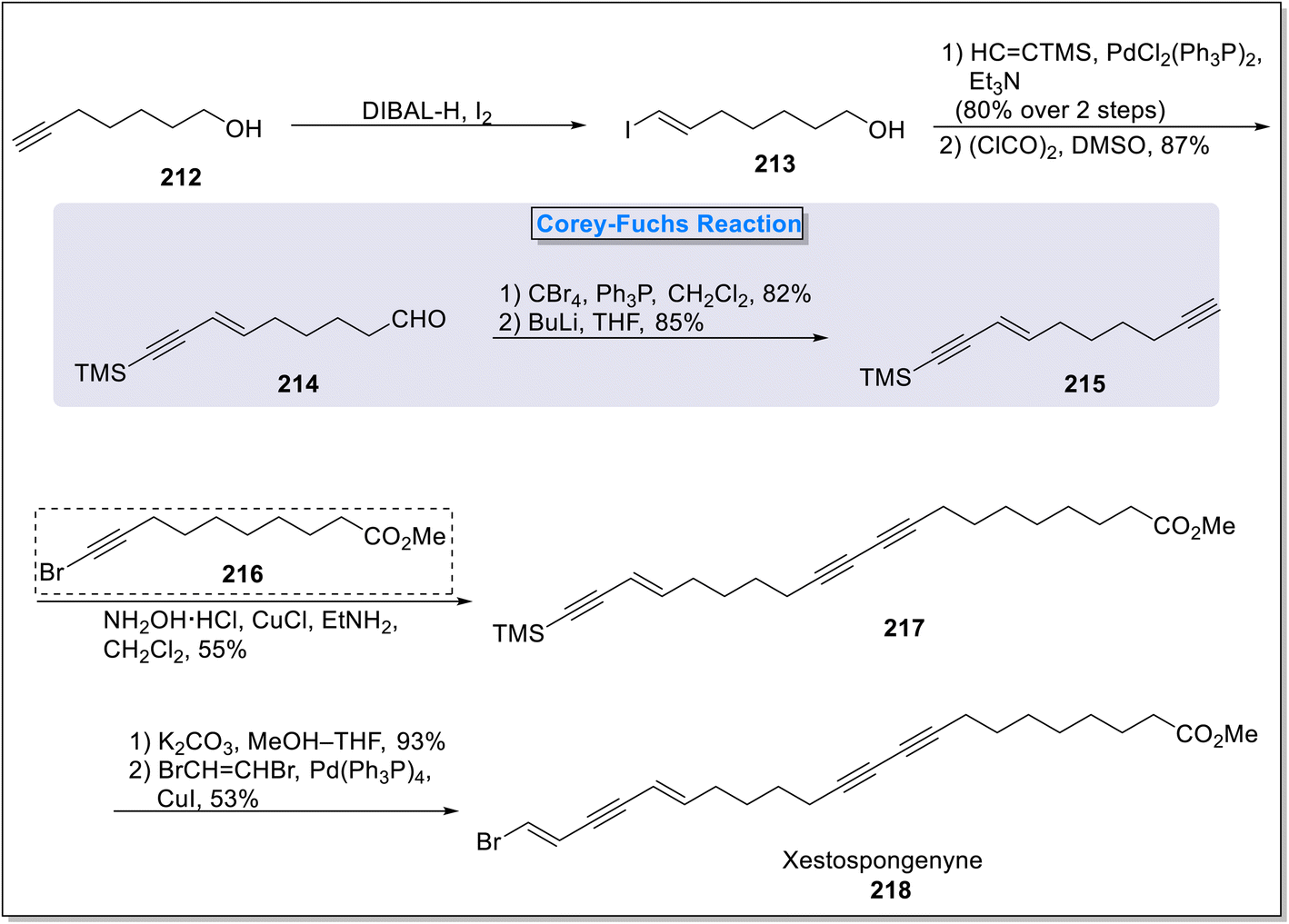
2.3.1. Xestospongenyne synthesis. Xestospongenyne is a marine brominated naturally occurring compound which belongs to the class of polyunsaturated lipids.84 It is considered as strong anti-obesity agent with exclusive pancreatic lipase inhibition potential with IC50 = 0.61 μM. Owing to the significant importance as potent pancreatic lipase inhibitor, the total synthesis of xestospongenyne attain much interest in organic synthesis.85 In the regard, Gong and coworkers86 in 2016, represented first total synthesis of xestospongenyne 218 in 13% overall yield. There were three key steps involved in the total synthesis of xestospongenyne 218 including Corey–Fuchs reaction, Cadiot–Choskiewicz cross-coupling and Sonogashira coupling. The total synthesis initiated with the synthesis of vinyl iodide 213 by the treatment of commercially available compound hept-6-yn-1-ol 212 with DIBAL-H in the presence of iodine. Further, the compound 213 was reacted in the presence of ethynyl trimethyl silane under the catalytic dichlorobis(triphenylphosphine)palladium in Et3N followed by Swern's oxidation to furnish enynal compound 214 (87%). In the next step, compound 214 was transformed into silylated enyne 215 (85%) via Corey–Fuchs reaction under the conditions of tetra-bromomethane and triphenylphosphine in conjugation with butyl lithium in THF. Next, the alkyne compound 215 was subjected to Cadiot–Choskiewicz cross-coupling with methyl 10-bromodec-9-ynoate 216 in the presence of hydroxylamine hydrochloride and copper chloride to furnish enyne ester 217 (55%). In the final step, subsequent potassium carbonate-mediated deprotection and Sonogashira coupling with dibromoethane under the presence of tetrakis(triphenylphosphine)palladium consequently gave xestospongenyne 218 in 53% yield (Scheme 20).
 |
| | Scheme 20 Total synthesis of xestospongenyne 218 by Gong and coworkers. | |
2.3.2. (5Z,9Z)-Eicosa-5,9-dienoic acid synthesis. (5Z,9Z)-Eicosa-5,9-dienoic acid 220 falls under the category of (5Z,9Z)-dienoic fatty acids which belongs to the class of non-methylene interrupted, very long chain fatty acids (VLCFA), mostly found in microorganisms, marine invertebrates and in some higher plants.87 As these fatty acids are amidst the primitive and simplest I and IIα inhibitors known so far and hence considered for the development of new antitumor agents.88,89 Owing to these properties, researchers are eager to develop new strategies towards the total synthesis of these structurally important natural products. In this prospective, Adrian and Stark90 in 2016, demonstrated the total synthesis of topoisomerase inhibitor, (5Z,9Z)-eicosa-5,9-dienoic acid 220, which is a potent human antitumor agent. The main steps towards the total synthesis involved Corey–Fuchs reaction, Lindlar reduction, TPAP-mediated oxidation of alcohol, and Arndt Eistert homologation. The synthesis commenced with precursor 17, which was converted to compound 23 (provided in Scheme 3). Further, Lindlar catalyst-mediated reduction of alkyne 23 resulted in (Z,Z)-alcohol 219 in 92% yield followed by TPAP/NMO·H2O catalyzed direct oxidation that led towards the efficient total synthesis of natural (5Z,9Z)-eicosa-5,9-dienoic acid 220. The straightforward total synthesis of compound 220 was achieved in 10 steps and with 20% overall yield (Scheme 21).
 |
| | Scheme 21 Total synthesis of (5Z,9Z)-eicosa-5,9-dienoic acid 220 by Adrian and Stark. | |
2.3.3. Resolvin D4 and its 17(R)-hydroxy-epimer synthesis. Resolvin D4 and its 17(R)-hydroxy-epimer are lipid based biosynthetic natural compounds derived from docosahexaenoic acid. These compounds have many biological active properties such as anti-inflammatory and pro resolution property which enhanced the activity of human macrophages.91 In this context, Nshimiyimana92 et al., in 2023 disclosed a convergent, stereo-controlled total synthesis of resolvin D4 232a and its epimer 17(R)-resolvin D4 232b. The synthesis was initiated by the generation of propargyl bromide 222 from D-erythrose 221 in multiple steps. The other key intermediates i.e., 229 & 230 was synthesized via a number of reaction protocols. In this regard, 1-butyne 223 was reacted with chiral glycidol 224 in the presence of boron trifluoride etherate followed by silylation in conjugation with mild deprotection to generate isomers of intermediate 225 (87–89%) in weak acidic medium. Next, Lindlar hydrogenation of 225 succeeded by Dess–Martin oxidation generated aldehyde 226 which on Wittig olefination with (triphenylphosphoranylidene)acetaldehyde 227 delivered elongated enol 228 (228a in 75% & 228b in 72% yield). This enol was converted to terminal alkyne 229 via two steps Corey–Fuchs alkyne synthesis reaction in the presence of CBr4 and PPh3 in CH2Cl2 in conjugation with dehydrohalogenation using LDA in tetrahydrofuran (THF). To generate second isomer i.e., 230, protection of 229 with less bulky silyl group was achieved which assisted in uniform desilylation in final step of the synthesis of AT-resolvin D4 232b. In the next step, the reactions between 222 & 229 and 222 & 230 were took place via copper-catalyzed coupling to form bis-acetylenic moieties followed by Lindlar catalyzed selective reduction to generate silyl ethers 231 & 233 respectively. Next, desilylation in the presence of TBAF succeeded by its reaction with diazomethane and alkaline hydrolysis in the presence of LiOH furnished resolvin D4 232a and its 17(R)-hydroxy-epimer 232b (Scheme 22).
 |
| | Scheme 22 Total synthesis of resolvin D4 232a and its 17(R)-hydroxy-epimer 232b by Nshimiyimana and coworkers. | |
2.4. Synthesis of polyacetylene-based natural products
2.4.1. (−)-Gummiferol synthesis. (−)-Gummiferol 242a is a naturally occurring polyacetylene compound extracted from Adenia gummifera by Wall et al., in 1995.93. (−)-Gummiferol 242a is characterized by excellent cytotoxic activities as it shows potential against P388 murine leukemia with ED50 values of 0.03 μg mL−1 and against U373 human glioma cell lines with the ED50 value 0.05 μg mL−1.94 Regarding the promising anti-cancer properties, the synthesis of different enantiomeric gummiferol is crucial in organic synthesis. In 2016, Sarabia and coworkers95 designed the total synthesis of natural and non-natural gummiferol utilizing the novel asymmetric approach of epoxide construction. The two epoxide rings, installed on natural product were formed via novel asymmetric sulfonium salts. The total synthesis commenced with the oxidation of commercially available alcohol 234 in the presence of PCC and sodium acetoacetate to form an aldehyde compound 235 followed by its reaction under the influence of sulfonium salt 236a in basic conditions to access epoxy amide 237 (54%). Compound 237 were efficiently converted to diepoxy compound 238 (95%) via Red-Al induced reduction followed by its reaction with novel sulfonium salt 236a. Subsequent reduction and Corey–Fuchs reaction delivered dibromo compound 239 in 40% yield. Next, the treatment of 239 with TBAF and coupling with dialkyne compound 240 in the presence of NH2OH·HCl and CuCl followed by acetylation that led to the synthesis of compound 241 (51%). Finally, compound 241 was subjected to silyl deprotection in the presence of HF·Py to furnish natural (−)-gummiferol 242a in overall 10% yield with 10 steps. Unnatural (+)-gummiferol 242b was also obtained via same synthetic protocol with the assistance of sulfonium salt 236b (Scheme 23).
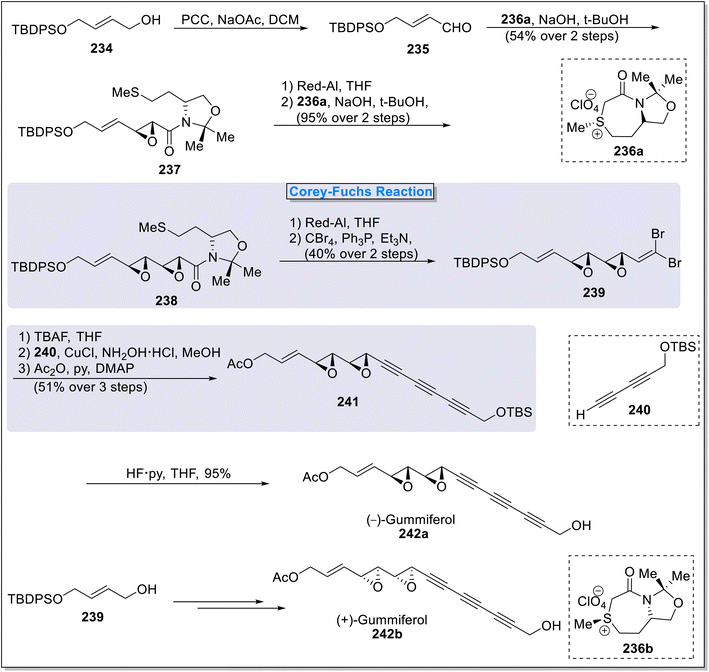 |
| | Scheme 23 Total synthesis of (−)-gummiferol 242a & (+)-gummiferol 242b by Sarabia and coworkers. | |
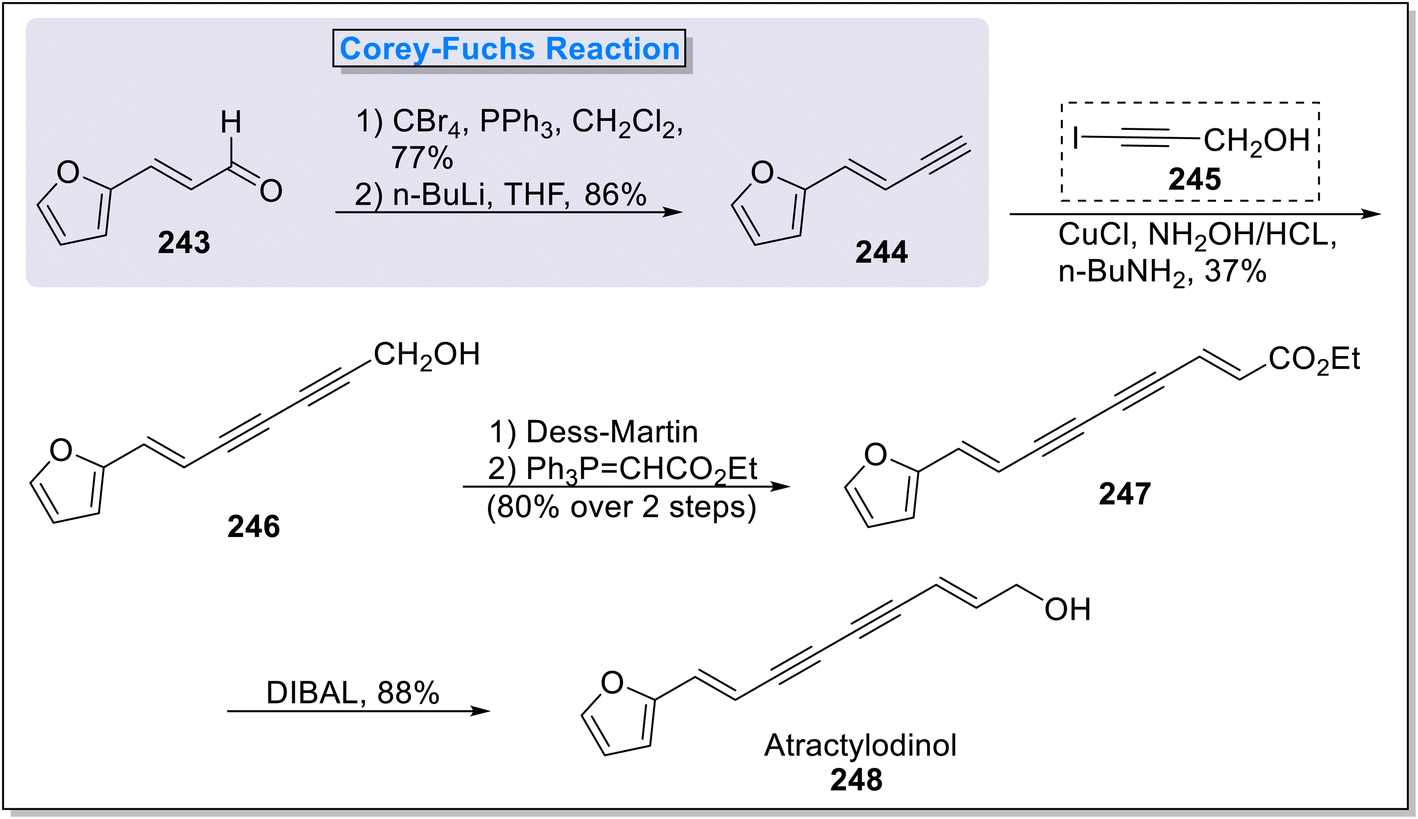
2.4.2. Atractylodinol synthesis. Atractylodinol 248 is an unsaturated natural product extracted from the rhizomes of Atractylodes lancea. A. lancea and known for its activity against porcine reproductive and respiratory syndrome virus (anti-PRRSV) potency with IC50 values of 7.9 μmol L−1.96 It was utilized as traditional Chinese and Japanese drug against various diseases including night blindness, influenza, rheumatic and digestive disorders.97 Owing to various biological activities, Kraus98 et al., in 2016, reported the total synthesis of atractylodinol 248 via utilizing a number of key synthetic procedure i.e., Corey–Fuchs alkyne synthesis, Dess–Martin oxidation of alcohol and Wittig reaction. The total synthesis began with the known two step Corey–Fuchs reaction in which 3-(2-furyl)acrolein 243 was transformed into terminal alkyne 244 (86% overall yield) under the presence of carbon tetrabromide and triphenylphosphine in DCM. Next, terminal alkyne 244 was coupled with 3-iodopropargyl alcohol 245 in the presence of copper chloride, sodium hydroxide in butyl amine to furnish compound 246, which was subjected to Dess–Martin oxidation in conjugation with Wittig reaction in the presence of phosphorane to form ester 247. In the final step, reduction using DIBAL led to the synthesis of final natural product atractylodinol 248 in 88% yield (Scheme 24).
 |
| | Scheme 24 Total synthesis of atractylodinol 248 by Kraus and coworkers. | |
2.4.3. Distaminolyne A synthesis. Distaminolyne A 258 is a first naturally occurring compound, featuring diacetylene 1-amino 2-alcohol core structure. It was extracted from the New Zealand ascidian Pseudodistoma opacum exhibiting anti-microbial activity against several bacterial species such as E. coli, S. aureus and M. tuberculosis.99 As M. tuberculosis remains a global health threat with unprecedented levels, spurring the need for effective inhibitors.100,101 Owing to potential biological property, Dumpala102 et al., in 2017 proposed the total synthesis of distaminolyne A 258 utilizing a number of key synthetic steps. These steps including Corey–Fuchs reaction, Cadiot–Chodkiewicz coupling and Witting olefination reaction. The total synthesis started with the formation of bromoalkynes analogue 251 in two steps involving the reaction of commercially available 4-pentyn-1-ol 249 under the presence of PMB-OH and amberlyst-15 to form PMB ether 250 (90%) followed by its conversion to bromoalkyne 251 (80%) in the presence of N-bromosuccinimide (NBS) and AgNO3 (as catalyst). Next, the second coupling partner 256 of Cadiot–Chodkiewicz reaction was synthesized. For this purpose, 9-dacene-1-ol 252 was transformed into compound 253 over few steps. Next, compound 253 was subjected to p-anisyl group deprotection in the presence of DDQ to furnish alcohol 254 in 89% yield followed by DMP catalyzed oxidation to form Corey–Fuchs reaction precursor. Subsequent reaction with CBr4 and triphenyl phosphine (TPP) yielded dibromide compound 255 (86%) succeeded by its treatment with n-BuLi in THF to afford alkyne 256 in 90% yield. Having the both coupling partners in hand, Cadiot–Chodkiewicz coupling of 251 & 256 led to the synthesis of diacetylene-1-amino-2-alcohol 257 (84%) under the influence of copper catalysis. Next, the desired natural product 258 was synthesized from compound 257 over few steps (Scheme 25).
 |
| | Scheme 25 Total synthesis of distaminolyne A 258 by Dumpala and coworkers. | |
2.4.4. Petrosiol B & D synthesis. Polyacetylenes are a large group of naturally occurring compounds featured with multitudinous double and triple bonds.84 Petrosiol B & D fits into the class of these polyacetylene natural products extracted from marine sponge named Petrosia strongylata. These types of compounds are emphasized by various medicinal properties including neuritogenic, anti-inflammatory, antiviral, antitumor and anti-microbial.103 Taking into account the significant potentials of these type of natural products Geng104 et al., in 2019 devised the total synthesis of petrosiol B 268 & D 269, reporting for the first time the total synthesis of petrosiol B 268 with 13 steps in 10% overall yield. The trajectory of the total synthesis involved Ohira–Bestmann homologation, Corey–Fuchs alkynes synthesis and Cadiot–Chodkiewicz reaction as key steps. The total synthesis commenced from unexpensive isopropylidene-α-D-mannofuranose 259 which was subjected to Ohira–Bestmann homologation in conjugation with its treatment with K2CO3 and MOM protection to furnish terminal alkyne 260 (93%). The synthesized alkyne 260 was then subjected to alkylation with bromide compound 261 under the influence of HMPA and butyl lithium followed by its hydrogenation in the presence of ethyl acetate and methanol to furnish compound 262 in 94% yield. Next, cis-olefin 263 was obtained from compound 263 over few steps. The regioselective cleavage of isopropylidene region of the compound 263 under the presence of periodic acid in conjugation with Corey–Fuchs reaction in assistance of CBr4 and PPh3 led to the formation of dibromoalkene 264 in 51% yield. The synthesized dibromoalkene 264 underwent sodium hydride catalyzed dehydrobromination to furnish mono-bromoalkyne compound 265. In the next step, compound 265 was subjected to Cadiot–Chodkiewicz coupling reaction with propargyl alcohol 266 in the presence of copper chloride, hydroxylamine, butylamine and diethyl ether to access diynes compound 267 in 68% yield. In the last step, the deprotection of terminal isopropylidene group of 267 in the presence of 3 N HCl led to the synthesis of natural product petrosiol B 268 in 97% yield. The other natural product i.e., petrosiol D 269 (98%) was also synthesized from the same precursor 259 over a few steps (Scheme 26).
 |
| | Scheme 26 Total synthesis of petrosiol B 268 & petrosiol D 269 by Geng and coworkers. | |
2.5. Synthesis of terpene-based natural products
2.5.1. Waihoensene synthesis. Waihoensene 279 is a naturally occurring tetracyclic diterpene, derived from New Zealand podocarp, Podocarpus totara var. waihoensis by Weavers et al., in 1997.105 Waihoensene has intriguing architectural features as it is decorated with four contiguous quaternary carbons and thus also recognized as structurally challenging compound for total synthesis. In 2017, Lee106 and coworkers reported the total of Waihoensene for the first time from commercially available reagents. The total synthesis commenced from a racemic keto ester 270 which was subjected to Lombardo–Takai olefination in the presence of Zn, TiCl4 and CH2I2 followed by lithium aluminum hydride (LAH) mediated reduction to furnish alcohol 271 (91%). Next, Swern's oxidation of compound 271 in conjugation with Horner Wadsworth–Emmons reaction yielded α,β-unsaturated ester 272 (93%). Selective reduction of compound 272 employing magnesium and then partial reduction using DIBAL generated aldehyde 273 in 97% yield. Aldehyde 273 was transformed into propargylic alcohol 274 (91%) utilizing Corey–Fuchs reaction conditions in conjugation with hydroxymethylation. Next, tetracyclic product 275 was accessed from compound 274 over a few steps. Compound 275 was further subjected to dihydroxylation in the presence of OsO4 succeeded by selective tosylation using TsCl to furnish compound 276 (39%). In the next step, the formation of enone 277 was achieved via elimination reaction in the presence of DBU followed by PDC mediated oxidation. Reaction of enone compound 277 with cyanocopper and boron trifluoride etherate in conjugation with LiHMDS and methyl iodide furnished methyl substituted compound 278 in 75% yield. In the final step, employing Petasis reagent (Cp2TiMe2) in toluene, natural product Waihoensene 279 was achieved in 32% yield (Scheme 27).
 |
| | Scheme 27 Total synthesis of Waihoensene 279 by Lee and coworkers. | |
2.5.2. Synthesis of key intermediate of shagene A. Shagene A 289 is a tricyclic sesquiterpenoid, extracted for the first time from soft corals of Scotia Sea by baker group in 2014.107 Shagene A showcase high toxicity against parasite Leishmania donovani (IC50 = 5 μM), which is a causative agent for serious visceral leishmaniasis.108 In 2024, Bai9 et al., demonstrated an efficient route leading towards the total synthesis of Shagene A 289 by employing ring closing cascade reaction of dienyne 286 constructed via Corey–Fuchs reaction, into 5/6 membered bicyclic compound. The ring closing metathesis was succeeded by the formation of diazoacetate 287 which was considered hypothetical key intermediate for the total synthesis of shagene A 289. The total synthesis was initiated with hydroxyl group protection of ester 280 in conjunction with hydrolysis followed by installation of chiral auxiliary 281 in the presence of DCC and DMAP to furnish amide 282 in 86% yield. Next, 282 was subjected to the subsequent aldol reaction by reacting it with acrolein followed by hydroxyl group protection with TBS and reductive cleavage of previously installed chiral auxiliaries in the presence of DIBAL-H to access compound 283 (in 86% yield). The conversion of aldehyde 283 to alkyne 285 took place by Corey–Fuchs reaction utilizing PPh3 and CBr4 and then n-BuLi and alkyl iodide over 2 steps reaction (resulting in 96% yield). The internal alkyne 285 was then subjected to selective deprotection of TBDPS followed by oxidation in the presence of DMP to form an aldehyde in 87% yield. Further, next step proceeded with the treatment of aldehyde with Grignard reagent via nucleophilic addition to generate two diastereomers 286a (53%) and 286b (31%). Both the isomers were transformed to the cyclic compound 287 over a few steps. The compound 286a was subjected to ring closing cascade reaction to deliver cyclic compound 287 (in 28% yield) catalyzed by Grubbs 2nd catalyst, 2-bromoacetic acid, DCC DMAP and N,N′-ditosylhydrazine (for the installation of diazo group) over three steps. Compound 286b was also transformed into compound 287 over several reaction steps. It was first reacted with Grubb's 2nd catalyst followed by its treatment with MnO2 and then sodium borohydride and CeCl3·7H2O. Next, its treatment with bromoacetic acid, DCC DMAP and N,N′-ditosylhydrazine led to the synthesis of 287 in 28%. The target compound 289 was assumed to achieve over few steps from compound 288. Several reaction conditions were applied for the efficient conversion of the intermediate 287 to 3-membered ring compound 288 via cyclopropanation reaction but the starting material was decomposed (Scheme 28).
 |
| | Scheme 28 Route leading towards the total synthesis of shagene A 289 by Bai and coworkers. | |
2.6. Synthesis of flavonoid-based natural products
2.6.1. Houttuynoid synthesis. Houttuynoid 301 is a naturally occurring flavonoid glycoside that belongs to the family of Sauruaceae and extracted from Chinese Houttuynia cordata by Yao et al., in 2012.109 It is considered as strong antiviral flavonoid having various viral inhibition properties such as inhibition of herpes simplex virus (with IC50 values of 57.7 μM).109 Owing to the importance of development of new antiviral agents, Kerl and coworkers110 in 2016 reported for the first the total synthesis of houttuynoid B 301 in overall 9 linear steps with 11% yield. The key steps involving the total synthesis include Corey–Fuchs alkyne synthesis reaction, Sonogashira coupling and Baker–Venkataraman rearrangement. The synthesis commenced from the unexpensive decanal 290 which was subjected to Corey–Fuchs reaction under the presence of carbon tetrabromide and triphenyl phosphine in conjugation with n-butyl lithium to furnish terminal alkyne 291 (70%). Next, alkyne 291 underwent Sonogashira coupling reaction with iodophenol 292 in the presence of Pd(PPh3)2Cl2, CuI and triethylamine to furnish 293 (51%) which was then rearranged to a benzofuran moiety. In the next step, the aldehydic functionality on 293 underwent Pinnick oxidation in the presence of NaH2PO4, NaClO2 and 2-methyl-2-butene2 294 to furnish carboxylic framework 295 in 71% yield. Further, to achieve HOBT activated ester 296 (87%), the carboxylic compound 295 was reacted with 1-hydroxybenzotriazole (HOBT) and ethyl-3-(3-dimethylaminopropyl)-carbodiimide in CH2Cl2. On the other side, the second precursor 298 was synthesized from commercially available chrysin 297 over a few steps. Having both the reaction partners i.e., 296 & 298 in hand, HOBT activated ester 296 was made to react with 298 in the presence of sodium hydride in THF to form arylester 299 (76%). Next, chromenone 300 (57%) was accessed from arylester 299 via Baker–Venkataraman rearrangement under the presence of K2CO3 and TBAB in toluene. Finally, the desired natural product houttuynoid B 301 was synthesized by deprotection of chromenone 300 under the presence of sodium methoxide in toluene with 99% yield (Scheme 29).
 |
| | Scheme 29 Total synthesis of houttuynoid B 301 by Kerl and coworkers. | |
2.7. Synthesis of polyphenol-based natural products
2.7.1. Nepetoidin B synthesis. Nepetoidin B 306 belongs to the class of phenolic compounds existing in two isomeric forms i.e., (Z, E)- and (E, E). It is extracted from P. frutescens,111 P. forsteri112 and S. miltiorrhiza113 and depicting remarkable bioactive properties including anti-viral, anti-fungal and anti-oxidant properties.114 It is also used for the production of nitric oxide and the inhibition of xanthine oxidase. Nepetoidin B was synthesized for the first time in 2018 by Timokhin and co-workers in 17% overall yield.115 Due to the ongoing research on the synthesis of bioactive phenolic compounds, Yao116 et al., in 2020 developed a three steps synthesis of nepetoidin B with an overall 52% yield. The key steps involved Corey–Fuchs alkyne synthesis reaction, Ru-catalyzed anti-Markovnikov addition and demethylation. The synthesis commenced from the commercially available 3,4-dimethoxybenzaldehyde 197 which underwent Corey–Fuchs reaction to form respective terminal alkyne 302 (95%). Next, the alkyne 302 was subjected to anti-Markovnikov addition with 3,4-dimethoxycinnamic acid 303 under the presence of Ru-catalyst 304 to afford tetramethylated nepetoidin B 305 in 90% yield. Subsequent demethylation of compound 305 in the presence of Me3SiI and pyridine afforded the mixture of (Z, E)/(E, E) isomers of nepetoidin B 306 with 2; 8:1 ratio and 61% yield (Scheme 30).
 |
| | Scheme 30 Total synthesis of nepetoidin B 306 by Yao and coworkers. | |
2.8. Synthesis of peptide-based natural products
2.8.1. Haliclamide synthesis. Haliclamide 315 is a naturally occurring compound, extracted from the marine sponge Haliclona sp. of Vanuatu island by Randazzo et al., in 2001.117 Haliclamide exhibited excellent in vitro anti-tumor properties against human lung carcinoma cell line. The first total synthesis of haliclamide was also done by the same group at the time of its extraction. Due to its significant anti-tumor properties, the total synthesis of haliclamide has gained much interest of synthetic chemists. In this perspective, Gahalawat118 et al., in 2016 published the total synthesis of haliclamide 315 employing MacMillan cross aldol reaction, Corey–Fuchs alkyne synthesis, Yamaguchi–Hirao Reaction, Steglich esterification reaction and macro-lactamization reaction as key steps. The total synthesis began with the commercially available ethylene glycol 307, which was subjected to MacMillan cross aldol strategy in the presence of L-proline followed by NaBH4-mediated reduction to furnish 1,3-diol 308 (85%). Next, compound 308 was easily transformed into monobenzylated derivative 309 (91%) via treating it with K2CO3 and BnBr followed by subsequent silyl deprotection, NaIO4 catalyzed oxidation and Corey–Fuchs alkyne synthesis reaction under the presence of carbon tetrabromide and phosphine triphenyl to furnish acetylene moiety 310 in 90% yield. Next, compound 310 was subjected to Yamaguchi–Hirao alkylation protocol with glycidol silyl ether 229b under the presence of boron trifluoride diethyl etherate to access homopropargylic alcohol 311 in 88% yield. Compound 311 was then transformed into compound 312 over a few steps. Next, LiOH mediated saponification of compound 312 took place succeeded by Steglich esterification reaction with alcohol 313 under the presence of DCC and DMAP to form compound 314 (89%). Next, subsequent Cbz deprotection, hydrogenation and HATU mediated macro-lactamization finally furnished haliclamide 315 in 71% yield (Scheme 31).
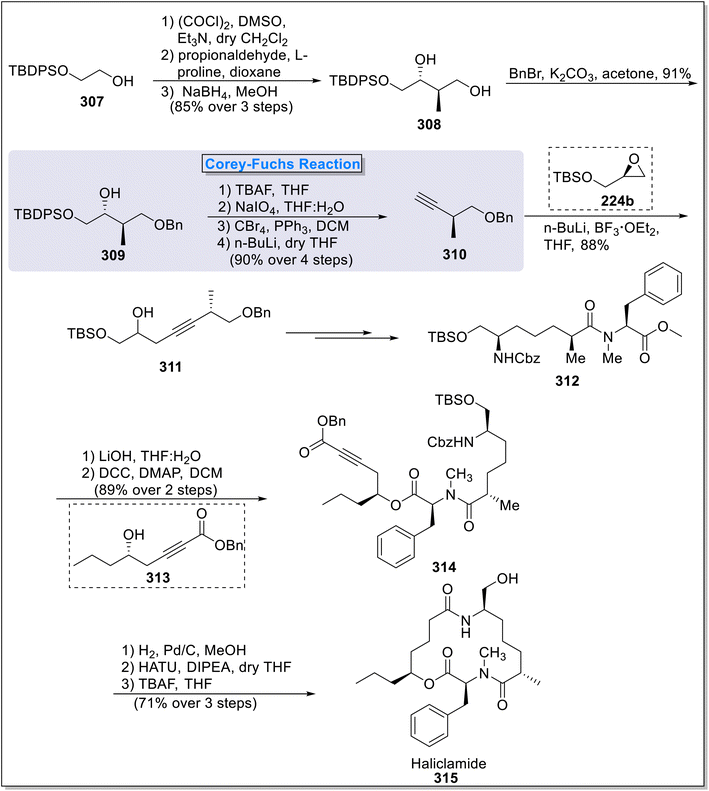 |
| | Scheme 31 Total synthesis of haliclamide 315 by Gahalawat and coworkers. | |
2.9. Miscellaneous natural products
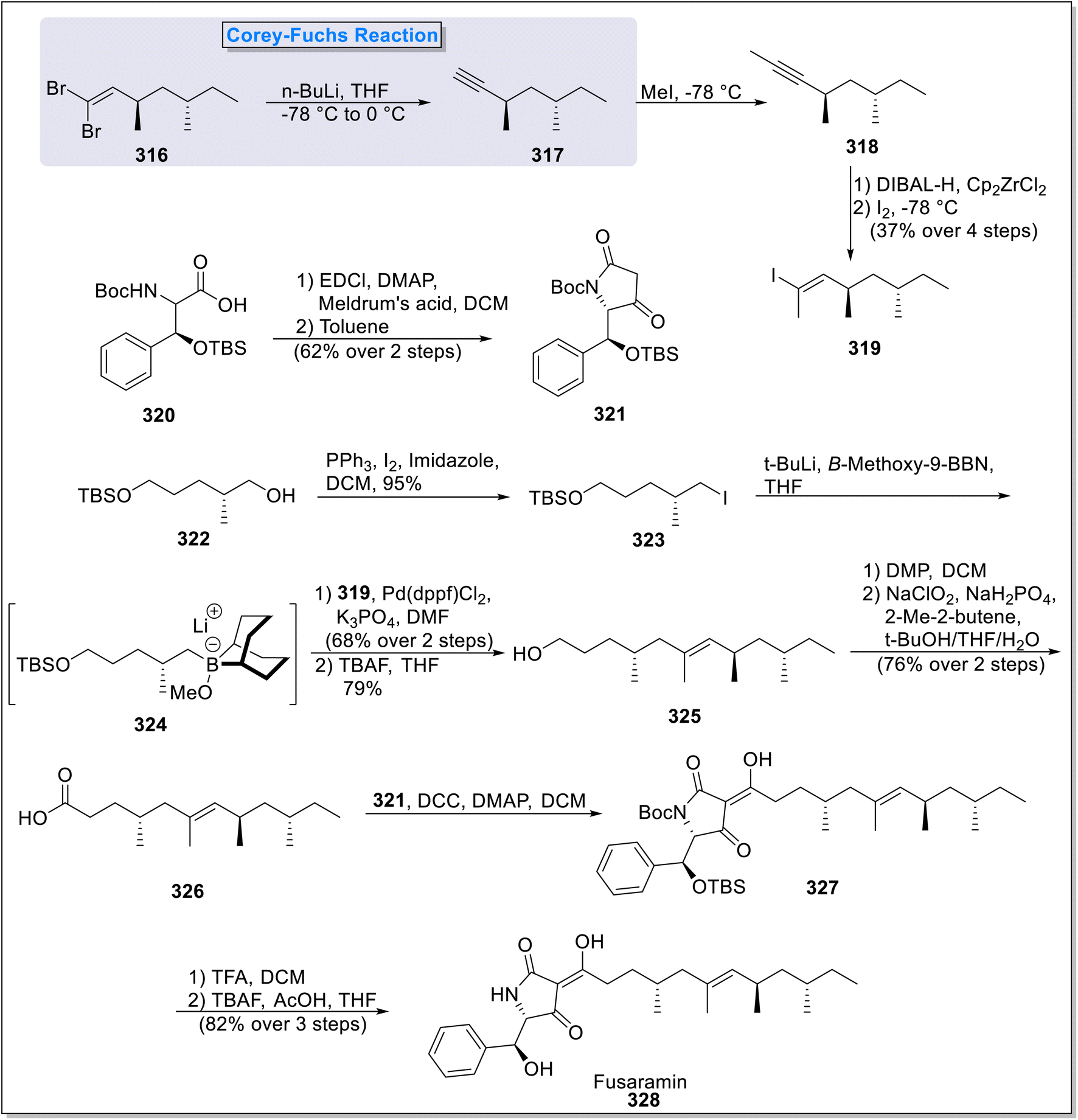
2.9.1. Fusaramin synthesis. Fusaramin 328 is a naturally occurring potent antimitochondrial compound obtained from agar broth of Fusarium sp. FKI-7550.119 This compound shows excellent anti-fungal activities against plant disease-causing fungi. Thus, in 2024, Kimishima120 et al., envisioned the total synthesis of Fusaramin 328 and enabled the elucidation of its stereochemical structure. The total synthesis commenced with the formation of vinyl iodide 319 (37% yield) from gem-dibromo alkene 316 via sequential Corey–Fuchs, methylation and Schwartz hydrozirconation reaction in the presence of n-BuLi, MeI, DIBAL-H, Cp2ZrCl2 and I2. Next, phenylalanine derivative 320 was condensed with Meldrum's acid followed by the ketene formation and attack of the Boc-amine group to access tetramic acid fragment 321 with 62% yield. The next step involved treatment of asymmetric silyl protected alcohol 322 under the conditions of PPh3, I2 and imidazole (Appel conditions) in DCM to afford alkyl iodo compound 323. This reaction was followed by the generation of an intermediate 324 via lithium–halogen exchange in the presence of n-BuLi which was then quenched with B-methoxy-9-BBN. The intermediate 324 underwent Suzuki–Miyaura coupling reaction with 319, followed by silyl deprotection in the presence of TBAF to deliver chiral alcohol 325 in 79% yield. Further, two steps oxidation of the compound 325 took place to form carboxylic acid compound 326 (76%) which was reacted with 321 to form Boc protected fusaramin 327. Finally, the deprotection of 327 in the presence of TFA, TBAF and AcOH led to the synthesis of desired product 328 with 82% yield (Scheme 32).
 |
| | Scheme 32 Total synthesis of fusaramin 328 by Kimishima and coworkers. | |
3. Conclusion
In conclusion, this manuscript summarizes the significance of Corey–Fuchs reaction in the total synthesis of various biologically important natural products as a key step. The protocol facilitates the direct and efficient pathway to alkynes from aldehydes and has been the area of extensive research and development. This review covers the total synthesis of various natural products i.e., polyketides, alkaloids, peptides and polyacetylene-based natural products utilizing the Corey–Fuchs reaction as one of the significant steps. Current breakthroughs and modifications of the reaction have further broadened its viability and practicality, and is expected to remain a cornerstone in the synthesis of complex compounds for the foreseeable future.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors are thankful to the facilities provided by Government College University Faisalabad, Pakistan. A. Irfan extends his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups Research Project under grant number (RGP2/172/45).
References
- N. Desai, N. McKelvie and F. Ramirez, J. Am. Chem. Soc., 1962, 84, 1745–1747 CrossRef.
- F. Pandolfi, I. Chiarotto and M. Feroci, Beilstein J. Org. Chem., 2018, 14, 891–899 CrossRef CAS.
- H. Okumura, N. I. Prakoso, T. Morozumi and T. Umezawa, Org. Lett., 2024, 26, 9817–9821 CrossRef CAS PubMed.
- A. K. Ghosh and Y. Wang, Tetrahedron Lett., 2000, 41, 4705–4708 CrossRef CAS.
- S. Das, S. Paul, K. K. Mandal, A. Anoop and S. Nanda, J. Org. Chem., 2024, 89, 15764–15776 CrossRef CAS PubMed.
- Y. Garg, S. Gahalawat and S. K. Pandey, RSC Adv., 2015, 5, 38846–38850 RSC.
- M. G. Jakobsen, A. Vik and T. V. Hansen, Tetrahedron Lett., 2014, 55, 2842–2844 CrossRef CAS.
- H. F. Sneddon, M. J. Gaunt and S. V. Ley, Org. Lett., 2003, 5, 1147–1150 CrossRef CAS PubMed.
- M. Bai, X. Wang and Q. Li, Tetrahedron, 2024, 154, 133846 CrossRef CAS.
- E. Corey and P. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
- D. Habrant, V. Rauhala and A. M. Koskinen, Chem. Soc. Rev., 2010, 39, 2007–2017 RSC.
- M. Dhameja and J. Pandey, Asian J. Org. Chem., 2018, 7, 1502–1523 CrossRef CAS.
- F. Eymery, B. Iorga and P. Savignac, Synthesis, 2000, 2000, 185–213 CrossRef.
- Y. Thummala, G. V. Karunakar and V. R. Doddi, Adv. Synth. Catal., 2019, 361, 611–616 CrossRef CAS.
- W. Doherty, N. Adler, A. Knox, D. Nolan, J. McGouran, A. P. Nikalje, D. Lokwani, A. Sarkate and P. Evans, Eur. J. Org Chem., 2017, 2017, 175–185 CrossRef CAS.
- S. Kumar, M.-T. Ho and Y.-T. Tao, Org. Lett., 2016, 18, 200–203 CrossRef CAS PubMed.
- A. Irfan, S. Faisal, S. Ahmad, S. A. Al-Hussain, S. Javed, A. F. Zahoor, B. Parveen and M. E. Zaki, pharmaceuticals, 2023, 16, 344 CrossRef CAS PubMed.
- S. Faiz, A. F. Zahoor, N. Rasool, M. Yousaf, A. Mansha, M. Zia-Ul-Haq and H. Z. Jaafar, Molecules, 2015, 20, 14699–14745 CrossRef CAS.
- P. J. Parsons, C. S. Penkett and A. J. Shell, Chem. Rev., 1996, 96, 195–206 CrossRef CAS.
- Y. Bai, X. Shen, Y. Li and M. Dai, J. Am. Chem. Soc., 2016, 138, 10838–10841 CrossRef CAS PubMed.
- D. L. Boger, S. Ichikawa and W. Zhong, J. Am. Chem. Soc., 2001, 123, 4161–4167 CrossRef CAS PubMed.
- H. Johansson and D. S. Pedersen, Eur. J. Org Chem., 2012, 2012, 4267–4281 CrossRef.
- M. Yao, W. Yang, J. Li, C. Huang, J. Fang, S. Qin, S. Liu and X. Yang, Chin. J. Chem., 2024, 42, 1509–1514 CrossRef CAS.
- A. Mushtaq, A. F. Zahoor, M. Bilal, S. M. Hussain, M. Irfan, R. Akhtar, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2023, 28, 2722 CrossRef CAS PubMed.
- J. S. Lee, J. Shin, H. J. Shin, H. S. Lee, Y. J. Lee, H. S. Lee and H. Won, Eur. J. Org Chem., 2014, 2014, 4472–4476 CrossRef CAS.
- T. Sengoku, S. Xu, K. Ogura, Y. Emori, K. Kitada, D. Uemura and H. Arimoto, Angew. Chem., 2014, 126, 4297–4300 CrossRef.
- S. Huang, S. Chen, G. Wang, J. Zhang, L. Tang, G. Du and X. Wang, Synthesis, 2015, 47, 1303–1308 CrossRef CAS.
- M. M Heravi, S. Asadi, N. Nazari and B. Malekzadeh Lashkariani, Curr. Org. Chem., 2015, 19, 2196–2219 CrossRef.
- N. Li, Z. Shi, Y. Tang, J. Chen and X. Li, Beilstein J. Org. Chem., 2008, 4, 48 Search PubMed.
- J. Adrian and C. B. Stark, J. Org. Chem., 2016, 81, 8175–8186 CrossRef CAS PubMed.
- K. Krohn and J. Rohr, Bioorganic Chemistry Deoxysugars, Polyketides and Related Classes: Synthesis, Biosynthesis, Enzymes, 2008, pp. 127–195 Search PubMed.
- P. M. Brown and R. H. Thomson, J. Chem. Soc., Perkin Trans. 1, 1976, 997–1000 RSC.
- D.-S. Hsu and J.-Y. Huang, J. Org. Chem., 2012, 77, 2659–2666 CrossRef CAS.
- K. J. Ngwira, A. L. Rousseau, M. M. Johnson and C. B. de Koning, Eur. J. Org Chem., 2017, 2017, 1479–1488 CrossRef CAS.
- S. M. Dalby and I. Paterson, Curr. Opin. Drug Discovery Dev., 2010, 13, 777–794 CAS.
- C. I. MacGregor, B. Y. Han, J. M. Goodman and I. Paterson, Chem. Commun., 2016, 52, 4632–4635 RSC.
- K.-H. Altmann and J. Gertsch, Nat. Prod. Rep., 2007, 24, 327–357 RSC.
- Y. Inahashi, M. Iwatsuki, A. Ishiyama, A. Matsumoto, T. Hirose, J. Oshita, T. Sunazuka, W. Panbangred, Y. Takahashi and M. Kaiser, Org. Lett., 2015, 17, 864–867 CrossRef CAS PubMed.
- J. Oshita, Y. Noguchi, A. Watanabe, G. Sennari, S. Sato, T. Hirose, D. Oikawa, Y. Inahashi, M. Iwatsuki and A. Ishiyama, Tetrahedron Lett., 2016, 57, 357–360 CrossRef CAS.
- F. Doraghi, L. Rezainia, M. H. Morshedsolouk, H. Navid, B. Larijani and M. Mahdavi, RSC Adv., 2025, 15, 1134–1151 RSC.
- R. Akhtar, A. F. Zahoor, M. Irfan, T. H. Bokhari and A. ul Haq, Chem. Pap., 2022, 76, 7275–7293 CrossRef CAS.
- Y.-Z. Shu, Q. Ye, H. Li, K. F. Kadow, R. A. Hussain, S. Huang, D. R. Gustavson, S. E. Lowe, L.-P. Chang and D. M. Pirnik, Bioorg. Med. Chem. Lett., 1997, 7, 2295–2298 CrossRef CAS.
- J. Peng, J. Jiao, J. Li, W. Wang, Q. Gu, T. Zhu and D. Li, Bioorg. Med. Chem. Lett., 2012, 22, 3188–3190 CrossRef CAS PubMed.
- J. Preindl, S. Schulthoff, C. Wirtz, J. Lingnau and A. Fürstner, Angew. Chem., Int. Ed., 2017, 56, 7525–7530 CrossRef CAS PubMed.
- A. Cutignano, I. Bruno, G. Bifulco, A. Casapullo, C. Debitus, L. Gomez-Paloma and R. Riccio, Eur. J. Org Chem., 2001, 2001, 775–778 CrossRef.
- T. Tanaka, Y. Murai, T. Kishi, H. Takamura and I. Kadota, Tetrahedron Lett., 2018, 59, 763–766 CrossRef CAS.
- C. Tanaka, J. Tanaka, R. F. Bolland, G. Marriott and T. Higa, Tetrahedron, 2006, 62, 3536–3542 CrossRef CAS.
- H. D. Arndt, S. Rizzo, C. Nöcker, V. N. Wakchaure, L. G. Milroy, V. Bieker, A. Calderon, T. T. Tran, S. Brand and L. Dehmelt, Chem.–Eur. J., 2015, 21, 5311–5316 CrossRef CAS PubMed.
- J. H. Lang and T. Lindel, Beilstein J. Org. Chem., 2019, 15, 577–583 CrossRef CAS.
- N. Oku, K. Takada, R. W. Fuller, J. A. Wilson, M. L. Peach, L. K. Pannell, J. B. McMahon and K. R. Gustafson, J. Am. Chem. Soc., 2010, 132, 10278–10285 CrossRef CAS PubMed.
- C. K. Skepper, T. Quach and T. F. Molinski, J. Am. Chem. Soc., 2010, 132, 10286–10292 CrossRef CAS.
- K. Sakurai, K. Sakamoto, M. Sasaki and H. Fuwa, Chem.–Asian J., 2020, 15, 3494–3502 CrossRef CAS PubMed.
- S. B. Krasnoff, U. Englich, P. G. Miller, M. L. Shuler, R. P. Glahn, B. G. Donzelli and D. M. Gibson, J. Nat. Prod., 2012, 75, 175–180 CrossRef CAS PubMed.
- M. Yoshida, Y. Okoshi and H. Kigoshi, Chem. Commun., 2023, 59, 9880–9883 RSC.
- A. Sharma, S. Athe, P. Ramesh, K. Vishali and S. Ghosh, Tetrahedron Lett., 2021, 82, 153374 CrossRef CAS.
- W. Zhou, H. A. Alharbi, E. Hummingbird, A. T. Keatinge-Clay and T. Mahmud, ACS Chem. Biol., 2022, 17, 2039–2045 CrossRef CAS.
- D. Dardić, N. Böhringer, A. Plaza, F. Zubeil, J. Pohl, S. Sommer, L. Padva, J. Becker, M. A. Patras and M.-K. Bill, Org. Chem. Front., 2022, 9, 1604–1615 RSC.
- S.-Z. Huang, F.-D. Kong, G. Chen, X.-H. Cai, L.-M. Zhou, Q.-Y. Ma, Q. Wang, W.-L. Mei, H.-F. Dai and Y.-X. Zhao, Phytochemistry, 2019, 159, 208–215 CrossRef CAS PubMed.
- L. Fang, X.-Q. Song, T.-T. He, K.-K. Zhu, J.-H. Yu, J.-T. Song, J. Zhou and H. Zhang, Fitoterapia, 2018, 129, 150–153 CrossRef CAS.
- M. C. Carson, B. J. Orzolek and M. C. Kozlowski, Org. Lett., 2022, 24, 7250–7254 CrossRef CAS.
- S. DGS, V. R. R. Ch, T. Syed, G. Sridhar and S. R. Alapati, Synth. Commun., 2024, 1–7 Search PubMed.
- X. Geng and S. J. Danishefsky, Org. Lett., 2004, 6, 413–416 CrossRef CAS PubMed.
- D. Sudhakar, V. R. R. Ch, T. Syed, G. Sridhar and S. R. Alapati, Synth. Commun., 2024, 54, 992–998 CrossRef.
- S. Faiz and A. F. Zahoor, Mol. Diversity, 2016, 20, 969–987 CrossRef CAS PubMed.
- S. Munawar, A. F. Zahoor, S. Ali, S. Javed, M. Irfan, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2022, 27, 6953 CrossRef CAS PubMed.
- U. Tillmann, M. Elbrächter, B. Krock, U. John and A. Cembella, Eur. J. Phycol., 2009, 44, 63–79 CrossRef CAS.
- E. Ito, M. Satake, K. Ofuji, N. Kurita, T. McMahon, K. James and T. Yasumoto, Toxicon, 2000, 38, 917–930 CrossRef CAS PubMed.
- Z. Zhang, Y. Chen, D. Adu-Ampratwum, A. A. Okumu, N. T. Kenton and C. J. Forsyth, Org. Lett., 2016, 18, 1824–1827 CrossRef CAS PubMed.
- M. Bader, T. D. Stark, C. Dawid, S. Lösch and T. Hofmann, J. Agric. Food Chem., 2014, 62, 2479–2488 CrossRef CAS PubMed.
- K. Aoki, Y. Igarashi, H. Nishimura, I. Morishita and K. Usui, Tetrahedron Lett., 2012, 53, 6000–6003 CrossRef CAS.
- C. Mugnaini and F. Corelli, Synthesis, 2016, 48, 2085–2092 CrossRef CAS.
- A. Irfan, S. Faiz, A. Rasul, R. Zafar, A. F. Zahoor, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2022, 27, 1023 CrossRef CAS PubMed.
- A. Kolodiazhna and O. Kolodiazhnyi, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 275–276 CrossRef CAS.
- A. Kolodyazhnaya and O. Kolodyazhny, Russ. J. Gen. Chem., 2019, 89, 1998–2004 CrossRef CAS.
- S. C. Bobzin and D. J. Faulkner, J. Org. Chem., 1991, 56, 4403–4407 CrossRef CAS.
- K. Nakagawa, Heterocycles, 1995, 41, 5 CrossRef.
- S. J. Gharpure, D. Anuradha, J. V. Prasad and P. Srinivasa Rao, Eur. J. Org Chem., 2015, 2015, 86–90 CrossRef CAS.
- M. Borah, U. Borthakur and A. K. Saikia, J. Org. Chem., 2017, 82, 1330–1339 CrossRef CAS.
- Y. Wang, W.-Y. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2228–2232 CrossRef CAS.
- N. Gunawan, M. J. Nutt, A. C. Bissember, J. A. Smith and S. G. Stewart, Synlett, 2023, 34, 1524–1528 CrossRef CAS.
- P. Krishnan, F.-K. Lee, V. A. Yap, Y.-Y. Low, T.-S. Kam, K.-T. Yong, K.-N. Ting and K.-H. Lim, J. Nat. Prod., 2020, 83, 152–158 CrossRef CAS PubMed.
- Y. Y. Mak, B. J. Loong, P. Millns, C. C. Bauer, R. S. Bon, Y. Mbaki, F. K. Lee, K. H. Lim, C. Kong and S. M. Then, Phytother. Res., 2022, 36, 2952–2963 CrossRef CAS PubMed.
- K. Annapurna and A. V. Narsaiah, Tetrahedron, 2024, 155, 133909 CrossRef CAS.
- Z.-F. Zhou, M. Menna, Y.-S. Cai and Y.-W. Guo, Chem. Rev., 2015, 115, 1543–1596 Search PubMed.
- M. Smietana, E. Benedetti, C. Bressy and S. Arseniyadis, Efficiency in Natural Product Total Synthesis, 2018, 319–344 Search PubMed.
- J.-X. Gong, H.-Y. Wang, L.-G. Yao, X.-W. Li and Y.-W. Guo, Synlett, 2016, 27, 391–394 CAS.
- C. Djerassi and W. K. Lam, Acc. Chem. Res., 1991, 24, 69–75 CrossRef CAS.
- K. Suzuki, F. Shono, H. Kai, T. Uno and M. Uyeda, J. Enzyme Inhib., 2000, 15, 357–366 CrossRef CAS PubMed.
- N. M. Carballeira, N. Montano, R. Alvarez-Velilla, C. F. Prada, R. M. Reguera and R. Balaña-Fouce, Mar. Drugs, 2013, 11, 3661–3675 CrossRef CAS PubMed.
- J. Adrian and C. B. Stark, Eur. J. Org Chem., 2016, 2016, 4607–4610 CrossRef.
- R. Nshimiyimana, T. F. Lam, S. Aggarwal, C. N. Serhan and N. A. Petasis, RSC Adv., 2022, 12, 11613–11618 RSC.
- R. Nshimiyimana, S. J. Glynn, C. N. Serhan and N. A. Petasis, Org. Biomol. Chem., 2023, 21, 1667–1673 RSC.
- F. Fullas, D. M. Brown, M. C. Wani, M. R. Wall, T. E. Chagwedera, N. R. Farnsworth, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 1995, 58, 1625–1628 CrossRef CAS PubMed.
- H. Takamura, H. Wada, N. Lu and I. Kadota, Org. Lett., 2011, 13, 3644–3647 CrossRef CAS PubMed.
- C. Garcia-Ruiz, I. Cheng-Sanchez and F. Sarabia, Synthesis, 2016, 48, 1655–1662 CrossRef CAS.
- W.-g. Li, F.-y. Dai, Y.-x. Cheng, G.-f. Yin, J.-l. Bi and D.-p. Li, Chem. Res. Chin. Univ., 2013, 29, 290–293 CrossRef CAS PubMed.
- Y. Nishikawa, I. Yasuda, Y. Watanabe and T. Seto, Yakugaku Zasshi, 1976, 96, 1322–1326 CrossRef CAS PubMed.
- G. A. Kraus, P. Dong, Y. Qu, A. Evans and S. Carpenter, Tetrahedron Lett., 2016, 57, 5185–5187 CrossRef CAS.
- J. Wang, A. N. Pearce, S. T. Chan, R. B. Taylor, M. J. Page, A. Valentin, M.-L. Bourguet-Kondracki, J. P. Dalton, S. Wiles and B. R. Copp, J. Nat. Prod., 2016, 79, 607–610 CrossRef CAS PubMed.
- A. Irfan, S. Faisal, A. F. Zahoor, R. Noreen, S. A. Al-Hussain, B. Tuzun, R. Javaid, A. A. Elhenawy, M. E. Zaki and S. Ahmad, Pharmaceuticals, 2023, 16, 829 CrossRef CAS PubMed.
- A. F. Zahoor, M. Yousaf, R. Siddique, S. Ahmad, S. A. R. Naqvi and S. M. A. Rizvi, Synth. Commun., 2017, 47, 1021–1039 CrossRef CAS.
- M. Dumpala, S. Theegala and R. K. Palakodety, Tetrahedron Lett., 2017, 58, 1273–1275 CrossRef CAS.
- R. Negri, Fitoterapia, 2015, 106, 92–109 CrossRef CAS PubMed.
- J. Geng, Q. Ren, C. Chang, X. Xie, J. Liu and Y. Du, RSC Adv., 2019, 9, 10253–10263 RSC.
- D. B. Clarke, S. F. Hinkley and R. T. Weavers, Tetrahedron Lett., 1997, 38, 4297–4300 CrossRef CAS.
- H. Lee, T. Kang and H. Y. Lee, Angew. Chem., 2017, 129, 8366–8369 CrossRef.
- J. L. von Salm, N. G. Wilson, B. A. Vesely, D. E. Kyle, J. Cuce and B. J. Baker, Org. Lett., 2014, 16, 2630–2633 CrossRef CAS PubMed.
- World Health Organization, Fourth WHO report on neglected tropical diseases, World Health Organization, Geneva, 2017 Search PubMed.
- S.-D. Chen, H. Gao, Q.-C. Zhu, Y.-Q. Wang, T. Li, Z.-Q. Mu, H.-L. Wu, T. Peng and X.-S. Yao, Org. Lett., 2012, 14, 1772–1775 CrossRef CAS PubMed.
- T. Kerl, F. Berger and H. G. Schmalz, Chem.–Eur. J., 2016, 22, 2935–2938 CrossRef CAS PubMed.
- T. Nakanishi, M. Nishi, A. Inada, H. Obata, N. Tanabe, S. Abe and M. Wakashiro, Chem. Pharm. Bull., 1990, 38, 1772–1774 CrossRef CAS PubMed.
- R. Kubínová, E. Švajdlenka, K. Schneiderová, Z. Hanáková, S. Dall'Acqua and O. Farsa, Biochem. Syst. Ecol., 2013, 49, 39–42 CrossRef.
- X. Wu, H. Gao, W. Sun, J. Yu, H. Hu, Q. Xu and X. Chen, Phytother. Res., 2017, 31, 1072–1077 CrossRef CAS PubMed.
- R. J. Grayer, M. R. Eckert, N. C. Veitch, G. C. Kite, P. D. Marin, T. Kokubun, M. S. Simmonds and A. J. Paton, Phytochemistry, 2003, 64, 519–528 CrossRef CAS PubMed.
- V. Timokhin, M. Regner, Y. Tsuji, J. Grabber and J. Ralph, Synlett, 2018, 29, 1229–1231 CrossRef CAS.
- M. Yao, J. Zhang, S. Yang and H. Xiong, ARKIVOC, 2020, 2020, 52–59 Search PubMed.
- A. Randazzo, C. Debitus and L. Gomez-Paloma, Tetrahedron, 2001, 57, 4443–4446 CrossRef CAS.
- S. Gahalawat and S. K. Pandey, Org. Biomol. Chem., 2016, 14, 9287–9293 RSC.
- K. Sakai, Y. Unten, M. Iwatsuki, H. Matsuo, W. Fukasawa, T. Hirose, T. Chinen, K. Nonaka, T. Nakashima and T. Sunazuka, J. Antibiot., 2019, 72, 645–652 CrossRef CAS PubMed.
- A. Kimishima, D. Hagimoto, M. Honsho, K. Sakai, S. Honma, S.-i. Fuji, M. Iwatsuki, T. Tokiwa, K. Nonaka and T. Chinen, Org. Lett., 2024, 26, 597–601 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Samreen Gul Khana,
Muhammad Jawwad Saif
*a,
Samreen Gul Khana,
Muhammad Jawwad Saif