
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting VEGFR-2 in breast cancer: synthesis and in silico and in vitro characterization of quinoxaline-based inhibitors†
Ibrahim H. Eissa *a,
Alaa Elwana,
Mustafa A. Al-Qadhib,
Dalal Z. Huseinc,
Fatma G. Amind,
Aisha A. Alsfouke,
Eslam B. Elkaeedf,
Hazem Elkady*a and
Ahmed M. Metwaly*g
*a,
Alaa Elwana,
Mustafa A. Al-Qadhib,
Dalal Z. Huseinc,
Fatma G. Amind,
Aisha A. Alsfouke,
Eslam B. Elkaeedf,
Hazem Elkady*a and
Ahmed M. Metwaly*g
aPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: Ibrahimeissa@azhar.edu.eg; Hazemelkady@azhar.edu.eg
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Sana'a University, 18084 Sana'a, Yemen
cChemistry Department, Faculty of Science, New Valley University, El-Kharja, 72511, Egypt
dPhysics Department, Faculty of Science, Alexandria University, Alexandria, Egypt
eDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah Bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia
fDepartment of Pharmaceutical Sciences, College of Pharmacy, AlMaarefa University, P.O. Box 71666, Riyadh 11597, Saudi Arabia
gPharmacognosy and Medicinal Plants Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: ametwaly@azhar.edu.eg
First published on 22nd April 2025
Abstract
A novel series of quinoxaline derivatives was designed and synthesized to target VEGFR-2, a receptor critical in cancer progression, with a focus on favorable pharmacophoric features. Among these derivatives, compound 11d emerged as a promising candidate, exhibiting potent cytotoxicity against MDA-MB-231 and MCF-7 cancer cell lines, with IC50 values of 21.68 μM and 35.81 μM, respectively, while displaying significantly reduced toxicity in normal cell lines WI-38 and WISH (IC50 values of 82.46 μM and 75.27 μM). Compared to standard treatments doxorubicin and sorafenib, compound 11d demonstrated a favorable therapeutic window. Inhibition assays showed that 11d inhibits VEGFR-2 with an IC50 of 62.26 nM ± 2.77, comparable to sorafenib. Mechanistically, treatment with 11d upregulated pro-apoptotic markers BAX, caspase-8, and caspase-9, while downregulating the anti-apoptotic marker Bcl-2, resulting in a significant BAX/Bcl-2 ratio increase (16.11). A wound healing assay confirmed 11d's anti-migratory effects, limiting wound closure in MDA-MB-231 cells to 27.51% compared to untreated cells. Additionally, flow cytometry revealed that 11d induced both early (46.43%) and late apoptosis (31.49%) in MDA-MB-231 cells, alongside G1 phase cell cycle arrest, reducing S and G2/M phase progression. Molecular docking and dynamics simulations over 200 ns demonstrated stable binding of compound 11d to VEGFR-2, with docking scores superior and comparable to sorafenib. Density Functional Theory (DFT) calculations underscored 11d's stability and reactivity, while in silico ADMET analysis predicted a favorable safety profile over sorafenib, particularly with respect to carcinogenic and chronic toxicity risks. These findings indicate that quinoxaline derivative 11d holds potential as a selective and effective VEGFR-2 inhibitor with promising antitumor and anti-metastatic properties, warranting further investigation.
1. Introduction
Cancer remains one of the leading causes of mortality worldwide,1 with breast cancer being one of the most prevalent and challenging forms to treat due to its potential for metastasis and resistance to standard therapies.2 According to the latest global cancer statistics, breast cancer accounted for approximately 2.26 million new cases in 2020 and remains the leading cause of cancer-related deaths among women globally.3 Among the therapeutic targets in cancer treatment, vascular endothelial growth factor receptor 2 (VEGFR-2) has gained significant attention for its role in promoting angiogenesis, which supports tumor growth and metastatic spread.4,5 Targeting VEGFR-2 with specific inhibitors can disrupt angiogenesis, thereby impeding tumor progression and offering a potential strategy for cancer management.6,7Quinoxaline derivatives have emerged as promising scaffolds in anti-cancer drug development,8 particularly in the context of breast cancer.9 Structurally, quinoxalines offer versatile chemical frameworks that can be modified to enhance selectivity and potency toward specific molecular targets, including VEGFR-2.10 Previous studies have shown that quinoxaline-based compounds can effectively inhibit cancer cell proliferation and metastasis, though further exploration is needed to optimize their efficacy and safety profiles.11
Building on these findings and our ongoing efforts to develop novel anticancer agents,12–17 particularly quinoxaline derivatives,18–22 we designed, synthesized, and evaluated a new series of quinoxaline derivatives targeting VEGFR-2 as potential anti-breast cancer agents. These compounds were designed based on key pharmacophoric features known to interact with VEGFR-2, aiming to maximize binding affinity and biological activity. Among the synthesized derivatives, compound 11d showed particular promise, exhibiting selective cytotoxicity in cancer cells. In addition, 11d demonstrated significant anti-migratory activity, induced apoptosis, and caused cell cycle arrest, indicating its potential as a multi-faceted anti-cancer agent.
To provide a comprehensive evaluation, we complemented our experimental findings with in silico analyses, including molecular docking, molecular dynamics (MD) simulations, density functional theory (DFT) studies, and ADMET profiling. These computational approaches allowed us to investigate the stability, reactivity, pharmacokinetic, and toxicological properties of compound 11d, further supporting its potential as a targeted cancer therapeutic. The promising results from this study underscore the therapeutic potential of quinoxaline derivatives in the development of new VEGFR-2 inhibitors and highlight the need for further preclinical evaluation of compound 11d as an anti-cancer candidate.
1.1. Rationale
Several VEGFR-2 inhibitors have received FDA approval for the clinical treatment of cancer.23–29 Fig. 1 shows some VEGFR-2 inhibitors as sorafenib I,30,31 regorafenib II,32 and lenvatinib III,33 which are urea-based derivatives.34–38 These drugs are approved for various cancers, including prostate cancer, thyroid cancer, melanoma, renal cell carcinoma, and hepatocellular carcinoma.39,40 However, their use is often associated with adverse effects such as cardiovascular complications, diarrhea, renal impairment, fatigue, hypertension, reduced appetite, nausea, thrombocytopenia, and proteinuria.41–45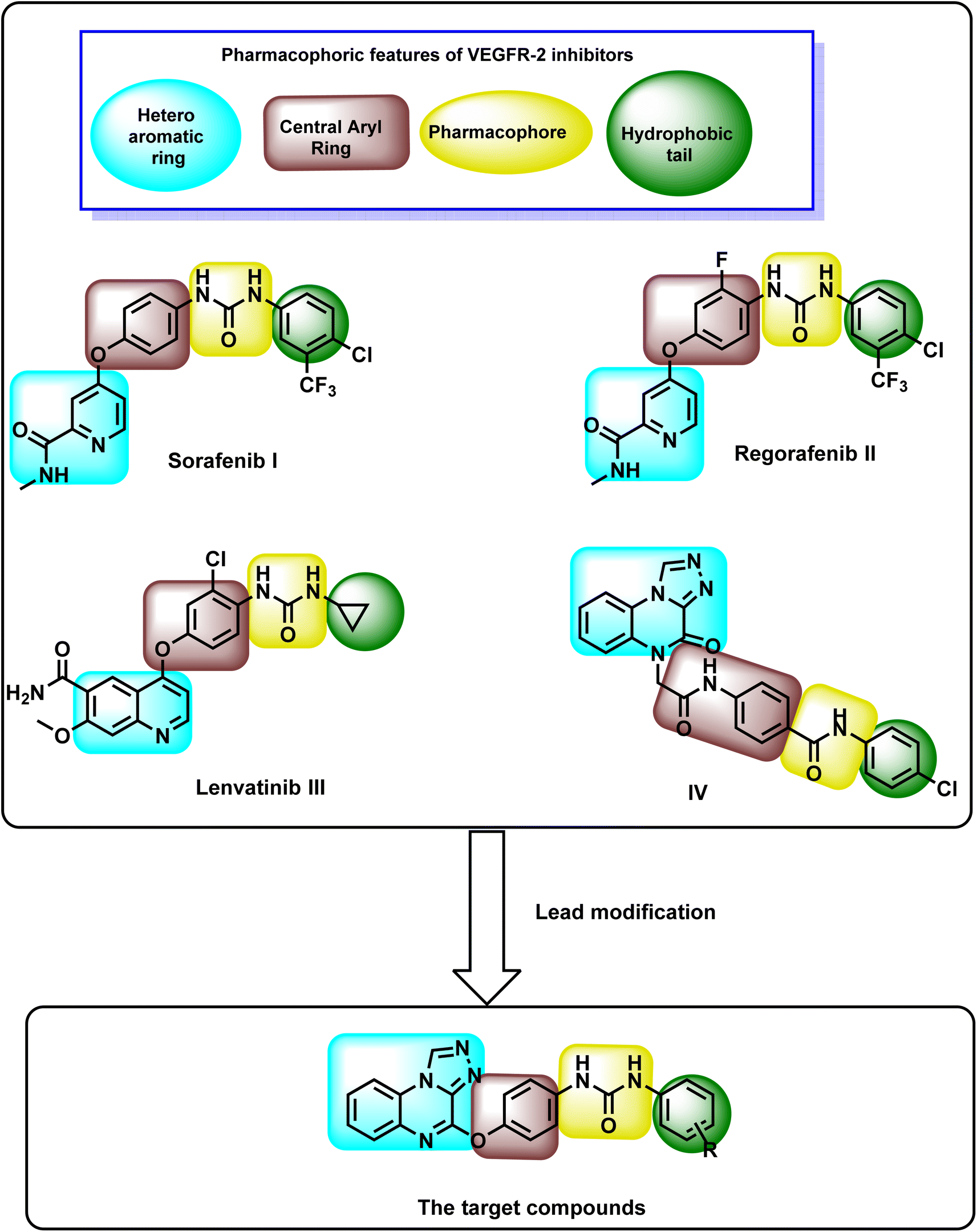 | ||
| Fig. 1 Design rationale for the synthesized quinoxaline derivatives. | ||
Our research group recently introduced compound IV, a bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivative, as a promising VEGFR-2 inhibitor with notable anti-proliferative activity against HepG-2 and MCF-7 cell lines. Compound IV effectively induced cell cycle arrest in HepG-2 cells at the G2/M phase and exhibited a strong pro-apoptotic effect. Additionally, it significantly increased the levels of caspase-3, caspase-9, and BAX while reducing the expression of Bcl-2 in treated cells.46
The chemical structure of VEGFR-2 inhibitors must include four key pharmacophoric features for effective binding at the VEGFR-2 active site (Fig. 1). (i) The first feature is a heteroaromatic moiety, essential for forming hydrogen bonds with Cys917 in the hinge region of the ATP-binding site.47 (ii) The second is a spacer group that occupies the space between the hinge region and the DFG domain (Asp–Phe–Gly motif located in the activation loop of protein kinases, which plays a critical role in regulating kinase activity and ATP binding).48 (iii) The third feature is a pharmacophore comprising hydrogen bond donor and acceptor groups, enabling interactions with Glu883 and Asp1044 in the DFG domain.49 (iv) The fourth is a terminal hydrophobic moiety, which engages in hydrophobic interactions with the allosteric hydrophobic pocket of the active site.50–52
The rationale in this research focused on modifying compound IV to develop new anti-proliferative agents targeting VEGFR-2 with enhanced apoptotic potential. The newly designed compounds incorporate bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline and urea functional groups. The design strategy preserved the bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety from compound IV as a heterocyclic core due to its established biological benefits. This moiety contains four nitrogen atoms that act as electron acceptors, facilitating hydrogen bonding in the hinge region. Additionally, its planar structure promotes hydrophobic interactions within the hinge region. Previous studies have highlighted its promising anti-proliferative activity.53–55 The second modification line included a phenoxy group, as observed in compounds I, II, and III, which served as a linker. The third modification line incorporated the urea group from compounds I, II, and III, functioning as a pharmacophore. Lastly, various substituted aromatic groups were introduced as hydrophobic tails to explore the structure–activity relationship (Fig. 1).
2. Results and discussions
2.1. Chemistry
The chemical processes for furnishing the target molecules are shown in Scheme 1. Initially, o-phenylenediamine 1 reacted with oxalic acid 2 in the presence of 4 N HCl to get 2,3-(1H,4H)-quinoxalinedione 3.56,57 Subsequent treatment of compound 3 with thionyl chloride yielded 2,3-dichloroquinoxaline 4. The reaction of compound 4 with hydrazine hydrate at ambient temperature yielded 2-chloro-3-hydrazinylquinoxaline 5.56,57 Subsequently, compound 5 was subjected to heating with triethyl orthoformate, resulting in the formation of 4-chloro[1,2,4]triazolo[4,3-a]quinoxaline 7.56,57 The subsequent step involved the reaction of isocyanate derivatives including phenyl isocyanate, 3-chlorophenyl isocyanate, 4-chlorophenyl isocyanate, and 3-methoxyphenyl isocyanate 9a–d, respectively with para-aminophenol 8 in acetonitrile, resulting in the formation of the key intermediates 10a–d.58 Finally, a nucleophilic substitution reaction of the formed intermediates 10a–d with compound 7 in the presence of triethylamine (TEA) in THF resulted in the synthesis of target compounds 11a–d, respectively with commendably high yield. | ||
| Scheme 1 Synthesis of the final compounds 11a–d. | ||
Spectral data were used to describe compounds 11a–d. These compounds' IR spectra revealed prominent NH bands at 3331–3103 cm−1. Additionally, it had significant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption bands between 1654 and 1682 cm−1. Moreover, the triazolo CH protons of the four derivatives were identified by singlet signals in 1H NMR spectra with δ 10.13 and 10.17 ppm. These results were supported by 13C NMR spectra, which revealed distinctive peaks for the corresponding carbons.
O absorption bands between 1654 and 1682 cm−1. Moreover, the triazolo CH protons of the four derivatives were identified by singlet signals in 1H NMR spectra with δ 10.13 and 10.17 ppm. These results were supported by 13C NMR spectra, which revealed distinctive peaks for the corresponding carbons.
2.2. Biological evaluation
| Comp. | In vitro cytotoxicity IC50a (μM) | |||
|---|---|---|---|---|
| MDA-231 | MCF-7 | WI-38 | WISH | |
| a The values are expressed as the mean ± SEM from three independent experiments. | ||||
| Sorafenib | 7.64 ± 0.4 | 7.26 ± 0.3 | 10.65 ± 0.8 | 13.45 ± 1.1 |
| 11a | 68.40 ± 3.7 | 77.98 ± 4.1 | — | — |
| 11c | 76.23 ± 3.9 | 69.82 ± 3.6 | — | — |
| 11d | 21.68 ± 1.5 | 35.81 ± 2.3 | 82.46 ± 4.2 | 75.27 ± 3.9 |
| 11b | 33.56 ± 2.1 | 46.53 ± 2.7 | — | — |
In normal cell lines WI-38 and WISH, 11d has significantly higher IC50 values (82.46 μM and 75.27 μM, respectively), indicating lower toxicity compared to sorafenib, which have IC50 values of 10.65 μM and 13.45 μM, respectively. The selectivity index (SI), calculated as the ratio of IC50 in normal cells (WI-38 or WISH) to that in cancer cells (MDA-231 or MCF-7), highlights the preferential cytotoxicity of compounds. For sorafenib, the SI ranges from 1.39 to 1.85, indicating limited selectivity between cancer and normal cells.
In contrast, compound 11d exhibited significantly higher SI values, ranging from 3.80 to 11.29, demonstrating greater selectivity for cancer cells over normal cells. This selective cytotoxicity of 11d suggests a promising therapeutic window, as it appears to be more effective against cancer cells than normal cells, making it a potentially safer candidate for further development.
Compound 11b also demonstrated moderate cytotoxicity, particularly in MDA-MB-231 cells (IC50 = 33.56 μM), while 11a and 11c exhibited weaker activity, with IC50 values above 68 μM in both cell lines. These findings suggested that 11d holds potential as a lead compound for further investigation, particularly due to its greater cytotoxic effect compared to the other compounds in the series.
| Comp. | VEGFR-2, IC50 (nM) ± SEM |
|---|---|
| a Values are given as mean ± SEM of three independent experiments. | |
| 11d | 62.26 ± 2.77 |
| Sorafenib | 53.32 ± 2.52 |
From cytotoxicity, selectivity, and VEGFR-2 assays, was can notice that compound 11d exhibited an IC50 value of 62.26 nM against VEGFR-2, while sorafenib showed a slightly lower IC50 of 53.32 nM. While this difference might seem small, its clinical relevance extends beyond mere numerical comparison.
A slight reduction in potency could necessitate a higher therapeutic dose of 11d compared to sorafenib to achieve the same level of VEGFR-2 inhibition. However, this must be weighed against the broader therapeutic window that 11d demonstrates, as indicated by its higher selectivity index (SI), meaning it spares normal cells more effectively than sorafenib. This could reduce the incidence of off-target toxicities, making dose escalation more feasible.
Sorafenib, while potent, is associated with several adverse effects, including hepatotoxicity, hypertension, and fatigue. The significantly lower toxicity of 11d in normal cells (WI-38 and WISH, IC50 = 82.46 μM and 75.27 μM, respectively) compared to sorafenib (IC50 = 10.65 μM and 13.45 μM) suggests that even if a slightly higher dose of 11d is required for equivalent VEGFR-2 inhibition, it may still offer a superior safety profile.
The balance between potency and selectivity is critical for drug development. Although 11d's IC50 is slightly higher than sorafenib's, its higher selectivity and reduced toxicity could make it a preferable candidate for long-term therapy, especially in patients susceptible to sorafenib's side effects. Additionally, further formulation strategies (e.g., prodrugs, nano-formulations) could enhance 11d's bioavailability and therapeutic efficacy, mitigating the impact of its slightly higher IC50.
 | ||
| Fig. 2 Microscopic images illustrating the inhibitory effect of compound 11d (11 μM) on the migration of the MDA-231 cell lines (B), compared to the control, untreated MDA-231 cell line, (A). | ||
| Sample | Quantitative closure (%) for scratched assay after 48 h |
|---|---|
| Control (MDA-231) | 58.95 |
| Compound 11d (11 μM) | 27.51 |
| Sample | Viableb (left bottom) | Apoptosisb | Necrosisb (left top) | |
|---|---|---|---|---|
| Early (right bottom) | Late (right top) | |||
| a Significant P value significant P value < 0.05 & by using one-way ANOVA followed by Tukey's post hoc multiple comparison tests.b Values represent stages of the cell death process in MDA-MB-231cells treated with or without 11d. | ||||
| MDA-MB-231 | 95.48 | 3.84 | 0.45 | 0.32 |
| 11d/MDA-MB-231 | 20.17a | 46.43a | 31.49a | 1.91a |
 | ||
| Fig. 3 The effect of compound 11d on the cell viability of the MDA-231 cell lines. | ||
| Sample | Cell cycle distributionb (%) | |||
|---|---|---|---|---|
| % sub-G1 | % G1 | % S | % G2/M | |
| a Significant P value significant P value < 0.05 & by using one-way ANOVA followed by Tukey's post hoc multiple comparison tests.b Cell cycle distribution (%) of MDA-MB-231 cells were treated with or without compound 11d. | ||||
| MDA-MB-231 | 19.48 | 44.98 | 24.95 | 10.59 |
| 11d/MDA-MB-231 | 15.75 | 64.06a | 16.45a | 3.74a |
 | ||
| Fig. 4 Impact of compound 11d on cell cycle progression in MDA-MB-231 cells following 48 hour treatment. | ||
| Sample | Gene expression (fold change)b | ||||
|---|---|---|---|---|---|
| BAX | Bcl-2 | BAX/Bcl-2 ratio | Caspases-8 | Caspases-9 | |
| a Significant P value significant P value < 0.05 & by using one-way ANOVA followed by Tukey's post hoc multiple comparison tests.b Values are given as changes from the corresponding control (MDA-231 cells) group. Data from three independent experiments represent the mean ± SEM, as the fold changes, with control is set to ‘1’. | |||||
| MDA-231 cells | 1.00 ± 0.08 | 1.00 ± 0.07 | 1.00 ± 0.13 | 1.00 ± 0.09 | 1.00 ± 0.11 |
| 11d | 5.48 ± 0.45a | 0.34 ± 0.01a | 16.11 ± 1.01 | 4.15 ± 0.48a | 5.58 ± 0.40a |
2.3. Computational evaluations
 | ||
| Fig. 5 (A) Overlay of the co-crystallized ligand (green) and the redocked ligand (turquoise) in the VEGFR-2 active site. 2D interaction of (B) sorafenib, (C) 11a, (D) 11b, (E) 11c, and (F) 11d, against VEGFR-2. | ||
The docking simulations evaluated the compounds' ability to interact with the active site of VEGFR-2. The docking scores for derivatives 11a–d ranged from −22.71 to −23.65 kcal mol−1, indicating strong binding affinities (Table 7). Notably, these scores were comparable and higher than those of sorafenib, a known VEGFR-2 inhibitor, suggesting that the new derivatives may exhibit enhanced binding efficiency. The binding score of sorafenib was determined to be −22.50 kcal mol−1. An examination of sorafenib's binding pattern indicated alignment with the reported findings, where the hydrophobic moiety (3 chloro-4-trifluoromethylphenyl) was positioned within the allosteric site. Additionally, two significant hydrogen bonds were identified between the urea linker and Glu883 and Asp1044. Also, Cys917 forms two hydrogen bonds with the N-methylpicolinamide arm (see Fig. 5B).
| Comp. | 11a | 11b | 11c | 11d | Sorafenib |
|---|---|---|---|---|---|
| Energy score (kcal mol−1) | −22.98 | −23.02 | −22.71 | −23.65 | −22.50 |
The docking results showed that the four synthesized derivatives nearly matched the orientation and position of sorafenib in the active binding region of the VEGFR-2 enzyme (Fig. 5). The derivatives' amide groups maintained two necessary hydrogen bonds with Glu883 and Asp1044 in the DFG domain. Interestingly, the hydrophobic moieties of the synthesized candidates (phenyl, 3-chlorophenyl, 4-chlorophenyl, and 3-methoxyphenyl) behaved similarly to sorafenib and occupied the VEGFR-2 enzyme's allosteric site. Such hydrophobic moieties formed several hydrophobic bonds with Leu886, Leu887, Leu1017, and Ile890. Additionally, in the hinge area, the [1,2,4]triazolo[4,3-a]quinoxaline moiety of compounds 11a–d established a crucial hydrogen bond with Cys917 and shared a number of hydrophobic contacts with Ala864, Leu1033, Phe1045, and Phe916. Moreover, the central phenyl group of the synthesized derivatives formed several hydrophobic interactions with Val846, Val914, and Lys866 besides one electrostatic interaction with Cys1043 (Fig. 5C–F).
Molecular docking results indicate that 11d has a docking score of −23.65 kcal mol−1, which is slightly better than sorafenib (−22.50 kcal mol−1), suggesting a strong binding affinity for VEGFR-2. However, a deeper structural comparison with other VEGFR-2 inhibitors will enhance our understanding of its potential advantages.
2.3.1.1. Key binding interactions. Compound 11d interacts with Cys917 (hinge region), Glu883 and Asp1044 (DFG domain), and Leu886, Leu887, and Ile890 (hydrophobic pocket). These interactions mirror those of known VEGFR-2 inhibitors like sorafenib and lenvatinib, reinforcing the rationale for its design.
This good binding mode is attributed to the high number of hydrophobic interaction caused by the planar bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety. This moiety contains four nitrogen atoms that act as electron acceptors, facilitating hydrogen bonding in the hinge region. Additionally, its planar structure promoted hydrophobic interactions within the hinge region.
 | ||
| Fig. 6 MD analysis parameters: (A) RMSD values from the trajectory for the VEGFR-2 protein in VEGFR-2_11d (red line) and VEGFR-2_sorafenib complex (dark green line), (B) shows the ligands RMSD values, (C) radius of gyration for the VEGFR-2 protein in VEGFR-2_11d (red line) and VEGFR-2_sorafenib complex (dark green line), (D) SASA for VEGFR-2 protein in VEGFR-2_11d (red line) and VEGFR-2_sorafenib complex (dark green line), (E) change in the number of hydrogen bonds between 11d (pink line) or sorafenib (brown line) and VEGFR-2, (F) RMSF for VEGFR-2 protein in VEGFR-2_11d (red line) and VEGFR-2_sorafenib complex (dark green line), (G) distance from the center of mass of sorafenib or 11d compound and VEGFR-2 protein. | ||
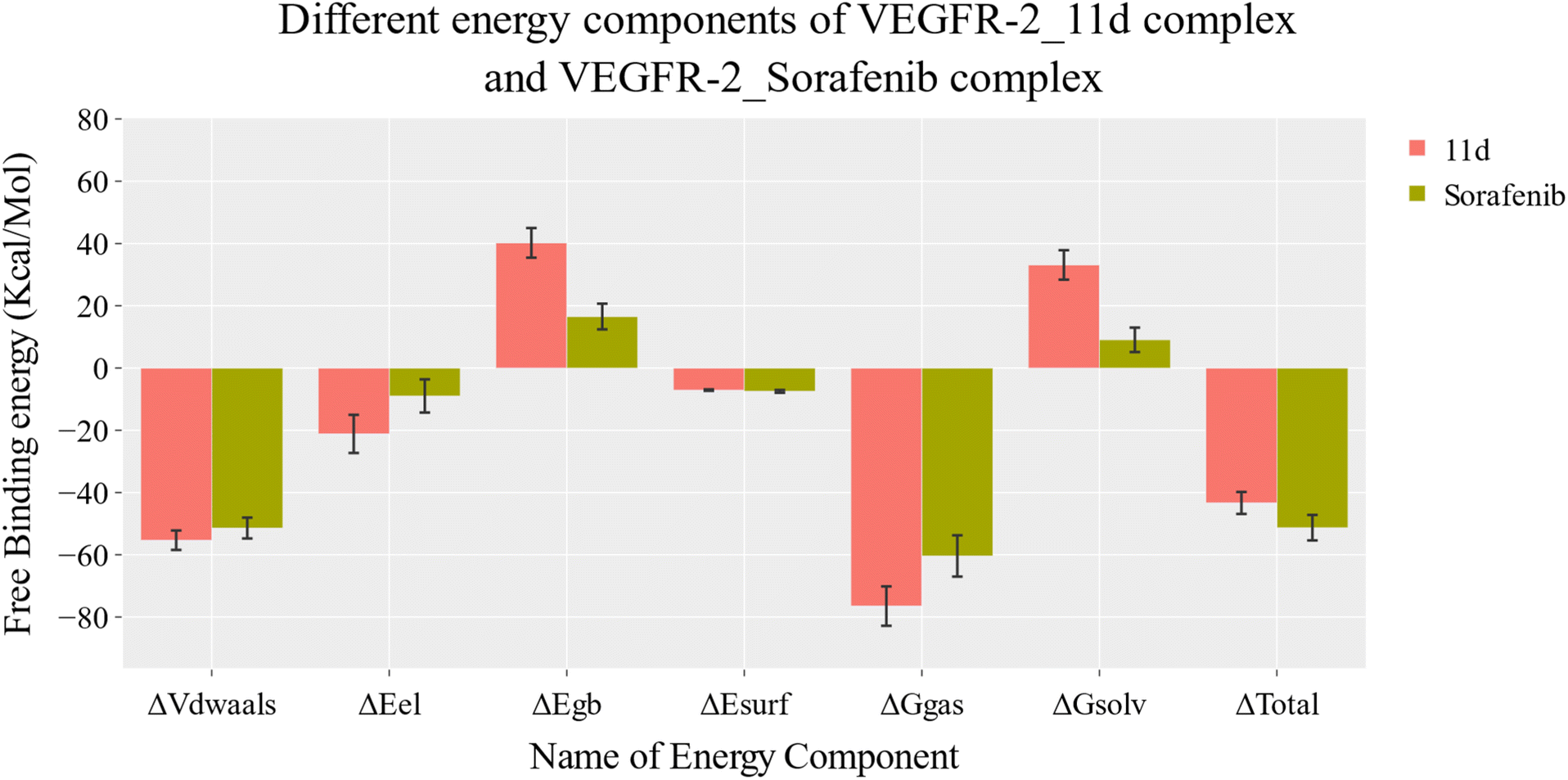 | ||
| Fig. 7 MM-GBSA analysis of the VEGFR-2-11d complex and VEGFR-2-sorafenib complex. Bars represent the standard deviations. | ||
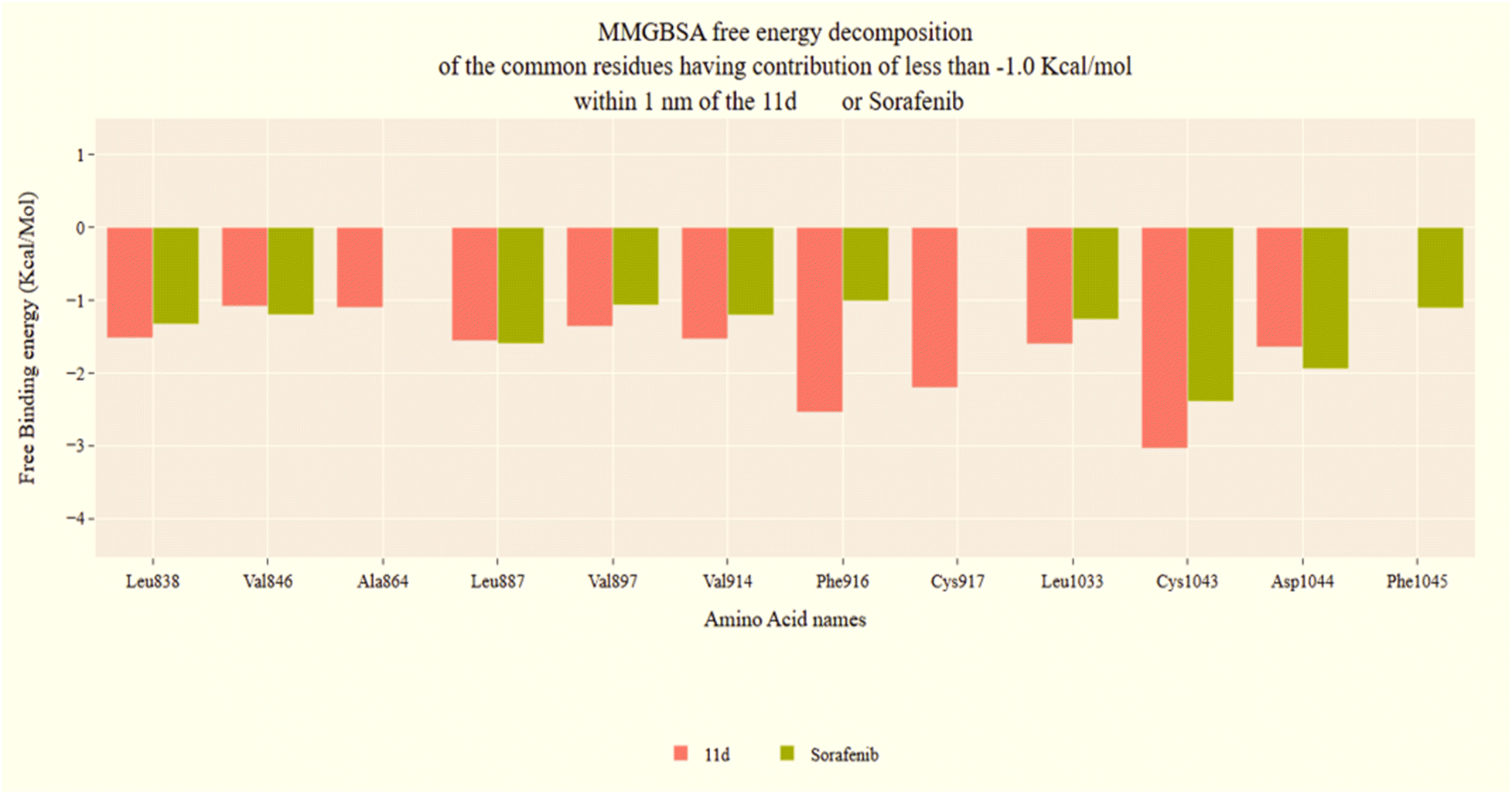 | ||
| Fig. 8 VEGFR-2-11d complex and VEGFR-2-sorafenib complex binding free energy decomposition. | ||
 | ||
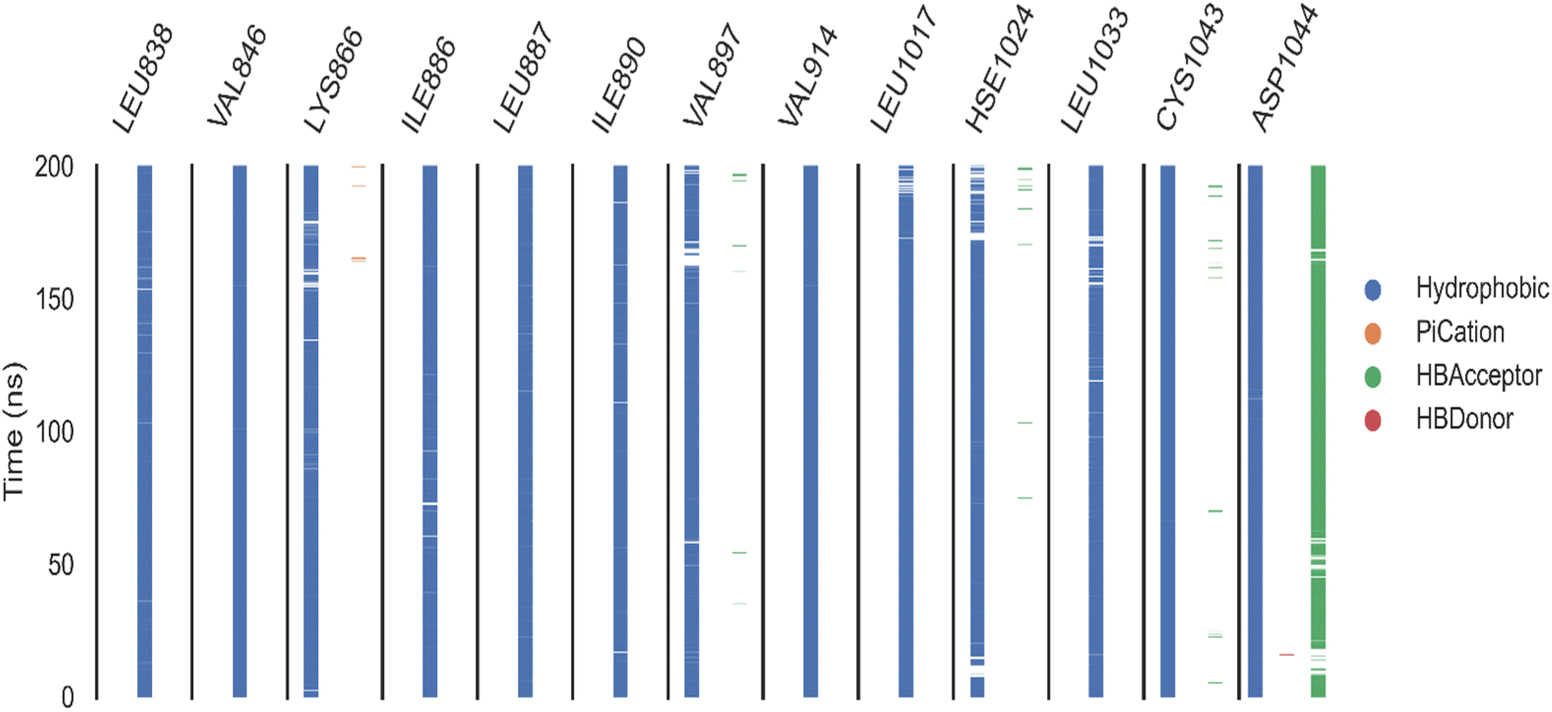
| Fig. 9 ProLIF assay: the ProLIF Python library was used to analyze the amino acids involved, the types of interactions within compound 11d-VEGFR-2 complex, and their frequency throughout the entire simulation. Panel (A) represents hydrophobic interactions (H.I.), panel (B) depicts van der Waals (VdW) contacts, and panel (C) shows hydrogen bond acceptor (HBA) interactions. | ||
 | ||
| Fig. 10 ProLIF assay: the ProLIF Python library was used to analyze the amino acids involved, the types of interactions within compound sorafenib-VEGFR-2 complex, and their frequency throughout the entire simulation. | ||
 | ||
| Fig. 11 Essential dynamics analysis for the first ten eigenvectors of VEGFER-2-11d complex and VEGFER-2-sorafenib complex. (A) Eigenvalues changing, (B) the eigenvectors' distribution, and (C) cosine values. | ||
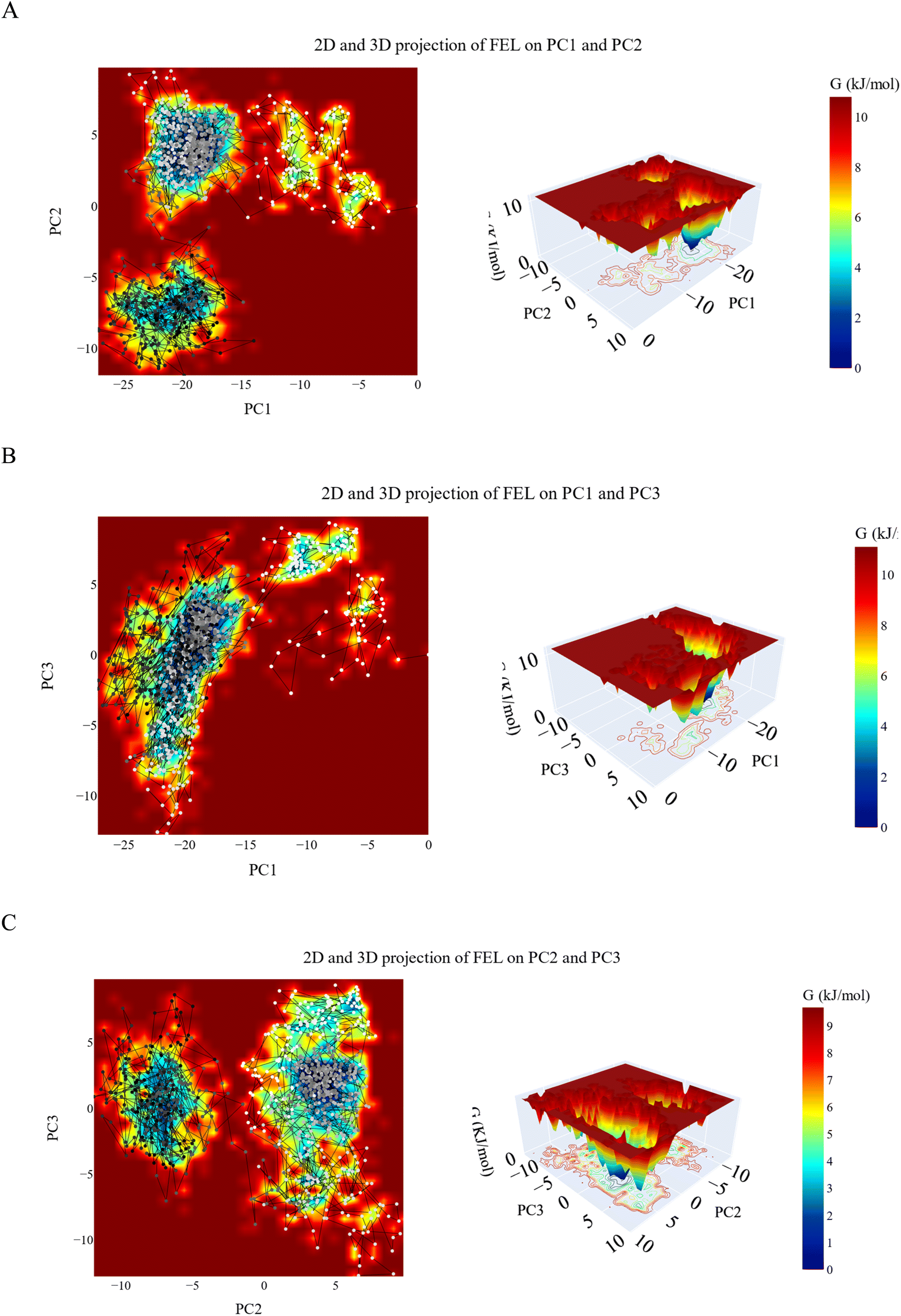 | ||
| Fig. 12 The 2D and 3D projections of the VEGFR-2_11d complex trajectories FEL on (A) the first two, (B) first and third and (C) second and third eigenvectors. | ||
 | ||
| Fig. 13 The optimized structure (A), the HOMO/LUMO distribution function and energy gap (B), molecular electrostatic potential map (C) and total density of state (D) at B3LYP/6-31+G(d,p) level for 11d. | ||
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | Dm (debye) | TE (eV) | ΔNmax | ΔE (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 5.904 | 2.335 | −4.120 | 4.120 | 1.784 | 0.560 | 15.142 | 4.279 | −39225.1 | 2.309 | −15.142 |
As seen in Fig. 13B, the density of the LUMO function is mostly dispersed across 11d's right side, but the distribution of the HOMO function is virtually evenly distributed over the left side. This unique distribution raises the possibility that directionality in the electron transfer routes through 11d from HOMO to LUMO which is important in electron transfer and charge separation reactions which are relevant in drug interaction. Additionally, as the HOMO designates the area where an electron can be transferred, the target will be able to approach the right side of 11d electrophilically. It was discovered that the HOMO/LUMO energy gap (Egap) was 3.569 eV. Table 8 displays the calculated global reactivity parameters. The electrophilicity index of 11d was determined to be 15.142 eV, indicating that it has a high electron-accepting capacity from biomolecules. Due to its high electrophilicity, the medication may interact effectively with protein nucleophilic sites. Also, 11d is soft (0.560 eV). The electrons' ability to escape is measured by chemical potential, which was found to be −4.12 eV. When a molecule loses electrons, its chemical potential is negative, indicating that it is relatively stable. A molecule is less able to donate electrons when its negative chemical potential is higher, which makes it more difficult to oxidize.60
To identify the nucleophilic and electrophilic active sites, Fig. 13C presents the analysis and presentation of the molecular electrostatic potential, or MESP. The electron-rich site's negative potential, as indicated by the MESP map, can form hydrogen bonds with the target's electrophilic active sites by spreading over the CO groups. The blue zones on hydrogens indicate sites that are electron-deficient have the ability to make hydrogen bonds with the target's nucleophilic sites. The target protein's hydrophobic regions can interact with the neutral green patches on the MESP map, Fig. 13C.
The overall density of states was examined using Multiwfn software, along with the number of states that might be created at each level. The HOMO energy line is shown as a dashed line in Fig. 13D. The electronic density of states is larger for orbitals higher than the LUMO, as can be seen from the total density chart. This suggests that 11d can readily receive more electrons from electron donors in biological settings. Because of its capacity to take electrons, 11d is more reactive in oxidative stress conditions, which are frequently present in cancer cells.
| Comp. | BBB level | Solubility level | Absorption level | CYP2D6 prediction | PPB prediction |
|---|---|---|---|---|---|
| 11a | High | Low | Good | Non inhibitor | More than 90% |
| 11b | Very low | Very low | |||
| 11c | |||||
| 11d | Low | ||||
| Sorafenib | Very low |
| Comp. | TD50 (rat)a | Ames prediction | Mouse-female FDA | MTD | Rat oral LD50b | Rat chronic LOAELb | SI | EI |
|---|---|---|---|---|---|---|---|---|
| a Unit: mg kg−1 body weight per day.b Unit: g kg−1 body weight. | ||||||||
| 11a | 38.7633 | Non-mutagen | Non-carcinogen | 0.12016 | 0.203002 | 0.0213632 | Non-irritant | Mild |
| 11b | 10.3763 | 0.147027 | 0.17387 | 0.017032 | ||||
| 11c | 10.3763 | 0.147027 | 0.275129 | 0.0158809 | ||||
| 11d | 3.12316 | 0.0655489 | 0.414131 | 0.0135571 | ||||
| Sorafenib | 14.2442 | Single-carcinogen | 0.088543 | 0.822583 | 0.00482816 | |||
In the aspect of chronic exposure, the lowest observed adverse effect levels (LOAEL) indicate that compounds 11a to 11d present lower chronic toxicity levels compared to sorafenib. For instance, compound 11a, with a LOAEL of 0.021 mg kg−1, is less toxic than sorafenib (LOAEL = 0.0048 mg kg−1), suggesting a potentially improved safety profile under long-term exposure. Skin and (SI) Eye Irritancy (EI): none of the 11-series compounds were found to be skin irritants, and all were categorized as mild ocular irritants. This aligns with sorafenib's profile, indicating a low likelihood of dermal toxicity.
In conclusion, the toxicological profile of the 11-series compounds shows potential advantages over sorafenib, particularly in terms of carcinogenic risk, chronic toxicity, and irritancy. However, additional studies, particularly in vivo, would be necessary to confirm the safety and efficacy of these compounds as therapeutic candidates.
The computational predictions suggest that 11d exhibits notable pharmacokinetic advantages over sorafenib, particularly in terms of absorption, safety, and toxicity as follows. (i) Sorafenib is known for its poor aqueous solubility, which limits its bioavailability and necessitates high oral doses (400 mg twice daily) to achieve therapeutic plasma concentrations. The ADMET results indicate that compound 11d has slightly better solubility than sorafenib, though it remains in the low-to-moderate range. Improved solubility enhances intestinal absorption, which could allow for lower dosing requirements, reducing systemic exposure to toxic metabolites. (ii) The ADMET predictions suggest that 11d has good intestinal permeability, similar to sorafenib. (iii) Unlike compound 11a, which has high BBB permeability, 11d has low BBB penetration, similar to sorafenib. This is a desirable feature for anticancer drugs targeting VEGFR-2, as it minimizes potential neurological side effects, such as cognitive impairment, that are often associated with tyrosine kinase inhibitors (TKIs) with high CNS exposure. (iv) Both sorafenib and 11d exhibit strong plasma protein binding (>90%), meaning that most of the drug remains bound to albumin and other plasma proteins. While high PPB can limit free drug availability, it also prolongs the half-life and may contribute to sustained drug action. The comparable PPB suggests that 11d could have a half-life similar to, or potentially longer than, sorafenib, though this needs to be validated with in vivo pharmacokinetic studies. (v) Sorafenib is classified as a single carcinogen, meaning it has some potential for long-term oncogenic effects. In contrast, compound 11d is predicted to be non-mutagenic and non-carcinogenic, making it a potentially safer long-term therapy. (vi) The Maximum Tolerated Dose (MTD) of 11d (0.065 mg kg−1) is slightly higher than sorafenib (0.0885 mg kg−1), suggesting that 11d has a better safety margin. (vii) The Lowest Observed Adverse Effect Level (LOAEL) of 11d (0.0136 mg kg−1) is higher than sorafenib (0.0048 mg kg−1), reinforcing its lower chronic toxicity potential. (viii) Unlike some kinase inhibitors that cause severe dermatological toxicity, 11d is predicted to be non-irritant to the skin and a mild ocular irritant, similar to sorafenib. This suggests that 11d may be better tolerated in long-term use.
3. Conclusion
In this study, we synthesized and evaluated a series of quinoxaline derivatives as potential VEGFR-2 inhibitors, focusing on their anti-cancer properties against breast cancer cells. Compound 11d demonstrated notable selectivity and efficacy, exhibiting potent cytotoxic effects on MDA-MB-231 and MCF-7 cancer cell lines while showing reduced toxicity in normal cell lines. Mechanistic studies revealed that 11d effectively inhibited VEGFR-2, induced apoptosis by upregulating pro-apoptotic genes, and significantly impaired cancer cell migration and cell cycle progression. Furthermore, in silico analyses, including molecular docking, molecular dynamics simulations, ProLIF, PCA, FEL and DFT calculations, indicated strong and stable binding of 11d to VEGFR-2, along with a favorable ADMET profile, positioning it as a safer alternative to sorafenib. Collectively, our findings highlight 11d as a promising candidate for targeted anti-angiogenic therapy. Further preclinical studies are warranted to validate these effects in vivo and assess the therapeutic potential of 11d in combination with current cancer treatments. For future directions, we hope to carry out the following tasks. In vivo efficacy studies to evaluate the tumor suppression ability of 11d. Further studies should assess half-life, metabolism, and biodistribution in vivo. Further modifications could improve solubility and oral bioavailability. Evaluating 11d in combination with standard chemotherapies could determine synergistic effects.4. Experimental
4.1. Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 9:1). Compounds 10a–d were obtained by filtering, washing with acetonitrile, and drying the precipitate.61
:methanol = 9:1). Compounds 10a–d were obtained by filtering, washing with acetonitrile, and drying the precipitate.614.1.2.1. 1-(4-([1,2,4]Triazolo[4,3-a]quinoxalin-4-yloxy)phenyl)-3-phenylurea 11a.
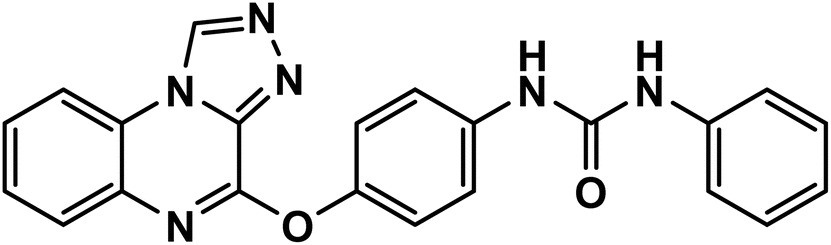 White powder (yield, 75%); mp = 213–215 °C. FT-IR (νmax, cm−1): 3421, 3119 (NH), 3060 (C–H aromatic), 2925, 2830 (C–H aliphatic), 1682 (CO); 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H, CH triazolo), 8.88 (s, 1H, NH), 8.85 (s, 1H, NH), 8.37 (d, J = 8.1 Hz, 1H, Ar–H), 7.76–7.48 (m, 8H, Ar–H), 7.35 (dd, J = 8.6, 6.1 Hz, 4H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 153.03, 151.72, 146.74, 139.19, 138.77, 138.73, 137.75, 134.34, 129.11, 128.30, 128.21, 127.78, 125.85, 124.54, 122.70, 120.23, 120.04, 116.92; mass (m/z): 396 (M+, 57.84%), 381 (100%, base peak); anal. calcd for C22H16N6O2 (396.41): C, 66.66; H, 4.07; N, 21.20. Found: C, 66.92; H, 4.21; N, 21.43%.
White powder (yield, 75%); mp = 213–215 °C. FT-IR (νmax, cm−1): 3421, 3119 (NH), 3060 (C–H aromatic), 2925, 2830 (C–H aliphatic), 1682 (CO); 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H, CH triazolo), 8.88 (s, 1H, NH), 8.85 (s, 1H, NH), 8.37 (d, J = 8.1 Hz, 1H, Ar–H), 7.76–7.48 (m, 8H, Ar–H), 7.35 (dd, J = 8.6, 6.1 Hz, 4H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 153.03, 151.72, 146.74, 139.19, 138.77, 138.73, 137.75, 134.34, 129.11, 128.30, 128.21, 127.78, 125.85, 124.54, 122.70, 120.23, 120.04, 116.92; mass (m/z): 396 (M+, 57.84%), 381 (100%, base peak); anal. calcd for C22H16N6O2 (396.41): C, 66.66; H, 4.07; N, 21.20. Found: C, 66.92; H, 4.21; N, 21.43%.
4.1.2.2. 1-(4-([1,2,4]Triazolo[4,3-a]quinoxalin-4-yloxy)phenyl)-3-(3-chlorophenyl)urea 11b.
 White powder (yield, 77%); mp = 207–209 °C. FT-IR (νmax, cm−1): 3331, 3103 (NH), 3067, 3014 (C–H aromatic), 1682 (CO); 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H, CH triazolo), 8.91 (s, 1H, NH), 8.86 (s, 1H, NH), 8.33 (dd, J = 8.1, 1.4 Hz, 1H, Ar–H), 7.69 (q, J = 1.5 Hz, 1H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.56–7.50 (m, 3H, Ar–H), 7.32 (d, J = 2.2 Hz, 1H, Ar–H), 7.31–7.29 (m, 1H, Ar–H), 7.28–7.24 (m, 2H, Ar–H), 6.98 (dt, J = 6.7, 2.3 Hz, 1H, Ar–H); 13C NMR (126 MHz, DMSO-d6) δ 153.06, 151.76, 147.00, 141.83, 138.81, 137.68, 134.44, 133.77, 130.91, 128.37, 128.31, 127.87, 124.64, 122.71, 122.04, 120.25, 118.23, 117.28, 116.99; mass (m/z): 430 (M+, 22.40%), 402 (100%, base peak); anal. calcd for C22H15ClN6O2 (430.85): C, 61.33; H, 3.51; N, 19.51. Found: C, 61.60; H, 3.79; N, 19.68%.
White powder (yield, 77%); mp = 207–209 °C. FT-IR (νmax, cm−1): 3331, 3103 (NH), 3067, 3014 (C–H aromatic), 1682 (CO); 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H, CH triazolo), 8.91 (s, 1H, NH), 8.86 (s, 1H, NH), 8.33 (dd, J = 8.1, 1.4 Hz, 1H, Ar–H), 7.69 (q, J = 1.5 Hz, 1H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.56–7.50 (m, 3H, Ar–H), 7.32 (d, J = 2.2 Hz, 1H, Ar–H), 7.31–7.29 (m, 1H, Ar–H), 7.28–7.24 (m, 2H, Ar–H), 6.98 (dt, J = 6.7, 2.3 Hz, 1H, Ar–H); 13C NMR (126 MHz, DMSO-d6) δ 153.06, 151.76, 147.00, 141.83, 138.81, 137.68, 134.44, 133.77, 130.91, 128.37, 128.31, 127.87, 124.64, 122.71, 122.04, 120.25, 118.23, 117.28, 116.99; mass (m/z): 430 (M+, 22.40%), 402 (100%, base peak); anal. calcd for C22H15ClN6O2 (430.85): C, 61.33; H, 3.51; N, 19.51. Found: C, 61.60; H, 3.79; N, 19.68%.
4.1.2.3. 1-(4-([1,2,4]Triazolo[4,3-a]quinoxalin-4-yloxy)phenyl)-3-(4-chlorophenyl)urea 11c.
 White powder (yield, 78%); mp = 200–202 °C. FT-IR (νmax, cm−1): 3320 (NH), 3056 (C–H aromatic), 2920 (C–H aliphatic), 1654 (CO); 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H, CH triazolo), 8.84 (s, 1H, NH), 8.81 (s, 1H, NH), 8.33 (dd, J = 8.2, 1.4 Hz, 1H, Ar–H), 7.65–7.58 (m, 2H, Ar–H), 7.56–7.50 (m, 3H, Ar–H), 7.49–7.45 (m, 2H, Ar–H), 7.32–7.28 (m, 4H, Ar–H); 13C NMR (126 MHz, DMSO-d6) δ 153.14, 151.73, 147.05, 139.27, 138.82, 138.76, 137.79, 134.47, 129.12, 128.35, 128.32, 127.86, 126.08, 124.66, 122.62, 120.47, 120.20, 116.97; mass (m/z): 430 (M+, 34.27%), 48 (100%, base peak); anal. calcd for C22H15ClN6O2 (430.85): C, 61.33; H, 3.51; N, 19.51. Found: C, 61.58; H, 3.75; N, 19.59%.
White powder (yield, 78%); mp = 200–202 °C. FT-IR (νmax, cm−1): 3320 (NH), 3056 (C–H aromatic), 2920 (C–H aliphatic), 1654 (CO); 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H, CH triazolo), 8.84 (s, 1H, NH), 8.81 (s, 1H, NH), 8.33 (dd, J = 8.2, 1.4 Hz, 1H, Ar–H), 7.65–7.58 (m, 2H, Ar–H), 7.56–7.50 (m, 3H, Ar–H), 7.49–7.45 (m, 2H, Ar–H), 7.32–7.28 (m, 4H, Ar–H); 13C NMR (126 MHz, DMSO-d6) δ 153.14, 151.73, 147.05, 139.27, 138.82, 138.76, 137.79, 134.47, 129.12, 128.35, 128.32, 127.86, 126.08, 124.66, 122.62, 120.47, 120.20, 116.97; mass (m/z): 430 (M+, 34.27%), 48 (100%, base peak); anal. calcd for C22H15ClN6O2 (430.85): C, 61.33; H, 3.51; N, 19.51. Found: C, 61.58; H, 3.75; N, 19.59%.
4.1.2.4. 1-(4-([1,2,4]Triazolo[4,3-a]quinoxalin-4-yloxy)phenyl)-3-(3-methoxyphenyl)urea 11d.
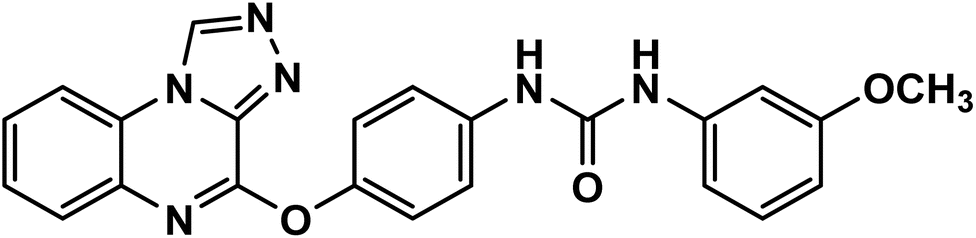 White powder (yield, 80%); mp = 208–210 °C. FT-IR (νmax, cm−1): 3327, 3103 (NH), 3067, 3014 (C–H aromatic), 2915 (C–H aliphatic), 1679 (CO); 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H, CH triazolo), 8.79 (s, 1H, NH), 8.74 (s, 1H, NH), 8.36 (d, J = 8.1 Hz, 1H, Ar–H), 7.68–7.54 (m, 6H, Ar–H), 7.34 (d, J = 8.5 Hz, 2H, Ar–H), 7.25–7.13 (m, 2H, Ar–H), 6.96 (d, J = 8.0 Hz, 1H, Ar–H), 6.61–6.52 (m, 1H, Ar–H), 3.74 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d6) δ 160.17, 153.06, 151.72, 146.66, 141.36, 138.76, 138.73, 137.84, 134.33, 130.05, 128.33, 128.21, 127.80, 124.52, 122.68, 119.98, 116.91, 111.04, 107.75, 104.49, 55.41; mass (m/z): 426 (M+, 33.73%), 350 (100%, base peak); anal. calcd for C23H18N6O3 (426.44): C, 64.78; H, 4.25; N, 19.71. Found: C, 64.67; H, 4.48; N, 19.95%.
White powder (yield, 80%); mp = 208–210 °C. FT-IR (νmax, cm−1): 3327, 3103 (NH), 3067, 3014 (C–H aromatic), 2915 (C–H aliphatic), 1679 (CO); 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H, CH triazolo), 8.79 (s, 1H, NH), 8.74 (s, 1H, NH), 8.36 (d, J = 8.1 Hz, 1H, Ar–H), 7.68–7.54 (m, 6H, Ar–H), 7.34 (d, J = 8.5 Hz, 2H, Ar–H), 7.25–7.13 (m, 2H, Ar–H), 6.96 (d, J = 8.0 Hz, 1H, Ar–H), 6.61–6.52 (m, 1H, Ar–H), 3.74 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO-d6) δ 160.17, 153.06, 151.72, 146.66, 141.36, 138.76, 138.73, 137.84, 134.33, 130.05, 128.33, 128.21, 127.80, 124.52, 122.68, 119.98, 116.91, 111.04, 107.75, 104.49, 55.41; mass (m/z): 426 (M+, 33.73%), 350 (100%, base peak); anal. calcd for C23H18N6O3 (426.44): C, 64.78; H, 4.25; N, 19.71. Found: C, 64.67; H, 4.48; N, 19.95%.
4.2. In vitro studies
4.3. Computational studies
Data availability
Data are available in the manuscript and the ESI.†Conflicts of interest
No conflict of interest to be declared.Acknowledgements
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2025R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors would like to thank Research Center at AlMaarefa University for funding this work.References
- S. M. Schwartz, Clin. Chem., 2024, 70, 140–149 CrossRef.
- D. Trapani, O. Ginsburg, T. Fadelu, N. U. Lin, M. Hassett, A. M. Ilbawi, B. O. Anderson and G. Curigliano, Cancer Treat Rev., 2022, 104, 102339 CrossRef PubMed.
- L. Wilkinson and T. Gathani, Br. J. Radiol., 2022, 95, 20211033 CrossRef PubMed.
- A. A. Shah, M. A. Kamal and S. Akhtar, Curr. Drug Metab., 2021, 22, 50–59 CAS.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, A. E. Abd El-Wahab, E. B. Elkaeed, B. A. Alsfouk, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dyn., 2023, 41, 11535–11550 CrossRef CAS.
- X. J. Liu, H. C. Zhao, S. J. Hou, H. J. Zhang, L. Cheng, S. Yuan, L. R. Zhang, J. Song, S. Y. Zhang and S. W. Chen, Bioorg. Chem., 2023, 133, 106425 CrossRef CAS.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. B. Mehany, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dyn., 2023, 41, 7986–8001 CrossRef CAS.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS.
- V. Montero, M. Montana, M. Carré and P. Vanelle, Eur. J. Med. Chem., 2024, 271, 116360 CrossRef CAS.
- M. Zayed, Chemistry, 2023, 5, 2566–2587 CrossRef CAS.
- M. S. Nafie, S. H. Kahwash, M. M. Youssef and K. M. Dawood, Arch. Pharm., 2024, 357, e2400225 CrossRef PubMed.
- M. A. Dahab, H. A. Mahdy, H. Elkady, M. S. Taghour, A. Elwan, M. A. Elkady, E. G. E. Elsakka, E. B. Elkaeed, A. A. Alsfouk, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dynam., 2024, 42, 4214–4233 CrossRef CAS PubMed.
- A. M. Metwaly, H. Abd-El-Azim, M. Zewail, A. A. Alsfouk, E. B. Elkaeed and I. H. Eissa, J. Comput. Biophys. Chem., 2024, 1–17 CAS.
- I. H. Eissa, R. G. Yousef, H. Elkady, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, N. El-Deeb, A. M. Kenawy, W. M. Eldehna, E. B. Elkaeed and A. M. Metwaly, Mol. Divers., 2024, 28, 1153–1173 CrossRef CAS PubMed.
- I. H. Eissa, R. G. Yousef, E. B. Elkaeed, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. Ismail, H. Elkady and A. M. Metwaly, ACS Omega, 2024, 9, 15861–15881 CrossRef CAS PubMed.
- E. A. Sobh, M. A. Dahab, E. B. Elkaeed, A. A. Alsfouk, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dynam., 2024, 42, 2369–2391 CrossRef CAS.
- I. H. Eissa, R. G. Yousef, E. B. Elkaeed, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, H. A. El-Mahdy, H. Elkady and A. M. Metwaly, Evol. Bioinf. Online, 2023, 19, 11769343231217916 CrossRef.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and I. H. Eissa, Bioorg. Chem., 2021, 114, 105105 CrossRef CAS.
- R. G. Yousef, H. M. Sakr, I. H. Eissa, A. B. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and K. El-Adl, New J. Chem., 2021, 45, 16949–16964 RSC.
- I. H. Eissa, A. M. Metwaly, A. Belal, A. B. M. Mehany, R. R. Ayyad, K. El-Adl, H. A. Mahdy, M. S. Taghour, K. M. A. El-Gamal, M. E. El-Sawah, S. A. Elmetwally, M. A. Elhendawy, M. M. Radwan and M. A. ElSohly, Arch. Pharm., 2019, 352, e1900123 CrossRef PubMed.
- M. K. Ibrahim, M. S. Taghour, A. M. Metwaly, A. Belal, A. B. M. Mehany, M. A. Elhendawy, M. M. Radwan, A. M. Yassin, N. M. El-Deeb, E. E. Hafez, M. A. ElSohly and I. H. Eissa, Eur. J. Med. Chem., 2018, 155, 117–134 CrossRef CAS.
- M. K. Ibrahim, I. H. Eissa, A. E. Abdallah, A. M. Metwaly, M. M. Radwan and M. A. ElSohly, Bioorg. Med. Chem., 2017, 25, 1496–1513 CrossRef CAS PubMed.
- Y. Liu, Y. Li, Y. Wang, C. Lin, D. Zhang, J. Chen, L. Ouyang, F. Wu, J. Zhang and L. Chen, J. Hematol. Oncol., 2022, 15, 89 CrossRef CAS.
- S. Fogli, C. Porta, M. Del Re, S. Crucitta, G. Gianfilippo, R. Danesi, B. I. Rini and M. Schmidinger, Cancer Treat Rev., 2020, 84, 101966 CrossRef CAS PubMed.
- E. S. Duke, A. K. Barone, S. Chatterjee, P. S. Mishra-Kalyani, Y.-L. Shen, E. Isikwei, H. Zhao, Y. Bi, J. Liu and N. A. Rahman, Clin. Cancer Res., 2022, 28, 4173–4177 CrossRef CAS PubMed.
- N. Agarwal, A. Azad, J. Carles, S. Chowdhury, B. McGregor, A. S. Merseburger, S. Oudard, F. Saad, A. Soares and F. Benzaghou, Future Oncol., 2022, 18, 1185–1198 CrossRef CAS PubMed.
- S. M. Ferrari, M. Centanni, C. Virili, M. Miccoli, P. Ferrari, I. Ruffilli, F. Ragusa, A. Antonelli and P. Fallahi, Curr. Med. Chem., 2019, 26, 963–972 CrossRef CAS.
- B. Chaudhari, H. Patel, S. Thakar, I. Ahmad and D. Bansode, In Silico Pharmacol., 2022, 10, 10 CrossRef PubMed.
- D. Pink, D. Andreou, S. Bauer, T. Brodowicz, B. Kasper, P. Reichardt, S. Richter, L. H. Lindner, J. Szkandera and V. Grünwald, Cancers, 2021, 13, 1223 CrossRef CAS PubMed.
- Y. Pang, A. Eresen, Z. Zhang, Q. Hou, Y. Wang, V. Yaghmai and Z. Zhang, Am. J. Cancer Res., 2022, 12, 2770 CAS.
- A.-L. Cheng, S. Qin, M. Ikeda, P. R. Galle, M. Ducreux, T.-Y. Kim, H. Y. Lim, M. Kudo, V. Breder and P. Merle, J. Hepatol., 2022, 76, 862–873 CrossRef CAS.
- B. Liang, M. Tang, C. Huang, Y. Yang, Y. He, S. Liao and W. Shen, J. Gastrointest. Cancer, 2025, 56, 36 CrossRef.
- J. Guo, J. Zhao, Q. Xu and D. Huang, Curr. Cancer Drug Targets, 2022, 22, 865–878 CrossRef CAS PubMed.
- L. J. Scott, Drugs, 2015, 75, 553–560 CrossRef CAS PubMed.
- P. Norman, Expert Opin. Orphan Drugs, 2015, 3, 445–455 CrossRef CAS.
- R. Elisei, M. J. Schlumberger, S. P. Müller, P. Schöffski, M. S. Brose, M. H. Shah, L. Licitra, B. Jarzab, V. Medvedev and M. C. Kreissl, J. Clin. Oncol., 2013, 31, 3639 CrossRef CAS PubMed.
- S. Wilhelm, C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz, R. Simantov and S. Kelley, Nat. Rev. Drug Discov., 2006, 5, 835–844 CrossRef CAS PubMed.
- S. M. Wilhelm, J. Dumas, L. Adnane, M. Lynch, C. A. Carter, G. Schütz, K. H. Thierauch and D. Zopf, Int. J. Cancer, 2011, 129, 245–255 CrossRef CAS PubMed.
- H. Y. Woo and J. Heo, Expet Opin. Pharmacother., 2012, 13, 1059–1067 CrossRef CAS PubMed.
- S. DiGiulio, Oncology Times, 2013 Search PubMed.
- Y. Li, Z. H. Gao and X. J. Qu, Basic Clin. Pharmacol. Toxicol., 2015, 116, 216–221 CrossRef CAS PubMed.
- S. K. Krishnamoorthy, V. Relias, S. Sebastian, V. Jayaraman and M. W. Saif, Ther. Adv. Gastroenterol., 2015, 8, 285–297 CrossRef CAS PubMed.
- C. Grüllich, in Small Molecules in Oncology, Springer, 2014, pp. 207–214 Search PubMed.
- C. Zhu, X. Ma, Y. Hu, L. Guo, B. Chen, K. Shen and Y. Xiao, Oncotarget, 2016, 7, 44545 CrossRef PubMed.
- F.-W. Peng, D.-K. Liu, Q.-W. Zhang, Y.-G. Xu and L. Shi, Expert Opin. Ther. Pat., 2017, 27, 987–1004 CrossRef CAS PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, J. Mol. Struct., 2021, 132220 Search PubMed.
- K. Lee, K.-W. Jeong, Y. Lee, J. Y. Song, M. S. Kim, G. S. Lee and Y. Kim, Eur. J. Med. Chem., 2010, 45, 5420–5427 CrossRef CAS PubMed.
- V. A. Machado, D. Peixoto, R. Costa, H. J. Froufe, R. C. Calhelha, R. M. Abreu, I. C. Ferreira, R. Soares and M.-J. R. Queiroz, Bioorg. Med. Chem., 2015, 23, 6497–6509 CrossRef CAS PubMed.
- Z. Wang, N. Wang, S. Han, D. Wang, S. Mo, L. Yu, H. Huang, K. Tsui, J. Shen and J. Chen, PLoS One, 2013, 8, e68566 CrossRef CAS PubMed.
- J. Dietrich, C. Hulme and L. H. Hurley, Bioorg. Med. Chem., 2010, 18, 5738–5748 CrossRef CAS PubMed.
- Q.-Q. Xie, H.-Z. Xie, J.-X. Ren, L.-L. Li and S.-Y. Yang, J. Mol. Graph. Model., 2009, 27, 751–758 CrossRef CAS PubMed.
- R. N. Eskander and K. S. Tewari, Gynecol. Oncol., 2014, 132, 496–505 CrossRef CAS PubMed.
- M. Ibrahim, M. Taghour, A. M. Metwaly, A. Belal, A. Mehany, M. Elhendawy, M. Radwan, A. Yassin, N. El-Deeb and E. Hafez, Eur. J. Med. Chem., 2018, 155, 117–134 CrossRef CAS PubMed.
- I. H. Eissa, A. M. Metwaly, A. Belal, A. B. Mehany, R. R. Ayyad, K. El-Adl, H. A. Mahdy, M. S. Taghour, K. M. El-Gamal and M. E. El-Sawah, Arch. Pharm., 2019, 352, 1900123 CrossRef CAS PubMed.
- E. M. Abbass, A. K. Khalil, M. M. Mohamed, I. H. Eissa and A. M. El-Naggar, Bioorg. Chem., 2020, 104, 104255 CrossRef CAS PubMed.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, A. Elwan and H. Elkady, J. Enzym. Inhib. Med. Chem., 2021, 36, 1093–1114 CrossRef CAS PubMed.
- F. Azimian, M. Hamzeh-Mivehroud, J. S. Mojarrad, S. Hemmati and S. Dastmalchi, Eur. J. Med. Chem., 2020, 201, 112461 CrossRef CAS PubMed.
- D. Z. Husein, R. Hassanien and M. Khamis, RSC Adv., 2021, 11, 27027–27041 RSC.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Gieling, M. Babur, L. Mamnani, N. Burrows, B. A. Telfer, F. Carta, J.-Y. Winum, A. Scozzafava, C. T. Supuran and K. J. Williams, J. Med. Chem., 2012, 55, 5591–5600 CrossRef CAS PubMed.
- A. R. Kotb, D. A. Bakhotmah, A. E. Abdallah, H. Elkady, M. S. Taghour, I. H. Eissa and M. A. El-Zahabi, RSC Adv., 2022, 12, 33525–33539 RSC.
- A. R. Kotb, A. E. Abdallah, H. Elkady, I. H. Eissa, M. S. Taghour, D. A. Bakhotmah, T. M. Abdelghany and M. A. El-Zahabi, RSC Adv., 2023, 13, 10488–10502 RSC.
- A. Saha, S. Mohapatra, P. Kurkute, B. Jana, J. Sarkar, P. Mondal and S. Ghosh, RSC Adv., 2015, 5, 92596–92601 RSC.
- H. A. Mahdy, H. Elkady, M. S. Taghour, A. Elwan, M. A. Dahab, M. A. Elkady, E. G. Elsakka, E. B. Elkaeed, B. A. Alsfouk and I. M. Ibrahim, Future Med. Chem., 2023, 15, 1233–1250 CrossRef CAS PubMed.
- A. E. Abdallah, R. R. Mabrouk, M. R. Elnagar, A. M. Farrag, M. H. Kalaba, M. H. Sharaf, E. M. El-Fakharany, D. A. Bakhotmah, E. B. Elkaeed and M. M. S. Al Ward, Drug Des., Dev. Ther., 2023, 587–606 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- Z. Darzynkiewicz, E. Bedner and P. Smolewski, Flow cytometry in analysis of cell cycle and apoptosis, Seminars in hematology, Elsevier, 2001, pp. 179–193 Search PubMed.
- D. Bhunia, A. Saha, A. Adak, G. Das and S. Ghosh, RSC Adv., 2016, 6, 113487–113491 RSC.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels and S. Boresch, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- S. Jo, X. Cheng, S. M. Islam, L. Huang, H. Rui, A. Zhu, H. S. Lee, Y. Qi, W. Han and K. Vanommeslaeghe, Adv. Protein Chem. Struct. Biol., 2014, 96, 235–265 CAS.
- T. Tuccinardi, Expet Opin. Drug Discov., 2021, 16, 1233–1237 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theor. Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins: Struct., Funct., Bioinf., 1993, 17, 412–425 CrossRef CAS PubMed.
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graph. Model., 2009, 27, 889–899 CrossRef CAS PubMed.
- A. M. Metwaly, E. B. Elkaeed, B. A. Alsfouk, A. M. Saleh, A. E. Mostafa and I. H. Eissa, Plants, 2022, 11, 1886 CrossRef CAS PubMed.
- D. S. Biovia, Discovery studio modeling environment, Release San Diego, CA, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00526d |
| This journal is © The Royal Society of Chemistry 2025 |