
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DFT study for hydrogen storage application on pristine magnesium dicarbide (MgC2) monolayer
Irfan Ahmeda,
Ukkasha Iqrara,
Ashir Saeeda,
Rajeh Alotaibib,
Syed Mansoor Alic,
Maida Anward and
Muhammad Isa Khan *a
*a
aDepartment of Physics, Rahim Yar Khan Campus, The Islamia University of Bahawalpur, Bahawalpur, Pakistan. E-mail: irfanjamm315@gmail.com; ashir.saeed@iub.edu.pk; ui939msc@gmail.com; muhammad.isa@iub.edu.pk
bDepartment of Chemistry, College of Science, King Saud University, P. O. BOX 145111, Riyadh, Saudi Arabia. E-mail: raalotaibi@ksu.edu.sa
cDepartment of Physics and Astronomy, College of Science, King Saud University, P. O. BOX 2455, Riyadh 11451, Saudi Arabia. E-mail: symali@ksu.edu.sa
dDepartment of Physics and Astronomy “Galileo Galilei”, University of Padua, Via VIII febbraio 2, 35122 Padua, Italy. E-mail: maidaanwar2021@gmail.com
First published on 11th April 2025
Abstract
The hydrogen storage potential of pure MgC2 was systematically investigated using density functional theory (DFT) calculations. The phonon dispersion and ab initio molecular dynamics (AIMD) simulations confirmed the dynamic and structural stability of MgC2, reinforcing its suitability as a promising hydrogen storage material. The electronic structure analysis revealed that pure MgC2 exhibits semiconducting behavior with a band gap of 0.25 eV, and transforms into a metallic state upon hydrogen adsorption. Hydrogen molecules were adsorbed onto the MgC2 surface via physisorption, with an average adsorption energy of 0.286 eV, indicating moderate binding strength suitable for reversible hydrogen storage. Hirshfeld charge analysis demonstrated that MgC2 transfers 0.041 e, 0.139 e, and 0.259 e to 1, 4, and 8 hydrogen molecules, respectively, highlighting charge redistribution upon adsorption. The calculated hydrogen storage capacity of 2.05% suggests a feasible adsorption mechanism. Additionally, AIMD simulations at 400 K confirmed that hydrogen adsorption does not induce significant distortions in the MgC2 framework, further validating its thermal and mechanical stability. These findings underscore the potential of MgC2 as an efficient hydrogen storage material for sustainable energy applications, offering a promising pathway for the development of next-generation clean energy technologies.
1. Introduction
One of the main and most efficient ways to address the enormous rise in energy consumption and environmental protection is the development of clean, sustainable energy sources like wind, solar, and hydrogen.1–4 However, since hydrogen is not readily available in nature, it must be stored for use in transportation, as well as other mobile and stationary applications.5,6 There are two types of hydrogen storage technologies: physical-based and material-based. The first category consists of currently in-use technologies that require specialized, big-volume containers to store hydrogen as compressed gas, cold/cryocompressed gas, and liquid.7 The second method involves chemical reactions within materials at the atomic or molecular level. It is still quite difficult to find a substance that can safely, effectively, and affordably store hydrogen for real-world applications.8,9 Recently, advancements in hydrogen storage technology have made it possible to store a substantial amount of hydrogen fuel safely, which has tremendously benefited the development of cars that run on hydrogen. Over the years, many efforts have been made to produce pure hydrogen (H2) without harming the environment. According to the standards of the U.S. Department of Energy (DOE), a material used for hydrogen storage must have a storage gravimetric density of more than 5.5 wt%.10In recent years, there has been enormous interest in nanostructured materials to store hydrogen by an adsorption mechanism.11,12 These factors have led to the proposal of many materials, including MgH2, Mg(BH4)2, and LiBH4, for the solid-state storage of hydrogen. Despite having a high gravimetric density of hydrogen, these materials exhibit significant dissociation temperatures and thermodynamic stability.13,14 The hydrogen storage capacity and recovery efficiency of carbon-based nanomaterials, such as graphene, are significantly influenced by lithium- and sodium-decorated inorganic graphenylene.
Although surface adsorption of H2 is inefficient for hydrogen release, it is appropriate for improving the quantity of storage. Additionally, to keep the reservoir pressure high once H2 has been retrieved, an absorbent gas, such as N2, CH4, or CO2, is required. This leads to competitive adsorption between H2 and the cushion gas, significantly affecting H2 recovery and purification in the recovered fluids.15 Consequently, none of them is a good option. On the other hand, magnesium hydride (MgH2) has attracted significant attention due to its high volumetric capacity (110 g L−1) and gravimetric density (7.6 wt%), non-toxicity, good reversibility, and vast natural reserves. Furthermore, when supplying mixed gases free of harmful components, materials like MgH2 show enormous potential for hydrogen filtration and selective storage.16,17 Qin et al. conducted first-principles calculations to investigate the structural, mechanical, electronic, dynamic, thermodynamic, optical, and hydrogen storage properties of XMoH3 (X = Na, K, Rb, Cs). Their findings confirm the thermodynamic, mechanical, and dynamic stability of these perovskites based on formation energy, elastic constants, and phonon spectra.18
Recent investigations have explored the potential of newly proposed 2D MXene materials, including Sc2C, Cr2C, Ti2C, Ti2N, and V2C for hydrogen storage applications. Liu et al. investigated the use of V2C (vanadium carbide) and Ti3C2 (titanium carbide) MXenes in combination to improve the capacity of MgH2 to absorb and desorb hydrogen. By incorporating 2V2C/Ti3C2, the activation energy for the hydrogen release process of magnesium hydride (MgH2) was successfully reduced by 36%, resulting in a reversible capacity of 6.3%.19
The SiB2 monolayer, recognized for its lightweight nature, high carrier mobility, and exceptional stability, holds significant potential as an electrode material. Its structure is similar to that of MgB2, which has been extensively studied.20,21 The MgB2 has been investigated for applications such as hydrogen storage and alkali metal ion batteries. The hydrogenation of bulk MgB2 requires high pressure (90–100 MPa) and temperature (390–400 °C) and is time-intensive. Li et al. studied nanoscale MgB2 hydrogenation via mechanical milling, though the reaction pathway and intermediate states remain unclear.22 Theoretical studies suggest that Li decoration can enhance hydrogen storage capacity to 7.26 wt%,23 while Na and Li addition improves H2 storage efficiency in C2O by 13.08 wt%.24 In our previous study, we employed DFT to investigate optimized structures, thermodynamic properties, and H2 storage capacities of MgB2. The results highlight a subtle charge transfer from Li/Na/K to the MgB2 monolayer, enhancing its electropositive nature and improving electrostatic interactions with H2 molecules. The maximum H2 adsorption occurs with nine H2 molecules for Li and eight H2 molecules for Na and K. The corresponding adsorption energies range from −0.24 to −0.21 eV for Li, −0.22 to −0.20 eV for Na, and −0.25 to −0.20 eV for K.25 Previously, we have investigated various 2D materials and transition metal chalcogenides (TMDs) for alkali and alkaline earth metal-ion batteries, focusing on their structural, adsorption, electronic properties, open-circuit voltage (OCV), theoretical capacity, and thermodynamic stability.26–31
From the above discussion, it is clear that a thorough analysis of pristine MgC2's capacity to store hydrogen, taking van der Waals correction parameters into account, has not yet been carried out. This gap inspired us to conduct a DFT calculation from the ground up to examine the hydrogen-adsorbed MgC2 system's electrical characteristics, binding/adsorption energies, and desorption temperatures.
2. Computational detail
The Amsterdam density functional (ADF-BAND) software was used in the DFT framework, and Slater Type Orbitals (STO) was primarily used to create molecular orbitals for every structure.32 Based on the framework offered by the Perdew–Burke–Ernzerhof (PBE) functional, the generalized gradient approximation (GGA) was used throughout the study to compute the exchange-correlation energies required for structure optimization. Given that van der Waals forces are the main interaction forces between H2 and the substrate material, we addressed the shortcomings of DFT by integrating the Grime semi-empirical correction technique (DFT-D3) throughout the calculations in handling dispersive interactions. Additionally, our method used double-zeta polarized (DZP) basis sets and excellent numerical quality, together with settings that permitted non-frozen cores.33 For fully relaxed geometries, we impose strict requirements, with an energy optimization threshold of 10−5 eV. We maintained 10−3 Å as the step convergence criterion and 0.02 eV Å−1 as the gradient convergence criterion. Hirshfeld charge analysis was used to probe the arrangement of electrical ions between the absorption setups and optimum substances.The adsorption energy of hydrogen molecules on pure MgC2 is calculated by following the formula.
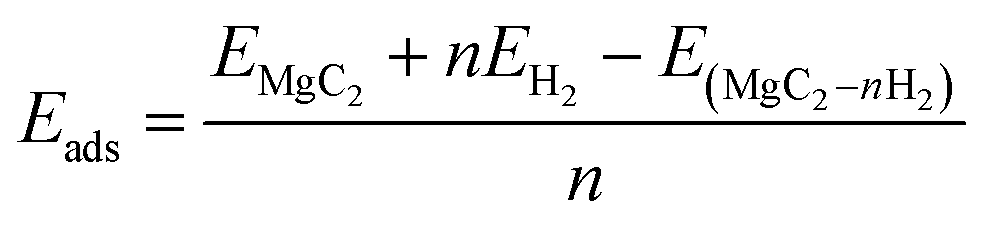 | (1) |
To determine hydrogen adsorption on the MgC2 nanosheet, we calculated the binding energy (EBE) of the H2 molecule using the following relation35
| EBE = EMgC2 + nEH2 − EnH2–MgC2 | (2) |
To determine the gravimetric storage capacity, the following formula was used:
 | (3) |
The desorption temperature (Td) of hydrogen-adsorbed systems is crucial for practical applications. We used the Van't Hoff equation to analyze the desorption temperature.35,36
 | (4) |
The desorption energy (Edes) is calculated as follows:
| Edes = EH2 + EMgC2+(n−1)H2 − EMgC2+nH2 | (5) |
3. Results and discussion
3.1 Structural and electronic properties of MgC2 monolayer
The relaxed crystal structure of the MgC2 monolayer within the P6mm space group is shown in Fig. 1(a). We have previously computationally designed MgB2 and analyzed its structural, electronic, and adsorption properties. This material was investigated for hydrogen storage applications after being decorated with alkali metals.25 Similarly, the MgC2 structure was designed computationally by replacing the B atoms in MgB2 with C atoms. MgB2 consists of two atomic layers and exhibits a geometry similar to that of BeB2, HfB2, TiB2, FeB2, and MgB2 monolayers.36–39 | ||
| Fig. 1 Optimized structure of (a) MgC2, (b) density of states (DOS), and (c) band structure (d) energy–time (e) temperature–time (f) phonon of MgC2. Green-colored atoms represent boron, while maroon-colored atoms represent magnesium. | ||
The upper atomic layer, composed of C atoms, follows a honeycomb arrangement similar to graphene, and the lower atomic layer consists of Mg atoms positioned at the center of the C–C honeycomb. The optimized lattice parameters of MgC2 are a = b = 3.181 Å, which are slightly larger than those of the BeB2 monolayer (a = b = 3.032 Å) and MgB2 (a = b = 3.017 Å). Additionally, the monolayer thickness of MgC2 is 1.460 Å, significantly greater than that of BeB2 (0.443 Å) and less than MgB2 (1.683 Å). A comparison of the lattice parameters is provided in Table 1.
| Lattice parameters (Å) | Bond length(Å) | Height (Å) | |
|---|---|---|---|
| MgC2(this work) | 3.181 | 2.14 | 1.46 |
| MgB2 | 3.017 | 2.42 | 1.68 |
| BeB2 | 3.032 | 1.84 | 0.44 |
| HfB2 | 3.165 | 2.32 | 1.41 |
| TiB2 | 3.083 | 1.73 | 1.19 |
Fig. 1(b and c) illustrates the band structure and density of states (DOS) of MgC2. The DOS results indicate that the material exhibits semiconducting behavior with a bandgap of 0.30 eV. The band structure indicates the direct bandgap of 0.3 eV at Γ point which is consistent with the DOS. The total DOS is mainly governed by the p-orbitals of carbon atoms, with only a minor contribution from the s-orbitals of magnesium atoms. Fig. 1(b) reveals that the s-orbital of Mg and the p-orbital of C overlap in the valence band.
MD calculations were conducted at 300 K for the pure MgC2 monolayer. As observed in Fig. 1d and e, the energy graph peaks at −62 eV, while the temperature graph reaches a maximum value of 460 K. The continuous fluctuations in both graphs indicate the structural stability of MgC2 at 300 K. The phonon calculations for the pure MgC2 monolayer reveal no imaginary frequencies throughout the Brillouin zone, indicating its dynamic stability as shown in Fig. 1f. The absence of imaginary peaks in the phonon dispersion spectrum confirms that the structure remains stable under small perturbations and does not undergo spontaneous distortions or soft modes. This result validates the robustness of the material and supports its potential for practical applications.
3.2 Adsorption of hydrogen molecule on pristine MgC2
This section examines the interaction of H2 molecules with MgC2, where a single H2 molecule was placed at various distinct locations, as shown in Fig. 1(a). The hydrogen adsorption sites include (i) the hollow site of the hexagonal boron ring (H), (ii) the top of a boron atom (T), and (iii) the bridge site between two boron atoms (B). The structures were sequentially optimized by varying the adsorption sites. The binding energy was calculated using the formula in eqn (2), and the corresponding values for different sites are presented in Table 2.35| Adsorption sites | TOP (T) | Bridge (B) | Hollow (H) |
|---|---|---|---|
| Binding energy (eV) | 0.23 | 0.25 | 0.25 |
The binding energy estimates for these adsorption sites are presented in Table 2, and all values fall within the DOE criteria range (0.2–0.8 eV per H2), making them suitable for hydrogen adsorption. The binding energy values for the H and B sites were found to be similar or higher. However, hydrogen initially placed at the B site migrated to the H site, indicating that the H site is more stable than the B site. Consequently, additional hydrogen molecules were sequentially adsorbed onto the H site, and their binding energies were calculated, as shown in Table 3.25,40,41 The 5.73 Å is the ideal upward distance for a deposited H2 molecule. In reference, the mean bond length of a free H2 molecule is approximately 0.74 Å. An H2 molecule adsorbed on the structure and caused a little increase in bond length to 0.75 Å.42 The retention of H2 molecular properties is indicated by the change in bond length that is seen throughout the procedure of adsorption and optimization. To determine the optimal adsorption site, adsorption energies were analyzed. Following structural optimization, the hexagonal hollow site emerged as the most favorable location for H2 adsorption, with an average binding energy of 0.24 eV.10,43
| Binding energy (eV) | Adsorption energy (eV per H2) | Weight percent (%) | Desorption energy (eV) | Desorption temperature (K) | |
|---|---|---|---|---|---|
| MgC2–1H2 | 0.25 | 0.290 | 0.26 | 0.29 | 370.52 |
| MgC2–2H2 | 0.40 | 0.242 | 0.52 | 0.20 | 309.19 |
| MgC2–3H2 | 0.62 | 0.234 | 0.77 | 0.21 | 298.97 |
| MgC2–4H2 | 0.82 | 0.226 | 1.03 | 0.20 | 288.75 |
| MgC2–5H2 | 1.03 | 0.223 | 1.29 | 0.21 | 284.92 |
| MgC2–6H2 | 1.24 | 0.220 | 1.54 | 0.20 | 281.08 |
| MgC2–7H2 | 1.44 | 0.218 | 1.80 | 0.20 | 278.53 |
| MgC2–8H2 | 1.62 | 0.640 | 2.05 | 0.17 | 817.71 |
3.3 Adsorption and electronic properties of H2 molecule adsorbed on MgC2
Based on the binding energy, it is concluded that the most favorable atom decoration occurs at the H site. Fig. 2 illustrates the optimized structures of hydrogenated MgC2. Hydrogen molecules were sequentially adsorbed onto the H site, up to a total of 8H2, and their binding energy, adsorption energy, weight percentage, desorption energy, and desorption temperature were calculated, as detailed in Table 3.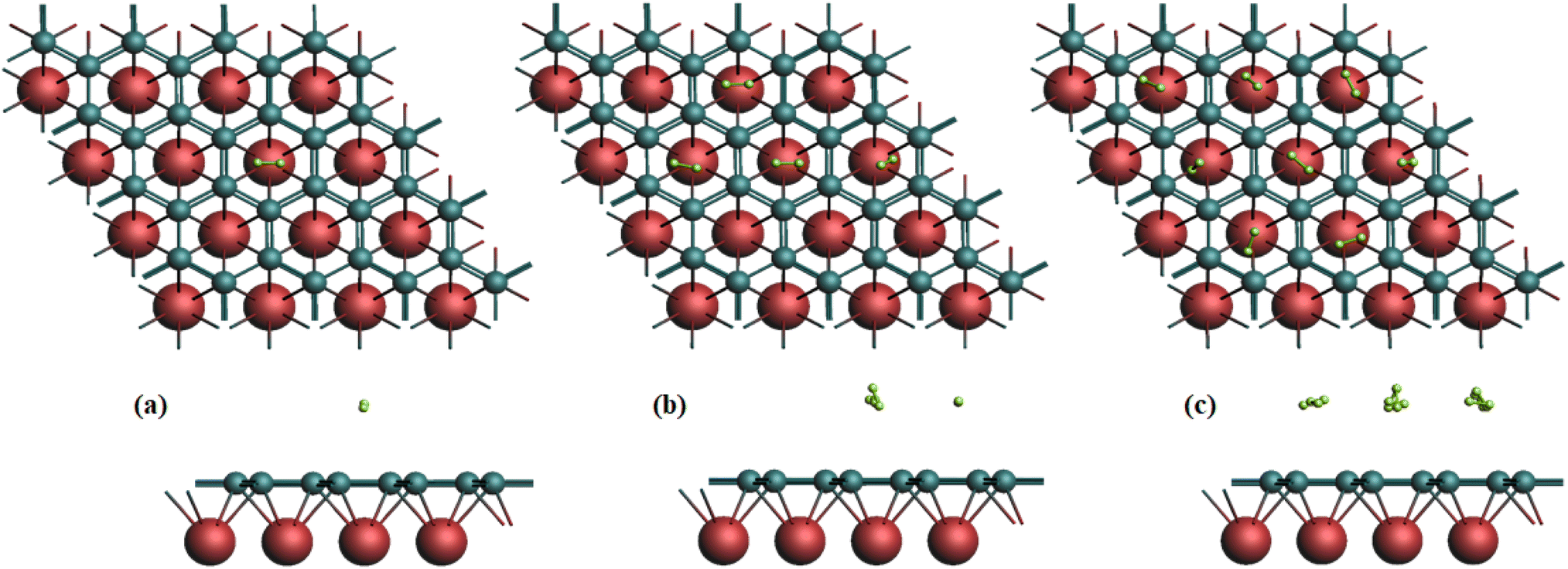 | ||
| Fig. 2 Optimized structures of (a) 1H2- (b) 4H2- (c) 8H2-adsorbed MgC2. | ||
Subsequently, each structure was optimized, and a vertical distance of 3.02 Å was determined to be optimal for the adsorbed H2 molecule. It is discovered that the free H2 molecule's bond length is 0.74 Å. The H–H bond length elongates to 0.78 Å when one H2 molecule is added to the system. The H–H bond length in the 2H2–8H2 molecule adsorption system is determined to be 0.78–0.80 Å. The H–H bond length determined throughout the adsorption and optimization procedures demonstrates that the molecule's structure remains intact.42
As shown in Fig. 3, the total and partial DOS were calculated for the Mg-s, C-p, H-s, and s orbitals of all metal elements to evaluate the system's electrochemical properties. Fig. 3(a) reveals that absorption of a single molecule of hydrogen transforms the behavior of MgC2 from semiconducting to metallic, resulting in a zero bandgap. The p orbital of the C atom significantly contributes to the total DOS, while the s orbital of hydrogen exhibits overlapping peaks at −4.5 eV in the valence band, and hybridized peaks between 6–8 eV in the conduction band. Similarly, Fig. 3(b) demonstrates that after the adsorption of four hydrogen molecules, MgC2 retains its metallic nature with a zero bandgap. The p orbital of the C atom remains the dominant contributor to total DOS, while the s orbital of hydrogen displays hybridized peaks within the ranges of −4 to −6 eV in the valence band and 6 to 8 eV in the conduction band.
 | ||
| Fig. 3 DOS of (a) 1H2, (b) 4H2, (c) 8H2 adsorbed MgC2. | ||
In Fig. 3(c), the total DOS of MgC2 after the adsorption of eight hydrogen molecules shows a reduction in the bandgap. The p orbital of the C atom remains the primary contributor to the total DOS, while the s orbital of hydrogen exhibits overlapping peaks at 7.1 eV in the conduction band and hybridized peaks within the ranges of −4 to −6 eV in the valence band and 6 to 8 eV in the conduction band. In summary, the adsorption between MgC2 and hydrogen is primarily driven by the polarizing effect of the localized voltage generated by charged particles and their spatial recombination.
After the adsorption of hydrogen molecules onto the MgC2 monolayer, charge analysis calculations were conducted. The results reveal that 1, 4, and 8 hydrogen molecules withdraw charges of 0.041 e, 0.139 e, and 0.259 e from the surface, respectively. For MgC2, the typical hydrogen desorption temperature ranged from 370 K to 496 K for 1H2. As illustrated in Fig. 4, these values were derived from adsorption energies calculated within a pressure range of 1–10 atm.
 | ||
| Fig. 4 Minimum, average, and maximum values for desorption temperature after hydrogen adsorption on MgC2. | ||
Furthermore, three critical temperatures related to hydrogen desorption were determined: the maximum temperature at which desorption takes place (max-TD), the minimum temperature necessary to start hydrogen release (min-TD), and the average desorption temperature (Av-TD). In our study, the average desorption temperature of MgC2 monolayers was calculated to range from 370 K to 496 K, indicating their suitability for fuel cell applications.44
3.4 Electron localization function (ELF)
The charge carrier density mechanism serves as a gauge of charge confinement and scattering and is connected to the kinetic energy density.45,46 Its values range from 0.5 to 1.0, indicating areas where electrons are present and engaged in bonding or nonbonding interactions, including lone pairs, covalent bonds, or atomic shells. On the other hand, values between 0.0 and 0.5 show that there are small intermolecular interactions, and electrons are dispersed throughout the atomic shells.47 The ELF map highlights regions of increased electron concentration and shows how charge carrier density changes on surfaces and adsorbents during complex creation.As seen in Fig. 5(a) a value near 0.5 is indicated by green, indicating the presence of covalent bonds, and a large charge density is indicated by the increased red coloration in the MgC2 layer after deposition of 1H2.48 Following the uptake of four hydrogen molecules, Fig. 5(b) shows a striking color shift with an increase in green and a decrease in red and yellow, suggesting covalent electron sharing between the hydrogen molecules and the MgC2 layer. The bright red hue represents a decrease in surface electrons, while the increasing green color and the presence of a light blue shade in Fig. 5(c) indicate charge transfer from the MgC2 layer to the hydrogen molecules, with up to eight hydrogen molecules adsorbed.
 | ||
| Fig. 5 (a) MgC2–1H2, (b) MgC2–4H2, (c) MgC2–8H2. | ||
3.5 Ab initio molecular dynamics (AIMD)
AIMD calculations were performed to assess the configurational stability of the system after hydrogen adsorption on MgC2 at elevated temperatures such as 400 K. Simulations were conducted with a time step of 0.25 femtoseconds for a total duration of approximately 4.5 picoseconds at a temperature of 400 K. Fig. 6 presents the temperature and energy fluctuation graphs at 400 K. | ||
| Fig. 6 Energy and temperature graph of (a1 and a2) MgC2–1H2 and (b1 and b2) MgC2–8H2. | ||
As can be seen from Fig. 6(a1), the system attained higher energy values of −1706.5 eV when one hydrogen molecule was adsorbed on MgC2. Fig. 6(a2) shows that the temperature peaks peaked at 5000 K. Based on Fig. 6(b1), it can be concluded that the system achieved higher energy values of −1855.9 eV when the eight hydrogen molecules were adsorbed on MgC2. Fig. 6(b2) shows the temperature peaks around 4500 K. The steady fluctuations of the peaks indicate the stability of the MgC2 sheet after hydrogen adsorption at 300 K.
4. Conclusion
In this study, the adsorption behavior of hydrogen on pure MgC2 was systematically analyzed using density functional theory (DFT) and Ab Initio Molecular Dynamics (AIMD) simulations. The structural stability of MgC2 was thoroughly evaluated using phonon dispersion calculations and AIMD simulations, confirming its robustness as a potential hydrogen storage material. MgC2 demonstrates a gravimetric hydrogen capacity of 2.05 wt%, capable of adsorbing up to eight hydrogen molecules. The hydrogen changes the behavior of the pure MgC2 to metallic. The average adsorption energy for MgC2 was calculated to be 0.286 eV. Hirshfeld charge analysis revealed charge transfers of 0.041, 0.139, and 0.259 e to 1, 4, and 8 hydrogen molecules to the pure MgB2 structures respectively. AIMD simulations confirmed that the pure MgC2 structure remains stable without any distortions upon hydrogen adsorption at 400 K. The comprehensive analysis of structural, electronic, and adsorption properties, along with binding energy calculations, desorption temperature and energy assessments, charge density analysis, and electron localization function (ELF) mapping, all indicate that MgC2 is a highly promising candidate for hydrogen storage applications. The strong interaction between MgC2 and hydrogen molecules, coupled with favorable adsorption and desorption characteristics, suggests its potential for practical hydrogen storage solutions. These findings provide a theoretical foundation for further experimental validation and the development of MgC2-based hydrogen storage materials.Data availability
Data is available on request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their sincere appreciation to the Researcher supporting program at King Saud University, Riyadh, for funding this work under project number (RSPD2025R644).References
- Y. Yong, Q. Hou, X. Yuan, H. Cui, X. Li and X. Li, Ultrahigh capacity and reversible hydrogen storage media based on Li-decorated T-BN monolayers, J. Energy Storage, 2023, 72, 108169 CrossRef.
- S. R. Sinsel, R. L. Riemke and V. H. Hoffmann, Challenges and solution technologies for the integration of variable renewable energy sources—a review, renewable energy, 2020, 145, 2271–2285 CrossRef.
- S. E. Hosseini and M. A. Wahid, Hydrogen from solar energy, a clean energy carrier from a sustainable source of energy, Int. J. Energy Res., 2020, 44(6), 4110–4131 CrossRef.
- J. O. Abe, A. Popoola, E. Ajenifuja and O. M. Popoola, Hydrogen energy, economy and storage: Review and recommendation, Int. J. Hydrogen Energy, 2019, 44(29), 15072–15086 CrossRef CAS.
- P. Banerjee, B. Pathak, R. Ahuja and G. Das, First principles design of Li functionalized hydrogenated h-BN nanosheet for hydrogen storage, Int. J. Hydrogen Energy, 2016, 41(32), 14437–14446 CrossRef CAS.
- M. Felderhoff, C. Weidenthaler, R. von Helmolt and U. Eberle, Hydrogen storage: the remaining scientific and technological challenges, Phys. Chem. Chem. Phys., 2007, 9(21), 2643–2653 RSC.
- S. Niaz, T. Manzoor and A. H. Pandith, Hydrogen storage: Materials, methods and perspectives, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef CAS.
- M. Jiang, J. Xu, P. Munroe, Z.-H. Xie and Z. Chen, Light metal decorated graphene-like Si2BN monolayers as hydrogen storage media: A DFT investigation, Int. J. Hydrogen Energy, 2024, 50, 865–878 CrossRef CAS.
- Y. Zhu, L. Ouyang, H. Zhong, J. Liu, H. Wang, H. Shao, Z. Huang and M. Zhu, Closing the loop for hydrogen storage: facile regeneration of NaBH4 from its hydrolytic product, Angew. Chem., 2020, 132(22), 8701–8707 CrossRef.
- X. Jin, P. Qi, H. Yang, Y. Zhang, J. Li and H. Chen, Enhanced hydrogen adsorption on Li-coated B12C6N6, J. Chem. Phys., 2016, 145(16), 164301 CrossRef PubMed.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Exceptional hydrogen storage achieved by screening nearly half a million metal-organic frameworks, Nat. Commun., 2019, 10(1), 1568 CrossRef PubMed.
- Y. Fu, Z. Yu, S. Guo, Y. Li, Q. Peng, L. Zhang, S. Wu and S. Han, Catalytic effect of bamboo-like carbon nanotubes loaded with NiFe nanoparticles on hydrogen storage properties of MgH2, Chem. Eng. J., 2023, 458, 141337 CrossRef CAS.
- M. Lakhal, M. Bhihi, A. Benyoussef, A. El Kenz, M. Loulidi and S. Naji, The hydrogen ab/desorption kinetic properties of doped magnesium hydride MgH2 systems by first principles calculations and kinetic Monte Carlo simulations, Int. J. Hydrogen Energy, 2015, 40(18), 6137–6144 CrossRef CAS.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Complex hydrides for hydrogen storage, Chem. Rev., 2007, 107(10), 4111–4132 CrossRef CAS PubMed.
- A. Ozarslan, Large-scale hydrogen energy storage in salt caverns, Int. J. Hydrogen Energy, 2012, 37(19), 14265–14277 CrossRef CAS.
- A. Fujisawa, S. Miura, Y. Mitsutake and M. Monde, Simulation study of hydrogen purification using metal hydride, J. Alloys Compd., 2013, 580, S423–S426 CrossRef CAS.
- M. D. Nashchekin, M. V. Minko, S. B. Morgunova and K. B. Minko, Enhancement of heat-and mass-transfer processes in a metal-hydride flow-through hydrogen-purification reactor, Int. J. Hydrogen Energy, 2020, 45(46), 25013–25029 CrossRef CAS.
- Y. Qin, R. Song, S. Chen, Y. Chen, J. Hou, N. Xu and W. Zhang, First-principles studies on the hydrogen storage properties of XMoH3 (X= Na, K, Rb, Cs) perovskite hydrides, Int. J. Hydrogen Energy, 2024, 88, 251–259 CrossRef CAS.
- Y. Liu, H. Du, X. Zhang, Y. Yang, M. Gao and H. Pan, Superior catalytic activity derived from a two-dimensional Ti 3 C 2 precursor towards the hydrogen storage reaction of magnesium hydride, Chem. Commun., 2016, 52(4), 705–708 RSC.
- S. Zhang, X. Feng, G. Yang and B. Wen, The hardness and electrical conduction in TiB2 and MgB2: Computational insights, Comput. Mater. Sci., 2024, 232, 112642 CrossRef CAS.
- T. Sruthi and V. Mathew, Computational investigation of quantum capacitance enhancement in Mg-substituted MgB2-based supercapacitor electrodes, Comput. Condens. Matter, 2024, 40, e00940 CrossRef.
- H.-W. Li, Y. Yan, S.-i. Orimo, A. Züttel and C. M. Jensen, Recent progress in metal borohydrides for hydrogen storage, Energies, 2011, 4(1), 185–214 CrossRef CAS.
- E. Beheshti, A. Nojeh and P. Servati, A first-principles study of calcium-decorated, boron-doped graphene for high capacity hydrogen storage, Carbon, 2011, 49(5), 1561–1567 CrossRef CAS.
- R. K. Sahoo, B. Chakraborty and S. Sahu, Reversible hydrogen storage on alkali metal (Li and Na) decorated C20 fullerene: a density functional study, Int. J. Hydrogen Energy, 2021, 46(80), 40251–40261 CrossRef CAS.
- M. I. Khan, A. Saeed, M. Shakil, G. Saira, A. Ahmad, F. Imam and S. S. Alarfaji, Computational exploration of high-capacity hydrogen storage in alkali metal-decorated MgB2 material, J. Power Sources, 2024, 613, 234881 CrossRef.
- M. I. Khan, M. Khurshid, S. S. Alarfaji and A. Majid, Bismuthene as a novel anode material of magnesium/zinc ion batteries with high capacity and stability: a DFT calculation, Phys. Chem. Chem. Phys., 2024, 26(42), 27007–27018 RSC.
- M. I. Khan, G. Nadeem, A. Majid and M. Shakil, A DFT study of bismuthene as anode material for alkali-metal (Li/Na/K)-ion batteries, Mater. Sci. Eng. B, 2021, 266, 115061 CrossRef CAS.
- M. I. Khan, A. Majid, N. Ashraf and I. Ullah, A DFT study on a borophene/boron nitride interface for its application as an electrode, Phys. Chem. Chem. Phys., 2020, 22(6), 3304–3313 RSC.
- M. I. Khan, I. Mehmood, S. S. Alarfaji, M. Junaid and T. Iqbal, Computational study of novel 2H chromium ditelluride as an anode material for Li/K-ion batteries, RSC Adv., 2024, 14(47), 34515–34525 RSC.
- M. I. Khan, M. Anwar, A. Majid, M. Shakil and M. Rizwan, Computational studies of super-B as anodes for AM (Li, Na, and K) ion batteries, J. Electrochem. Soc., 2022, 169(9), 090514 CrossRef CAS.
- M. I. Khan, S. Aslam, A. Majid and S. S. A. Gillani, Intercalation of Lithium inside bilayer buckled borophene: a first principles prospective, J. Electrochem. Soc., 2021, 168(7), 070535 CrossRef CAS.
- G. Te Velde, F. Bickelhaupt, E. Baerends, C. Fonseca Guerra, S. Van Gisbergen, J. G. snijders and t. ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22(9), 931–967 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- F. Fan, J. Ren, Y. He and X. Chen, DFT study of Mg decorated on the planar B2N as a novel hydrogen storage media, Results Phys., 2023, 46, 106263 CrossRef.
- R. Varunaa and P. Ravindran, Potential hydrogen storage materials from metal decorated 2D-C 2 N: an ab initio study, Phys. Chem. Chem. Phys., 2019, 21(45), 25311–25322 RSC.
- Z. Liu, P. Wang, Q. Cui, G. Yang, S. Jin and K. Xiong, Theoretical prediction of HfB 2 monolayer, a two-dimensional Dirac cone material with remarkable Fermi velocity, RSC Adv., 2019, 9(5), 2740–2745 RSC.
- L. Zhang, Z. Wang, S. Du, H.-J. Gao and F. Liu, Prediction of a Dirac state in monolayer TiB 2, Phys. Rev. B, 2014, 90(16), 161402 CrossRef.
- M. Wan, S. Zhao, Z. Zhang and N. Zhou, Two-dimensional BeB2 and MgB2 as high capacity Dirac anodes for Li-ion batteries: A DFT study, J. Phys. Chem. C, 2022, 126(23), 9642–9651 CrossRef CAS.
- H. Zhang, Y. Li, J. Hou, A. Du and Z. Chen, Dirac state in the FeB2 monolayer with graphene-like boron sheet, Nano Lett., 2016, 16(10), 6124–6129 CrossRef CAS PubMed.
- B. Chettri, P. Patra, N. N. Hieu and D. Rai, Hexagonal boron nitride (h-BN) nanosheet as a potential hydrogen adsorption material: A density functional theory (DFT) study, Surf. Interfaces, 2021, 24, 101043 CrossRef CAS.
- G. Saira, S. M. Zaigam, M. I. Khan, M. B. Tahir, M. Rafique and S. S. Alarfaji, Computational study of Be/Mg-decorated hexagonal chromium ditelluride (CrTe2) for hydrogen storage applications, J. Energy Storage, 2024, 103, 114221 CrossRef.
- I. López-Corral, E. Germán, A. Juan, M. A. Volpe and G. P. Brizuela, DFT study of hydrogen adsorption on palladium decorated graphene, J. Phys. Chem. C, 2011, 115(10), 4315–4323 CrossRef.
- I. K. Petrushenko and K. B. Petrushenko, Hydrogen adsorption on graphene, hexagonal boron nitride, and graphene-like boron nitride-carbon heterostructures: A comparative theoretical study, Int. J. Hydrogen Energy, 2018, 43(2), 801–808 CrossRef CAS.
- A. N. Sosa, B. J. Cid, Á. Miranda, L. A. Pérez, G. H. Cocoletzi and M. Cruz-Irisson, A DFT investigation: high-capacity hydrogen storage in metal-decorated doped germanene, J. Energy Storage, 2023, 73, 108913 CrossRef.
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371(6499), 683–686 CrossRef CAS.
- C. S. Abraham, S. Muthu, J. C. Prasana, S. Armaković, S. J. Armaković and B. Geoffrey, Computational evaluation of the reactivity and pharmaceutical potential of an organic amine: A DFT, molecular dynamics simulations and molecular docking approach, Spectrochim. Acta, Part A, 2019, 222, 117188 CrossRef CAS PubMed.
- M. Vatanparast and A.-R. Nekoei, RAHB concept and σ-skeleton in some oximes of 3-hydroxy fulvene; DFT, AIM, ELF and NBO studies, Struct. Chem., 2015, 26, 1039–1048 CrossRef CAS.
- M. I. Khan, M. I. Akber, M. Gul, T. Iqbal, S. S. Alarfaji and A. Mahmood, Exploring the sensing potential of Fe-decorated h-BN toward harmful gases: a DFT study, RSC Adv., 2024, 14(10), 7040–7051 RSC.
| This journal is © The Royal Society of Chemistry 2025 |