
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxylation and sulfidation for nickel recovery from a spent catalyst containing the chelating agent, 2,2′-bipyridine†
Hisanori Iwai a,
Mauricio Cordovab,
Yutaro Takayac,
Naoki Yokotad,
Yuko Takahashid and
Chiharu Tokoro*bc
a,
Mauricio Cordovab,
Yutaro Takayac,
Naoki Yokotad,
Yuko Takahashid and
Chiharu Tokoro*bc
aSustainable Energy & Environmental Society Open Innovation Research Organization, Waseda University, 169-8555 Tokyo, Japan
bDepartment of Resources and Environmental Engineering, Faculty of Science and Engineering, Waseda University, 169-8555 Tokyo, Japan. E-mail: tokoro@waseda.jp
cDepartment of System Innovation, Faculty of Engineering, The University of Tokyo, 113-8656 Tokyo, Japan
dNext-Generation Technology Design and Creation, TAKAHATA PRECISION Co., Ltd, 160-0023 Tokyo, Japan
First published on 11th March 2025
Abstract
Techniques for recovering nickel (Ni) from various Ni-containing products are needed for resource circulation. In this study, Ni recovery from a spent catalyst containing 2,2′-bipyridine (bpy) was conducted by precipitation using hydroxylation and sulfidation. In the absence of bpy, both methods completely precipitated Ni as estimated in chemical equilibrium calculations. For an actual spent catalyst with a bpy/Ni molar ratio of one, the recovery rates were reduced to approximately 70% and 90% for the hydroxylation and sulfidation methods, respectively. Similar values were obtained for a simulated spent catalyst with a bpy/Ni molar ratio of one. Precipitation was inhibited in both methods for simulated spent catalyst with an initial bpy/Ni molar ratio of three. Ultraviolet-visible spectroscopy revealed that the bpy/Ni molar ratio increased with Ni precipitation, and Ni that remained in the solution was converted from Ni(bpy)1 to Ni(bpy)3. Fourier transform infrared spectra showed that the precipitates obtained by the sulfidation method contained bpy in a complex with Ni, and thermogravimetry-differential thermal analysis curves showed different proportions from those of the simulated spent catalysts. The precipitates formed in the presence of bpy were thin film fragments. It is known that S2− forms an ion bridge with the Ni(bpy)1 complex, and in the sulfidation method, Ni precipitated as a S–Ni–bpy cluster. These findings establish the chemical composition of Ni recovered from spent catalysts and show that the Ni recoverability depends on the bpy/Ni molar ratio.
1. Introduction
Nickel (Ni) is a valuable element found in several regions around the world, where it is commonly obtained from sulfidic and lateritic deposits, and it is mainly used for industrial purposes in the stainless steel and electroplating industries.1 Because of its chemical reactivity, Ni has recently been used as a cathode material for lithium-ion batteries,2 and as a nickel complex-based catalyst for polymerization and reduction of CO2.3–5 Continuation of Ni extraction from raw minerals is unsustainable because of market price increases (Fig. S1†).6 Furthermore, traditional mineral processing has a large environmental impact caused by the massive amounts of reagents required.2,7 Consequently, the recovery of Ni from secondary sources has become an important alternative in recent years.1 Several techniques have been developed for Ni recovery by adsorption, selective precipitation, ion exchange, and solvent extraction.8–11Spent Ni catalyst has emerged as a suitable candidate for Ni recovery from recycled materials because a typical spent catalyst can contain 15–45% of Ni by mass.12 The easiest technique to recover Ni in a scalable manner is precipitation, which forms solid Ni species. Ni can be precipitated as a hydroxide or carbonate under high pH conditions or as a sulfide in the presence of S2− ions.8,12–14 These precipitation techniques are very useful for the recovery of transition elements because the obtained solids have extremely low solubility. NiO is used as an inorganic Ni catalyst, and techniques for recovering Ni from inorganic catalysts have already been reported,15,16 however, Ni recovery technology from Ni complex catalysts containing organic chelating agents has not yet been established. Chelating agent contained in the spent Ni catalyst, which is an essential component for the catalytic effect of Ni,5,17 makes it difficult to separate or precipitate Ni.18 2,2′-Bipyridine (bpy) is widely used as a chelating agent for Ni complex-based catalysts and reportedly generates stable complexes under various pH conditions.19,20 Stable complexation of Ni with bpy will interfere with the formation of a Ni precipitate in the recovery process. However, there has been little research on the effect of the coexisting bpy on the precipitation and recovery of Ni. Investigation of the precipitation recovery of Ni from spent catalysts containing Ni–bpy complexes will provide practical knowledge for future resource circulation of Ni.
The aim of the present study was to clarify the influence of bpy on the recoverability of Ni from spent catalysts for recycling. The hydroxylation and sulfidation precipitation methods were investigated for treatment of an actual spent catalyst and simulated spent catalysts of Ni solutions containing bpy. The generated precipitates were characterized using spectroscopic methods and thermogravimetry differential thermal analysis.
2. Materials and methods
2.1. Chemicals
We obtained bpy (98% purity) from TCI (Tokyo, Japan). All other all chemicals were from FUJIFILM Wako Pure Chemical Industry Ltd (Osaka, Japan) and were of analytical reagent grade. These chemicals were used without any treatment.An actual spent Ni complex-based containing Ni (SpC-A), which had been used for polymerization, was obtained from TAKAHATA PRECISION Co., Ltd (Tokyo, Japan). A mixture of 50% MeOH with 3 M HCl was prepared as a medium. Solutions of 40 mM Ni2+ containing 0, 40, and 120 mM of bpy were prepared by dissolving NiCl2 and bpy in the medium. The resulting solutions were used as simulated spent Ni–bpy complex catalysts with the initial bpy/Ni molar ratios of 0, 1, and 3, and the resulting mixtures were labeled as SpC-0, SpC-1, and SpC-3, according to the number of bpy/Ni ratios. The chemical compositions of the SpC solutions are summarized in Table 1.
A 0.1 M Na2S aqueous was prepared fresh each day by dissolving Na2S in ultrapure water purged with N2 gas for approximately 10 min. The resulting solution was used as a sulfur agent.
Standards of Ni(bpy)1Cl2 and Ni(bpy)3Cl2 complexes were synthesized according to previous report,5,17 detailed as follows. NiCl2 and bpy were dissolved separately in MeOH and the solutions were then mixed to obtain separate solutions with bpy/Ni molar ratios of 1 and 3. After evaporated the solvent in a draft chamber at room temperature (20–25 °C) for 1 day, the resulting pastes were stored in vacuum desiccator at room temperature (20–25 °C) for more than 3 days. The turquoise and pink colored powders were obtained as crystalized Ni(bpy)1Cl2 and Ni(bpy)3Cl2, respectively.18
2.2. Hydroxylation and sulfidation processes for Ni recovery
In hydroxylation, Ni was removed as Ni(OH)2 precipitate as follows. First, 100 mL of SpC solution was stirred in a 300 mL glass beaker and the pH was adjusted to 7–14 by adding 12.5 M or 1 M NaOH aqueous solution while monitoring the pH value. The volume of NaOH added here was recorded and used as a factor to correct the total Ni concentration. At arbitrary pH values, 10–30 mL of the sample solution was removed into a 50 mL centrifuge tube that was then sealed with a cap and incubated at room temperature (20–25 °C) for 24 h. The resulting suspension was centrifuged (5072 g, 10 °C, and 10 min) and the supernatant was filtered through a polytetrafluoroethylene (PTFE) filter (pore size: 0.45 μm). The resulting filtrate and the precipitate trapped on the filter were collected for analysis.Sulfidation for Ni precipitation was conducted according to an established method.21 First, 100 mL of the SpC solution was stirred in a 300 mL glass beaker. To avoid H2S generation and to safety experiment, the pH was adjusted to 3–4 using 12.5 M aqueous NaOH. Aliquots (10–30 mL) of the solution were removed and placed in centrifuge tubes, sulfurization was then carried out by adding 0.1 M Na2S to adjust the S/Ni ratio to between 0.2 and 1.2. The sample volume in all tubes was adjusted to 30 mL by adding ultrapure water, the tubes were sealed, and the solutions were incubated at room temperature (20–25 °C) for 24 h. The resulting suspensions were separated into the supernatant and precipitate as described for the hydroxylation above, and the % Ni-precipitation by the sulfidation method was calculated based on the Ni concentration without a sulfur agent.
2.3. Qualification and quantification of bpy and Ni in the supernatants
A 100 μL aliquot of the filtrate (PTFE filter, <0.45 μm) was diluted 100 times with 1% aqueous HNO3 and the dissolved Ni concentration (<0.45 μm) was determined by inductively coupled plasma atomic emission spectroscopy (Thermo Fisher Science, Waltham, MA USA). The precipitation behavior of Ni was evaluated using thermodynamic calculations performed by geochemical modeling using PHREEQC Version 3 (United States Geological Survey, Reston, VA, USA) and the accompanying llnl database.22Adsorption spectra of the filtrates were measured in the visible range (450–700 nm) using an ultraviolet-visible spectrometer (GENESYS 180, Thermo Fisher Scientific) with a quartz cell (path length: 10 cm). A portion of the filtrate was diluted 1000 times with 0.01 M aqueous HCl and the spectrum was recorded in the range of 200–350 nm using a quartz cell (path length: 10 cm). The adsorption spectrum of bpy depends on the sample conditions, such as the pH, solvents, and complexation with metals (Fig. S2†).23 We found that the peaks were more distinct under acidic conditions than at other pH values, and the spectra had two isosbestic points at 263.5 and 307.5 nm. Accordingly, we quantified bpy under acidic conditions (0.01 M HCl) and used the peak at 307.5 nm, which was larger.24
2.4. Characterization of the precipitates
Fourier transform infrared (FT-IR) spectra of the freeze-dried precipitates were recorded directly using the attenuated total reflection method using a 4500a FTIR type FT-IR spectrometer (Agilent, CA, USA). All spectra were recorded in the region of 650–4000 cm−1 at 1 cm−1 resolution, and 32 times scanning.The thermal stabilities of the precipitates were measured using a synchronous thermal analyzer (STA 2500 Regulus, NETZSCH, Germany). Freeze-dried precipitate (10–15 mg) was placed in an aluminum cup and thermogravimetry (TG)-differential thermal analysis (DTA) was carried out under air. The temperature was increased from room temperature (approximately 20–23 °C) to 600 °C at 10 °C min−1 and maintained at 600 °C for 30 min.
3. Results and discussion
3.1. Removal of Ni from the spent catalysts
Fig. 1 shows the percentage of Ni2+ remaining in the SpC-0 solution (% Ni) after hydroxylation or sulfidation. In hydroxylation, the % Ni decreased when the pH was increased above 8, and it was almost zero at pH values above 10. In sulfidation, the % Ni decreased as the ratio of the amount of sulfur to the initial amount of dissolved Ni increased. Theoretical values were calculated using PHREEQC Version 3 and the solubility products (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ksp) of Ni(OH)2 and NiS for the following reactions:
Ksp) of Ni(OH)2 and NiS for the following reactions:| Ni2+ + 2OH− ⇄ Ni(OH)2↓, Ksp = 2.0 × 10−15, |
| Ni2+ + S2− ⇄ NiS↓, Ksp = 3.0 × 10−19. |
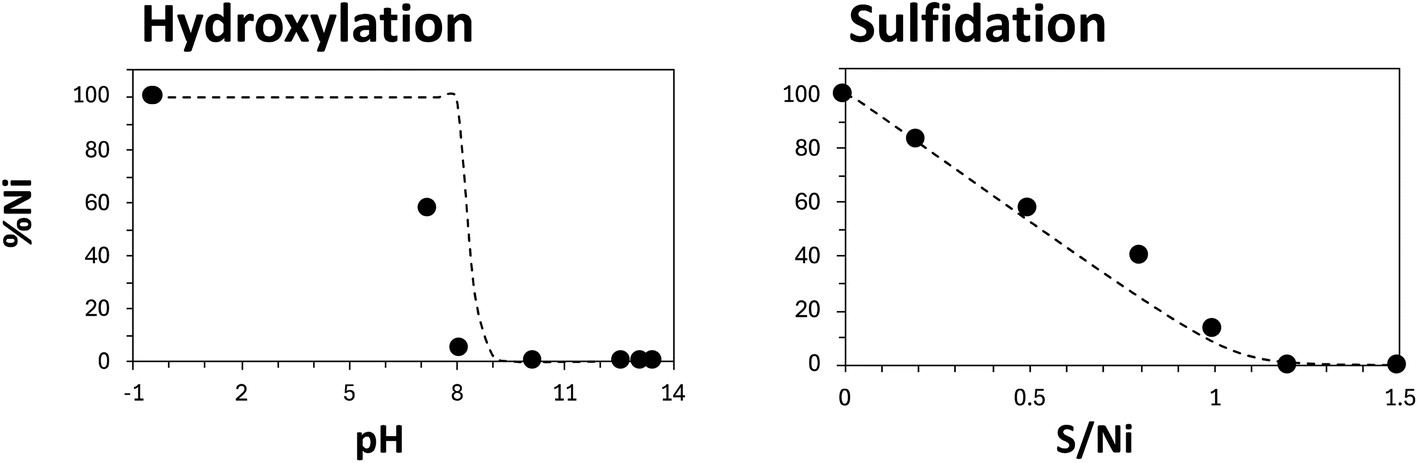 | ||
| Fig. 1 Ni precipitation from SpC-0 by hydroxylation and sulfidation at an initial pH of 3–4. Broken lines indicate simulated removal as Ni(OH)2 and NiS (PHREEQC version 3, llnl database), respectively. | ||
The theoretical values were consistent with the experimental values (Tables S1 and S2†), which showed that recovery of Ni by hydroxylation or sulfidation was effective, even in the selected solvent. For SpC-A, the % Ni decreased as the pH was increased from 8 to 9. However, the % Ni value did not fall below 26.7% even in the pH 13.8 (Fig. 1). In the sulfidation process, when the S/Ni molar ratio was less than 0.75, the decrease in % Ni with addition of sulfur was consistent with the theoretical results. When the S/Ni molar ratio was 1.0, the % Ni decreased to 15%. For SpC-1, the decreases in % Ni were similar to those for SpC-A, the % Ni decreased to 13% at S/Ni of 1.0. Even when excess S was added (S/Ni = 1.2), the % Ni values for SpC-1 and SpC-A did not decrease further, and H2S was released, indicating that S did not react with Ni. For SpC-3, Ni was not recovered by hydroxylation, and a small amount of Ni was precipitated by sulfidation.
Interestingly, no large changes were observed in the percentage of dissolved bpy (% bpy) with decreases in the % Ni in hydroxylation. By contrast, in the sulfidation process, % bpy decreased with decreases in % Ni (Fig. 2). The change in the bpy/Ni ratio in the residual solution during hydroxide and sulfidation is shown in Fig. 3. During hydroxide, the ratios of SpC-1 and SpC-A increased with the precipitation of Ni (Fig. 3). It was caused by Ni being removed as a precipitate, but bpy was still dissolved (Fig. 2). In the case of sulfidation, the ratios gradually increased during the Ni precipitation reaction. At an S/Ni of 1, the ratios for SpC-1 and SpC-A reached 2.8 and 3.0, respectively (Fig. 3). In SpC-A, the bpy/Ni ratios when maximum Ni precipitation occurred increased to 3.6 (pH 13.8) and 3.0 (S/Ni 1.0) in the hydroxylation and the sulfidation, respectively. Similarly, in SpC-1, the bpy/Ni ratios at maximum Ni precipitation increased to 3.7 (pH 13.6) and 3.0 (S/Ni 1.2), respectively. The relationship between % Ni and bpy/Ni ratio indicated that the precipitation is limited when the bpy/Ni ratio exceeds 3 in both precipitation processes. The Ni–bpy complex has a specific absorbance peak in the visible range and this peak shifts from approximately 600 nm for Ni(bpy)1 to approximately 510 nm for Ni(bpy)3 (Fig. S3†).25 In the present study, the spectra of SPC-A and SPC-1 blue-shifted as Ni precipitation progressed, which was evidence of an increase in the bpy coordination number of the Ni–bpy complex (Fig. 4). The precipitation stopped when the peak shifted to 510 nm for Ni(bpy)3. The stability constants of Ni(bpy)1 and Ni(bpy)3 are 6.9 and 19.9, respectively.23 The Ni(bpy)3 complex is more stable than Ni(OH)2 and NiS, indicating that Ni does not form Ni(OH)2 and NiS in the presence of bpy. Therefore, precipitation is limited by Ni(bpy)3 formation and the recovery performance will largely depend on the bpy/Ni molar ratio; this also explains why almost no Ni was precipitated by either precipitation method in SpC-3.
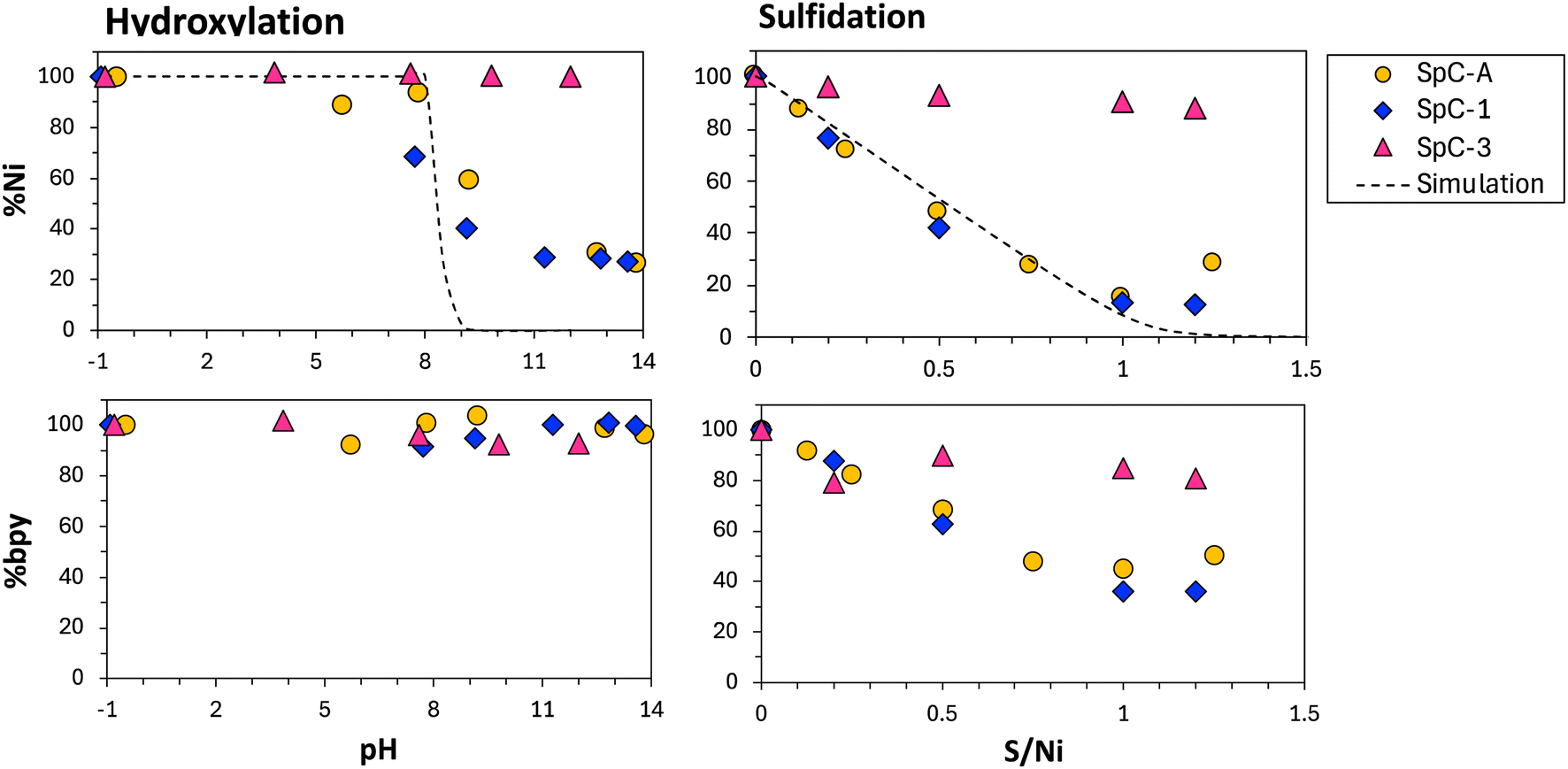 | ||
| Fig. 2 Percentage of dissolved Ni and bpy in SpC-A, SpC-1, and SpC-3 during hydroxylation and sulfidation at an initial pH 3 of 4. Broken lines indicate simulated values of Ni removal as Ni(OH)2 or NiS (PHREEQC version 3). | ||
 | ||
| Fig. 3 Changes in the bpy/Ni molar ratio during the precipitation processes. | ||
 | ||
| Fig. 4 Visible range absorption spectra of the SpC-A and SpC-1 filtrates (<0.45 μm) after the precipitation processes. | ||
3.2. Structural properties of the Ni precipitates
Attenuated total reflection FT-IR spectra were obtained for bpy and synthesized complexes as reference compounds and for the precipitates generated by hydroxylation and sulfidation with SpC-A, SpC-1, and SpC-0 (Fig. 5). The peaks were assigned according to previous reports.26–28 For samples precipitated by hydroxylation (Fig. 5b), a small peak at approximately 3640 cm−1 and a broad peak at around 3340 cm−1 were observed for non-hydrogen bonded and hydrogen-bonded hydroxyl groups, respectively. A prominent peak for in-plane vibration of Ni–O–H occurred at 600 cm−1.27 The rising peak that appears below 1000 cm−1 indicated that the precipitate was characterized as Ni(OH)2. Two peaks at approximately 1450 and 1360 cm−1 were attributed to symmetric and asymmetric vibrations of intercalated CO32−, respectively.28 Exposure to air converts hydroxylates to carbonates, and the CO32− peaks were attributed to NiCO3 generated from Ni(OH)2. The FT-IR spectral analyses showed that no organic contamination, such as bpy, was detected, indicating that Ni(OH)2 precipitates were generated even in bpy containing conditions by the hydroxylation method as anticipated.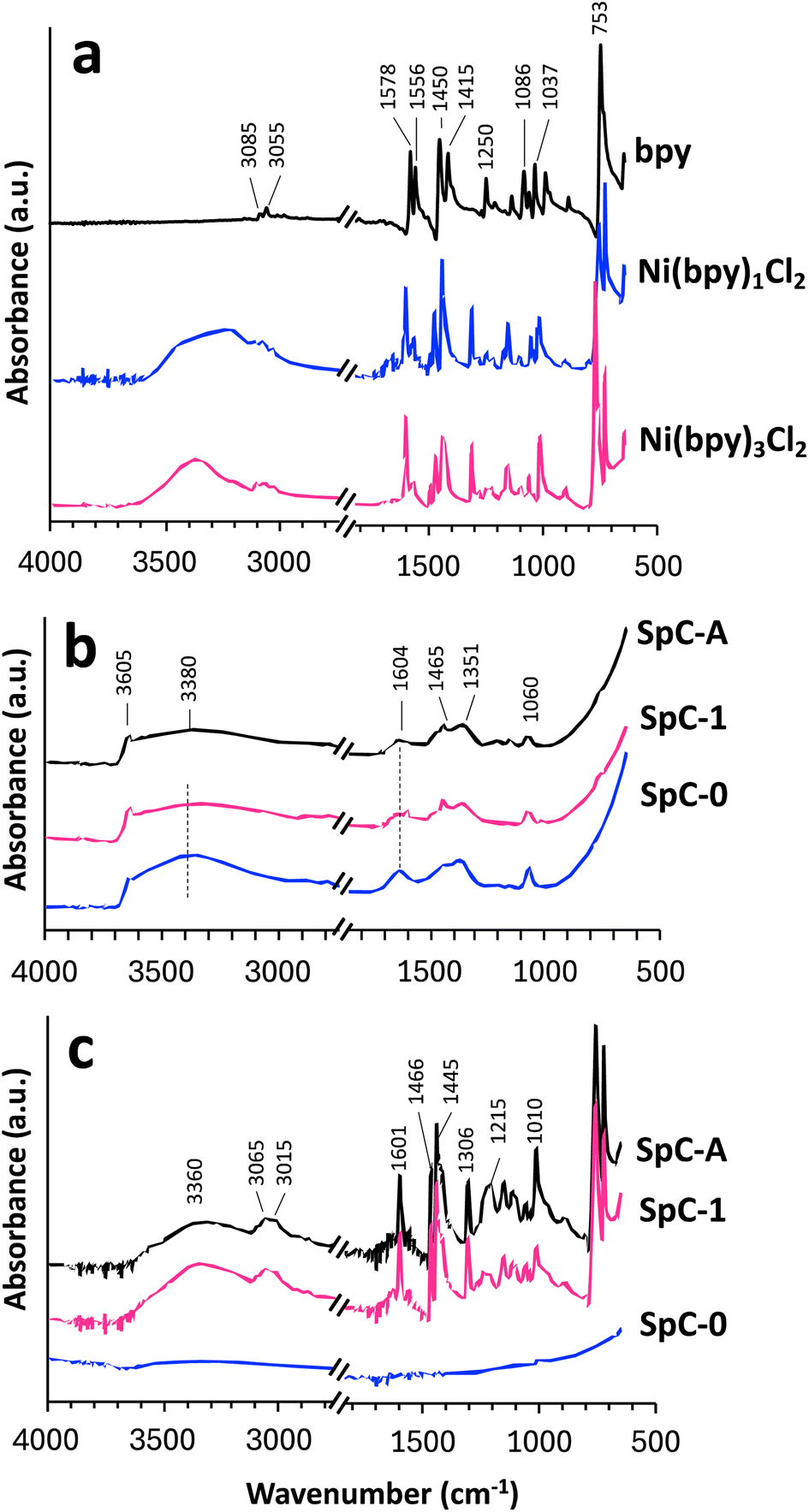 | ||
| Fig. 5 ATR FT-IR spectra of standard materials (a), the hydroxide precipitates (b), and the precipitates generated by the sulfidation (c). | ||
For the sulfidation samples (Fig. 5c), no remarkable peaks were observed for the precipitate of SpC-0. The background of NiS was negligible in the FT-IR spectra of the sulfidation precipitates. The spectrum for bpy (Fig. 5a) showed weak peaks at 3064–3049 cm−1, four skeletal peaks at 1578–1414 cm−1, and a large peak at 753 cm−1, which were assigned to heteroaromatic C–H stretching, ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching, and aromatic C–H bending, respectively. Minor peaks at 1250 and 1134–1041 cm−1 were attributed to C–N stretching and aromatic C–C stretching or aromatic C–H bending, respectively. These peaks were characteristic of bpy.29 The spectra for SpC-A and 1 after sulfidation were similar to that of bpy; although, the skeletal peaks shifted to higher frequencies. Peak shifts like this occur when pyridine is coordinated to a metal.30 Pyridine coordinated with elements other than hydrogen can be qualitatively distinguished from free pyridine by a shift in the intense peak at 1578 cm−1 to 1600 cm−1.30 A similar peak shift also occurs when bpy complexes with metals. Comparison of the skeletal peaks for bpy appeared at approximately 1578–1414 cm−1 and peaks in sulfidation-precipitated SpC-A showed that the peaks at 1556 and 1578 cm−1 weakened and shifted to 1600 cm−1. Furthermore, two peaks at 1414 and 1455 cm−1 shifted to slightly higher frequencies at 1443, and 1473 cm−1 for the SpC-A and 1 samples. The broad peak at around 3340 cm−1 was attributed to intermolecular hydrogen-bonded O–H stretching. Specific FT-IR peaks for SpC-A and SpC-1 were caused by the presence of bpy. In addition, these peak shifts observed for the sulfidation precipitates in SpC-A and SpC-1 were consistent with the peak properties in the complexed bpy reported in other studies.5,29,30 The FT-IR spectra thus showed that bpy in the precipitate was complexed with Ni.
C and CN stretching, and aromatic C–H bending, respectively. Minor peaks at 1250 and 1134–1041 cm−1 were attributed to C–N stretching and aromatic C–C stretching or aromatic C–H bending, respectively. These peaks were characteristic of bpy.29 The spectra for SpC-A and 1 after sulfidation were similar to that of bpy; although, the skeletal peaks shifted to higher frequencies. Peak shifts like this occur when pyridine is coordinated to a metal.30 Pyridine coordinated with elements other than hydrogen can be qualitatively distinguished from free pyridine by a shift in the intense peak at 1578 cm−1 to 1600 cm−1.30 A similar peak shift also occurs when bpy complexes with metals. Comparison of the skeletal peaks for bpy appeared at approximately 1578–1414 cm−1 and peaks in sulfidation-precipitated SpC-A showed that the peaks at 1556 and 1578 cm−1 weakened and shifted to 1600 cm−1. Furthermore, two peaks at 1414 and 1455 cm−1 shifted to slightly higher frequencies at 1443, and 1473 cm−1 for the SpC-A and 1 samples. The broad peak at around 3340 cm−1 was attributed to intermolecular hydrogen-bonded O–H stretching. Specific FT-IR peaks for SpC-A and SpC-1 were caused by the presence of bpy. In addition, these peak shifts observed for the sulfidation precipitates in SpC-A and SpC-1 were consistent with the peak properties in the complexed bpy reported in other studies.5,29,30 The FT-IR spectra thus showed that bpy in the precipitate was complexed with Ni.
3.3. Thermal stability of the Ni precipitate from sulfidation
The thermal stabilities of standards and Ni precipitates recovered by sulfidation were measured (Fig. 6a and b). For bpy, mass loss occurred at approximately 210 °C and the peak was endothermic, which showed that the mass loss occurred with evaporation (Fig. 6a). The TG curves for the synthesized Ni(bpy)1Cl2 and Ni(bpy)3Cl2 complexes showed stepwise decreases in the mass. Some of the mass of Ni(bpy)1Cl2 was lost at 100–120 °C in an endothermic reaction with the remaining solvent (MeOH), which was consistent with the FT-IR spectrum of Ni(bpy)1Cl2. Evaporation of two Cl atoms produced a small endothermic peak at 250 °C. Thermal decomposition of the complexed bpy then occurred at >250 °C with a peak at 470 °C. For Ni(bpy)3Cl2, the first mass loss appeared at approximately 150–210 °C. This stage was attributed to evaporation of one of the bpy units from the complex. In the next stage, the mass gradually decreased, which was attributed to thermal decomposition of the remaining two bpy units.31,32 The TG curve of the NiS standard slightly increased by approximately 550 °C, which suggested that NiS was converted to NiO (Fig. 6a). | ||
| Fig. 6 TG-DTA curves for standards (a) and Ni precipitates generated by sulfidation (b). | ||
The sulfidation precipitate obtained from SpC-0 (Fig. 6b) was identified as NiS, and some of this was oxidized in air to form NiSO4.21 The mass decreased at approximately 440 °C because of the elimination of SO4.33 Gradual mass loss of the sulfide precipitates from SpC-1 and SpC-3 occurred exothermically at temperatures above 240 °C. Stepped, large decreases were observed at temperatures up to 600 °C, and an exothermic peak centered at approximately 465 °C appeared (Fig. 6b). The SpC-A TG curve was similar to the curves of SpC-1 and SpC-3, except for the exothermic peak occurring at a higher temperature of approximately 460 °C. The precipitate from SpC-A could be contaminated with other organic compounds in the medium (Table 1), and this peak was associated with the thermal degradation of bpy and contaminants. Although the bpy in the sulfidation precipitates of SpC-1 and SpC-3 was likely in a complex with Ni (Fig. 5c), the TG curves of the synthesized complexes showed decreasing concave shapes. By contrast, the precipitates of the SpC samples showed decreasing convex shapes. These results indicate that the binding state of bpy differs between the synthetic complexes and the sulfidation precipitates.
From the results in Fig. 2, the bpy/Ni ratios in the precipitates in SpC-A and SpC-1 at S/Ni 1 were estimated to be 0.64 and 0.74, respectively. In a previous report, 84% of precipitated Ni in SpC-A under the same conditions was bound to S, and 16% was Ni(OH)2, analyzed by XAFS spectra.21 Because hydroxides do not contain bpy, 5–15% of precipitated Ni was precipitated as sulfides, and most of the Ni was precipitated bonding to both S and bpy. Metal complexes with S-bridging are well-known.34,35 In these complexes, S atoms donate two or four electrons to between two and four metal atoms to form a bridge between metals.34 In the sulfidation of spent catalyst containing bpy, S–Ni–bpy cluster formed by S bridging. This complexation is thought to be the reason for the decrease in bpy with Ni precipitation during sulfidation and the different thermochemical properties compared with hydroxylation. Consequently, compared with the sulfidation process, the bpy/Ni ratio in the remaining solution in the hydroxylation process increased more quickly and the recovery of Ni was low.
4. Conclusions
The effect of bpy on the formation of Ni precipitates was evaluated in Ni recovery from spent catalysts by hydroxylation and sulfidation. In hydroxylation, the presence of bpy decreased the precipitation rate by approximately 30% in spent catalyst with a bpy/Ni molar ratio of one. In sulfidation, the precipitation rate decreased by approximately 10%, which showed that sulfidation was more effective than hydroxylation for recovering Ni by precipitation from spent catalyst containing bpy. The bpy/Ni molar ratio increased with precipitate formation and Ni was stabilized as a soluble Ni(bpy)3 complex. This phenomenon limited recovery of Ni by precipitation when the bpy/Ni molar ratio was more than three. Some bpy also precipitated in the sulfidation. FTIR spectra showed that the bpy in the sulfidation precipitate was complexed with Ni through sulfur bridging as a S–Ni–bpy cluster rather than a soluble Ni–bpy complex. This increased the thermal decomposition temperature of bpy from S–Ni–bpy slightly compared with that of Ni–bpy complexes. These findings are critical for planning recovery of Ni and will improve the sustainability of industrial activities.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gabrielle David, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.References
- V. Coman, B. Robotin and P. Ilea, Resour., Conserv. Recycl., 2013, 73, 229–238 CrossRef.
- G. M. Mudd, Ore Geol. Rev., 2010, 38, 9–26 CrossRef.
- I. N. Buffoni, F. Pompeo, G. F. Santori and N. N. Nichio, Catal. Commun., 2009, 10, 1656–1660 CrossRef.
- C. Wu and P. T. Williams, Appl. Catal., B, 2009, 87, 152–161 CrossRef.
- J. Lin, B. Qin and Z. Fang, Catal. Lett., 2019, 149, 25–33 CrossRef.
- O. B. Wani, S. Khan, M. Shoaib, C. da Costa Goçalves, Z. Chen, H. Zeng and E. Bobicki, Miner. Eng., 2024, 218, 108976 CrossRef.
- K. Quast, J. N. Connor, W. Skinner, D. J. Robinson and J. Addai-Mensah, Miner. Eng., 2015, 79, 261–268 CrossRef.
- S. Krishnan, N. S. Zulkapli, H. Kamyab, S. M. Taib, M. F. B. M. Din, Z. A. Majid, S. Chaiprapat, I. Kenzo, Y. Ichikawa, M. Nasrullah, S. Chelliapen and N. Othman, Environ. Technol. Innovation, 2021, 22, 101525 CrossRef.
- N. P. Raval, P. U. Shah and N. K. Shah, J. Environ. Manage., 2016, 179, 1–20 CrossRef PubMed.
- X. Yang, X. Peng, L. Kong and X. Hu, J. Environ. Sci., 2021, 104, 365–375 CrossRef.
- I. Van De Voorde, L. Pinoy and P. F. De Ketelaere, J. Membr. Sci., 2024, 234, 11–21 CrossRef.
- Q. Z. Yang, G. J. Qi, H. C. Low and B. Song, J. Cleaner Prod., 2011, 19, 365–375 CrossRef.
- J. Y. Lee, S. V. Rao, B. N. Kumar, D. J. Kang and B. R. Reddy, J. Hazard. Mater., 2010, 176, 1122–1125 CrossRef PubMed.
- S. Jerroumi, M. Amarine, H. Nour, B. Lekhlif and J. E. Jamal, Water Qual. Res. J., 2020, 55, 345–357 Search PubMed.
- K. K. Sahu, A. Agarwal and B. D. Pandey, Waste Manage. Res., 2005, 23, 148–154 CrossRef PubMed.
- N. M. Al-Mansi and N. M. A. Monem, Waste Manage., 2002, 22, 85–90 CrossRef PubMed.
- C. Ruiz-Pérez, P. A. L. Luis, F. Lloret and M. Julve, Inorg. Chim. Acta, 2002, 336, 131–136 CrossRef.
- B. Brewer, N. R. Brooks, S. Abdul-Halim and A. G. Sykes, J. Chem. Crystallogr., 2003, 33, 651–662 Search PubMed.
- Y. Zhu, W. Fan, T. Zhou and X. Li, Sci. Total Environ., 2019, 678, 253–266 CrossRef PubMed.
- C. Ruiz-Pérez, P. A. Lorenzo Luis, F. Lloret and M. Julve, Inorg. Chim. Acta, 2002, 336, 131–136 CrossRef.
- H. Iwai, S. Ishii, K. Oyama, S. Fuchida, N. Yokota, Y. Takahashi and C. Tokoro, J. Soc. Powder Technol., Jpn., 2023, 60, 600–606 CrossRef.
- D. L. Parkhurst and C. A. J. Appelo, Description of Input and Examples for PHREEQC Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations, U.S. Geological Survey Water-Resources Investigations, 2013 Search PubMed.
- K. Nakamoto, J. Phys. Chem., 1960, 64, 1420–1425 CrossRef.
- G. Atkinson and E. Bauman Jr, Inorg. Chem., 1962, 1(4), 900–904 CrossRef.
- D. A. Vander Griend, D. K. Bediako, M. J. DeVries, N. A. DeJong and L. P. Heeringa, Inorg. Chem., 2008, 47, 656–662 CrossRef PubMed.
- R. M. Silverstein and F. X. Webster, Spectrometric Identification of Organic Compounds, John Wiley & Sons, New York, 6th edn, 1998 Search PubMed.
- C. C. Miao, Y. J. Zhu, L. G. Huang and T. Q. Zhao, J. Power Sources, 2015, 274, 186–193 CrossRef.
- R. S. Jayashree and P. V. Kamath, J. Appl. Electrochem., 2011, 31, 1315–1320 CrossRef.
- T. P. Gerasimova and S. A. Katysuba, Dalton Trans., 2013, 42, 1787–17897 RSC.
- N. S. Gill, R. H. Nuttall, D. E. Scaife and D. W. A. Shapp, J. Inorg. Nucl. Chem., 1961, 18, 79–87 Search PubMed.
- R. H. Lee, E. Griswold and J. Kleinberg, Inorg. Chem., 1964, 3, 1278–1283 Search PubMed.
- N. Parveen, R. Nazir and M. Mazhar, J. Therm. Anal. Calorim., 2012, 111, 93–99 CrossRef.
- E. Tomaszewicz and M. Kotfica, J. Therm. Anal. Calorim., 2004, 77, 25–31 CrossRef.
- H. Vahrenkamp, Angew. Chem., Int. Ed., 1975, 14, 322–329 Search PubMed.
- M. A. Greaney, C. L. Coyle, M. A. Harmer, A. Jordan and E. I. Stiefel, Inorg. Chem., 1989, 28, 912–920 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00470e |
| This journal is © The Royal Society of Chemistry 2025 |