DOI:
10.1039/D5RA00461F
(Paper)
RSC Adv., 2025,
15, 10170-10182
Discovery of novel azole derivatives with benzanilide-containing hydrophobic side chains for the treatment of drug-resistant fungal infections†
Received
19th January 2025
, Accepted 26th March 2025
First published on 2nd April 2025
Abstract
As fungal resistance to existing antifungal drugs continues to rise, there is an urgent need for new drugs with anti-resistance activity. In this study, a series of newly designed and synthesized benzanilide-containing azoles exhibited promising antifungal activity against fluconazole-sensitive Candida albicans. Importantly, the newly synthesized compounds also displayed potent activity against azole-resistant strains, surpassing the performance of the positive control fluconazole. This suggests that these compounds may have the potential to combat drug-resistant fungal infections. Subsequent studies on the antifungal mechanisms revealed that the compound can inhibit fungal CYP51, thereby blocking ergosterol biosynthesis. Morphological observations of fungal cells further confirmed CYP51 as the target of action. Resistance mechanisms elucidated that these compounds can inhibit biofilm formation and the expression of resistance-related genes ERG11 and efflux pump gene CDR1, thereby reversing resistance. Meanwhile, the most potent compound A11 demonstrated the ability to stimulate reactive oxygen species, thereby exhibiting potent fungicidal activity. Furthermore, the compound A11 also showed good stability in liver microsomes and plasma metabolism. Cytotoxicity studies demonstrated low toxicity of the compounds against MRC-5 cells, indicating their potential safety for therapeutic use. In vivo experimental results indicated that the representative compound A11 significantly inhibited fungal infections caused by resistant strains. Molecular docking studies further supported the efficacy of compound A11, showing its ability to bind to Candida albicans CYP51. These findings highlight the promising antifungal activity and minimal cytotoxicity of the benzanilide-containing azoles, making them potential candidates for the treatment of drug-resistant fungal infections.
1. Introduction
With the increasing number of human immunodeficiency disease patients and the use of anti-tumor chemotherapy drugs, the incidence of fungal infection has shown an increasing trend year by year and the invasive infection has become one of the important causes of clinical death.1–4 At present, antifungal drugs are mainly composed of polyenes (e.g., amphotericin B), echinococcus (e.g., caspofungin), allylamines (e.g., terbinafine) and azole drugs (Fig. 1).5 Azole drugs such as imidazole (e.g., miconazole, MCZ), triazole (e.g., fluconazole, FLC) and tetrazoles (e.g., oteseconazole) are widely used in clinic because of their excellent therapeutic effect. However, the overuse of azole drugs has led to the emergence of drug-resistant strains, making the treatment of fungal infections more difficult.6 It is urgent to accelerate the development of new antifungal drugs to deal with the increasing number of drug-resistant fungi. Considering the clinical therapeutic advantages of azole drugs, we still focused on the modification of azole drugs in this study.
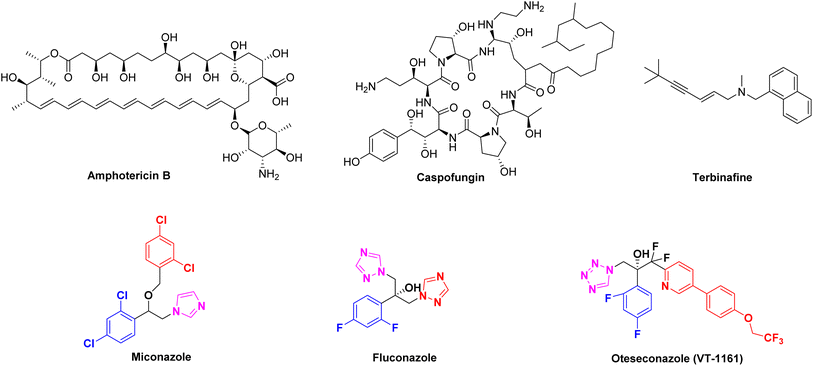 |
| | Fig. 1 The chemical structures of clinical antifungal drugs. | |
2. Results and discussion
2.1. The design of benzamide-containing azoles compounds
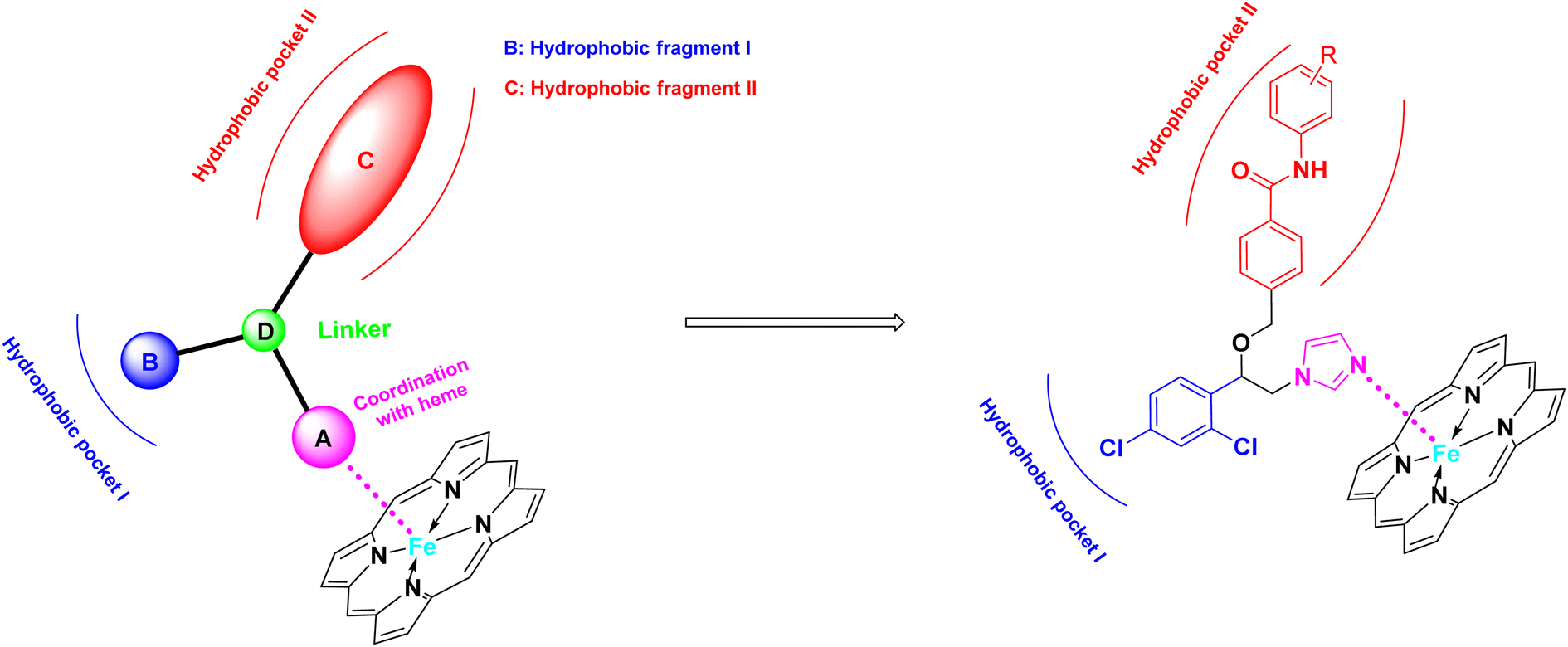
The azole class of antifungal drugs inhibits fungal lanosterol 14-α-demethylase (CYP51) activity through competitive bind to the enzyme's heme cofactor.7,8 Therefore, it is necessary to analyze the structural characteristics of CYP51 inhibitors. As is well known, the CYP51 protein, in addition to the heme ring, contains two hydrophobic channels in its protein cavity, namely hydrophobic channel I and II as shown in the Fig. 2.9,10 In general, we can use the nitrogen-containing heterocyclic to bind to the heme, and the two hydrophobic segments to bind to the two hydrophobic channels. Particularly, hydrophobic channel II is relatively narrow, making it difficult for larger fragments to penetrate into this channel.11 The benzanilide fragment, being relatively flat, has the potential to extend into this channel. Therefore, in this study, the benzanilide fragment was chosen as hydrophobic fragment II for the first time, with the expectation that it would extend into the narrow hydrophobic channel II, thereby enhancing antifungal activity (Fig. 2). Based on this design rationale, compounds A01–A15 were synthesized and evaluated for their antifungal activity using microdilution methods. Further, the mechanism, cytotoxicity, metabolic stability and in vivo efficiency were studied.
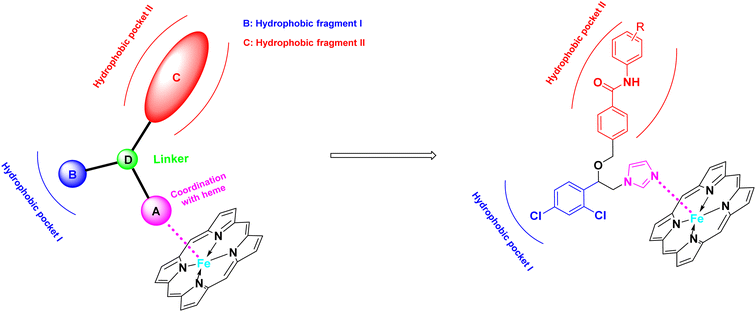 |
| | Fig. 2 Design strategies for target compounds. | |
2.2. Chemistry
The general synthetic routes of target compounds A01–A15 were outlined in Scheme 1. Firstly, intermediate 3 were prepared from substituted α-bromoacetophenone 1 upon treatment with imidazole in the presence of triethylamine. Then, the carbonyl group of intermediate 3 was reduced to intermediate 4 using sodium borohydride. Compound 4 reacted with methyl 4-(bromomethyl) benzoate to generate intermediate 5. After hydrolysis of intermediate 5, it was condensed with various substituted anilines to yield the final products A01–A15.
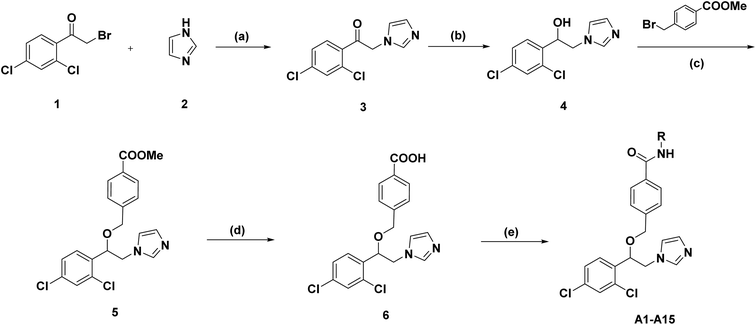 |
| | Scheme 1 Synthetic route of target compounds A01–A15. Reagents and conditions: (a) Et3N, MeOH, 65 °C, 4 h, 90%; (b) NaBH4, MeOH, 25 °C, 2 h, 80%. (c) NaH, tetrahydrofuran, 70 °C, 4 h, 65.5%; (d) KOH, MeOH, reflux, 1 h (without purification); (e) R-NH2, DCM, DMAP, EDCI, 25 °C, 1 h. | |
2.3. In vitro antifungal activity against fluconazole-sensitive Candida albicans strains
The antifungal activity of these compounds was assessed against five species of C. alb (5314),C. par, C. gla, C. kru and C. neo. The minimum inhibitory concentrations (MIC80) were determined to quantify the degree of growth inhibition, with fluconazole and miconazole being used as positive controls for comparison. Interestingly, compounds A01–A15 exhibited moderate to strong antifungal activity against all of the tested fungi. Moreover, there was a clear structure–activity relationship observed among these compounds, suggesting that specific structural features are responsible for their antifungal activity (Table 1). When R is substituted monocycle, compounds A1–A11 exhibit good antifungal activity. The MIC80 values of these compounds range from 0.03 to 0.5 μg mL−1, which are superior to those of the positive control drug fluconazole. As analogs of miconazole, these compounds exhibited better inhibitory activity against both C. alb (5314) and C. kru compared to miconazole. In contrast, when R is substituted bicyclic, compounds A12, A13 and A14 exhibit decreased antifungal activity (MIC = 2–64 μg mL−1). This could be due to the larger size of the bicyclic substituent, which may form unfavorable interactions in the narrow channel II. The antifungal activity significantly decreases when the R group contains a hydrogen bond donor, such as compounds A15 (MIC = 32–>128 μg mL−1). This may be because channel II is hydrophobic and cannot accommodate hydrogen bond donor groups within this hydrophobic channel.
Table 1 The antifungal activity of the target compounds A01–A15
2.4. Analysis of sterol composition in C. albicans (SC5314)
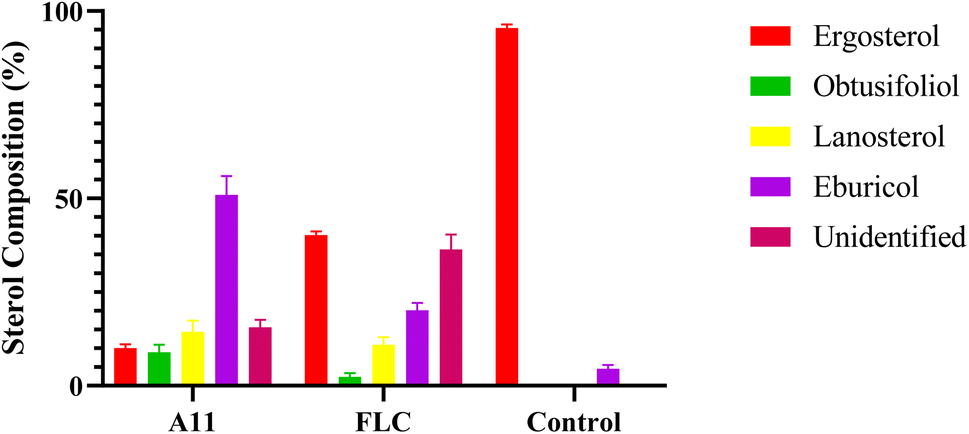
GC-MS was employed to investigate the antifungal mechanism of the target compounds by determining the changes in sterol content in fungal cell membrane.12 Fluconazole was used as a positive control in this study. The model strain C. alb 5314 was incubated with the drugs for 24 hours to detect the alterations in sterol content. The results, presented in Fig. 3, demonstrate that fluconazole and compound A11 effectively suppresses ergosterol biosynthesis in fungal cell membranes, leading to significant accumulations of obtusifoliol, lanosterol, and eburicol. These findings suggest that our compounds exert their antifungal activity by inhibiting the enzyme CYP51, involved in the biosynthesis of ergosterol.
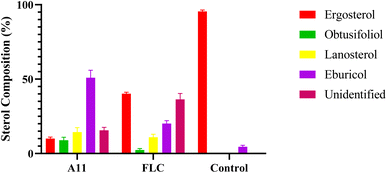 |
| | Fig. 3 Sterol composition analysis. FLC and A11 were co-cultured with C. alb SC5314 for 24 h at 0.03 μg mL−1. Bars represent the mean ± SD (n = 3). | |
2.5. Morphological observation of fungal cells
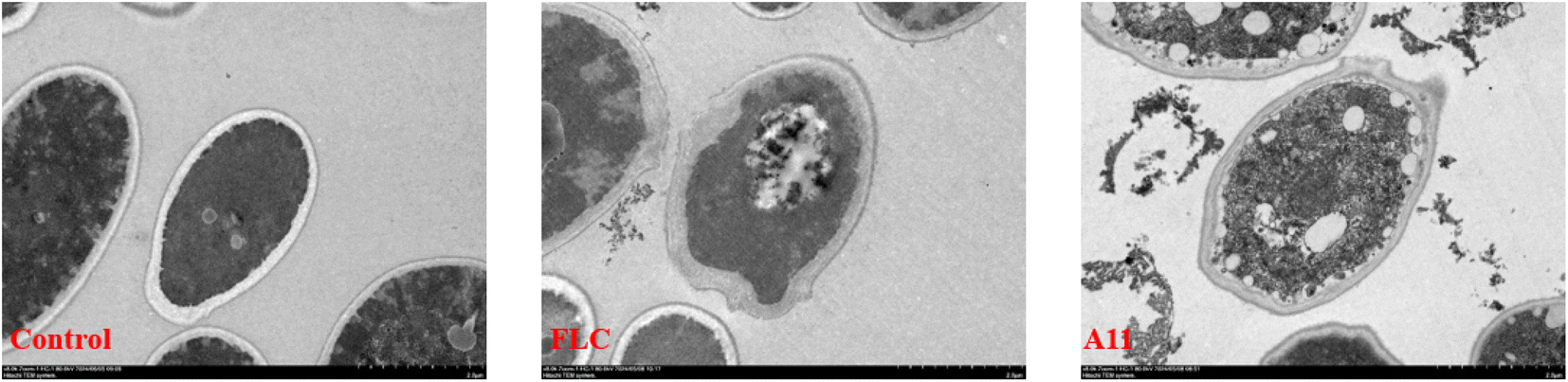
The GC-MS analysis experiment preliminarily proved that the target of our compounds is CYP51. It is well known that the azole drugs inhibit the biosynthesis of ergosterol by inhibiting the activity of CYP51, thereby increasing the permeability of cell membranes and leading to fungal death. Therefore, the observation of cell membrane morphology can further directly prove that the target of our compound is CYP51. To complete this experiment, we chosen C. alb 5314 as the test strain and applied transmission electron microscopy (TEM) to observe the morphology changes of the fungal cell membrane after the drug was added. The experimental results were shown in Fig. 4. It can be observed that the fungal cells in the control group presented a regular and complete structure. After the addition of drugs or fluconazole, the cell membrane was damaged and the contents leak out into the surrounding area. That said, compound A11 could indeed disrupt the structural integrity of the cells, ultimately resulting in the breakdown and death of the fungus. This result is consistent with the physiological function of CYP51, which further proves that the target of our compounds is CYP51.
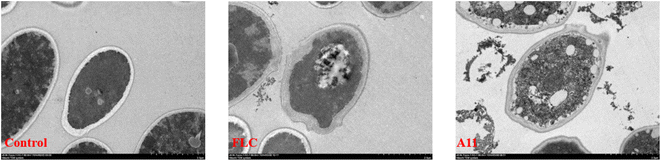 |
| | Fig. 4 Morphological observation of fungal cells (C. alb SC5314). FLC and A11 were co-cultured with C. alb SC5314 for 24 h at 2 μg mL−1. | |
2.6. In vitro antifungal activity against fluconazole-resistant Candida albicans strains
The widespread occurrence of fungal resistance exacerbates the difficulty of treatment of fungal infection.13–15 For the new compounds, if they have anti-drug resistance activity, it will have further development value. Thus, we further determined the antifungal activity of all the target compounds against five fluconazole-resistant strains obtained from clinical isolation. Fluconazole was used as a reference drug in this study. As indicated in Table 2, all target compounds exhibited significant antifungal activity against the tested resistant strains. Among these compounds, A11 also displayed the most potent effects with MIC80 values of 0.125–2 μg mL−1 on the tested strains. In contrast, fluconazole was completely inactive. These findings suggest that compound A11 has the ability to effectively overcome fluconazole resistance. Subsequently, we also conducted preliminary studies on the mechanisms of drug resistance.
Table 2 The antifungal activity of the target compounds A01–A15
2.7. Prevented the morphological transition of C. albicans and inhibited fungal biofilm formation
The biofilm is a polysaccharide–protein complex secreted by fungi after forming a community and enveloping the fungal surface. Due to its natural barrier effect, it reduces drug uptake, leading to fungal drug resistance. Therefore, we investigated whether our target compounds have biofilm inhibition effects, thereby confirming the compound's anti-resistance mechanism. The yeast-to-hypha transition, also known as the filamentation of microbial colonies, is a crucial step in the formation of fungal biofilms. Consequently, we investigated the impact of the target molecule A11 on fungal morphological transition, particularly during the yeast-to-hypha transition. This analysis focused on understanding the underlying mechanisms involved in this transition. The experimental results were shown in Fig. 5.
 |
| | Fig. 5 DIC microscope photographs on the filamentation of azole-resistant strain 904. FLC and A11 were co-cultured with strain 904 for 5 h at 0.25 μg mL−1. | |
The results of the study demonstrated that fluconazole did not inhibit the morphological transition in azole-resistant strain 904. Conversely, the introduction of the target compound A11 at a concentration of 0.25 μg mL−1 completely hindered the morphogenetic transition of azole-resistant strain 904 from yeast to hyphae (as depicted in Fig. 5).
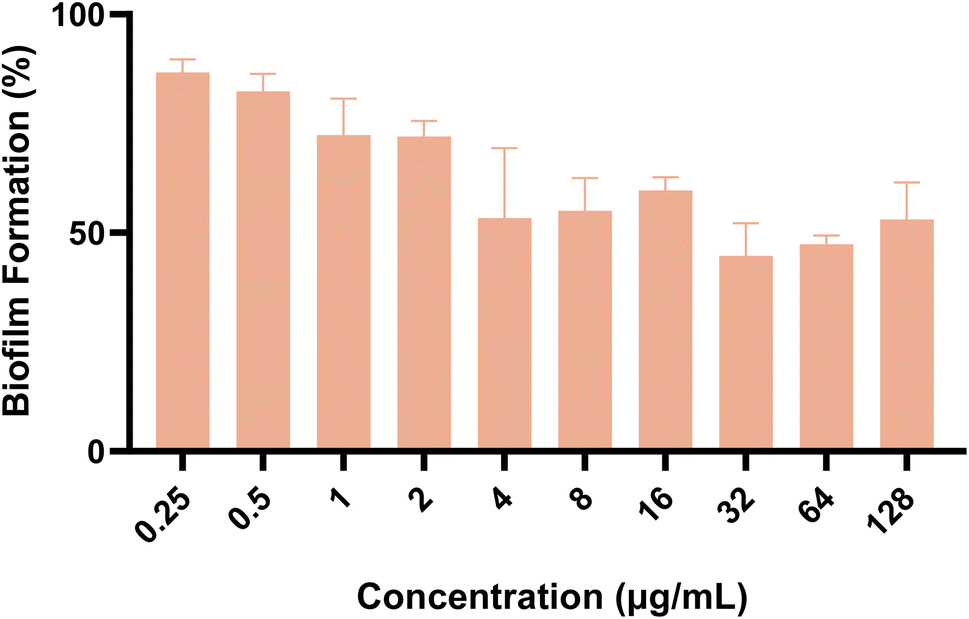
Subsequently, we also investigated the impact of the target compound on the formation of mature biofilms. As shown in Fig. 6, compound A11 showed dose-dependent destruction of mature fungal biofilms of azole-resistant strain 904. These two experiments showed that A11 can inhibit the initial transformation of yeast into mycelium and destroy mature biofilms.
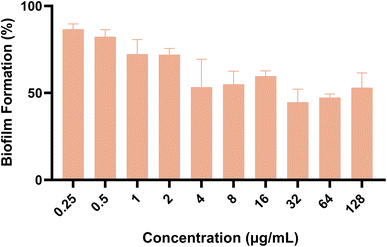 |
| | Fig. 6 The effect of compound A11 on the biofilm formation (%) of azole-resistant strain 904. The outcomes were expressed as the average ± standard deviation via three separate trials. | |
2.8. Downregulated drug resistant gene
Our target compound showed good anti-resistance activity, which prompted us to investigate whether this was related to the expression of resistance genes. The resistance of Candida albicans to azole has been shown to be related to a variety of factors, such as the overexpression of ERG11 or the upregulation of efflux pumps.16,17 To investigate whether the anti-drug resistance mechanism of our target compounds is related to the two factors, the expression of the resistant gene ERG11 and CDR1 treated with drugs was determined by the real-time reverse transcriptase polymerase chain reaction (RT-PCR).
From the obtained experimental results (Fig. 7), there was overexpression of drug resistance gene ERG11 and CDR1 after fluconazole treatment, which may be the cause of fluconazole resistance. When treated with the target compounds A01, A07 and A11, the expression of these genes was reduced to basic levels. Overall, the resistance mechanism of our compounds is related to these two genes.
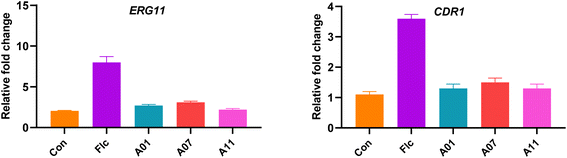 |
| | Fig. 7 Expression of resistant gene ERG11 and CDR1 of azole-resistant strain of Candida albicans (strain 904). The concentration of compounds and FLC was 0.25 μg mL−1 and DMSO as control. The outcomes were expressed as the average ± standard deviation via three separate trials. | |
2.9. Minimum fungicidal concentration and time-kill tests
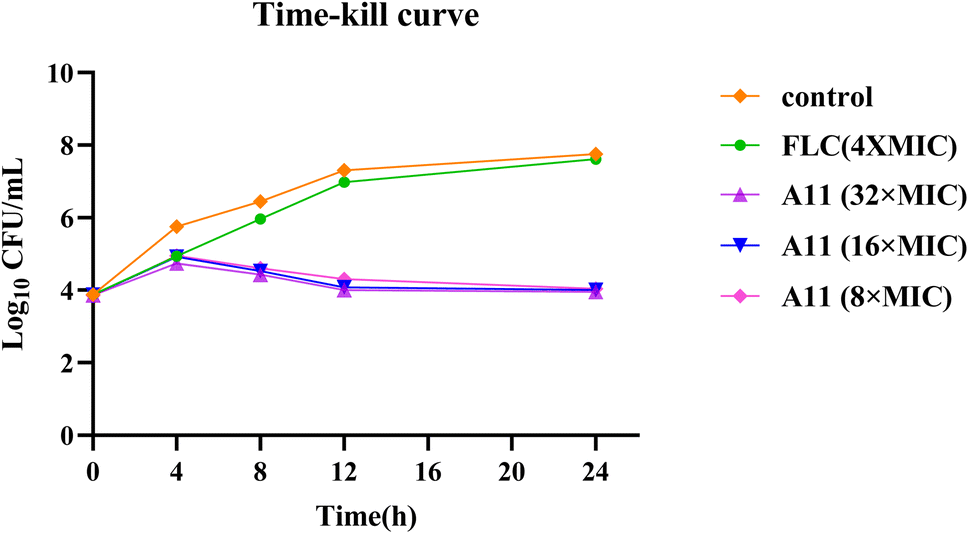
If a compound can completely eradicate fungi, especially resistant strains, it would be more valuable for development. Therefore, we have also conducted minimum fungicidal concentration testing and concentration-dependent time-kill assays for compound A11 against strain 904.
As shown in Table 3, compound A11 demonstrated significant fungicidal activity in this study, with MFC = 2 μg mL−1. In the time-kill curve assay, gradient doses of compound A11 (8 × MIC, 16 × MIC, 32 × MIC) and fluconazole (16 μg mL−1) were tested. Compound A11 exhibited potent fungicidal effects at three concentrations. Consequently, compound A11 is expected to shorten the duration of treatment for invasive fungal infections (Fig. 8).
Table 3 The minimum fungicidal concentration of compound A11a
| Compd |
MIC against C. alb (μg mL−1) |
MFC against C. alb (μg mL−1) |
MFC/MIC |
| Abbreviations: C. alb, strain 904, fluconazole-resistant strain of C. albicans. |
| A11 |
0.125 |
2 |
16 |
| Fluconazole |
>128 |
>128 |
— |
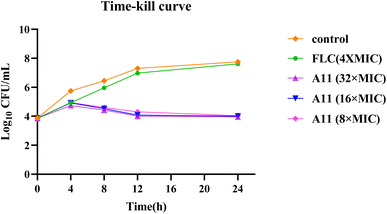 |
| | Fig. 8 The time-kill curves of compound A11. | |
2.10. Analysis of changes in ROS level
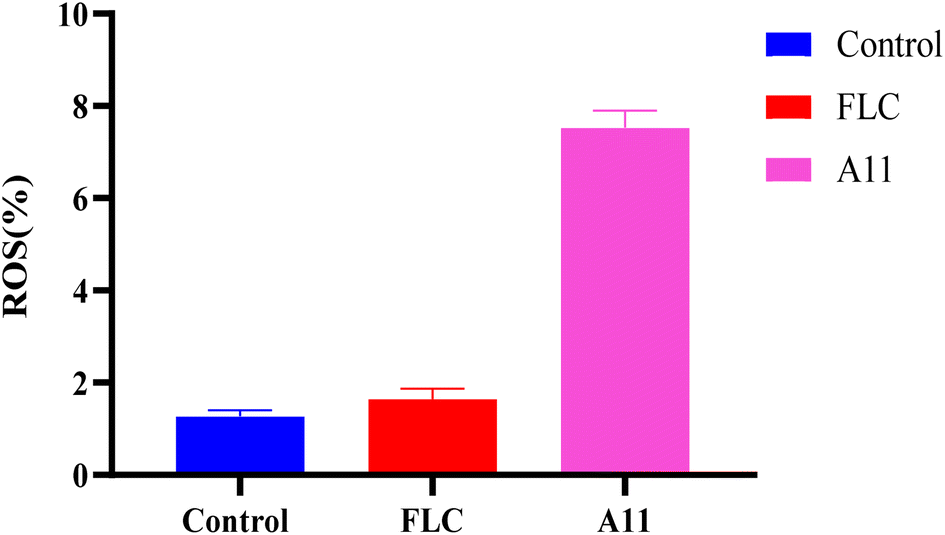
Our target compounds not only exhibit fungistatic activity but also fungicidal activity. This result prompted us to investigate whether the compounds have a new mechanism of action. The generation of reactive oxygen species (ROS) has been reported to cause mitochondrial damage in fungi, leading to fungal death.17 Therefore, we also investigated whether the target compounds can stimulate the production of reactive oxygen species, thereby demonstrating fungicidal activity. In detail, we employed the DCFH-DA method to examine the variations in ROS levels in strain 904 when exposed to compound A11.
Fig. 9 demonstrates a significant increase in intracellular ROS levels when exposed to 0.25 μg mL−1 of A11, as compared to the blank and fluconazole experimental groups. The results showed the target compounds could enhance their antifungal effectiveness by inducing the substantial production of reactive oxygen species (ROS) within fungal cells.
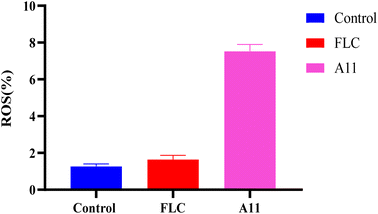 |
| | Fig. 9 Effect of compound A11 on ROS in strain 904. | |
2.11. Metabolic stability assay of liver microsomes
During the drug discovery process, the metabolic stability of lead compounds is a critical factor influencing their druggability. Therefore, we first evaluated the metabolic stability of the representative compound A11 in liver microsomes, with miconazole selected as the positive control. The results are shown in the Table 4. Miconazole had a T1/2 of 8.3 min, while the representative compound A11 exhibited greater metabolic stability with a T1/2 of 15.5 min.
Table 4 Metabolic stability of the representative compound A11
| Compd |
T1/2 (min) |
CL (mL min−1 mg−1) |
| A11 |
15.5 |
0.0894 |
| Miconazole |
8.3 |
0.6601 |
| Fluconazole |
>120 |
0.002 |
2.12. Plasma stability assay
Despite the hepatic metabolic stability of a compound being widely regarded as one of the major challenges in the drug discovery process, the plasma stability of the compound remains a significant influencing factor in new drug development. Since the metabolic enzymes in the liver differ from those in the blood, stability in liver microsomes in vitro does not necessarily indicate stability in plasma. Therefore, we also evaluated the plasma stability of the representative compound A11. The results are shown in the Table 5. The representative compound A11 and positive drugs such as miconazole and fluconazole all demonstrated excellent plasma stability, with T1/2 values exceeding 360 min. The results of liver microsomal metabolic stability and plasma metabolic stability indicated that the target compounds were valuable for further research and development.
Table 5 Plasma stability of the representative compound A11
| Compd |
Time point (min) |
% remaining |
T1/2 (min) |
| A11 |
180 |
73.9 |
>360 |
| Miconazole |
180 |
94.9 |
>360 |
| Fluconazole |
180 |
102 |
>360 |
2.13. In vitro cytotoxical effects on normal cells
Toxicity study on normal human cells is an important part of drug development. Therefore, we selected the human embryonic lung fibroblast cell (MRC-5) as model cell to evaluate the cytotoxicity of our compounds. The cells were treated with our compounds for 72 hours, and the results are presented in Table 6. We observed that the IC50 values of compounds A01–A15 were all greater than 100 μM. This suggests that compounds A01–A15 did not exhibit any toxicity towards MRC-5 cells, indicating their safety in normal human cells.
Table 6 The IC50 of compounds A01-A15 on MRC-5 cell lines
| Compd |
IC50 (μM) |
Compd |
IC50 (μM) |
| A01 |
>100 |
A10 |
>100 |
| A02 |
>100 |
A11 |
>100 |
| A03 |
>100 |
A12 |
>100 |
| A04 |
>100 |
A13 |
>100 |
| A05 |
>100 |
A14 |
>100 |
| A06 |
>100 |
A15 |
>100 |
| A07 |
>100 |
Miconazole |
>100 |
| A08 |
>100 |
Fluconazole |
>100 |
| A09 |
>100 |
cis-Pt |
78.3 |
2.14. In vivo efficiency of compound A11
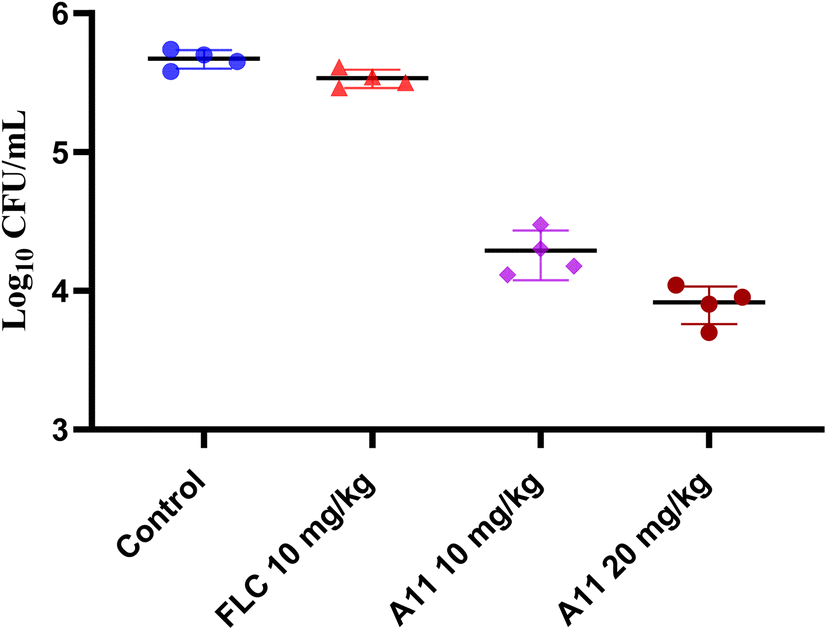
Given the compound A11's good in vitro antifungal activity, especially its significant inhibitory effect on resistant strains, we subsequently conducted in vivo activity studies on compound A11, with fluconazole being used as a positive control. In this experiment, a mouse model of azole-resistant strain 904 infection was established by injecting the resistant strain into the tail vein of mice. The effectiveness of compound A11 in combating the infection was evaluated by assessing the colony-forming units (CFU) of the fungal burden in the kidneys of mice on the fifth day after infection. The results are shown in the Fig. 10.
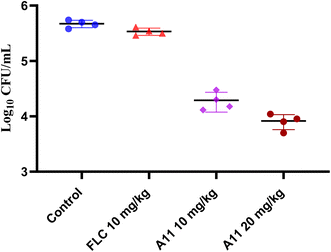 |
| | Fig. 10 Therapeutic efficacies of compound A11 and fluconazole in the azole-resistant strain 904. | |
The results showed that compound A11 also exhibited potent efficacy against the azole-resistant strain 904 in vivo (Fig. 10). In contrast, fluconazole was ineffective against azole-resistant strains in vivo. This result suggests that our target compound holds promise for treating infections caused by resistant fungi.
2.15. Molecular docking studies
Design strategies for target compounds were validated through molecular docking studies using Candida albicans CYP51 crystal structure (PDB code: 5TZ1) with compound A11, conducted via Discovery Studio 3.0. Miconazole, an imidazole drug, served as a control (Fig. 11). Results illustrated interactions between the imidazole group of compound A11 and miconazole with the iron of the heme group. Moreover, the chlorophenyl group of compound A11 fit snugly within hydrophobic pocket I formed by ILE131 and TYR132. Furthermore, the other dichlorobenzene of miconazole interacts solely with Met508 at the entrance of hydrophobic channel II. However, the benzanilide fragment of target compound A11 can deeply insert into the narrow hydrophobic channel II, which is composed of MET508, LEU121 and PRO230.
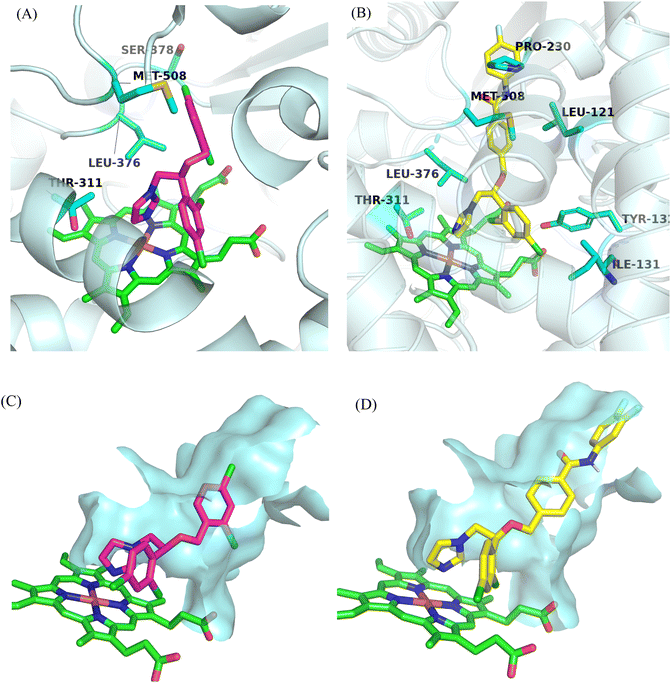 |
| | Fig. 11 Predicted binding mode of (A) MCZ, (B) A11 with Candida albicans CYP51. Binding conformation of (C) MCZ, (D) A11 in the active pocket of Candida albicans CYP51. | |
3. Conclusion
Severe fungal infections pose a significant threat to human health, and the global incidence of deep fungal infections is on the rise. Furthermore, the increasing use of antifungal medications has contributed to the emergence of drug-resistant strains, exacerbating the problem. Consequently, there is an urgent need to develop novel antifungal drugs with unique structures to effectively combat drug-resistant fungal infections. In this study, a series of novel benzanilide-containing azoles have been designed. In particular, the hydrophobic benzanilide rings was expected to penetrate deep into narrow hydrophobic channels II of CYP51, thereby enhancing antifungal activity. Then, the new derivatives were synthesized and their structures were analyzed by 1H NMR, 13C-NMR, and HRMS spectral. And the compounds A01–A15 were screened for in vitro antifungal activity against Candida albicans (SC5314), Candida parapsilosis, Candida glabrata, Candida krusei and Cryptococcus neoformans. Intriguingly, all the target compounds showed moderate to exceptional efficacy against the five fluconazole-sensitive Candida albicans. Further investigation demonstrated that compound A11, the most potent among the synthesized derivatives, exhibits significant antifungal activity by inhibiting the fungal CYP51 enzyme and suppressing fungal ergosterol production. Meantime, the direct observation of cell membrane damage further confirmed that the target of our compound is CYP51. Most interesting is that the target compounds displayed notable activity against fluconazole-resistant strains, surpassing the efficacy of the positive control drug fluconazole. Meanwhile, investigations into the mechanisms of drug resistance unveiled that the compounds were capable of suppressing the formation of biofilm and the expression of the resistance-related gene ERG11 and the efflux pumps gene CDR1, consequently reversing drug resistance. Moreover, compound A11 exhibited fungicide effects against fluconazole-resistant strain and notably increased the intracellular ROS level, leading to cellular harm. Compound A11 also demonstrated good liver microsomal stability (T1/2 = 15.5 min) and plasma stability (T1/2 > 360 min). In addition, these compounds also showed low toxicity towards human MRC-5 cell with IC50 >100 μM, indicating their potential as safe and effective antifungal agents. In a mouse infection model, mice treated with A11 for infection showed a significantly reduced fungal burden compared to mice treated with FLC. Molecular docking experiment demonstrated compound A11 can form a strong binding interaction with Candida albicans CYP51. Specifically, the benzanilide fragment can penetrate deeply into the narrow hydrophobic channel II, thereby achieving the intended design goal. These findings suggest that the novel benzanilide-containing azoles could be promising candidates for the treatment of drug-resistant fungal infections.
4. Experimental
4.1. Chemistry
All reagents and solvents used in the study were purchased from commercial suppliers and used as received without further purification. Silica gel (200–300 mesh) was used for column chromatography, and silica gel precoated GF254 plates were used for TLC analysis. Melting points were measured using an X-5 melting point equipment from Beijing Tech Instrument Co., Ltd, Beijing, China. High-resolution mass spectra (HRMS) were recorded using an Agilent Accurate-Mass Q-TOF 6530 instrument (Santa Clara, CA, USA) in ESI mode. 1H NMR and 13C NMR spectra were recorded in DMSO-d6 solvent on a 400 (100)-MHz Bruker AV-400 spectrometer (Bruker Bioscience, Billerica, MA, USA).
4.2. General procedure for the synthesis of target compounds A01–A15
The synthesis of target compound 4-((1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-phenylbenzamide (A01) was chosen as the example: 2-bromo-1-(2,4-dichlorophenyl)ethan-1-one (5.32 g, 20 mmol) and imidazole (1.50 g, 22 mmol) are added to a solution of methanol (20 mL), followed by the addition of triethylamine (2.02 g, 20 mmol), and refluxed for 4 hours. The solvent was removed under reduced pressure, and 20 mL of water was added. The mixture was then extracted with ethyl acetate (3 × 10 mL), and the combined organic phase was sequentially washed with water (3 × 10 mL) and saturated brine (20 mL). After drying over anhydrous sodium sulfate and filtering the drying agent, the crude product was obtained by vacuum concentration. The crude product was purified by column chromatography (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 2:1) to afford intermediate 3 (4.12 g) as a yellow solid with a yield of 90.0%. Subsequently, compound 3 (4.12 g, 18 mmol) was dissolved in methanol (30 mL), followed by the portion-wise addition of sodium borohydride (0.68 g, 18 mmol), and the mixture was stirred at room temperature for 2 hours. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 20 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 4:1) to obtain the key intermediate 4 (3.67 g) with a yield of 80.0%. Intermediate 4 (3.67 g, 14.4 mmol) and 4-(bromomethyl) benzoate (3.28 g, 14.4 mmol) were added to tetrahydrofuran (30 mL), followed by the portion-wise addition of sodium hydride (0.5 g), and the reaction was carried out at 70 °C for 4 hours. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 20 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 8:1) to obtain the key intermediate 5 (3.81 g) with a yield of 65.5%. Intermediate 5 (0.38 g, 0.94 mmol) was dissolved in methanol (10 mL), and potassium hydroxide (0.05 g, 0.94 mmol) was added, followed by heating under reflux for 1 hour. After the reaction was completed, the reaction mixture was concentrated and, without further purification, directly dissolved in dichloromethane. EDCI (0.18 g, 0.94 mmol) and DMAP (0.06 g, 0.47 mmol) were then added, followed by the addition of aniline (0.09 g, 0.94 mmol), and the reaction was allowed to proceed at room temperature for 1 hour. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 10 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 8:1) to obtain the final product A01 (0.36 g) by column chromatography with a two-step yield of 82.0%. m.p. 132.4–134.3 °C. HRMS (ESI): calcd for C25H21N3O2Cl2 [M + H]+: 466.1084, found 466.1100 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.80–7.77 (m, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.53 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.38–7.33 (m, 2H), 7.31 (d, J = 8.3 Hz, 2H), 7.12–7.08 (m, 2H), 6.90 (s, 1H), 5.05 (dd, J = 7.0, 3.8 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.7, 141.7, 139.6, 138.4, 135.2, 134.6, 134.1, 133.7, 129.8, 129.5, 129.1, 128.7, 128.4, 128.2, 127.4, 124.1, 120.9, 120.6, 77.1, 70.3, 50.5. The preparation methods of target compounds A02–A15 were the same as that of target compound A01.
:ethyl acetate = 2:1) to afford intermediate 3 (4.12 g) as a yellow solid with a yield of 90.0%. Subsequently, compound 3 (4.12 g, 18 mmol) was dissolved in methanol (30 mL), followed by the portion-wise addition of sodium borohydride (0.68 g, 18 mmol), and the mixture was stirred at room temperature for 2 hours. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 20 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 4:1) to obtain the key intermediate 4 (3.67 g) with a yield of 80.0%. Intermediate 4 (3.67 g, 14.4 mmol) and 4-(bromomethyl) benzoate (3.28 g, 14.4 mmol) were added to tetrahydrofuran (30 mL), followed by the portion-wise addition of sodium hydride (0.5 g), and the reaction was carried out at 70 °C for 4 hours. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 20 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 8:1) to obtain the key intermediate 5 (3.81 g) with a yield of 65.5%. Intermediate 5 (0.38 g, 0.94 mmol) was dissolved in methanol (10 mL), and potassium hydroxide (0.05 g, 0.94 mmol) was added, followed by heating under reflux for 1 hour. After the reaction was completed, the reaction mixture was concentrated and, without further purification, directly dissolved in dichloromethane. EDCI (0.18 g, 0.94 mmol) and DMAP (0.06 g, 0.47 mmol) were then added, followed by the addition of aniline (0.09 g, 0.94 mmol), and the reaction was allowed to proceed at room temperature for 1 hour. After the reaction was completed, water (50 mL) was added, and the reaction mixture was extracted three times with ethyl acetate (3 × 10 mL). The combined organic layers were concentrated under reduced pressure. The crude product was purified by column chromatography (petroleum ether:ethyl acetate = 8:1) to obtain the final product A01 (0.36 g) by column chromatography with a two-step yield of 82.0%. m.p. 132.4–134.3 °C. HRMS (ESI): calcd for C25H21N3O2Cl2 [M + H]+: 466.1084, found 466.1100 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.80–7.77 (m, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.53 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.38–7.33 (m, 2H), 7.31 (d, J = 8.3 Hz, 2H), 7.12–7.08 (m, 2H), 6.90 (s, 1H), 5.05 (dd, J = 7.0, 3.8 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.7, 141.7, 139.6, 138.4, 135.2, 134.6, 134.1, 133.7, 129.8, 129.5, 129.1, 128.7, 128.4, 128.2, 127.4, 124.1, 120.9, 120.6, 77.1, 70.3, 50.5. The preparation methods of target compounds A02–A15 were the same as that of target compound A01.
4.2.1. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(4-fluorophenyl)benzamide (A02). White solid (88.1% yield); m.p. 139.1–141.3 °C. HRMS (ESI): calcd for C25H20N3O2Cl2F [M + Na]+: 506.0809, found 506.0837 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.80 (dd, J = 9.2, 5.1 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.31 (d, J = 8.3 Hz, 2H), 7.20 (t, J = 8.9 Hz, 2H), 7.08 (s, 1H), 6.90 (s, 1H), 5.04 (dd, J = 7.0, 3.8 Hz, 1H), 4.55–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.6, 160.0, 157.6, 141.8, 138.4, 136.0, 135.9, 135.2, 134.4, 134.1, 133.7, 129.8, 129.5, 128.7, 128.4, 128.2, 127.4, 122.7, 122.6, 120.6, 115.8, 115.5, 77.1, 70.3, 50.5.
4.2.2. N-(4-Chlorophenyl)-4-((1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)benzamide (A03). White solid (86.3% yield); m.p. 141.4–143.4 °C. HRMS (ESI): calcd for C25H20N3O2Cl3 [M + Na]+: 522.0513, found 522.0539 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.90 (d, J = 8.3 Hz, 2H), 7.83 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.44–7.41 (m, 2H), 7.40 (d, J = 2.1 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 7.09 (s, 1H), 6.90 (s, 1H), 5.04 (dd, J = 6.9, 3.8 Hz, 1H), 4.55–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.8, 141.9, 138.6, 135.2, 134.3, 134.1, 133.7, 129.8, 129.5, 129.0, 128.7, 128.4, 128.2, 127.7, 127.4, 122.3, 77.1, 70.3, 50.5.
4.2.3. N-(4-Bromophenyl)-4-((1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)benzamide (A04). White solid (83.2% yield); m.p. 139.1–141.3 °C. HRMS (ESI): calcd for C25H20N3O2Cl2Br [M + Na]+: 566.0008, found 566.0018 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.90 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.54–7.52 (m, 2H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 7.08 (s, 1H), 6.90 (s, 1H), 5.04 (dd, J = 7.0, 3.9 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.8, 141.9, 139.01, 138.4, 135.2, 134.3, 134.1, 133.7, 131.9, 129.8, 129.5, 128.7, 128.4, 128.2, 127.4, 122.7, 120.6, 115.8, 77.1, 70.3, 50.5.
4.2.4. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(p-tolyl)benzamide (A05). White solid (84.5% yield); m.p. 134.7–136.4 °C. HRMS (ESI): calcd for C26H23N3O2Cl2 [M + Na]+: 502.1060, found 502.1080 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 2.1 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.53 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 8.3 Hz, 2H), 7.08 (s, 1H), 6.90 (s, 1H), 5.04 (dd, J = 7.0, 3.8 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.25 (m, 2H), 2.28 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.4, 141.6, 138.4, 137.1, 135.3, 134.6, 134.1, 133.7, 133.1, 129.8, 129.5, 129.4, 128.7, 128.4, 128.1, 127.4, 120.9, 120.6, 77.1, 70.3, 50.5, 21.0.
4.2.5. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(4-(trifluoromethyl)phenyl)benzamide (A06). White solid (86.9% yield); m.p. 137.6–139.2 °C. HRMS (ESI): calcd for C26H20N3O2Cl2F3 [M + Na]+: 556.0777, found 556.0809 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.03 (d, J = 8.5 Hz, 2H), 7.93 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 8.3 Hz, 2H), 7.09 (s, 1H), 6.90 (s, 1H), 5.05 (dd, J = 7.0, 3.8 Hz, 1H), 4.57–4.38 (m, 2H), 4.39–4.24 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.2, 143.3, 142.1, 138.4, 135.2, 134.1, 133.7, 129.8, 129.5, 128.4, 127.5, 126.4, 126.4, 124.3, 123.9, 123.5, 120.6, 77.1, 70.3, 50.5.
4.2.6. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(4-methoxyphenyl)benzamide (A07). White solid (81.5% yield); m.p. 144.6–146.3 °C. HRMS (ESI): calcd for C26H23N3O3Cl2 [M + Na]+: 518.1009, found 518.1030 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 1.9 Hz, 1H), 7.68 (d, J = 5.3 Hz, 2H), 7.53 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.08 (s, 1H), 6.93 (d, J = 9.0 Hz, 2H), 6.90 (s, 1H), 5.04 (dd, J = 7.0, 3.9 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.25 (m, 2H), 3.75 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.2, 156.0, 141.5, 138.4, 135.2, 134.6, 134.1, 133.7, 132.7, 129.8, 129.5, 128.7, 128.4, 128.1, 127.4, 122.4, 120.6, 114.2, 77.1, 70.3, 55.6, 50.5.
4.2.7. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(3-fluorophenyl)benzamide (A08). White solid (86.2% yield); m.p. 137.8–139.5 °C. HRMS (ESI): calcd for C25H20N3O2Cl2F [M + Na]+: 506.0809, found 506.0824 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 7.92–7.88 (m, 2H), 7.77 (dt, J = 11.8, 2.3 Hz, 1H), 7.69 (d, J = 2.1 Hz, 1H), 7.57 (dd, J = 8.2, 1.1 Hz, 1H), 7.53 (d, J = 1.1 Hz, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.44–7.36 (m, 2H), 7.33 (d, J = 8.3 Hz, 2H), 7.09 (s, 1H), 6.90 (d, J = 1.1 Hz, 1H), 5.05 (dd, J = 7.0, 3.9 Hz, 1H), 4.56–4.39 (m, 2H), 4.36–4.24 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 163.7, 161.4, 142.0, 141.5, 141.4, 138.4, 135.2, 134.2, 134.1, 133.7, 130.7, 130.7, 129.8, 129.5, 128.7, 128.4, 128.2, 127.5, 120.6, 116.5, 116.4, 110.7, 110.5, 107.6, 107.3, 77.1, 70.3, 50.5.
4.2.8. N-(3-Chlorophenyl)-4-((1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)benzamide (A09). White solid (88.0% yield); m.p. 129.6–131.8 °C. HRMS (ESI): calcd for C25H20N3O2Cl3 [M + Na]+: 522.0513, found 522.0507 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 7.98 (t, J = 2.1 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.72 (dd, J = 7.7, 1.8 Hz, 1H), 7.69 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.40 (dd, J = 20.4, 8.3 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 7.16 (dd, J = 8.0, 1.2 Hz, 1H), 7.09 (s, 1H), 6.90 (s, 1H), 5.05 (dd, J = 7.0, 3.8 Hz, 1H), 4.55–4.39 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 142.0, 141.1, 138.4, 135.2, 134.1, 134.1, 133.7, 133.4, 130.8, 129.8, 129.5, 128.7, 128.4, 128.3, 127.5, 123.8, 120.6, 120.2, 119.1, 77.1, 70.3, 50.5.
4.2.9. N-(3-Bromophenyl)-4-((1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)benzamide (A10). White solid (82.4% yield); m.p. 136.1–138.3 °C. HRMS (ESI): calcd for C25H20N3O2Cl2Br [M + Na]+: 566.0008, found 566.0020 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.12 (t, J = 2.0 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.77 (dt, J = 7.7, 1.8 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 7.56–7.53 (m, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.34–7.30 (m, 4H), 7.09 (s, 1H), 6.90 (s, 1H), 5.05 (dd, J = 7.0, 3.8 Hz, 1H), 4.55–4.38 (m, 2H), 4.36–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 142.0, 141.3, 138.4, 135.2, 134.1, 134.1, 133.7, 131.1, 129.8, 129.5, 128.7, 128.4, 128.3, 127.4, 126.7, 123.0, 121.9, 120.6, 119.5, 77.1, 70.3, 50.5.
4.2.10. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(2,4-difluorophenyl)benzamide (A11). White solid (81.7% yield); m.p. 141.0–143.4 °C. HRMS (ESI): calcd for C25H19N3O2Cl2F2 [M + Na]+: 524.0715, found 524.0722 [M + Na]+. 1H NMR (600 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.59 (td, J = 8.9, 6.2 Hz, 1H), 7.53 (d, J = 1.1 Hz, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.36 (ddd, J = 10.6, 9.1, 2.8 Hz, 1H), 7.31 (d, J = 8.2 Hz, 2H), 7.16–7.10 (m, 1H), 7.08 (t, J = 1.2 Hz, 1H), 6.89 (d, J = 1.1 Hz, 1H), 5.04 (dd, J = 7.0, 3.9 Hz, 1H), 4.56–4.38 (m, 2H), 4.35–4.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.6, 161.3, 159.0, 158.9, 157.9, 157.8, 155.4, 155.3, 142.1, 138.4, 135.2, 134.1, 133.7, 133.3, 129.9, 129.5, 129.1, 129.1, 129.0, 128.9, 128.7, 128.4, 128.3, 127.5, 122.8, 122.7, 122.7, 120.6, 111.8, 111.8, 111.6, 111.5, 105.1, 104.9, 104.6, 77.1, 70.3, 50.5.
4.2.11. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(quinolin-7-yl)benzamide (A12). White solid (87.6% yield); m.p. 133.6–136.1 °C. HRMS (ESI): calcd for C28H22N4O2Cl2 [M + Na]+: 539.1012, found 539.1028 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.82 (dd, J = 4.2, 1.7 Hz, 1H), 8.56 (d, J = 2.3 Hz, 1H), 8.33 (dd, J = 8.5, 1.7 Hz, 1H), 8.08–8.00 (m, 2H), 7.99–7.96 (m, 2H), 7.70 (d, J = 2.1 Hz, 1H), 7.55 (d, J = 1.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.44 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.10 (s, 1H), 6.91 (s, 1H), 5.06 (dd, J = 7.0, 3.9 Hz, 1H), 4.56–4.41 (m, 2H), 4.37–4.27 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 149.7, 145.4, 142.0, 138.4, 137.6, 136.1, 135.2, 134.3, 134.1, 133.7, 132.2, 132.0, 129.8, 129.5, 129.1, 128.7, 128.4, 128.3, 127.5, 124.8, 122.2, 120.6, 116.8, 77.1, 70.3, 50.5.
4.2.12. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(naphthalen-1-yl)benzamide (A13). White solid (84.1% yield); m.p. 143.1–145.1 °C. HRMS (ESI): calcd for C29H23N3O2Cl2 [M + Na]+: 538.1060, found 538.1075 [M + Na]+. 1H NMR (400 MHz,) δ 10.42 (s, 1H), 8.04 (d, J = 8.2 Hz, 2H), 8.00–7.96 (m, 2H), 7.87 (d, J = 7.5 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.63–7.60 (m, 1H), 7.58–7.54 (m, 4H), 7.51 (dd, J = 8.4, 2.1 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.10 (t, J = 1.2 Hz, 1H), 6.91 (t, J = 1.1 Hz, 1H), 5.07 (dd, J = 6.9, 3.9 Hz, 1H), 4.57–4.41 (m, 2H), 4.37–4.26 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.3, 141.8, 138.4, 135.3, 134.3, 134.3, 134.1, 133.7, 129.9, 129.7, 129.5, 128.7, 128.6, 128.4, 128.3, 127.5, 126.8, 126.6, 126.5, 126.0, 124.4, 123.8, 120.6, 77.1, 70.3, 50.5.
4.2.13. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(naphthalen-2-yl)benzamide (A14). White solid (85.4% yield); m.p. 144.4–146.5 °C. HRMS (ESI): calcd for C29H23N3O2Cl2 [M + Na]+: 538.1066, found 538.1074 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.92–7.83 (m, 4H), 7.69 (d, J = 2.1 Hz, 1H), 7.55 (s, 1H), 7.50 (td, J = 7.7, 7.1, 1.7 Hz, 2H), 7.46–7.41 (m, 2H), 7.34 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 1.3 Hz, 1H), 6.91 (d, J = 1.1 Hz, 1H), 5.06 (dd, J = 7.0, 3.9 Hz, 1H), 4.56–4.40 (m, 2H), 4.37–4.26 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 141.8, 138.4, 137.3, 135.3, 134.5, 134.1, 133.8, 133.7, 130.5, 129.8, 129.5, 128.7, 128.6, 128.4, 128.3, 127.9, 127.9, 127.5, 126.9, 125.3, 121.5, 120.6, 117.1, 77.1, 70.3, 50.5.
4.2.14. 4-((1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethoxy)methyl)-N-(1H-indol-5-yl)benzamide (A15). White solid (83.3% yield); m.p. 128.7–130.8 °C. HRMS (ESI): calcd for C27H22N4O2Cl2 [M + Na]+: 527.1012, found 527.1033 [M + Na]+. 1H NMR (400 MHz, DMSO-d6) 11.04 (s, 1H), 10.06 (s, 1H), 7.99 (d, J = 1.9 Hz, 1H), 7.93 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.54 (s, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.45–7.39 (m, 2H), 7.37–7.32 (m, 2H), 7.31 (d, J = 8.2 Hz, 2H), 7.09 (s, 1H), 6.90 (s, 1H), 6.44–6.39 (m, 1H), 5.05 (dd, J = 7.0, 3.9 Hz, 1H), 4.54–4.38 (m, 2H), 4.36–4.26 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.2, 141.2, 138.4, 135.3, 135.1, 134.1, 133.7, 133.5, 131.4, 129.8, 129.5, 128.7, 128.4, 128.1, 127.9, 127.4, 126.4, 120.6, 116.6, 112.7, 111.5, 101.6, 77.1, 70.4, 50.5.
4.3. In vitro antifungal assay
For antifungal assessment, the CLSI M27-A3 micro broth dilution assay was utilized. Compounds and controls were dissolved in DMSO, with RPM1640 medium added to 96-well plates. Tween-20 was used for stabilization. Each well received specific volumes of stock solutions, with fungal suspensions (1 × 103–5 × 103 CFU mL−1) added before incubation at 35 °C to determine MIC.
4.4. GC-MS analysis of sterol composition
GC-MS analysis of sterol composition utilized Candida albicans (SC5314) in YEPD medium. After incubation and activation, fungal cells were saponified using a 15% sodium hydroxide solution in ethanol/water, followed by extraction with petroleum ether. Samples were then dissolved in cyclohexane and analyzed by GC-MS.
4.5. Experiment of transmission electron microscopy (TEM)
For transmission electron microscopy (TEM), Candida albicans SC5314 was treated with compounds and controls, incubated, fixed, and observed under TEM as per established methods.
4.6. RT-PCR assay
The RT-PCR analyses were performed following the methods outlined in earlier studies.18
4.7. Metabolic stabilities assay
The metabolic stability of the target compounds, encompassing liver microsomal and plasma stability, was evaluated using established experimental protocols from previous reports.18 The liver microsomes were extracted from mouse. The plasma was collected from adult male SD rats.
4.8. In vivo antifungal potency
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Zhengzhou University and approved by the Animal Ethics Committee of Zhengzhou University. To establish the Candida albicans infection model, mice were intravenously injected with a suspension of C. alb 904 (1 × 105 CFU mL−1, 100 μL) into the tail vein. Intraperitoneal administration of the compounds followed 2 hours post-infection. After 5 days, the left kidneys of the mice were aseptically excised, pulverized, and spread on PDA plates to quantify colony-forming units (CFUs), assessing the fungal burden.
4.9. Cytotoxicity assay
The MRC-5 cell line used in this study was obtained from the American Type Culture Collection (ATCC). The cytotoxicity assay was performed following the methods outlined in earlier studies.18
4.10. Docking study
The crystal structure of CYP51 (PDB: 5TZ1) was obtained from the Protein Data Bank and optimized. Molecular docking was performed to simulate the interaction of the target molecule with the protein by using Discovery Studio 3.0 software. Docking results were analyzed graphically using PyMOL software.
Data availability
All relevant data are within the manuscript and its additional files.
Conflicts of interest
The authors have stated that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
Acknowledgements
This work was financially supported by the Science and Technology Department of Henan Province (No. 232102310370).
References
- C. F. Neoh, W. Jeong, D. C. Kong and M. A. Slavin, Expert Rev. Anti-Infect. Ther., 2023, 21, 577–594 Search PubMed.
- D. A. Enoch, H. Yang, S. H Aliyu and C. Micallef, Methods Mol. Biol., 2017, 1508, 17–65 CrossRef CAS PubMed.
- K. C. Howard, E. K. Dennis, D. S. Watt and S. Garneau-Tsodikova, Chem. Soc. Rev., 2020, 49, 2426–2480 RSC.
- D. Allen, D. Wilson, R. Drew and J. Perfect, Expert Rev. Anti-Infect. Ther., 2015, 13, 787–798 Search PubMed.
- S. Seyedmousavi, H. Rafati, M. Ilkit, A. Tolooe, M. T. Hedayati and P. Verweij, Methods Mol. Biol., 2017, 1508, 107–139 Search PubMed.
- X. Cui, L. Wang, Y. Lü and C. Yue, J Infect Public Health, 2022, 15, 986–1000 Search PubMed.
- T. Y. Hargrove, L. Friggeri, Z. Wawrzak, A. Qi, W. J. Hoekstra, R. J. Schotzinger, J. D. York, F. P Guengerich and G. I. Lepesheva, J. Biol. Chem., 2017, 292, 6728–6743 CAS.
- N. Liu, J. Tu, G. Dong, Y. Wang and C. Sheng, J. Med. Chem., 2018, 61, 5484–5511 CAS.
- S. Emami, E. Ghobadi, S. Saednia and S. M. Hashemi, Eur. J. Med. Chem., 2019, 170, 173–194 CAS.
- Z. Z. Yan, Z. B. Lv, D. Z. Zhao, M. B. Guo, Y. T. Wang, Z. Hou and C. Guo, J. Mol. Struct., 2023, 1291, 135997 CAS.
- Z. Z. Yan, Q. Li, X. Y. Li, H. L. Wang, D. Z. Zhao, H. Yu, M. B. Guo, Y. T. Wang, X. Wang, H. Xu, Y. H. Mou, Z. Hou and C. Guo, J. Med. Chem., 2024, 67, 12601–12617 CAS.
- H. Xu, C. Cao, X. Wang, M. B. Guo, Z. Z. Yan, R. An, R. Zhang, E. H. Dong, Y. H. Mou, Z. Hou and C. Guo, Bioorg. Chem., 2021, 115, 105182 CAS.
- G. Han, N. Liu, C. Li, J. Tu, Z. Li and C. Q. Sheng, J. Med. Chem., 2020, 63, 5341–5359 CAS.
- R. Pereira, R. O. Dos Santos Fontenelle, E. H. S. de Brito and S. M. de Morais, J. Appl. Microbiol., 2021, 131, 11–22 CAS.
- X. Cui, L. Wang, Y. Lü and C. Yue, J Infect Public Health, 2022, 15, 986–1000 CrossRef.
- P. Marichal, L. Koymans, S. Willemsens, D. Bellens, P. Verhasselt, W. Luyten, M. Borgers, F. C. S. F. C. Odds and H. Vanden Bossche, Microbiology, 1999, 145, 2701–2713 CrossRef CAS.
- Z. Li, J. Tu, G. Han, C. Sheng and N. Liu, J. Med. Chem., 2021, 64, 1116–1126 CrossRef CAS.
- Z. Z. Yan, Y. X. Huang, D. Z. Zhao, Z. Y. Li, X. Wang, M. B. Guo, Y. Wei, Y. T. Wang, Y. H. Mou, Z. Hou and C. Guo, J. Med. Chem., 2023, 66, 13247–13265 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sha-sha Gonga,
Yong-xin Zhanga,
Ying-xia Xie*ab and
Naying Chu*a
a,
Sha-sha Gonga,
Yong-xin Zhanga,
Ying-xia Xie*ab and
Naying Chu*a