
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA 2,6-diamidopyridine-based macrocyclic aromatic amide receptor with cascade ion pair recognition†
Xinguo Maoa,
Rui Zhang a,
Yulong Sund,
Xuping Wang*a,
Qinggang Lia,
Han Zuilhof*b,
Leyong Wangc and
Qiang Shi*a
a,
Yulong Sund,
Xuping Wang*a,
Qinggang Lia,
Han Zuilhof*b,
Leyong Wangc and
Qiang Shi*a
aKey Laboratory of Light Conversion Materials and Technology, Shandong Provincial Key Laboratory of High Strength Light Weight Metallic Materials, Advanced Materials Institute, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250014, China. E-mail: shiqiang@sdas.org; wangxp@sdas.org
bLaboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: Han.Zuilhof@wur.nl
cKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
dShigatse Science and Technology Bureau, Shigatse, Tibet 857000, China
First published on 20th February 2025
Abstract
Ion-pair receptors constitute an important class of synthetic receptors within the realm of host–guest and supramolecular chemistry. Their unique ability to simultaneously recognize and accommodate both cations and anions has rendered them invaluable across various applications. In this study, we have synthesized a cascade macrocyclic ion-pair receptor, composed of three 2,6-amidopyridine building blocks bridged by aromatic spacers. Notably, the diamide binding sites of this receptor exhibit a high degree of selectivity for fluoride ions. Furthermore, despite lacking any dedicated cation-binding sites within its macrocyclic structure, this receptor is capable of selectively binding tetraethylammonium cations through a series of cascade electrostatic interactions facilitated by the bound flouride ions.
Introduction
Artificial macrocyclic receptors stand as the cornerstone of supramolecular chemistry,1 featuring pre-organized cyclic skeletons and central cavities. Typically, these receptors are constructed from fundamental building blocks, which serve as the basic structural units within a symmetric molecular host.2 The intrinsic properties, recognition capabilities, and diverse applications3,4 of these macrocycles are largely dictated by the composition of these building blocks. For instance, the electron-dense p-dimethoxybenzene constitutes the backbone of pillar[n]arenes (n = 5, 6), enabling their cavities to host electron-deficient guests like ammonium or pyridinium salts.5–7 Conversely, the blue box, employing paraquat as its foundational unit, possesses an electron-deficient cavity tailored for recognizing electron-rich guests.8 Thanks to the variety of building blocks, a plenty of artificial macrocyclic receptors have been synthesized and investigated.9–12Traditionally, macrocycles have predominantly been designed as monotopic receptors, tailored to bind specific guest species. In contrast, ion-pair receptors13–17 exhibit a unique ability to simultaneously capture both cations and anions. To date, two primary types of ion-pair receptors have been devised: heterotopic18,19 and cascade13 receptors. Heterotopic receptors integrate anion and cationic recognition sites within a single macrocyclic skeleton, whereas cascade receptors could host ion pairs through relay interactions like the form of [H⋯A−(+)⋯B+(–)], whereas H is the cascade macrocyclic receptor with only binding sites for A−(+) guest, B+(−) guest is attracted by encapsulated A−(+) counterion through noncovalent interactions of them such as electrostatic interaction, rather than weak interactions with the macrocycle. The recognition mechanism endows ion-pair receptors with enhanced selectivity for separating specific cations and anions.20,21 Consequently, ion-pair receptors, particularly heterotopic ones, are garnering significant attention due to their promising applications in salt extraction,22 membrane transport,23,24 sensing,25 and beyond.26 Currently, there is a surge of research focusing on the design, synthesis, and functionality of these receptors. However, cascade ion-pair receptors, despite their advantages of straightforward design, easy synthesis and broader applicability, have received comparatively less attention.27
Amide, a ubiquitous and versatile building block, finds extensive use not only in polypeptides and proteins but also in artificial macrocycles,28 owing to its exceptional properties as a hydrogen bonding donor or acceptor. 2,6-Diamidopyridine, a distinctive diamide fragment, adopts a predominantly fixed V-shape conformation due to intramolecular hydrogen bonding between the pyridine ring's nitrogen atom and the amide group's hydrogen atoms.29 This conformation enables it to function as a chelating hydrogen bond donor, capturing hydrogen bond acceptor molecules such as anions.30 Furthermore, 2,6-diamidopyridine has been established as a ligand for certain transition metal complexes.31,32 Leveraging its directional hydrogen bonding sites, derivatives of 2,6-diamidopyridine emerge as exceptional building blocks for constructing macrocyclic receptors capable of capturing anionic guests.33,34
In this study, capitalizing on the 120° angle formed by the two amide groups in 2,6-diamidopyridine derivatives, we report herein the synthesis of a novel class of hexagonal macrocyclic aromatic amide (MAA, Fig. 1), interlinked by aromatic spacers. This macrocycle, equipped with three diamide binding sites, demonstrated remarkable selectivity for fluoride ions35,36 and formed a cascade complex with tetraethylammonium cations upon fluoride binding, without requiring any cationic binding sites within the macrocyclic structure.
 | ||
| Fig. 1 Structure of the macrocyclic aromatic amide (MAA) receptor. | ||
Results and discussion
Macrocycle synthesis
The synthetic route of macrocyclic aromatic amide receptor MAA was shown in Scheme 1. The intermediate meso-triphenylenediamine derivative 1 was first efficiently synthesized from 1,3-dibromo-5-methoxybenzene and 2 equivalents of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline by a Suzuki–Miyaura coupling reaction (86% yield, typically 4.86 mmol scale). In this experiment, a mixed solvent system of 1,4-dioxane and H2O was used to facilitate the complete dissolution of the reactants and the separation of the product. Next, MAA was prepared by the condensation reaction of intermediate 1 with equal equivalents of 2,6-pyridinedicarboxylic acid in THF using EDC and HOBT as condensation reagents (typical yield ca. 5% on 8 × 10−2 mmol scale). In this study, the two molecules, 1 and 2,6-pyridinedicarboxylic acid, were chosen as the fragments for the synthesis of MAA because they had the precisely right bond angles to allow the 3 + 3 amide condensation to form an amide macrocycle with three pyridines and three diamides. Compared with some low polar solvents such as dichloromethane (DCM), the use of THF as the reaction solvent improved both the solubility of the reactants and the yield of MAA. Meanwhile, it should be noted that some precipitate was produced during the reaction, which likely was due to the formation of non-cyclic oligomers and polymers. After MAA was isolated using column chromatography, recrystallizations were performed to further improve the purity of the product. All products were characterized by 1H NMR, 13C NMR and HRMS (Fig. S1 to S4†). | ||
| Scheme 1 Synthetic route towards MAA. | ||
Recognition ability and selectivity to anions
In order to investigate the recognition properties of MAA, equal amounts (2.97 × 10−2 M) of different species of Et4N+X− (X− = F−, Cl−, Br−, I−, HSO4−, CF3SO3−) were, in separate experiments, added to a DMSO-d6 solution of acceptor MAA at 298 K, and the changes in the 1H NMR spectrum observed (Fig. 2). Upon the addition of the F− salt, the 1H NMR spectra showed obvious chemical shifts. Most strongly, the amide –(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)N–H proton peak showed a large downfield shift from 11.2 to 14.8 ppm (more precisely: Δδ = 3.68 ppm). In addition, the 1H peaks in the aromatic region also changed significantly, with partial overlapping of the A, B peaks shifted upfield (A: Δδ = −0.13 ppm; B: Δδ = −0.18 ppm and the D peak shifted to downfield (Δδ = 0.29 ppm). Smaller, but still significant changes were observed for the C, E, and F peaks, which also showed upfield shifts (Δδ = −0.15 ppm, −0.08 ppm, and −0.07 ppm, respectively). The results clearly demonstrated the formation of stable complexes of MAA with F− in DMSO-d6. In sharp contrast, the addition of anions such as Cl−, Br−, I−, HSO4−, and CF3SO3− hardly induced any significant chemical shifts in the 1H NMR spectrum of MAA. This indicates the absence of any significant complex formation in DMSO-d6 of MAA with these anions, and thus a remarkably strong and selective binding of fluoride to MAA. In addition, the binding ratio of MAA with F− was detected to 1
O)N–H proton peak showed a large downfield shift from 11.2 to 14.8 ppm (more precisely: Δδ = 3.68 ppm). In addition, the 1H peaks in the aromatic region also changed significantly, with partial overlapping of the A, B peaks shifted upfield (A: Δδ = −0.13 ppm; B: Δδ = −0.18 ppm and the D peak shifted to downfield (Δδ = 0.29 ppm). Smaller, but still significant changes were observed for the C, E, and F peaks, which also showed upfield shifts (Δδ = −0.15 ppm, −0.08 ppm, and −0.07 ppm, respectively). The results clearly demonstrated the formation of stable complexes of MAA with F− in DMSO-d6. In sharp contrast, the addition of anions such as Cl−, Br−, I−, HSO4−, and CF3SO3− hardly induced any significant chemical shifts in the 1H NMR spectrum of MAA. This indicates the absence of any significant complex formation in DMSO-d6 of MAA with these anions, and thus a remarkably strong and selective binding of fluoride to MAA. In addition, the binding ratio of MAA with F− was detected to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. S5 and S6†).
:2 (Fig. S5 and S6†).
 | ||
| Fig. 2 1H NMR (400 MHz, DMSO-d6, 298 K) of (a) receptor MAA; (b) MAA + 5.0 equiv. of tetraethylammonium fluoride; (c) MAA + 5.0 equiv. of tetraethylammonium chloride; (d) MAA + 5.0 equiv. of tetraethylammonium bromide; (e) MAA + 5.0 equiv. of tetraethylammonium iodide; (f) MAA + 5.0 equiv. of tetraethylammonium trifluoromethanesulfonate; (g) MAA + 5.0 equiv. of tetraethylammonium hydrogensulfate. | ||
In order to quantify the complexation ability of the receptor MAA to F−, 1H NMR titration experiments were carried out (DMSO-d6, 298 K; Fig. S7†), in which F− was added to MAA and the step sized changes in the corresponding 1H peaks observed. Based on these NMR titration data, the MAA and F− binding constants could be calculated by the nonlinear curve fitting method (Fig. S8†), which were Ka1 = (1.47 ± 0.40) × 104 M−1 and Ka2 = (7.15 ± 0.51) × 103 M−1, respectively.
UV-vis spectrophotometric studies
During the study of recognition studies of receptor MAA with the tetraethylammonium salts of F−, Cl−, Br−, I−, HSO4− and CF3SO3−, it was found that after the addition of tetraethylammonium fluoride (Et4NF) to the DMSO solution of MAA, a drastic color change from colorless to yellow could be observed by the naked eye. In contrast, the addition of several other anions did not result in any noticeable color change, as shown in Fig. 3. This distinct color change also reflected the highly selective recognition of F− by receptor MAA, making it useful for rapid detection and recognition of F−. | ||
| Fig. 3 Visible color changes of MAA observed before and after addition of equal amounts of various tetraethylammonium salts to DMSO solution (1.0 × 10−4 M): from left to right, DMSO, free MAA, MAA + F−, MAA + Cl−, MAA + Br−, MAA + I−, MAA + HSO4−, and MAA + CF3SO3−. | ||
In order to further investigate the phenomenon of color change after the addition of F−, UV-vis spectrophotometric experiments were carried out in a DMSO solution of MAA (1.98 × 10−4 M). Fig. 4 shows the UV-vis absorption spectra after the addition of 5 equivalents of the selected anions, all of which resulted in an increase in the absorption intensity in the visible band due to the fact that the selected anions all carry lone pairs of electrons, which acted as a color-booster in the UV-vis spectrophotometric experiments. However, after the addition of F−, the UV-vis absorption spectrum of MAA not only showed an increase in absorbance, but also an obviously redshift maximum (Δλ = 8 nm) and a new shoulder peak formed at 367 nm which correspond to the deprotonation37 of –NH– groups by F− ions (Fig. S10†). Combined with the chemical shift changes of the –CONH– group and the peaks of the aromatic region upon the addition of F− (Fig. 2), this further indicates the formation of a stable complex between MAA and F− in DMSO solution.
 | ||
| Fig. 4 UV-vis absorption spectra of MAA (in DMSO) to which various salts was added. | ||
Selective ion-pair recognition
As shown in Fig. 5, it was found from 1H NMR that there were different binding patterns of MAA with different cations including smaller tetraethylammonium cation (Et4N+) and larger tetrabutylammonium ion (n-Bu4N+). The addition of the receptor MAA to the solution of Et4NF resulted in a pronounced upfield shift (CH2 peak: Δδ = 0.04 ppm; CH3 peak: Δδ = 0.02 ppm) of Et4N+ as shown in (b) and (c) in Fig. 5, implying that the small Et4N+ has “fallen into” the cavity of the complex of MAA with F− ions, and therefore the formation of ternary complex between MAA with two F− ions and Et4N+ counter cation. The cross peaks (Fig. S11†) between H atoms on the alkyl chains of Et4N+ cations with aromatic rings of MAA in the ROESY spectrum of the host-guest mixture in DMSO-d6 solution further confirmed the formation of the stable complex between MAA with F− and Et4N+ ion-pairs. In fact, the electrostatic interaction between tetraethylammonium cation with F− ions is the main driving force to bind Et4N+ ions in the cavity of the MAA receptor. In addition, the size matching effect of Et4N+ with the cavity of MAA is another factor to allow the binding of the guest ions.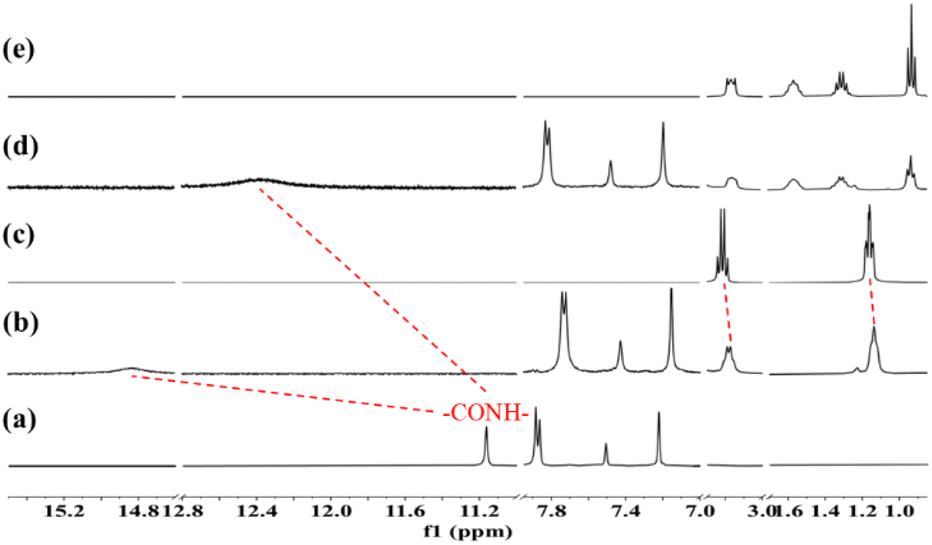 | ||
| Fig. 5 1H NMR (400 MHz, DMSO-d6, 298 K) of (a) receptor MAA; (b) MAA + 5.0 equiv. of tetraethylammonium fluoride; (c) tetraethylammonium fluoride; (d) MAA + 5.0 equiv. of tetrabutylammonium fluoride; and (e) tetrabutylammonium fluoride. | ||
Compared with Et4N+, the peaks of n-Bu4N+ showed almost no chemical shifts after mixing with MAA as shown in Fig. 5(d) and (e). This indicates that the oversized n-Bu4N+ is hard to bind into the macrocyclic cavity of MAA: it is much harder to fit into the cavity (and then only one can fit, with significant loss of entropy), and the C4 alkyl chains does display their larger shielding of the positive charge on the central N atom, yields weaker interactions with the F− anions (and thus with all proton's 1H NMR shift is influenced by changes in the F− binding). In addition, the smaller binding constants (Ka1 = 1.18 × 103 M and Ka2 = 2.32 × 102 M) between F− and MAA in the presence of larger cation indicated that the association of cation in the cavity could facilitate the binding of F− with the macrocycle.
DFT calculations
To further understand the spatial configuration of MAA, the binding mode of MAA with F− ions and Et4N+ counter cation, and the source of its binding selectivity, we optimized the structures of MAA and its complexes with related ion-pairs by density functional theory (DFT) calculations at the B3LPY/6-31G level of theory using a PCM solvent model for DMSO. This yields the structures as shown in Fig. 6. From Fig. 6(a), it can be seen that macrocycle MAA presents a saddle-like planar structure in DMSO, which not only has a huge internal cavity (15.15 Å × 13.34 Å), but also displays three effective anion-binding sites due to the presence of the pyridine N atoms that made the hydrogen atoms of the two –(CO)NH− groups attached to it locked tightly inside the molecule, and thus tune these H atoms towards divalent binding of anions. | ||
| Fig. 6 The optimized structure of (a) MAA and (b) MAA + 2F− + Et4N+ in DMSO. | ||
For the 2F−@MAA complex, the computationally optimized model was shown in Fig. S13,† the receptor MAA maintained a saddle-shaped steric structure upon binding to F−. Within the molecule of the receptor MAA, there were not only strong N–H⋯F hydrogen bonds of lengths from 1.65 Å to 1.69 Å formed by the two –(CO)NH− groups and F−, but also C–H⋯F weak hydrogen bonds of lengths from 2.14 Å to 2.36 Å formed by the benzene ring directly connected to the –(CO)NH− group and F−. As can be seen in the optimized structures, F− really fits in the plane of the macrocyclic moiety that binds it, yielding strong interactions. In comparison, the complex of Cl− with MAA was similarly yielding the structure shown in Fig. S14.† In this complex, Cl− can only be bound above (or below) the plane of that moiety, and the H atoms of the –(CO)NH− groups have to bend outwards to interact optimally with the Cl−. This size effect (Cl− too big to fit, all other anions under study even bigger) clarifies why there is such selectivity towards binding fluoride anions.
The calculation also revealed further binding behavior of 2F−@MAA with counter cation. As shown in Fig. 6(b), the complex 2F−@MAA still possess a huge cavity which could easily fit an Et4N+. As expected, the cation bond into the cavity in a MAA–F−–Et4N+ cascade form. In the optimized model, it is clearly that two or more Et4N+ cations could not fit in the rest cavity of MAA after binding F− ions not only due to the size effect but also the strong electrostatic repulsion. In fact, the saddle-shape allows all two of the F− to simultaneously interact with an Et4N+ cation, and thus form a relatively stable [2F−@MAA][Et4N+] complex. In this complex, the main driven force is the electrostatic attraction between the positively charged N atom and F−. The model also showed that the multiple C–H⋯F hydrogen bonds (range from 2.14 to 2.19 Å) between the H atoms of alkyl chains in Et4N+ with F−, and the C–H⋯π interactions between the alkyl chain H atoms with the aromatic rings (2.79 Å) of MAA play crucial roles to the formation of the quaternary complex. In this aspect, the formation of C–H⋯π interactions in the optimized model is in good agreement with the results of ROESY spectrum (Fig. S11†). According to these results, the binding of the counter cation Et4N+ partially counteracts the electrostatic repulsion between the two bonded fluoride ions, which further increased the binding strength and recognition selectivity between MAA and fluoride ions.
This changes for the much bigger and much more shielded n-Bu4N+ cation (Fig. S15†): to fit one n-Bu4N+ cation, the macrocycle the macrocyclic skeleton needs to undergo a very large distortion, noting that this would require a significant entropic penalty, but the overall electrostatic interactions are, of course, much weaker. Positioning two of such bulky Bu4N+ cations interacting with the 2F−@MAA structure requires them to move out considerably. As a result, the average electronic influence of n-Bu4N+ is much smaller than that of Et4N+, in line with experiment.
Conclusions
We designed and synthesized a 2,6-diamidopyridine-based macrocyclic aromatic amide receptor –– MAA, which combines a strong binding to fluoride anions with a high selectivity (very less binding to Cl−, no binding to Br−, I−, HSO4−, and CF3SO3−), which was confirmed by the 1H NMR and UV-vis spectra. NMR titration experiments showed that MAA acts as a receptor to form a 1:2 complex with F− in a large polar solvent, with binding constants of Ka1 = (1.47 ± 0.4) × 104 M−1 and Ka2 = (7.15 ± 0.51) × 103 M−1, respectively. This complexation with F− could be detected by the naked eye (color changes of DMSO solution from colorless to yellow). Importantly, MAA is also a cascade ion-pair recognition receptor without any cations binding sites in its macrocyclic structure. MAA could selectively host a Et4N+ rather than a larger n-Bu4N+ cation in the cavity after binding F− ions in the amide sites by the cascade MAA–F−–Et4N+ pattern. The strong binding selectivity of MAA to F− and Et4N+ rather than other studied ions was also explained through using DFT calculations upon B3LYP/6-31G level.
Experimental section
General information
Unless otherwise stated, all reagents were obtained from commercial sources and were used without further purification. The solvents used were dried over 4A molecular sieves except for H2O. 1H NMR and 13C NMR spectra were recorded on a Bruker DMX300 NMR and a Bruker® Avance III 400 MHz NMR spectrometer. HRMS was recorded on a Thermo Fisher. UV-vis absorption spectra were recorded on an Agilent Cary 7000 UV-vis-NIR spectrometer.Synthesis
:1, v/v) solution system, then K2CO3 (4.67 g, 33.84 mmol) was added and heated to 100 °C in an argon atmosphere and the catalyst Pd(PPh3)4 (0.10 g, 0.09 mmol) was added and reacted for 12 h. At the end of the reaction the pH of the mixture was adjusted to weak acidity, water was added, and the organic solvent was removed under vacuum, then the organic layer was extracted with water and DCM and collected and dried with anhydrous MgSO4. Purification by silica gel column chromatography (eluent: DCM/MeOH = 100/1 to DCM/MeOH = 50/1) afforded a yellow oily product 1 (1.41 g, yield 86.11%). 1H NMR (400 MHz, DMSO-d6) δ 7.44–7.38 (m, 4H), 7.23 (t, J = 1.6 Hz, 1H), 6.90 (d, J = 1.5 Hz, 2H), 6.66–6.60 (m, 4H), 5.23 (s, 4H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.05, 148.37, 142.37, 127.62, 127.27, 115.27, 114.03, 108.44, 54.98. HRMS (ESI) for C19H18N2O, m/z, [M + H]+, calculated: 290.3660, found: 291.1491.Data availability
The data that support the findings of this study are available from the corresponding author, [Q. Shi], upon reasonable request.Author contributions
X. Mao and Q. Shi did the synthesis work and the experiments of 1H NMR, 13C NMR, UV-vis spectra. R. Zhang did the HRMS experiments. X. Mao, Q. Shi, Y. Sun, and Q. Li written the manuscript. H. Zuilhof calculated the structures of receptor and its complexes. H. Zuilhof, L. Wang and X. Wang checked and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Project for Shandong Provincial Natural Science Foundation (ZR2021QB196, ZR2021QE199), the Talent research project of Qilu University of Technology (Shandong Academy of Sciences) (2023RCKY022, 2023RCKY015) and the basic research projects in science, education and production of Qilu University of Technology (Shandong Academy of Sciences) (2023PX065, 2023PX040).References
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459 RSC
.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed
- L.-M. Bai, H. Yao, L.-P. Yang, W. Zhang and W. Jiang, Chin. Chem. Lett., 2019, 30, 881–884 CrossRef CAS
- K. Jie, Y. Zhou, E. Li, R. Zhao and F. Huang, Angew. Chem., Int. Ed., 2018, 57, 12845 CrossRef CAS
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed
- T. Ogoshi, T. Kakuta and T. Yamagishi, Angew. Chem., Int. Ed., 2019, 58, 2197–2206 CrossRef CAS PubMed
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721 CrossRef CAS PubMed
- X.-Y. Chen, H. Chen and J. F. Stoddart, Angew. Chem., Int. Ed., 2023, 62, e202211387 CrossRef CAS
- S. Toyota and E. Tsurumaki, Chem.–Eur. J., 2019, 25, 6878–6890 CrossRef CAS PubMed
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC
- Y.-D. Yang, X.-L. Chen, J.-L. Sessler and H.-Y. Gong, J. Am. Chem. Soc., 2021, 143, 2315 CrossRef CAS
- M.-L. Tan, Q.-H. Guo, X.-Y. Wang, T.-H. Shi, Q. Zhang, S. K. Hou, S. Tong, J. S. You and M.-X. Wang, Angew. Chem., Int. Ed., 2020, 59, 23649 CrossRef CAS PubMed
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC
- S. J. Nicholson, S. R. Barlow and N. H. Evans, Chemistry, 2023, 5, 106–118 CrossRef CAS
- A. J. McConnell, A. Docker and P. D. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS PubMed
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed
- S. K. Kim, J. L. Sessler, D. E. Gross, C.-H. Lee, J. S. Kim, V. M. Lynch, L. H. Delmau and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 5827–5836 CrossRef CAS PubMed
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed
- D. Jagleniec, S. Siennicka, L. Dobrzycki, M. Karbarz and J. Romański, Inorg. Chem., 2018, 57, 12941–12952 CrossRef CAS PubMed
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed
- X. D. Chi, G. M. Peters, F. Hammel, C. Brockman and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 9124–9127 CrossRef CAS PubMed
- P. Breccia, M. Van Gool, R. Pérez-Fernández, S. Martín-Santamaria, F. Gago, P. Prados and J. C. de Mendoza, J. Am. Chem. Soc., 2003, 125, 8270–8284 CrossRef CAS PubMed
- X. L. Ni, X. Zeng, C. Redshaw and T. Yamato, J. Org. Chem., 2011, 76, 5696–5702 CrossRef CAS PubMed
- D. S. Kim, V. M. Lynch, J. S. Park and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 14889–14894 CrossRef CAS PubMed
- V. Valderrey, E. C. Escudero-Adan and P. Ballester, Angew. Chem., Int. Ed., 2013, 52, 6898–6902 CrossRef CAS PubMed
- Z. Liu, Y. Zhou and L. Yuan, Org. Biomol. Chem., 2022, 20, 9023 RSC
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894 CrossRef CAS
- M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936–2937 CrossRef CAS PubMed
- Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Inorg. Chem., 2012, 51, 760–762 CrossRef CAS PubMed
- J. Lohrman, H. Telikepalli, T. S. Johnson, T. A. Jackson, V. W. Day and K. Bowman-James, Inorg. Chem., 2016, 55, 5098–5100 CrossRef CAS
- Q.-Q. Wang, V. W. Day and K. Bowman-James, Org. Lett., 2014, 16, 3982–3985 CrossRef CAS PubMed
- S. O. Kang, D. Powell and K. Bowman-James, J. Am. Chem. Soc., 2005, 127, 13478–13479 CrossRef CAS
- S. Xiong, M. V. N. Kishore, W. Zhou and Q. He, Coord. Chem. Rev., 2022, 461, 214480 CrossRef CAS
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and L. Mosca, J. Am. Chem. Soc., 2013, 135, 6345–6355 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR of compounds, as well as the details of the experiments. See DOI: https://doi.org/10.1039/d5ra00434a |
| This journal is © The Royal Society of Chemistry 2025 |