
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCryo-EM reveals conformational variability in the SARS-CoV-2 spike protein RBD induced by two broadly neutralizing monoclonal antibodies†
Clayton Fernando Rencilin‡
 a,
Arnab Chatterjee‡a,
Mohammad Yousuf Ansarib,
Suprit Deshpandebc,
Sohini Mukherjeebef,
Randhir Singhd,
Sowrabha B. Jayatheerthad,
Poorvi M. Reddyd,
Nitin Hingankarb,
Raghavan Varadarajanad,
Jayanta Bhattacharyabc and
Somnath Dutta*a
a,
Arnab Chatterjee‡a,
Mohammad Yousuf Ansarib,
Suprit Deshpandebc,
Sohini Mukherjeebef,
Randhir Singhd,
Sowrabha B. Jayatheerthad,
Poorvi M. Reddyd,
Nitin Hingankarb,
Raghavan Varadarajanad,
Jayanta Bhattacharyabc and
Somnath Dutta*a
aMolecular Biophysics Unit, Indian Institute of Science, Bengaluru 560012, India. E-mail: somnath@iisc.ac.in
bAntibody Translational Research Program, Translational Health Science & Technology Institute, Faridabad, Haryana 121001, India
cBRIC-Translational Health Science & Technology Institute, Faridabad, Haryana 121001, India
dMynvax Private Limited, Vani Vilas Road, Basavanagudi, Bengaluru 560004, India
eIAVI, Gurugram, Haryana 122022, India
fIAVI, New York, NY 10004, USA
First published on 6th May 2025
Abstract
SARS-CoV-2 spike proteins play a critical role in infection by interacting with the ACE2 receptors. Their receptor-binding domains and N-terminal domains exhibit remarkable flexibility and can adopt various conformations that facilitate receptor engagement. Previous structural studies have reported the RBD of the spike protein in “up”, “down”, and various intermediate states, as well as its different conformational changes during ACE2 binding. This flexibility also influences its interactions with the neutralizing antibodies, yet its role in the antibody complexes remains understudied. In this study, we used cryo-electron microscopy to investigate the structural properties of two broadly neutralizing monoclonal antibodies, THSC20.HVTR04 and THSC20.HVTR26. These antibodies were isolated from an unvaccinated individual and demonstrated potent neutralization of multiple SARS-CoV-2 variants. Our analysis revealed distinct binding characteristics and conformational changes in the spike RBD upon binding with the monoclonal antibodies. The structural characterization of the spike protein-monoclonal antibody complexes provided valuable insights into the structural variability of the spike protein and the possible mechanisms for antibody-mediated neutralization.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected over 700 million people globally and caused more than 7.7 million deaths (https://covid19.who.int/). The trimeric spike (S) protein of SARS-CoV-2 is a homotrimeric type I transmembrane fusion glycoprotein that facilitates the initiation of the virus infection by binding to the human angiotensin-converting enzyme 2 (ACE2) receptor found on the epithelial cells.1 The S protein consists of two subunits: S1 and S2. S1 contains the N-terminal domain (NTD) and the receptor binding domain (RBD). The receptor binding domain (RBD) is responsible for interacting with the host ACE2 receptor and initiates the infection. Thus, the RBD is the principal target for neutralizing antibodies to inhibit the interactions between RBD and the ACE2 receptors, as well as the involvement of the S2 subunit in membrane fusion. The receptor-binding motif (RBM) within the S1/RBD region is responsible for extensive binding with the N-terminal alpha-helix of the ACE2 receptor.2,3 The S1/RBD-ACE2 binding allows S2 to undergo conformational changes that further promote the fusion of the viral and host membranes. This enables the viral RNA to enter the host cell and establish infection.4 The RBD region of the S protein can switch between closed and open forms based on its engagement with the receptor, and both forms can co-exist at equilibrium.3 The receptor initially binds to the “one RBD-up” conformation, which facilitates a sequential movement of the other RBDs to the “up” conformation. Cryo-EM-based structural studies have reported different conformational states of ACE2 bound to the RBD.5,6Additionally, so far, several neutralizing antibodies have been reported to broadly target three specific sites in the S1 subunit: (a) the NTD region,7,8 (b) the ACE2 binding surface of the RBD (blocking the S-ACE2 interaction)9,10 and (c) rarely the N-linked glycans in RBD (without any influence on S-ACE2 interaction).11 However, the last two processes have demonstrated destabilization of RBD binding with the ACE2 receptor.12–14 Due to the conserved nature of the S2 stem-helix region of the S protein, several broadly neutralizing antibodies (bnAbs) have been isolated against this region to block the viral fusion machinery. Along with bnAbs, several potent fusion inhibitors against heptad repeat 1 (HR1) and heptad repeat 2 (HR2) have also been designed.14–16 While the binding of ACE2 to the RBD induces various conformational states, antibodies bound to the RBD may also stabilize or induce several intermediate states. However, most studies have not focused on the conformational flexibility of the RBD during monoclonal antibody (mAb)-mediated neutralization despite its potential importance in determining the neutralization efficacy.
Recently, we reported several SARS-CoV-2 RBD-reactive mAbs isolated from an unvaccinated convalescent donor who was infected with ancestral SARS-CoV-2 in early 2020.17,18 Of these antibodies, two (THSC20.HVTR04 & THSC20.HVTR26) demonstrated potent neutralization capability towards several SARS-CoV-2 variants of concern (VOCs), such as Alpha, Beta, Gamma, and Delta, and variants of interest (VOIs), such as Kappa and Delta Plus. However, only THSC20.HVTR26 neutralized Omicron B.1.1.529 (BA.1) variant.17 Conversely, we found that THSC20.HVTR04 could potently neutralize the Omicron BA.2 and BA.4/BA.5 variants but not the BA.1 variant.18 Previous studies have suggested that the antibody-bound conformational states of the RBD region are crucial for neutralization and inhibiting ACE2 receptor binding.19,20 However, antigen–antibody binding can induce allosteric conformational changes in the antigen.21 Additionally, previous studies have characterized the conformational changes of IgG when bound to small antigens22 and its conformational variabilities both in the presence and absence of the interacting partners.23 Therefore, understanding the conformational selectivity associated with antigen–antibody complexes is crucial. Only a few studies have described the structural characteristics of antibody-mediated dynamics.24 Here, we have characterized the high-resolution structure of the S-protein bound to the two broadest mAbs at a resolution of 4.5 Å using cryo-EM. The structural analysis showed the conformational flexibility of the two Fabs bound to the S protein, which adopts several conformations. Furthermore, we demonstrate that mAb/Fab can modulate the position of the RBD domain, resulting in a more open conformation of the S protein RBD. More interestingly, our study shows that the RBD-Fab complex may adopt new conformations that have not been demonstrated previously, but the interaction of the epitope residues is conserved in different conformations of RBD-Fab. These structural insights will be beneficial to designing antibody-based drugs in the future.
Results
Structural properties of S-protein-bound THSC20.HVTR04 (Fab4) and THSC20.HVTR04 (Fab26) complexes based on cryo-EM analysis
Neutralization efficiency was exhibited by both mAbs, namely, THSC20.HVTR04 and THSC20.HVTR26, indicating epitope conservation in most of the variants tested17,18. Next, we performed single-particle cryo-electron micrography (cryo-EM) to decipher the neutralization mechanism of both THSC20.HVTR04 and THSC20.HVTR26 antibodies. For cryo-EM-based structural analysis, the SARS-CoV-2 S protein was incubated with the Fab fragments of both mAbs. Previous studies have demonstrated that both purified Fabs (referred to as Fab4 and Fab26 here on) could strongly bind to the RBD.17,18 However, we also carried out negative stain TEM analysis to first predict the frequency of the Fab-bound forms of the S protein complexes. For TEM analysis, the SARS-CoV-2 S protein was incubated separately with the two Fabs (Fab4/THSC20.HVTR04 and Fab26/THSC20.HVTR26). Negative staining (NS) TEM imaging and reference-free 2D classifications were performed to investigate the sample quality and visualize the dynamic nature of the S protein-antibody interactions. Our TEM studies showed that most of the S proteins were highly homogeneous triangular cone-shaped molecules with some extra densities associated with the RBDs in the S1 region (Fig. S1†), which were completely absent in previously reported negative staining images of the intact S protein at different pH conditions.2 These extra densities clearly represent the binding of Fabs to the S protein in different proportions. Additionally, no tendency of S protein aggregation was observed in the presence of either Fab4 or F26 (Fig. S1A and B†). The reference-free 2D class averages of the S protein with Fabs indicated that the extra densities were related to the RBDs. In 2D class averages, only one Fab, two Fab, and three Fab densities were clearly visible in different class averages. Overall, the NS-TEM analysis indicated that the majority of the S protein formed a stable complex with the Fab fragment (Fig. S1†). These results encouraged us to proceed with the structural characterization of the S protein complexes with Fab4 and Fab26 using cryo-EM.Thus, to achieve our targets, we examined the conformational changes of the S protein protomers by complexing both the Fabs with the S trimer under physiological and cryogenic conditions. The Fab fragments Fab4 and Fab26 were incubated with the SARS-CoV-2 S protein ectodomain and imaged at cryogenic temperature to analyze the structural properties. In the cryo-EM analysis, we were able to see extra densities at the RBDs in the S1 region of the S protein in reference-free 2D class averages (Fig. S2†). Most of the 2D class averages indicated that the S protein either interacts with two Fabs or three Fabs to form complexes, which was noticeable for both the Fabs (Fig. S2A and B†). Therefore, 3D classification and refinement were performed to characterize the high-resolution 3D structures of S protein complexes with Fab4 and Fab26. For this, a low-resolution (40 Å) S protein trimer was used as the initial model (EMD-31096). Interestingly, a Fab-like density appeared near the RBD region after 3D classification (Fig. S3 and S4†) for both Fabs, whereas the initial model demonstrated only the S protein with no Fab density. The Fab densities were clearly visible and firmly connected with RBDs, and this was supported by the negative staining TEM results. As a result, the high-resolution cryo-EM maps of both Fabs with S protein in the “up” state were resolved, and the different projections are shown in Fig. 1. Furthermore, we captured the structural variabilities of RBDs bound to the Fab. Amongst them, the best classes were targeted to obtain the high-resolution cryo-EM maps of the Fab4 complexes. Finally, we obtained three different conformations of the Fab4-S complexes with global resolutions of 4.54 Å, 5.15 Å, and 4.90 Å (Fig. 2A–F and S6†), respectively. The three different states of the Fab4-S trimer complex were as follows: two Fabs bound to the S protein protomer (state I-initial stage) and three Fabs bound to the S protein (state II – intermediate stage and state III – final stage) (Fig. 2A–F). From this study, we identified the actual Fab4-RBD binding propensity from these 3D classification studies, which indicated that Fab4 adopts two-Fab and three-Fab binding conformations (Fig. 2L). However, a lesser proportion of the Fab4-S protein complex was observed to be in the three-Fab binding states, in which three Fabs interacted with three RBDs, and all three RBDs were in the up and partial open conformation (Fig. 2A–F, S3, and S4†). A major population was observed to exist in the two-Fab state i.e., two Fab4 tightly interacted with two RBDs in the up conformation, whereas the remaining one RBD existed in the down and closed conformation (Fig. 2L). Similarly, conformational variabilities were observed in Fab26 bound to the S protein (Fig. 2G–J); two Fab26 tightly interacted with two up-conformation RBDs – state I (Fig. 2G and I) and another state II, in which each protomer accommodated a partially opened RBD of the S trimer (Fig. 2H and J). The proportion of the two- and three-Fab binders was equivalent in the case of the Fab26 complexes (Fig. 2K). These results suggest that both antibodies interact strongly with the S protein RBD. However, significant conformational heterogeneities exist in the RBDs and Fab structures. This observation prompted us further to identify the conformational variabilities of the S-trimer-Fab complexes.
 | ||
| Fig. 1 Cryo-EM maps of the spike (S) protein-Fab complex: (A) high-resolution EM map showing the two RBDs (in the up conformation) of the S trimer bound to two independent Fab4 units. Top and side views of the complexes are shown. (B) High-resolution EM map of one-up RBD of the S trimer bound to one Fab26; the top and side views of the complexes are shown. S protein protomers (Pro 1, 2, 3) are colored gold, magenta, and cyan, respectively, while the Fab is colored orange-red. | ||
 | ||
| Fig. 2 Outline of different conformational states captured from the Fab4- and Fab26-bound S trimer complexes: (A) cryo-EM map represents state I where two Fab4 units are bound to two “up” RBD conformation protomers. The colors of the first, second, and third protomers are gold, magenta, and cyan, respectively. Fab is colored orange-red. (B) Cryo-EM map of state II; two Fab4 units bound to “up” RBD and one Fab4 bound to partially open RBD. (C) State III represents an atomic model of three Fab4 units bound to the open form of the protomers. Shifting of all the RBDs from one state to another upon binding to Fab4 is shown by arrows. (D–F) Atomic models of the different conformations of the captured Fab4-S complexes (states I, II and III, respectively). (G) Cryo-EM map of state I represents two Fab26 units bound to two “up” RBD conformation protomers. The shift of one RBD while binding to Fab26 is shown by the arrow. (H) Cryo-EM map of state II represents three Fab26 units bound to the partial open form of the protomers. To accommodate the binding of Fabs to the protomers, all RBDs undergo conformational changes upon binding to Fab26 and adopt a partially open form, which is shown by the arrows. (I and J) Atomic models of the two different captured conformations of the Fab26-S complexes (states I and II respectively). (K and L) Bar graphs represent the number of particles present in the 2-Fab and 3-Fab binding conformations of Fab4 and Fab26 in the S protein complexes, respectively. | ||
Conformational dynamics of the SARS-CoV-2 S protein complexed with Fab4
To analyze the conformational dynamics of the S protein-bound Fab4 and Fab26 complexes, we initially analyzed the Fab4-S trimer complex and captured three different conformations: the first with two bound fab units and the second with three bound Fabs, and the binding of three Fabs to the S protein adopts two different conformations (Fig. 2A–F). As mentioned earlier, we determined several cryo-EM maps of the S-trimer-Fab complexes at different resolutions. However, we considered the most high-resolution EM-map of the Fab4-S trimer complex to analyze the Fab4-RBD interaction (Fig. 3A and B) at atomic resolution. The available crystal structure (PDB ID: 7KMS) was docked into the EM maps, which were further refined using real-space refinement to calculate the atomic models of all the conformational states and identify the detailed conformational changes (Fig. 2D–F) in the Fab4-S trimer complexes. This atomic model of S protein with two Fab4 complexes allowed us to characterize the epitopes for each protomer from each state, which are represented as a heatmap in Fig. 3C. A total of 16 amino acid residues interacting with Fab4 were identified. The strong interacting residues were identified as N440, K444, V445, G446, G447, P499, and T500 based on their involvement in each protomer from different states (Fig. 3C), which agrees with our previous observation.17 Fab4 belongs to the family of IGHV3-53 antibodies, and based on the interaction studies, we concluded that Fab4 comes under the most immunodominant Class1 mAbs targeting RBD.18 The interfacial area of Fab4 in the complex with S trimer was mapped at 729 Å2, with 598 and 131 Å2 for the heavy and light chains, respectively. We observed that 11 residues participated in non-hydrogen binding with Fab4. Amino acid residues E107 and N31 from the CDRH3 region of the Fab4 heavy chain were involved in H-bonding with N440 and N437, respectively (Fig. S6B†). In the hydrogen bonding network, N440 established a stronger interaction with paratope E107 than others with a distance of 2.69 Å. Simultaneously, paratope N31 formed a hydrogen bonding with epitope N437 with a distance of 3.20 Å. Paratopes Y51, P57, V105, and P106 were found to be involved in hydrophobic contacts with the epitope V445 (Fig. 3E). This hydrophobic pocket was created by the combination of the light and heavy chains of Fab4. Paratopes Y51 and P57 were found in the CDRL2 region, while the V105 and P106 residues in the CDRH3 region constructed the stable hydrophobic pocket for interaction with the epitope (Fig. 3E). These structural observations demonstrate that two of the CDRHs (CRDH1 and CDRH3) and one of the light chains (CDRL2) of Fab4 play a major role in potentially neutralizing SARS-CoV-2 by targeting the S protein (Fig. 3D). As mentioned above, we observed three different conformations of the Fab4-S trimer complex, i.e., two Fabs bound to the S protein (state I-initial stage) and three Fab units binding to the S protein (via two states, namely, state II – intermediate stage and state III – final stage) (Fig. 2A–F). To understand the changes in the binding of Fabs to each protomer, the complexes were further superimposed, and we observed a shift in the angle of RBD binding to Fab4 by 16.4° from each other. In the subsequent stage, when the third Fab binds to the free protomer 3 of the S protein, conformational changes happen to accommodate the Fab. Furthermore, the 3D structures suggested that in state II, two RBD-Fabs were in the open-up conformation and very close proximity, and one RBD-Fab was in the down and closed conformation (Fig. 2B and E); however, in state III, two RBD-Fabs was in the open and up conformation, and one RBD-Fab seemed to be in a partially open intermediate conformation (Fig. 2C, F and 5H). This phenomenon is only possible if RBD adopts different intermediate conformations, as previously shown by Pramanik et al. in 2021.2 Therefore, our study strongly suggests that RBD adopts various flexible conformations and may accommodate the neutralizing antibodies. The structural similarity of the protomer 2 and 3 complexes was found to be very high, which signifies that all protomer complexes were comparably analogous. However, the interaction angles of protomers 2 and 3 showed a considerable change of 6.9° and 9.8°, respectively, with respect to protomer 1 (Fig. 5F and G). Interestingly, the common amino acid residues that form epitopes in THSC20.HVTR04 (K444, P499, S443, T500, and V445) were found in protomers 2 and 3, which did not lose any interaction with protomer 1. Besides, epitopes G446 and Y449 were found not to participate in the protomer 3 complex (Fig. S7A†).3 The protomer 3 complex adopted the same conformation as protomer 1, which stabilized the conformation of the S-trimer-Fab complex. Additionally, the interaction of Fabs with protomers 1 and 2 was identical, and the RMSD score of the individual protomers between the states was 0.321 Å and 0.372 Å, respectively. On account of the third Fab4 binding in the intermediate stage, major structural changes happened in the pre-existing protomer complexes (Fig. 5A and B). The conformational change of protomers 1 and 2 between states I and II were 0.951 Å and 1.113 Å, respectively, as calculated by their RMSD scores. A minor angle shift of protomers 1 and 2 was observed between states I and II. Besides, protomer 3 was shifted to 25.6° to accommodate the third Fab in a stable complex (Fig. 4A–C). From our structural studies, we predicted that around 21 amino acid residues of S protein were involved in the interaction with Fab4, and these interacting amino acid residues were consistent throughout all the conformations of the RBD of the S protein (Fig. 3C). However, we noticed that a few amino acid residues (L441, N437, N448, N450, Q506, S443) of the S protein epitopes were different for promoters 1 and 2 in states I and II (ESI Fig. S7A†). Generally, the interacting amino acid residues of the epitope should be fixed for an antibody. However, this inconsistency was observed due to the drastic conformational changes in RBD-Fab4 complexes. We hypothesized that some poorly interacting amino acid residues change the interacting partner during conformation changes. However, the common epitopes in the interaction network of the S protein-Fab4 complex in the steady state were determined to be G446, G447, K444, N440, P499, T500, and V445 (Fig. S11A†). | ||
| Fig. 3 Structural basis for the accommodation of Fab4 and Fab26 in the SARS-CoV2 S protein: cryo-EM maps fitted to the atomic model, generated for the highest-resolution EM maps obtained from the (A) S protein-Fab4 complex and (B) S protein-Fab26 complex. The colors of the first, second, and third protomers are gold, magenta, and cyan, respectively, and Fab4 and 26 are colored orange-red. (C) Heat map represents epitope residue selectivity and their instances in the different conformations captured. Accordingly, the frequency of epitope involvement in antibody interaction is colored linearly in green, whereas zero involvement is shown in yellow. It demonstrates the potent epitope residues in all the binding modes/conformational states. (D–G) Details of the RBD interactions with Fab4. (D and F) Interfacial area shows the CDR regions of Fab4 and 26 with RBD, respectively. CDRH1, CDRH2, CDRH3, CDRL1, CDRL2, and CDRL3 are highlighted in light salmon, Indian red, maroon, cornflower blue, medium blue, and dark blue respectively, and the RBD region is colored hot pink. (E) Important interacting residue V445 of the S protein makes contact with a hydrophobic pocket of Fab4 comprising the residues P57, V105, and P106. (G) F486 is shielded in a hydrophobic pocket of Fab26 comprising L34 and G112 residues. | ||
 | ||
| Fig. 4 Distinct binding features of Fab4 and Fab26 in the RBD region of the S protein with respect to ACE2: (A) Licorice diagram of the S protein bound to Fab4 and ACE2, and the magnified view of the conformational variable region of RBD bound to Fab4 and ACE2. ACE2 is shown in lime green, Fab4 is shown in hot pink, Fab4-bound S protein is shown as a black ribbon, and the ACE2-bound S protein is displayed in gold. (B) Licorice diagram of the conformationally variable region and the clashing region of Fab26- and ACE2-bound RBD. (C and D) Overlapping region of Fab4- and Fab26-bound RBD with ACE2-bound RBD is shown in the silhouette form. (E) High-resolution 3D structure obtained for the S protein binding loop along with Fab4 and Fab26 in the RBD region highlights the distinct targeting ability. | ||
Conformational dynamics of SARS-CoV-2 S protein complexed with Fab26
For the complex of Fab26-S trimer, we found two conformational states: state I, with an asymmetric S trimer and two ‘‘up’’ RBDs forming a complex, each with one Fab (Fig. 2G and I); state II consists of a symmetric trimer bound to three RBDs in the same ‘‘up’’ conformation, where each RBD participated in forming a complex with each of the Fab26 units (Fig. 2H and J). We refined the state I EM map using masking to achieve a high-resolution Fab26-S protein complex map, resulting in 4.4 Å, and the atomic fitted model used for detailed structural analysis (pro1) is shown in Fig. 3B. The epitope residues involved in interaction with Fab26 for states I and II are represented in the heat map (Fig. 2C).The structural analysis of the pro1 map generated an atomic model map of Fab26 bound to the S protein complex, which revealed 11 amino acid residues in S protein in the interacting network with Fab26, and the overall interacting interface area found was 636 Å2 (361 Å2 on the heavy chain and 275 Å2 on the light chain) (Fig. 3F). The number of interfacial residues involved in interactions with the heavy and light chains were 7 and 5, respectively, with 11 epitopes likely forming the basis for the potent neutralization capability conferred by THSC20.HVTR26 Fab (Fig. 3C). The epitope residue F486 interacts with the hydrophobic pocket formed by paratopes L34, Y93, and G112. The RBD interaction is stabilized by this hydrophobic pocket formed in the region between CDRH3 and CDRL3 (Fig. 3G). To pause the flexible movement of the RBD, one of the aromatic residues (F486) in the epitope region, protrudes towards the pocket formed by the β-sheets of the CDRL1, CDRL3, and CDRH3 regions. Overall, the epitopes for Fab26 on the S protein were T478, N481, V483, E484, F486, C488, and Y489. These residues were observed to frequently participate in the interaction of the S protein-Fab complexes (Fig. 3C). The amino acid residues N481 and Y489 of the epitopes formed hydrogen bonding with paratopes Y32 and A101, respectively, from the heavy chain regions (CDHR3) (Fig. S6C†). Our analysis further revealed that residues Y32 and N33 from the CDRL1 region interacted with N481, while T478 and S97 of the CDRL3 region interacted with the epitope V483. Other potential paratopes, such as Y32, S96, S97, and D110, also interacted with the same loop of epitope region (Fig. S6C and D†). Taken together, our data indicates that for the neutralization of SARS-CoV-2, Fab26 utilized the heavy chain CDR (CDRH3) and two of the light chain CDRs (CDRL1 and CDRL3) to stall the receptor-binding ridge of the S protein (Fig. 3F). Similar to Fab4, Fab26 also belongs to the IGHV3-53 antibody germline and the identification of the potent epitopes suggests that Fab26 belongs to most immunodominant Class1 mAbs targeting RBD. Apart from that, we observed another state of conformation in which the third Fab26 was found to bind to the free RBD in the S protein. To accommodate the third Fab26, all the protomers of the S protein adopted a partially open conformation, likely for stabilization (Fig. 2G–J). The number of two-Fab26-bound S trimers was found to be significantly higher than the three-Fab26-bound forms (Fig. 2K).
As stated above, among the two states of Fab26-S protein complexes, the 7.3 Å resolution state I showed two RBDs in the up conformation bound to Fab and another RBD found in the partially open form, whereas the 4.8 Å resolution EM map of state II exhibited three RBDs interacting with Fabs in the partially open conformation (Fig. 2G–J). The crystal structure of the open-conformation S protein (PDB ID: 6ZGG) was then fitted into the auto-sharpened EM map to generate atomic models for both complex states (Fig. 3B and 2I, J). The change in states I and II of protomer I of the Fab26 complexes was found to be 12.1° (Fig. 5D) and an angle shift of 26.1° was observed between protomers 1 and 2 of state I (Fig. 5I), whereas no change was observed between the RBD region of both the CDR-RBD interfaces across the state II complex of all three protomers (Fig. 5J). The common interacting residues A475, V483, E484, F486, and N487 were identified from our structural studies (Fig. 3C and S7B†). To accommodate the third Fab26, a clear shift from “up” RBD to a “partially open” conformation was observed in the superimposed model of protomer 1 of both states, whereas the shift was minimal in the case of the protomer 2 in both the captured conformational states of Fab26 complexes (Fig. 5D and E).
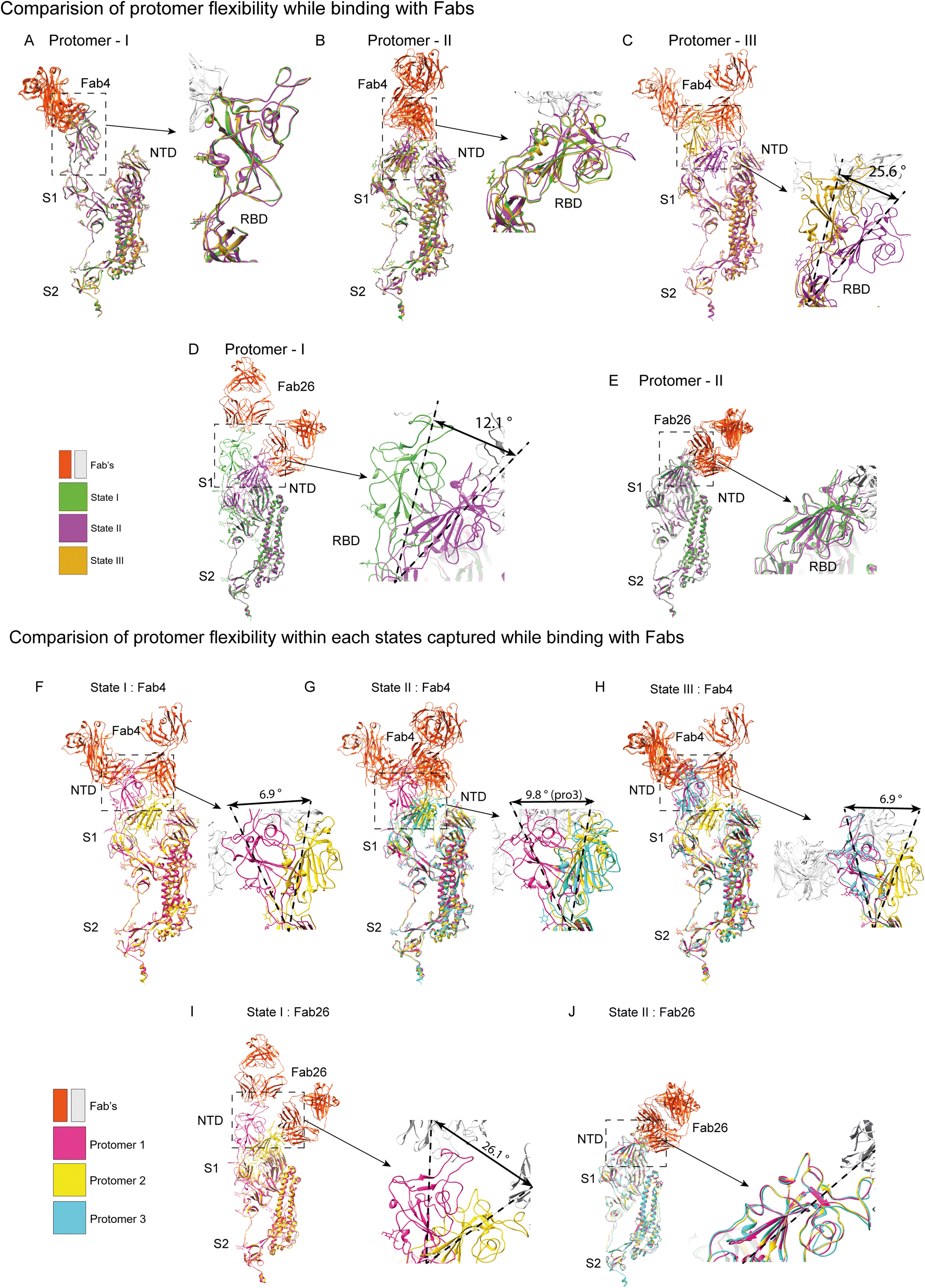 | ||
| Fig. 5 Comparison of the conformational changes in the S protein during the formation of Fab4- and Fab26-bound complexes: (A–C) superimposition of individual protomer 1 (F), protomer 2 (G), and protomer 3 (H) from three different states of the Fab4-S protein complex provides insights into the dynamics of each protomer across the states. A magnified view of the RBD region is shown in the right corner of each image. (D–E) Similarly, a comparison of protomer 1 (I), and protomer 2 (J) in states I and II of the Fab26-S protein complexes is shown. Apart from minor angle shifts, significant angles are highlighted in the figures. (F–J) Protomer conformations captured within each state are compared, showing the structural changes in the RBD domain of each complex state. Protomer superimposition shows the protomers for (F–H) states I, II and III of the Fab4 complexes and (I and J) states I and II of the Fab26 complexes. Protomers 1, 2 and 3 are coloured magenta, gold and cyan, respectively. | ||
In state I, protomer 1 is likely to adopt the up conformation, while protomer 2 adopts the partially open form. The angle of shift observed between protomers 1 to 2 was 26.1° in state I (Fig. 5I), whereas no change was observed between the RBD-Fab bound regions of the state II protomers (Fig. 5J). This suggests that all the protomers adopted the same partially open conformation, which might be due to the stabilized Fab26 fragment. This inward movement of RBD in two protomers permitted the third protomer to bind with the third Fab. These conformational movements facilitate the partially open conformation-mediated binding of three Fab26s. The cryo-EM-based structural studies elucidate that RBDs are flexible and can adopt open conformations, as well as partially open conformations, during their interaction with antibodies. The flexibility of RBD is also supported by our previous studies.17 In addition, we identified the interacting residues responsible for Fab-RBD stabilization to understand whether these two antibodies have an affinity for other SARS-CoV-2 variants. The change in dynamics of the epitope interaction residues with Fab26 is shown as a heat map according to each protomer in different states (Fig. S7B†). The potent epitope residues observed to very frequently interact in all the states captured were T478, N481, Y489, V483, and F486 (Fig. S8D†).
Antibody binding alters the conformational states of the SARS-CoV-2 S trimer in its apo- and hACE-2-bound forms
To understand conformational variations that occur in the RBD region during binding to Fab4 or Fab26 concerning its apo form and in the ACE2-bound complex, we compared our Fab4- and Fab26-S protein complexes with the hACE2 attached and apo forms of the S protomer. For this analysis, we used the first protomer of the S trimer-Fab4 (Fig. 2A and 3A) and S trimer-Fab26 (Fig. 2G; masked 3B) high-resolution atomic models. In the structural analysis, the superimposition of both Fab-bound complexes demonstrated independent distal targeting of the RBD region (Fig. 4E). The region containing the N439-V445 and V483-C488 residues formed a primary hotspot for the antigen–antibody interactions of Fab4 and Fab26, respectively, validating the independent nature of these antibodies in targeting the S protein RBD. In addition, the structural analysis also proved its independent nature without any clashes (Fig. 4E). For Fab4, a 7.1° shift of the plane was observed compared to the hACE2-bound protomer, and an average shift of 8.2 Å was found in the ACE2-binding loop of the RBD spanning from N440 to V445 (Fig. 4A and B). On the other hand, another loop containing two interacting residues, namely, P499 and T500, moved upward by 9.6 Å to form contacts with Fab4. As for Fab26, a minor shift of 1.2° from the hACE2-bound structure and an average shift of 6.3 Å in the ACE2 binding RBD ridge were observed (Fig. 4C and D). Q493 and Y489 are known to be critical residues in the RBD region for interaction with the ACE2 receptor.25 Interestingly, these two residues fell in the region of Fab26-interacting residues. Likewise, we observed 8.2 Å and 7.3 Å shifts of the respective residues Q493 and Y489, along with a 2.3° outward shift of the entire RBD core. Interfacial area of the ACE2 and Fab4 was 3791 Å2, where 53 residues of the heavy chain overlapped with 46 residues of the ACE2 covering 1505 Å2 and 61 light chain residues covering 2286 Å2 interface area. Therefore, our analysis showed the overlapping interfacial area of the RBD with ACE2, which emphasizes the inhibition of ACE2-S trimer binding (Fig. 4C and D).Discussion
Most biological macromolecules are structurally flexible, and the S protein of SARS-CoV-2 is one of the most dynamic proteins, exhibiting highly flexible and dynamic receptor-binding domains (RBDs) and N-terminal domains (NTDs). These dynamic regions play a crucial role in viral infection by facilitating interactions with host cell receptors, such as ACE2. Recent advances in cryo-EM have enabled capturing various intermediate states of these dynamic and flexible RBDs and NTDs, revealing their conformational diversity.2,26–28 The S protein exists in different conformations with the RBDs in either a “down” closed state or open states with one, two, or three RBDs “up”. These open conformations are critical for initiating viral infection. Previous studies have shown that open RBD conformations are the most active forms for interaction with the ACE2 receptor.5,29 However, other reports have described the continuous motion of RBD, forming intermediate conformations between the open and closed states.2,5,26–28 Given that RBDs are critical for receptor binding and infection initiation, researchers have targeted them for antibody and inhibitor development to block viral interactions.30–32 Notably, the flexibility of RBDs, which is important for ACE2 interactions, may also influence antibody binding.5,6 The SARS-CoV-2 S protein exhibits intrinsic flexibility, which plays a crucial role in viral entry.28 Pramanick et al. demonstrated the conformational dynamics of the S protein under varying pH conditions using cryo-EM and also demonstrated various intermediate conformations of RBDs possible on the S protein.2 Additionally, continuous mutations in the S protein influence its ACE2 binding affinity and flexibility. Maroli et al., 2023 reported restricted RBD flexibility in the antibody-bound states33 compared with unbound conditions. However, to our knowledge, very few studies have explored the antibody-induced conformational flexibility of the SARS-CoV-2 S protein so far. Most of the existing studies focus on the up or down states of the S protein, but the intermediate conformations introduce cryptic pockets, which are significant for therapeutic development as they provide insights into additional sites for inhibitors to potentially bind.28A recent study investigated the structural dynamics of the S protein during ACE2 recognition, providing insights into the intermediate RBD-down and -up transition states.5 Similarly, during the antibody neutralization process, the RBDs may adopt various conformations when binding to antibodies. While some reports have described different RBD-antibody binding modes,19,20 the intermediate conformations of RBD-antibody complexes have mostly been overlooked. Understanding these dynamics is essential for explaining the antibody-mediated neutralizing mechanisms and their function in immune response. Many previous studies have mainly focused on characterizing static interactions between the S protein and antibodies or inhibitors. As a result, the importance of the structural flexibility of the RBD during these interactions might have been overlooked. Here, we explored the broad neutralizing features associated with two potent antibodies originating from a single patient.17 Using single-particle cryo-EM, we captured the dynamic RBD-Fab interactions. During these interactions, various intermediate RBD-Fab states were observed, demonstrating ways in which all three RBDs accommodate large antibodies. This study exclusively captures several intermediate RBD-antibody complex states and highlights S protein conformational flexibility during neutralization (Fig. 2F).
Solvent-accessible surface area (SASA) may be variable and depends on the conformational flexibility of the protein. Thus, solvent-accessible surface area (SASA) variations across different transition states play a key role in determining epitope accessibility of the antigen and therapeutic effectiveness of the antibody.2,28 Notably, each state of the S protein can be targeted with distinct antibody classes, emphasizing the importance of designing conformation-dependent epitopes for therapeutic efficiency.28 This study successfully demonstrates the existence of various intermediate conformational states of the RBD-Fab complex during interactions, contrary to previous studies that describe antibody-bound S protein states as rigid.33 Possible explanations for the wide range of observed conformational states include: (i) these antibodies target the S protein across multiple transitional states and (ii) antibody-induced flexibility upon binding. While several studies have discussed the antibody-mediated conformational flexibility of many other systems, only a few studies have specifically targeted the S protein from SARS-CoV-2. These investigations demonstrate how antibody binding can modulate conformational behaviour and reduce the flexibility of S protein. Beyond the direct neutralization mechanism by competing with receptor binding, certain antibodies may act as allosteric regulators, influencing the flexible motion of the S protein and its interactions.34
Our study highlights key interaction features and sequential binding steps of the two novel mAbs THSC20.HVTR04 and THSC20.HVTR26 isolated from a single individual. Thus, our study also opens up the possibility of structure-guided paratope modification to combat future SARS-CoV-2 variants. This study aligns with previous findings that demonstrated antibody-induced conformational flexibility of S protein from the SARS-CoV-2 Delta variant.35 Notably, these conformational changes explain variations in the neutralization efficiency and binding affinity across emerging variants. Additionally, the degree of exposure of the aromatic amino acids plays a pivotal role in conformational flexibility.36 Moreover, certain antibodies that induce long-range conformational changes in the antigen may cause positive or negative allosteric effects.37
Furthermore, through cryo-EM analysis of the TAU-2212 complex, a recent study revealed a unique flexible binding mode across five distinct conformational states, rationalizing the broad neutralization potential of this antibody.35 Similar to the S protein-Fab complexes of TAU-2212,35 we also observed the immense structural flexibility of RBD in the THSC20.HVTR04/Fab4 and THSC20.HVTR26/Fab26 complexes. The structural dynamics of these three complexes were compared (ESI Fig. S9 A–C†). Sequence analysis revealed distinct interaction residues in all Fabs, except for a common epitope F486 in TAU-2212 and THSC20.HVTR26/Fab26 (ESI Fig. S9D†). Further investigation of these interacting residues is necessary for the development of therapeutics that can target restricting the flexible motions and mitigating resistance from new mutational variants.38 For instance, the D614G mutation shifts the conformational equilibrium toward the up-state, increasing antibody accessibility.39
These findings highlight the importance of studying antibody-induced conformational changes and identifying conserved allosteric hotspots for the development of next-generation therapeutic antibodies. Our cryo-EM study highlights the potential of antibody-induced structural dynamics of the RBD of the S protein. Understanding these mechanisms will assist in designing potent antibodies that consider the structural flexibility of the S protein to combat emerging variants. As monoclonal antibody therapy remains one of the most effective treatments for viral infections, our findings offer crucial insights for the development of potent therapeutic mAbs to block viral entry. This study emphasizes the value of antibody cocktails that target diverse antigen conformations to prevent host cell fusion and enhance therapeutic efficacy.
Materials and methods
Preparation of IgG and Fabs
IgGs were prepared according to the reported protocols using the Expi293 cell line.17 The fragment antigen binding domain (Fab) of each IgG was prepared by mixing ∼500 μL of the monoclonal antibody (mAb) (1–5 mg mL−1) with 1200 μL of 100 mM sodium acetate (sodium acetate anhydrous from Sigma Aldrich) containing 1 mM ehylenediaminetetraacetic acid disodium salt dihydrate (EDTA for electrophoresis, for molecular biology, 99.0–101.0% from Sigma), 100 μL of 50 mM cysteine (L-cysteine for biochemistry from Merck) and 200 μL of papain (10 μg per mg mAb concentration; papain from papaya Latex, 89% pure lyophilized powder from Sigma). The mixture was then kept in an incubator shaker at 37 degrees Celsius for 8–10 hours. After 10 hours, 75 mM (∼150 μL) iodoacetamide (Iodoacetamide BioUltra from Sigma) was added to the mixture to stop papain activity and incubated at room temperature for 30–40 min. Then, the sample was added to protein A beads (Immobilized Protein A resin from G Biosciences), washed 2–3 times with PBS to remove the residual ethanol and incubated for another 3–4 hours at room temperature on the rocker. After ∼4 hours, the mixture was poured into the gravity column, and the flow through was collected in a 15 mL Falcon tube. The flowthrough contained the Fab region, whereas the FC region was bound to the beads. The beads were then washed with PBS twice to collect the remaining Fab fractions from the column. The column was then eluted with 100 mM glycine (G Biosciences) at pH = 2.2 to remove the bound Fc fractions and then washed with PBS 3–4 times. Finally, the flowthrough and the PBS wash containing the residual Fab fractions were pooled together and brought to a final concentration of 0.5–0.8 mg mL−1.Spike 6P protein expression and purification
The SARS-CoV-2 S HexaPro plasmid was a gift from Jason McLellan (Addgene plasmid # 154754). This plasmid contains a CMF promotor that drives the expression of the SARS-COV-2 Spike-B.1 ectodomain (1–1208 AAs) with a mutated furin site (682–685 GSAS) and hexa proline mutations F817P, A892P, A899P, A942P, K986P, V987P as fold on. The HRV 3C cleavage site was placed before 8× His tags followed by a 2× Strep-Tag II at the C terminal.40 This plasmid was transfected into Expi293F cells (Thermo Fisher Scientific, Cat # A14527, USA) using the Expifectamine 293 Transfection Kit (Gibco, Thermo Fisher, Cat # A14524) according to the manufacturer's instruction. In brief, 24 h prior to transfection, 100% viable Expi293F cells were passaged at a density of 2 million per mL. On the next day, at 4 million cells per mL (100 mL), the cells were transiently transfected with the expifectamine-plasmid-DNA complex (100 μg plasmid, complexed with 270 μl of expiFectamine293). After 20 h, Enhancer 1 and Enhancer 2 were added according to the manufacturer's protocol. Five days after the day of transfection, the culture supernatant without cells was collected and used for the purification of the 6× histidine-tagged protein. The supernatant media were affinity-purified by immobilized metal affinity chromatography using the Ni-NTA resin (G Biosciences, Cat # 786940). 100 mL of the supernatant medium was mixed with PBS (pH 7.4) to equilibrate the Ni-NTA resin (5 mL) and incubated for 4 h at 4 °C for protein immobilization under gentle rotation. The supernatant-resin complex was gently applied to the column. After the unbound fraction was collected, the column was washed with a ten-column volume of 1× PBS (pH 7.4) supplemented with 25 mM imidazole. Finally, the bound protein was eluted with a gradient of 200 to 500 mM imidazole in PBS (pH 7.4). The eluted fractions were pooled and dialyzed against 1× PBS buffer (pH 7.4) twice using dialysis tubing cellulose membrane [avg. flat width 43 mm (1.7 in.), 14 kDa MWCO, Sigma-Aldrich, Cat # D9527]. The protein concentration was determined based on absorbance (A280) using a Biophotometer D30 (Eppendorf) and a theoretical molar extinction coefficient of 146720 M−1 cm−1 calculated using the ProtParam tool (ExPASy). The protein was analyzed by 8% SDS PAGE for purity and homogeneity under reducing and non-reducing conditions.Size exclusion chromatography (SEC)
The aggregation tendency of the IgGs that represent the mAbs was characterized by SEC. For SEC, the samples were passed through Superdex™ 200 Increase 10/300 GL size exclusion column (GE, Inc.) and eluted with degassed PBS buffer (pH 7.4) at a 0.3 mL min−1 flow rate using AKTApurifier™ 100 (GE, Inc.).Sample preparation for negative staining TEM and data processing
To analyze the binding of Fab to the S protein and its homogeneity, we performed conventional negative staining TEM. The diluted samples of S protein (1 mg mL−1, 10×) and the Fabs (1.4 mg mL−1, 300× times) were mixed and incubated for 2 minutes. Then, 3.5 μL of each sample mixture was applied on freshly glow-discharged carbon-coated Cu grids (30 s) (EM grid, 300 mesh, Electron Microscopy Sciences). After 1 min, the excess solvent was blotted and 1% uranyl acetate (Uranyl Acetate 98%, ACS Reagent, Polysciences, Inc.) was added to the grid. After air drying, the grids were used for data acquisition at room temperature and 120 kV using a Talos L120C electron microscope. Data collection was performed using a 4k × 4k Ceta camera at a magnification of 73k× and calibrated at 3.84 Å per pixel. The collected micrographs were processed in EMAN 2.1.41 Automated and manual modes of particle picking were employed, and the coordinates were extracted using e2boxer.py in EMAN 2.1. Subsequently, the reference-free 2D class averages allowed us to visualize various projections of the Fab-bound S protein complexes. The cleaned dataset of the samples was used for reference-free 2D classification, and the reference-free 2D class averages of different particle projections were calculated using simple_prime2D of the SIMPLE 2.1 software42 with a mask diameter of 30 pixels at 3.84 Å per pix.Cryo-EM sample preparation
R1.2/1.3 300 mesh gold grids (Quantifoil) (Electron Microscopy Sciences) were glow-discharged for 90 seconds at 20 mA before freezing. Equal volumes of the SARS-CoV 2 S protein solution (1 mg mL−1) and 30× diluted solution of the respective Fab (∼1.4 mg mL−1) were mixed and incubated at room temperature for 2 minutes. Three microliters of the final sample were applied to the freshly glow-discharged grids, incubated for 10 s and immediately blotted for 8.5 s at a blot force of zero in a pre-equilibrated chamber of the FEI Vitrobot Mark IV plunger. Immediately after blotting, the grid was plunged into liquid ethane.Cryo-EM data collection
Cryo-EM data were acquired using a 200 kV Talos Arctica transmission electron microscope (Thermo Scientific™) equipped with a Gatan K2 Summit Direct Electron Detector. Movies were recorded automatically using Latitude-S (DigitalMicrograph – GMS 3.5) at a nominal magnification of 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× and an effective pixel size of 0.92 Å.43 The micrographs were collected in the counting mode with a total dose of 50 e− per Å2 and an exposure time of 8 s distributed over 20 frames. A total of 3789 and 2102 movies were acquired for the Fab4- and Fab26-S protein complexes, respectively.
000× and an effective pixel size of 0.92 Å.43 The micrographs were collected in the counting mode with a total dose of 50 e− per Å2 and an exposure time of 8 s distributed over 20 frames. A total of 3789 and 2102 movies were acquired for the Fab4- and Fab26-S protein complexes, respectively.
Cryo-EM data processing
Single-Particle Analysis (SPA) of the acquired cryo-EM data was performed using Relion version 3.1.44 Initially, the drift and gain corrections of the individual movies were corrected using MotionCor2.45 Subsequently, the motion-corrected micrographs were subjected to screening using cisTEM to identify bad micrographs,46 and the detected fit resolution threshold chosen for screening was 7 Å. The best micrographs were chosen for the estimation of Contrast Transfer Function (CTF) parameters using CTFFIND 4.1.13.47 Then, particles were picked using the Laplacian picker in Relion and extracted using the box sizes of 336 and 360 Å for the S:Fab4 and S:Fab26 complexes, respectively. The well-defined classes of S protein with Fab4 and Fab26 complexes were found to be 418672 and 255039 particles, respectively, after six rounds of 2D classification. These particles were selected for 3D classification without imposing symmetry (C1). The selected particles of Fab4 and Fab26 with the S protein were classified into 8 and 5 classes, respectively (ESI Fig. S3 and S4†). Then, the best classes showing the same conformations were merged to improve the resolution. For the S protein-Fab4 complex, classes 1 and 5 (93959 particles), as well as classes 2 and 8 (163492 particles), were merged together. On the other hand, the S protein-Fab26 complex was classified into five classes, of which three good classes, namely, class-2 (26719 particles), class-3 (48584 particles), and class-5 (109596 particles), showed proper 3D structures. To achieve high resolution, all particles belonging to the best classes of the Fab complexes were subjected to movie refinement, which included the estimation of beam tilt, anisotropic magnification, and per-particle CTF refinement for defocus and astigmatism. The soft-edged mask was applied during the 3D-refinement process to remove the solvent noise surrounding the molecule. Sharpening of the 3D auto-refined maps was performed using Relion 3.1 (ref. 44) and PHENIX.48,49 The overviews of cryo-EM data processing are shown in Fig. S4 and S5.† The global resolution of Fourier shell correlation (FSC) was estimated at a threshold of 0.143, and the estimation of the local resolution was performed with ResMap using auto-refined half maps.
Model building and structure refinement
Automated model building was iteratively performed using Phenix Real Space Refinement. Based on the S protein conformations, the SARS-S protein PDB: 7kms – 3 RBD up; 6zwv – 3 RBD down; 7akd – 1 partially open RBD were docked with the cryo-EM maps using the UCSF Chimera “Fit in map” tool. To build the Fab models, the query sequences of both chains were submitted to the Swiss Model, and the resultant models were also docked to the EM maps. The fitted models were used as the initial models and refined against the sharpened EM maps. The structural statistics of the Cryo-EM map and atomic model were analyzed using Phenix, EMringer, Molprobity, and UCSF chimera.50Analysis and visualization
The cryo-EM maps and atomic models were visualized using UCSF Chimera. PDBsum was used to identify the interacting residues of the S protein and Fab complexes.51 Surface coloring based on the Kyte-Doolittle hydrophobicity scale was applied using UCSF ChimeraX.52 The angles of chain rotation were estimated by creating planes between the three terminal residues (N370, F490, P1140) of the spike (S protein) protomer and its shift between the planes was calculated. The RMSD values were calculated using the UCSF Chimera “MatchMaker” tool.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
SD designed the TEM and cryo-EM structural studies. AC performed negative staining sample preparation and image processing. CFR carried out the cryo-EM sample preparation, data acquisition, data processing, and structural analysis. CFR and AC carried out manuscript writing, editing, and reviewing. NH and MYA helped in the cloning and purification of the mAbs. RS, SJ, and PMR worked on spike protein expression and purification.Conflicts of interest
A patent application (PCT/IB2022/057923) was filed on the invention of novel monoclonal antibodies with the following inventors: Jayanta Bhattacharya, Nitin Hingankar, Payel Das, Suprit Deshpande, Pallavi Kshetrapal, Ramachandran Thiruvengadam, Amit Awasthi, Zaigham Abbas Rizvi. Authors Randhir Singh and Sowrabha Jayatheertha are employed by Mynvax Private Limited, Basavanagudi, Bengaluru 560004, India, and author Poorvi M. Reddy was employed by Mynvax Private Limited, Basavanagudi, Bengaluru 560004, India. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
We thank Mynvax Private Limited for the support with SARS-CoV-2 spike protein expression and characterization. We acknowledge the Department of Biotechnology, Department of Science and Technology (DST) and Science, and Ministry of Human Resource Development, India for funding the cryo-EM facility at IISc-Bangalore. We acknowledge the DBT-BUILDER Program (BT/INF/22/SP22844/2017) and DST-FIST (SR/FST/LSII-039/2015) for the National Cryo-EM facility at IISc, Bangalore. This research was supported by the STARS (SERB-STR/2022/000006) and SERB CRG/2022/002674. We thank Penny Moore and Jinal Bhiman, NICD, Johannesburg, South Africa for kindly providing the omicron spike plasmid constructs, Jason McLellan, Texas, USA for kindly providing the SARS-CoV-2 RBD construct used for the purification of the RBD protein. This study was supported by funding from the Research Council of Norway (Project ID: 285136), Bill and Melinda Gates Foundation supported Global Immunology & Immune Sequencing for Epidemic Responses (GIISER-South Asia) project at THSTI, (INV-030592). JB is supported by the DBT-Wellcome Trust India Alliance Team Science Grant (IA/TSG/19/1/600019). CFR and AC are graduate students funded by the Indian Institute of Science (MHRD and CSIR) fellowship.References
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- I. Pramanick, N. Sengupta, S. Mishra, S. Pandey, N. Girish, A. Das and S. Dutta, Conformational flexibility and structural variability of SARS-CoV2 S protein, Structure, 2021, 29, 834–845 CrossRef CAS PubMed.
- J. Fedry, D. L. Hurdiss, C. Wang, W. Li, G. Obal, I. Drulyte, W. Du, S. C. Howes, F. J. M. van Kuppeveld, F. Förster and B.-J. Bosch, Structural insights into the cross-neutralization of SARS-CoV and SARS-CoV-2 by the human monoclonal antibody 47D11, Sci. Adv., 2021, 7, eabf5632 CrossRef CAS PubMed.
- A. C. Walls, M. A. Tortorici, J. Snijder, X. Xiong, B.-J. Bosch, F. A. Rey and D. Veesler, Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11157–11162 CrossRef CAS PubMed.
- H. Yajima, Y. Anraku, Y. Kaku, K. T. Kimura, A. Plianchaisuk, K. Okumura, Y. Nakada-Nakura, Y. Atarashi, T. Hemmi, D. Kuroda, Y. Takahashi, S. Kita, J. Sasaki, H. Sumita, J. Ito, K. Maenaka, K. Sato and T. Hashiguchi, Structural basis for receptor-binding domain mobility of the spike in SARS-CoV-2 BA.2.86 and JN.1, Nat. Commun., 2024, 15, 8574 CrossRef CAS PubMed.
- D. J. Benton, A. G. Wrobel, P. Xu, C. Roustan, S. R. Martin, P. B. Rosenthal, J. J. Skehel and S. J. Gamblin, Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion, Nature, 2020, 588, 327–330 CrossRef CAS PubMed.
- G. Cerutti, Y. Guo, T. Zhou, J. Gorman, M. Lee, M. Rapp, E. R. Reddem, J. Yu, F. Bahna, J. Bimela, Y. Huang, P. S. Katsamba, L. Liu, M. S. Nair, R. Rawi, A. S. Olia, P. Wang, B. Zhang, G.-Y. Chuang, D. D. Ho, Z. Sheng, P. D. Kwong and L. Shapiro, Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite, Cell Host Microbe, 2021, 29, 819–833 CrossRef CAS PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong, Y. Yang, Z. Chen, Y. Guo, J. Zhang, Y. Li, X. Song, Y. Chen, L. Xia, L. Fu, L. Hou, J. Xu, C. Yu, J. Li, Q. Zhou and W. Chen, A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2, Science, 2020, 369, 650–655 Search PubMed.
- W. Dejnirattisai, D. Zhou, H. M. Ginn, H. M. E. Duyvesteyn, P. Supasa, J. B. Case, Y. Zhao, T. S. Walter, A. J. Mentzer, C. Liu, B. Wang, G. C. Paesen, J. Slon-Campos, C. López-Camacho, N. M. Kafai, A. L. Bailey, R. E. Chen, B. Ying, C. Thompson, J. Bolton, A. Fyfe, S. Gupta, T. K. Tan, J. Gilbert-Jaramillo, W. James, M. Knight, M. W. Carroll, D. Skelly, C. Dold, Y. Peng, R. Levin, T. Dong, A. J. Pollard, J. C. Knight, P. Klenerman, N. Temperton, D. R. Hall, M. A. Williams, N. G. Paterson, F. K. R. Bertram, C. A. Siebert, D. K. Clare, A. Howe, J. Radecke, Y. Song, A. R. Townsend, K.-Y. A. Huang, E. E. Fry, J. Mongkolsapaya, M. S. Diamond, J. Ren, D. I. Stuart and G. R. Screaton, The antigenic anatomy of SARS-CoV-2 receptor binding domain, Cell, 2021, 184, 2183–2200 Search PubMed.
- M. Yuan, H. Liu, N. C. Wu, C.-C. D. Lee, X. Zhu, F. Zhao, D. Huang, W. Yu, Y. Hua, H. Tien, T. F. Rogers, E. Landais, D. Sok, J. G. Jardine, D. R. Burton and I. A. Wilson, Structural basis of a shared antibody response to SARS-CoV-2, Science, 2020, 369, 1119–1123 Search PubMed.
- D. Pinto, Y.-J. Park, M. Beltramello, A. C. Walls, M. A. Tortorici, S. Bianchi, S. Jaconi, K. Culap, F. Zatta, A. De Marco, A. Peter, B. Guarino, R. Spreafico, E. Cameroni, J. B. Case, R. E. Chen, C. Havenar-Daughton, G. Snell, A. Telenti, H. W. Virgin, A. Lanzavecchia, M. S. Diamond, K. Fink, D. Veesler and D. Corti, Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody, Nature, 2020, 583, 290–295 CrossRef CAS PubMed.
- J. Huo, Y. Zhao, J. Ren, D. Zhou, H. M. E. Duyvesteyn, H. M. Ginn, L. Carrique, T. Malinauskas, R. R. Ruza, P. N. M. Shah, T. K. Tan, P. Rijal, N. Coombes, K. R. Bewley, J. A. Tree, J. Radecke, N. G. Paterson, P. Supasa, J. Mongkolsapaya, G. R. Screaton, M. Carroll, A. Townsend, E. E. Fry, R. J. Owens and D. I. Stuart, Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike, Cell Host Microbe, 2020, 28, 445–454 CrossRef CAS PubMed.
- M. Yuan, N. C. Wu, X. Zhu, C.-C. D. Lee, R. T. Y. So, H. Lv, C. K. P. Mok and I. A. Wilson, A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV, Science, 2020, 368, 630–633 CrossRef CAS PubMed.
- P. Zhou, G. Song, H. Liu, M. Yuan, W. He, N. Beutler, X. Zhu, L. V. Tse, D. R. Martinez, A. Schäfer, F. Anzanello, P. Yong, L. Peng, K. Dueker, R. Musharrafieh, S. Callaghan, T. Capozzola, O. Limbo, M. Parren, E. Garcia, S. A. Rawlings, D. M. Smith, D. Nemazee, J. G. Jardine, Y. Safonova, B. Briney, T. F. Rogers, I. A. Wilson, R. S. Baric, L. E. Gralinski, D. R. Burton and R. Andrabi, Broadly neutralizing anti-S2 antibodies protect against all three human betacoronaviruses that cause deadly disease, Immunity, 2023, 56, 669–686 Search PubMed.
- K. W. Ng, N. Faulkner, K. Finsterbusch, M. Wu, R. Harvey, S. Hussain, M. Greco, Y. Liu, S. Kjaer, C. Swanton, S. Gandhi, R. Beale, S. J. Gamblin, P. Cherepanov, J. McCauley, R. Daniels, M. Howell, H. Arase, A. Wack, D. L. V. Bauer and G. Kassiotis, SARS-CoV-2 S2–targeted vaccination elicits broadly neutralizing antibodies, Sci. Transl. Med., 2022, 14, eabn3715 CrossRef CAS PubMed.
- S. Xia, Y. Zhu, M. Liu, Q. Lan, W. Xu, Y. Wu, T. Ying, S. Liu, Z. Shi, S. Jiang and L. Lu, Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein, Cell. Mol. Immunol., 2020, 17, 765–767 Search PubMed.
- N. Hingankar, S. Deshpande, P. Das, Z. A. Rizvi, C. K. Wibmer, P. Mashilo, M. Y. Ansari, A. Burns, S. Barman, F. Zhao, S. Mukherjee, J. L. Torres, S. Chattopadhyay, F. Mehdi, J. Sutar, D. K. Rathore, K. Pargai, J. Singh, S. Sonar, K. Jakhar, J. Dandotiya, S. Bhattacharyya, S. Mani, S. Samal, S. Singh, P. Kshetrapal, R. Thiruvengadam, G. Batra, G. Medigeshi, A. B. Ward, S. Bhatnagar, A. Awasthi, D. Sok and J. Bhattacharya, A combination of potently neutralizing monoclonal antibodies isolated from an Indian convalescent donor protects against the SARS-CoV-2 Delta variant, PLoS Pathog., 2022, 18, e1010465 CrossRef CAS PubMed.
- S. Deshpande, M. Y. Ansari, J. Sutar, P. Das, N. Hingankar, S. Mukherjee, P. Jayal, S. Singh, A. Anantharaj, J. Singh, S. Chattopadhyay, S. Raghavan, M. Gosain, S. Chauhan, S. Shrivas, C. Prasad, S. Chauhan, N. Sharma, P. Jana, R. Thiruvengadam, P. Kshetrapal, N. Wadhwa, B. Das, G. Batra, G. Medigeshi, D. Sok, S. Bhatnagar, P. K. Garg and J. Bhattacharya, Ancestral SARS-CoV-2-Driven Antibody Repertoire Diversity in an Unvaccinated Individual Correlates with Expanded Neutralization Breadth, Microbiol. Spectrum, 2023, 11, e0433222 Search PubMed.
- E. N. Lyukmanova, E. B. Pichkur, D. E. Nolde, M. V. Kocharovskaya, V. A. Manuvera, D. A. Shirokov, D. D. Kharlampieva, E. N. Grafskaia, J. I. Svetlova, V. N. Lazarev, A. M. Varizhuk, M. P. Kirpichnikov and Z. O. Shenkarev, Structure and dynamics of the interaction of Delta and Omicron BA.1 SARS-CoV-2 variants with REGN10987 Fab reveal mechanism of antibody action, Commun. Biol., 2024, 7, 1–18 CrossRef PubMed.
- Z. Zhao, J. Zhou, M. Tian, M. Huang, S. Liu, Y. Xie, P. Han, C. Bai, P. Han, A. Zheng, L. Fu, Y. Gao, Q. Peng, Y. Li, Y. Chai, Z. Zhang, X. Zhao, H. Song, J. Qi, Q. Wang, P. Wang and G. F. Gao, Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape, Nat. Commun., 2022, 13, 4958 CrossRef CAS PubMed.
- M. Oda, H. Kozono, H. Morii and T. Azuma, Evidence of allosteric conformational changes in the antibody constant region upon antigen binding, Int. Immunol., 2003, 15, 417–426 CrossRef CAS PubMed.
- M. Galanti, D. Fanelli and F. Piazza, Conformation-controlled binding kinetics of antibodies, Sci. Rep., 2016, 6, 18976 CrossRef CAS PubMed.
- P. C. C. da Silva and L. Martinez, Extended Conformational Selection in the Antigen–Antibody Interaction of the PfAMA1 Protein, J. Phys. Chem. B, 2024, 128, 8400–8408 CrossRef CAS PubMed.
- A. H. Moraes, L. Simonelli, M. Pedotti, F. C. L. Almeida, L. Varani and A. P. Valente, Antibody Binding Modulates Conformational Exchange in Domain III of Dengue Virus E Protein, J. Virol., 2016, 90, 1802–1811 CrossRef CAS PubMed.
- P. Han, L. Li, S. Liu, Q. Wang, D. Zhang, Z. Xu, P. Han, X. Li, Q. Peng, C. Su, B. Huang, D. Li, R. Zhang, M. Tian, L. Fu, Y. Gao, X. Zhao, K. Liu, J. Qi, G. F. Gao and P. Wang, Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2, Cell, 2022, 185, 630–640 CrossRef CAS PubMed.
- C. L. Pierri, SARS-CoV-2 spike protein: flexibility as a new target for fighting infection, Signal Transduction Targeted Ther., 2020, 5, 1–3 CrossRef PubMed.
- R. A. Römer, N. S. Römer and A. K. Wallis, Flexibility and mobility of SARS-CoV-2-related protein structures, Sci. Rep., 2021, 11, 4257 CrossRef PubMed.
- H. M. Dokainish, S. Re, T. Mori, C. Kobayashi, J. Jung and Y. Sugita, The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein, eLife, 2022, 11, e75720 Search PubMed.
- M. Valério, L. Borges-Araújo, M. N. Melo, D. Lousa and C. M. Soares, SARS-CoV-2 variants impact RBD conformational dynamics and ACE2 accessibility, Front. Biomed. Biotechnol., 2022, 4 DOI:10.3389/fmedt.2022.1009451.
- S. Pomplun, Targeting the SARS-CoV-2-spike protein: from antibodies to miniproteins and peptides, RSC Med. Chem., 2021, 12, 197–202 Search PubMed.
- S. Xiaojie, L. Yu, Y. lei, Y. Guang and Q. Min, Neutralizing antibodies targeting SARS-CoV-2 spike protein, Stem Cell Res., 2021, 50, 102125 CrossRef CAS PubMed.
- J. Yang, W. Wang, Z. Chen, S. Lu, F. Yang, Z. Bi, L. Bao, F. Mo, X. Li, Y. Huang, W. Hong, Y. Yang, Y. Zhao, F. Ye, S. Lin, W. Deng, H. Chen, H. Lei, Z. Zhang, M. Luo, H. Gao, Y. Zheng, Y. Gong, X. Jiang, Y. Xu, Q. Lv, D. Li, M. Wang, F. Li, S. Wang, G. Wang, P. Yu, Y. Qu, L. Yang, H. Deng, A. Tong, J. Li, Z. Wang, J. Yang, G. Shen, Z. Zhao, Y. Li, J. Luo, H. Liu, W. Yu, M. Yang, J. Xu, J. Wang, H. Li, H. Wang, D. Kuang, P. Lin, Z. Hu, W. Guo, W. Cheng, Y. He, X. Song, C. Chen, Z. Xue, S. Yao, L. Chen, X. Ma, S. Chen, M. Gou, W. Huang, Y. Wang, C. Fan, Z. Tian, M. Shi, F.-S. Wang, L. Dai, M. Wu, G. Li, G. Wang, Y. Peng, Z. Qian, C. Huang, J. Y.-N. Lau, Z. Yang, Y. Wei, X. Cen, X. Peng, C. Qin, K. Zhang, G. Lu and X. Wei, A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity, Nature, 2020, 586, 572–577 CrossRef CAS PubMed.
- N. Maroli, Riding the Wave: Unveiling the Conformational Waves from RBD of SARS-CoV-2 Spike Protein to ACE2, J. Phys. Chem. B, 2023, 127, 8525–8536 CrossRef CAS PubMed.
- G. M. Verkhivker and L. Di Paola, Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies, J. Phys. Chem. B, 2021, 125, 4596–4619 CrossRef CAS PubMed.
- R. Li, M. Mor, B. Ma, A. E. Clark, J. Alter, M. Werbner, J. C. Lee, S. L. Leibel, A. F. Carlin, M. Dessau, M. Gal-Tanamy, B. A. Croker, Y. Xiang and N. T. Freund, Conformational flexibility in neutralization of SARS-CoV-2 by naturally elicited anti-SARS-CoV-2 antibodies, Commun. Biol., 2022, 5, 1–15 CrossRef PubMed.
- L. M. Pérez, A. d. l. C. R. Taño, L. R. M. Márquez, J. A. G. Pérez, A. V. Garay and R. B. Santana, Conformational characterization of a novel anti-HER2 candidate antibody, PLoS One, 2019, 14, e0215442 CrossRef PubMed.
- L. P. Roguin and L. A. Retegui, Monoclonal Antibodies Inducing Conformational Changes on the Antigen Molecule, Scand. J. Immunol., 2003, 58, 387–394 CrossRef CAS PubMed.
- G. Verkhivker, S. Agajanian, D. Oztas and G. Gupta, Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots, J. Chem. Theory Comput., 2021, 17, 4578–4598 Search PubMed.
- M. A. Díaz-Salinas, Q. Li, M. Ejemel, L. Yurkovetskiy, J. Luban, K. Shen, Y. Wang and J. B. Munro, Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike, eLife, 2022, 11, e75433 Search PubMed.
- C.-L. Hsieh, J. A. Goldsmith, J. M. Schaub, A. M. DiVenere, H.-C. Kuo, K. Javanmardi, K. C. Le, D. Wrapp, A. G. Lee, Y. Liu, C.-W. Chou, P. O. Byrne, C. K. Hjorth, N. V. Johnson, J. Ludes-Meyers, A. W. Nguyen, J. Park, N. Wang, D. Amengor, J. J. Lavinder, G. C. Ippolito, J. A. Maynard, I. J. Finkelstein and J. S. McLellan, Structure-based design of prefusion-stabilized SARS-CoV-2 spikes, Science, 2020, 369, 1501–1505 CrossRef CAS PubMed.
- G. Tang, L. Peng, P. R. Baldwin, D. S. Mann, W. Jiang, I. Rees and S. J. Ludtke, EMAN2: An extensible image processing suite for electron microscopy, J. Struct. Biol., 2007, 157, 38–46 Search PubMed.
- C. F. Reboul, M. Eager, D. Elmlund and H. Elmlund, Single-particle cryo-EM—Improved ab initio 3D reconstruction with SIMPLE/PRIME, Protein Sci., 2018, 27, 51–61 Search PubMed.
- A. Kumar, S. P, S. Gulati and S. Dutta, User-friendly, High-throughput, and Fully Automated Data Acquisition Software for Single-particle Cryo-electron Microscopy, J. Visualized Exp., 2021, 173 DOI:10.3791/62832.
- S. H. W. Scheres, RELION: Implementation of a Bayesian approach to cryo-EM structure determination, J. Struct. Biol., 2012, 180, 519–530 CrossRef CAS PubMed.
- S. Q. Zheng, E. Palovcak, J.-P. Armache, K. A. Verba, Y. Cheng and D. A. Agard, MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy, Nat. Methods, 2017, 14, 331–332 Search PubMed.
- T. Grant, A. Rohou and N. Grigorieff, cisTEM, user-friendly software for single-particle image processing, eLife, 2018, 7, e35383 CrossRef PubMed.
- A. Rohou and N. Grigorieff, CTFFIND4: Fast and accurate defocus estimation from electron micrographs, J. Struct. Biol., 2015, 192, 216–221 CrossRef PubMed.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L.-W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix, Acta Crystallogr., Sect. D:Struct. Biol., 2019, 75, 861–877 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr., Sect. D: Struct. Biol., 2010, 66, 213–221 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera—A visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- R. A. Laskowski, J. Jabłońska, L. Pravda, R. S. Vařeková and J. M. Thornton, PDBsum: Structural summaries of PDB entries, Protein Sci., 2018, 27, 129–134 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, UCSF ChimeraX: Meeting modern challenges in visualization and analysis, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00373c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |