
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevolutionizing silver quantification: a novel ternary surfactant system with 2-nitro-6-(thiazol-2-yl-diazenyl)phenol and Triton X-100 for enhanced spectrophotometric analysis
Muneera Alrasheedia,
Salah M. El-Bahyb,
Refat El-Sayedcd,
Khaled F. Debbabide and
Alaa S. Amin *c
*c
aDepartment of Chemistry, College of Science, Qassim University, Buraidah, 51452, Saudi Arabia
bDepartment of Chemistry, Turabah University College, Taif University, Taif, Saudi Arabia
cChemistry Department, Faculty of Science, Benha University, Benha, Egypt. E-mail: asamin2005@hotmail.com
dDepartment of Chemistry, Univ. College in Al-Jamoum, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
eDepartment of Chemistry, High Institute of Applied Science & Technology of Monastir, Monastir, Tunisia
First published on 7th April 2025
Abstract
Although modern reported methods, such as AAS, ICP-AES, ICP-MS, have good sensitivity, the high cost of equipment, the need for sophisticated instruments, separation and preconcentration steps and experienced technicians along with lack of precise methods make them cumbersome. Solid phase extraction (SPE) emerges as an attractive technique that reduces solvent consumption, minimizes exposure, shortens extraction time, and lowers disposal costs. Herein, a pioneering methodology for the quantification of minute amounts of silver is introduced, using 2-nitro-6-(thiazol-2-yl-diazenyl)phenol (NTDP) as a complexing agent and Triton X-100 as a nonionic surfactant within a ternary surfactant system at a pH of 5.3. This novel extraction strategy demonstrated selective preconcentration. The enriched solution was subjected to spectrophotometric analysis for the quantification of the analyte. After refining extraction and complexation parameters, a remarkable 250-fold increase in the enrichment factor was attained, highlighting a sensitivity boost of 509 times compared with traditional extraction approaches relying solely on nonionic surfactants. The key parameters of molar absorptivity and Sandell sensitivity were determined to be 6.04 × 106 L mol−1 cm−1 and 0.0018 ng cm−2, respectively. The calibration plot was observed from 5.0–175 ng mL−1, whereas Ringbom optimum concentrations ranged from 15–160 ng mL−1. The detection and quantification limits were 1.63 and 4.95 ng mL−1, respectively. The relative standard deviation (RSD) of the complex was 2.27. The suggested method was efficiently utilized for assessing the Ag+ concentration in real samples, producing acceptable outcomes.
Introduction
The presence of heavy metals in water raises substantial health, environmental, and economic apprehensions, thereby emphasizing the central role of their remediation in water conservation initiatives.1 Analyzing trace metals is a crucial process for analytical chemists. To overcome the interference of matrices and accurately determine low levels of trace metal ions in water samples through spectrophotometry, an effective preconcentration step is often necessary. Employing separation and enrichment techniques typically enhances the sensitivity and selectivity of trace metal analysis.2–5Silver, which is a non-essential component in the human body, can be deposited on the skin and mucous membranes following ingestion or prolonged topical use. This accumulation may result in a persistent blue-gray discoloration and, in extreme instances, may lead to sudden death.6 Hence, the determination of silver has become a pivotal process in environmental surveillance and for the prevention of health epidemics.
Moreover, silver holds significance as a valuable precious metal7 owing to its outstanding thermal and electrical conductivities. However, there are concerns related to the interaction of silver with vital nutrients, especially selenium, vitamins E and B12, and copper, emphasizing its potential harm.8 Consequently, precise and accurate measurement of silver in diverse matrices mandate an approach of heightened sensitivity.
Globally, substantial amounts of silver are annually discarded as waste by galvanizing or photographic facilities and via engineering, manufacturing, and medical processes.9 Considering the exceedingly low concentrations of numerous elements in environmental samples (including silver), the precise determination and separation of these elements require the utilization of preconcentration or trace enrichment techniques.10–12
In the contemporary era, various analytical techniques are available for the direct identification of silver in authentic samples, including spectrophotometry,13–17 flame atomic absorption spectrometry (FAAS),18 spectrofluorometry,19,20 inductively coupled plasma atomic emission spectrometry (ICP-AES),21 electrothermal atomic absorption spectrometry (ETAAS),22,23 graphite furnace atomic absorption spectrometry (GFAAS),24,25 and inductively coupled plasma mass spectrometry (ICP-MS).26,27 These different approaches have been created for evaluating silver levels in various environmental samples. However, prior to precisely measuring low concentrations of silver in complex sample analyses, it is crucial to execute separation and preconcentration steps.
Solid-phase extraction (SPE) emerges as an attractive technique that reduces solvent consumption, minimizes exposure, shortens extraction time, and lowers disposal costs.28–37 Cloud point methodology has been successfully utilized for the preconcentration and extraction of metal ions following the formation of sparingly water–soluble complexes. Spectrophotometric determination of various elements, including iron,28 vanadium,29 gold,30 nickel,31 uranium,32 cobalt,33 bismuth,34 boron,35 gallium,36 and palladium,37 has been accomplished following solid-phase extraction utilizing complexing agents.
Various methods encompassing electrochemical and spectrometric techniques have been suggested for the assessment of silver in diverse environmental samples.38–42 Nevertheless, except for spectrophotometry, these methods generally involve higher expenses and greater instrument complexity, restricting their broad application for routine analytical tasks. Directly detecting trace metal ions in specific samples via spectrophotometry proves challenging due to their low sensitivity. Consequently, preconcentration procedures are often necessary. Different techniques have been employed for the enrichment of silver(I) ions and their separation from potential interferences, including liquid–liquid extraction,43 cloud point extraction,44 solid-phase extraction,45–48 and dispersive liquid–liquid microextraction.49–51
Until now, nonionic surfactants have been predominantly utilized in cloud point extraction (CPE), although zwitterionic surfactants and combinations of nonionic and ionic surfactants have also found application.52,53 The occurrence of clouding is ascribed to the effective dehydration of the hydrophilic segment of micelles under elevated temperature conditions. Additionally, there have been indications of several substances causing phase separation in aqueous solutions of bile salts, such as sodium cholate (NaC), even at room temperature.54 Conversely, among cationic surfactants, cetyltrimethyl ammonium bromide (CTAB) undeniably serves as an example of a self-assembled ordered medium with micelles, along with other structures and phases. CTAB has been extensively utilized in analytical chemistry for various purposes.55–58
Our literature survey did not reveal any instances of the application of NTDP as a complexing agent for metal ions in SPE. The current study is mainly focused on the suitability of SPE combined with UV-vis spectrophotometry to determine Ag+ ions. The effects of various experimental factors on the complex formation, enrichment, and extraction processes were thoroughly examined. To assess the feasibility of the developed method, it was applied to the quantification of Ag+ in samples of water, medical radiology waste, blood, food, and urine.
Experimental
Apparatus
To facilitate the separation of phases, a water bath equipped with precise temperature control and a centrifuge with 25 mL calibrated tubes (Superior, Germany) were employed. The analysis was conducted using a Flame Atomic Absorption Spectroscopy (FAAS) instrument (PerkinElmer model Analyst 100, USA). The pH of the solutions was tracked using an Orion Research Model 601 A/digital ion analyzer pH meter. Absorbance spectra were captured using a PerkinElmer Lambda 12 UV/Vis spectrometer, utilizing a 1.0 cm quartz cell.Reagents
Merck (E-Merck, Darmstadt, Germany) provided stock standard solutions containing 1000 mg L−1 of Ag+, formulated by dissolving AgNO3 in 2% (v/v) HNO3. Each day, working standard solutions were concocted through the stepwise dilution of the original stock solution using double distilled water (DDW). The non-ionic surfactant Triton X-100, sourced from Aladdin, underwent dilution to achieve a concentration of 5.0 mg mL−1 before being applied in this study.Diverse solutions covering a pH spectrum from 2.75 to 10.63, including universal, phosphate, acetate, and thiel buffers, were created using the method outlined previously.59 Acetonitrile solvent and potassium iodide salt were sourced from Merck. The NTDP employed in this investigation was synthesized following the method detailed in a prior report.60 A specific quantity was dissolved in 100 mL of absolute ethanol (2.5 × 10−3 M). The resulting solution exhibited stability for a period exceeding one month.
General procedure
A portion of the frigid Ag+ standard solution was transferred into a 100 mL polypropylene tube. Subsequently, 3.0 mL of the 2.5 × 10−3 M NTDP solution and 12.5 mL of acetate buffer solution with a pH of 5.3 were added. Following this, 2.5 mL of a 5.0% Triton X-100 solution and 5.0 mL of a 0.4 M KI solution were introduced. The entire system was then subjected to a thermostatic bath at 40 °C for a duration of 5.0 minutes. The separation of phases was achieved through centrifugation for 5 minutes at 3800 rpm using 25 mL graduated centrifuge tubes. To enhance the viscosity of the surfactant-rich phase, the samples were cooled in an ice bath. Afterward, the concentrated surfactant phase was diluted to 0.4 mL with acetonitrile and transferred into a 5.0 mm quartz cuvette. The absorbance of the final solution was measured at 623 nm and compared to a reference sample that contained no Ag+ ions but underwent the same procedure.Samples treatment
A mixture weighing 10 g underwent a heating process for 3.0 hours in a silicon crucible on a heated plate. The resulting charred material was treated according to our previous research.64 The obtained material was then placed in a furnace and heated overnight at 650 °C. Once cooled, the remaining substance was mixed with 3.0 mL of 30% H2O2 and 10 mL of concentrated HNO3.
This was followed by an additional 2.0 hours furnace treatment at the same temperature to ensure the complete elimination of any remaining traces of organic compounds. The definitive residue encountered treatment with 3.0 mL of concentrated hydrochloric acid and 2.0–4.0 mL of 70% perchloric acid, and heated to vaporize the fumes, ensuring the conversion of all metals into their corresponding ions.65 The compact residue was dissolved in water, sieved, and adjusted to 25 mL in a volumetric flask, maintaining a pH of 5.3 through the introduction of diluted KOH. Blank digestions were also executed. In sequence, the previously delineated preconcentration methodology was implemented.
Assessment of silver by FAAS18
Silver content in the aforementioned samples was quantified using the PerkinElmer Analyst 100 flame atomic absorption spectroscopy (FAAS) instrument (USA).18 Both the samples and standard solutions were required to have a 5.0% (v/v) HNO3 concentration to ensure the solubility of Ag+. To mitigate interferences arising from chromate, bromide, iodide, iodate, chloride, and permanganate precipitates of silver, a 5.0% CH3COOH solution was employed. Conventional atomic absorption parameters were employed for the assessment of silver, employing a wavelength of 328.1 nm and an air-acetylene flame. A calibration chart covering concentrations from 1.0 to 15 mg L−1 of Ag+ was established and employed to ascertain the silver levels in the samples.Interferences study
Varied amounts of ions, conceivably present in the specimens, were added at different ratios of Ag+/interferent (1/1, 1/100, 1/200, 1/500, 1/1000, 1/5000, 1/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 1/50000, 1/100000 and 1/120000) to the experimental solution containing 100 ng mL−1 of Ag+. The detailed methodology described previously was subsequently applied to these experimental solutions. Analyses of interference were carried out in specimens without the inclusion of masking or anticoagulant substances.
000, 1/50000, 1/100000 and 1/120000) to the experimental solution containing 100 ng mL−1 of Ag+. The detailed methodology described previously was subsequently applied to these experimental solutions. Analyses of interference were carried out in specimens without the inclusion of masking or anticoagulant substances.
Accuracy study
A significant portion of each specimen was spiked with progressively higher concentrations of Ag+ (15 and 25 ng mL−1). The target analyte levels were measured utilizing the proposed methodology.Results and discussion
NTDP is frequently used as a chromogenic reagent to identify the presence of Cu2+.60 Silver(I) reacts gradually with NTDP in an aqueous medium. To accelerate this process, the mixture required heating in a water bath for at least 5 minutes. However, it was observed that the resulting complex precipitated slowly after heating, making it impossible to extract with an organic solvent. Additionally, the quantity of precipitate formed during slow cooling was greater than when cooled rapidly in an ice bath. To improve component interaction and maintain the stability of the complex in a micellar solution, the non-ionic surfactant Triton X-100 was introduced. This modification facilitated a strong interaction between Ag+ ions and NTDP, even without the need for heating.In an aqueous medium containing Triton X-100, Ag+ forms a complex with NTDP, displaying its peak absorbance at 608 nm. The addition of iodide ions induces turbidity in the solution, facilitating extraction via the solid phase extraction (SPE) technique. The ternary complex developed in the phase rich in surfactant displays a maximum absorbance at 623 nm [Fig. 1], as opposed to the absorbance of NTPD at 486 nm. Absorbance measurements were taken at 623 nm, with a reagent blank serving as the reference after separation of the surfactant-rich phase.
 | ||
| Fig. 1 Absorption spectra for NTDP and its complex with 10 mg mL−1 Ag+ without SPE and 100 ng mL−1 Ag+ with SPE at optimum conditions. | ||
Optimization of the system
To maximize the effectiveness of the approach, it is crucial to fine-tune and optimize the reaction conditions. Various parameters were explored to attain the most favorable experimental conditions. The optimization process involved maintaining all parameters constant while optimizing one parameter at a time.The impact of NTDP concentration on the preconcentration, determination, and extraction of Ag+ was investigated within the 0.5–15 × 10−5 M range, as illustrated in Fig. 2. The complex that was formed exhibited an increase with rising NTDP concentration up to 7.5 × 10−5 M, followed by a decline at higher concentrations. It was anticipated that an elevation in NTDP concentration would lead to an augmentation in the complex's absorbance. Nevertheless, for concentrations ≥ 9.0 × 10−5 M, a significant rise in the uncomplexed NTDP concentration was observed in the phase rich in surfactant. As a result, the reduction in absorbance change at concentrations ≥ 9.0 × 10−5 M is probably due to increased competition between unbound NTDP and the complexes during extraction into the surfactant-rich phase. The optimal NTDP concentration was identified as 7.5 × 10−5 M.
 | ||
| Fig. 2 Effect of NTDP concentration on complexation with 100 ng mL−1 Ag+ at optimum conditions. | ||
The influence of pH (2.75–10.63) on the generated hue was tested at a consistent concentration of the complex within the surfactant-rich phase. Various buffer solutions (universal, borate, acetate, phosphate, and thiel buffers) were employed. Optimal outcomes were observed with acetate buffers, displaying high conformity and stable results. The absorbance of the Ag+–NTDP-Triton X-100 system at 623 nm within the surfactant-rich phase was assessed in relation to the reagent blank. Within the 5.0–5.6 range, the absorbance remained relatively unchanged. Consequently, pH 5.3 was identified as the most suitable [Fig. 3]. To ascertain the optimal volume, the amount at pH 5.3 was assessed, and the peak absorbance value was achieved with the addition of 11–14 mL. Consequently, for all subsequent examinations, a volume of 12.5 mL of pH 5.3 per 100 mL was utilized.
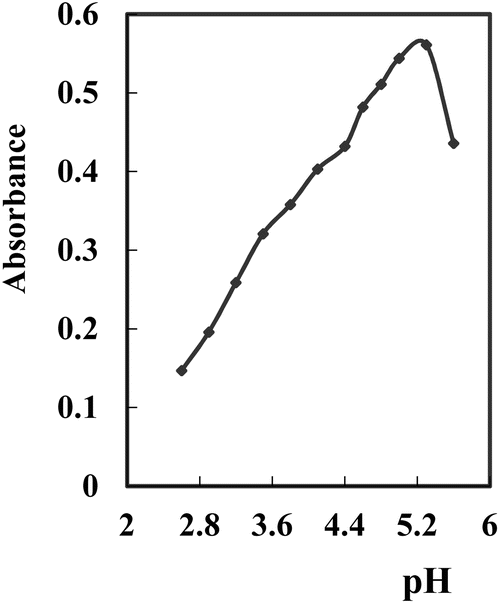 | ||
| Fig. 3 Effect of pH on the SPE of 100 ng mL−1 Ag+ complexed with NTDP at optimum conditions. | ||
The impact of 5.0% Triton X-100 concentration on the complexation of Ag+ was explored within the volume range of 0.5–5.0 mL. Absorbance demonstrated an increase with escalating Triton X-100 concentration, reaching a peak at 2.5 mL of 5.0%, followed by a decline at higher concentrations. Simultaneously, the absorbance of the blank also rose with an increasing concentration of Triton X-100. This phenomenon is attributed to the heightened extraction of NTDP due to the increased Triton X-100 concentration. However, the disparity between the sample and blank (ΔA) showed an escalation up to 2.5 mL of 5.0% Triton X-100 and diminished at higher concentrations (Fig. 4). Hence, 2.5 mL of 5.0% Triton X-100 was considered as the optimal concentration.
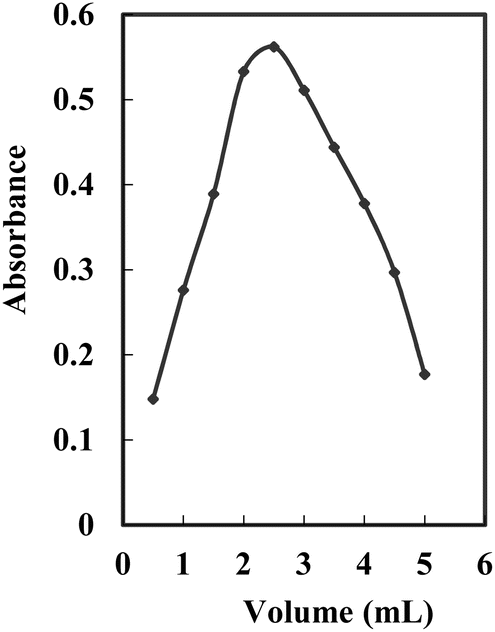 | ||
| Fig. 4 Effect of 5.0% Triton X-100 on the complexation of 100 ng mL−1 Ag+ at optimum conditions. | ||
Conversely, progressively increasing the concentration of Triton X-100 leads to a gradual decline in the absorbance of the tested solution. This effect can be attributed to the expansion of the micellar phase, which in turn causes dilution after the surfactant dissolves in the organic solvent. Therefore, the Triton X-100 concentration was kept constant at 0.1% for all subsequent analyses to ensure consistency in sample preparation.
The introduction of salt has the potential to induce the separation of non-ionic surfactant solutions into immiscible surfactant-rich and surfactant-poor phases. A variety of inorganic salts underwent examination, including KBr, NaCl, KI, KNO3, and NaF, with KI emerging as the optimal choice. Consequently, iodide was incorporated to facilitate the extraction of the complex and stimulate the growth of micelle. The impact of iodide concentration was examined within the 0.005–0.08 M range. The introduction of 5.0 mL of 0.4 M iodide into the prepared 100 mL solution significantly enhanced the efficiency of complex extraction. However, an increase in iodide concentration led to a noticeable decline in absorbance. As a result, a final iodide concentration of 0.02 M within the 100 mL solution was determined to be optimal for subsequent experiments.
Another important factor that affected the complex formation yield was the heating temperature in the water bath. Determining the optimal incubation duration and equilibration temperature played a pivotal role in ensuring the culmination of the reaction and attaining maximum efficiency for seamless phase separation and Ag+ preconcentration. The influence of the temperature of equilibration on Ag+ extraction recovery was explored across the 20–70 °C spectrum. A discernible increase in extraction recovery was noted within the 35–45 °C range, stabilizing up to 50 °C. Consequently, 40 °C was singled out as the temperature for achieving peak absorbance. Subsequently, the experiment proceeded with a fixed equilibration temperature of 40 °C, and the impact of time of incubation on solid phase extraction (SPE) was scrutinized within the 1.0–15 min range. The results demonstrated that a 5.0 min incubation period was adequate for the separation process. Additionally, the time of centrifugation and cooling was also checked to complete the optimization process of the method for Ag+ determination. Centrifugation at 3800 rpm for 5.0 min was established as satisfactory for ensuring a successful SPE.
In relation to sensitivity, different solvents were explored to pinpoint the most appropriate one for achieving optimal outcomes, given the tendency of the surfactant-rich phase to precipitate. Between methanol, acetonitrile, ethanol, acetone, and DMF, acetonitrile produced the most advantageous results because of its elevated sensitivity and limited overlap of spectral elements. As a result, acetonitrile was selected to guarantee a suitable quantity of the specimen for transfer and absorbance measurements, along with an ideal preconcentration factor. A volume of 0.4 mL of acetonitrile was determined to be adequate for dissolving the precipitated ternary complex. Thus, the proposed procedure achieved a preconcentration factor of 250.
The absorbance of acetone-based solutions exhibited a continuous increase, likely due to solvent evaporation caused by its high volatility, leading to a rise in complex concentration within the container. In contrast, monoatomic alcohols caused the gradual degradation of the NTDP-Ag+ complex. Specifically, the ethanol solution became completely colorless after 12 hours, whereas the methanol solution lost its color within 2.0 hours. Meanwhile, the solution prepared in DMF remained stable over time; however, the blank sample also absorbed light at the selected wavelength, which was likely linked to the solvent's basic nature and the conversion of the ligand into its anionic form. Among the tested solvents, acetonitrile exhibited the lowest volatility, ensuring the highest stability of the complex over time. Furthermore, acetonitrile significantly reduced the viscosity of the surfactant-rich phase while maintaining minimal absorbance in the blank solution. Due to these advantages, acetonitrile was chosen for further experimentation.
Stoichiometric ratio
The nature of the compound was determined using the optimal conditions outlined earlier, utilizing both the molar ratio and continuous variation techniques. The graph illustrating absorbance against the molar ratio of NTDP to Ag+, manipulated by altering the NTDP concentration, displayed a turning point at a molar ratio of 1.0, indicating the existence of three NTDP molecules in the resulting complex. Furthermore, the Job technique noted a proportion of NTDP to Ag+ at 1.0. As a result, the results suggested a stoichiometric proportion of (1:1) [NTDP:Ag+]. The computed conditional formation constant (log K) using information from the two previously mentioned approaches via the Harvey and Manning formula was established at 5.54, whereas the actual constant was 5.35.
Regarding the ternary compound with Triton X-100, the results indicated the creation of a 1:1 complex between the [(NTDP)Ag] compound and Triton X-100. Consequently, the findings pointed to a stoichiometric balance of 1:1:1 [(NTDP)Ag][Triton X-100], as illustrated in the following equations. The computed conditional formation constant (logK) utilizing the Harvey and Manning formula with data derived from the two previously mentioned techniques was established at 5.17, whereas the actual constant was 5.05.


Selectivity
The selectivity of the developed approach in distinguishing the target analyte was assessed. To evaluate potential interferences, various ions were introduced in excess into a reference solution containing 100 ng mL−1 of Ag+. The influence of various anion and cation concentrations (alkali metals, transition metals, heavy elements, lanthanides and/or actinides) on the assessment of 100 ng mL−1 Ag+ using the suggested method was investigated. An ion was considered disruptive if it led to a change in absorbance exceeding ± 5.0%. Table 1 outlines the permissible concentrations of foreign ions {various anions and cations (alkali metals, transition metals, heavy elements, lanthanides and/or actinides)} for the analysis of 100 ng mL−1 Ag+. NTDP exhibits the capacity to create stable complexes with diverse ions, encompassing transition metal ions. The majority of the examined anions and cations did not hinder the extraction and quantification of Ag+.| Ion added | Tolerated, mg |
|---|---|
| K+, Na+, tartaric acid, acetate | 12.0 |
| Li+, Al3+, N, P, Cl, S, oxalic acid | 10.0 |
| Ca2+, Mg2+, S, Sr2+, Ba2+, Br− | 9.0 |
| Ce4+, Mn2+, UO22+, W6+ | 7.5 |
| F−, Cr6+, B3+, ClO3− | 6.0 |
| Bi3+, Ti4+, V5+ | 5.0 |
| Cr6+, Mo6+ | 3.8 |
| Cd2+, Tl3+, Sn4+, Pd2+ | 3.2 |
| Ru3+, Pb2+, Hg2+ | 2.75 |
| Os8+, Zr4+ | |
| Cr3+, La3+, Sb3+, Co2+, Ni2+ | 2.25 |
| Se4+, Te4+, Au3+, Sn2+ | 1.5 |
| Rh3+, Ir3+, Th4+, Ru3+ | 0.75 |
| Pt4+, Cl−, Zn2 | 0.20 |
| I−, CN−, SCN− | 0.08 |
| Fe2+, Fe3+, Cu2+ | 0.02 |
The results suggest that increased concentrations of specific common anions and cations do not interfere with the determinations of the analyte, underscoring the satisfactory selectivity of the developed approach. However, Fe2+, Fe3+ and Cu2+ can cause interference in the assessment of Ag+, even at a ratio of 20:1. To enhance the specificity of Ag+ detection in the presence of Fe2+, Fe3+, and Cu2+ ions, the potential use of masking agents—including sodium thiosulfate, potassium thiocyanate, ascorbic acid, o-phenanthroline, and thiourea—was investigated. The results indicated that a 200-fold excess of Fe2+ and Fe3+ did not interfere with Ag+ analysis when o-phenanthroline and thiocyanate were utilized as masking agents. Similarly, a 2.0% ascorbic acid solution effectively masked the presence of Cu2+ at the same concentration ratio. Additionally, EDTA, along with sodium salts of tartrate and thiosulfate, demonstrated the ability to suppress interference from various ions; however, they also contributed to a reduction in solution absorbance.
Analytical characteristics
Table 2 presents the performance parameters of both the traditional extractive spectrophotometric approach for Ag+ detection using NTDP and the solid-phase extraction (SPE) technique incorporating NTDP and Triton X-100. The data revealed that the microextraction method offers significantly improved analytical precision and sensitivity. Moreover, this approach aligns with green chemistry principles, as it eliminates the need for hazardous organic solvents, making it a more environmentally friendly alternative. Table 2 furnishes a thorough summary of the analytical features of the refined approach, incorporating the linear range, regression equation, reproducibility, limit of detection, preconcentration, and improvement factor.| Parameters | After CPP | Before CPP |
|---|---|---|
| a Average of sex determination. | ||
| Amount of acetonitrile | 0.4 | — |
| pH | 5.3 | 5.3 |
| Optimum [CPAHPD] (M) | 7.5 × 10−5 | 7.5 × 10−4 |
| Reaction time (min) | 5.0 | 5.0 |
| Stirring time (min) | 5.0 | — |
| Beer's range (ng mL−1) | 5.0–175 | 500–15000 |
| Ringbom range (ng mL−1) | 15–160 | 800–14400 |
| Molar absorptivity (L mol−1 cm−1) | 6.04 × 106 | 1.19 × 103 |
| Sandell sensitivity (ng cm−2) | 0.0018 | 9.09 |
| Intercept | ||
| Slope | 5.6 | 0.011 |
| Intercept | −0.006 | 0.009 |
| Correlation coefficient (r) | 0.9995 | 0.9980 |
| RSDa (%) | 2.27 | 3.76 |
| Detection limits (ng mL−1) | 1.63 | 175 |
| Quantification limits (ng mL−1) | 4.95 | 515 |
| Preconcentration factor | 250 | — |
| Improvement factor | 509 | — |
The detection limit,66 computed as CL = 3SB/m (where CL, SB, and m denote the limit of detection, standard deviation of the blank, and slope of the calibration graph, respectively), was found to be 1.63 ng mL−1. When analyzing Ag+ in a 100 mL sample solution that undergoes preconcentration into a final volume of 0.4 mL acetonitrile, the concentration is amplified by a factor of 250. The enhancement factor was found to be 509, calculated as the ratio between the slope of the calibration curve obtained through the CPE technique and the slope of the calibration curve in micellar media without preconcentration.
The relative error and RSD were calculated for six repeated analyses of 100 ng mL−1 of Ag+, resulting in values of 2.43% and 2.14%, respectively. Similarly, for 150 ng mL−1 of Ag+, the RSD and relative error were found to be 2.54%, and 2.27%, respectively.
The attributes of the recommended method have been compared with those of alternative approaches. Table 3 and 4 contrast the analytical quality parameters of the proposed method with those previously reported for Ag+ determination. The comparison reveals that the recommended technique exemplifies analytical characteristics on par with previous studies focused on Ag+ determination. As a result, the combination of solid-phase extraction (SPE) with spectrophotometric detection stands out as a straightforward, sensitive, and selective approach for the determination and preconcentration of Ag+.
| Reagent | Characteristicsa | Ref. |
|---|---|---|
| a Remarks: ε/L mol−1 cm−1; linear range μg mL−1. | ||
| 5-[p-Dimethylamino) benzylidene]rhodanine | Absorbance coefficient (ε): 3.5 × 104. Measurable range: 10–40. The reagent cost is high. Addition of poly(vinyl alcohol)-200 is necessary for enhanced ε, and color development takes 15 min. Restricted aqueous phase volume. Interference observed from a few metal ions | 67 |
| 4-(2-Hydroxy-4-substituted-azobenzene)-2-methylquinoline | Measurable range: 2.5–23.0. Restricted aqueous phase volume. Significant interference observed from certain metal ions. Primarily employed in photographic fixing solutions | 63 |
| Dithizone | Absorbance coefficient (ε): 3.45 × 104. Measurable range: 0.1–6.0. Extraction into polyurethane and elution with Me2CO. Restricted aqueous phase volume. Interference observed from certain metal ions. Utilized in glass analysis | 68 |
| Dithizone immobilized in a polymethacrylate matrix | Solid phase spectrophotometric determination of silver; with a detection limit of 0.01 μg L−1. The methodology was employed for the analysis of mineral waters and protargol medication | 69 |
| 5-[4-(2-Methyl-3-hydroxy-5-hydroxy-methyl)pyridylene] rhodanine | ε: 1.5 × 104. Linear range: 0.25–4.0. The reagent is costly. There is a constraint on the volume of the aqueous phase, and interference is observed from Au3+, Hg2+, I−, Pd2+, Br−, and S2O32−. The determination of silver was carried out in drug and ore samples | 70 |
| 2-Carboxybenzalaldehyde thiosemicarbazoneoctylmethyl-ammonium chloride | Linear range: 10–70. The method is time-consuming, and there is interference from common metal ions. It was applied to ore samples | 71 |
| 4,7-Dimethyl-2-thiol-2-thion-1,3,2-dioxophosphorinan (DOPh111) | Linear range: 1.0–18.0. The method includes replacing Cu2+ from Cu(DOPh111)2 with silver and measuring the decrease in the absorbance of the Cu(DOPh111)2 toluene solution. SCN−, F−, S2O32−, and Hg2+ seriously interfered. It was applied to some standard samples | 72 |
| 2-Nitroso-1-naphthol -4-sulfonic acid | ε: 6.47 × 103. Dynamic range: 0.2–30. Preconcentration factor: 80, sensitivity: 0.198 (d4A dλ4)/μg mL−1; detection limit: 0.15 μg mL−1. The method exhibits high selectivity for Ag+, and the utilization of derivative spectrophotometry significantly enhances both sensitivity and selectivity. Applied successfully to standard alloys and biological samples | 73 |
| 2-(8-Hydroxyquinolin -5-ylazo)benzoic acid | ε: 3.65 × 104. Dynamic range: 0.05–0.65. The reagent is costly. Separation of various metal ions as hydroxide is necessary due to interference. Applied to specific geological samples | 74 |
| Bromopyrogallol red cetylpyridinium chloride | ε: 3.2 × 103. Linear range: 2.15–8.6. Limited aqueous volume, a few metal ions interfered. Applied to silver amalgam and dental prosthesis | 75 |
| NTDP | ε: 6.04 × 106. Linear range: 5.0–175 ng mL−1. Limited aqueous volume, a few metal ions interfered. Applied to silver in environmental samples | This work |
| Reagent | Medium/solvent | Interfering ions | λmax (nm) | ε (×104) L mol−1 cm−1 | Linear range (μg mL−1) | Ref. |
|---|---|---|---|---|---|---|
| Dithizonate | Extraction with Chloroform | Hg2+, Cd2+, Pb2+, CN−, I− | 565 | 5.5 | 0–1.0 | 75 |
| 2-(2-Quinolylazo)-5-diethylaminophenol | Aqueous (SDS), pH = 5.0 | I−, CN−, Pt4+, Br− | 590 | 13.3 | 0.01–0.6 | 76 |
| 2-Carboxybenzaldehyde thiosemicarbazone | Extraction with toluene | Hg2+, Cu2+, CN−, I− | 350 | 1.7 | 0.1–0.7 | 71 |
| 2-(3,5-Dibromo-2-pyridylazo)-5-diethylaminophenol | Aqueous (pH = 5), SDS | Zn2+, Co2+, Fe3+, Cu2+, Pb2+, Hg2+ | 565 | 6.4 | 0.02–0.48 | 77 |
| 1,10-Phenanthroline, tetrabromophenolphthalein ethyl ester | Extraction with 1,2-dichloroethane | I−, Sn4+, Hg2+, Co2+, Ni2+, Fe2+, Zn2+,Cd2+ | 610 | 36.5 | 0.0004–0.032 | 78 |
| o-Carboxylbenzene diazoaminoazobenzene | Aqueous (pH = 11), OP | Fe3+, Cd2+, Ni2+ Cu2+, Zn2+ | 540 | 8.2 | 0–0.48 | 79 |
| Sulfochlorophenolazothio-rhodanine | Aqueous pH = 2.8, Triton X-100 | Hg2+, Pd2+, Pt4+, Au3+ | 540 | 6.3 | 0–0.8 | 80 |
| Thio-Michler's ketone | Aqueous (pH = 5), SDS | Au3+, Pd2+, Hg2+, Pt4+, Ir2+ | 535 | 9.4 | 0–0.4 | 81 |
| Meloxicam | Aqueous pH 4.6 and Triton X-100 | Fe3+, Cd2+, Ni2+, Cu2+, Zn2+ | 412 | 1.1 | 1.0–15.0 | 82 |
| 2-(2-Quinolylazo)-5-diethylaminoaniline | Aqueous (pH = 6.5), SDS | 580 | 13.9 | 0.01–0.6 | 83 | |
| NTDP-SPE | Aqueous pH = 5.3, Triton X-100 | Fe3+, Cu2+ | 623 | 604 | 0.005–0.175 | This work |
Analytical applications
In an effort to showcase the effectiveness of the recommended system, a series of samples were subjected to analysis, including various natural water samples. The system operated under the refined parameters specified in Table 2. The results of the sample analysis are presented in Table 5. Accuracy was evaluated by comparing the outcomes with those obtained using FAAS. The paired F-value and t-test84 were applied, revealing no significant difference at a 95% confidence level.| Sample | Added ng mL−1 | Founda (ng mL−1) | Recovery (%) | t-test | F-value | |
|---|---|---|---|---|---|---|
| Proposed | FAAS | |||||
| a Mean of five extractions.b From a rinse water of photography.c From Sewa mineral water.d Not detected.e Collected at Zagazig city, Egypt (Dec. 2024).f From drinking water system of Beha, Egypt.g From Benha river water (Nile river).h Mediterranean sea water. | ||||||
| Waste waterb | — | 63.0 ± 1.8 | 62.5 ± 1.6 | — | 1.32 | 2.71 |
| 10 | 72.4 ± 1.6 | 72.9 ± 1.7 | 99.18 | 1.19 | 2.39 | |
| 20 | 84.1 ± 1.4 | 83.3 ± 1.8 | 101.33 | 1.06 | 2.25 | |
| Mineral waterc | — | NDd | — | |||
| 50 | 49.3 ± 0.24 | 65.7 ± 0.9 | 98.60 | 1.82 | 3.33 | |
| 100 | 100.8 ± 0.47 | 98.7 ± 0.8 | 100.80 | 1.17 | 2.36 | |
| Rain watere | — | ND | — | |||
| 60 | 59.3 ± 0.22 | 60.8 ± 0.7 | 98.83 | 1.37 | 2.98 | |
| 120 | 121.7 ± 0.45 | 118.7 ± 1.0 | 101.42 | 1.25 | 2.81 | |
| Tap waterf | — | ND | — | |||
| 75 | 76.2 ± 0.21 | 73.9 ± 0.9 | 101.60 | 1.87 | 3.26 | |
| 150 | 149.1 ± 0.43 | 151.6 ± 1.0 | 99.40 | 1.77 | 3.06 | |
| River waterg | — | ND | — | |||
| 40 | 40.8 ± 0.23 | 39.0 ± 0.8 | 102.00 | 1.31 | 2.65 | |
| 80 | 78.8 ± 0.45 | 81.5 ± 0.9 | 98.50 | 1.13 | 2.39 | |
| Sea waterh | — | ND | — | |||
| 70 | 69.2 ± 0.26 | 71.5 ± 1.1 | 98.86 | 0.96 | 2.18 | |
| 140 | 138.8 ± 0.48 | 141.7 ± 1.3 | 99.14 | 1.11 | 2.34 | |
To gauge the reliability of the proposed technique, the procedure was employed to quantify trace amounts of Ag+ in various biological and food samples (Table 6). To confirm the precision of the established protocol, recovery experiments were conducted by adding different quantities of silver ions to the samples prior to any pre-processing. The outcomes are presented in Table 6, indicating recoveries ranging between 98.0% and 102.0% (±2.0% due to the interference of some diverse ions), affirming the precision of the suggested approach. The method was successfully employed for the determination of Ag+ ions in samples of radiological film and sulphadiazine cream (Table 7). The analysis of Ag+ in both samples was carried out using the cloud point precipitation with spectrophotometric analysis employing the standard additions method, as detailed in Table 3. The results were in good agreement with the data obtained from flame atomic absorption spectrometry (FAAS).
| Sample | Added (ng mL−1) | Founda (μg mL−1) | Recovery (%) | |
|---|---|---|---|---|
| Proposed | FAAS | |||
| a Mean ± SD (n = 6). | ||||
| Human blood (ng mL−1) | — | 0.00 | 0.00 | — |
| 10 | 10.2 ± 0.20 | 9.7 ± 0.56 | 102.00 | |
| 20 | 19.7 ± 0.15 | 21.0 ± 0.43 | 98.50 | |
| 30 | 29.6 ± 0.33 | 31.2 ± 0.68 | 98.67 | |
| Human urine (ng mL−1) | — | 0.00 | 0.161 | — |
| 20 | 19.6 ± 0.26 | 19.3 ± 0.71 | 98.00 | |
| 40 | 40.2 ± 0.55 | 39.2 ± 0.39 | 100.50 | |
| 60 | 59.4 ± 0.19 | 61.1 ± 0.46 | 99.00 | |
| Green tea sample (ng g−1) | — | 0.00 | 0.00 | — |
| 25 | 25.1 ± 0.32 | 24.8 ± 0.55 | 100.40 | |
| 50 | 49.7 ± 0.41 | 51.6 ± 0.67 | 99.40 | |
| 75 | 76.2 ± 0.28 | 74.3 ± 0.74 | 101.60 | |
| Rice (ng g−1) | — | — | ||
| 40 | 40.6 ± 0.51 | 41.0 ± 0.63 | 101.50 | |
| 80 | 79.6 ± 0.72 | 81.6 ± 0.63 | 99.50 | |
| 120 | 119.0 ± 0.31 | 122.2 ± 0.63 | 99.17 | |
| Lentils (ng g−1) | — | — | ||
| 30 | 29.7 ± 0.32 | 30.7 ± 0.63 | 99.00 | |
| 60 | 59.1 ± 0.47 | 61.9 ± 0.63 | 98.50 | |
| 90 | 91.4 ± 0.52 | 88.8 ± 0.63 | 101.55 | |
| Wheat (ng g−1) | — | — | ||
| 50 | 50.7 ± 0.55 | 49.1 ± 0.63 | 101.40 | |
| 100 | 101.2 ± 0.62 | 99.0 ± 0.63 | 101.20 | |
| 150 | 148.3 ± 0.76 | 152.2 ± 0.63 | 98.87 | |
| Spices (ng g−1) | — | — | ||
| 45 | 44.7 ± 0.37 | 45.8 ± 0.63 | 99.33 | |
| 90 | 91.0 ± 0.51 | 89.0 ± 0.63 | 101.11 | |
| 135 | 133.8 ± 0.82 | 137.5 ± 0.63 | 99.11 | |
| Corn (ng g−1) | — | — | ||
| 55 | 55.9 ± 0.56 | 54.0 ± 0.63 | 101.64 | |
| 110 | 111.5 ± 0.63 | 108.7 ± 0.63 | 101.36 | |
| 165 | 163.7 ± 0.87 | 167.5 ± 0.63 | 99.21 | |
| Sample | Added ng mL−1 | Founda (ng mL−1) | Recovery% | t-Testb | F-Valueb | |
|---|---|---|---|---|---|---|
| Proposed | FAAS | |||||
| a Results average of six consecutive measurements.b Theoretical values for t and F at 95% confidence limit are 2.57 and 5.05, respectively. | ||||||
| Silver sulphadiazine | 0.00 | 7.3 ± 0.35 | 7.6 ± 0.60 | — | 2.82 | 1.57 |
| 15.0 | 22.5 ± 0.25 | 22.2 ± 0.75 | 100.90 | 3.46 | 1.81 | |
| 30.0 | 37.1 ± 0.40 | 38.0 ± 0.95 | 99.46 | 2.94 | 1.63 | |
| 40.0 | 47.7 ± 0.50 | 47.3 ± 0.95 | 100.85 | 2.54 | 1.37 | |
| Radiological film | 0.00 | 12.5 ± 0.55 | 12.7 ± 0.95 | — | 2.89 | 1.61 |
| 10.0 | 22.4 ± 0.35 | 23.0 ± 0.80 | 99.56 | 2.47 | 1.38 | |
| 20.0 | 32.6 ± 0.20 | 33.0 ± 0.76 | 100.31 | 3.26 | 1.71 | |
| 30.0 | 42.7 ± 0.25 | 42.6 ± 0.85 | 100.47 | 3.71 | 1.85 | |
| Photographic plate | 0.00 | 45.5 ± 0.60 | 45.3 ± 1.30 | — | 2.57 | 1.41 |
| 20.0 | 66.4 ± 0.45 | 64.6 ± 1.10 | 101.37 | 3.62 | 1.82 | |
| 40.0 | 84.6 ± 0.30 | 87.1 ± 0.80 | 98.95 | 2.67 | 1.48 | |
| 60.0 | 106.8 ± 0.35 | 104.2 ± 0.95 | 101.23 | 3.51 | 1.78 | |
Conclusion
The suggested approach offers a straightforward, highly sensitive, and cost-effective colorimetric method for Ag+ ion determination applicable to real samples. The utilization of surfactant for Ag+ preconcentration in water circumvents the need for toxic solvent extraction. Comparative analysis with previously reported methods employing various spectrophotometric techniques reveals that this method exhibits superior sensitivity, making it a safe, convenient, rapid, simple, and economical option for tracing Ag+ quantities in real samples.Ethical statement
All experiments were performed in accordance with the Guidelines of “Laboratory Animal Ethics Committee of Egypt”, and experiments were approved by the ethics committee at “BUFSC 201254” university. Informed consents were obtained from human participants of this study.Data availability
The authors declare that the data supporting the findings of this study are available within the article.Author contributions
Muneera Alrasheedi and Salah El-Bahy: conceptualization, investigation, data curation, methodology, visualization, validation, writing – original draft, writing– review & editing. Refat El-Sayed and Khaled Debbabi: conceptualization, data curation, investigation, methodology, validation, writing–original draft, writing–review & editing. Alaa Amin: conceptualization, methodology, data curation, investigation, supervision, validation, writing – original draft, writing – review & editing.Conflicts of interest
There is no conflict of interest.Acknowledgements
The authors extend their appreciation to Taif University, Saudi Arabia for supporting this work through project number TU-DSPP-2024-20.References
- G. I. Edo, P. O. Samuel, G. O. Oloni, G. O. Ezekiel, V. O. Ikpekoro, P. Obasohan, J. Ongulu, C. F. Otunuya, A. R. Opiti, R. S. Ajakaye, A. E. A. Essaghah and J. J. Agbo, Chem. Ecol., 2024, 40, 322–349 CrossRef CAS.
- A. Duran, M. Tuzen and M. Soylak, J. Hazard. Mater., 2009, 169, 466–471 CrossRef CAS PubMed.
- M. Miro and E. H. Hansen, Anal. Chim. Acta, 2013, 782, 1–11 CrossRef CAS PubMed.
- M. Tuzen, K. O. Saygi and M. Soylak, J. Hazard. Mater., 2008, 159, 632–639 CrossRef PubMed.
- L. El Hosry, N. Sok, R. Richa, L. Al Mashtoub, P. Cayot and E. Bou-Maroun, Foods, 2023, 12, 895 CrossRef CAS PubMed.
- H. Renner, Ulmanns Encyklopedie Der Technischen-Chemie, Verlag Chemie, Weinheim, vol. 21. 1982 Search PubMed.
- H. Sverdrup, D. Koca and K. V. Ragnarsdottir, Resour. Conserv. Recycl., 2014, 83, 121–140 CrossRef.
- C. S. F. Gomes and E. A. F. Silva, Health benefits and risks of minerals: bioavailability, bio-essentiality, toxicity, and pathologies. Minerals Latu Sensu And Human Health: Benefits, Springer, 2021 Search PubMed.
- Z. Hubicki and H. Hubicka, Hydrometallurgy, 1995, 37, 207–219 Search PubMed.
- M. N. Oviedo, C. E. Luján, A. A. Lemos, M. B. Botella, M. Llaver and R. G. Wuilloud, Anal. Bioanal. Chem., 2024, 416, 2641–2656 Search PubMed.
- C. C. Huang and M. H. Yang, Anal. Chem., 1997, 69, 3930–3939 CrossRef CAS PubMed.
- Q. Yang, Y. Zhao, H. Yu, X. Xiong and K. Huang, Appl. Spectrosc. Rev., 2024, 59, 652–677 CrossRef CAS.
- X. Wen, L. Kong, M. Chen, Q. Deng, X. Zhao and J. Guo, Spectrochim. Acta, Part A, 2012, 97, 782–787 Search PubMed.
- Q. Hu, Y. Guangyu, Z. Huang and J. Yin, Talanta, 2002, 58, 467–473 CrossRef CAS PubMed.
- V. V. Divarova, D. D. Kiradzhiyska and K. B. Gavazov, J. Chem. Soc. Pak., 2023, 45, 501–507 CAS.
- X. J. Guo, Q. L. Deng, B. Peng, J. Z. Gao and J. W. Kang, Chin. J. Chem., 2002, 20, 39–44 CrossRef CAS.
- M. A. Kassem, Anal. Methods, 2015, 7, 6747–6754 CAS.
- X. Yang, Z. Jia, X. Yang, G. Li and X. Liao, Saudi J. Biol. Sci., 2017, 24, 589–594 CAS.
- L. D. Phuc, V. H. Thien and N. V. Dong, Vietnam J. Chem., 2023, 61, 109–117 CrossRef.
- V. N. Losev, S. I. Metelitsa, S. L. Didukh, A. I. Kashkevich, A. K. Trofimchuk and E. A. Siryk, J. Anal. Chem., 2018, 73, 50–57 CrossRef CAS.
- A. A. Argekar, M. J. Kulkarni, J. N. Mathur, A. Page and G. R. H. Iyer, Talanta, 1995, 42, 1937–1942 CAS.
- J. Medved, P. Matus, M. Bujdos and J. Kubova, Chem. Pap., 2006, 60, 27–31 CAS.
- I. Lopez-García, M. J. Munoz-Sandoval and M. Hernandez-Cordoba, Spectrochim. Acta, Part B, 2023, 202, 106643 Search PubMed.
- J. A. López-López, B. Herce-Sesa and C. Moreno, Talanta, 2016, 159, 117–121 CrossRef PubMed.
- P. Liang and L. Peng, Microchim. Acta, 2010, 168, 45–50 CAS.
- M. Zhang, H. Wang, Y. Wu and X. Yu, Anal. Chim. Acta, 2023, 1279, 341846 CAS.
- J. Zhao, X. Wang, B. Gao, X. Xia and Y. Li, J. Hazard. Mater., 2024, 466, 1336 Search PubMed.
- A. S. Amin and A. A. Gouda, Talanta, 2008, 76, 1241–1245 CAS.
- A. S. Amin, A. L. Saber and T. Y. Mohammed, Spectrochim. Acta, Part A, 2009, 73, 195–200 CrossRef PubMed.
- A. S. Amin, Spectrochim. Acta, Part A, 2010, 77, 1054–1058 CrossRef PubMed.
- A. S. Amin and A. S. AL-Attas, J. Saudi Chem. Soc., 2012, 16, 451–459 CrossRef.
- A. S. Amin, Arabian J. Chem., 2014, 7, 715–721 CrossRef CAS.
- A. S. Amin, RSC Adv., 2015, 5, 66975–66980 RSC.
- A. S. Amin and S. M. N. Moalla, RSC Adv., 2016, 6, 1938–1944 RSC.
- Y. Bazel, A. Tupys, Y. Ostapiuk, O. Tymoshuk and V. Matiychuk, J. Mol. Liq., 2017, 242, 471–477 CrossRef CAS.
- R. Kurrey, M. K. Deb and K. Shrivas, New J. Chem., 2019, 43, 8109–8121 RSC.
- Y.-S. Lu, W.-Y. Pan, T.-C. Hung and Y.-T. Hsieh, Langmuir, 2020, 36(38), 11358–11365 CrossRef CAS PubMed.
- M. R. H. Nezhad, M. A. Karimi and F. Shahheydari, Sci. Iran., Trans. F, 2010, 17, 148–153 CAS.
- Y. Luo, W. Xiang, X. Zhang, L. Hu and Y. Dong, New J. Chem., 2022, 46, 5026–5033 RSC.
- M. Javanbakht, F. Divsar, A. Badiei, F. Fatollahi, Y. Khaniani, M. R. Ganjali, P. Norouzi, M. Chaloosi and G. M. Ziarani, Electrochim. Acta, 2009, 54, 5381–5386 CrossRef CAS.
- M. A. Hussein, K. A. Alamry, Q. A. Alsulami, E. A. Elshehy and W. A. El-Said, Spectrochim. Acta, Part A, 2022, 272, 120938 Search PubMed.
- E. A. Azooza, F. A. Wannas, R. K. Ridha, S. K. Jawad and E. A. J. Al-Mulla, Anal. Bioanal. Chem. Res., 2022, 9, 133–140 Search PubMed.
- M. S. El-Shahawi, A. S. Bashammakh and S. O. Bahaffi, Talanta, 2007, 72, 1494–1499 CrossRef CAS PubMed.
- A. Shokrollahi, M. Ghaedi, O. Hossaini, N. Khanjari and M. Soylak, J. Hazard. Mater., 2008, 160, 435–440 CrossRef CAS PubMed.
- A. A. Hill, R. J. Lipert and M. D. Porter, Talanta, 2010, 80, 1606–1610 CrossRef CAS PubMed.
- M. R. Fathi, N. Pourreza and S. Purweis, J. Chin. Chem. Soc., 2009, 56, 725–728 CrossRef CAS.
- M. K. Rofouei, M. Payehghadr, M. Shamsipur and A. Ahmadalinezhad, J. Hazard. Mater., 2009, 168, 1184–1187 CrossRef CAS PubMed.
- G. Azimi, J. Zolgharnein, M. R. Sangi and S. Ebrahimi, Anal. Sci., 2009, 25, 711–716 CrossRef CAS.
- P. Liang and L. Peng, Microchim. Acta, 2010, 168, 45–50 CrossRef CAS.
- S. Jafarvand, A. Bidari, P. Hemmatkhah, M. R. Milani Hosseini and Y. Assadi, Anal. Lett., 2009, 42, 2214–2231 CrossRef CAS.
- S. Z. Mohammadi, D. Afzali, M. A. Taher and Y. M. Baghelani, Talanta, 2009, 80, 875–879 CrossRef CAS PubMed.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Anal. Bioanal. Chem., 2012, 404, 1539–1548 CrossRef CAS PubMed.
- N. Pourreza, S. Rastegarzadeh and A. Larki, Food Chem., 2011, 126, 1465–1469 CrossRef CAS.
- M. Acosta, M. Talio, M. Luconi, W. Hinze and L. Fernández, Talanta, 2014, 129, 516–522 CrossRef PubMed.
- H. L. Su, M. T. Lan and Y. Z. Hsieh, J. Chromatogr. A, 2009, 1216, 5313–5319 CrossRef CAS PubMed.
- Y. Xia, X. Zhi, X. Wang, M. Chen and J. Cheng, Anal. Bioanal. Chem., 2012, 402, 1241–1247 CrossRef CAS PubMed.
- E. Feitosa, K. T. Catelam, F. A. Hasmann, H. O. Johansson, I. C. Roberto and A. Pessoa Jr, J. Chromatogr. B:Biomed. Sci. Appl., 2008, 862, 58–63 CrossRef CAS PubMed.
- M. C. Talio, M. Kaplan, M. Acosta, R. A. Gil, M. O. Luconi and L. P. Fernández, Microchem. J., 2015, 23, 237–242 CrossRef.
- H. T. S. Britton, Hydrogen Ions, Chapman and Hall, 4th edn, London, pp. 1168, 1952 Search PubMed.
- A. S. Amin, R. El-Sheikh and M. I. Shaltout, Can. Chem. Trans., 2014, 2, 296–305 Search PubMed.
- H. H. El-Feky, A. M. Askar and A. S. Amin, RSC Adv., 2021, 11, 35300–35310 RSC.
- A. Abbaspour, A. Izadyar and H. Sharghi, Anal. Chim. Acta, 2004, 525, 91–96 CrossRef CAS.
- E. H. El-Mossalamy and A. S. Amin, Monatsh. Chem., 1997, 128, 23–26 CrossRef CAS.
- M. A. Ali, A. O. Babalghith, A. A. Gouda and A. S. Amin, Int. J. Environ. Anal. Chem., 2024, 1–22 Search PubMed.
- M. Ghaedi, A. Shokrollahi and F. Ahmadi, J. Hazard. Mater., 2007, 142, 272–278 CrossRef CAS PubMed.
- IUPAC, Spectrochim. Acta, Part B, 1978, 33, 241–245 CrossRef.
- Z. Nan, Z. Min, H. Chun-xiang, J. Zhao-qiang and L. Li-fen, Talanta, 1990, 37, 531–533 CrossRef CAS PubMed.
- S. K. Roy and D. Kundu, Anal. Lett., 1991, 24, 139–153 CrossRef CAS.
- N. A. Gavrilenko and N. V. Saranchina, J. Anal. Chem., 2010, 65, 148–152 CrossRef CAS.
- R. E. Godoy and A. G. Perez, Analyst, 1986, 111, 1297–1299 RSC.
- F. Salinas, A. Espinosa-Mansila and P. L. Lopez-de-Alba, Analyst, 1995, 120, 2857–2860 RSC.
- D. Atanassova and A. N. Shishkov, Talanta, 1990, 37, 527–529 CrossRef CAS PubMed.
- M. A. Taher, B. K. Puri and A. K. Malik, Croat. Chem. Acta, 2002, 75, 25–39 CAS.
- N. R. Saran, V. Umashanker and G. V. Ramanaiah, Bull. Chem. Soc. Jpn., 1992, 65, 2291–2293 CrossRef CAS.
- M. Benamor, K. Lahbiben and Z. Hanouti, Proc. 5th Inter. Symp. Met. Ions Biol. Med. 1999, 2, 28–32, Chem. Abstr., 1998, 130, 331974h Search PubMed.
- G. Yang, Q. Hu, J. Yang and J. Yin, Anal. Bioanal. Chem., 2002, 374, 1325–1329 CrossRef CAS PubMed.
- L. H. Chai and X. J. Zhang, Acta Fudan Univ., 1992, 31, 115–120 Search PubMed.
- T. Koh and T. Sugimoto, Anal. Chim. Acta, 1996, 333, 167–173 CrossRef CAS.
- Z. X. Ge, L. Yang and S. Y. Zhang, Lihua Jianyan, Huaxue Fence, 1995, 31, 274–276 Search PubMed.
- Y. L. Yang, G. Y. Yang, J. Y. Yin and Q. H. Xu, Lihua Jianyan, Huaxue Fence, 2000, 36, 157–158 CAS.
- G. Xue, Huangjin, 1996, 17, 43–44 Search PubMed.
- R. K. Shah, Orient. J. Chem., 2016, 32, 499–507 CrossRef CAS.
- Q. Hua, Y. Guangyu, Z. Huang and J. Yin, Talanta, 2002, 58, 467–473 CrossRef PubMed.
- J. N. Miller and J. C. Miller, Statistics And Chemometrics For Analytical Chemistry, Prentice-Hall, 5th edn, London, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |