
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic approaches for novel 3-heteroaryl-4-hydroxy-1-methylquinoline-2(1H)one: spectroscopic characterization, molecular docking and DFT investigations†
Mai A. Mostafa ,
Magdy A. Ibrahim,
S. S. Ibrahim,
Nada Mohamed and
Al-Shimaa Badran*
,
Magdy A. Ibrahim,
S. S. Ibrahim,
Nada Mohamed and
Al-Shimaa Badran*
Department of Chemistry, Faculty of Education, Ain Shams University, Roxy, Cairo, 11711, Egypt. E-mail: badran.shimaa@yahoo.com; Fax: +2 022581243; Tel: +2 01011444940
First published on 28th February 2025
Abstract
Ring opening and recyclization reactions with 4-hydroxy-6-methyl-3-nitro-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (1) was examined towards some carbon nucleophilic reagents. Treatment of key precursor 1 with cyanoacetamide, malononitrile dimer and 1H-benzimidazol-2-ylacetonitrile afforded pyridines 2, 3 and pyrido[1,2-a]benzimidazole 4, respectively. Reaction of compound 1 with 5-amino-2,4-dihydro-3H-pyrazol-3-one and 5-amino-3-methyl-1H-pyrazole furnished pyrazolo[3,4-b]pyridine derivatives 5 and 6. Further, pyrido[2,3-d]pyrimidines 7–9 were synthesized from recyclization of compound 1 with some 6-aminouracil derivatives. Compounds 2–4 demonstrated favorable efficacy in HepG-2 liver cancer cells comparable to reference drug (cisplatin), exhibiting IC50 values of 8.79–17.78 μM L−1. The optimized geometrical configurations of the synthesized compounds were determined, using DFT calculations conducted at the B3LYP/6-311++G(d,p) level, for computing various parameters including molecular electrostatic potential (MEP), global reactivity indices, frontier molecular orbital (FMO) analysis, and nonlinear optical (NLO) characteristics. The obtained results revealed that, compound 3 exhibits the smallest energy gap (ΔE = 2.783 eV), while compound 9 presents the largest energy gap (ΔE = 3.995 eV). Also, compound 9 with a hardness value (η) of 1.998 eV demonstrated greater hardness and stability compared to other compounds, whereas compound 3 with a softness value (S) of 0.719 eV−1, is comparatively softer and more reactive. The experimental infrared (IR) and nuclear magnetic resonance (NMR) spectra of the current compounds were compared with the simulated spectra derived from DFT computations, revealing a good agreement. SwissADME analyses suggested that all prepared compounds adhere to the Lipinski, Ghose, and Veber principles fitting to drug likeness. Utilizing the topoisomerase IIβ protein (PDB ID: 4G0U) as a receptor, molecular docking analyses were conducted to investigate the binding interactions of the synthesized compounds and correlated with their anticancer efficacy. Further, MEP surfaces of the studied compounds were analyzed to ascertain the reactive sites appropriated to electrophilic and nucleophilic attacks. The theoretical investigation suggests that the synthesized compounds possess potential applicability for future NLO applications.
1. Introduction
Nitrogen-containing heterocyclic compounds, particularly quinolines, are of significant interest in medicinal and pharmaceutical chemistry due to their diverse biological activities.1–3 Basic building component of many natural alkaloids is the quinoline ring structure and widely employed as bioactive scaffolds.4,5 Additionally, they are commonly utilized as colorants and dyes in a wide range of commercial industrial field.6 The majority of bioactive quinolines, both synthetic and natural, are crucial for the creation of novel pharmacological drugs such as insecticidal,7 anthelmintic,8 antimicrobial,9 anti-inflammatory,10 anti-viral,11 antioxidant,3 antimalarial,12 antihypertensive,13 antitumor,14 antiproliferative,15 and antibiotic agents.16 Luminescent, fluorescent, thin films, optoelectronic, solvatochromic, optical properties, photodiode photovoltaic fabrication as well as corrosion inhibitors and other applications were recently examined for some quinolines.17–19 Pyrano[3,2-c]quinoline derivatives are active substrates due to the availability of α-pyrone moiety and were employed as flexible intermediates for building a range of both heteroannulated pyrano[3,2-c]quinolines and 3-heteroarylquinolines; through reactions with a variety of carbon and nitrogen nucleophiles.20–23 Density Functional Theory (DFT) is one of modeling techniques that is notable for its capacity to precisely compute the physicochemical characteristics of molecules at a cheap computing cost.24,25 DFT technique is employed to optimize geometrical configurations of organic compounds and identify nucleophilic and electrophilic sites.26 This method makes it easier to fully comprehend how the molecule behaves as an electrophile or nucleophile.27,28 DFT is utilized to compute Frontier Molecular Orbitals (FMO), which characterize the electronic characteristic and provide quantum reactivity descriptors. Further, it enables the simulation of infrared red (IR) and NMR spectra.29In this study, various heterocyclic compounds bearing quinoline moiety were synthesized from 4-hydroxy-6-methyl-3-nitro-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (1) with some carbon nucleophilic reagents. The current compounds were optimized using the DFT/B3LYP/6-311++(d,p) basis set and ascertain their global reactivity descriptors. To investigate the structural properties of these molecules, infrared spectroscopy (IR) and nuclear magnetic resonance (NMR) spectroscopy were computed and compared with experimental results. Bioavailability of synthesized molecules was investigated by ADME analysis and evaluated as potential inhibitors for the topoisomerase IIβ protein (PDB ID: 4G0U). Additionally, MEP maps and the nonlinear optical (NLO) characteristics were calculated for the studied molecules.
2. Experimental
Melting points were measured using the digital Stuart SMP3 device. Using KBr discs, infrared spectra were obtained by FTIR Nicolet IS10 spectrophotometer (cm−1). Mercury-400BB (400 MHz) was utilized to measure the 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra. The solvent used was DMSO-d6, and the internal standard was TMS (δ). The GC-2010 Shimadzu Gas Chromatography Instrument mass spectrometer (70 eV) was used to obtain mass spectra. A PerkinElmer CHN-2400 analyzer was used to conduct elemental microanalyses. 4-Hydroxy-6-methyl-3-nitro-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (1) was synthesized according to literature.302.1. Synthesis and characterization of compounds
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1667 (C
N), 1667 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Opyridone), 1639 (COquinolone) and 1617 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.68 (s, 3H, CH3), 7.40 (t, 1H, J = 8.0 Hz, H-6), 7.62 (d, 1H, J = 8.4 Hz, H-8), 7.81 (t, 1H, J = 8.4 Hz, H-7), 8.11 (d, 1H, J = 8.0 Hz, H-5), 11.34 (bs, 1H, NH exchangeable with D2O), 12.32 (bs, 1H, OH exchangeable with D2O), 12.61 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir %): 354 (M+, 100), 326 (42), 310 (29), 280 (49), 259 (55), 237 (21), 213 (40), 175 (53), 159 (26), 133 (12), 105 (18), 93 (40), 77 (32), 64 (11). Anal. calcd. for C16H10N4O6 (354.27); C, 54.24; H, 2.85; N, 15.81%. Found: C, 53.96; H, 2.74; N, 15.63%.
Opyridone), 1639 (COquinolone) and 1617 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.68 (s, 3H, CH3), 7.40 (t, 1H, J = 8.0 Hz, H-6), 7.62 (d, 1H, J = 8.4 Hz, H-8), 7.81 (t, 1H, J = 8.4 Hz, H-7), 8.11 (d, 1H, J = 8.0 Hz, H-5), 11.34 (bs, 1H, NH exchangeable with D2O), 12.32 (bs, 1H, OH exchangeable with D2O), 12.61 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir %): 354 (M+, 100), 326 (42), 310 (29), 280 (49), 259 (55), 237 (21), 213 (40), 175 (53), 159 (26), 133 (12), 105 (18), 93 (40), 77 (32), 64 (11). Anal. calcd. for C16H10N4O6 (354.27); C, 54.24; H, 2.85; N, 15.81%. Found: C, 53.96; H, 2.74; N, 15.63%.
 | ||
| Scheme 1 Reaction of precursor 1 with cyanoacetamide. | ||
N), 1651 (COquinolone), 1594 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.54 (s, 3H, CH3), 7.13 (t, 1H, J = 7.6 Hz, H-6), 7.33 (d, 1H, J = 7.6 Hz, H-8), 7.54 (t, 1H, J = 7.6 Hz, H-7), 8.14 (d, 1H, J = 7.6 Hz, H-5), 11.46 (bs, 1H, NH exchangeable with D2O), 12.39 (bs, 1H, OH exchangeable with D2O), 12.79 (bs, 1H, OH exchangeable with D2O). Anal. calcd. for C19H10N6O5 (402.32); C, 56.72; H, 2.51; N, 20.89%. Found: 56.49; H, 2.32; N, 20.83%.
 | ||
| Scheme 2 Formation of pyridine 3 and pyrido[1,2-a]benzimidazole 4. | ||
N), 1647 (COquinolone), 1610 (CN), 1566 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.62 (s, 3H, CH3), 7.23 (t, 1H, J = 7.2 Hz, H-6), 7.26–7.34 (m, 2H, 2Ar–H), 7.39 (d, 1H, J = 7.2 Hz, H-8), 7.62–7.69 (m, 2H, 2Ar–H), 8.04 (t, 1H, J = 8.0 Hz, H-7), 8.21 (d, 1H, J = 8.4 Hz, H-5), 12.59 (bs, 1H, OH exchangeable with D2O), 13.31 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir %): 427 (M+, 66), 381 (100), 150 (18), 306 (33), 252 (26), 208 (28), 174 (56), 159 (13), 144 (46), 132 (19), 117 (12), 90 (53), 77 (42), 64 (16). Anal. calcd. for C22H13N5O5 (427.37); C, 61.83; H, 3.07; N, 16.39%. Found: C, 61.75; H, 3.02; N, 16.23%.Opyrazonone), 1657 (COquinolone), 1614 (CN) and 1574 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.62 (s, 3H, CH3), 7.24 (t, 1H, J = 8.0 Hz, H-6), 7.45 (d, 1H, J = 8.0 Hz, H-8), 7.64 (t, 1H, J = 8.0 Hz, H-7), 8.21 (d, 1H, J = 8.0 Hz, H-5), 12.18 (bs, 1H, NHpyrazole exchangeable with D2O), 12.58 (bs, 1H, NHpyrazole exchangeable with D2O), 13.12 (bs, 1H, OH exchangeable with D2O), 13.67 (bs, 1H, OH exchangeable with D2O). Anal. calcd. for C16H11N5O6 (369.29); C, 52.04; H, 3.00; N, 18.96%. Found: 51.86; H, 2.93; N, 18.96%.
 | ||
| Scheme 3 Formation of pyrazolo[3,4-b]pyridine derivatives 5 and 6. | ||
Oquinolone), 1616 (CN), 1575 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 2.25 (s, 3H, CH3pyrazole), 3.64 (s, 3H, CH3), 7.42 (t, 1H, J = 7.2 Hz, H-6), 7.67 (d, 1H, J = 7.2 Hz, H-8), 7.85 (t, 1H, J = 7.2 Hz, H-7), 8.13 (d, 1H, J = 7.2 Hz, H-5), 11.28 (bs, 1H, NHpyrazole exchangeable with D2O), 12.33 (bs, 1H, OH exchangeable with D2O), 12.61 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir %): 367 (M+, 100), 337 (38), 292 (31), 251 (26), 209 (23), 195 (38), 175 (44), 159 (14), 133 (12), 105 (28), 91 (57), 77 (41), 64 (20). Anal. calcd. for C17H13N5O5 (367.32); C, 55.59; H, 3.57; N, 19.07%. Found: C, 55.46; H, 3.51; N, 18.93%.Opyrimidine), 1667 (COpyrimidine), 1648 (COquinolone), 1619 (CN), 1567 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.53 (s, 3H, CH3), 7.23 (t, 1H, J = 7.2 Hz, H-6), 7.46 (d, 1H, J = 7.2 Hz, H-8), 7.62 (t, 1H, J = 7.2 Hz, H-7), 8.15 (d, 1H, J = 7.2 Hz, H-5), 10.09 (bs, 2H, 2NHpyrimidine exchangeable with D2O), 12.78 (bs, 1H, OH exchangeable with D2O), 13.39 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir %): 397 (M+, 100), 369 (69), 326 (38), 298 (27), 253 (30), 223 (43), 195 (24), 174 (39), 150 (28), 133 (31), 104 (17), 92 (57), 77 (33), 64 (16). Anal. calcd. for C17H11N5O7 (397.30); C, 51.39; H, 2.79; N, 17.63%. Found: C, 51.18; H, 2.52; N, 17.58%.
 | ||
| Scheme 4 Formation of pyrido[2,3-d]pyrimidines 7–9. | ||
Opyrimidine), 1651 (COquinolone), 1613 (CN), 1571 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.61 (s, 3H, CH3), 7.37 (t, 1H, J = 7.6 Hz, H-6), 7.60 (d, 1H, J = 7.6 Hz, H-8), 7.78 (t, 1H, J = 7.6 Hz, H-7), 8.07 (d, 1H, J = 7.6 Hz, H-5), 10.32 (bs, 1H, NHpyrimidine exchangeable with D2O), 10.99 (bs, 1H, NHpyrimidine exchangeable with D2O), 12.11 (bs, 1H, OH exchangeable with D2O), 12.87 (bs, 1H, OH exchangeable with D2O). Anal. calcd. for C17H11N5O6S (413.36); C, 49.40; H, 2.68; N, 16.94; S, 7.76%. Found: C, 49.23; H, 2.46; N, 16.72; S, 7.65%.Opyrimidine), 1646 (COquinolone), 1614 (CN) and 1577 (CC). 1H NMR (DMSO-d6, δ, 400 MHz): 3.16 (s, 3H, CH3), 3.47 (s, 3H, CH3), 3.63 (s, 3H, CH3), 7.39 (t, 1H, J = 8.0 Hz, H-6), 7.59 (d, 1H, J = 7.6 Hz, H-8), 7.99–8.11 (m, 2H, H-7 and H-5), 12.72 (bs, 1H, OH exchangeable with D2O), 13.26 (bs, 1H, OH exchangeable with D2O). Anal. calcd. for C19H15N5O7 (425.35); C, 53.65; H, 3.55; N, 16.46%. Found: C, 53.39; H, 3.46; N, 16.35%.2.2. Computational methods
The GAUSSIAN 09 W program was used to do the computational chemistry calculations for the produced compounds using DFT with B3LYP level at standard 6-311G++(d,p). This method is appropriate for assessing the stability and reactivity of molecules similar to those studied in the present work.31–33 Gauss View 5.0 was used to make visual presentations.34,35 During the geometry optimization process, there were no restrictions on symmetry. Molecule electrostatic potential (MEP), and optimized geometries associated with MOs energies were among the outcomes of quantum chemistry computations. Moreover, using the 6-311++G(d,p) basis set and the Gauge-Invariant Atomic Orbital (GIAO) technique,36 the chemical shift calculations of 1H and 13C NMR were performed at the B3LYP level as well as these results were compared with experimental values. The polarizability, the first hyperpolarizability, and electronic dipole moment were all characterized at the same level of the DFT. The NLO characteristics of the synthesized compounds are calculated using quantum chemistry. The NLO computations were performed in gas phase at level B3LYP/6-311++G(d,p).372.3. Biological evaluation
These ligands were then converted to PDBQT format. A grid box was defined for the 4G0U receptor with coordinates (X = 32.127, Y = 30.108, Z = 10.345) and dimensions (X = 14.687 Å, Y = 12.254 Å, Z = 22.254 Å). The binding affinities of the compounds were subsequently evaluated based on their binding energies (kcal mol−1).50 This computational study offers a comprehensive analysis of the interactions between the ligands and the 4G0U receptor.
3. Results and discussion
3.1. Characterization of the synthesized compounds
The starting substrate 1 possess an electron deficient α-pyrone ring which structure is expected to generate potentially interesting interactions with nucleophilic reagents. To the best of our knowledge, substrate 1 did not reacted previously with carbon nucleophiles. Consequently, we are motivated to investigate its chemical behavior towards some carbon nucleophiles.Reaction of 4-hydroxy-6-methyl-3-nitro-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (1) with cyanoacetamide, in boiling butanol containing TEA, afforded the novel pyridine-3-carbonitrile derivative 2 (Scheme 1).51,52 The reaction initially proceeds through deprotonation of cyanoacetamide followed by nucleophilic attack at C-2 position of compound 1 (intermediate A) with subsequent α-pyrone ring opening (intermediate B), cylocondensation (intermediate C) and proton transfer. The mass spectrum for compound 2 exhibited the parent ion peak, as the base peak, at m/z 354, which is coincident well with the suggested molecular formula C16H10N4O6 (354.27). Its IR spectrum presented characteristic absorption bands at ν 3445 (OH), 3248 (NH), 2224 (CN), 1667 (COpyridone) and 1639 cm−1 (COquinolone). The 1H NMR spectrum presented definite singlet in the upfield region due to NCH3 protons at δ 3.68, in addition to four aromatic protons at δ 7.40 (triplet, H-6), 7.62 (doublet, H-8), 7.81 (triplet, H-7) and 8.11 (doublet, H-5). The spectrum also revealed D2O-exchangeable signals in the downfield region at δ 11.34, 12.32 and 12.61 due to NH and 2OH protons, respectively.
Similar to the previous mechanism, reaction of compound 1 with 2-aminoprop-1-ene-1,1,3-tricarbonitrile and 1H-benzimidazol-2-ylacetonitrile, in refluxing butanol/TEA, furnished 4-hydroxyquinolinone linked pyridine-3-carbonitrile 3 and pyrido[1,2-a]benzimidazole-4-carbonitrile 4, respectively (Scheme 2). The IR spectrum of compound 3 presented distinctive absorption bands due to 3CN functions at ν 2201, 2194 and 2174, in addition to COquinolone at ν 1651 cm−1. While the IR spectrum of compound 4 present definite absorption bands due to CN, COquinolone and CN at ν 2211, 1647 and 1610 cm−1. Structure 4 was further established through mass spectrum which presented the molecular ion peak at m/z 427 which matches well with the assigned molecular formula C22H13N5O5 (427.37).
Then, the reactivity of compound 1 was examined towards some pyrazoles including activated nucleophilic centers such as 5-amino-2,4-dihydro-3H-pyrazol-3-one and 5-amino-3-methyl-1H-pyrazole giving the corresponding 4-hydroxy-1-methylquinolin-2(1H)-one tethered pyrazolo[3,4-b]pyridines 5 and 6 (Scheme 3). Specific absorption bands were seen in the IR spectrum of compound 5 at ν 3202, 3179 (2NH), 1678 (COpyrazonone) and 1657 cm−1 (COquinolone), while the spectrum of compound 6 displayed absorption bands at ν 3332 (NH) and 1645 cm−1 (COquinolone). Structure 6 was also confirmed by mass spectrum which recorded the parent ion peak at m/z 367 that coincided well with the proposed formula weight C17H13N5O5 (367.32). The 1H NMR spectrum of compound 6 showed distinctive singlet signals in the aliphatic region at δ 2.25 (CH3pyrazole) and 3.64 (CH3quinoline), as well as three D2O-vanished signals at δ 11.28 (NHpyrazole), 12.33 (OH) and 12.61 (OH).
After that, the behavior of compound 1 was investigated towards some uracils namely 6-aminouracil, 6-aminothiouracil and 6-amino-1,3-dimethyluracil producing the novel 4-hydroxy 1-methylquinolin-2(1H)-one tethered pyrido[2,3-d]pyrimidines 7–9, respectively (Scheme 4). The mass spectrum of compound 7 displayed the molecular ion peak, as the base peak, at m/z 397 supporting the identity of the suggested molecular formula C17H11N5O7 (397.30). Its 1H NMR spectrum displayed the NCH3 protons as typical singlet at δ 3.53, meanwhile the aromatic protons appeared the expected splitting patterns as triplet (δ 7.23), doublet (δ 7.46), triplet (δ 7.62) and doublet (δ 8.15). Three exchangeable signals were recorded at δ 10.09 (2NHpyrimidine), 12.78 (OH) and 13.39 (OH).
3.2. Theoretical studies
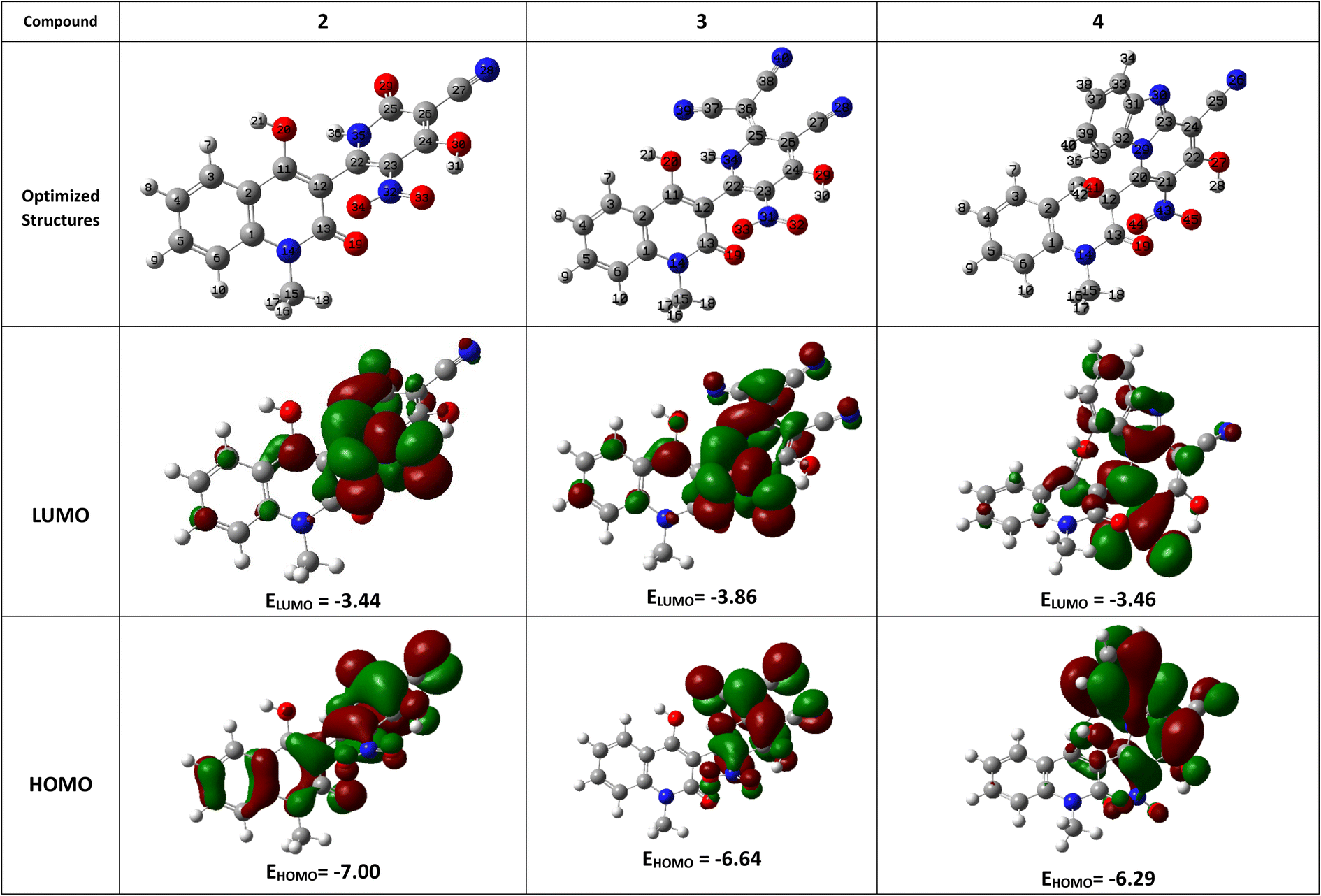 | ||
| Fig. 1 Molecular modeling and the electron density of HOMO and LUMO of compounds 2–4. | ||
The lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) make up these FMOs. The ability of a compound to give electrons is shown by its EHOMO value, whereas a component's ELUMO value shows its capacity to receive electrons (Table 1).
| Compound | ET (au) | EHOMO (eV) | ELUMO (eV) | IP (eV) | EA (eV) | ΔE (eV) | χ (eV) | μ (eV) | η (eV) | S (eV−1) | ω (eV) | ε (eV−1) | ΔN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −1061.27 | −7.042 | −3.418 | 7.042 | 3.418 | 3.624 | 5.230 | −5.230 | 1.812 | 0.552 | 7.547 | 0.133 | 2.886 |
| 2 | −1286.35 | −7.003 | −3.437 | 7.003 | 3.437 | 3.566 | 5.220 | −5.220 | 1.783 | 0.561 | 7.642 | 0.131 | 2.928 |
| 3 | −1434.92 | −6.647 | −3.864 | 6.647 | 3.864 | 2.783 | 5.255 | −5.255 | 1.391 | 0.719 | 9.925 | 0.101 | 3.777 |
| 4 | −1496.35 | −6.286 | −3.457 | 6.286 | 3.457 | 2.829 | 4.871 | −4.871 | 1.414 | 0.707 | 8.390 | 0.119 | 3.444 |
| 5 | −1341.71 | −6.563 | −3.008 | 6.563 | 3.008 | 3.555 | 4.785 | −4.785 | 1.778 | 0.563 | 6.440 | 0.155 | 2.692 |
| 6 | −1305.80 | −6.392 | −2.822 | 6.392 | 2.822 | 3.570 | 4.607 | −4.607 | 1.785 | 0.560 | 5.944 | 0.168 | 2.581 |
| 7 | −1455.15 | −6.625 | −2.716 | 6.625 | 2.716 | 3.909 | 4.671 | −4.671 | 1.955 | 0.512 | 5.581 | 0.179 | 2.389 |
| 8 | −1778.11 | −6.646 | −3.071 | 6.646 | 3.071 | 3.575 | 4.859 | −4.859 | 1.788 | 0.559 | 6.603 | 0.151 | 2.718 |
| 9 | −1533.79 | −6.550 | −2.554 | 6.550 | 2.554 | 3.995 | 4.552 | −4.552 | 1.998 | 0.501 | 5.186 | 0.193 | 2.279 |
Energy gap (ΔE, eV) is a measure of a molecule's chemical stability and reactivity; a larger ΔE suggests higher stability. The compound 9 has a ΔE of 3.995 eV, which is the largest value. This implies that the compound 9 is more stable than other compounds.
Additionally, the quantum chemical parameters as chemical potential (μ), electronegativity (χ), chemical hardness (η), softness (S), electrophilicity (ω), nucleophilicity (ε), and additional electronic charge (ΔNmax) were calculated using the following equations56–58
| I = −EHOMO | (1) |
| Y = −ELUMO | (2) |
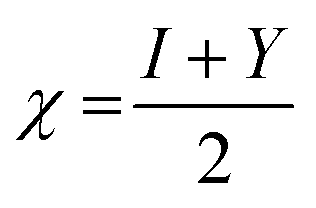 | (3) |
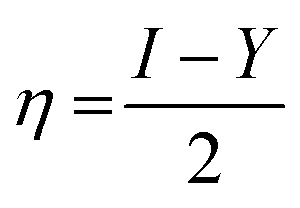 | (4) |
 | (5) |
 | (6) |
 | (7) |
| μ = −χ | (8) |
 | (9) |
The energy required to remove an electron from a component is indicated by the ionization potential (I, eV).50 The energy shift that occurs when an electron is added to a component is measured by electron affinity (Y, eV). In addition, chemical resistance to shifts in electron distribution is measured by hardness (η, eV), whereas chemical softness (S, eV−1) is the opposite of hardness (η).59 Compound 9 has a hardness value (η) of 1.998 eV which is the hardest compound, hence the less reactive one and comparatively hard. On the other hand, compound 3 has a softness value (S) of 0.719 eV −1 and therefore the more reactive and softer than the others. A molecule's propensity to draw electrons is indicated by its electronegativity (χ, eV).60 Compared to other compounds, compound 3 has a greater χ of 5.255 eV. Further, the tendency of electrons to escape from a molecule is indicated by chemical potentials (μ, eV). The prepared compounds have a negative chemical potential, indicating their stability. The ability of a molecule to accept electrons is measured by its electrophilicity index (ω, eV). Compound 3 exhibits a stronger electrophile than the others, as evidenced by its ω value of 9.925 eV. A component's ability to donate electrons is known as its nucleophilicity (ε, eV−1).61 Compared to other compounds, compound 7 has a greater ε of 0.179 eV−1, so the strongest nucleophile.
| Compound 2 | Compound 3 | Compound 4 | Compound 5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Atoms | Calculated | Experimental | Atoms | Calculated | Experimental | Atoms | Calculated | Experimental | Atoms | Calculated | Experimental |
| 17-H | 1.58 | 3.68 | 17-H | 1.63 | 3.54 | 17-H | 3.16 | 3.62 | 16-H | 2.92 | 3.62 |
| 18-H | 2.76 | 3.68 | 18-H | 2.77 | 3.54 | 16-H | 3.18 | 3.62 | 17-H | 2.94 | 3.62 |
| 16-H | 3.41 | 3.68 | 16-H | 3.49 | 3.54 | 18-H | 3.45 | 3.62 | 18-H | 3.80 | 3.62 |
| 8-H | 7.57 | 7.4 | 8-H | 7.51 | 7.13 | 40-H | 6.53 | 7.23 | 8-H | 7.62 | 7.24 |
| 7-H | 7.61 | 7.62 | 7-H | 7.54 | 7.33 | 34-H | 7.48 | 7.26 | 10-H | 7.63 | 7.45 |
| 9-H | 7.927 | 7.81 | 9-H | 7.91 | 7.54 | 8-H | 7.49 | 7.34 | 7-H | 7.76 | 7.64 |
| 10-H | 8.18 | 8.11 | 10-H | 8.13 | 8.14 | 38-H | 7.58 | 7.39 | 9-H | 8.04 | 8.21 |
| 36-H | 10.11 | 11.34 | 35-H | 9.85 | 11.46 | 36-H | 7.68 | 7.62 | 37-H | 11.58 | 12.18 |
| 31-H | 13.29 | 12.32 | 30-H | 12.99 | 12.39 | 10-H | 7.74 | 7.69 | 35-H | 11.69 | 12.58 |
| 21-H | 13.57 | 12.61 | 21-H | 13.05 | 12.79 | 7-H | 7.74 | 8.04 | 28-H | 12.78 | 13.12 |
| 9-H | 8.09 | 8.21 | 21-H | 13.01 | 13.67 | ||||||
| 42-H | 12.52 | 12.59 | |||||||||
| 28-H | 13.44 | 13.31 | |||||||||
| Compound 6 | Compound 7 | Compound 8 | Compound 9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Atoms | Calculated | Experimental | Atoms | Calculated | Experimental | Atoms | Calculated | Experimental | Atoms | Calculated | Experimental |
| 39-H | 2.69 | 2.25 | 16-H | 2.95 | 3.53 | 16-H | 2.95 | 3.61 | 40-H | 2.88 | 3.16 |
| 37-H | 2.92 | 2.25 | 17-H | 2.97 | 3.53 | 17-H | 2.96 | 3.61 | 17-H | 2.94 | 3.63 |
| 16-H | 2.93 | 3.64 | 18-H | 3.90 | 3.53 | 18-H | 4.04 | 3.61 | 16-H | 2.96 | 3.63 |
| 17-H | 2.94 | 3.64 | 8-H | 7.53 | 7.23 | 10-H | 7.56 | 7.37 | 42-H | 2.98 | 3.16 |
| 38-H | 2.95 | 2.25 | 10-H | 7.55 | 7.46 | 8-H | 7.57 | 7.6 | 44-H | 3.193 | 3.47 |
| 18-H | 5.08 | 3.64 | 7-H | 7.83 | 7.62 | 7-H | 7.80 | 7.78 | 45-H | 3.19 | 3.47 |
| 8-H | 7.63 | 7.42 | 9-H | 7.92 | 8.15 | 9-H | 7.94 | 8.07 | 46-H | 3.45 | 3.47 |
| 10-H | 7.64 | 7.67 | 38-H | 10.43 | 10.09 | 35-H | 10.48 | 10.32 | 41-H | 3.49 | 3.16 |
| 7-H | 7.78 | 7.85 | 35-H | 10.46 | 10.09 | 38-H | 11.05 | 10.99 | 18-H | 3.91 | 3.63 |
| 9-H | 8.04 | 8.13 | 27-H | 12.09 | 12.78 | 21-H | 12.31 | 12.11 | 8-H | 7.51 | 7.39 |
| 35-H | 10.43 | 11.28 | 21-H | 13.08 | 13.39 | 27-H | 13.63 | 12.87 | 10-H | 7.52 | 7.59 |
| 21-H | 12.92 | 12.33 | 7-H | 7.83 | 7.99 | ||||||
| 28-H | 13.20 | 12.61 | 9-H | 7.86 | 8.11 | ||||||
| 21-H | 12.08 | 12.72 | |||||||||
| 27-H | 12.92 | 13.26 | |||||||||
In compound 2, the experimental 1H NMR chemical shift of the two OH groups were found at δ 12.32 and 12.61 ppm, while the computed signals were determined at δ 13.29 and 13.57 ppm. The 1H NMR signals of benzene ring of compound 3 were recorded experimentally in the range δ 7.13–8.14 ppm, whereas theoretically calculated at δ 7.51–8.13 ppm. The 1H NMR chemical shift of OH protons of compound 4 were observed experimentally at δ 12.59 and δ 13.31 ppm, while computed at δ 12.52 and 13.44 ppm.
In compound 5, the two singlet signals for NH protons were calculated at δ 11.58 and 11.69 ppm, while the signals in the experimental spectrum were identified at δ 12.18 and 12.58 ppm. The calculated chemical shifts of methyl protons were computed at δ 2.69, 2.92 and 2.95 ppm; showing good agreement with the experimental value at δ 2.25 ppm (singlet).
Two signals of NH groups were detected at δ 10.44 and 10.46 for compound 7 as well as at δ 10.49 and 11.05 ppm for compound 8. These signals were identified experimentally at δ 10.09 and 10.09 for compound 7, and at δ 10.32 and 10.99 ppm for compound 8. Additionally, 1H NMR chemical shift of the two N–Me groups of pyrimidine ring in compound 9 were recorded experimentally at δ 3.16 and 3.47 ppm, while their theoretically values were observed in the range δ 2.88–3.49 ppm.
The experimental 1H NMR chemical shifts were plotted with the theoretical 1H NMR chemical shifts and correlation coefficient (R2) is found in the range 0.98–0.99 as shown in Fig. 2 and S3–S5.† Thus, these computed values are good agreement with experimental values.
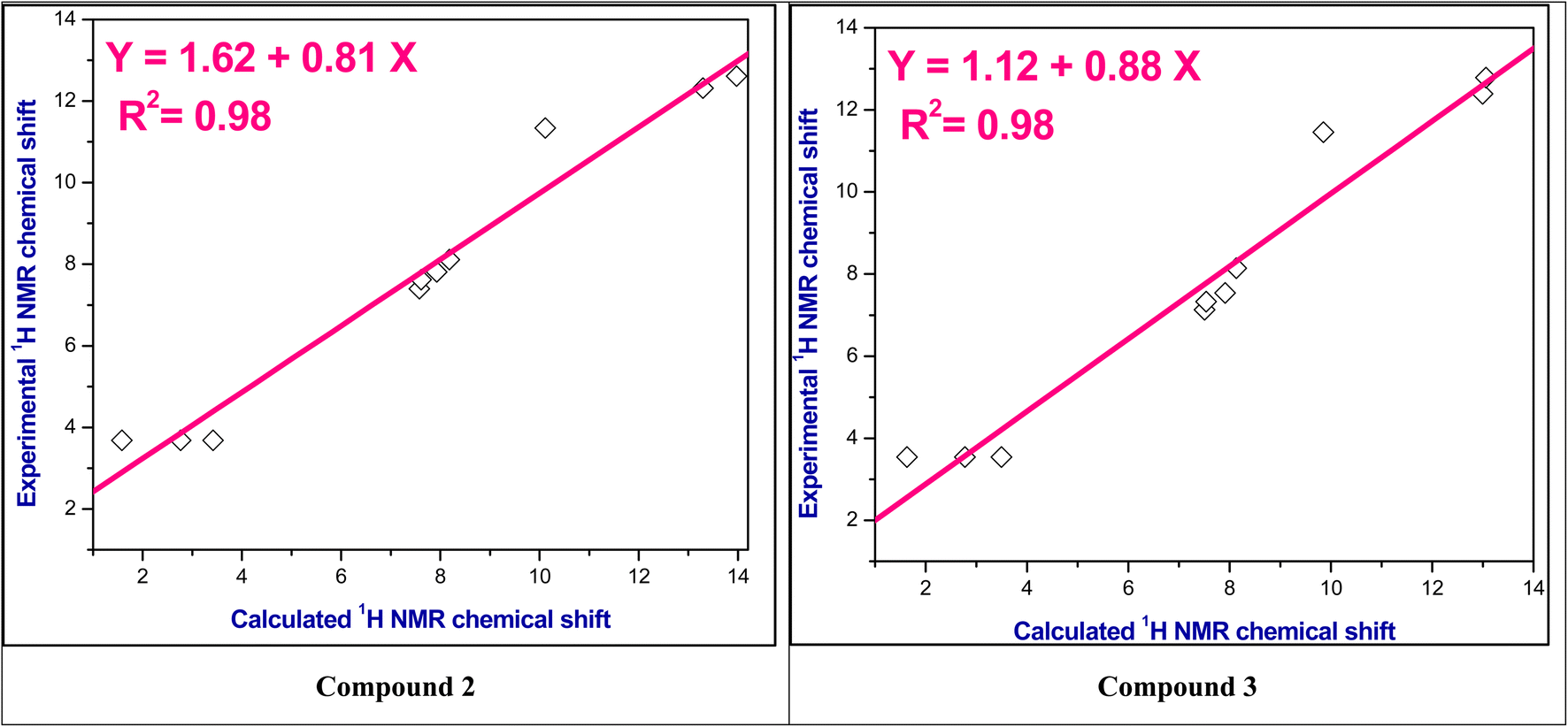 | ||
| Fig. 2 The correlation relationships of the experimental versus calculated 1H NMR chemical shifts of compounds 2 and 3. | ||
| Compound 2 | Compound 3 | Compound 4 | ||||||
|---|---|---|---|---|---|---|---|---|
| υexp. (cm−1) | υthe. (cm−1) | Assignment | υexp. (cm−1) | υthe. (cm−1) | Assignment | υexp. (cm−1) | υthe. (cm−1) | Assignment |
| 3445 | 3414 | OH | 3440 | 3477 | OH | 3445 | 3420 | OH |
| 3248 | 3321 | NH | 3281 | 3362 | NH | 3042 | 3173 | CHaromatic |
| 3028 | 3178 | CHaromatic | 3014 | 3061 | CHaromatic | 2981, 2943 | 3100, 2985 | CHaliphatic |
| 2966, 2931 | 3012, 2932 | CHaliphatic | 2982, 2936 | 3009, 2973 | CHaliphatic | 2211 | 2218 | CN |
| 2224 | 2173 | CN |
2201, 2194, 2174 | 2214, 2181, 2162 | CN |
1647 | 1672 | COquinolone |
| 1667 | 1685 | COpyridone |
1651 | 1656 | COquinolone |
1610 | 1604 | CN |
| 1639 | 1646 | COquinolone |
1594 | 1596 | CC |
1566 | 1563 | CC |
| 1617 | 1602 | CC |
||||||
| Compound 5 | Compound 6 | Compound 7 | ||||||
|---|---|---|---|---|---|---|---|---|
| υexp. (cm−1) | υthe. (cm−1) | Assignment | υexp. (cm−1) | υthe. (cm−1) | Assignment | υexp. (cm−1) | υthe. (cm−1) | Assignment |
| 3446 | 3428 | OH | 3406 | 3442 | OH | 3445 | 3430 | OH |
| 3202, 3179 | 3347, 3282 | NH | 3332 | 3297 | NH | 3242, 3218 | 3344, 3280 | NH |
| 3055 | 3064 | CHaromatic | 3046 | 3125 | CHaromatic | 3045 | 3129 | CHaromatic |
| 2966, 2935 | 3036, 2911 | CHaliphatic | 2955, 2927 | 3032, 2977 | CHaliphatic | 2983, 2948 | 3023, 2983 | CHaliphatic |
| 1678 | 1685 | COpyrazolone |
1645 | 1660 | COquinolone |
1685, 1667 | 1690, 1669 | COpyrimidine |
| 1657 | 1646 | COquinolone |
1616 | 1606 | CN |
1648 | 1649 | COquinolone |
| 1614 | 1603 | CN |
1575 | 1592 | CC |
1619 | 1605 | CN |
| 1574 | 1595 | CC |
1567 | 1583 | CC |
|||
| Compound 8 | Compound 9 | ||||
|---|---|---|---|---|---|
| υexp. (cm−1) | υthe. (cm−1) | Assignment | υexp. (cm−1) | υthe. (cm−1) | Assignment |
| 3438 | 3442 | OH | 3446 | 3390 | OH |
| 3372, 3262 | 3365, 3338 | NH | 3084 | 3120 | CHaromatic |
| 3074 | 3104 | CHaromatic | 2992, 2953, 2864 | 3067, 3039, 2984 | CHaliphatic |
| 2989, 2928 | 3032, 2994 | CHaliphatic | 1687 | 1690, 1678 | COpyrimidine |
| 1677 | 1670 | COpyrimidine |
1646 | 1650 | COquinolone |
| 1651 | 1641 | COquinolone |
1614 | 1622 | CN |
| 1613 | 1614 | CN |
1577 | 1595 | CC |
| 1571 | 1576 | CC |
|||
In compound 2, the OH and NH stretching vibrations were recorded experimentally at ν 3445 and 3248 cm−1, which found theoretically at ν 3414 and 3321 cm−1, respectively. In addition, the experimental IR spectrum of compound 3 showed three CN stretching modes at ν 2201, 2194 and 2174 cm−1, while the theoretical values were observed at ν 2214, 2181 and 2162 cm−1. Moreover, the measured values for CN and COquinolone stretching modes in compound 4 were seen at ν 2211 and 1647 cm−1, which matched with the theoretical values at ν 2218 and 1672 cm−1.
Further, in compound 5, the measured result for the stretching vibrations of COpyrazole and COquinolone were seen at ν 1678 and 1657 cm−1, whereas the calculated values observed at ν 1685 and 1646 cm−1, respectively. In compound 6, the experimental findings of the stretching modes showed COquinolone at ν 1645 cm−1, which is consistent with the theoretical value at ν 1660 cm−1.
Additionally, the stretching vibrations of 2COpyrimidine and COquinolone in compounds 7 and 9 were found at ν 1685/1687, 1667/1687 and 1648/1646 cm−1, respectively, which were theoretically predicted at ν 1690/1690, 1669/1678 and 1649/1650 cm−1. The theoretical stretching values for COpyrimidine and COquinolone in compound 8 were seen at ν 1670 and 1641 cm−1, whereas these values were observed at ν 1677 and 1651 cm−1, respectively.
For the studied compounds 2–9, the stretching modes of CC in the benzene rings were observed in the range ν 1566–1617 cm−1 which falls within the theoretical spectra in the range at ν 1563–1602 cm−1. Furthermore, the actual aliphatic C–H stretching vibrations were in the range ν 2864–2992 cm−1 which consistent with the predicted value of ν 3100–2911 cm−1. It was found that the aromatic C–H stretching modes had values in the range at ν 3014–3084 cm−1, whereas the theoretical values found in the range at ν 3061–3178 cm−1. Fig. 3 and S6–S8† showed a relationship between the wavenumbers derived from experimental observations and those computed theoretically for these functional groups; and exhibit a correlation coefficient (R2) of 0.99.
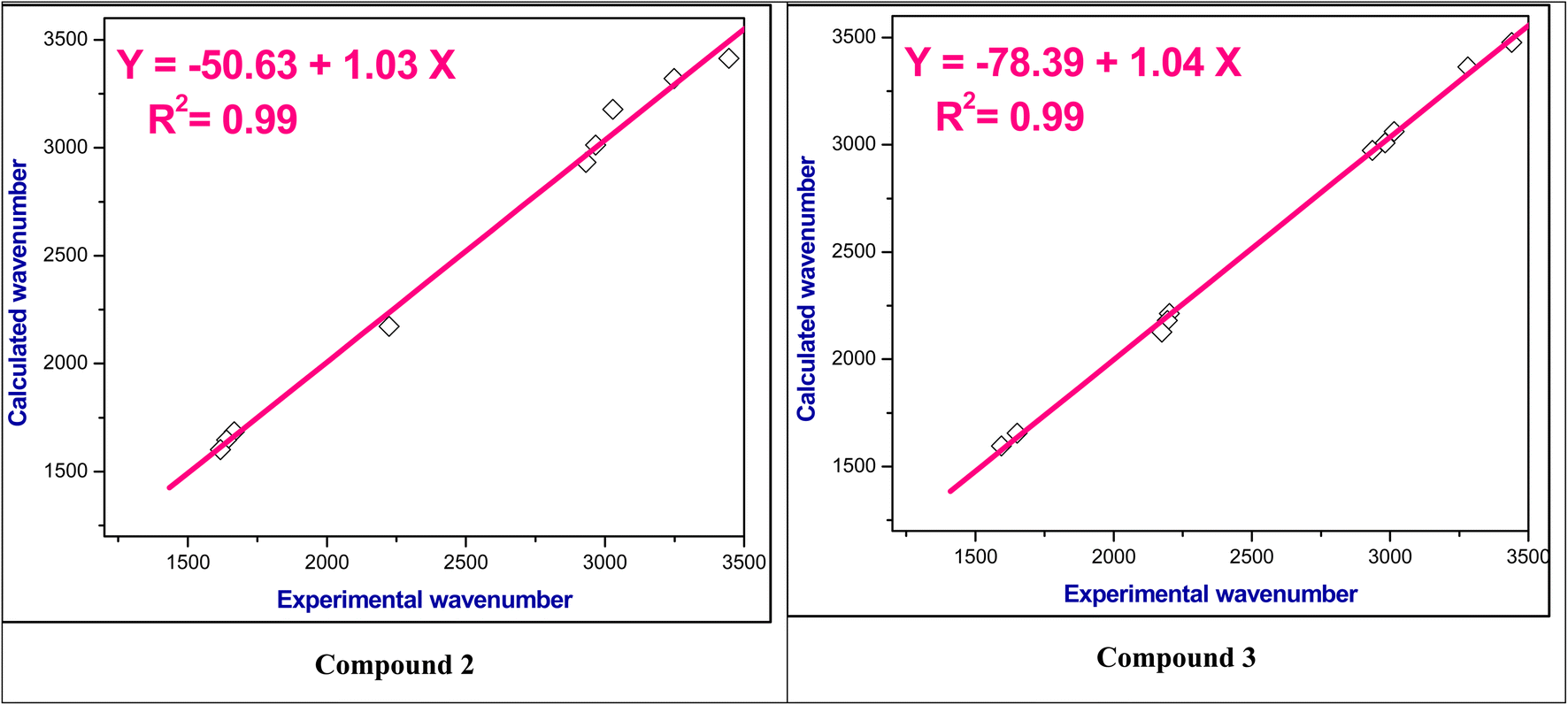 | ||
| Fig. 3 The correlation relationships of the experimental versus calculated IR wavenumbers of compounds 2 and 3. | ||
 | ||
| Fig. 4 Molecular electrostatic potential of compounds 2–4. | ||
The MEP map displays various electrostatic potentials in color. The electrostatic potential in the MEP map's color scheme is arranged as follows: blue > green > yellow > orange > red. As a result, the blue regions on the map symbolized areas with the strongest attraction (lower electron density), while the red regions showed areas with the strongest repulsion (higher electron density).69 Compounds' strong electron-attracting capabilities are indicated by the deep blue representation of their nucleophilic attack sites, which are primarily found on hydrogen and carbon atoms.
On the other hand, areas of significant electron repulsion are shown by the deep yellow and red coloration of the electrophilic attack sites, which are mostly connected to the nitrogen, sulphur and oxygen atoms. Where, the lone pairs of electrons on oxygen, sulphur and nitrogen atoms create areas of negative electrostatic potential that confirm their nucleophilic capabilities.70
| μ = (μx2 + μy2 + μz2)1/2 | (10) |
| α = (αxx + αyy + αzz)/3 | (11) |
| Δα = (2)−0.5[(αxx − αyy)2 + (αyy − αzz)2 + (αzz − αxx)2 + 6(αyz)2 + 6(αxy)2 + 6(αxz)2]0.5 | (12) |
| βtot = [(βxxx + βxyy + βxzz)2 + (βyyy + βyzz + βyxx)2 + (βzzz + βzxx + βzyy)2]0.5 | (13) |
Table 7 displays the outcomes of these computations. The dipole moment (μ) of products 1–9 was calculated to fall between 5.59 and 15.55 D. Furthermore, values of first hyperpolarizability of these compounds are 3–18 times higher than urea.73 Consequently, these compounds are the most promising and effective NLO materials, suitable materials for NLO applications and attractive objects for additional research into nonlinear optical properties.
| Compound no. | μx | μy | μz | μtotal | 〈α〉 (au) | 〈α〉 (esu) ×10−23 | Δα (au) | Δα (esu) ×10−24 | βtotal (au) | βtotal (esu) ×10−30 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9.14 | 0.21 | −0.001 | 9.15 | −123.51 | −1.83 | 16.85 | 2.50 | 203.50 | 1.76 |
| 2 | −14.50 | −1.08 | −1.59 | 14.63 | −158.14 | −2.34 | 40.25 | 5.97 | 605.64 | 5.23 |
| 3 | 15.45 | −0.07 | −1.73 | 15.55 | −186.52 | −2.76 | 48.38 | 7.17 | 816.91 | 7.06 |
| 4 | −11.80 | 4.68 | 0.19 | 12.70 | −188.40 | −2.79 | 27.34 | 4.05 | 553.71 | 4.78 |
| 5 | 7.32 | −5.94 | −0.52 | 9.44 | −149.66 | −2.22 | 19.33 | 2.86 | 299.15 | 2.58 |
| 6 | 4.28 | −3.56 | 0.52 | 5.59 | −146.33 | −2.17 | 28.66 | 4.25 | 147.27 | 1.27 |
| 7 | −5.27 | −5.23 | −0.75 | 7.46 | −161.68 | −2.40 | 45.35 | 6.72 | 244.71 | 2.11 |
| 8 | 5.75 | 5.05 | −0.91 | 7.71 | −169.19 | −2.51 | 45.09 | 6.68 | 316.89 | 2.74 |
| 9 | −3.96 | −6.28 | −0.81 | 7.47 | −171.01 | −2.53 | 52.35 | 7.76 | 188.75 | 1.63 |
3.3. Biological evaluation
 | ||
| Fig. 5 Relation between cell viability and concentration of all synthesized compounds on the proliferation of (HepG2) cell line (cisplatin as a standard drug). | ||
The results are provided as IC50 values and compared with that of cisplatin. The IC50 values are summarized in Fig. 6 and Table 8. The target compounds 2–9 showed high to moderate antiproliferative efficacy against hepatocellular carcinoma (HepG-2) cell. Interestingly, compound 3 with IC50 8.79 μM L−1 has the strongest potency effect against HepG-2, as well as is more effective than cisplatin with IC50 17.86 μM L−1. Additionally, compounds 2, 4 and 5 exerted more potent effect than cisplatin as a reference with IC50 values 17.78 ± 0.83, 10.95 ± 0.76 and 16.56 ± 0.80 μM L−1, respectively. Finally, compounds 6–9 demonstrated moderate inhibitory effect compared to the positive drug. The investigation into the structural activity relationship (SAR) of the designed compounds focused on their efficacy against the HepG-2 cell line revealed that: high anticancer activity of compounds 2 and 3 may attribute to the presence of pyridine moiety. By substituting oxygen atom of the pyridine ring in compound 2 by propanedinitrile in compound 3 led to more potent activity. Moreover, compound 4 with IC50 = 10.95 μM L−1 has stronger potency against HepG-2 than cisplatin. This may be due to fusion of pyridine moiety with benzimidazole ring.
 | ||
| Fig. 6 Virtual IC50 values of the studied compounds and cisplatin as a standard drug against HepG-2 cell lines. | ||
| Compound no. | (HepG-2 cells) IC50 (μM L−1) |
|---|---|
| 2 | 17.78 ± 0.83 |
| 3 | 8.79 ± 0.68 |
| 4 | 10.95 ± 0.76 |
| 5 | 16.56 ± 0.80 |
| 6 | 20.26 ± 0.96 |
| 7 | 28.67 ± 1.12 |
| 8 | 22.98 ± 0.99 |
| 9 | 32.49 ± 1.23 |
| Cisplatin | 17.86 ± 0.92 |
Additionally, compounds 5 and 6 have IC50 of 16.56 ± 0.80 and 20.26 ± 0.96 μM L−1, respectively. This may be attribute to presence of pyrazolo[3,4-b]pyridine moieties. Where, compound 5 has similar potency to cisplatin, while compound 6 has moderate activity. Replacing C–Me at position 3 in compound 6 with CO in compound 5 led to increasing activity against the HepG-2 cell lines. Moreover, pyrido[2,3-d]pyrimidines 7–9, in which the pyridine ring fused with pyrimidine moiety, showed moderated potency relative to cisplatin (IC50 = 17.86 ± 0.92 μM L−1), with IC50 values ranging from 22.98 ± 0.99 to 32.49 ± 1.23 μM L−1.
Linear regression analysis was performed using the experimental inhibitory activity (IC50) as the dependent variable and DFT-based global reactivity descriptors as the independent variables for the gas phase (Table 1). From the linear regression, the following are the main interest points:
(1) The positive slope of linear correlation IC50 = −39.47 + 17.07 ΔE/eV, r = 0.93, n = 8, indicated lowering energy gap (ΔE/eV) of the present compounds led to decrease the half-maximal inhibitory concentration (IC50) and therefore enhance the anticancer activity (Fig. 7).
 | ||
| Fig. 7 The linear correlation between Egap versus IC50. | ||
(2) In addition, the positive slope of IC50 and hardness, IC50 = −39.37 + 34.08 η/eV, r = 0.93, n = 8, indicating IC50 falls, as well increasing inhibitory action, with decreasing hardness (Fig. 8). Additionally, IC50 and softness have an inverse relationship where the softness versus IC50 showed a negative slope as follows IC50 = 71.99–89.16 S/eV, r = 0.91, n = 8 points (Fig. 9).
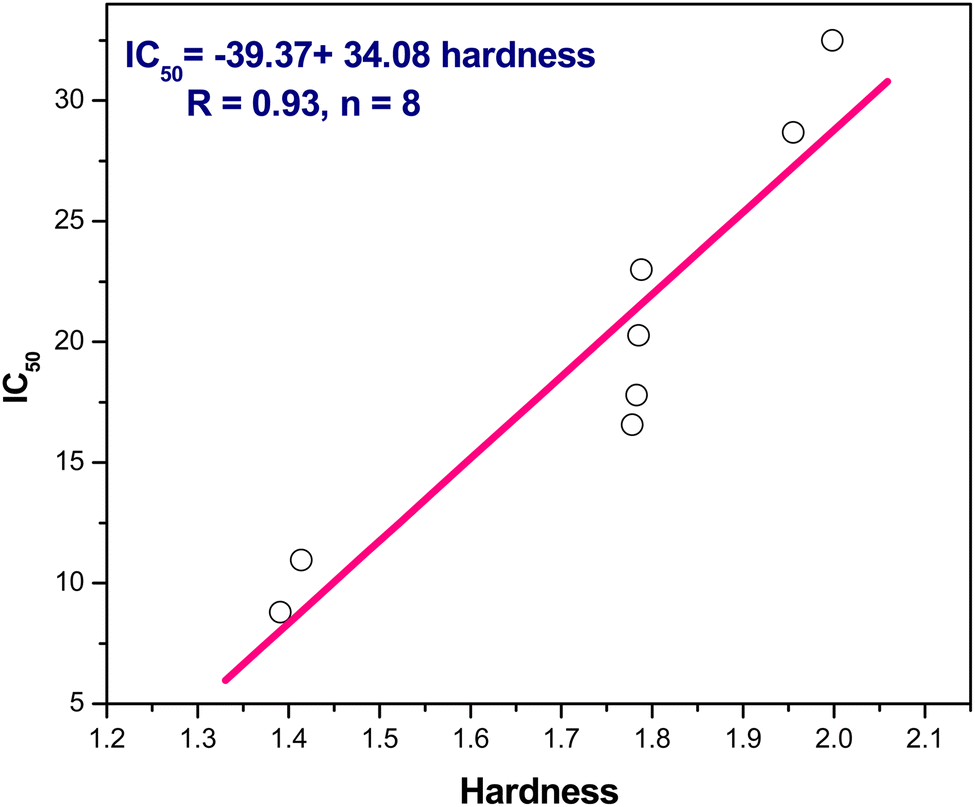 | ||
| Fig. 8 The linear correlation between hardness versus IC50. | ||
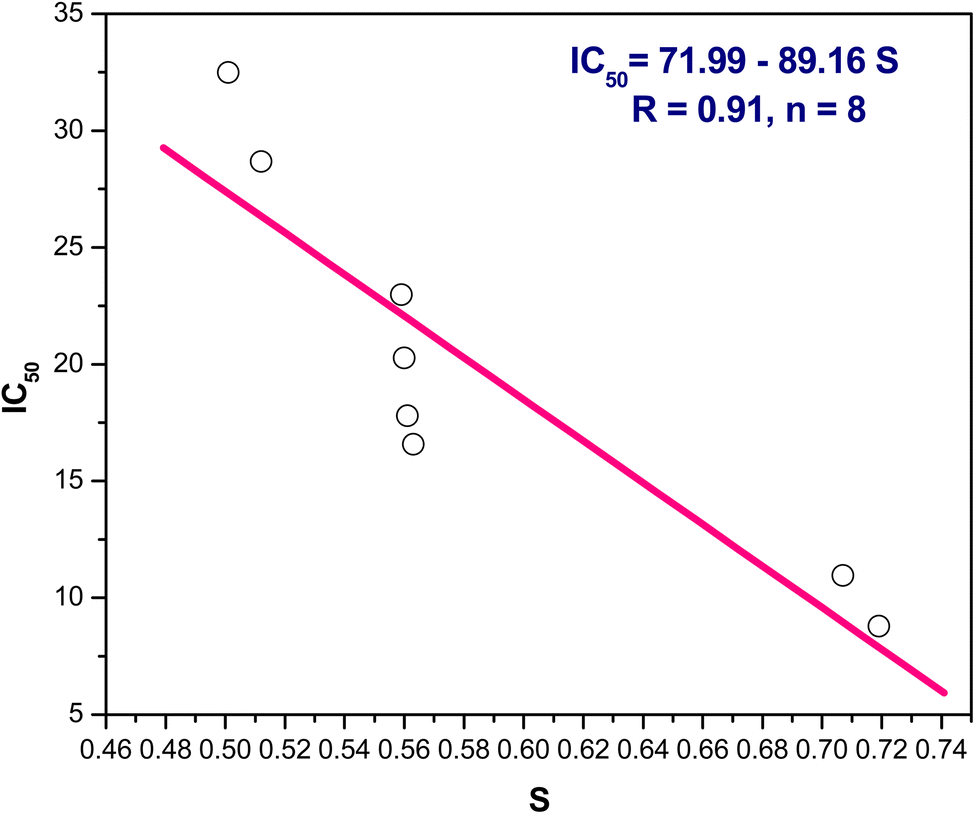 | ||
| Fig. 9 The linear correlation between softness (S) versus IC50. | ||
From the above results we can conclude that, lowering the energy gap indicates more susceptibility to accept electron from donor species. The molecule with a small energy gap which is a soft molecule was more polarized and reactive than hard ones due to its easily offering of the electrons to an acceptor. The energy gap of compounds 3 and 4 are smaller than other compounds indicating the ease of charge transfer and therefore enhanced the biological activity.74
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP should be less than 5, hydrogen bond donor (HBD) should not be greater than 5, hydrogen bond acceptor (HBA) should not be greater than 10, and MW should be less than 500 amu. Molecular weight of compounds is between 290 and 425 g mol−1, which is the permissible range. Compounds have an HBA between 6 and 8 and HBD between 1 and 4, both of which are within acceptable bounds. Moreover, the estimated values of the log octanol/water partition coefficient (MlogP) range from −1.48 to 1.06 (Table 9). Additionally, Veber's rule outlines the following restrictions on drug likeness: (a) the total polar surface area (TPSA) is associated with drug bioavailability and should not exceed 140 Å; the values for these compounds fall between 112.66 and −139.83 Å. (b) The number of rotatable bonds that is less than 10 and the produced compounds 1–2's rotatable bond count (Table 9). Additionally, the Ghose filter covers the following ranges: MW ranges from 160 to 480, MR ranges from 40 to 130, WlogP ranges from −0.4 to 5.6, and the total number of atoms ranges from 20 to 70 (Table 9). Additionally, all compounds exhibit favorable qualities for oral bioavailability by adhering to Lipinski's, Ghose's, and Veber's criteria.
logP should be less than 5, hydrogen bond donor (HBD) should not be greater than 5, hydrogen bond acceptor (HBA) should not be greater than 10, and MW should be less than 500 amu. Molecular weight of compounds is between 290 and 425 g mol−1, which is the permissible range. Compounds have an HBA between 6 and 8 and HBD between 1 and 4, both of which are within acceptable bounds. Moreover, the estimated values of the log octanol/water partition coefficient (MlogP) range from −1.48 to 1.06 (Table 9). Additionally, Veber's rule outlines the following restrictions on drug likeness: (a) the total polar surface area (TPSA) is associated with drug bioavailability and should not exceed 140 Å; the values for these compounds fall between 112.66 and −139.83 Å. (b) The number of rotatable bonds that is less than 10 and the produced compounds 1–2's rotatable bond count (Table 9). Additionally, the Ghose filter covers the following ranges: MW ranges from 160 to 480, MR ranges from 40 to 130, WlogP ranges from −0.4 to 5.6, and the total number of atoms ranges from 20 to 70 (Table 9). Additionally, all compounds exhibit favorable qualities for oral bioavailability by adhering to Lipinski's, Ghose's, and Veber's criteria.
| Compound no. | MW | MlogP |
HBA | HBD | TPSA | nRotB | WlogP |
MR | Lipinski violations | Ghose violations | Veber violations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 290.23 | −0.51 | 6 | 1 | 112.66 | 1 | 0.22 | 74.24 | 0 | 0 | 0 |
| 2 | 354.27 | −0.49 | 7 | 3 | 134.93 | 2 | 1.08 | 93.11 | 0 | 0 | 0 |
| 3 | 402.32 | −1.48 | 8 | 3 | 135.44 | 2 | 1.2 | 105.51 | 0 | 0 | 0 |
| 4 | 427.37 | 1.06 | 7 | 2 | 131.37 | 2 | 3.2 | 118.75 | 0 | 0 | 0 |
| 5 | 369.29 | 0.17 | 7 | 4 | 139.82 | 2 | 1.09 | 98.05 | 0 | 0 | 0 |
| 6 | 367.32 | 0.43 | 7 | 3 | 133.85 | 2 | 2.1 | 100.19 | 0 | 0 | 0 |
| 7 | 397.3 | −0.04 | 8 | 4 | 132.89 | 2 | 0.45 | 104.32 | 0 | 0 | 0 |
| 8 | 413.36 | −0.1 | 7 | 4 | 131.91 | 2 | 1.82 | 108.88 | 0 | 0 | 0 |
| 9 | 425.35 | 0.02 | 8 | 2 | 135.17 | 2 | 0.47 | 114.12 | 0 | 0 | 0 |
Initially, docking methodology had been validated by re-docking and superimposition of the native ligand, N-[4-(acridin-9-ylamino)-3-methoxyphenyl] methanesulfonamide, which was co-crystallized with the 4G0U active site. The binding pattern of the co-crystallized ligand was successfully reproduced during the re-docking validation stage, yielding a binding energy of −9.74 kcal mol−1. The superimposition between the re-docked ligand and the native co-crystallized ligand showed a close alignment with the re-docked and native co-crystallized ligand, further confirming the accuracy of the docking methodology, Fig. S30.†
The molecular docking of the studied compounds against the crystal structure of the Human topoisomerase II beta in complex with DNA and amacrine (PDB ID: 4G0U) was performed and the best binding mode with docked compounds are shown in Fig. 10 and S31† while the interactions with amino acids residues were shown in Table 10. The binding energy of the standard etoposide is −8.54 kcal mol−1, while that of the current compounds are ranged between −7.41 and −8.44 kcal mol−1. The percent (%) of the compounds' binding energy to that of the standard etoposide (−8.54 kcal mol−1) had been calculated and reported in Table 10.
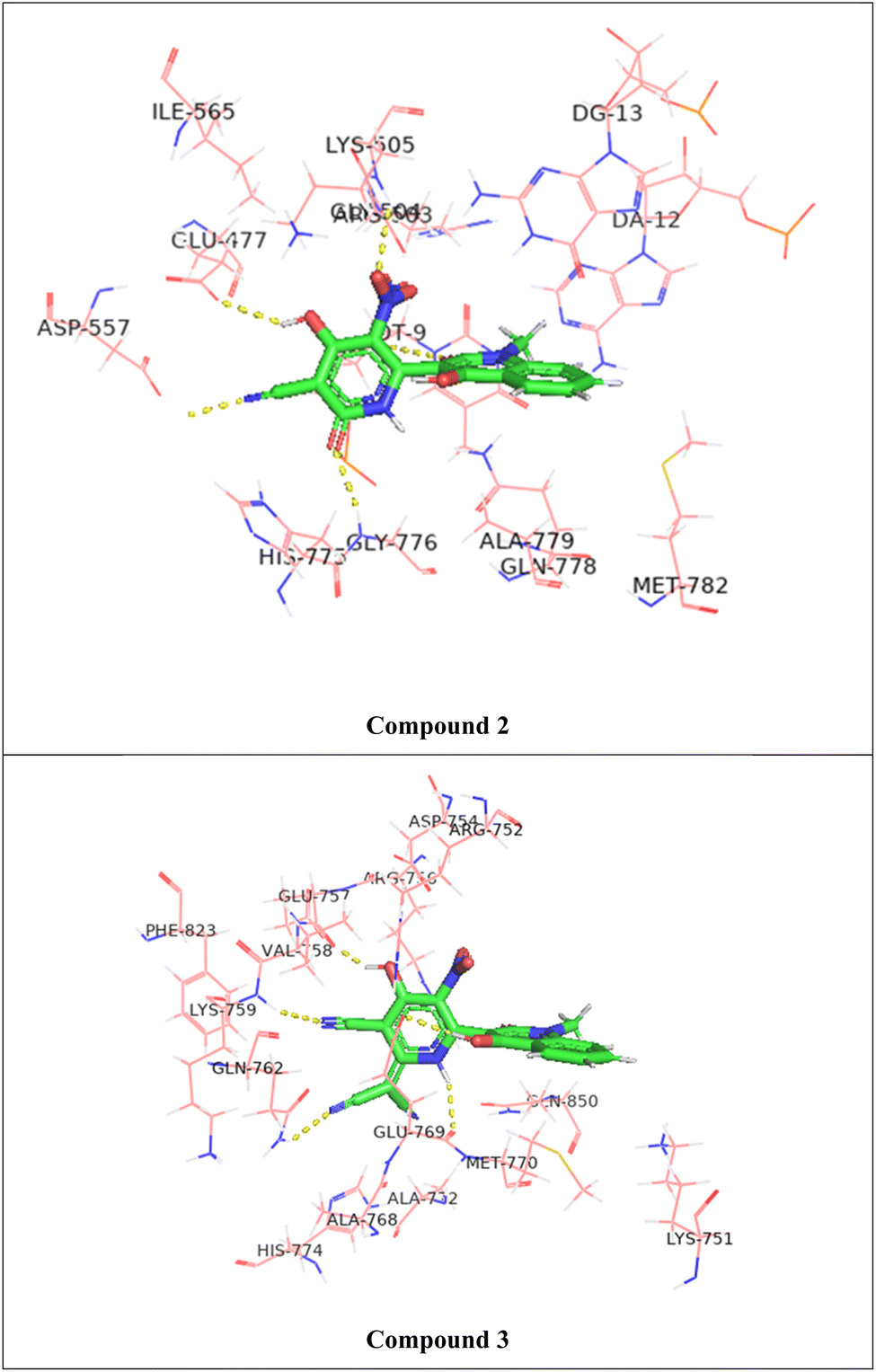 | ||
| Fig. 10 3D representation of the hydrogen bonding between the studied compounds (2 and 3) and the amino acids residues of the target protein (PDB ID: 4G0U). | ||
| Compound | Ligand | Receptor | Distance | Binding energy (kcal mol−1) | % (standard, etoposide) |
|---|---|---|---|---|---|
| 2 | O 25 | LYS 505 | 3.36 | −8.23 | 96.37 |
| H 35 | GLU 477 | 3.14 | |||
| O 22 | GLY 776 | 3.04 | |||
| O 22 | DT 9 | 3.11 | |||
| O 13 | DT 9 | 3.15 | |||
| 3 | H 39 | GLU 757 | 3.03 | −8.44 | 98.83 |
| N 24 | LYS 759 | 2.76 | |||
| N 29 | GLN 762 | 3.14 | |||
| O 11 | GLU 769 | 3.56 | |||
| H 40 | GLU 769 | 3.47 | |||
| 4 | O 13 | ARG 729 | 3.1 | −8.13 | 95.20 |
| O 31 | HIS 775 | 3.46 | |||
| O 32 | HIS 775 | 3.58 | |||
| O 14 | GLU 477 | 3.48 | |||
| N 22 | LYS 505 | 3.24 | |||
| 5 | O 11 | GLN 850 | 2.91 | −8.09 | 94.73 |
| H 38 | ALA 768 | 2.89 | |||
| O 27 | LYS 759 | 3.25 | |||
| H 37 | TYR 773 | 3.65 | |||
| 6 | O 11 | GLN 762 | 3.07 | −7.41 | 86.77 |
| O 26 | LYS 759 | 3.06 | |||
| O 36 | GLU 769 | 3.22 | |||
| H 37 | GLU 769 | 3.18 | |||
| 7 | O 13 | GLN 762 | 2.76 | −7.75 | 90.75 |
| H 38 | GLU 769 | 2.91 | |||
| H 38 | GLN 850 | 3.27 | |||
| O 29 | GLN 850 | 3.11 | |||
| O 29 | GLU 769 | 3.24 | |||
| O 28 | GLU 757 | 3.38 | |||
| H 40 | GLU 757 | 3.18 | |||
| 8 | O 11 | DC 14 | 3.35 | −8.02 | 93.91 |
| O 23 | GLN 778 | 3.17 | |||
| O 20 | LYS 505 | 4.47 | |||
| O 14 | DC 13 | 3.79 | |||
| 9 | O 28 | LYS 505 | 3.51 | −7.74 | 90.63 |
| N 16 | DC 13 | 3.21 | |||
| O 13 | GLN 778 | 3.52 | |||
| O 14 | GLN 778 | 3.44 |
Compound 2 exhibits a binding affinity of −8.23 kcal mol−1, forming hydrogen bonds with several residues. The oxygen atom at position 25 interacts with LYS 505 at a distance of 3.36 Å. Additionally, compound 2 forms hydrogen bonds with GLU 477 (3.14 Å), GLY 776 (3.04 Å), and two bonds with the DT9 residue (3.11 Å and 3.15 Å).
Compound 3 shows a slightly higher binding affinity of −8.44 kcal mol−1. This compound forms hydrogen bonds with GLU 757 (3.03 Å), LYS 759 (2.76 Å), GLN 762 (3.14 Å), and two bonds with GLU 769 (3.56 Å and 3.47 Å).
Compound 4 demonstrates a binding affinity of −8.13 kcal mol−1, with hydrogen bonds formed between O13 and ARG 729 (3.1 Å), O31 and HIS 775 (3.46 Å), O32 and HIS 775 (3.58 Å), O14 and GLU 477 (3.48 Å), and N22 and LYS 505 (3.24 Å).
Compound 5 has a binding affinity of −8.09 kcal mol−1 and forms hydrogen bonds with GLN 850 (2.91 Å), ALA 768 (2.89 Å), LYS 759 (3.25 Å), and TYR 773 (3.65 Å).
Compound 6 exhibits a lower binding affinity of −7.41 kcal mol−1, with hydrogen bonds formed with GLN 762 (3.07 Å), LYS 759 (3.06 Å), GLU 769 (3.22 Å), and another bond with GLU 769 (3.18 Å).
Compound 7 shows a binding affinity of −7.75 kcal mol−1, with interactions with GLN 762 (2.76 Å), GLU 769 (2.91 Å), GLN 850 (3.27 Å), GLU 769 (3.24 Å), GLU 757 (3.38 Å), and another bond with GLU 757 (3.18 Å).
Compound 8 has a binding affinity of −8.02 kcal mol−1, forming hydrogen bonds with DC 14 (3.35 Å), GLN 778 (3.17 Å), LYS 505 (4.47 Å), and DC 13 (3.79 Å).
Compound 9 exhibits a binding affinity of −7.74 kcal mol−1, with hydrogen bonds formed with LYS 505 (3.51 Å), DC 13 (3.21 Å), and two bonds with GLN 778 (3.52 Å and 3.44 Å).
In general, compounds 2–4 show the highest binding affinities, indicating their stronger interactions with the target protein. The order of binding affinities, from highest to lowest, is as follows: compound 3 > compound 2 > compound 4 > compound 5 > compound 8 > compound 7 > compound 9 > compound 6.
Compound 3, with the highest binding affinity, demonstrates the most favorable interactions, particularly with residues such as LYS 759 and GLU 769. Conversely, compound 6, with the lowest binding affinity, shows weaker interactions, suggesting it may be less effective in binding to the target protein.
4. Conclusion
• A diversity of 4-hydroxy-1-methylquinolin-2(1H)-one tethered pyridines, pyrido[1,2-a]benzimidazole, pyrazolo[3,4-b]pyridines and pyrido[2,3-d]pyrimidines were efficiently synthesized from recyclization reactions of the starting substrate 1 with some carbon nucleophilic reagents.• In quantum chemical calculations, the B3LYP/6-311++G (d,p) basis set was utilized to optimize the prepared compounds. From FMO, compound 3 displayed lower energy gap and lower hardness which coupled with higher softness; suggesting higher reactivity and reduced kinetic stability compared to other compounds.
• A good agreement was found for both computational and experimental infrared (IR) and nuclear magnetic resonance (NMR) spectra, where the correlation coefficient (R2) is found in the range 0.98–0.99 for 1H NMR spectra and 0.99 for IR spectra.
• According to in silico ADME investigations, these compounds showed promise drug-likeness without violating Lipinski's, Ghose's, or Veber's criteria.
• The studied molecules were evaluated for hepatocellular carcinoma (HepG-2) cell lines revealing that, compound 3 demonstrated the most anticancer potential (IC50 = 8.79 μM L−1). Moreover, the binding energy computations have confirmed the experimental data in anticancer evaluation.
• MEP analysis identified the most favorable sites for nucleophilic and electrophilic attacks. The prepared compounds have a first-order hyperpolarizability greater that urea by about 3–18 times. Thus, these compounds may have future applications in developing novel NLO materials.
Data availability
Data are available upon request from the authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- N. Omidkhah, J. Chamani, F. Fatemi, F. Hadizadeh, P. Lavaee and R. Ghodsi, Synthesis, cytotoxicity, docking, MD simulation, drug-likeness, ADMET prediction and multi spectroscopic studies of some novel quinoline-4-carboxamide derivatives as DNA intercalating and anticancer agents, J. Mol. Struct., 2025, 1322, 140334, DOI:10.1016/j.molstruc.2024.140334.
- S. D. K. Alzamili, H. S. Mohammed and T. M. Muslim, Biological activity of azo quinoline dye and its palladium (II) complex, Bull. Chem. Soc. Ethiop., 2025, 39, 91–100, DOI:10.4314/bcse.v39i1.7.
- A. Xing, P. Zhu, B. Zhang, J. Lu, Y. Zhang, D. Zeng, X. Li and J. Yuan, Synthesis, anticancer and antioxidant activities of novel heterocyclic phenolic hydrazone based derivatives: Investigation of DFT calculation, molecular docking and drug-likeness studies, J. Mol. Struct., 2025, 1319, 139523, DOI:10.1016/j.molstruc.2024.139523.
- S. Rajendran, K. Sivalingam, R. P. Karnam Jayarampillai, W. L. Wang and C. O. Salas, Friedlander's synthesis of quinolines as a pivotal step in the development of bioactive heterocyclic derivatives in the current era of medicinal chemistry, Chem. Biol. Drug Des., 2022, 100, 1042–1085, DOI:10.1111/cbdd.14044.
- H. Chen and H. A. Aisa, Alkaloids from Anacyclus pyrethrum, Phytochemistry, 2025, 229, 114255, DOI:10.1016/j.phytochem.2024.114255.
- P. l. Lam, C. w. Kan, M. C. w. Yuen, S. y. Cheung, R. Gambari, K. h. Lam, J. C. O. Tang and C. h. Chui, Studies on quinoline type dyes and their characterisation studies on acrylic fabric, Color. Technol., 2012, 128, 192–198, DOI:10.1111/j.1478-4408.2012.00363.x.
- E. A. El-Helw, E. M. Hosni, M. Kamal, A. I. Hashem and S. K. Ramadan, Synthesis, insecticidal Activity, and molecular docking analysis of some benzo [h] quinoline derivatives against Culex pipiens L. Larvae, Bioorg. Chem., 2024, 150, 107591, DOI:10.1016/j.bioorg.2024.107591.
- P. Kumar, R. Bhatia and N. K. Rangra, Scaffolds imparting anthelmintic activity: recent advancements and SAR studies, Mol. Diversity, 2024, 1–34, DOI:10.1007/s11030-024-10869-x.
- R. T. Kumah, R. O. Omondi, G. S. Smith and S. O. Ojwach, Molecular structures, DFT calculations, in silico molecular docking, predicted pharmacokinetics and biological activities of N-(quinolin-8-yl)-2-pyridine/pyrazine carboxamide compounds, J. Mol. Struct., 2025, 1319, 139425, DOI:10.1016/j.molstruc.2024.139425.
- Y. Zhao, B. Han, Z. Wei, Y. Li, Y. Yao, C. Song and Y. Duan, Discovery of a potent, Highly selective, and In vivo anti-inflammatory Efficacious, P2Y6R antagonist with a novel quinoline-pyrazole scaffold, Eur. J. Med. Chem., 2024, 279, 116890, DOI:10.1016/j.ejmech.2024.116890.
- V. K. Singh, S. Rai, A. S. Parihar, I. Ahmad, H. Patel, D. Schols and R. K. Singh, Design, synthesis, molecular docking, dynamics simulations and antiviral activities of quinoline derivatives, J. Mol. Struct., 2025, 1319, 139531, DOI:10.1016/j.molstruc.2024.139531.
- S. Khanikar, P. Joshi, A. Sharma, L. B. Marpna, T. R. A. Sangma, R. B. Browne, S. Kaping, P. Helissey, R. Tripathi and J. N. Vishwakarma, Ultrasound-assisted synthesis and structure elucidation of novel quinoline-pyrazolo[1,5-a] pyrimidine hybrids for anti-malarial potential against drug-sensitive and drug-resistant malaria parasites and molecular docking, J. Chem. Sci., 2024, 136, 1–19, DOI:10.1007/s12039-024-02294-2.
- H. Kumar, V. Devaraji, R. Joshi, M. Jadhao, P. Ahirkar, R. Prasath, P. Bhavana and S. K. Ghosh, Antihypertensive activity of a quinoline appended chalcone derivative and its site specific binding interaction with a relevant target carrier protein, RSC Adv., 2015, 5, 65496–65513, 10.1039/C5RA08778C.
- Q. Xu, Y. Tu, Y. Zhang, Y. Xiu, Z. Yu, H. Jiang and C. Wang, Discovery and biological evaluation of 6-aryl-4-(3, 4, 5-trimethoxyphenyl) quinoline derivatives with promising antitumor activities as novel colchicine-binding site inhibitors, Eur. J. Med. Chem., 2024, 279, 116869, DOI:10.1016/j.ejmech.2024.116869.
- M. Piškor, I. Ćorić, B. Perić, K. M. Špoljarić, S. I. Kirin, L. Glavaš-Obrovac and S. Raić-Malić, Quinoline-and coumarin-based ligands and their rhenium (I) tricarbonyl complexes: synthesis, spectral characterization and antiproliferative activity on T-cell lymphoma, J. Inorg. Biochem., 2025, 262, 112770, DOI:10.1016/j.jinorgbio.2024.112770.
- M. Ghasemi, A. Iraji, M. Dehghan, Y. L. Nosood, C. Irajie, N. B. Khouzani, S. Mojtabavi, M. A. Faramarzi, M. Mahdavi and A. Al-Harrasi, Rational design of new quinoline-benzimidazole scaffold bearing piperazine acetamide derivatives as antidiabetic agents, Bioorg. Chem., 2024, 153, 107908, DOI:10.1016/j.bioorg.2024.107908.
- M. A. Ibrahim, A.-S. Badran, S. A. Halim, N. Roushdy and A. Farag, Enhanced structural and optical performance of the novel 3-[(5-amino-1-phenyl-1 H-pyrazol-4-yl) carbonyl]-1-ethyl-4-hydroxyquinolin-2 (1H)-one heterojunction: experimental and DFT modeling, Opt. Quantum Electron., 2024, 56, 257, DOI:10.1007/s11082-023-05797-3.
- M. A. Ibrahim, A.-S. Badran, S. A. Halim, N. Roushdy, A. Atta and A. Farag, Examination of structural and spectrophotometric optical characteristics of nano-like flower quinolinyl carbonyl pyrazole-1-carbodithioate films: a new trend for optoelectronic applications, J. Mol. Struct., 2024, 1314, 138692, DOI:10.1016/j.molstruc.2024.138692.
- M. A. Ibrahim, N. Roushdy, A. Atta, A.-S. Badran and A. Farag, Comprehensive study on pyrano [3, 2-c] quinoline-based indole: Synthesis, characterization, and potential for optoelectronic and photovoltaic applications, J. Mol. Struct., 2024, 1312, 138660, DOI:10.1016/j.molstruc.2024.138660.
- A.-S. Badran, A. Ahmed, A. I. Nabeel and M. A. Ibrahim, Ring opening ring closure reactions with 5, 9-diethyl-7-(chromon-3-yl)-7-hydroquinolino [3′, 4′: 5, 6] pyrano [3, 2-c] quinoline-6, 8 (5H, 9H)-dione with some 1, 2-binucleophiles: Synthesis, characterization, DFT study and biological activity, J. Mol. Struct., 2024, 1298, 137030, DOI:10.1016/j.molstruc.2023.137030.
- Y. A. Alnamer, M. A. Ibrahim, A. Ahmed, N. M. Ali and A.-S. Badran, Synthetic Approaches for Annulated Quinolines at Face a, Polycyclic Aromat. Compd., 2024, 44, 2863–2886, DOI:10.1080/10406638.2023.2225674.
- A. Farag, N. Roushdy, A.-S. Badran, A. Atta, A. A. Alkathiri and M. A. Ibrahim, A comprehensive investigation of the synthesis, spectral, DFT, and optical properties of a novel oxadiazolyl-pyrano [3, 2-c] quinoline for photosensor applications, J. Mol. Struct., 2024, 1318, 139387, DOI:10.1016/j.molstruc.2024.139387.
- N. Mohamed, N. A. Alshaye, M. A. Mostafa, A.-S. Badran, Z. Hussain and M. A. Ibrahim, Synthetic approaches for quinoline heterocycles fused at face b: A review, Synth. Commun., 2024, 54, 1629–1651, DOI:10.1080/00397911.2024.2390172.
- M. M. Marinho, F. W. Q. Almeida-Neto, E. M. Marinho, L. P. da Silva, R. R. Menezes, R. P. Dos Santos, E. S. Marinho, P. de Lima-Neto and A. M. Martins, Quantum computational investigations and molecular docking studies on amentoflavone, Heliyon, 2021, 7, e06079, DOI:10.1016/j.heliyon.2021.e06079.
- A. S. Al-Barwari, A. S. Faihan, M. Ashfaq, R. Behjatmanesh-Ardakani, R. H. Al-Shammari, M. N. Tahir, M. R. Hatshan and A. S. Al-Janabi, Bis (diphenyl) phosphinomethane Platinum (II) complex of 1-(benzo[d]thiazol-2-yl)-3-phenylthiourea: Crystal structure and DFT study, J. Mol. Struct., 2024, 1304, 137624, DOI:10.1016/j.molstruc.2024.137624.
- P. Koparır, R. Omer, M. Karatepe and L. Ahmed, Synthesis, Characterization, and theoretical inhibitor study for (1E, 1'E)-2, 2'-thiobis (1-(3-mesityl-3-methylcyclobutyl) ethan-1-one) dioxime, El-Cezeri, 2020, 8, 1495–1510, DOI:10.31202/ecjse.951527.
- E. J. Braga, B. T. Corpe, M. M. Marinho and E. S. Marinho, Molecular electrostatic potential surface, HOMO–LUMO, and computational analysis of synthetic drug Rilpivirine, Int. J. Sci. Eng. Res., 2016, 7, 315–319 Search PubMed.
- M. E. I. Khan, M. Ashfaq, M. N. Tahir, K. S. Munawar, K. Ayub, F. Nawaz, M. Yar and K. Husnain, Synthesis, Characterization, Single Crystal XRD, Solid State Assembly Inspection by Hirshfeld Surface Analysis, and Theoretical Studies of Pb (II) complex with Thiourea Derivative, J. Mol. Struct., 2024, 1304, 137689, DOI:10.1016/j.molstruc.2024.137689.
- R. Satheeshkumar, R. Montecinos, A. Vera, K. J. R. Prasad, W. Kaminsky and C. O. Salas, Experimental and theoretical physicochemical study of a new dispirocompound: 4′-(4-fluorophenyl)-2′,7-dimethyl-1,4-dihydro-3H-dispiro [cyclopent[b]indol-2,5′-[1,2] oxazinan-6′, 3′-indolin]-2′, 3-dione, J. Mol. Struct., 2021, 1227, 129431, DOI:10.1016/j.molstruc.2020.129431.
- M. A. Ibrahim, H. M. Hassanin, Y. A. Gabr and Y. A. Alnamer, Studies on the chemical behavior of 3-(nitroacetyl)-1-ethyl-4-hydroxyquinolin-2(1H)-one towards some electrophilic and nucleophilic reagents, J. Braz. Chem. Soc., 2012, 23, 905–912, DOI:10.1590/S0103-50532012000500016.
- Y. El Bakri, D. A. Safin, S. K. Mohamed, I. S. Marae, E. A. Bakhite, E. Khamies, A. A. E. Soliman, S. Abuelhassan, H. A. Abuelizzg, R. Al-Salahi, J. T. Mague and C. H. Lai, A highly substituted isoquinolinethione: Synthesis, crystal structure, DFT analysis and molecular docking studies against a series of the SARS-CoV-2 proteins, J. Mol. Struct., 2025, 1331, 141527, DOI:10.1016/j.molstruc.2025.141527.
- S. Celik, DFT investigations and molecular docking as potent inhibitors of SARS- CoV-2 main protease of 4-phenylpyrimidine, J. Mol. Struct., 2023, 1277, 134895, DOI:10.1016/j.molstruc.2022.134895.
- V. Shalini, D. C. V. Kumar, D. Gowda, B. S. Chethan, K. B. Harsha, S. M. Rajesh and K. S. Rangappa, Unveiling the structural and theoretical properties of 6-(2-fluoro-3-methylpyridin-4-yl)-2-(4-methoxyphenyl)-N-phenylquinoline-4-carboxamide compound as Sonic Hedgehog protein inhibitor: Synthesis, SCXRD, HSA, DFT, Docking and ADMET studies, J. Mol. Struct., 2025, 1330, 141495, DOI:10.1016/j.molstruc.2025.141495.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, B. E, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. J. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, D. 0109, Revision D. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, 5.0. 8, Gaussian Inc., 2008 Search PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112, 8251–8260, DOI:10.1021/ja00179a005.
- F. Hussain, R. Hussain, M. Adnan, S. Muhammad, Z. Irshad, M. U. Khan, J. Yaqoob and K. Ayub, Insights into the nonlinear optical (NLO) response of pure Aum (2 ≥ m ≤ 7) and copper-doped Aum–xCux clusters, RSC Adv., 2022, 12, 25143–25153, 10.1039/D2RA03664A.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- A. Sabt, W. M. Eldehna, T. Al-Warhi, O. J. Alotaibi, M. M. Elaasser, H. Suliman and H. A. Abdel-Aziz, Discovery of 3, 6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: synthesis, biological evaluation and in silico insights, J. Enzyme Inhib. Med. Chem., 2020, 35, 1616–1630, DOI:10.1080/14756366.2020.1806259.
- S. M. Riyadh, S. M. Gomha, E. A. Mahmmoud and M. M. Elaasser, Synthesis and anticancer activities of thiazoles, 1, 3-thiazines, and thiazolidine using chitosan-grafted-poly (vinylpyridine) as basic catalyst, Heterocycles, 2015, 91, 1227, DOI:10.1002/chin.201538150.
- S. Mandal, A. Pan, R. Bhaduri, S. K. Tarai, B. S. Kapoor and S. C. Moi, Theoretical investigation on hydrolysis mechanism of cis-platin analogous Pt (II)/Pd (II) complex by DFT calculation and molecular docking approach for their interaction with DNA & HSA, J. Mol. Graphics Modell., 2022, 117, 108314, DOI:10.1016/J.JMGM.2022.108314.
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Computational methods in drug discovery, Pharmacol. Rev., 2014, 66, 334–395, DOI:10.1124/pr.112.007336.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17, DOI:10.1016/j.addr.2012.09.019.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases, J. Comb. Chem., 1999, 1, 55–68, DOI:10.1021/cc9800071.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623, DOI:10.1021/jm020017n.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461, DOI:10.1002/jcc.21334.
- N. C. Suresh, B. A. Kumar, H. D. Preetham, S. M. Srinivasa, M. S. Ali, H. A. Al-Lohedan, K. S. S. Kumar, C. Shivamallu, A. Jain and S. Rangappa, Synthesis, molecular docking and pharmacological studies of novel quinoline derivative as anticancer agent that targets topoisomerase IIB, J. Mol. Struct., 2024, 1312, 138519, DOI:10.1016/j.molstruc.2024.138519.
- A. Zahirović, S. Fetahović, M. Feizi-Dehnayebi, R. Bešta-Gajević, M. Dizdar, J. Ostojić and S. Roca, Substituent effect in salicylaldehyde 2-furoic acid hydrazones: Theoretical and experimental insights into DNA/BSA affinity modulation, antimicrobial and antioxidant activity, J. Mol. Struct., 2024, 1312, 138628, DOI:10.1016/j.molstruc.2024.138628.
- S. Asghar, N. Mushtaq, A. Ahmed, L. Anwar, R. Munawar and S. Akhtar, Potential of Tryptamine Derivatives as Multi-Target Directed Ligands for Alzheimer's Disease: AChE, MAO-B, and COX-2 as Molecular Targets, Molecules, 2024, 29(2), 490, DOI:10.3390/molecules29020490.
- R. K. Mohapatra, A. Mahal, A. Ansari, M. Kumar, J. P. Guru, A. K. Sarangi, A. Abdou, S. Mishra, M. Aljeldah and B. M. AlShehail, Comparison of the binding energies of approved mpox drugs and phytochemicals through molecular docking, molecular dynamics simulation, and ADMET studies: An in silico approach, J. Biosaf. Biosecur., 2023, 5, 118–132, DOI:10.1016/j.jobb.2023.09.001.
- H. M. Hassanin and D. Abdel-Kader, Synthesis of some novel binuclear heterocyclic compounds from 6-ethyl-3-nitropyrano[3,2-c]quinoline-4,5(6H)-dione, Heterocycles, 2013, 87(2), 369–380, DOI:10.3987/com-12-12639.
- A. Kumar and K. R. Prabhu, Rhodium(III)-Catalyzed C−H Activation: A Cascade Approach for the Regioselective Synthesis of Fused Heterocyclic Lactone Scaffolds, J. Org. Chem., 2020, 85, 3548–3559, DOI:10.1021/acs.joc.9b03266.
- C. K. Ozer, U. Solmaz and H. Arslan, Crystal structure, Hirshfeld surface analysis, and DFT studies of N-(2-chlorophenylcarbamothioyl) cyclohexanecarboxamide, Eur. J. Chem., 2021, 12, 439–449, DOI:10.5155/eurjchem.12.4.439-449.2196.
- M. M. Khalaf, H. M. Abd El-Lateef, M. Gouda, A. A. Amer, A. A. Abdelhamid, M. F. Abou Taleb, A. Alfarsi, T. M. A. Ibrahim, H. El-Shamy and A. Abdou, Synthesis, characterization, DFT, biological activity evaluation, and molecular docking analysis of new 8-[(2-hydroxynaphthalen-1-yl) diazenyl] naphthalene-1, 3-disulfonic acid based complexes, J. Mol. Struct., 2024, 1300, 137175, DOI:10.1016/j.molstruc.2023.137175.
- S. S. Ramasamy, K. Adhigaman, V. Nandakumar, A. Sundarasamy, S. Jagadeesan, M. Saravanakumar, J. G. Malecki, N. Easwaran and S. Thangaraj, In-Silico exploration: Unraveling the anti-cancer potential of 8-Nitroquinoline hydrazides, J. Mol. Struct., 2025, 1321, 140218, DOI:10.1016/j.molstruc.2024.140218.
- C. Kucuk, S. Celik, S. Yurdakul and E. Coteli, A new Ag(I)-complex of 5-chloroquinolin-8-ol ligand: Synthesis, spectroscopic characterization, and DFT investigations, in vitro antioxidant (DPPH and ABTS), α-glucosidase, α-amylase inhibitory activities with protein-binding analysis, J. Mol. Struct., 2025, 1325, 141285, DOI:10.1016/j.molstruc.2024.141285.
- L. Feng, H. Yang, X. Cui, D. Chen and G. Li, Experimental and theoretical investigation on corrosion inhibitive properties of steel rebar by a newly designed environmentally friendly inhibitor formula, RSC Adv., 2018, 8, 6507–6518, 10.1039/c7ra13045g.
- N. A. Alshaye, M. A. Ibrahim and A. Badran, Nucleophilic transformation of 3-substituted-6,8-dimethylchromones with phenylhydrazine under various reaction conditions: Theoretical, Spectroscopic characterization and in silico ADME studies, J. Mol. Struct., 2025, 1297, 37006, DOI:10.1016/j.molstruc.2023.137006.
- H. M. Abd El-Lateef, A. M. Ali, M. M. Khalaf and A. Abdou, New iron (III), cobalt (II), nickel (II), copper (II), zinc (II) mixed-ligand complexes: Synthesis, structural, DFT, molecular docking and antimicrobial analysis, Bull. Chem. Soc. Ethiop., 2024, 38, 147–166, DOI:10.4314/bcse.v38i1.12.
- H. M. Abd El-Lateef, M. M. Khalaf, F. E. T. Heakal and A. Abdou, Fe (III), Ni (II), and Cu (II)-moxifloxacin-tri-substituted imidazole mixed ligand complexes: Synthesis, structural, DFT, biological, and protein-binding analysis, Inorg. Chem. Commun., 2023, 158, 111486, DOI:10.1016/j.inoche.2023.111486.
- H. M. Abd El-Lateef, M. M. Khalaf, A. A. Amer, A. A. Abdelhamid and A. Abdou, Antibacterial, antifungal, anti-inflammatory evaluation, molecular docking, and density functional theory exploration of 2-(1 H-benzimidazol-2-yl) guanidine mixed-ligand complexes: Synthesis and characterization, Appl. Organomet. Chem., 2024, 38, e7299, DOI:10.1002/aoc.7299.
- R. Suja, A. Rathika, V. S. J. Reeda, A. A. kumar and P. Divya, Synthesis, spectroscopic analysis (FT-IR, FT-Raman, UV, NMR), non-covalent interactions (RDG, IGM) and dynamic simulation on Bis (8-hydroxyquinoline)salicylate salicylic acid, J. Mol. Struct., 2024, 1310, 138231, DOI:10.1016/j.molstruc.2024.138231.
- M. Bazrafshan, M. Vakili, S. F. Tayyari, F. S. Kamounah, P. E. Hansen and A. Shiri, Synthesis, molecular structure, conformational, and intramolecular hydrogen bond strength of ethyl 3-amino-2-butenoate and its N-Me, N-Ph, and N-Bn analogs; An experimental and theoretical study, J. Mol. Struct., 2023, 1274, 134479, DOI:10.1016/j.molstruc.2022.134479.
- H. Tiernan, B. Byrne and S. G. Kazarian, ATR-FTIR spectroscopy and spectroscopic imaging for the analysis of biopharmaceuticals, Spectrochim. Acta, Part A, 2020, 241, 118636, DOI:10.1016/j.saa.2020.118636.
- R. A. Omer, K. M. Ahmed, K. A. Omar, W. M. Hamad and D. M. Mamad, N, N-Bis (2, 4-dihydroxy benzaldehyde) benzidine: Synthesis, characterization, DFT, and theoretical corrosion study, J. Mol. Struct., 2024, 1300, 137279, DOI:10.1016/j.molstruc.2023.137279.
- S. Gul, A. Alam, M. Assad, A. A. Elhenawy, M. S. Islam, S. A. A. Shah, Z. Parveen, T. A. Shah and M. Ahmad, Exploring the synthesis, molecular structure and biological activities of novel Bis-Schiff base derivatives: A combined theoretical and experimental approach, J. Mol. Struct., 2024, 1306, 137828, DOI:10.1016/j.molstruc.2024.137828.
- Y. Sert, M. R. Albayati, F. Şen and N. Dege, The DFT and in-silico analysis of 2, 2′-((1e, 1′e)-((3, 3′-dimethyl-[1, 1′-biphenyl]− 4, 4′ diyl) bis (azanylylidene)) bis (methanylylidene)) diphenol molecule, Colloids Surf., A, 2024, 687, 133444, DOI:10.1016/j.colsurfa.2024.133444.
- M. S. Elkotamy, M. K. Elgohary, S. T. Al-Rashood, H. Almahli, W. M. Eldehna and H. A. Abdel-Aziz, Novel imidazo [2, 1-b] thiazoles and imidazo [1, 2-a] pyridines tethered with indolinone motif as VEGFR-2 inhibitors and apoptotic inducers: Design, synthesis and biological evaluations, Bioorg. Chem., 2024, 151, 107644, DOI:10.1016/j.bioorg.2024.107644.
- N. Uludağ and G. Serdaroğlu, An improved synthesis, spectroscopic (FT-IR, NMR) study and DFT computational analysis (IR, NMR, UV–Vis, MEP diagrams, NBO, NLO, FMO) of the 1,5-methanoazocino [4,3-b]indole core structure, J. Mol. Struct., 2018, 1155, 548–560, DOI:10.1016/j.molstruc.2017.11.032.
- C. F. Matta, Modeling biophysical and biological properties from the characteristics of the molecular electron density, electron localization and delocalization matrices, and the electrostatic potential, J. Comput. Chem., 2014, 35, 1165–1198, DOI:10.1002/jcc.23608.
- A. M. Abu-Dief, R. M. El-Khatib, T. El-Dabea, S. A. Abdel-Latif, I. O. Barnawi, F. S. Aljohani, K. Al-Ghamdi and M. A. E. A. A. A. El-Remaily, Design, synthesize, physicochemical characterization, nonlinear optical properties structural elucidation, biomedical studies, and DNA interaction of some new mixed ligand complexes incorporating 4,6-dimethylpyrimidine derivative and imidazole ligand, Appl. Organomet. Chem., 2024, 38, e7463, DOI:10.1002/aoc.7463.
- T. Rajamani and S. Muthu, Electronic absorption, vibrational spectra, non-linear optical properties, NBO analysis and thermodynamic properties of 9-[(2-hydroxyethoxy) methyl] guanine molecule by density functional method, Solid State Sci., 2013, 16, 90–101, DOI:10.1016/j.solidstatesciences.2012.10.023.
- C. Adant, M. Dupuis and J. Bredas, Ab initio study of the nonlinear optical properties of urea: electron correlation and dispersion effects, Int. J. Quantum Chem., 1995, 56, 497–507, DOI:10.1002/qua.560560853.
- Z. Akbari, C. Stagno, N. Iraci, T. Efferth, E. A. Omer, A. Piperno, M. Montazerozohori, M. Feizi-Dehnayebi and N. Micale, Biological evaluation, DFT, MEP, HOMO-LUMO analysis and ensemble docking studies of Zn(II) complexes of bidentate and tetradentate Schiff base ligands as antileukemia agents, J. Mol. Struct., 2024, 1301, 137400, DOI:10.1016/j.molstruc.2023.137400.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- V. Pogaku, K. Gangarapu, S. Basavoju, K. K. Tatapudi and S. B. Katragadda, Design, synthesis, molecular modelling, ADME prediction and anti-hyperglycemic evaluation of new pyrazole-triazolopyrimidine hybrids as potent α-glucosidase inhibitors, Bioorg. Chem., 2019, 93, 103307, DOI:10.1016/j.bioorg.2019.103307.
- T. Lanez, M. Feizi-Dehnayebi and E. Lanez, Assessment of the electrostatic binding of ferrocenylmethyl-nitroaniline derivatives to DNA: A combined experimental and theoretical study, J. Mol. Struct., 2024, 1308, 138386, DOI:10.1016/j.molstruc.2024.138386.
- A. M. Abu-Dief, O. A. Omran, M. Feizi-Dehnayebi, A. Alqurashi, I. Omar, D. Alhashmialameer and A. D. M. Mohamad, Fabrication, structural elucidation, and DFT calculation of some new hydrophilic metal chelates based on NN′-(1-methyl-2-oxoindolin-3-ylidene) benzohydrazide ligand: Pharmaceutical studies and molecular docking approach, Appl. Organomet. Chem., 2024, 38, e7593, DOI:10.1002/aoc.7593.
- A. M. El-Saghier, S. S. Enaili, A. M. Kadry, A. Abdou and M. A. Gad, Green synthesis, biological and molecular docking of some novel sulfonamide thiadiazole derivatives as potential insecticidal against Spodoptera littoralis, Sci. Rep., 2023, 13, 19142, DOI:10.1038/s41598-023-46602-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00325c |
| This journal is © The Royal Society of Chemistry 2025 |