
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum(II) complexes of aryl guanidine-like derivatives as potential anticancer agents: between coordination and cyclometallation†
Patrick O'Sullivana,
Viola Previtalia,
Brendan Twamleya,
Celine J. Marmion b,
Aidan R. McDonalda and
Isabel Rozas*a
b,
Aidan R. McDonalda and
Isabel Rozas*a
aSchool of Chemistry, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: rozasi@tcd.ie; Tel: +353 1 896 3731
bDepartment of Chemistry, RCSI University of Medicine and Health Sciences, 123 St. Stephen's Green, Dublin 2, Ireland
First published on 4th February 2025
Abstract
The preparation of a wide variety of Pt(II) complexes with aryl guanidines and their potential application as anticancer agents have been explored. A relatively facile synthesis of cyclometallated Pt(II) complexes of arylguanidines, preparation of Pt(II) guanidine coordination complexes and an in situ activation of platinum arylguanidine complexes with acetonitrile to create a bidentate aryl iminoguanidine Pt(II) complex were achieved. Cyclometallation methodology was extended to create a water-stable conjugate incorporating two Pt(II) ions and a diaryl bis-guanidine DNA minor groove binder. Several crystal structures were obtained confirming these complexation modes. The cyclometallated Pt(II) complexes were particularly stable to aqueous environments and were tested for Reactive Oxygen Species generation and anticancer activity in a leukaemia cancer cell line.
Introduction
It is well known that platinum complexes like cis-platin and carboplatin play a crucial role in the treatment of several cancers. They exert their therapeutic activity by forming DNA adducts within cancer cells, thus inhibiting DNA replication and transcription and ultimately leading to cancer cell apoptosis or cell death.1,2 Platinum drugs are also widely employed in combination therapy regimens.3,4 Notwithstanding their success, platinum-based therapies also exhibit secondary adverse effects (myelosuppression, nephrotoxicity, ototoxicity, thrombocytopenia or extensive plasma protein binding) that limit their use and/or dose.5 The development of cancer drug resistance is also a major challenge. To overcome these unwanted shortcomings, new platinum-based derivatives are being continuously developed. These include but are not limited to non-classical Pt(II) complexes, polynuclear Pt(II) compounds, photoactivatable Pt(IV) complexes6 or multi-targeted Pt(II) and Pt(IV) complexes, as summarised in recent comprehensive reviews.7 The multi-targeted approach has led to highly efficacious Pt(II) complexes which not only target cancer cell DNA but also, amongst others, enzymes such as histone deacetylases, kinases, reductases, matrix metalloproteases and DNA topoisomerases, peptides and intracellular proteins such as STAT3 and tubulin. Examples also include Pt-DNA targeting agents that carry vectors such as sugars and hormone or integrin receptors to enable them selectively target cancer cells.Among the non-classical Pt-based derivatives, Pt(II) complexes of guanidine-like systems are particularly interesting. Guanidines have been shown to possess versatile biological properties with numerous reports of guanidine derivatives possessing anticancer, antibacterial, antifungal, antiprotozoal and antiviral activities and some guanidine-based drugs are already in clinical use, all of which are eloquently summarised by Roleira et al. in a 2023 comprehensive review.8 Guanidines are versatile ligands possessing a rich coordination chemistry. Different metal complexes with neutral guanidine, anionic guanidinate and dianionic guanidinate are known.9,10 For example, Jakupec and Keppler reported novel trans Pt complexes of guanidine synthesized by the nucleophilic addition of methylamine to di-alkyl cyanamide ligands of push–pull trans Pt nitrile complexes. Their in vitro assessment in different cancer cell lines indicated that the cytotoxicity of several trans-3,6 complexes was higher than that of cis-3,6 analogues. DNA interaction studies with some of these guanidine complexes confirmed that these compounds alter the DNA secondary structure, suggesting DNA as their possible target.10 Additionally, Carrillo-Hermosilla et al. published a very thorough review on the organometallic chemistry of guanidinate compounds, examining particular modes of coordination, reactivity and applications in catalysis or materials science.11 Thirupathi et al. reported in 2013 a series of interesting cis, trans and cycloplatinated guanidine Pt(II) complexes. They achieved complexation of symmetrically tri-substituted aromatic guanidines using cis-[Cl2Pt(S(O)Me2)2] and NaOAc in refluxing methanol. Depending on the reflux time and nature of the substituents in the aryl systems, they achieved guanidine complexation or cycloplatination.12 Furthermore, Nieto et al. reported the reaction of a Pt(II) reagent with ferrocene derivatives of substituted guanidines that produced the corresponding heterometallic complexes; further they proved that guanidine-based ferrocene–Pt complexes were active against different human cancer cells.13
Given the known propensity of the guanidium cation to form non-covalent interactions with the phosphate residues of DNA's minor groove helix and the fact that clinically used Pt(II) drugs also target DNA albeit at a different site, namely by Pt binding to DNA nucleobases, we sought to explore the chemistry and biological activity of novel guanidine Pt(II) complexes as a new class of anticancer agent. We had previously studied their suitability from a theoretical point of view,14 and found that the most favourable Pt(II) complexes with guanidine derivatives were monodentate coordinated systems. Informed by these theoretical studies, our goal was to develop Pt(II) complexes of aryl guanidine-like systems with the final aim to produce cytotoxic agents.
Results and discussion
Preparation of guanidine-like ligands
To explore the best Pt complexation approach, mono-guanidine aryl systems (phenyl, pyridine, benzyl) with different substituents (p-OMe, o-NH2) were selected, not only to probe the electron density effect on the aryl ring, but also to obtain a diverse range of possible metal-binding domains, (e.g. pyridine N or o-NH2 vs. guanidine/2-aminoimidazoline N atoms). Since complexation of guanidines with Pt may be carried out by direct reaction of the corresponding ligands with a Pt source, the corresponding aryl guanidine-based ligands were first synthesised (see details in ESI†). Thus, aryl guanidinium salts (1–5), 2-(iminophenyl)imidazolidinium (6) and benzylguanidinium (7) were prepared following standard methods (Fig. 1). | ||
| Fig. 1 Structures of the guanidinium-like hydrochlorides 1–7, prepared as models to explore the conditions for the preparation of guanidine-Pt(II) complexes. | ||
To prepare the corresponding Pt complexes, the conditions reported by Aitken et al.15 for the aminoguanidine complexation were followed by treating guanidinium salts 1 and 2 (5 eq.) with K2PtCl4 (1 eq.) in water in the absence of base. Precipitates, that were only soluble in DMSO, instantly formed corresponding to the tetrachloroplatinate salts of 1 and 2, clearly indicating that the use of the free base of these arylguanidinium salts was required in order to achieve Pt complexation. Accordingly, different bases (NH4OH, KOtBu, KOH) in solutions of dichloromethane/water were explored to isolate the free base of 1 used as a model (Table 1). In all cases, the yields obtained were not ideal (0–66%, entries 1–3, Table 1) because the neutral guanidine was soluble in water. Considering that Yamada et al. had described the formation of a guanidine derivative in ethanol using sodium ethoxide as a base,16 and after adapting the procedure, the free base 8 was quantitatively obtained (entries 4–5, Table 1).

With these optimal conditions in hand (i.e., 1.1 equiv. of sodium ethoxide in ethanol), conversion of the corresponding aryl guanidinium (1–5), 2-aminoimidazolinium (6) and benzyl guanidinium (7) salts to the corresponding free bases 8–14, was achieved with quantitative yields (Scheme 1).
 | ||
| Scheme 1 Aryl guanidinium/2-aminoimidazolinium derivatives converted to the corresponding aryl guanidine free base. | ||
Thus, seven guanidine-like ligands were prepared with the aim of forming the corresponding Pt(II) complexes.
Preparation of cyclometallated Pt(II) guanidine complexes
Once these mono-guanidines were prepared, different approaches for their complexation with Pt(II) were explored. First, commercial K2PtCl4 was reacted with phenylguanidine 8 in the presence of aqueous KOH at pH = 9 and pH = 13, at room temperature and at reflux, yielding in all cases a black precipitate of Pt(0) as the major product. Similarly, reaction of the free bases 8, 9 and 13 with K2PtCl4 in MeOH or H2O at room temperature in subdued lighting conditions led to decomposition to Pt(0). Next, considering that cis-[PtCl2(DMSO)2] had been reported to form Pt(II) complexes with guanidine N atoms,13,17,18 this agent was tested to achieve complexation of the aryl guanidines prepared.Complex cis-[PtCl2(DMSO)2] was prepared as previously described in the literature.19 Initial attempts of complexation using two equivalents of 8 and cis-[PtCl2(DMSO)2] in THF or chloroform at reflux resulted in the recovery of starting materials. When the reaction was carried out with one equivalent of 8 in methanol at reflux, two products were obtained, the cationic precursor of the starting material (i.e. compound 1) and a new derivative 15. After filtration and solvent removal in vacuo, the product was dissolved in the minimum amount of DMSO and precipitated out by addition of water. The solid obtained was characterised by NMR and IR spectroscopy. TOCSY and NOESY experiments, together with high temperature 1H NMR experiments of 15, showed Pt-complexation as indicated by satellite peaks at 6.3 ppm (NH) and 8.0 ppm (CH) (Fig. S1†). Additionally, the 13C NMR spectrum indicated coordination with DMSO and the IR spectrum showed an S–O stretch at 1091 cm−1. Furthermore, low resolution mass spectrometry revealed an [M + H]+ peak at 443.02, with a characteristic isotope splitting pattern for Pt and matching a calculated formula of [C9H15ClN3OPtS]+, thus suggesting that the fourth ligand must be Cl. Although there was no direct evidence of the arrangement of the Cl and DMSO ligands relative to the aryl guanidine, it was reasoned that DMSO would not bind trans due to the strong trans-effect of the cyclometallated group. Taking all this data into account a structure for the cyclometallated compound 15 was proposed (see Scheme 2). As mentioned, the reaction of one equivalent of free base 8 with excess of cis-[PtCl2(DMSO)2] produced cyclometallated product 15 and guanidinium salt 1. However, since a second equivalent of free 8 could act as a base and then generate a new molecule of free base 8 from the in situ formed salt 1, an equivalent of NaOMe was added to optimise the synthetic procedure, yielding exclusively the cyclometallated derivative 15 (Scheme 2). The reaction mixture was purified by repeated precipitation from a concentrated DMSO mixture with water and crystals of suitable quality for X-ray crystallography were grown in darkness at room temperature over a period of three months by slow evaporation of water into a concentrated DMSO solution of 15. Gratifyingly, the structure confirmed our solution phase assignment (see Scheme 2, Table S1 and details in ESI†).
 | ||
| Scheme 2 Preparation of cyclometallated Pt(II) complexes 15 and 16 and crystal structures of the asymmetric unit in the corresponding complexes. Displacement ellipsoids are at 50% probability. | ||
Complex 15 crystallised in an orthorhombic unit cell (0.71 Å resolution); the relative configuration of the ligands was confirmed to be SP-4-4 (square planar 4-coordinate with the highest priority ligand, in this case Cl, trans to the fourth priority ligand, C). The strong trans-effect of C was demonstrated by the long Pt–Cl bond (2.4095(6) Å). The distances around the central guanidine C atom (C8) indicate that the double bond is localised between N10–C8 (1.306(3) Å) and that the other C–N bonds are single bonds in nature (i.e. N7–C8, 1.354(3) Å; and N9–C8, 1.350(3) Å). The N–Pt–C and Cl–Pt–S angles around Pt were 89.13(10)° and 92.57(2)° respectively, close to the ideal bond angle of 90° for a square planar complex. The crystal is stabilised by intermolecular hydrogen bonds (HBs) between the DMSO's O atom of one molecule and a H atom on the guanidine NH2 of another molecule.
A similar reaction using 2-aminoimidazolidine 13 produced the corresponding cyclometallated complex 16 (Scheme 2, Table S1 and details in ESI†) and, under identical conditions for crystallisation as those used with 15, suitable crystals for X-ray crystallography were formed and resolved (see Scheme 2). The crystal structure of 16 was very similar to that of 15; the Pt–Cl bond length was 2.4117(4) Å, and the N–Pt–C and Cl–Pt–S angles were 86.81(6)° and 87.785(15)°, and the unit cell was orthorhombic (0.71 Å resolution), in the space group Pbca. Like in complex 15, the distances around the central guanidine C atom (C8) indicate a localised double bond between N9–C8 (1.312(2) Å) while N7–C8 (1.345(2) Å), and N12–C8 (1.365(2) Å) seem to be single bonds. The main difference in the crystal packing was that 16 contained an intermolecular HB between the DMSO's O1 atom and the aniline-type N7′H of another molecule.
When reactions of cis-[PtCl2(DMSO)2] with p-methoxy phenylguanidine 9, pyridine guanidines 11 and 12, and benzylguanidine 14, were attempted in similar conditions, no products were obtained. However, the reaction with the o-amino phenylguanidine 10 yielded a bimetallic species (17, Fig. 2) In this complex, a combination of cyclometallated and non-cyclometallated interactions with each Pt(II) centre is observed. Small purple crystals of suitable quality for X-ray diffraction were grown from a mixture of DMSO and H2O over a period of six weeks. Complex 17 crystallised in a monoclinic unit cell (P21/n space group, 0.80 Å resolution, Fig. 2, Table S1 and details in ESI†). One of the Pt–Cl bonds is long, (2.421(3) Å) whereas the other is shorter (2.329(3) Å), again showing the strong trans-effect of C, with N–Pt–C and Cl–Pt–S angles of 85.4(4)° and 93.38(11)° respectively. Contrary to what was observed in complexes 15 and 16, distances around the central guanidine C atom (C8) point toward a delocalised double bond between N1–C7 (1.338(15) Å) and N3–C7 (1.313(14) Å), but this delocalization does not extend to N4–C7 (1.395(14) Å). When 17 was put in a solution of DMSO-d6 at room temperature, the compound degraded and was insoluble in any other solvent.
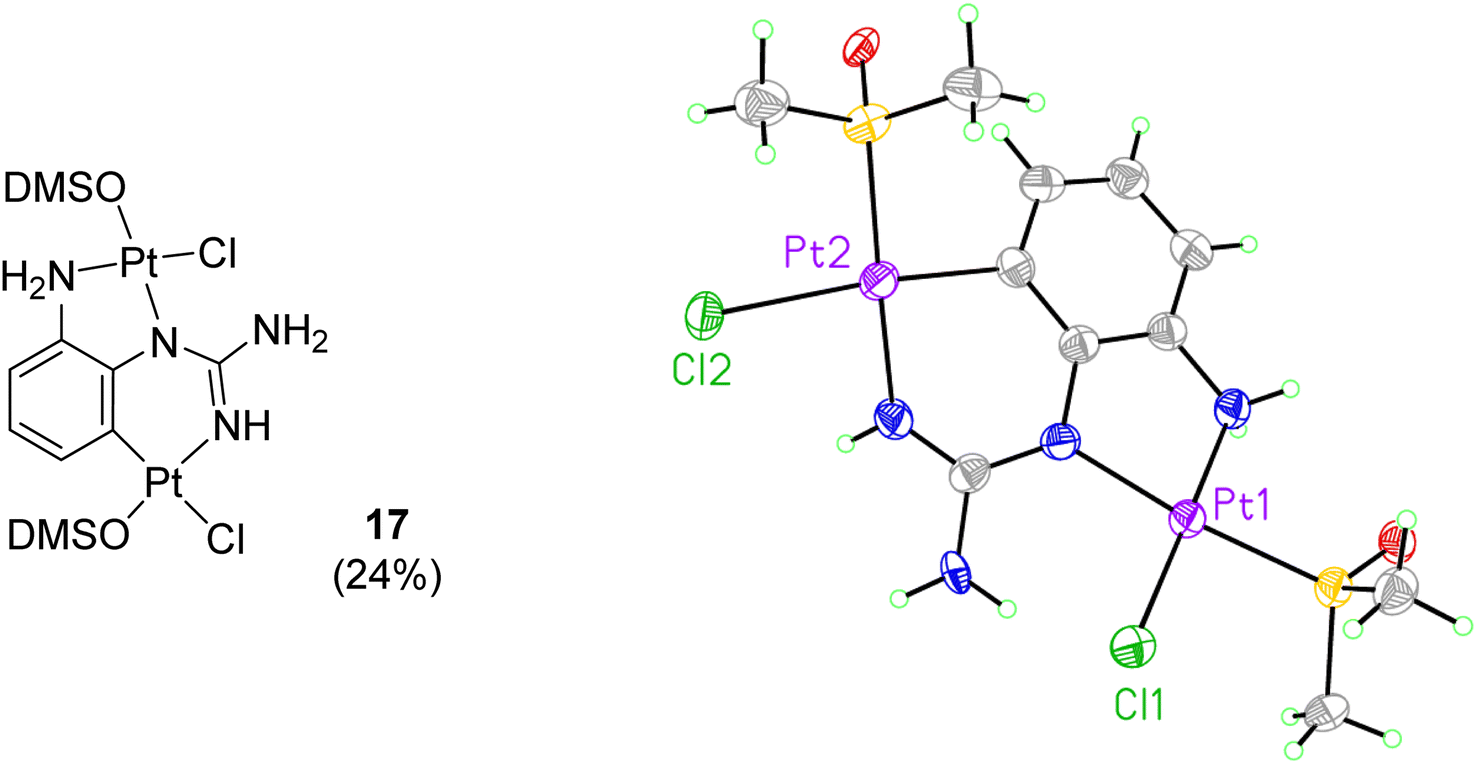 | ||
| Fig. 2 Crystal structure of bimetallic complex 17. Displacement ellipsoids are at 50% probability. | ||
Once suitable conditions were found for the preparation of some phenyl guanidine-like Pt(II) cyclometallated complexes we proceed to explore the requirements to prepare non-cyclometallated complexes.
Preparation of coordinated Pt(II) guanidine complexes
Aiming to prepare non-cyclometallated Pt(II) arylguanidine complexes, other Pt precursors were investigated. Thus, as in the synthesis of cis-platin, K2PtI4 was tested with the electron-rich p-methoxyphenyl guanidine 9. Accordingly, KI (6 eq.) was added to a solution of K2PtCl4 in H2O at 55 °C for 30 min to generate K2PtI4 in situ.20 Then, free base 9 (2 eq.) was added at room temperature and the reaction stirred for 15 min. A brown precipitate was isolated by filtration and washed with water, dissolved subsequently in hot EtOAc, purified by slow diffusion of Et2O and then filtered. Crystals suitable for X-ray diffraction were grown from a standing solution of the filtrate to give 18 (Scheme 3, Table S1 and details in ESI†). The crystal structure of 18 (P21/c, monoclinic, 0.78 Å resolution) was not the expected coordination complex and contained a mixture of cyclometallated and non-cyclometalated guanidine Pt interactions. This structure was notable for many features (see Scheme 3). First, the dinuclear structure [Pt2(p-OMe-PhGua)3I3] contained a Pt–Pt bond (2.6149(3) Å), with each Pt bound to one axial I atom (Pt–I = 2.7690(4) Å and 2.7412(4) Å) and one bridging I atom (Pt–I = 2.7967(4) Å and 2.8230(4) Å, Pt–I–Pt = 55.459(9)°). Moreover, each Pt atom was forming a cyclometallated complex with two different molecules of 9 (C–Pt–N = 91.97(19)° and 87.41(19)°), and there was also a bridging guanidinate group of a third 9 molecule (each Pt–N bond = 2.021(4), 2.024(4) Å). | ||
| Scheme 3 Preparation of dinuclear Pt(III) complex 18 and crystal structure of the asymmetric unit. Displacement ellipsoids are at 50% probability. | ||
There are two solvent pores in the crystal (Et2O and H2O). The three negatively charged guanidine and iodine ligands require a +6 overall charge; hence, considering the almost symmetrical nature of the complex, we can assume these two Pt centres to be Pt(III). Most Pt(III) complexes are bimetallic, stabilising the unpaired radicals in a Pt–Pt bond; besides, reported bimetallic Pt(III) complexes have a distorted octahedral geometry, similar to that of 18.21–23 The 1H NMR in DMSO-d6 of 18 suggested that in solution the complex either decomposes or exists as a mixture of species.
To prevent cyclometallation, milder conditions based on work by Kukushkin were attempted.24,25 Thus, K[PtCl3(DMSO)] was synthesised in situ, followed by addition of water-soluble ligand 13 and CH2Cl2 at room temperature to obtain trans-dichloro coordination/non-cyclometallated complex 19 after evaporation of the organic layer at room temperature and quick chromatography on silica (Scheme 4). Since pure 19 decomposed in DMSO, all 1H NMR experiments were carried out in DMF-d7 in which the complex is stable for at least seven days. The NOE experiments of compound 19 in DMF-d7 showed that no such effect existed between the DMSO protons and any other protons on the molecule, suggesting that DMSO is trans to the 2-aminoimidazoline ligand. High resolution mass spectrometry (ESI−) confirmed a molecular mass in agreement with the molecular formula C11H17Cl2N3OPtS. Crystals suitable for X-ray crystallography were grown over 12 h by slow diffusion of Et2O into a concentrated solution of 19 in CH2Cl2 (see Scheme 4, Table S1 and details in ESI†). The crystal structure of complex 19 again demonstrated that DMSO binds to Pt trans to the guanidine-like N. The compound crystallised in an orthorhombic unit cell (resolution = 0.71 Å) and P21 21 21 space group. The crystal packing is stabilised by an intermolecular HB between the DMSO O1 of one molecule and the aniline-type N6H of another molecule. The Pt–Cl bond lengths are both 2.3101(10) and 2.3130(10) Å and the Cl–Pt–Cl angle is 174.65(4)° whereas the N–Pt–S angle is 179.19(10)°.
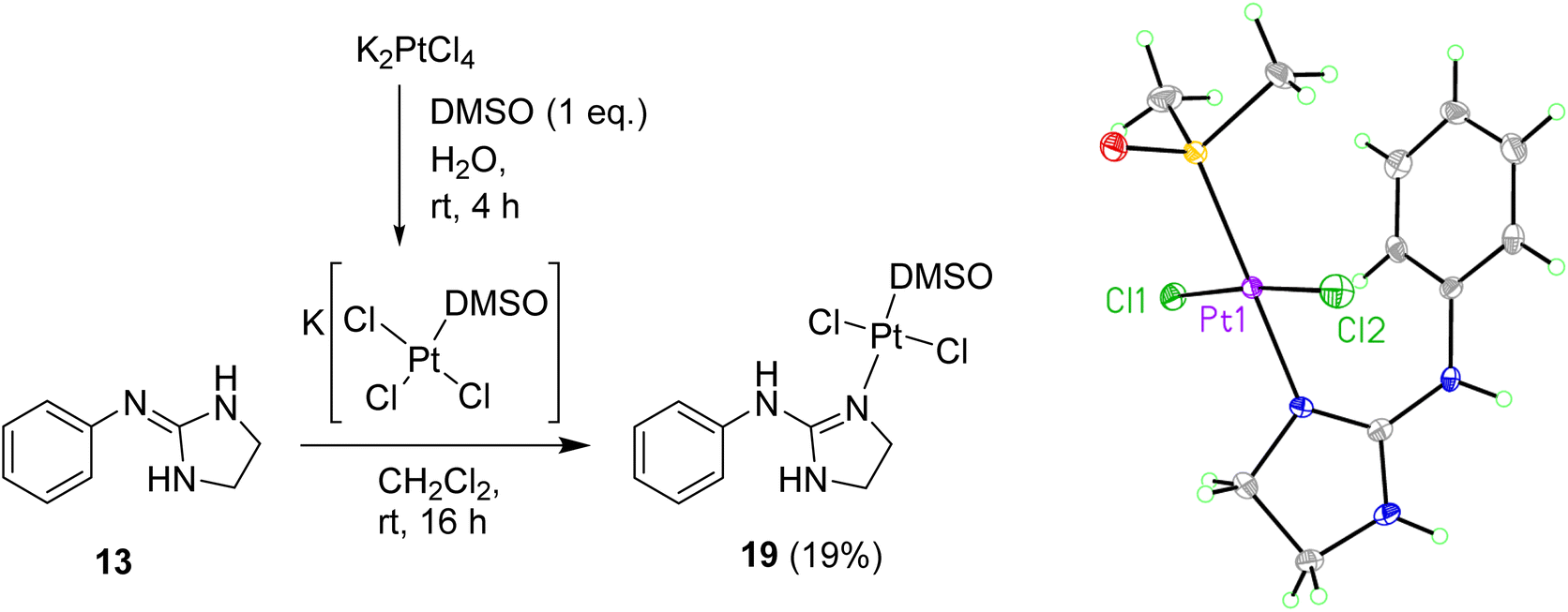 | ||
| Scheme 4 Preparation of Pt complex 19 and crystal structure of the asymmetric unit. Displacement ellipsoids are at 50% probability. | ||
Exploring other conditions for the obtention of 19 in better yields, two equivalents of free base 13 were reacted with K[PtCl3(DMSO)] (Scheme 5), and when the crude reaction mixture from the organic layer was evaporated and re-dissolved in acetonitrile, crystals suitable for X-ray crystallography were isolated showing that, surprisingly, a reaction with a molecule of acetonitrile had taken place forming complex 20 (see Scheme 5, Table S1 and details in ESI†), which incorporates a methylimine into the structure.
 | ||
| Scheme 5 Preparation of Pt complex 20 with the insertion of an acetonitrile molecule and crystal structure of the asymmetric unit. Displacement ellipsoids are at 50% probability. | ||
The crystal structure of 20 is in a monoclinic unit cell (0.66 Å resolution), with space group P21/c. The Pt–Cl and Pt–N bond lengths are 2.3104(5), 2.3105(5) Å and 1.9749(17), 1.9794(17) Å, and the N–Pt–N and Cl–Pt–Cl angles are 88.36(7)° and 88.959(18)°, respectively. The three C–N bond lengths around the guanidine C atom are 1.303(3) Å (N1–C5), 1.352(3) Å (N4–C5), 1.385(3) Å (C5–N6), and the C–N bond lengths around the amidine C atom are 1.385(3) Å (N6–C7) and 1.284(3) Å (C7–N8). Clearly, the C–N bonds coordinated to Pt are the shortest, implying that the guanidine/amidine double bonds are more localised on these atoms (the CN triple bond in the acetonitrile solvate is much shorter, 1.13 Å). The other three C–N bonds to the guanidine or amidine C atom are intermediate in length between single and double bonds, demonstrating some degree of resonance. The C–N bonds to the imidazole CH2 groups (1.453(3) Å and 1.476(3) Å) and to the aryl ring (1.452(2) Å) are clearly single bonds.
Regarding the possible mechanisms for the acetonitrile insertion, it is not clear whether the acetonitrile first coordinates to K[PtCl3(DMSO)] and then undergoes nucleophilic addition to coordinated 2-aminoimidazoline, if the coordinated acetonitrile is attacked by free 2-aminoimidazoline or if coordination of both ligands first takes place followed by C–N bond formation. Attack of coordinated 2-aminoimidazoline on uncoordinated acetonitrile was ruled out for steric reasons and the lack of precedence in the literature for nucleophilic attack on inactivated nitriles. In contrast, the strongly electrophilic properties of metal-acetonitrile complexes have been comprehensively reviewed.26
Further attempts to prepare more non-cyclometallated complexes like 19 with the rest of the free bases were unsuccessful as these compounds did not dissolve in the biphasic H2O/CH2Cl2 solvent system at room temperature as used in Scheme 4. Thus, a single solvent system was attempted using either MeOH or THF with isolated K[PtCl3(DMSO)]; however, these reactions gave a mixture of many compounds as adjudged by 1H NMR.
Considering the results obtained so far, the cyclometallation of in-house diaryl bis-guanidine systems known to be good DNA minor groove binders was next attempted.
Preparation of cyclometallated complexes of bis-guanidine diaryl systems
Once the conditions for Pt cyclometallation of mono-guanidine-like systems 8 and 13 were established, the corresponding complexation of two bis-guanidinium systems, previously prepared in our lab showing good binding to the minor groove of DNA, was attempted. Thus, the free bases 21 and 22 were prepared and exposed to the corresponding cyclometallation conditions (i.e. cis-[PtCl2(DMSO)2], NaOCH3, CH3OH, 70 °C, 2 h; Scheme 6); however, only an insoluble yellow precipitate was formed in both cases instead of cyclometallation. Free bases 21 and 22 have two N atoms capable of forming bis-Pt complexes by coordinating with one N atom of another ligand and, hence, these potential oligomers could keep polymerising yielding the precipitate observed. To stop this hypothesised polymerisation and to identify the monomer complex, NaOCH3 was removed, the solvent changed to DMSO-d6, and the reaction with 21 was followed by 1H NMR as indicated in the spectra shown in Fig. S2† confirming the formation of a mixture of the hydrochloride salt of the starting material, mono- and di-cyclometallation product.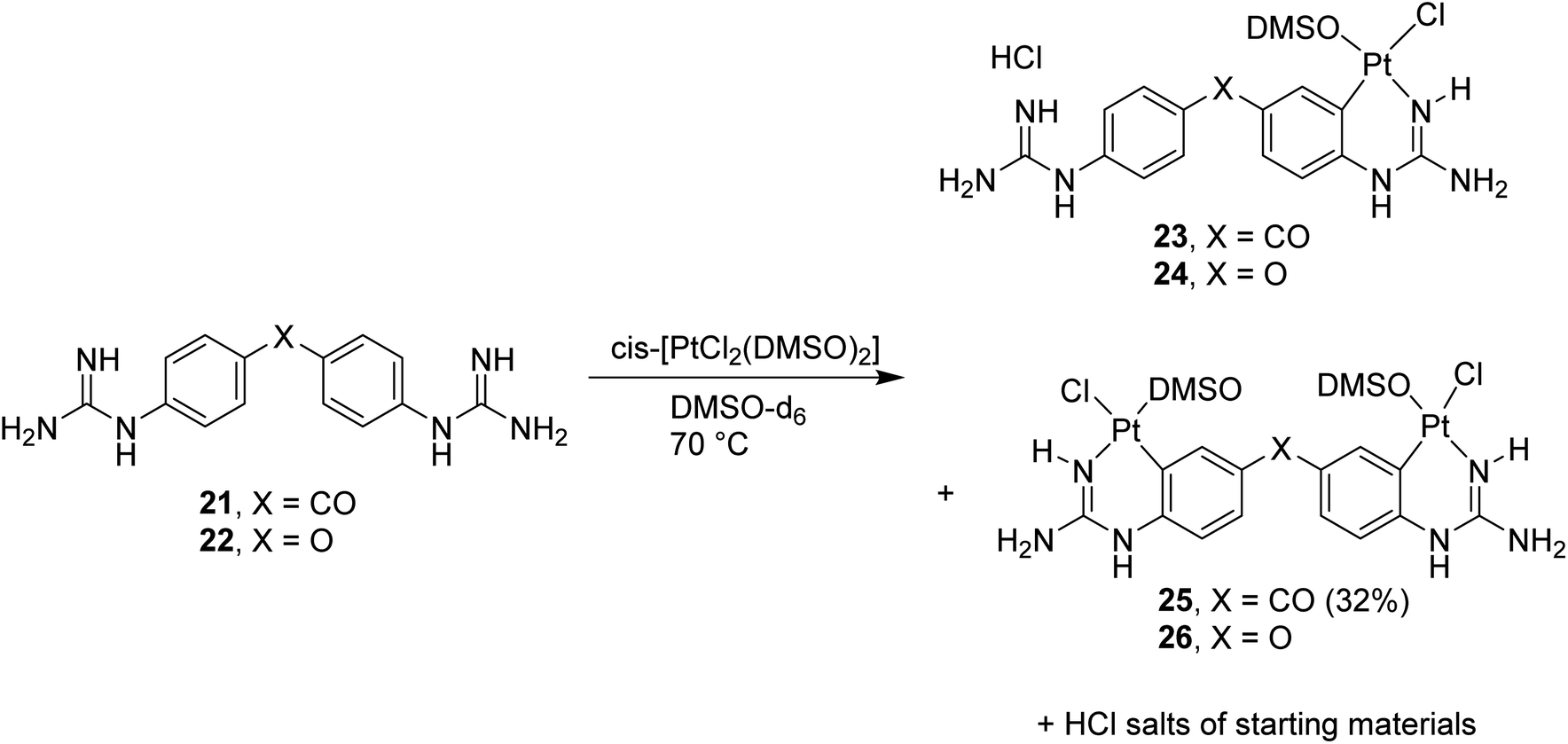 | ||
| Scheme 6 Cyclometallation reactions with bis-guanidine diaryl systems 21 (X = CO) and 22 (X = O) followed by 1H NMR. | ||
Thus, in the absence of MeOH or NaOCH3 both reactions yielded the mentioned mixture as detected by NMR; however, isolation of the complexes proved difficult since all three species for each reaction were water soluble except for compound 25 (Scheme 6). Repeated dissolution of the crude mixture in DMSO, followed by precipitation with H2O and filtration gave a filtrate from which carbonyl-linked 25 crystallised. Despite repeated attempts, purification of 23, 24 or 26 did not yield adequate material for characterisation.
The X-ray structure of 25 (Fig. 3a, Table S1 and details in ESI†) was in agreement with the solution structure. The binuclear complex crystallised in an orthorhombic unit cell (0.80 Å resolution, Pna21 space group). Restraints and constraints were used to model the disorder in one of the Pt–Cl positions (72%/28% occupancy). There are three and a half water molecules per asymmetric unit.
 | ||
| Fig. 3 (a) Asymmetric unit of the first crystal structure obtained of 25 showing the majority occupied disordered moiety (Pt2, Cl2A). (b) asymmetric unit of the second crystal structure obtained of 25. In all structures displacement ellipsoids are at 50% probability. See ESI† for structures including solvents/disorder. | ||
Aiming to improve the preparation of cyclometallated Pt complexes of bis-guanidines 21 and 22, many variations of the biphasic reaction from Scheme 5 were attempted, but no product could be isolated. Switching to a mono-phasic system using MeOH with K[PtCl3(DMSO)] and free base 21, yielded again cyclometallated 25 after crystallisation from aqueous solution and a new sample suitable for X-ray diffraction was solved (Fig. 3b, Table S1 and details in ESI†). In this case, 25 crystallised in a triclinic unit cell (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, 0.78 Å resolution) with two water molecules per complex. The Pt–N bonds were 1.995(4) and 2.008(4) Å. The Pt atoms were both square planar. Within the platinacycles, the C–Pt–N angles were both 89° (89.12(18) and 88.79(17)°). However, the C–Pt–S angle was 98° (98.44(14) and 97.91(13)°) in both cases. The overall molecule showed a helical twist. The Pt–Pt distance was ca. 6.09 Å and the angle between the planes of the square planar Pt geometries was 37.3(1)°. The dihedral angle between the planes of each benzene ring was 49.3(2)°.
space group, 0.78 Å resolution) with two water molecules per complex. The Pt–N bonds were 1.995(4) and 2.008(4) Å. The Pt atoms were both square planar. Within the platinacycles, the C–Pt–N angles were both 89° (89.12(18) and 88.79(17)°). However, the C–Pt–S angle was 98° (98.44(14) and 97.91(13)°) in both cases. The overall molecule showed a helical twist. The Pt–Pt distance was ca. 6.09 Å and the angle between the planes of the square planar Pt geometries was 37.3(1)°. The dihedral angle between the planes of each benzene ring was 49.3(2)°.
In summary, bis-guanidine diaryl systems are likely to cyclometallate, even at room temperature and, therefore, non-cyclometallated Pt-complexes bound via a guanidine, even if isolated from a reaction mixture, would likely decompose under biological conditions.
Assessment of cytotoxic potential of the cyclometallated complexes
 | ||
| Fig. 4 Graph of change in DPBF absorbance vs. time in the presence of DMSO (blue dots, slope = −0.0033 min−1), with 15 (red dots, slope = −0.0031 min−1) and with 16 (green dots, slope = −0.0037 min−1). All linear fits (R2 = 0.999). Each data point is an average of two runs. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 40000 cells per well are shown in Table 2. Binuclear complex 25 had weak but still measurable cytotoxicity with 10000 cells per well assays (IC50 = 101 μM); however, this is lost when running the experiments with 40000 cells per well (IC50 > 100 μM). The cycloplatinated 2-aminoimidazoline complex 16 was the most active (IC50 = 55 μM in 10000 cells per well and IC50 = 65 μM in 40000 cells per well) indicating that the five-membered 2-aminoimidazoline ring may be important for activity. Furthermore, the cytotoxicity of Pt complex 16 compares well to the bioactive mono- and bis-isouronium and bis-hydroxyguanidinium families previously tested in our group.30
000 and 40000 cells per well are shown in Table 2. Binuclear complex 25 had weak but still measurable cytotoxicity with 10000 cells per well assays (IC50 = 101 μM); however, this is lost when running the experiments with 40000 cells per well (IC50 > 100 μM). The cycloplatinated 2-aminoimidazoline complex 16 was the most active (IC50 = 55 μM in 10000 cells per well and IC50 = 65 μM in 40000 cells per well) indicating that the five-membered 2-aminoimidazoline ring may be important for activity. Furthermore, the cytotoxicity of Pt complex 16 compares well to the bioactive mono- and bis-isouronium and bis-hydroxyguanidinium families previously tested in our group.30
|
||
|---|---|---|
| Compd | IC50 ± SEMa (μM) 10000 cells per well |
IC50 ± SEMb (μM) 40000 cells per well |
| a Cells were seeded at a density of 5 × 104 cells per mL in a 96-well plate and treated with the compounds dissolved in 0.1% DMSO in ddH2O at 10 μM, 25 μM, 50 μM, 75 μM and 100 μM. Carboplatin (dissolved in ddH2O) was used as a reference and tested in the same manner. Once treated, cells were incubated for 72 h at 37 °C after which they were treated with AlamarBlue® and left in darkness in an incubator for 5 h. The resulting fluorescence was read using a plate reader from which percentage viability was calculated. IC50 values were calculated using Prism GraphPad 5 software from at least three independent experiments performed in triplicate.b As note (a) but cells were seeded at a density of 2 × 105 cells per mL. | ||
| 15 | >100 | >100 |
| 16 | 55.29 ± 3.0 | 66.82 ± 5.9 |
| 17 | 100.81 ± 3.4 | >100 |
| 25 | >100 | >100 |
| Carboplatin | 12.62 ± 1.7 | N/A |
It is interesting to note that the free ligand in complex 25 (i.e. compound 21) had previously been shown to have no activity in HL-60 cells;31 however, complexation with Pt resulted in a weak though measurable effect on cell viability.
Experimental
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). δPt (86 MHz, DMSO-d6): −3582. νmax (ATR)/cm−1: 3407 (NH), 3310 (N), 3193 (NH), 2352, 1645 (CN), 1602, 1551, 1479, 1399, 1300, 1091 (S–O), 1022, 758, 721. % Calculated for C9H14N3OPtClS·0.5NaCl·1.5H2O·0.5DMSO: C 22.32, H 3.28, N 7.81. % Found: C 22.12, H 3.01, N 8.01. LRMS (m/z ESI+) found: 444.2 ([M + H]+. C9H15N3OSCl2Pt requires 444.03).N). δPt (86 MHz, DMSO-d6) −3633. νmax (ATR)/cm−1: 3339, 3282 (NH), 3185 (NH), 2994 (CH), 1606, 1578, 1465, 1413, 1288, 1094, 1017, 743. % Calculated for C11H17N3OPtClS·H2O·0.5DMSO: C 27.35, H 3.73, N 7.97. % Found: C 27.16, H 3.45, N 8.16.N). δPt (86 MHz, DMF-d7): −3033. νmax (ATR)/cm−1: 3289 (NH), 3090 (NH), 1617 (CN), 1093 (S–O), 3018, 1588, 1571, 1478, 1432, 1267, 1022, 730. HRMS (m/z ESI−) Found: 503.0063 ([M − H]−. C11H16N3OSCl2Pt requires 503.0039).N), 1576, 1436, 1271, 1098, 1030, 730.N), 194.4 (q CO). δPt (86 MHz, DMSO-d6): −3556, −3559. νmax (ATR)/cm−1: 3356 (NH), 3206 (NH), 3000, 2917 (CH), 1649, 1619, 1535, 1477, 1304, 1288, 1249, 1093, 1017, 1002, 947, 823, 748, 677. % Calculated for C19H26N6O3Pt2Cl2S2·6NaCl·6H2O·4DMSO: C 19.27, H 2.55, N 4.99. % Found: C 19.32, H 2.90, N 5.21.
N). δPt (86 MHz, DMSO-d6): −3582. νmax (ATR)/cm−1: 3407 (NH), 3310 (N), 3193 (NH), 2352, 1645 (CN), 1602, 1551, 1479, 1399, 1300, 1091 (S–O), 1022, 758, 721. % Calculated for C9H14N3OPtClS·0.5NaCl·1.5H2O·0.5DMSO: C 22.32, H 3.28, N 7.81. % Found: C 22.12, H 3.01, N 8.01. LRMS (m/z ESI+) found: 444.2 ([M + H]+. C9H15N3OSCl2Pt requires 444.03).N). δPt (86 MHz, DMSO-d6) −3633. νmax (ATR)/cm−1: 3339, 3282 (NH), 3185 (NH), 2994 (CH), 1606, 1578, 1465, 1413, 1288, 1094, 1017, 743. % Calculated for C11H17N3OPtClS·H2O·0.5DMSO: C 27.35, H 3.73, N 7.97. % Found: C 27.16, H 3.45, N 8.16.N). δPt (86 MHz, DMF-d7): −3033. νmax (ATR)/cm−1: 3289 (NH), 3090 (NH), 1617 (CN), 1093 (S–O), 3018, 1588, 1571, 1478, 1432, 1267, 1022, 730. HRMS (m/z ESI−) Found: 503.0063 ([M − H]−. C11H16N3OSCl2Pt requires 503.0039).N), 1576, 1436, 1271, 1098, 1030, 730.N), 194.4 (q CO). δPt (86 MHz, DMSO-d6): −3556, −3559. νmax (ATR)/cm−1: 3356 (NH), 3206 (NH), 3000, 2917 (CH), 1649, 1619, 1535, 1477, 1304, 1288, 1249, 1093, 1017, 1002, 947, 823, 748, 677. % Calculated for C19H26N6O3Pt2Cl2S2·6NaCl·6H2O·4DMSO: C 19.27, H 2.55, N 4.99. % Found: C 19.32, H 2.90, N 5.21.ROS generation experimental details
For the evaluation of the singlet oxygen production, solutions the 1O2 trap 1,3-diphenylisobenzofuran (DPBF) in DMSO were employed (20 mg in 50 mL). Calibration was achieved using 50:50 of a 112 μM DPBF solution to get absorption 1 at λmax = 418 nm (in DMSO). Next, a fresh solution of each Pt complex (15, or 16) in DMSO was prepared at high concentration (150 μM), added to the cuvette and its absorbance was adjusted to around 0.01 at wavelength of irradiation. Potential singlet oxygen production was assessed looking at the decay absorption spectra of DPBF at time points 0, 2, 5, 10, 20, and 30 min. Since no clear effect was observed in the presence of the DMSO Pt(II) complexes solutions, no further measurements at a lower concentrations were carried out. The slope of plots of absorbance of DPBF at 418 nm vs. irradiation time for each photosensitizer was calculated.
Cell viability experimental details
The HL-60 (human caucasian promyelocytic leukemia) cell line was maintained between 200000–2000000 cells per mL in Roswell Park Memorial Institute (RPMI) 1640 medium with stable glutamate (GlutaMax I) supplemented with 10% (v/v) foetal bovine serum (FBS) and 50 μg per mL penicillin/streptomycin (pen-strep). The growth medium was stored in the fridge at 4 °C and heated to 37 °C prior to culture work. Cells were grown at 37 °C in a humidified environment maintained at 95% O2 and 5% CO2 and passaged at least three times weekly depending on their levels of confluency. When required for sub-culturing, cells were transferred to a sterile tube and centrifuged at 1296 rpm for 5 min. The supernatant was discarded, and the cell pellet was resuspended in fresh medium. Cells were then counted using a haemocytometer slide and seeded at the required density.
HL-60 cells in the log phase of growth were seeded in 96-well plates at a density of 50000 cells per mL (200 μL per well or 10000 cells per well) in complete RPMI medium the same day of the experiment. Compounds 13, 16, 17 and 25 were dissolved in DMSO to obtain a starting 100 mM stock solution. The cells were then treated with either 2 μL of a 1:100 dilution of stock concentrations of drugs or ddH2O as vehicle control, or 0.2 μL of a 1:1000 dilution of stock concentrations of drugs or DMSO as vehicle control. All experiment were repeated in triplicate for at least three times. Three wells containing 200 μL RPMI with no cells were also set up as blanks.
After 72 h incubation, 20 μL AlamarBlue® was added to each well. The plates were incubated in darkness at 37 °C for 5 hours. Using a Molecular Devices microplate reader, the fluorescence (F) was then read at an excitation wavelength of 544 nm and an emission wavelength of 590 nm. Cell viability was then determined by subtracting the mean blank fluorescence (Fb) from the treated sample fluorescence (Fs) and expressing this as a percentage of the fluorescence of the blanked vehicle control (Fc). This is demonstrated in the equation below. The results were then plotted as nonlinear regression, sigmoidal dose–response curves on Prism GraphPad 5 software, from which the IC50 value for each drug was determined.

Conclusions
The preparation of Pt(II) complexes of aryl guanidines has been explored. Suitable conditions have been found for the preparation of unsubstituted phenyl guanidine-like Pt(II) cyclometallated complexes. However, replacement of the phenyl system by a pyridine, separation of the guanidine and phenyl moieties by a methylene group or p-substitution of the phenyl ring with –OCH3 did not result in the corresponding cyclometallated complexes. Only o-substitution of the phenyl ring with a NH2 provided a bimetallic Pt(II) complex (17) combining cyclometallation and coordination with each of the Pt centres. Additionally, an in situ activation of Pt-arylguanidine complexes with acetonitrile to create a bidentate aryl iminoguanidine Pt(II) complex are reported. The cyclometallation methodology was extended to create a water-stable conjugate incorporating two Pt(II) ions and a diaryl bis-guanidine DNA minor groove binder previously reported by us. The cyclometallated Pt(II) complexes were particularly stable to aqueous environments and some of them were tested for ROS generation and anticancer activity in HL-60 cells.Data availability
The following ESI† is available: high temperature 1H NMR experiments for the formation of complex 15; preparation of mono-guanidinium-like derivatives used as precursors of the ligands used to explore Pt complexation conditions (i.e. aryl guanidinium salts 1–7 and the corresponding precursors bis-Boc-protected guanidines S6, S7–S8, S9–S10, S13–S14); experimental details of the methods utilised; reaction of compound 21 with cis-[PtCl2(DMSO)2] followed by 1H NMR; graphs showing the experiments with complexes 15 and 16 searching for ROS generation; biochemical details (i.e. cell viability assays); X-ray crystallographic data for compounds 15, 16, 17, 18, 19, 20, and 25.Author contributions
Conceptualization, I. R. and C. J. M.; methodology, I. R., C. J. M. and A. R. McD.; data analysis, P. O'. S., V. P., B. T., C. J. M., A. R. McD., and I. R.; experiments, P. O'. S., V. P., and B. T.; writing and editing, P. O'. S., V. P., B. T., C. J. M., A. R. McD., and I. R.; supervision, I. R., C. J. M. and A. R. McD. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by Science Foundation Ireland (SFI-RFP project: CH3060). P. O'. S. thanks SFI (SFI-RFP, project: CH3060) and V. P. thanks the School of Chemistry at Trinity College Dublin for postgraduate funding. We thank Dr John O'Brien for expert NMR studies and Dr Aaron Keogh for preliminary work in Pt-guanidine complexation. We also would like to thank Carsten Lenczyk, Bruker AXS for his insight and assistance.References
- S. Rottenberg, C. Disler and P. Perego, The rediscovery of platinum-based cancer therapy, Nat. Rev. Cancer, 2021, 21(1), 37–50 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs, Chem. Rev., 2016, 116(5), 3436–3486 CrossRef CAS PubMed.
- T. Zhong, J. Yu, Y. Pan, N. Zhang, Y. Qi and Y. Huang, Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy, Adv. Healthcare Mater., 2023, 12(22), e2300253 CrossRef PubMed.
- S. Alassadi, M. J. Pisani and N. J. Wheate, A chemical perspective on the clinical use of platinum-based anticancer drugs, Dalton Trans., 2022, 51(29), 10835–10846 RSC.
- Drug Monitoring and Clinical Chemistry, ed. Hempel G., Universität Münster, Germany: Institut für Pharmazeutische und Medizinische Chemie, 2004, Chapter 7: Hempel G, Dose and therapy individualisation in cancer chemotherapy Search PubMed.
- (a) T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV), Prodrugs Chem Rev., 2016, 116(5), 3436–3486 CrossRef CAS PubMed; (b) T. C. Johnstone, J. J. Wilson and S. J. Lippard, Monofunctional and Higher-Valent Platinum Anticancer Agents, Inorg. Chem., 2013, 52(21), 12234–12249 CrossRef CAS PubMed.
- (a) R. G. Kenny and C. J. Marmion, Toward Multi-Targeted Platinum and Ruthenium Drugs- A New Paradigm in Cancer Drug Treatment Regimens?, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed; (b) R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Platinum(IV) Prodrugs – A Step Closer to Ehrlich's Vision?, Eur. J. Inorg. Chem., 2017, 2017(12), 1596–1612 CrossRef CAS; (c) A. Khoury, K. M. Deo and J. R. Aldrich-Wright, Recent advances in platinum-based chemotherapeutics that exhibit inhibitory and targeted mechanisms of action, J. Inorg. Biochem., 2020, 207, 111070 CrossRef CAS PubMed.
- A. R. Gomes, C. L. Varela, A. S. Pires, E. J. Tavares-da-Silva and M. F. Roleira, Synthetic and natural guanidine derivatives as antitumor and antimicrobial agents: a review, Bioorg. Chem., 2023, 138, 106600 CrossRef CAS PubMed.
- P. J. Bailey and S. Pace, The coordination chemistry of guanidines and guanidinates, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS.
- A. A. Legin, M. A. Jakupec, N. A. Bokach, M. R. Tyan, V. Y. Kukushkin and B. K. Keppler, Guanidine platinum(II) complexes: synthesis, in vitro antitumor activity, and DNA interactions, J. Inorg. Biochem., 2014, 133, 33–39 CrossRef CAS PubMed.
- F. Carrillo-Hermosilla, R. Fernández-Galán, A. Ramos and D. Elorriaga, Guanidinates as Alternative Ligands for Organometallic Complexes, Molecules, 2022, 27, 5962 CrossRef CAS PubMed.
- P. Elumalai, N. Thirupathi and M. Nethaji, Dual Role of Acetate as a Nucleophile and as an Internal Base in Cycloplatination Reaction of sym N,N′,N′′-Triarylguanidines, Inorg. Chem., 2013, 52(4), 1883–1894 CrossRef CAS PubMed.
- D. Nieto, S. Bruña, A. M. González-Vadillo, J. Perles, F. Carrillo-Hermosilla, A. Antiñolo, J. M. Padrón, G. B. Plata and I. Cuadrado, Catalytically generated ferrocene-containing Guanidines as efficient precursors for new redox-active heterometallic Platinum(II) complexes with anticancer activity, Organometallics, 2015, 34, 5407–5417 CrossRef CAS.
- M. Marin-Luna, G. Sanchez-Sanz, P. O'Sullivan and I. Rozas, Guanidine Complexes of Platinum: A Theoretical Study, J. Phys. Chem. A, 2014, 118(29), 5540–5547 CrossRef CAS PubMed.
- D. Aitken, A. Albinati, A. Gautier, H. P. Husson, G. Morgant, D. Nguyen-Huy, J. Kozelka, P. Lemoine, S. Ongeri, S. Rizzato and B. Voissat, Platinum(II) and Palladium(II) complexes with N-aminoguanidine, Eur. J. Inorg. Chem., 2007, 2007(21), 3327–3334 CrossRef.
- T. Yamada, X. Liu, U. Englert, H. Yamane and R. Dronskowski, Solid-state structure of free base guanidine achieved at last, Chem.–Eur J., 2009, 15(23), 5651–5655 CrossRef CAS PubMed.
- U. Wild, P. Roquette, E. Kaifer, J. Mautz, O. Hübner, H. Wadepohl and H. J. Himmel, Synthesis and Structural Characterisation of cis- and trans-[(hpp H) 2 Pt Cl 2 ], [(hpp H) 3 Pt Cl] + Cl – and Some new Salts of the [hpp H 2 ] + Cation (hpp H = 1,3,4,6,7,8-Hexahydro-2H-pyrimido[1,2-a]pyrimidine): the Importance of Hydrogen Bonding, Eur. J. Inorg. Chem., 2008, 8, 1248–1257 CrossRef.
- P. Elumalai, N. Thirupathi and M. Nethaji, Dual role of acetate as a nucleophile and as an internal base in cycloplatination reaction of sym-N,N’,N”-triarylguanidines, Inorg. Chem., 2013, 52(4), 1883–1894 CrossRef CAS PubMed.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Palladium(II) and platinum(II) alkyl sulfoxide complexes. Examples of sulfur-bonded, mixed sulfur- and oxygen-bonded, and totally oxygen-bonded complexes, Inorg. Chem., 1972, 11(6), 1280–1284 CrossRef CAS.
- D. Griffith, M. P. Morgan and C. J. Marmion, A novel anti-cancer bifunctional platinum drug candidate with dual DNA binding and histone deacetylase inhibitory activity, Chem. Commun., 2009, 2009(44), 6735–6737 RSC.
- V. Sicilia, M. Baya, P. Borja and A. Martín, Oxidation of half-lantern Pt2(II,II) compounds by halocarbons. Evidence of dioxygen insertion into a Pt(III)-CH3 bond, Inorg. Chem., 2015, 54(15), 7316–7324 CrossRef CAS PubMed.
- E. Anger, M. Rudolph, C. Shen, N. Vanthuyne, L. Toupet, C. Roussel, J. Autschbach, J. Crassous and R. Réau, From hetero- to homochiral bis(metallahelicene)s based on Pt(III)-Pt(III) bonded scaffold: isomerization, structure, and chiroptical properties, J. Am. Chem. Soc., 2011, 133(11), 3800–3803 CrossRef CAS PubMed.
- K. Matsumoto and M. Ochiai, Organometallic chemistry of platinum-blue derived platinum III dinuclear complexes, Coord. Chem. Rev., 2002, 231(1–2), 229–238 CrossRef CAS.
- S. U. Pandya, K. C. Moss, M. R. Bryce, A. S. Batsanov, M. A. Fox, V. Jankus, H. A. Al Attar and A. P. Monkman, Luminescent platinum (II) complexes containing cyclometallated diaryl ketimine ligands: synthesis, photophysical and computational properties, Eur. J. Inorg. Chem., 2010, 2010(13), 1963–1972 CrossRef.
- Y. Y. Scaffidi-Domianello, A. A. Nazarov, M. Haukka, M. Galanski, B. K. Keppler, J. Schneider, P. Du, R. Eisenberg and V. Y. Kukushkin, First example of the solid-state thermal cyclometallation of ligated benzophenone imine giving novel luminescent platinum(II) species, Inorg. Chem., 2007, 46(11), 4469–4482 CrossRef CAS PubMed.
- V. Y. Kukushkin and A. J. L. Pombeiro, Additions to metal-activated organonitriles, Chem. Rev., 2002, 102(5), 1771–1802 CrossRef CAS PubMed.
- D. Ashen-Garry and M. Selke, Singlet Oxygen Generation by cyclometalated complexes and applications, Photochem. Photobiol., 2014, 90(2), 257–274 CrossRef CAS PubMed.
- W. Spiller, H. Kliesch, D. Wöhrle, S. Hackbarth, B. Röder and G. Schnurpfeil, Singlet oxygen quantum yields of different photosensitizers in polar solvents and micellar solutions, J. Porphyrins Phthalocyanines, 1998, 2(2), 145–158 CrossRef CAS.
- P. R. Ogilby, Singlet oxygen: there is indeed something new under the sun, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- A. Kahvedžic, S. M. Nathwani, D. M. Zisterer and I. Rozas, Aromatic bis-N-hydroxyguanidinium derivatives: synthesis, biophysical and biochemical evaluations, J. Med. Chem., 2013, 56(2), 451–459 CrossRef PubMed.
- A. Kahvedzic, PhD thesis, University of Dublin, 2011.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2391910–2391917. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra00310e |
| This journal is © The Royal Society of Chemistry 2025 |