DOI:
10.1039/D4RA07163H
(Paper)
RSC Adv., 2025,
15, 3416-3426
An insight into the structure of acebutolol tetraphenylborate: crystal structure and quantum chemical calculations†
Received
5th October 2024
, Accepted 25th January 2025
First published on 4th February 2025
Abstract
Reaction between Acebutolol hydrochloride and sodium tetraphenylborate in water afforded an Acebutolol-tetraphenyl-borate complex. This ion-pair was characterized by a variety of analytical and spectroscopic tools including X-ray crystallographic analysis. Comprehensive theoretical studies including ground-state geometry optimization, Mulliken atomic charges, and vibrational analysis were executed to get an insight into the nature of the charge transfer between the donor and acceptor ions. The crystal structure demonstrated that three inter- and intramolecular hydrogen-bonds stabilize the molecular packing in the solid state. Natural bond orbital analysis confirmed the presence of many interactions between B(Ph)4− and particular Acebutolol functional groups, such as NH and OH groups. The positive Mulliken atomic charges of the acidic protons of the NH and OH groups of Acebutolol were increased upon the formation of the ion-pair. This has been experimentally confirmed by IR and NMR spectroscopies. Through the use of frontier molecular orbital models, time-dependent density functional theory calculations have provided more insight into the existence of two electronic transitions, beginning at B(Ph)4− and terminating at the π* system of mono-protonated Acebutolol ions.
1. Introduction
Acebutolol (Scheme 1) is a medication that selectively binds to β1 receptors and is used to treat arrhythmia in the heart and hypertension.1 Acebutolol functions by obstructing β-receptors present in both the heart and blood arteries, so causing the latter to remain relaxed. On the other hand, significant adverse effects from acebutolol's overdosage include low blood pressure, low heart rate, erectile dysfunction, as well as liver damage.2 A search of the literature shows that several methods for analysing Acebutolol hydrochloride and its metabolites have been published. These techniques include chromatographic,3,4 electrochemical techniques,5–8 as well as spectrophotometry, through the reactions with chromogens e.g., benzoquinone derivatives9 or with iodine.10 Modified carbon-paste electrodes were employed as electrochemical sensors to determine the concentration of Acebutolol hydrochloride (DH+Cl−).5,11 Two potentiometric PVC sensors based on ion-pair complexes of Acebutolol with tetraphenylborate and phosphomolybdate were fabricated for the determination of the title drug with a μM detection limit.12 Although the tetraphenylborate sensor exhibited generic features including long-stability and fast response time, no information was available on the structure of the ionophore or the functional groups that were involved in its formation.12 On the other hand, the fabricated Acebutolol-phosphotungstate sensors exhibited Nernstian responses over wide concentration ranges with lower detection limits.6 Based on the ion-pair formation between Acebutolol and two dyes (bromocresol green and bromothymol blue), a direct, extraction-free spectrophotometric method was established for the determination of DH+Cl− in pharmaceutical formulations.13
 |
| | Scheme 1 Synthesis of the Acebutolol–tetraphenyl borate ion-pair complex. | |
Herein, the absence of any crystal structure and quantum chemical calculations for any of the previously reported ion-pair complexes of Acebutolol in the literature motivated us to synthesize an Acebutolol–tetraphenyl-borate (DH–B(Ph)4) complex (Scheme 1) to identify the functional groups involved in the formation of such a class of complexes. Density functional theory calculations were used to conduct extensive theoretical and experimental investigations on Acebutolol (D), and its mono-protonated form (DH+), tetraphenyl borate ion (B(Ph)4−), as well as DH–B(Ph)4 to precisely and fully determine the structural properties of the title ion-pair as well as to get an insight into the nature of the observed electronic transitions. The charge transfer and/or conjugative contact, intra- and intermolecular interactions, bond hybridization, natural charges, occupancy, and bond polarization were all investigated using the natural bond orbital (NBO) approach, developed by Weinhold and co-workers.14
2. Results and discussion
2.1. Crystal structure
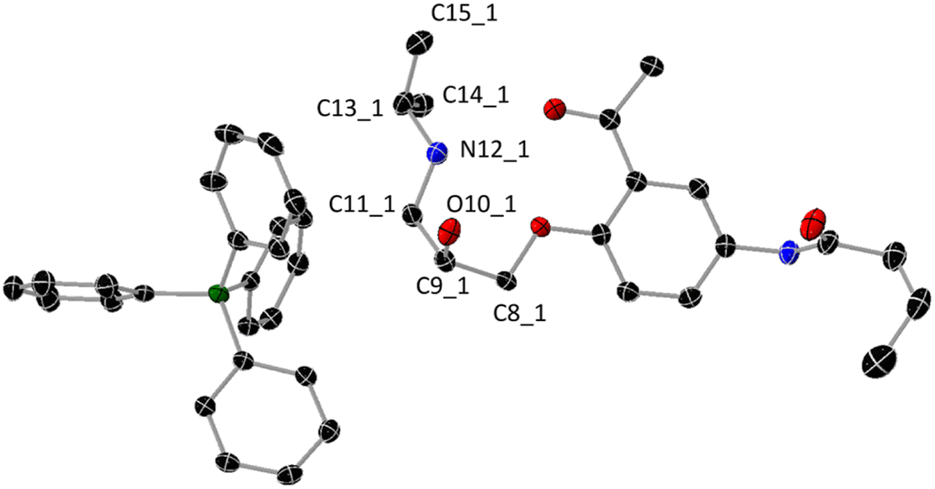
As shown in Scheme 1, the reaction between Acebutolol hydrochloride and sodium tetraphenyl borate in water afforded an Acebutolol–tetraphenyl borate ion-pair complex in a pure form and good yield (73%). The complex has been structurally characterized using X-ray crystallographic analysis, IR, NMR, and mass spectrometry. Appropriate single crystals for X-ray crystallographic analysis have been formed following the slow evaporation of the methanolic solution of the ion-pair. A view of the molecular structure of the ion-pair is shown in Fig. 1, and its crystal data (bond lengths and bond angles) are tabulated in Table S1.† The relevant crystallographic parameters of the complex are listed in Table S2.† The ion-pair crystallizes in a monoclinic crystal system of P21/c space group with a = 12.45670 (10) Å, b = 23.0603 (3) Å, c = 12.78140 (10) Å and β = 91.2060 (10) °. As shown in Fig. 2, the unit-cell is built by four molecules.
 |
| | Fig. 1 Molecular structure of the ion-pair complex (thermal ellipsoids are shown at the 50% probability level). Selected bond lengths (Å): C8_1–C9_1 1.511(2); C9_1–O10_1 1.423(1); C9_1–C11_1 1.525(2); C11_1–N12_1 1.488(2); N12_1–C13_1 1.510(2); C13_1–C14_1 1.514(2); C13_1–C15_1 1.515(2). Hydrogen atoms were omitted for the clarity. Two H-atoms are attached to N12_1 atom. | |
 |
| | Fig. 2 The unit cell of the Acebutolol–tetraphenyl borate ion-pair complex. | |
Because hydrogen bonds are essential to many chemical and biological processes, there has been a growing amount of computational and experimental research on the topic.15–17 The most well-known type of the hydrogen-bond is the traditional one, which is described as A–H⋯B, where A and B are electro-negative atoms such as nitrogen, oxygen, or halogen. The blue-shifting H-bond is an additional form of H-bonding. It is typically represented as C–H⋯B, where B might be an electronegative atom carrying one or more electron lone pairs or an area of excess electron density (π-electron of an atomic system). The packing diagram of the synthesized ion-pair complex shows both intra- and intermolecular H-bond interactions (Fig. 3).
 |
| | Fig. 3 Intra- and intermolecular hydrogen-bonding stabilizing the crystal packing of the Acebutolol–tetraphenyl borate ion-pair complex. | |
As shown in Fig. 3, there are two intramolecular H-bonds, where the NH (N = N12_1) group (Fig. 1) interacts with the ether and ester groups at O⋯N distances of 2.786 and 2.810 Å, and angles of 127.0° and 150.7°, respectively. On the other hand, an intermolecular H-bond exists, in which the amide group interacts with the OH (O = O10_1) of the next molecule at an O⋯O distance of 2.782 Å and angle of 164.1°. It is commonly recognized that hydrogen-bonds that approach or are linear are typically stronger than those whose X–H⋯Y angle deviates from linearity.18,19 As a result, due to its shorter interaction length and closer bond angle to linearity, the intermolecular H-bond is stronger than the intramolecular contacts of the ion-pair.
2.2. Hirshfeld surface analysis
Hirshfeld surface analysis20 was performed on the title complex to investigate intermolecular interactions using CrystalExplorer 17.5 software.21 The white surface on the Hirshfeld surface shown over d-norm (Fig. 4) represents contacts that are separated by the sum of the van der Waals radii. The red and blue colors, on the other hand, represent distances that are shorter (near contact) or longer (far contact) than the sum of the van der Waals radii. The overall two-dimensional fingerprint plots for DH–B(Ph)4 are shown in Fig. 4a, and those demonstrating H⋯H, O⋯H/H⋯O, and N⋯H/H⋯N contacts are presented in Fig. 4b–d, together with their relative sharing to the Hirshfeld surface. The presence of red around the OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the monoprotonated Acebutolol cation as well as the specific CH groups of B(Ph)4−, shown in the d-norm surface of DH–B(Ph)4 (Fig. 4a), indicates that these groups are involved in H-bonding interactions. The participation in π-stacking interactions is suggested by the growing number of red and blue triangular patches surrounding the aromatic ring. As shown in Fig. 4b, the most noteworthy contacts in DH–B(Ph)4 are H⋯H (68.6%). These contacts have a major effect on the molecular packing and crystal structure stabilization. They are shown as widely scattered points of high density with tips at de = di = 2 Å. Given that they make up 7.4% of the surface, the H⋯O/O⋯H interactions (Fig. 4c) highlight the importance of H-bonds involving oxygen atoms in controlling the stability and other physical properties. The H⋯N/N⋯H contacts account for 0.1% to the stabilization of the crystal structure.
O groups of the monoprotonated Acebutolol cation as well as the specific CH groups of B(Ph)4−, shown in the d-norm surface of DH–B(Ph)4 (Fig. 4a), indicates that these groups are involved in H-bonding interactions. The participation in π-stacking interactions is suggested by the growing number of red and blue triangular patches surrounding the aromatic ring. As shown in Fig. 4b, the most noteworthy contacts in DH–B(Ph)4 are H⋯H (68.6%). These contacts have a major effect on the molecular packing and crystal structure stabilization. They are shown as widely scattered points of high density with tips at de = di = 2 Å. Given that they make up 7.4% of the surface, the H⋯O/O⋯H interactions (Fig. 4c) highlight the importance of H-bonds involving oxygen atoms in controlling the stability and other physical properties. The H⋯N/N⋯H contacts account for 0.1% to the stabilization of the crystal structure.
 |
| | Fig. 4 The full two-dimensional fingerprint plots and their relative Hirshfeld surface for the most abundant interactions in DH–B(Ph)4, showing (a) all interactions, and delineated into (b) H⋯H, (c) O⋯H/H⋯O, (d) N⋯H/H⋯N interactions. | |
2.3. Density functional theory calculations
2.3.1. Geometry optimization. In the ground-state, full geometry optimizations of D, DH+, B(Ph)4− and DH–B(Ph)4 were executed with Becke 3-parameter (exchange) Lee–Yang–Parr functional,22,23 and a split-valence double-zeta polarized basis set, 6-31G(d).24 From the crystal data of DH–B(Ph)4, the initial coordinates for each optimization run were obtained. Through frequency calculations, the optimized structures were verified as a minimum on the potential energy surfaces. Vibrational analysis was obtained at the same theoretical level as the optimization method. The geometries obtained are thought to represent the local minimal structures as imaginary vibrations are absent. The local minimal structures and their atomic coordinates are tabulated in Tables S3–S6.† When the bond lengths of Acebutolol and its mono-protonated form were compared (Table S7†), it was found that there were no appreciable variations between the two forms—the exception being the bonds produced from the electronegative atoms, where the protonation causes the bond lengths to drop by 0.01–0.05 Å. The significant variation was observed for the N–C bonds of the protonated nitrogen, N12_1 (Fig. 1). A comparison of the bond lengths of DH+ and DH–B(Ph)4 revealed no significant differences between the two forms, except for C9_1–O10_1 (Fig. 1), where the ion-pair formation results in a 0.01 Å elongation of the bond (Table S7†). Alternatively, to determine whether the applied calculation approach (B3LYP/6-31G(d)) was appropriate for this size of compound, we then compared the optimized data with the crystal data. Except for the bonds that participate in intermolecular hydrogen bonding, most of the optimized bond lengths are somewhat longer than the experimental values, agreeing within 0.01–0.03 Å. This occurs because of the computations being carried out in a gaseous state, whereas the X-ray crystallographic analysis handle packed molecules with intra- and intermolecular interactions. Nonetheless, the bond lengths between the crystal and optimized structures show a root mean square deviation of 0.012, indicating that the computation approach used was appropriate for size and type of molecule. The experimental and calculated bond lengths were found to fit linearly (dcalcu = 1.0166dexp − 0.0163) with small regression coefficient (R2 = 0.9877).
2.3.2. Mulliken atomic charges. When applying quantum mechanical computations to molecular systems, the computation of effective Mulliken atomic charge is crucial. In molecular or ionic species, the total (σ + π) charge distribution is a crucial metric that can be used to discuss several significant features. These comprise hydrogen bond proton–acceptor ability and nature of the studied species (dipolar vs. zwitterionic). Despite being widely used in the literature, Mulliken's charge population notion has a conceptual flaw in that it does not describe the probability distribution of the electron on the constituent atoms; small negative population values can also arise.25 Certain practical applications are also prohibited by this fact. For comparison, Mulliken atomic charges of selected atoms of D, DH+, B(Ph)4− and DH−B(Ph)4 are shown in Fig. 5. Due to the σ withdrawing nature of O10_1 and N12_1, both C9_1 and C13_1 (Fig. 1) exhibit positive Mulliken values, in contrast to all the other selected carbon atoms (C11_1, C14_1, and C15_1). When Acebutolol was protonated and ion-pair complex formation ensued, a rise in Mulliken value was observed for the negatively charged carbon atoms. On the other hand, the positively charged Mulliken charges of C9_1 and C13_1 decreased after the drug was protonated and a complex formed. This could indicate that there are interactions between these C–OH and –NH2+– groups of Acebutolol and B(Ph)4−. An increase in the positively Mulliken charges of the acidic protons of the O10_1 and N12_1 atoms (Fig. 1) lends more evidence to this. However, once an ion-pair forms and facilitates the charge transfer of B(Ph)4− to DH+, the positively charged boron atom of B(Ph)4− has slightly increased. Based on Mulliken atomic charges in the ground and excited states (TDDFT) data (vide infra), the B3LYP/6-31G(d) method has used to provide answers to a number of questions about the charge transfer process. How long is the charge transfer between DH+ and B(Ph)4−, for example? Do the ions stated above overlap with one another?26 Ciofini and coworkers paper provides a spreadsheet26 to calculate the distance of charge transfer (dCT) and amount of charge transferred (qCT) of the title ion pair. The dCT and qCT are 1.35 Å and 0.54e, respectively. The HCT index can be calculated for rod-like systems where the CT is mostly along the x-axis. The HCT value refers to the electron transfer direction (B(Ph)4− → DH+) and half of the two centroid axes.27 The HCT index of our ion pair is 5.78 Å. On the other hand, the t-index, calculated as the difference between dCT and HCT, indicates the degree of separation between ρ+ and ρ−. If the t-index is < 0, it indicates that the ρ+ and ρ− are not much separated due to charge transfer. The t value of our ion-pair is −4.42 Å. Thus, the overlap between the centroids along B(Ph)4− → DH+ direction is expected. The dipole moment changes during the S0 → S1 transition is caused by intramolecular charge transfer or rearrangement. μCT is used to define the variation of dipole moment between the ground and excited states. The μCT of our ion-pair is 3.51 debye.
 |
| | Fig. 5 Comparison of Mulliken atomic charges of selected atoms of D, DH+, B(Ph)4− and DH–B(Ph)4 (numbering of atoms is according to that given in Fig. 1). | |
2.3.3. Natural bond orbital analysis. The natural bond orbital (NBO) analysis, developed by Weinhold and co-workers14 is a useful framework for studying charge transfer or conjugative contact in molecular systems and has been widely applied in the assessment of intra- and inter-molecular interactions. To determine the contribution of atomic orbitals to NBO σ and π hybrid orbitals for bonded atom pairs, NBO analysis was carried out. There are three distinct NBO hybrid orbitals: the core (CR), lone pair (LP), and bonding orbital (BO). Lewis σ-type (donor) NBOs combined with non-Lewis σ-type (acceptor) NBOs form the ideal Lewis structure picture. While the anti-bonds reflect vacant valence-shell capacity and span regions of the atomic valence space that are formally unsaturated by covalent electrons, the full NBOs indicate covalency effects in molecules. Truly “delocalization effects” are indicated by weak occupancies of the valence anti-bonds, which indicate irreducible departure from an idealized localized Lewis picture. An overlap between an occupied and another orbital deficient in electrons results in hyperconjugation, which is described as a stabilizing action. The second-order perturbation approach of the Fock Matrix in the NBO basis between the donor and acceptor orbitals was used to calculate the hyper-conjugative interaction energy. The more strongly electron donors and acceptors interact, or the more inclined they are to donate electrons to one another, and the more strongly the system is conjugated, the higher the value of the second order interaction energy (E2). A rise in the electron density (ED) of the anti-bonding orbital that weakens the corresponding bonds can be used to identify these interactions. NBO study showed that, in terms of hybridization, natural charges, occupancy, and bond polarization, there are no appreciable differences between DH+ and its ion-pair complex (see Tables S8 & S9†). However, the NBO analysis verified the existence of several interactions between B(Ph)4− and specific DH+ functional groups, including C9_1–H, O10_1–H and N12_1–H (Fig. 1), which might be interpreted as proof of charge transfer between the two molecules. It was found that the σ(C9–H15) orbital is formed from sp3.03d0.00 hybrid (C9_1) [which is a mixture of 24.81% s, 75.11% p and 0.07% d], with occupancy of 0.98478, while the σ(O10–H) orbital is formed from sp3.11d0.00 hybrid (O10_1) [which is a mixture of 24.31% s, 75.59% p and 0.10% d], with occupancy of 0.99366. With comparable E2 values (0.21 kcal mol−1), the most important interactions beginning at DH+ and ending at B(Ph)4− are σ(C9–H15) → σ*(C–H)B(Ph)4 and σ(O10–H) → σ*(C–H)B(Ph)4. As expected, several interactions have been seen originating from σ(C–H) and π(C–H) of B(Ph)4− and terminating at C9_1–H, O10_1–H and N12_1–H σ*-orbitals of DH+. The most two significant interactions are originated from π(C–C) of B(Ph)4− and terminated at σ*(O10_1–H) and σ*(C9_1–H) with E2 values of 1.56 and 1.15 kcal mol−1, respectively. Hence, we can conclude that both C9_1–O10_1–H and N12_1–H have significant role in the formation of the studied ion-pair complex.To shed further insight on the two intramolecular H-bonds that were previously described (Fig. 3), NBO analysis has been carried out. The first intramolecular H-bond between NH (N = N12_1) and ether oxygen atom (LP(1)O) has E2 value of 1.60 kcal mol−1, while the second intramolecular H-bond including the same NH group and carbonyl group (LP(2)O) has E2 value of 5.18 kcal mol−1. In addition, the O10 atom (LP(2)O) participates with N12_1–H in another intramolecular H-bond with E2 value of 1.96 kcal mol−1. Fig. 6 illustrates that every atom that was chosen has a negative natural charge, except for the C9_1 atom of DH+ and boron atom of B(Ph)4−. A small increase in the natural charges of all negatively charged carbon atoms, except for N12_1, was noted during the protonation of Acebutolol and the subsequent formation of the ion-pair. As anticipated, the protonation of Acebutolol resulted in a large decrease in the natural charge of N12_1, which was subsequently marginally reduced by the creation of the ion-pair. This might be explained by the presence of interactions between the NH group and B(Ph)4−.
 |
| | Fig. 6 Comparison of the natural charges of selected atoms of D, DH+, B(Ph)4− and DH–B(Ph)4 (numbering of atoms is according to that given in Fig. 1). | |
2.3.4. Molecular electrostatic potential (MEP). When describing the H-bonding interactions, the locations for electrophilic attack, and nucleophilic reactions, the molecular electrostatic potential (MEP) map, which is correlated with electronic density, is a highly helpful feature. This has an association with the molecules' dipole moments, partial charges, electronegativity, and chemical reactivity. We can see distinctively charged areas of a molecule with these maps. Understanding the distribution of charges allows one to ascertain the interactions between molecules. Colors are used to depict different values of the electrostatic potential: green denotes zero potential, blue indicates regions of the highest positive potential, and red indicates regions of the greatest negative potential. The potential increases are as follows: red, orange, yellow, green, blue. The same methodology and basis set (B3LYP/6-31G(d)) utilized for geometry optimization were applied to this calculation of MEP. As shown in Fig. 7, a strong negative potential zone is spread over the π-system of B(Ph)4− ion. In the case of D, the oxygen atoms are primarily covered by negative charge areas. Positive charge spreads throughout the entire ion upon the protonation of D, particularly in the NH2+ area, which experiences electron deficiency. The interaction between B(Ph)4− and DH+ results in a decrease in the positively charged area surrounding the NH and OH groups. Besides, the negatively charged π-system of B(Ph)4− is lower than that of B(Ph)4− alone, indicating a charge transfer process from the donor to the acceptor ion.
 |
| | Fig. 7 Molecular electrostatic potential maps of B(Ph)4−, D, DH+, and DH–B(Ph)4. The electron density isosurface is 0.004 a.u. | |
The electric and optical characteristics of a particular molecule are largely determined by its frontier molecular orbitals. Generally, HOMO and LUMO stand for a given molecule's ability to lose and gain electrons, respectively. Moreover, the HOMO–LUMO energy gap provide excellent insight into a substance's optical polarizability, chemical hardness, softness, and chemical reactivity. From the results of ground-state DFT calculations, the energies of HOMO, LUMO, energy gap, chemical potential (μ), electro-affinity (A), ionization potential (I), Mulliken electronegativity (χ), chemical hardness (η), softness (S) and electrophilicity index (ω) of D, DH+, B(Ph)4−, and DH–B(Ph)4 were calculated,28–30 and tabulated in Table 1. One can often infer the chemical reactivity of a given molecule from its chemical hardness and softness. The energy gap can be used to determine the molecule's hardness or softness. Because they require less energy to excite than hard molecules, soft molecules are more polarizable than hard ones. As shown in Table 1, B(Ph)4− has the highest EHOMO value, softest HOMO, and hence lowest ionization potential demonstrating its high ability to donate electrons. On the other hand, D has the highest ELUMO value (except for B(Ph)4−), and hardest LUMO, and hence it has the lowest electron affinity value. Besides, the protonation of D boosts the reactivity by reducing the energy gap by approximately 0.32 eV. Comparing the reactivity to both drug forms, the formation of the ion-pair further increases it. The data of Mulliken electronegativity (χ) discloses that DH+ has the greatest ability to gain electrons, which matches the softness of its LUMO and its large electron affinity. The electrophilicity indexes (ω) of the investigated species reflect the absolute value of the electronic potential and the global hardness. Here, ω is lowest for B(Ph)4−. This is consistent with B(Ph)4− having the hardest LUMO and the least inclination to accept extra electronic charge. Alternatively, DH+ has the highest ω among the species under investigation, indicating its higher LUMO softness.
Table 1 The computed quantum chemical descriptors based on DFT and TDDFT calculations
| |
HOMO (eV) |
LUMO (eV) |
E-gap (eV) |
1st excited state (eV) |
Ionization potential (I) |
Electro- affinity (A) |
Chemical potential (μ) |
Mulliken electro-negativity (χ) |
Hardness (η) |
Softness (S) |
Electrophilicity index (ω) |
| D |
−5.80 |
−1.55 |
4.24 |
3.74 |
5.80 |
1.55 |
−3.68 |
3.68 |
2.12 |
0.47 |
14.37 |
| DH+ |
−8.47 |
−4.54 |
3.92 |
3.49 |
8.47 |
4.55 |
−6.51 |
6.51 |
1.96 |
0.51 |
41.55 |
| B(Ph)4− |
−2.53 |
3.18 |
5.71 |
4.99 |
2.53 |
−3.18 |
0.32 |
−0.32 |
2.85 |
0.35 |
0.15 |
| DH–B(Ph)4 |
−4.84 |
−2.51 |
2.33 |
3.56 |
4.84 |
2.51 |
−3.68 |
3.68 |
1.17 |
0.86 |
7.89 |
2.3.5. Vibrational assignment. Given the difficulty of assigning experimental frequencies, particularly the bending vibrational modes, the computation of vibrational frequencies is a useful tool in helping with spectrum assignment. Additionally, the gas phase IR spectra have some advantages over the solution or solid phase spectra because most vibrational coordinates' symmetry could be easily ascertained from the IR bands' rotational profiles. Moreover, the influence of H-bond formation and other intermolecular interactions are largely avoided. Due to a few issues, including basis set limitations and anharmonicity,31,32 the vibrational frequencies derived by quantum chemical calculations are typically greater than the equivalent experimental ones. Scale factors were included to address these drawbacks.33–35 In fact, the calculation method and basis sets determine the scaling factors to use. The mean deviation between the calculated and experimental frequencies can be used to calculate them. The current study introduces the uniform scaling approach, wherein vibrational frequencies are scaled as 0.958 for B3LYP/6-31G(d) in the wavenumber range above 1700 cm−1 and 0.983 in the wavenumber range below 1700 cm−1. The experimental IR spectra of Acebutolol hydrochloride and its ion-pair are shown in Fig. S1 and S2.† Furthermore, Fig. S3† displays the computed IR spectra of the same compounds, in addition to B(Ph)4− and the free hydrochloride form of Acebutolol. Three stretching bands at 3291, 1673, and 1662 cm−1, which correspond to the amide v(NH) and ν(CO) as well as the keto ν(CO), respectively, are observed in the experimental IR spectrum (Fig. S1†) of Acebutolol hydrochloride. When the keto group participated in an intra-molecular H-bond with the NH group, the v(CO) shifted to lower wavenumbers and the protonated NH group disappeared. Theoretically, the scaled wavenumbers of the latter stretching modes in Acebutolol are 3457, 1701, and 1687 cm−1, respectively. Upon protonation, the latter mentioned scaled vibrational modes are found at 3456, 1710, and 1642 cm−1, in that order. The IR spectrum of the ion-pair complex (Fig. S2†) is characterized by four stretching bands at 3347, 3291, 3240 and 1661 cm−1. The bands at 3347, 3291 and 3240 cm−1 may be assigned to the NH and NH2+ groups. The overlapped stretching modes of the amide CO and keto groups are represented by the band at 1661 cm−1. Theoretically, the scaled IR spectrum of the ion-pair exhibits the v(NH2+), amide v(NH), amide v(CO) and keto v(CO) at 3456, 3211, 1704, and 1665 cm−1. In summary, the position of the keto v(CO) group reflects the disruption of intramolecular connections generated by the electrostatic attraction between the protonated form of acebutolol, namely NH and OH groups, and tetraphenylborate ion.
2.3.6. NMR spectroscopy. To obtain a clear assignment of the 1H NMR spectrum of the ion-pair complex, we performed a set of NMR calculations using the gauge-invariant atomic orbital (GIAO),36 at two different computational levels; DFT/B3LYP/6-311+G(2d,p) and HF/6-31G(d). The values for the 1H isotropic were compared to TMS, which was also calculated at the same level of theory. The influence of the solvent on the calculated NMR resonances was explored by comprising one of the solvation models, the polarizable continuum model (PCM),37 and universal solvation models (SMD).38 Experimentally, the 1H and 13C NMR spectra (Fig. S4†) of the ion-pair were recorded. Unfortunately, tiny signals were detected in the 1H NMR spectrum revealing that the ion pair was partially decomposed due to the proton transfer from the DH+ to DMSO molecules. The acidic protons of NH2+ and OH as well as their closer CH-protons, for example, whose 1H chemical shifts are important in the formation of complexes, were compared between the theoretical and actual values (Table S10†). The results acquired using DFT and HF are generally similar. Additionally, our findings do not significantly alter when we change the solvation model used in our computations. All the experimental and theoretical 1H chemical shifts of the CH protons coincide well, except for the one near the NH2+ group. The chemical shift of the NH2+ group found experimentally and that obtained theoretically differ slightly. On the other hand, an unsatisfactory theoretical value for the OH group indicates the existence of particular solute–solvent interactions19 or intermolecular H-bonds, both of which are seen in the solid state.
2.3.7. Electronic structure. To get an insight into the nature of the observed electronic transitions, time-dependent density functional theory (TDDFT) calculations were carried out using two different computational level of theories; B3LYP22,23/6-31G(d) and a Coulomb-attenuated hybrid exchange-correlation functional, CAM-B3LYP39/6-31G(d). To account for the solvent influence in the computations, the PCM model was used. It has been demonstrated that molecular charge-transfer spectra can be accurately predicted by CAM-B3LYP with a long-range correction term.40,41 To calculate the electronic absorption spectra of D, DH+, B(Ph)4− and DH–B(Ph)4, the lowest 30 spin-allowed excitation states were considered. We performed all the computations to get insight into how the complex formation and protonation affect the electronic structures of the drug under study. Gauss-Sum was used to model the calculated electronic spectra. In every excited state, a Gaussian convolution with a full width at half maximum of 3000 cm−1 was used. The computed excitation state energies with transition strength (f > 0.05) were considered. The B3LYP spectrum of B(Ph)4− (Fig. S5†) is characterized by a main band and a shoulder in the range of 200–250 nm. Four transitions at 215, 222, 228, and 232 nm corresponding to H−4 → L, H−1 → L+4/H → L+3, H−1 → L+1/H → L+2 and H → L+1 (H: HOMO and L: LUMO) are the main features of the two electronic bands of B(Ph)4−. On the other hand, the key characteristic of the CAM-B3LYP spectrum of B(Ph)4− includes three main electronic transitions at 185 (H−4 → L/H → L+1), 210 (H → L/H → L+2) and 230 nm (H → L). Regarding B3LYP in comparison to CAM-B3LYP, it appears that the highest energy bands occur at longer wavelengths, whereas the lowest energy band is coincident in both electronic spectra.The computed electronic absorption spectrum (Fig. S6†) of Acebutolol, obtained by B3LYP functional, shows two main bands at 234 and 340 nm as well as two shoulders at 222 and 250 nm due to H−5 → L, H → L, H−6 → L, and H+1 → L, respectively. Although the CAM-B3LYP and B3LYP spectra of Acebutolol are similar (Fig. S6†), the CAM-B3LYP spectrum has the primary bands at shorter wavelengths (219 (H−3 → L/H → L+1) and 304 nm (H → L)). Fig. 8, shows the computed B3LYP electronic spectrum of Acebutolol, the descriptions and energies of selected frontiers molecular orbitals. As shown in Fig. 8, the lowest energy transition at 304 nm is mainly π–π* within the phenyl ring with a minor contribution of intramolecular transition initiating from the ether side-chain and terminating at the same phenyl ring.
 |
| | Fig. 8 The calculated electronic absorption spectrum of acebutolol at CAM-B3LYP/6-31G(d) level of theory, selected electronic transitions as well as the energies and frontier orbitals obtained by B3LYP/6-31G(d) method. | |
The electronic transitions of Acebutolol, obtained by either B3LYP or CAM-B3LYP functional (Fig. S6†), move to longer wavelengths after protonation by about 10–15 nm using the same level of computations. The B3LYP electronic spectrum of the mono-protonated form shows two main bands at 239 and 355 nm corresponding to H−4 → L and H → L, respectively. Two other interested transitions are found at 192 and 239 nm due to H−3 → L+1 and H−3 → L/H → L+1, respectively. However, the two primary bands in the CAM-B3LYP spectrum (Fig. 9) of the same species are visible at 223 and 214, which represent H−2 → L/H → L+1 and H → L, respectively. By contrast, it appears that the protonation prevents the protonated ether side chain from contributing in any way and makes the HOMO orbital's electron density concentrated entirely on the aromatic ring.
 |
| | Fig. 9 The calculated electronic absorption spectrum of mono-protonated acebutolol at CAM-B3LYP/6-31G(d) level of theory, selected electronic transitions as well as the energies and frontier orbitals obtained by B3LYP/6-31G(d) method. | |
Experimentally, the electronic absorption spectrum (Fig. S7†) of the ion-pair in methanol is characterized by two bands at 231 and 326 nm. This electronic spectrum matches the comparable one generated theoretically using CAM-B3LYP functional (vide infra). The electronic absorption spectra (Fig. S7†) of the ion-pair, in solvents of different polarity and hydrogen-bond strength e.g., DMSO, DMF, acetonitrile, methanol, dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, and toluene, were recorded to gain an understanding of the nature of the observed electronic transitions as well as the solvatochromism properties. The complex is insoluble in toluene. When the solvent polarity was changed, the lowest energy transition of the ion-pair did not exhibit any discernible spectrum shift. This might be seen as proof that the lowest energy transition does not represent a charge transfer band from the donor to the acceptor ion. On the other hand, because of the solvent cutoff, it was difficult to compare the spectrum changes even though the position of the highest energy band changed significantly.
The electronic absorption spectrum of Acebutolol–tetraphenyl borate complex, obtained by B3LYP functional (Fig. S6†), displays two main bands at around 238 and 348 nm corresponding to H−14 → L/H−2 → L+1 and H−2 → L, respectively. Alternatively, the CAM-B3LYP spectrum is characterized by three main transitions at 210, 221, and 310 nm due to H−1 → L+3, H−11 → L/H−2 → L+1 and H−2 → L, in that order. In contrast to those calculated by CAM-B3LYP, the electronic transitions computed by B3LYP shift to longer wavelengths. Fig. 10, shows the computed B3LYP electronic spectrum of the ion-pair complex, the descriptions and energies of selected frontiers molecular orbitals. As shown in Fig. 10, the selected HOMOs orbitals (HOMO−11, HOMO−2 and HOMO−1) are mainly composed of π-character of B(Ph)4−, whereas the LUMOs orbitals (LUMO and LUMO+1) are of π* character of mono-protonated Acebutolol drug. The LUMO+3 is of π*-character of B(Ph)4−. Hence, the band at 210 nm is mainly π–π* within the B(Ph)4− part. Two more transitions, at 221 and 310 nm, show how the charge is transferred from B(Ph)4− to the mono-protonated Acebutolol species. The two computational approaches anticipate spectra that are generally equivalent, yet at shorter wavelength the lowest energy band created by CAM-B3LYP functional is more evident. Comparing CAM-B3LYP to the other calculation method, the molecular charge transfer transitions in the ion-pair are better predicted in relation to the observed spectrum (Fig. S8†).
 |
| | Fig. 10 The calculated electronic absorption spectrum of Acebutolol–tetraphenyl borate complex at CAM-B3LYP/6-31G(d) level of theory, selected electronic transitions as well as the energies and frontier orbitals obtained by B3LYP/6-31G(d) method. | |
3. Conclusions
Tetraphenylborate ion pairs with a variety of pharmaceuticals were extensively and successfully utilized in the development of ion-selective electrodes for recognizing of the medications across a broad concentration range. Recently, potentiometric sensor was fabricated using acebutolol–tetraphenylborate for the determination of Acebutolol. The tetraphenylborate sensor demonstrated general characteristics such as extended stability and quick reaction time, but no details about the ionophore's structure or the functional groups that contributed to its formation were known. In this contribution, single crystals of Acebutolol–tetraphenylborate ion-pair appropriate for X-ray crystallographic analysis were successfully obtained. The packing diagram of the ion-pair shows both the intra- and intermolecular H-bond interactions. Owing to its shorter interaction length and closer bond angle to linearity, the intermolecular H-bond is stronger than the intramolecular contacts. Mulliken charges, natural bond-orbital analysis (NBO) and molecular electrostatic potential maps were executed to shed mor light on the functional groups participated in the complex formation as well as the probability of charge transfer between the donor and the acceptor ions. For example, NBO analysis showed that the OH and NH2+ have significant role in the formation of the studied ion-pair complex. With the help of quantum chemical calculations, the vibrational spectra of the free drug and its ion-pair were interpreted, and it was discovered that the keto v(CO) group is a key band whose position is changed because of the electrostatic attraction between the tetraphenylborate ion and the protonated form of acebutolol, specifically the NH and OH groups. The position of the charge transfer bands, and the orbitals involved in the process are strongly supported by TDDFT, which is made possible by the descriptions of Frontier molecular orbitals.
4. Experimental
4.1. Materials and instruments
Sodium tetraphenyl borate, Acebutolol hydrochloride, as well as organic solvents were purchased from reputable commercial suppliers and used precisely as directed. On JASCO, FT/IR-6600 Fourier Transform Infrared spectrometer, the IR spectra of Acebutolol hydrochloride and its ion-pair complex recorded. The electronic absorption spectra of the ion-pair were recorded on a Shimadzu 1800 UV double-beam spectrophotometer. On an Agilent triple-quadrupole 6410 QQQ LC/MS with an electrospray ionization source, the mass spectrum of the same complex was recorded. On a Bruker-Advance 500 (1H, 700.17 MHz; 13C{1H}, 176.06 MHz) spectrometer, the 1H and 13C NMR resonances of the ion-pair were recorded using DMSO-d6.
4.2. Synthesis12
Aqueous solutions (10 mL) of one mmol of sodium tetraphenyl borate (0.3422 g) and Acebutolol hydrochloride were mixed, and the reaction mixture was stirring for 20 min at the room temperature. During the stirring, a white precipitate formed. The precipitate was filtrated off, washed with water, and diethyl ether (3 × 5 mL) and finally dried under vacuum. Yield: 73% (0.523 g). IR (ATR): ν = 3347, 2959, 1661, 1507, 1214, 707 cm−1. 1H NMR (700.17 MHz, DMSO-d6) δ = 9.88 (s, 1H, amide NH), 7.80 (m, 2H, phenyl-H), 7.17 (m, 5H, B(Ph)4−), 7.11 (d, 2H, B(Ph)4−), 6.92 (m, 5H, B(Ph)4−), 6.78 (m, 3H, NH2+ and phenyl-H), 6.03–5.78 (br, 1H, OH), 4.14–4.07 (m, 3H, CH–OH and O–CH2–CH), 3.27 (m, 1H, CH(CH3)2), 3.11 (m, 1H, CH–CH2–NH2+), 2.96 (m, 1H, CH–CH2–NH2+), 2.61–2.57 (m, 3H, CH3), 2.25 (m, 2H, CH2–CH2), 1.60 (m, 2H, CH2–CH2), 1.20–1.03 (m, 6H, CH3), 0.91 (m, 3H, CH3) ppm (The complex is partially decomposed in DMSO-d6). 13C NMR (176.05 MHz, DMSO-d6) δ = 198.6, 198.5, 171.0, 163.8, 163.2, 162.9, 153.3, 135.6, 135.4, 132.7, 127.5, 125.3, 124.8, 124.7, 121.5, 120.5, 120.4, 113.8, 113.6, 70.8, 70.1, 49.7, 40.5, 40.4, 40.3, 40.2, 40.1, 40.0, 38.2, 32.1, 31.6, 18.9, 18.6, 13.7, 13.6 ppm. ESI-MS (positive mode, acetone): m/z = 337.21 {DH+} (Fig. S9†). ESI-MS (negative mode, acetone): m/z = 319.16 {B(Ph)4−} (Fig. S9†).
4.3. Single crystal X-ray diffraction analysis
A solution of the ion-pair in methanol gradually evaporated over a few weeks, producing yellow crystals appropriate for single crystal X-ray diffraction analysis. The crystal data of the ion-pair were collected on a Rigaku XtaLAB Synergy-R diffractometer equipped with a semiconductor HPA-detector (HyPix-6000) and multi-layer mirror mono-chromated Cu-Kα radiation. The intrinsic phasing approach (SHELXT programme) was used to solve the structure of the ion-pair,42 refined with SHELXL program43 and expanded using Fourier techniques. The non H-atoms were refined using an anisotropic approximation, whereas H-atoms were ‘riding’ on idealised locations. Starting with C-carbonyl, the N(CO)-propyl chain displayed rotational disorder. To properly position amide H-atoms, the nitrogen atoms were included in disorder with constrained position (EXYZ) and constrained displacement parameters (EADP) in both disordered parts. The geometrical restraint SAME was applied to the rest of the chain. Atomic displacement parameters of all disordered atoms were restraint with RIGU keyword in SHELXT input (‘enhanced rigid bond’ restraint), similarity restraint SIMU, and to approximate isotropic behaviour with ISOR. Crystal data: C42H49BN2O4, Mr = 657.63, colourless block, 0.230 × 0.170 × 0.080 mm3, monoclinic space group P21/c, a = 12.45670(10) Å, b = 23.0603(3) Å, c = 12.78140(10) Å, b = 91.2060(10)°, V = 3670.71(6) Å3, Z = 4, rcalcd = 1.190 g cm−3, m = 0.599 mm−1, F(000) = 1408, T = 100(2) K, R1 = 0.0429, wR2 = 0.1044, 7220 independent reflections [2q ≤ 149.736°] and 496 parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC- 2388718. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/.
4.4. Density functional theory calculations
Full-unconstrained geometry optimizations of Acebutolol and its protonated form as well as the ion-pair complex have been carried out with Becke 3-parameter (exchange) Lee–Yang–Parr functional,22,23 and a split-valence double-zeta polarized basis set, 6-31G(d).24 The starting coordinates for all optimization runs were acquired from crystal data of the ion-pair complex. The vibrational analysis was obtained at the same level of theory. The absence of imaginary vibrations suggests that the obtained geometry represents the local minimum structure. In fact, the computed harmonic wavenumbers at this theoretical level were found to be greater than the equivalent experimental values because of basis set limitations, anharmonicity effects, and the approximate treatment of electron correlation.31,32 The wave-number range above 1700 cm−1 is scaled at 0.958 vibrational frequencies, whereas the range below 1700 cm−1 is scaled at 0.983.18 Using the gauge-invariant atomic orbital (GIAO),36 the NMR chemical shifts of the ion-pair were computed at the B3LYP/6-311+G(2d,p) and HF/6-31G(d) levels of theory. The values for the 1H isotropic were compared to TMS, which was also computed at the same level of theory. The impact of the solvent on the calculated NMR resonances was investigated by including one of the solvation models, the polarizable continuum model (PCM),37 and universal solvation models (SMD).38 According to Koopman's theorem,28 the ionization (I = −EHOMO), electron affinity (A = −ELUMO) and Mulliken electronegativity (χ = (I + A)/2) were calculated. The molecule's softness (S) is a property that quantifies its chemical reactivity. Definitionally, softness is the hardness (η = (I − A)/2) reciprocal.29 The energy reduction brought about by the maximum electron flow between the donor and acceptor is measured by the electrophilicity index, ω = (−χ2/2η).30 Natural bonding orbitals (NBO) analysis of Weinhold and co-workers were carried out at the same level of theory of the optimization process14 Time-dependent density functional theory (TDDFT) computations were carried out utilizing two distinct level of theories; B3LYP22,23/6-31G(d) and a Coulomb-attenuated hybrid exchange-correlation functional, CAM-B3LYP39/6-31G(d), with PCM solvation model. All the calculations have been done on Gaussian03,44 Visualization of the electronic spectra and frontier molecular orbitals was achieved using Gaussview03.45
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Ahmed Mansour: conceptualization, investigation, validation, formal analysis, software, writing – original draft, writing – review & editing. Gamal A. E. Mostafa: conceptualization, investigation, validation, resources, formal analysis, writing – review & editing. Krzysztof Radacki: methodology, investigation, resources. Essam A. Ali, writing – review & editing, resources. Ola Shehab: investigation, writing – review & editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research work was supported by researchers supporting project number (RSPD2025R1000) King Saud University, Riyadh, Saudi Arabia. The authors extend their appreciation to the researchers Supporting Project Number (RSPD2025R1000) at King Saud University Riyadh Saudi Arabia, for financial support. The authors wish to thank United Arab Emirates University, Al Ain, UAE, for research support.
Notes and references
- C. R. Kumana, M. Leighton, C. M. Kaye, P. Turner and J. Hamer, Cardiac And Pulmonary Effects Of Acebutolol, Lancet, 1975, 306, 89–93 CrossRef PubMed.
- M. Lewis, D. Jones, A. Dart and A. Henderson, The psychological side effects of acebutolol and atenolol, Br. J. Clin. Pharmacol., 1984, 17, 364–366 CrossRef CAS PubMed.
- I. Rapado-Martínez, M. C. García-Alvarez-Coque and R. M. Villanueva-Camañas, Liquid chromatographic procedure for the evaluation of β-blockers in pharmaceuticals using hybrid micellar mobile phases, J. Chromatogr. A, 1997, 765, 221–231 CrossRef.
- M. Delamoye, C. Duverneuil, F. Paraire, P. d. Mazancourt and J.-C. Alvarez, Simultaneous determination of thirteen β-blockers and one metabolite by gradient high-performance liquid chromatography with photodiode-array UV detection, Forensic Sci. Int., 2004, 141, 23–31 CrossRef CAS PubMed.
- M. Silva, S. Morante-Zarcero, D. Pérez-Quintanilla and I. Sierra, Simultaneous determination of pindolol, acebutolol and metoprolol in waters by differential-pulse voltammetry using an efficient sensor based on carbon paste electrode modified with amino-functionalized mesostructured silica, Sens. Actuators, B, 2019, 283, 434–442 CrossRef CAS.
- N. A. Alarfaj and M. F. El-Tohamy, Construction and validation of new electrochemical carbon nanotubes sensors for determination of acebutolol hydrochloride in pharmaceuticals and biological fluids, J. Chin. Chem. Soc., 2014, 61, 910–920 CrossRef CAS.
- A. Yamuna, P. Sundaresan and S.-M. Chen, Ethylcellulose assisted exfoliation of graphite by the ultrasound emulsification: an application in electrochemical acebutolol sensor, Ultrason. Sonochem., 2019, 59, 104720 CrossRef CAS PubMed.
- S. V. Selvi, N. Nataraj, T.-W. Chen, S.-M. Chen, P. Balu and X. Liu, Disposable cerium oxide/graphene nanosheets based sensor for monitoring acebutolol in environmental samples and bio-fluids, J. Environ. Chem. Eng., 2022, 10, 107182 CrossRef CAS.
- M. A.-H. Elsayed, M. Barary, M. Abdel-Salam and S. Mohamed, Spectrophotometric assay of certain cardiovascular drugs through charge transfer reactions, Anal. Lett., 1989, 22, 1665–1684 CrossRef CAS.
- C. Sastry, T. T. Rao, A. Sailaja and J. V. Rao, Micro-determination of warfarin sodium, nicoumalone and acebutolol hydrochloride in pharmaceutical preparations, Talanta, 1991, 38, 1107–1109 CrossRef CAS PubMed.
- A. M. Al-Mohaimeed, S. Y. Al Omar and M. F. El-Tohamy, Fast and novel multiwalled carbon nanotubes decorated with metal oxide nanoparticles for potentiometric detection of a prohibited medication in sports acebutolol hydrochloride, Heliyon, 2023, 9, e20997 CrossRef CAS.
- G. A.-H. Mostafa, M. M. Hefnawy and A. Al-Majed, PVC membrane sensors for potentiometric determination of acebutolol, Sensors, 2007, 7, 3272–3286 CrossRef PubMed.
- D. Manjunatha, S. Shaikh, K. Harikrishna, R. Sudhirkumar, P. B. Kandagal and J. Seetharamappa, Simple and sensitive spectrophotometric methods for the determination of acebutolol hydrochloride in bulk sample and pharmaceutical preparations, Ecletica Quim., 2008, 33, 37–40 CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- G. A. Jeffrey, Hydrogen-Bonding: An Update, Crystallogr. Rev., 2003, 9(2–3), 135–176 CrossRef CAS.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Science & Business Media, 2012 Search PubMed.
- S. Scheiner, Hydrogen Bonding: a Theoretical Perspective, Oxford University Press, USA, 1997 Search PubMed.
- A. M. Mansour and N. T. A. Ghani, Hydrogen-bond effect, spectroscopic and molecular structure investigation of sulfamethazine Schiff-base: Experimental and quantum chemical calculations, J. Mol. Struct., 2013, 1040, 226–237 CrossRef CAS.
- N. T. A. Ghani, A. M. Mansour and B. Spectroscopy, Molecular structure of 2-chloromethyl-1H-benzimidazole hydrochloride: Single crystal, spectral, biological studies, and DFT calculations, Spectrochim. Acta, Part A, 2012, 86, 605–613 CrossRef.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- A. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, 6-31G* basis set for third-row atoms, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- J. Pipek, Unique positive definite extension of Mulliken's charge populations of non-orthogonal atomic basis functions, J. Mol. Struct., 2000, 501–502, 395–401 CrossRef CAS.
- D. Jacquemin, T. Le Bahers, C. Adamo and I. Ciofini, What is the “best” atomic charge model to describe through-space charge-transfer excitations?, Phys. Chem. Chem. Phys., 2012, 14, 5383–5388 RSC.
- S. Nadeem, M. U. Khan, R. Hussain, F. Shafiq and N. Alhokbany, Novel cyano functional small molecule photosensitizers with amplified electron injection and photovoltaic attributes: molecular modelling towards efficient dye-sensitized solar cells, J. Chem. Technol. Biotechnol., 2024, 99, 902–921 CrossRef CAS.
- T. Koopmans, Ordering of wave functions and eigenenergies to the individual electrons of an atom, Physica, 1933, 1, 104–113 CrossRef CAS.
- H. Chermette, Chemical reactivity indexes in density functional theory, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- R. G. Parr and W. Yang, Density functional approach to the frontier-electron theory of chemical reactivity, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS.
- J. Pople, H. B. Schlegel, R. Krishnan, D. Defrees, J. Binkley, M. Frisch, R. Whiteside, R. Hout and W. Hehre, Molecular orbital studies of vibrational frequencies, Int. J. Quantum Chem., 1981, 20, 269–278 CrossRef.
- G. Rauhut and P. Pulay, Transferable scaling factors for density functional derived vibrational force fields, J. Phys. Chem., 1995, 99, 3093–3100 CrossRef CAS.
- N. T. A. Ghani and A. M. Mansour, 2-[(1H-Benzimidazol-2-ylmethyl)-amino]-benzoic acid methyl ester: Crystal structure, DFT calculations and biological activity evaluation, Spectrochim. Acta Mol. Biomol. Spectrosc., 2011, 81, 754–763 CrossRef PubMed.
- N. T. Abdel Ghani and A. M. Mansour, Palladium(II) and platinum(II) complexes containing benzimidazole ligands: Molecular structures, vibrational frequencies and cytotoxicity, J. Mol. Struct., 2011, 991, 108–126 CrossRef CAS.
- O. R. Shehab and A. M. Mansour, Charge transfer complexes of 2-arylaminomethyl-1H-benzimidazole with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone: Experimental and DFT studies, J. Mol. Struct., 2013, 1047, 121–135 CrossRef CAS.
- S. Wolff and T. Ziegler, Calculation of DFT-GIAO NMR shifts with the inclusion of spin-orbit coupling, J. Chem. Phys., 1998, 109, 895–905 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- D. A. Habashy, R. M. Khaled, A. Y. Ahmed, K. Radacki, S. K. Ahmed, E. K. Tharwat, H. Magdy, A. Zeinhom and A. M. Mansour, Cytotoxicity of fac-Mn(CO)3 complexes with a bidentate quinoline ligand towards triple negative breast cancer, Dalton Trans., 2022, 51, 14041–14048 RSC.
- A. M. Mansour, R. M. Khaled, K. Radacki, Z. Younes, M. Gamal, B. Guirguis, G. A. E. Mostafa, E. A. Ali and O. R. Shehab, In vitro cytotoxicity of Mn(i) and Ru(ii) carbonyls with a diphenyl pyridyl phosphine coligand towards leukaemia, Dalton Trans., 2023, 52, 10286–10293 RSC.
- G. M. Sheldrick, SHELXT–Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, J. C. B. R. E. Stratmann, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, A. L. G. Liu, I. K. P. Piskorz, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 03 (Revision A.9), Gaussian, Inc., Pittsburgh, 2003 Search PubMed.
- A. Frisch, A. Nielson and A. J. G. I. Holder, Gaussview User Manual, Pittsburgh, PA, 2000, p. 556 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2388718. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07163h |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Gamal A. E. Mostafa‡c,
Krzysztof Radacki
*ab,
Gamal A. E. Mostafa‡c,
Krzysztof Radacki