
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydrophosphorylation of alkynes for the synthesis of (E)-vinylphosphonates†
Babak Kaboudin *a,
Hesam Esfandiaria,
Nematollah Arshadi*b and
Haruhiko Fukayac
*a,
Hesam Esfandiaria,
Nematollah Arshadi*b and
Haruhiko Fukayac
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences, Gava Zang, Zanjan, Iran. E-mail: kaboudin@iasbs.ac.ir; kaboudin@gmail.com; Fax: +98 24 33153232; Tel: +98 24 33153220
bDepartment of Chemistry, Faculty of Sciences, University of Zanjan, Zanjan, Iran
cSchool of Pharmacy, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan
First published on 17th February 2025
Abstract
Hydrophosphorylation of alkynes with dialkylphosphites in the various copper catalysts was investigated. The reactions provided the regio- and stereoselective E-vinylphosphonates under commercially available copper chloride catalyst in the presence of ethylene diamine as an efficient ligand. The impact of solvents, temperature, and diamine ligands are included in this report. In addition, the DFT calculations provided insight into the regio- and stereoselectivity of the reaction. It is suggested that the reaction proceeded via an in situ generated Cu(AN)4+ complex. The reaction of phenylacetylene with diethyl phosphite in the presence of EDA and the (CH3CN)4CuBF4 complex as a catalyst also gave the corresponding E-vinylphosphonates in good yield.
Introduction
Transition metal-catalyzed carbon–phosphorus (C–P) bond formation is one of the most straightforward ways for the synthesis of various organophosphorus compounds.1–5 One of the important and atom-economical approaches for the carbon–phosphorus bond formation is transition metal-catalyzed addition of P–H bonds to alkynes for the synthesis of alkenylphosphine oxides and vinylphosphonates, which have wide applications in organic synthesis and industrial processes.6–10 Several metal-catalyzed C–P bond formation methods have been reported for the synthesis of alkenylphosphine oxides via addition reaction of R2P(O)H compounds to alkynes, including palladium,11 nickel,12 rhodium,13 ytterbium complex,14 and copper salts.15 However, in contrast to R2P(O)H, hydrophosphorylation of alkynes with dialkylphosphite [(RO)2P(O)H] for the synthesis of highly selective vinylphosphonates (Markovnikov and anti-Markovnikov addition and also E/Z-selectivity) is still hot research topic. The first palladium and nickel-catalyzed addition of dialkyl phosphite [(RO)2P(O)H] to alkynes was reported by Tanaka and Han to give a mixture of non-selective corresponding vinylphosphonates.16 In spite some significant achievements in the palladium- and nickel-catalyzed addition of dialkylphosphite to alkynes, the drawbacks of the catalyst systems, such as sensitivity of complexes to air, high cost, low selectivity and toxicity, limit their applications.Three different research groups developed copper-catalyzed additions of R2P(O)H compounds to alkynes for the synthesis of alkenylphosphine oxides under copper catalyst system.15,17,18
In continuing our efforts for the synthesis of organophosphorus compounds,19 we recently reported a convenient preparation of vinylphosphonates via chemo- and stereoselective Knoevenagel condensation reaction of carbonyl compounds with cyanomethylphosphonate.20 Herein, a simple copper-catalyzed hydrophosphorylation of alkynes with dialkylphosphite was reported for the regio- and stereoselective synthesis of E-vinylphosphonates. In addition, in order to reveal the details of the regio- and stereoselectivity of the reaction, the proposed mechanism was also studied using DFT method of calculation.
Results and discussion
Initially, the reaction of phenylacetylene 1a with diethyl phosphite was examined as the model reaction. The results screening of various reaction conditions are shown in Table 1. No reaction was observed when Cu(OAc)2, Pd(OAc)2, and FeCl3 were used as a catalyst in the presence of ethylenediamine (EDA) in acetonitrile at reflux for 24 h (entries 1–3). Upon using CuCl2 a mixture of products was obtained in a low yield after 24 h (entry 4). When the reaction was conducted with CuCl (10 mol%) in acetonitrile for 24 h, the compound 2a was obtained in 42% yield (entry 5). No catalytic activities were observed with CuBr and CuI (entries 6 and 7). It seems that CuCl has better activity in the presence of EDA. The results for the reactions conducted in various solvents showed that no reaction was observed when CH2Cl2, DMF, or DMSO were used as a solvent (entries 8–10), while using dioxane, the compound 2a was obtained in only 24% in moderate selectivity (entry 11). To improve the reaction yield, the effects of the amount of CuCl, the reaction time, ligand, and temperatures were investigated (entries 12–17). Interestingly, when the reaction was conducted with increasing the amount of the CuCl to 20 mol%, the compound 2a was obtained in 89% yield (entry12). It is worthy to note that the reaction proceeds to give the product 2a in 89% yield for 12 h (entry 13). When the reaction was carried out at reflux for 4 h, the reaction yield was decreased to 35% (entry 14). No reaction was observed when the reaction was carried out in the presence of EDA in acetonitrile at rt for 48 h (entries 15). When 1,2-diamino cyclohexane was taken in place of ethylenediamine during the reaction, the compound 2a was obtained in 53% yield with lower selectivity (entry 16). The reaction in the presence of ammonia solution also proceeded and the compound 2a was obtained in 41% yield (entry 17). When the reaction was carried out at reflux for 24 h under Ar, the reaction yield was slightly decreased to 83% (entry 18).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Time (h) | Yield of 2a (%) |
a Yield refers to the isolated pure product 2a after short column chromatography on silica-gel for the reaction of 1a (2.0 mmol) with diethylphosphite (1 mmol) in the presence of EDA (0.5 mmol) and catalyst (10 mol%).b Mixture of products were detected.c Has an E/Z ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20.d CuCl (20 mol%).e 1,2-Cyclohexyldiamine (0.5 mmol) as a ligand.f Ammonia solution (0.5 mL, 25%) was added instead of EDA.g Reaction carried-out under Ar. :20.d CuCl (20 mol%).e 1,2-Cyclohexyldiamine (0.5 mmol) as a ligand.f Ammonia solution (0.5 mL, 25%) was added instead of EDA.g Reaction carried-out under Ar. |
|||||
| 1 | Cu(OAc)2 | MeCN | Reflux | 24 | — |
| 2 | Pd(OAc)2 | MeCN | Reflux | 24 | — |
| 3 | FeCl3 | MeCN | Reflux | 24 | — |
| 4 | CuCl2 | MeCN | Reflux | 24 | —b |
| 5 | CuCl | MeCN | Reflux | 24 | 42 |
| 6 | CuBr | MeCN | Reflux | 24 | — |
| 7 | CuI | MeCN | Reflux | 24 | — |
| 8 | CuCl | CH2Cl2 | Reflux | 24 | — |
| 9 | CuCl | DMSO | 90 | 24 | — |
| 10 | CuCl | DMF | 90 | 24 | — |
| 11 | CuCl | Dioxane | 90 | 24 | 24c |
| 12 | CuCl | MeCN | Reflux | 24 | 89d |
| 13 | CuCl | MeCN | Reflux | 12 | 89d |
| 14 | CuCl | MeCN | Reflux | 4 | 35d |
| 15 | CuCl | MeCN | rt | 48 | —d |
| 16 | CuCl | MeCN | Reflux | 24 | 53d,e |
| 17 | CuCl | MeCN | Reflux | 24 | 41d,f |
| 18 | CuCl | MeCN | Reflux | 24 | 83d,g |
With the optimized conditions in hands, next we examined the synthetic scope of the reaction. As shown in Table 2, the present reaction was successfully applied to a wide range of alkynes. The results showed that the electronic nature of the aryl group in aromatic alkyne 1 did not affect the reaction outcome significantly (2a–2w). It should be noted that alkynes with amide (1k), ester (1n), and nitrile (1p), group gave the desired products without any side products. The reaction of 2-naphthylacetylene 1r with diethylphosphite in the presence of EDA and CuCl (20 mol%) gave the desired product 2r in moderate yield. Treatment of heterocyclic alkyne 1s with a mixture of diphenylphosphine oxide, EDA and CuCl (20 mol%) for 12 h gave the corresponding product 2t in good yield with 90:10 of E/Z ratio based on 31P NMR analysis. The reactions of aliphatic substrate 1u also gave the desired product 2v in good yield. Treatment of alkyne 1y, an aminophosphonate derivative, with a mixture of diethyl phosphite, EDA and CuCl (20 mol%) for 12 h gave the corresponding product 2y in good yield. It should be noted that the internal alkyne, diphenylacetylene, failed to give any addition product (2z) after 24 h.

To gain insights into the mechanism, several control experiments were carried out. The reaction of alkyne with diethylphosphite in the presence of CuCl without the addition of EDA was conducted; however, no product was observed. In other experiment, when the reaction was carried out in the absence of diethylphosphite, an alkyne homo-coupling product 3 was obtained in 90% yield (Scheme 1). In the next trial, the influence of EDA on the reaction efficiency was also examined in the absence of diethylphosphite. As a result, the yield of 3 was decreased to 10% in the absence of EDA. Thus, it was confirmed that the EDA has major role in the conversion.
 | ||
| Scheme 1 Control reactions. | ||
Furthermore, the nature of reaction mixture is revealed by NMR-spectroscopy. The 1H NMR spectrum of the reaction mixture without further purification was shown in Fig. 1. As shown in Fig. 1, the olefinic proton signal appearing as doublets at 5.95 and 6.72 ppm with a J-value of 12.0 Hz confirmed the presence of Z or cis-configuration of double bonds and doublets at 6.41 and 7.08 ppm with a J-value of 16.0 Hz confirmed the presence of E or trans-configuration of self-addition adduct of alkyne 1a in the reaction condition.21 The isolation as the individual pure alkene side products by chromatography was not unfortunately successful due to their very similar polarities on a stationary phase of silica-gel.
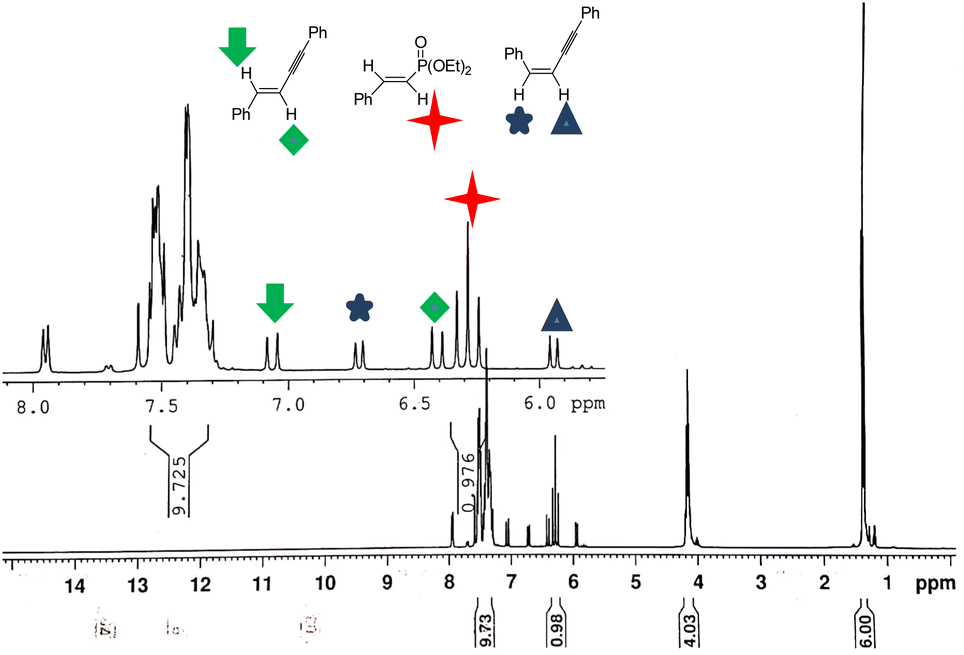 | ||
| Fig. 1 1H NMR spectrum of the reaction mixture of 1a in CDCl3. | ||
To find insights for the reaction outcome, we evaluated the reaction paths from cationic species bearing a dimethyl phosphonate moiety to yield E- and Z-products by the DFT calculations at B3LYP/LACVP++** level of theory (Fig. 2 and 3). In the first step, phenylacetylene, A, and tetrakis(acetonitrile) copper(I) complex, AN4Cu, produces a copper alkyne complex, AN3CuA. Due to low electrostatic and Mulliken charges on the Cu atom and the steric effect of the coordinated dimethylphosphite, P′, of the AN3CuP′ complex, the reaction is preferred to start by the attack of A as nucleophile at the electrophilic Cu atom of the AN4Cu complex (Fig. 2). The main tetrahedral Cu(I) complex, AN2CuAP′, that contains both reactive species is formed by the attack of P′ on the centre of copper π-complex, AN3CuA. P′ that is 3.1 kcal mol−1 less stable than its tautomer, P, can act as a nucleophile. The complex formation constants for aqueous copper(I) ion with halide ions and acetonitrile (AN) as ligand are as follow: I− > Br− > Cl− > AN. Therefore, the formation of Cu(AN)4+ complex in the presence of I− and Br− ions is more inhibited than that of Cl− ion.22 It should be noted that the reaction of 1a with diethylphosphite in the presence of EDA and the (CH3CN)4CuBF4 complex as a catalyst gave the compound 2a in 85% yield (see Fig. S74†). Many of the dialkyl phosphite reactions appear to progress via its minor tautomer.23 The reaction is proceded by the formation of a metal σ-complex, σ-Cpx, via π-σrearrangement of the organocopper π-complex, π-Cpx. The reaction occurred by the addition of ethylene diamine, EDA. During the reaction process, the σ-Cpx intermediate is converted into the E-Pdt via an intramolecular proton transfer reaction. The syn-periplanar conformation of the hydroxyl group with Cu is required for this 1,4-prototropic mechanism (Scheme 2). The Z-σ-Cpx intermediate is only formed from the rearrangement of π-Cpx to σ-Cpx via the stereospecific syn-addition of Cu–P bond to the C–C triple bond. The E-Pdt is created in the rate determining step of the reaction. An energy barrier of about 22.6 kcal mol−1 limits the formation of the product at the fourth step (Fig. 3). Based-on this result, the reaction is expected to proceed at temperatures a little above 25 °C. Therefore, the reaction is enthalpically favoured and E-Pdt is the only product of the highly regioselective reaction.
 | ||
| Fig. 2 The electrostatic (ESP) and Mulliken charges of the Cu atom in the starting complexes; from left to right: AN4Cu, AN3CuA and AN3CuP′. | ||
 | ||
| Fig. 3 Energy profile for the reaction in gas phase and in acetonitrile solution (the number in parentheses), calculated at the B3LYP/LACVP++** level of theory at 298.1. | ||
 | ||
| Scheme 2 Plausible reaction mechanism for the highly regio- and stereoselective synthesis of E-Pdt. | ||
The solvent effect is addressed by the comparison between the energy profile of the reaction in the gas and acetonitrile phases determined at the same level of theory (Fig. 3). The results obviously show that the reaction speed down in the solution phase at room temperature.
In summary, herein we reported a novel approach for the regio and stereoselective synthesis of E-vinylphosphonates via a selective hydrophosphorylation of alkynes in copper chloride catalyst in the presence of EDA as an efficient ligand at reflux. Results showed that the CuClin acetonitrile in the presence of EDA is a convenient and effective catalyst for the selective hydrophosphorylation of alkynes. It is suggested that the reaction proceeded via an AN3CuP′ complex in EDA media. Stereochemistry of the E-isomer was determined by careful NMR analysis. To understand the selectivity of the reaction, DFT calculations have provided insight into the basis of these reactivity and selectivity.
Experimental
General methods
All chemicals were commercial products. CuCl (Merck, Art. No. 102739) was purchased from commercial distributors and used as received. NMR spectra were obtained with a 400 MHz Bruker Avance instrument with the chemical shifts being reported as δ ppm and couplings expressed in hertz. The chemical shift data for each signal on 1H NMR are given in units of δ relative to CHCl3 (δ = 7.26) for CDCl3 solution. For 13C NMR spectra, the chemical shifts in CDCl3 and DMSO are recorded relative to the CDCl3 resonance (δ = 77.0) and DMSO resonance (δ = 40.45). Silica gel column chromatography was carried out with silica gel 100 (Merck No. 10184). Merck silica-gel 60 F254 plates (No. 5744) were used for the preparative TLC.N-(3-Ethynylphenyl)benzamide (1k). The product was obtained as white solid 95% yield; 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.91–7.78 (m, 3H), 7.75–7.68 (m, 1H), 7.61–7.42 (m, 3H), 7.31 (dd, J = 7.8, 4.4 Hz, 2H), 3.11 (s, 1H). 13C{1H} NMR (101 MHz, CDCl3) δ 166.1, 138.0, 134.7, 132.0, 129.10, 128.8, 128.3, 127.1, 123.8, 122.9, 121.0, 83.2.
1,3-Dimethyl-7-(prop-2-yn-1-yl)-3,7-dihydro-1H-purine-2,6-dione (1s). 1H NMR (400 MHz, DMSO): δ 8.20 (s, 1H), 5.19 (d, J = 2.6 Hz, 2H), 3.58 (t, J = 2.6 Hz, 1H), 3.44 (s, 3H), 3.25 (s, 3H); 13C{1H} NMR (101 MHz, DMSO): δ 154.7, 151.5, 148.8, 142.6, 106.2, 79.7, 78.4, 77.3, 36.2, 29.9.
:1 to 1:5) to give the compound 2 as the viscous oil or white solid. All products gave satisfactory spectral data in accord with the assigned structures.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is based upon research funded by Iran National Science Foundation (INSF) under Project No. 4038081. The authors gratefully acknowledge partial support by the Institute for Advanced Studies in Basic Sciences (IASBS).References
- F. M. J. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 2010, 3037–3062 CrossRef.
- J. Yang, T. Chen and L. B. Han, J. Am. Chem. Soc., 2015, 137, 1782–1785 CrossRef CAS PubMed.
- I. Hicks, J. McTague, T. Hapatsha, R. Teriak and P. Kaur, Molecules, 2020, 25, 290 CrossRef CAS PubMed.
- D. S. Glueck, Topics in Organometallic Chemistry, Springer, Berlin, Heidelberg, 2010, vol 31 Search PubMed.
- S. Hore, A. Srivastava and R. P. Singh, J. Org. Chem., 2019, 84, 6868–6878 CrossRef CAS PubMed.
- S. Ben-Valid, A. A. A. Quntar and M. Srebnik, J. Org. Chem., 2005, 70, 3554–3563 CrossRef CAS PubMed.
- K. Hara, S.-Y. Park, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Chem.–Asian J., 2008, 3, 1500–1504 CrossRef CAS PubMed.
- N. S. Goulioukina, N. N. Makykhin, E. Shinkarev, Y. K. Grishin, A. Roznyatovskya and I. P. Beletskaya, Org. Biomol. Chem., 2016, 14, 10000–10010 RSC.
- (a) J. Parvolea and D. Janasch, Macromolecules, 2008, 41, 3893–3903 CrossRef; (b) G. J. Schlichting, J. L. Horan, J. D. Jessop, S. D. Nelson, S. Seifert, Y. Yang and A. M. Herring, Macromolecules, 2012, 45, 3874–3882 CrossRef CAS.
- (a) R. K. Haynes, S. C. Vonwiller and T. W. Hambley, J. Org. Chem., 1989, 54, 5162–5170 CrossRef CAS; (b) R. K. Haynes, W. A. Loughlin and T. W. Hambley, J. Org. Chem., 1991, 56, 5785–5790 CrossRef CAS.
- (a) L.-B. Han, R. Hua and M. Tanaka, Angew. Chem., Int. Ed., 1998, 37, 94–96 CrossRef CAS; (b) T. Chen, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2018, 140, 3139–3155 CrossRef CAS PubMed.
- (a) L.-B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080–5081 CrossRef CAS PubMed; (b) Y. Wu, L. Liu, K. Yang, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, 8118–8127 CrossRef CAS PubMed.
- (a) L.-B. Han, C.-Q. Zhao and M. Tanaka, J. Org. Chem., 2001, 66, 5929–5932 CrossRef CAS PubMed; (b) A. Kondoh, H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2008, 81, 502–505 CrossRef CAS; (c) Y. Huang, W. Hao, G. Ding and M.-Z. Cai, J. Organomet. Chem., 2012, 715, 141–146 CrossRef CAS.
- K. Takaki, G. Koshoji, K. Komeyama, M. Takeda, T. Shishido, A. Kitani and K. Takehira, J. Org. Chem., 2003, 68, 6554–6565 CrossRef CAS PubMed.
- (a) J. Li, Z. Gao, Y. Guo, H. Liu, P. Zhao, X. Bi, E. Shi and J. Xiao, RSC Adv., 2022, 12, 18889 RSC; (b) I. G. Trostyanskaya and I. P. Beletskaya, Tetrahedron, 2014, 70, 2556–2562 CrossRef CAS.
- L.-B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571–1572 CrossRef CAS.
- M. Niu, H. Fu, Y. Jiang and Y. Zhao, Chem. Commun., 2007, 272–274 RSC.
- Y. Gao, G. Wang, L. Chen, P. Xu, Y. Zhao, Y. Zhou and L.-B. Han, J. Am. Chem. Soc., 2009, 131, 7956–7957 CrossRef CAS PubMed.
- B. Kaboudin, M. Ghashghaee, H. Fukaya and H. Yanai, Chem. Commun., 2023, 59, 7076–7079 RSC.
- B. Kaboudin, A. Moradi, H. Esfandiari, P. Daliri, F. Kazemi, H. Yanai and H. Aoyama, Org. Biomol. Chem., 2022, 20, 2500–2507 RSC.
- Q. Liang, K. Hayashi and D. Song, ACS Catal., 2020, 10, 4895–4905 CrossRef CAS.
- P. Kamau and R. B. Jordan, Inorg. Chem., 2001, 40, 3879–3883 CrossRef CAS PubMed.
- G. O. Doak and L. D. Freedman, Chem. Rev., 1961, 61, 31–44 CrossRef CAS.
- X. Wang, S. O. de Silva, J. N. Reed, R. Billadeau, E. J. Griffen, A. Chan and V. Snieckus, Org. Synth., 1995, 72, 163 CrossRef CAS.
- S. Keskin and M. Balci, Org. Lett., 2015, 17, 964–967 CrossRef CAS PubMed.
- R. R. Ruddarraju, A. C. Murugulla, R. Kotla, M. C. B. Tirumalasetty, R. Wudayagiri, S. Donthabakthuni, R. Maroju, K. Baburao and L. S. Parasa, Eur. J. Med. Chem., 2016, 123, 379–396 CrossRef CAS PubMed.
- B. Kaboudin, S. Faghih, S. Alavi, M. R. Naimi-Jamal and A. Fattahi, Synthesis, 2023, 55, 121–130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for all the compounds 2a–2y and copies of 1H NMR and 13C NMR, and 31P NMR of all products. See DOI: https://doi.org/10.1039/d5ra00300h |
| This journal is © The Royal Society of Chemistry 2025 |