DOI:
10.1039/D5RA00250H
(Paper)
RSC Adv., 2025,
15, 4829-4846
Novel ethyl 2-hydrazineylidenethiazolidin-5-ylidene acetate clubbed with coumarinylthiazolyl pyrazole system as potential VEGFR-2 inhibitors and apoptosis inducer: synthesis, cytotoxic evaluation, cell cycle, autophagy, in silico ADMET and molecular docking studies†
Received
10th January 2025
, Accepted 9th February 2025
First published on 13th February 2025
Abstract
Novel derivatives of ethyl 3-substituted-2-{4-oxo-2-(2-((3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazol-4-yl)methylene)hydrazineyl)thiazol-5(4H)-ylidene}acetate (5a–h) were synthesized and assessed for their cytotoxic potential against the liver cancer cell lines Huh-7 and HepG-2. Among these, compounds 5d and 5g demonstrated notable antiproliferative effects, which were benchmarked against the standard drug doxorubicin. To further understand the mechanisms behind their antiproliferative activity, compounds 5d and 5g were investigated for their impact on the cell cycle and their ability to induce apoptosis. They were found to induce significant cellular cycle arrest at the G1 phase. Besides, they potentially enhanced the cellular late apoptosis and reduced the cellular viability. In consent with the apoptosis results, compounds 5d and 5g displayed significant potential autophagic induction against the studied cancer cell lines. Further, both compounds 5d and 5g showed strong interactions with the VEGFR-2 receptor when they were studied using molecular docking. The ADMET prediction indicated that these bioactive compounds have the potential to serve as effective to fight liver cancer.
1. Introduction
The thiazole ring is a five-membered heterocyclic structure that occurs naturally in various substances, such as vitamins, pigments, alkaloids, and components of both plant and animal cells.1 The presence of the –S–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moiety in the thiazole system has been demonstrated to enhance lipid solubility and hydrophilicity, which are critical factors enabling the structure to be efficiently metabolized through standard biochemical pathways.2 Thiazole and its thiazolidinone derivatives, particularly those featuring a carbonyl group at the 2-, 4-, or 5-position, have been shown to possess significant pharmaceutical potential.3,4 Notably, thiazolidin-4-one derivatives have displayed broad-spectrum anticancer activity against breast cancer (MCF-7) and lung cancer (A549) cell lines, as well as promising EGFR inhibitory effects5,6 (Fig. 1).
N– moiety in the thiazole system has been demonstrated to enhance lipid solubility and hydrophilicity, which are critical factors enabling the structure to be efficiently metabolized through standard biochemical pathways.2 Thiazole and its thiazolidinone derivatives, particularly those featuring a carbonyl group at the 2-, 4-, or 5-position, have been shown to possess significant pharmaceutical potential.3,4 Notably, thiazolidin-4-one derivatives have displayed broad-spectrum anticancer activity against breast cancer (MCF-7) and lung cancer (A549) cell lines, as well as promising EGFR inhibitory effects5,6 (Fig. 1).
 |
| | Fig. 1 Some thiazolidinone compounds exhibited anticancer effects against MCF-7 and A549 cell lines. | |
The thiazolidin-4-one containing ylidene acetate fragment at 5-position was synthesized by reaction of thioamides with dialkyl acetylene dicarboxylate (DAAD) under mild conditions.7 These compounds recorded pharmacological activities such as leukotriene B4 inhibitors,8 antidiabetic,9 antiparasitic,10 antibacterial11 and anticancer.12 On the other hand, numerous 2-hydrazinyl-thiazolidinone derivatives have been identified as potential chemotherapeutic agents, demonstrating a range of biological activities, including antimicrobial,13,14 antiviral,15 antiplasmodial,16 and antitumoral effects.17 In particular, arylidenehydrazinothiazole pharmacophore structures have shown potent inhibitory activity against tyrosyl-DNA phosphodiesterase 1 (TDP1) in the nanomolar range. By inhibiting DNA repair mechanisms, these compounds serve as adjuvant therapies, enhancing the efficacy of chemotherapy drugs, especially in the treatment of drug-resistant tumors.18,19
The coumarin ring as one of the most significant heterocyclic compounds exhibited numerous biological actions, such as anticancer,20 antituberculosis,21 anticholinesterase,22 antiviral,23 anticoagulant,24 antidepressant,25 antioxidant26 and antibacterial agent.27 In the same manner, five-membered heterocyclic scaffolds containing pyrazole and thiazole, are the most important ones.28–30 Chemists have been interested in pyrazole and thiazole scaffolds because of their wide range of biological actions, including anti-inflammatory,31 antiviral,32 anticancer,33 antibacterial34 and antituberculosis.35 Several commercial drugs containing coumarin, pyrazole, or thiazole rings are illustrated in Fig. 2.
 |
| | Fig. 2 Marketed drugs containing coumarin, pyrazole or thiazole scaffold. | |
Cancer is one of the most serious diseases. After heart disease, cancer diseases are thought to be the second most common cause of mortality worldwide.36,37 One of the most important facets of cancer treatment remains chemotherapy. The most significant issue with the use of chemotherapy is its negative effects. Therefore, finding novel compounds as safe anticancer agents is a challenging goal that will help not only treat cancer but also overcome drug-resistant malignancy and prevent adverse medication reactions.
Depending on the previous and continuing our efforts to discover novel polyheterocyclic frames as a new antiproliferative agents to inhibit of cancer cell growth with targeting inhibitory action,38–42 we have now designed a new series of ethyl 2-hydrazineylidenethiazolidin-5-ylidene acetate clubbed with coumarinylthiazolyl pyrazole system (Fig. 3). Using the SBR assay, the synthesized compounds were tested on a panel of liver cancer cell lines, namely Huh-7 and HepG2 and the results were compared with standard drug doxorubicin.
 |
| | Fig. 3 The designed molecular frame for the title compounds. | |
2. Results and discussion
A new series of coumarinyl-thiazolyl-pyrazole clubbed with hydrazineylidene-thiazolidin-5-ylidene acetate derivatives 5a–h were synthesized by two-step reaction: first, a mixture of 3-acetylcoumarin (1), thiosemicarbazide and phenacyl bromide in DMF at 70 °C temperature was heated for 1 hour to give the corresponding coumarinyl-thiazolyl hydrazone 2. Then, the latter hydrazone reacted with the dimethylformamide (DMF) in the presence of phosphoryl chloride under Vilsmeier–Haack reaction conditions to form 3-(2-oxo-2H-chromen-3-yl)-1-(4-phenyl-thiazol-2-yl)-1H-pyrazole-4-carboxaldehyde (3) (Scheme 1).43 In the next step, one-pot three-component reaction of the aldehyde 3 with a series of 4-substituted thiosemicarbazide and diethyl acetylenedicarboxylate in glacial acetic acid afforded the corresponding ethyl 3-substituted-2-{4-oxo-2-(2-((3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazol-4-yl)methylene)hydrazineyl)thiazol-5(4H)-ylidene}acetate (5a–h) (Scheme 2 and Fig. 4). The progress of the reaction was monitored at regular intervals using thin layer chromatography (TLC). The structures of the synthesized compounds (5a–h) were confirmed and characterized through various spectroscopic methods, including IR, MS and NMR spectroscopy, specifically 1H-NMR and 13C-NMR spectra.
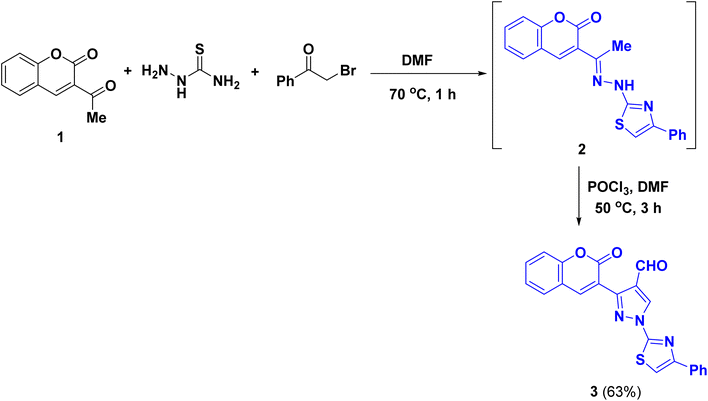 |
| | Scheme 1 The synthetic pathway for the synthesis of the starting material 3. | |
 |
| | Scheme 2 One-pot multicomponent strategy for the synthesis of the target compounds 5a–h. | |
 |
| | Fig. 4 The chemical structures for the synthesized compounds and their yields. | |
The IR spectroscopic analysis of target compounds 5a–h, featured four absorption bands at 1706–1720, 1688–1704, 1716–1754 and 1626–1652 cm−1 corresponded to COcoumarin, COester, COthiazolidinone and CN stretching, respectively. Also, there is band for NH stretching band observed for the product 5a at 3116 cm−1. The 1H-NMR spectra of target compounds 5a–h present a similar pattern for the ylidene acetate unit. The most upfield signals, between δ 1.20–1.29 and 4.19–4.25 ppm, corresponds to the ethoxy moiety (OCH2CH3). The singlets around δ 6.54–6.71 ppm were assigned to the olefinic protons (CH). In addition, the other singlets in range δ 8.02–8.08, 8.45–8.64, 8.36–8.41 and 9.06–9.26 ppm due to the characteristic protons H–5thiazole, H–4coumarin, CHN and H–5pyrazole, respectively.44 In the 13C-NMR spectra, the signals belonging to CHCOOCH2CH3 moiety were observed at δ 13.6–15.1 (CH3), 61.2–62.3 (OCH2), 122.1–122.5 (CH) and 160.2–161.2 (CO) ppm.44 Also, the carbon atoms of the CHN, COthiazolidinone, COcoumarin and C–2thiazolidinone appeared in the regions δ 137.0–138.7, 159.0–159.8, 159.4–160.2 and 166.2–167.6 ppm, respectively. The expected molecular ions (M+) were found in the mass spectra of all target compounds 5a–h.
3. Antiproliferative properties
3.1. Assessment of cytotoxicity effects
To assess the impact of the synthesized 3-substituted-2-{4-oxo-2-(2-((3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazol-4-yl)methylene)hydrazineyl)thiazol-5(4H)-ylidene}acetate (5a–h) on the viability of cancer cells, the SRB assay45 was conducted in two liver cancer cell lines such as Huh-7 and HepG2 and the results were compared with standard drug doxorubicin. Huh-7 and HepG2 are well-established human hepatoma cell lines employed extensively in hepatic research. They possess distinct characteristics that influence their utility in various scientific applications. HepG2 cancer cells originated from a hepatoblastoma, an embryonic liver tumor primarily affecting children, while Huh-7 cancer cells derived from a well-differentiated hepatocellular carcinoma (HCC), the most common form of adult liver cancer. The results were presented in Table 1 and provided valuable insights into the compounds' half-maximal inhibitory concentration (IC50) values for each tested cell line. According to the National Cancer Institute (NCI) guidelines, compounds exhibiting an IC50 value of ≤10 μg mL−1 are regarded as demonstrating highly significant inhibitory activity. This threshold serves as a key benchmark for evaluating the potency of potential anticancer agents in vitro. Among the compounds tested, compounds 5a, 5c, 5d and 5g demonstrated significant inhibitory effects on the proliferation of in Huh-7 cells, indicating their potential in treating liver human hepatoma cancer cell lines compared to doxorubicin. Also, compound 5f exhibited notable antiproliferation activity, while compounds 5b, 5e and 5h displayed low inhibitory effects on this type of cancer cell line. With respect to HepG2 cancer cell lines, both compounds 5d and 5g showed considerable cell cytotoxicity at a concentration of 4.2 ± 0.4 and 2.3 ± 0.3 μg mL−1, respectively, whereas compounds 5b, 5c, 5e, 5f and 5h showed weak activity. Both compounds 5d and 5g may serve as potential candidates for further optimization to treat hepatoma cancer.
Table 1 In vitro anticancer activity of synthesized compounds 5a–h against Huh-7 and HepG2 cells
| Compounds |
IC50a (μg mL−1) |
| Huh-7 |
HepG2 |
| IC50 values are the mean ± SD of three separate experiments. |
| 5a |
0.8 ± 0.1 |
28.9 ± 1.7 |
| 5b |
≥100 |
≥100 |
| 5c |
0.5 ± 0.8 |
≥100 |
| 5d |
0.2 ± 0.01 |
4.2 ± 0.4 |
| 5e |
≥100 |
≥100 |
| 5f |
3.2 ± 0.8 |
≥100 |
| 5g |
0.3 ± 0.03 |
2.3 ± 0.3 |
| 5h |
≥100 |
≥100 |
| Doxorubicin |
1.5 ± 0.6 |
1.3 ± 0.4 |
3.2. Study of the structure–activity relationship (SAR)
The Structure–Activity Relationship (SAR) analysis of the prepared compounds against HepG2 and Huh-7 cancer cell lines revealed key trends. The unsubstituted compound 5a (R = H) exhibited moderate activity against HepG2 but was highly potent against Huh-7, indicating greater sensitivity of Huh-7 to this compound. Introduction of a methyl group in 5b resulted in complete loss of activity against both cell lines, suggesting that small alkyl groups like methyl hinder activity. Similarly, the ethyl group in 5c led to inactivity against HepG2 but retained high potency against Huh-7, indicating that Huh-7 tolerates larger alkyl groups better than HepG2. The allyl group in 5d significantly enhanced activity against both cell lines, particularly against Huh-7, suggesting that the presence of a double bond improves binding or cellular uptake. In contrast, the bulky adamantanyl group in 5e caused complete inactivity against both cell lines, highlighting the detrimental effect of steric hindrance. The phenyl group in 5f resulted in inactivity against HepG2 but retained moderate activity against Huh-7, indicating that aromatic groups are better tolerated by Huh-7. The benzyl group in 5g further enhanced activity against both cell lines, especially Huh-7, suggesting that a phenyl ring with a flexible methylene linker improves potency. However, the 4-chlorophenyl group in 5h led to complete inactivity, indicating that electron-withdrawing groups like chlorine negatively impact activity. In summary, the Huh-7 cell line is generally more sensitive to these compounds than HepG2. Small alkyl groups reduce activity, while unsaturated or aromatic groups with flexible linkers (e.g., allyl, benzyl) enhance potency. Bulky or electron-withdrawing groups (e.g., adamantanyl, 4-chlorophenyl) are detrimental, likely due to steric hindrance or unfavorable electronic effects. These insights can guide further optimization, focusing on substituents that enhance activity against both cell lines while avoiding sterically hindered or electron-withdrawing groups.
3.3. Apoptosis analysis
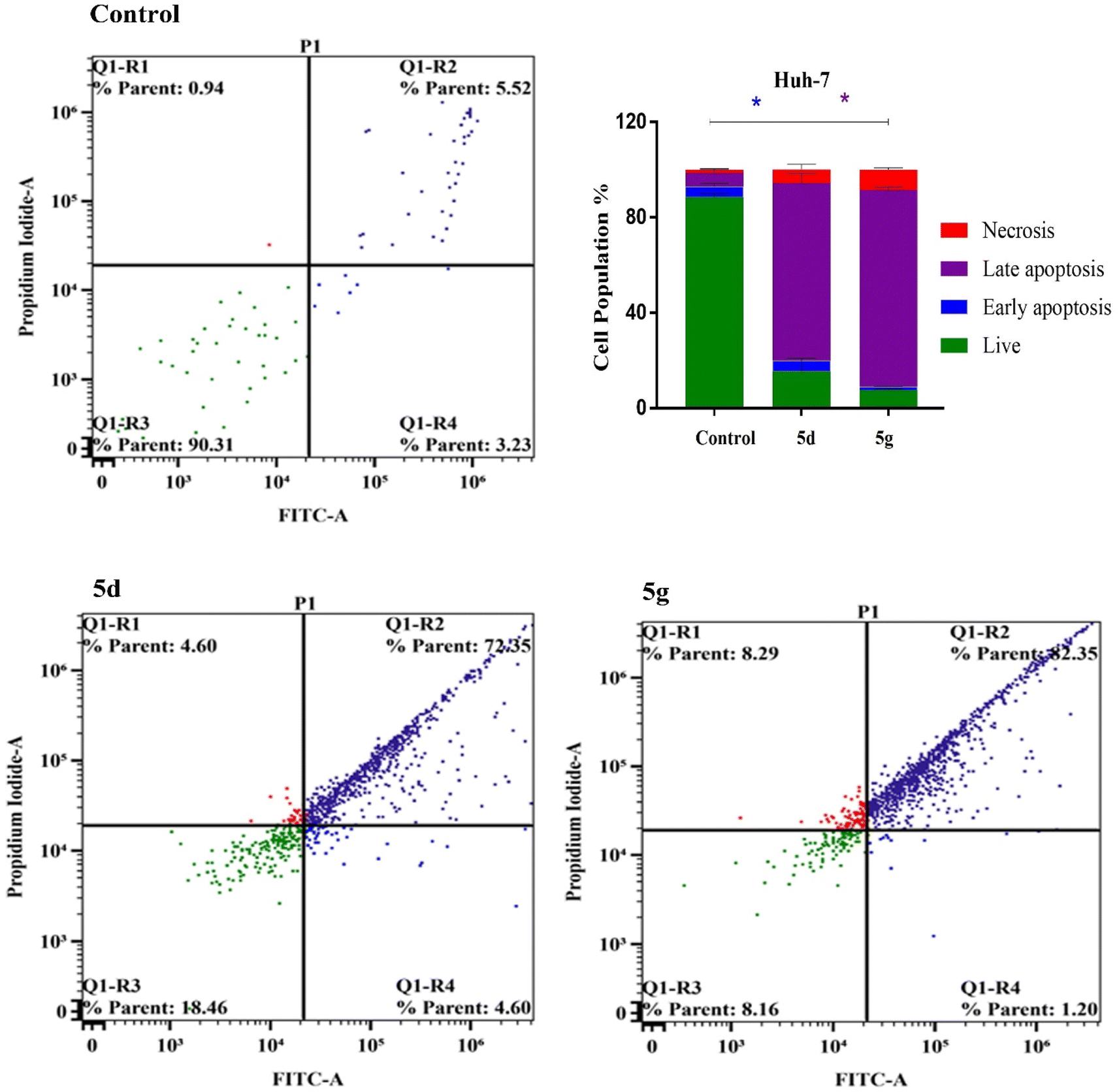
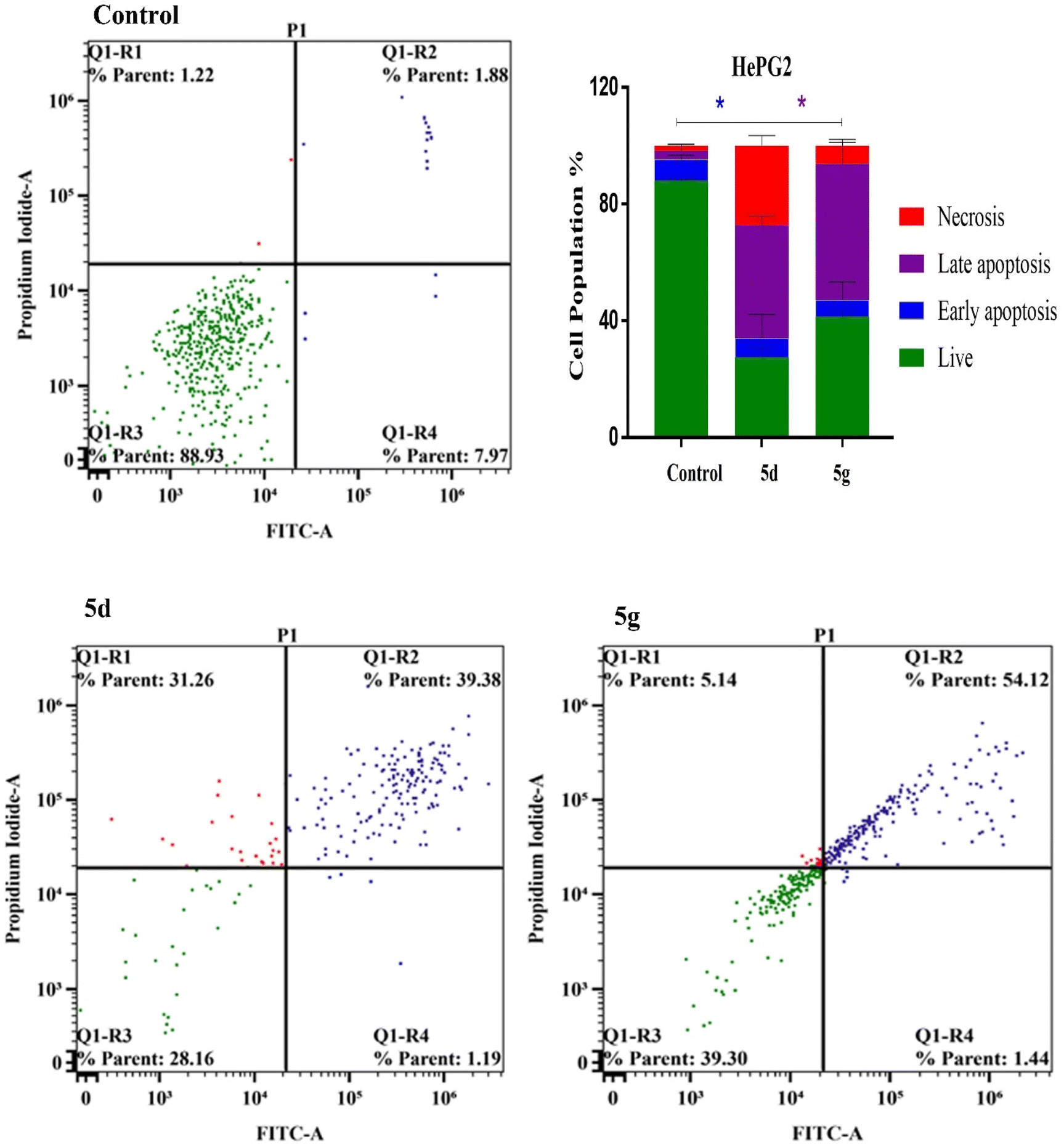
To further characterize the apoptotic activity of the studied compounds 5d and 5g, we examined and quantified the percentage of apoptotic cells that was determined by Alexa Fluor-488/PI staining, and the stained cells were subsequently detected by flow cytometric analysis.46 Our splitting data depicted that the quantification of annexin-V was remarkably increased in the late apoptosis quadrants of Huh-7 cells subjected to 5d and 5g (72.35% and 82.35%, respectively) compared to that of control (5.52%). Also, we observed no significant changes for both 5d and 5g compounds in the early apoptosis. Furthermore, the cellular viability of Huh-7 cells was significantly decreased by treatments with compounds 5d and 5g (18.46% and 8.16%, respectively) compared to that of control (90.31%). Consistently, our results showed increased levels of necrotic cells in Huh-7 cells subjected to 5d and 5g (4.60% and 8.29%, respectively) compared with control (0.94%) (Fig. 5). Concurrently, our investigations of cellular late apoptosis within the HePG2 hepatic cells was remarkably increased using compounds 5d and 5g (39.38% and 54.14%, respectively) versus control (1.88%). Furthermore, compound 5d induced a significant increase in the populations of necrotic cells (31.26%) compared to control (1.22%). Together with increased late apoptotic activity, the early apoptosis did not change for both 5d and 5g compounds. However, they successfully lower the viability rates of HePG2 cells (28.16% and 39.30%, respectively) compared to control (88.93%) (Fig. 6). These results supported that these compounds exert their cytotoxic activities against both Huh-7 and HePG2 hepatic cancer cells and potentially enhance the cellular late apoptosis and reduce the cellular viability.
 |
| | Fig. 5 Apoptosis and necrosis evaluation of compounds 5d and 5g in Huh-7 cancer cells. | |
 |
| | Fig. 6 Apoptosis and necrosis evaluation of compounds 5d and 5g in HepG2 cancer cells. | |
3.4. Cell-cycle distribution
To determine the potentials of activity of the bioactive compounds 5d and 5g, we analyzed their influence on the cell cycle of Huh-7 and HePG2 hepatic cancer cells using the standard flow cytometry technique.47 In case of Huh-7 cancer cells, our records showed that both compounds 5d and 5g arrested the cell cycle at the G1 phase with average percentages of (50.47% and 34.12%) with moderate significant changes compared to that of control (43.36%). Also, compound 5g caused significant increase in the cellular distribution at the S and G2 phases (50.11% and 15.77%) compared to control (43.36% and 13.27%), respectively. However, compound 5d did not cause significant changes compared to that of control at the S and G2 phases (Fig. 7). With respect to the HePG2 cells, our analyses showed that both compounds 5d and 5g significantly arrested the cell cycles at the G1 phase (55.13% and 57.13%, respectively) compared to that of control (46.43%). However, they displayed non-significant increase for cellular distribution at S and G2 phases in comparison with control (Fig. 8). Therefore, both compounds 5d and 5g showed promising high ability to arrest the cell cycle generally at G1 phase in Huh-7 and HepG2 hepatic cancer cells.
 |
| | Fig. 7 Cell cycle distribution of compounds 5d and 5g was analyzed using DNA cytometry in Huh-7 cell lines. | |
 |
| | Fig. 8 Cell cycle distribution of compounds 5d and 5g was analyzed using DNA cytometry in HepG2 cell lines. | |
3.5. Autophagy assessment
We investigated the effect of bioactive compounds 5d and 5g on autophagy, a well-characterized cellular self-degradation pathway, in colon cancer cell lines. Acridine orange staining coupled with flow cytometry served as a quantitative measure for autophagic vacuole (AVO) formation, a hallmark of autophagy induction.48 The net fluorescence intensity (NFI) percentage change in AVO population was determined following treatment with pre-established half-maximal inhibitory concentration (IC50) doses of compounds 5d and 5g, alongside doxorubicin, a positive control for autophagy induction. Compounds 5d and 5g significantly elevated AVO formation in both Huh-7 and HepG2 liver cancer cell lines compared to the control group at the tested concentration. Notably, 42.08% and 45.46% increases in AVO formation were observed in Huh-7 cells treated with 5d and 5g, respectively, compared to the control (26.42%) (Fig. 9). Similarly, HepG2 cell lines displayed a substantial increase in autophagic signal upon treatment with 5d and 5g, with NFI percentage changes of 36.53% and 18.42%, respectively, compared to the control (16.69%) (Fig. 10). These findings collectively suggest that compounds 5d and 5g have the potential to induce autophagic cell death in liver cancer cells.
 |
| | Fig. 9 Autophagic cell death evaluation of compounds 5d and 5g in Huh-7 cancer cells. | |
 |
| | Fig. 10 Autophagic cell death evaluation of compounds 5d and 5g in HepG2 cancer cells. | |
3.6. Molecular docking
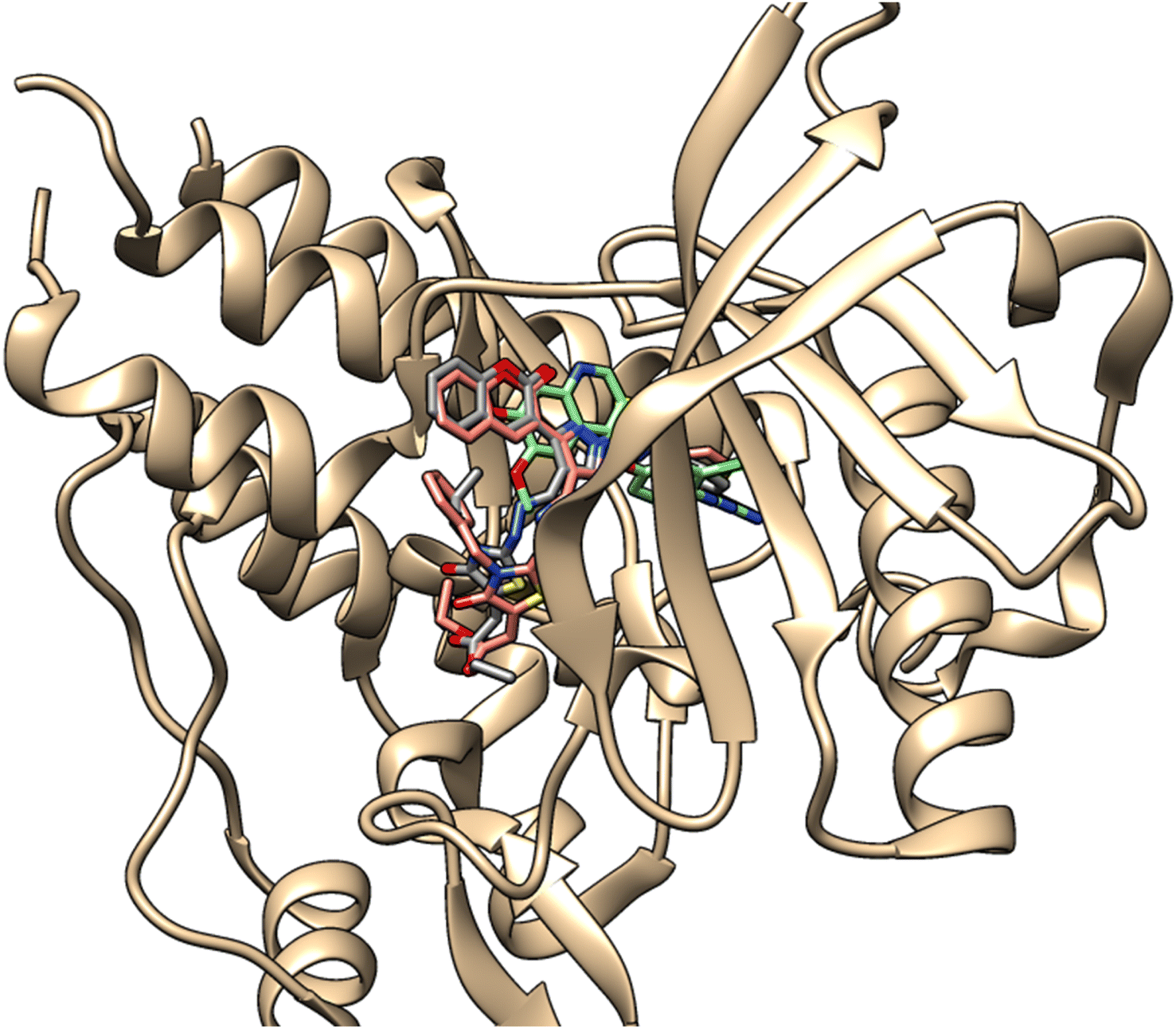
Vascular Endothelial Growth Factor Receptor (VEGFR) plays an important role in the process of angiogenesis. VEGFR-2 is critical in angiogenesis, a mechanism required for tumor growth and metastasis. Its overexpression has been linked to several malignancies, including breast, cervical, non-small cell lung, hepatocellular, and renal carcinoma. As a result, blocking VEGFR-2 has emerged as a possible anticancer therapy strategy.49–56 To identify achievable VEGFR-2 inhibitors, the bioactive compounds 5d and 5g were docked against VEGFR-2 (Juxtamembrane and Kinase Domains) (PDB ID:4ASE). These compounds were compared to tivozanib (AV-951), a recognized VEGFR-2 inhibitor. Table S1 (ESI†) displayed the binding affinity (kcal mol−1) along with the specific interactions of each product. Tivozanib (AV-951), as VEGFR-2 inhibitor, was able to form strong interactions by forming three hydrogen bonds with CYS 919, GLU 885, and ASP 1046 with distances 1.96, 3.23 and 2.06 Å, respectively. Further, it formed a carbon H-bond interaction with CYS 1045 with distance 2.54 Å, with a binding energy of −11.4 kcal mol−1, as shown in Fig. 11. Pi interactions also observed with the key amino acids through planar aromatic rings, including π-alkyl (LEU 1035, ALA 866), π-sigma (LEU 840), and π–π T-shaped (PHE 1047). The products compounds 5d and 5g showed similar docking interactions, which pointed to tivozanib (AV-951) having a similar binding pocket.57–62 The product 5d demonstrated a binding mode with an affinity score of −8.4 kcal mol−1 against VEGFR-2 enzyme. It established three hydrogen bonds with ARG 1051 (COester), ASN 923 (COthiazolidinone) and CYS 919 (COcoumarin), with distances measuring 2.76, 2.82 and 2.48 Å, respectively, as well as carbon hydrogen bond with the key amino acid residue GLY 841 through CN bond with distance measuring 2.37 Å (Fig. 12). In addition, it established several hydrophobic interactions, specifically π-cation (LYS 868), π-sulfur (CYS 1045), π-sigma (LEU 840), π–π stacked (PHE 1047) and π-alkyl (LEU 1035, ALA 866, VAL 848, 899, 916) interactions. In the same manner, product 5g had a binding energy of −7.5 kcal mol−1 towards VEGFR-2 enzyme. The compound had three hydrophobic π–π stacked (PHE 918) and π-alkyl (VAL 848, VAL 916, VAL 899, ALA 866 and CYS 1045) interactions. Additionally, it formed two hydrogen bonds and one carbon hydrogen bond with ARG 1051 (Oester), CYS 919 (COcoumarin), and LEU 840 (pyrazole), with distances of 3.71, 2.50 and 4.94 Å, respectively. Moreover, it formed π-doner hydrogen bond with ASN 923 through benzyl ring with distance 3.08 Å (Fig. 13). The molecular docking study supported that both compounds 5d and 5g may be promising antiproliferative agents for further development as VEGFR-2 inhibitors. Their interactions with key amino acid residues of the receptors agreed with the cytotoxicity values, confirming their potential as anticancer agents (Fig. 14 and 15).
 |
| | Fig. 11 The 2D and 3D interactions of tivozanib (AV-951) with VEGFR-2 receptor (PDB ID:4ASE). | |
 |
| | Fig. 12 The 2D interactions of compound 5d with VEGFR-2 receptor (PDB ID:4ASE). | |
 |
| | Fig. 13 The 2D interactions of compound 5g with VEGFR-2 receptor (PDB ID:4ASE). | |
 |
| | Fig. 14 Superimposition of compounds 5d and 5g inside the binding pocket of VEGFR-2 receptor (PDB ID:4ASE). | |
 |
| | Fig. 15 Superimposition of compounds 5d and 5g with AV-951 inside the binding pocket of VEGFR-2 receptor (PDB ID:4ASE). | |
3.7. In silico ADMET prediction study
Analyzing the ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of targeted drugs can offer valuable insights into the most appropriate therapeutic options. SwissADME, a freely accessible online tool, facilitated the efficient completion of this study.63–66 The selection of the most suitable drug for oral administration was guided by Lipinski's rule (molecular weight ≤500, hydrogen bond acceptors ≤10, hydrogen bond donors ≤5, and MlogP ≤ 4.15) and Veber's rule (topological polar surface area <140 Å2, rotatable bonds ≤10). Based on these criteria, the bioactive compounds 5d and 5g were found to comply with most rules, exhibiting only two violations (Table S2, ESI†). The synthesized compounds 5d and 5g were observed to fall within the optimal range (pink area) for six key parameters-lipophilicity, size, polarity, solubility, saturation, and flexibility as indicated by the bioavailability radar chart (Fig. 16). This suggested a favorable prediction for their oral bioavailability, with the exception of the saturation parameter.
 |
| | Fig. 16 Bioavailability radar charts for the synthesized compounds 5d, 5g and doxorubicin. | |
The pharmacokinetic characteristics analysis for the synthesized compounds 5d and 5g are provided in Table S3 (ESI†) and Fig. 17. All products were away from both the white and yellow zones of the boiled-egg chart (Fig. 17). Therefore, these products can lead to a weak anticipated absorption in the gut and used to treat peripheral infections and are expected to have a negative impact on the central nervous system. In addition, all products had a high bioavailability rating of 0.17, which is almost not expected to trigger a PAIN warning. The drug efflux transporter P-glycoprotein (P-gp), which is known to move drugs out of cells, may be a factor in drug tolerance. According to the SwissADME website's prognosis (red dots in Fig. 17), these products under study are P-gp non-substrates, indicating that there is a small possibility of them effluxing out of the cell at maximal activity. The pharmacokinetic properties of the synthesized compounds 5d and 5g are detailed in Table S3† and Fig. 17. All compounds were located outside the white and yellow zones of the boiled-egg chart (Fig. 17), suggesting poor anticipated absorption in the gastrointestinal tract. As a result, these compounds are more suitable for treating peripheral infections and are likely to have a limited effect on the central nervous system. Additionally, all compounds exhibited a high bioavailability score of 0.17, which is unlikely to trigger a PAIN (pan-assay interference compounds) alert. The drug efflux transporter P-glycoprotein (P-gp), known for expelling drugs from cells, may contribute to drug resistance. However, according to the SwissADME prediction (represented by red dots in Fig. 17), the studied compounds are classified as P-gp non-substrates, indicating a low probability of being effluxed out of cells at peak activity levels.
 |
| | Fig. 17 A boiled-egg of the synthesized 5d, 5g and doxorubicin. | |
The synthesized compounds 5d and 5g did not display toxicity characteristics associated with the inhibition of the human ether-a-go-go-related gene (hERG) (Table S4, ESI†), indicating no risk of cardiotoxicity or adverse cardiac effects. This is a critical factor in the clinical evaluation of drug candidates. Additionally, none of the tested compounds showed Ames toxicity, a significant finding in early drug discovery as it helps assess the potential genotoxicity of the target molecules. The acute oral toxicity calculations for all synthesized compounds yielded values ranging from 381 to 487 mg kg−1, placing them in the third toxicity category (500 mg kg−1 < LD50 ≤ 5000 mg kg−1), which classifies them as relatively harmless. Furthermore, the carcinogenicity descriptor (CARC) values for these compounds ranged between 768 and 839 mg kg−1 of body weight per day, suggesting they are likely non-carcinogenic and do not require further carcinogenicity testing.
4. Conclusion
In conclusions, a new set of ethyl 3-substituted-2-{4-oxo-2-(2-((3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazol-4-yl)methylene)hydrazineyl)thiazol-5(4H)-ylidene}acetate (5a–h) was synthesized and assessed for their in vitro antiproliferative activity against Huh-7 and HepG-2 liver carcinoma cell line. Most tested hybrids revealed significant antiproliferative activity. Among them, two products 5d and 5g presented potent antiproliferative activity against the examined cell lines. The cell cycle analysis showed evidence that 5d and 5g halted the Huh-7 and HepG2 at the G1 phase. Besides, they induced a significant increase in the populations of the cellular late apoptosis and necrotic cells with reducing the cellular viability. Additionally, compounds 5d and 5g demonstrated promising autophagic induction potential compared control. Finally, both products demonstrated favorable binding affinities with the active sites of VEGFR-2 enzyme, as shown by molecular docking. ADMET profiling studies indicated that these bioactive compounds are likely to be safe and hold significant promise as anticancer agents. Based on the data, compounds 5d and 5g are promising inhibitor candidates for advancing innovative liver cancer treatments.
5. Experimental
5.1. The synthesis of 3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazole-4-carboxaldehyde (3)
A mixture of 3-acetylcoumarin (1) (0.94 g, 5 mmol), thiosemicarbazide (0.46 g, 5 mmol) and phenacyl bromide (5 mmol) in absolute ethanol (20 ml) containing a few drops of glacial acetic acid, was heated under reflux for 4 h. The formed hydrazone 2 was filtered off and dried. A solution of DMF (15 ml) and POCl3 (1.5 ml, 16 mmol) was allowed to stir at 0–5 °C for 15 minutes. After that the hydrazones 2 (4 mmol) was added at room temperature. The reaction mixture was warmed under stirring at 50 °C for 6 hours. The reaction mixture was poured into beakers containing crushed ice or ice-cold water and neutralized with sodium acetate. The formed solid was filtered off and washed several times with water. The crude product was dried and crystallized from acetic acid to afford the target aldehyde 3 as beige solid in 77% yield, mp 226–228 °C.43 IR (KBr), (vmax, cm−1): 3093 (C–Harom), 2987, 2899 (C–Haldehyde), 1716 (COcoumarin), 1694 (COaldehyde), 1605, 1538 (CC), 1572 (CN). 1H-NMR (400 MHz, DMSO-d6): δ 7.38–7.52 (m, 5H, Ph–H, H–6coumarin and H–8coumarin), 7.71 (t, 1H, J = 8.0 Hz, H–7coumarin), 7.90 (d, 1H, J = 7.2 Hz, H–5coumarin), 8.02 (d, 2H, J = 8.0 Hz, Ph–H), 8.10 (s, 1H, H–5thiazole), 8.44 (s, 1H, H–4coumarin), 9.37 (s, 1H, H–5pyrazole), 9.98 (s, 1H, CHO). 13C-NMR (100 MHz, DMSO-d6): δ 113.5 (C–5thiazole), 119.2 (C–8coumarin), 119.7 (C–3coumarin), 120.8 (C–4acoumarin), 124.4 (C–4pyrazole), 125.5 (C–6coumarin), 126.5 (C–4phenyl), 127.7 (C–2,6phenyl), 128.6 (C–5coumarin), 129.1 (C–1phenyl), 129.5 (C–3,5phenyl), 133.9 (C–7coumarin), 146.9 (C–4coumarin), 149.1 (C–5pyrazole), 150.0 (C–4thiazole), 152.3 (C–3pyrazole), 154.2 (C–8acoumarin), 159.5 (COcoumarin), 166.9 (C–2thiazole), 189.9 (COaldehyde). MS (m/z, I %): 399 (M+, 17%). Anal. calcd for C22H13N3O3S (399.42): C, 66.16%; H, 3.28%; N, 10.52%; S, 8.03%. Found: C, 66.03%; H, 3.16%; N, 10.35%; S, 7.89%.
5.1.1. General procedure for the synthesis of products 5a–h. A mixture of 3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazole-4-carboxaldehyde (3) (0.39 g, 1.0 mmol), thiosemicarbazide derivative 4a–h (1 mmol) and diethyl acetylene dicarboxylate (1.1 mmol) in acetic acid (20 ml) was heated under reflux for 5 h. The reaction mixtures were cooled to room temperature. The formed solids were filtered off and crystallized from ethanol to afford the target compounds.
5.1.1.1. Ethyl 2-{4-oxo-2-(2-((3-(2-oxo-2H-chromen-3-yl)-1-(4-phenylthiazol-2-yl)-1H-pyrazol-4-yl)methylene)hydrazineyl)thiazol-5(4H)-ylidene}acetate (5a). Yellow solid in 67% yield, mp > 300 °C. IR (KBr), (vmax, cm−1): 3116 (NH), 3002 (C–Harom), 2946, 2938 (C–Haliph), 1726 (COthiazolidinone), 1716 (COcoumarin), 1691 (COester), 1644 (CHNexocyclic), 1609, 1578, 1544 (CN, CC). 1H-NMR (400 MHz, DMSO-d6): δ 1.25 (t, 3H, J = 6.8 Hz, CH3), 4.19 (q, 2H, J = 6.8 Hz, OCH2), 6.54 (s, 1H, CHexocyclic), 7.40 (t, 2H, J = 6.8 Hz, Ph–H), 7.47–7.52 (m, 3H, Ph–H, H–6coumarin and H–8coumarin), 7.67 (t, 1H, J = 7.2 Hz, H–7coumarin), 7.86 (d, 1H, J = 7.2 Hz, H–5coumarin), 8.00–8.02 (m, 3H, Ph–H and H–5thiazole), 8.36 (s, 1H, CHNexocyclic), 8.54 (s, 1H, H–4coumarin), 9.09 (s, 1H, H–5pyrazole), 12.69 (brs, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ 14.5 (CH3), 61.6 (CH2), 112.7 (C–5thiazole), 117.2 (C–8coumarin), 119.0 (C–3coumarin), 119.8 (C–4acoumarin), 120.7 (C–4pyrazole), 122.5 (CHexocyclic), 125.1 (C–4phenyl), 126.4 (C–2,6phenyl), 128.9 (C–6coumarin), 129.3 (C–3,5phenyl), 129.9 (C–5coumarin), 130.4 (C–1phenyl), 133.8 (C–7coumarin), 137.4 (CHNexocyclic), 143.2 (C–5thiazolidinone), 143.6 (C–4coumarin), 148.6 (C–5pyrazole), 151.3 (C–4thiazole), 152.2 (C–3pyrazole), 154.2 (C–8acoumarin), 159.2 (COthiazolidinone), 159.6 (COcoumarin), 160.2 (COester), 165.5 (C–2thiazole), 166.2 (C–2thiazolidinone). MS (m/z, I%): 596 (M+, 25%). Anal. calcd for C29H20N6O5S2 (596.64): C, 58.38%, H, 3.38%, N, 14.09%, S, 10.75%. Found: C, 58.30%, H, 3.24%, N, 14.01%, S, 10.66%.
Data availability
All data are available in the manuscript and ESI.†
Author contributions
Data curation: TEA and AAS. Formal analysis: AAS and AKA. Investigation: TEA, WAB, AKA, MYA and SEIE. Writing – original draft: TEA, MAA and WAB. Conceptualization: TEA, MAA and WAB. Supervision: TEA. Resources: TEA, WAB and SEIE. Software: TEA and SEIE. Methodology: WAB, AKA and SEIE. Writing – review & editing: TEA, MAA, WAB and SEIE. All authors read and approved the final version of the manuscript.
Conflicts of interest
Authors declare that they have no conflict of interest.
Acknowledgements
The authors extend their appreciation to University Higher Education Fund for funding this research work under Research Support Program for Central labs at King Khalid University through the Project number CL/PRI/B/6.
References
- A. Faritha, A. J. A. Nasser and S. S. Sathish, Synthesis of Aryl Substituted Hydrazinothiazolyl Pyrazole Derivatives and Evaluation of Their Biological Activity, J. Chem. Pharm. Res., 2014, 6, 808–813 Search PubMed.
- S. Bondock, O. Albormani, A. M. Fouda and K. A. Abu Safieh, Progress in the Chemistry of 5-Acetylthiazoles, Synth. Commun., 2016, 46, 1081–1117 Search PubMed.
- M. Haroun, C. Tratrat, K. Kositzi, E. Tsolaki, A. Petrou, B. Aldhubiab, M. Attimarad, S. Harsha, A. Geronikaki and K. N. Venugopala, et al., New benzothiazole-based thiazolidinones as potent antimicrobial agents. Design, synthesis and biological evaluation, Curr. Top. Med. Chem., 2018, 18, 75–87 Search PubMed.
- B. Sadek, M. M. Al-Tabakha and K. M. S. Fahelelbom, Antimicrobial prospect of newly synthesized 1,3-thiazole derivatives, Molecules, 2011, 16, 9386–9396 Search PubMed.
- D. Mech, A. Kurowska and N. Trotsko, The Bioactivity of Thiazolidin-4-Ones: A Short Review of the Most Recent Studies, Int. J. Mol. Sci., 2021, 22, 11533 Search PubMed.
- M. W. Aziz, A. M. Kamal, K. O. Mohamed and A. A. Elgendy, Design, synthesis, and assessment of new series of quinazolinone derivatives as EGFR inhibitors along with their cytotoxic evaluation against MCF7 and A549 cancer cell lines, Bioorg. Med. Chem. Lett., 2021, 41, 127987 Search PubMed.
- V. S. Berseneva, A. V. Tkachev, Y. Morzherin, W. Dehaen, I. Luyten, S. Toppet and V. Bakulev, J. Chem. Soc. Perkin Trans. 1, 1998,(15), 2133 RSC.
- A. A. Aly, M. A. A. Ibrahim, E. M. Shehata, A. A. M. Hassan and A. B. Brown, Prospective new amidinothiazoles as leukotriene B4 inhibitors, J. Mol. Struct., 2019, 1175, 414–427 CrossRef CAS.
- S. P. Singh, S. S. Parmar, K. Raman and V. I. Stenberg, Chemistry and biological activity of thiazolidinones, Chem. Rev., 1981, 81, 175–203 CrossRef CAS.
- N. Trotsko, A. Bekier, A. Paneth, M. Wujec and K. Dzitko, Synthesis and in vitro anti-Toxoplasma gondii activity of novel thiazolidin-4-one derivatives, Molecules, 2019, 24, 3029 CrossRef PubMed.
- N. Trotsko, A. Przekora, J. Zalewska, G. Ginalska, A. Paneth and M. Wujec, Synthesis and in vitro antiproliferative and antibacterial activity of new thiazolidine-2,4-dione derivatives, J. Enzyme Inhib. Med. Chem., 2018, 33, 17–24 CrossRef CAS PubMed.
- M. B. Alshammari, A. A. Aly, B. G. M. Youssif, S. Bräse, A. Ahmad, A. B. Brown, M. A. A. Ibrahim and A. H. Mohamed, Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E, Front. Chem., 2022, 10, 1076383 CrossRef CAS PubMed.
- A. A. Hassan, Y. R. Ibrahim, E. M. El-Sheref, M. Abdel-Aziz, S. Brase and M. Nieger, Synthesis and Antibacterial Activity of 4-Aryl-2-(1-Substitutedethylidene) Thiazoles, Arch. Pharm. Chem. Life Sci., 2013, 346, 562–570 CrossRef CAS PubMed.
- M. M. Alsharekh, I. I. Althagafi, M. R. Shaaban and T. A. Farghaly, Microwave-Assisted and Thermal Synthesis of Nanosized Thiazolyl-Phenothiazine Derivatives and Their Biological Activities, Res. Chem. Intermed., 2019, 45, 127–154 Search PubMed.
- S. Singh Jadav, S. Kaptein, A. Timiri, T. De Burghgraeve, V. N. Badavath, R. Ganesan, B. N. Sinha, J. Neyts, P. Leyssen and V. Jayaprakash, Design, Synthesis, Optimization and Antiviral Activity of a Class of Hybrid Dengue Virus E Protein Inhibitors, Bioorg. Med. Chem. Lett., 2015, 25, 1747–1752 Search PubMed.
- P. Makam, P. K. Thakur and T. Kannan, In Vitro and in Silico Antimalarial Activity of 2-(2-Hydrazinyl) Thiazole Derivatives, Eur. J. Pharm. Sci., 2014, 52, 138–145 Search PubMed.
- A. Ignat, T. Lovasz, M. Vasilescu, E. Fischer-Fodor, C. B. Tatomir, C. Cristea, L. Silaghi-Dumitrescu and V. Zaharia, Heterocycles 27. Microwave Assisted Synthesis and Antitumor Activity of Novel Phenothiazinyl-Thiazolyl-Hydrazine Derivatives, Arch. Pharm. Pharm. Med. Chem., 2012, 345, 574–583 CrossRef CAS PubMed.
- A. S. Filimonov, A. A. Chepanova, O. A. Luzina, A. L. Zakharenko, O. D. Zakharova, E. S. Ilina, N. S. Dyrkheeva, M. S. Kuprushkin, A. V. Kolotaev, D. S. Khachatryan, J. Patel, I. K. H. Leung, R. Chand, D. M. Ayine-Tora, J. Reynisson, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, New Hydrazinothiazole Derivatives of Usnic Acid as Potent Tdp1 Inhibitors, Molecules, 2019, 24, 3711 Search PubMed.
- A. L. Zakharenko, O. A. Luzina, D. N. Sokolov, V. I. Kaledin, V. P. Nikolin, N. A. Popova, J. Patel, O. D. Zakharova, A. A. Chepanova and A. Zafar, et al., Novel tyrosyl-DNA Phosphodiesterase 1 Inhibitors Enhance the Therapeutic Impact of Topotecan on in Vivo Tumor Models, Eur. J. Med. Chem., 2019, 161, 581–593 CrossRef CAS PubMed.
- M. Basanagouda, V. B. Jambagi, N. N. Barigidad, S. S. Laxmeshwar and V. Devaru, Synthesis, structure-activity relationship of iodinated4-aryloxymethyl-coumarins as potential anti-cancer and anti-mycobacterial agents, Eur. J. Med. Chem., 2014, 74, 225–233 CrossRef CAS PubMed.
- A. Manvar, A. Bavishi, A. Radadiya, J. Patel, V. Vora, N. Dodia, K. Rawal and A. Shah, Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37Rv strains, Bioorg. Med. Chem. Lett., 2011, 21, 4728–4731 Search PubMed.
- M. Alipour, M. Khoobi, A. Moradi, H. Nadri, F. H. Moghadam, S. Emami, Z. Hasanpour, A. Foroumadi and A. Shafiee, Synthesis and anticholinesterase activity of new 7-hydroxy-coumarin derivatives, Eur. J. Med. Chem., 2014, 82, 536–544 CrossRef CAS PubMed.
- Y. Shikishima, Y. Takaishi, G. Honda, M. Ito, Y. Takeda, O. K. Kodzhimatov, O. Ashurmetov and K. H. Lee, Chemical Constituents of Prangos tschimganica; Structure Elucidation and Absolute Configuration of Coumarin and Furanocoumarin Derivatives with Anti-HIV Activity, Chem. Pharm. Bull., 2001, 49, 877–880 CrossRef CAS PubMed.
- M. Manjunatha, V. H. Naik, A. D. Kulkarni and S. A. Patil, DNA cleavage, antimicrobial, anti-inflammatory anthelmintic activities, and spectroscopic studies of Co(II), Ni(II), and Cu(II) complexes of biologically potential coumarin Schiff bases, J. Coord. Chem., 2011, 64, 4264–4275 CrossRef CAS.
- K. V. Sashidhara, A. Kumar, M. Chatterjee, K. B. Rao, S. Singh, A. K. Verma and G. Palit, Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents, Bioorg. Med. Chem. Lett., 2011, 21, 1937–1941 CrossRef CAS PubMed.
- M. Khoobi, A. Foroumadi, S. Emami, M. Safavi, G. Dehghan, B. H. Alizadeh, A. Ramazani, S. K. Ardestani and A. Shafiee, Coumarin-Based Bioactive Compounds: Facile Synthesis and Biological Evaluation of Coumarin-Fused 1,4-Thiazines, Chem. Biol. Drug Des., 2011, 78, 580–586 CrossRef CAS PubMed.
- S. Mamidala, S. R. Peddi, R. K. Aravilli, P. C. Jilloju, V. Manga and R. R. Vedula, Microwave irradiated one pot, three component synthesis of a new series of hybrid coumarin based thiazoles: antibacterial evaluation and molecular docking studies, J. Mol. Struct., 2021, 1225, 129114 CrossRef CAS.
- M. M. Fakhry, A. A. Mattar, M. Alsulaimany, E. M. Al-Olayan, S. T. Al-Rashood and H. A. Abdel-Aziz, New Thiazolyl-Pyrazoline Derivatives as Potential
Dual EGFR/HER2 Inhibitors: Design, Synthesis, Anticancer Activity Evaluation and In Silico Study, Molecules, 2023, 28, 7455 CrossRef CAS PubMed.
- I. Koca, M. Gumus, A. Ozgur, A. Disli and Y. Tutar, A novel approach to inhibit heat shock response as anticancer strategy by coumarin compounds containing thiazole skeleton, Adv. Anticancer Agents Med. Chem., 2015, 15, 916–930 CrossRef CAS PubMed.
- X. H. Liu, H. F. Liu, J. Chen, Y. Yang, B. A. Song, L. S. Bai, J. X. Liu, H. L. Zhu and X. B. Qi, Synthesis and molecular docking study of novel coumarin derivatives containing 4,5-dihydropyrazole moiety as potential antitumor agents, Bioorg. Med. Chem. Lett., 2010, 20, 5705–5708 CrossRef CAS PubMed.
- M. A. Chowdhury, K. R. A. Abdellatif, Y. Dong, D. Das, M. R. Suresh and E. E. Knaus, Synthesis of Celecoxib Analogues Possessing a N-Difluoromethyl-1,2-dihydropyrid-2-one 5-Lipoxygenase Pharmacophore: Biological Evaluation as Dual Inhibitors of Cyclooxygenases and 5-Lipoxygenase with Anti-inflammatory Activity, J. Med. Chem., 2009, 52, 1525–1529 CrossRef CAS PubMed.
- A. E. Rashad, M. I. Hegab, R. E. Abdel-Megeid, N. Fathalla and F. M. E. Abdel-Megeid, Synthesis and anti-HSV-1 evaluation of some pyrazoles and fused pyrazolopyrimidines, Eur. J. Med. Chem., 2009, 44, 3285–3292 CrossRef CAS PubMed.
- M. Popsavin, S. Spaic, M. Svircev, V. Kojic, G. Bogdanovic and V. Popsavin, Synthesis and antitumour activity of new tiazofurin analogues bearing a 2,3-anhydro functionality in the furanose ring, Bioorg. Med. Chem. Lett., 2007, 17, 4123–4127 CrossRef CAS PubMed.
- S. Ningaiah, U. K. Bhadraiah, S. D. Doddaramappa, S. Keshavamurthy and C. Javarasetty, Novel pyrazole integrated 1,3,4-oxadiazoles: synthesis, characterization and antimicrobial evaluation, Bioorg. Med. Chem. Lett., 2014, 24, 245–248 CrossRef CAS PubMed.
- Z. Xu, C. Gao, Q. C. Ren, X. F. Song, L. S. Feng and Z. S. Lv, Recent advances of pyrazole-containing derivatives as anti-tubercular agents, Eur. J. Med. Chem., 2017, 139, 429–440 CrossRef CAS PubMed.
- J. Y. Zhang, Apoptosis-based anticancer drugs, Nat. Rev. Drug. Discov., 2002, 1, 101–102 CrossRef CAS PubMed.
- J. K. Buolamwini, Novel anticancer drug discovery, Curr. Opin. Chem. Biol., 1999, 3, 500–509 CrossRef CAS PubMed.
- M. A. Assiri, T. E. Ali, M. N. Alqahtani, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Synthesis, Cytotoxic Evaluation, Apoptosis, Cell cycle and Molecular docking Studies of Some New 5-(Arylidene/heteroarylidene)-2-(Morpholinoimino)-3-Phenylthiazolidin-4-ones, Synth. Commun., 2023, 23, 1240–1261 CrossRef.
- M. A. Assiri, T. E. Ali, M. N. Alqahtani, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Recyclization of Morpholinochromonylidene-Thiazolidinone Using Nucleophiles: Facile Synthesis, Cytotoxic Evaluation, Apoptosis, Cell cycle and Molecular docking Studies of a Novel Series of Azole, Azine, Azepine and Pyran Derivatives, RSC Adv., 2023, 13, 18658–18675 RSC.
- M. Rana, H. Hungyo, P. Parashar, S. Ahmad, R. Mehandi, V. Tandon, K. Raza, M. A. Assiri, T. E. Ali, Z. M. El-Bahy and R. Uddin, Design, synthesis, X-ray crystal structures, anticancer, DNA binding and modeling studies of pyrazole–pyrazoline hybrid derivatives, RSC Adv., 2023, 13, 26766–26779 RSC.
- M. A. Assiri, T. E. Ali, M. N. Alqahtani, I. A. Shaaban, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, New Functionalized Morpholinothiazole Derivatives: Regioselective Synthesis, Computational Studies, Anticancer Activity Evaluation, and Molecular Docking Studies, Curr. Org. Chem., 2023, 27, 1985–1998 CrossRef CAS.
- N. M. Hassanin, T. E. Ali and S. M. Abdel-Kariem, Synthesis, Cytotoxic Evaluation, Molecular Docking Studies and Drug-likeness Analysis of Some Novel 2-[(9-Ethyl-9H-carbazol-3-yl)imino]thiazoles/thiazolidinones, J. Sulfur Chem., 2024, 45, 272–292 CrossRef CAS.
- S. Mamidala, R. K. Aravilli, G. Ramesh, S. Khajavali, R. Chedupaka, V. Manga and R. R. Vedula, A facile one-pot, three-component synthesis of a new series of thiazolyl pyrazole carbaldehydes: in vitro anticancer evaluation, in silico ADME/T, and molecular docking studies, J. Mol. Struct., 2021, 1236, 130356 CrossRef CAS.
- A. A. Aly, M. A. A. Ibrahim, E. M. El-Sheref, A. M. A. Hassan and A. B. Brown, Prospective new amidinothiazoles as leukotriene B4 inhibitors, J. Mol. Struct., 2019, 1175, 414–427 CrossRef CAS.
- A. M. Mahmoud, A. M. Al-Abd, D. A. Lightfoot and H. A. El-Shemy, Anticancer characteristics of mevinolin against three different solid tumor cell lines was not solely p53-dependent, J. Enzyme Inhib. Med. Chem., 2012, 27, 673–679 CrossRef CAS PubMed.
- H. A. Bashmail, A. A. Alamoudi, A. Noorwali, G. A. Hegazy, G. Ajabnoor, H. Choudhry and A. M. Al-Abd, Thymoquinone synergizes gemcitabine anti-breast cancer activity via modulating its apoptotic and autophagic activities, Sci. Rep., 2018, 8, 11674–11685 CrossRef PubMed.
- R. Nunez, DNA measurement and cell cycle analysis by flow cytometry, Curr. Issues Mol. Biol., 2001, 3, 67–70 CAS.
- P. Czarny, E. Pawlowska, J. Bialkowska-Warzecha, K. Kaarniranta and J. Blasiak, Autophagy in DNA Damage Response, Int. J. Mol. Sci., 2015, 16, 2641–2662 CrossRef CAS PubMed.
- D. Shweiki, A. Itin, D. Soffer and E. Keshet, Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis, Nature, 1992, 359, 843–845 CrossRef CAS PubMed.
- G. McMahon, VEGF receptor signaling in tumor angiogenesis, Oncologist, 2000, 5, 3–10 CrossRef CAS PubMed.
- J. A. Forsythe, B. H. Jiang, N. V. Iyer, F. Agani, S. W. Leung, R. D. Koos and G. L. Semenza, Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1, Mol. Cell. Biol., 1996, 16, 4604–4613 CrossRef CAS PubMed.
- E. Ikeda, M. G. Achen, G. Breier and W. Risau, Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells, J. Biol. Chem., 1995, 270, 19761–19766 CrossRef CAS PubMed.
- S. E. Abdullah and R. Perez-Soler, Mechanisms of resistance to vascular endothelial growth factor blockade, Cancer, 2012, 118, 3455–3467 CrossRef CAS PubMed.
- C. Yang, Y. Guo, C. C. Jadlowiec, X. Li, W. Lv, L. S. Model, M. J. Collins, Y. Kondo, A. Muto, C. Shu and A. Dardik, Vascular endothelial growth factor-A inhibits EphB4 and stimulates delta-like ligand 4 expression in adult endothelial cells, J. Surg. Res., 2013, 183, 478–486 CrossRef CAS PubMed.
- S. J. Modi and V. M. Kulkarni, Vascular Endothelial Growth Factor Receptor (VEGFR-2)-KDR Inhibitors: Medicinal Chemistry Perspective, Med. Drug Discov., 2019, 2, 100009 CrossRef.
- V. J. Cee, A. C. Cheng, K. Romero, S. Bellon, C. Mohr, D. A. Whittington, A. Bak, S. Caenepeel, A. Coxon, H. L. Deak, J. Fretland, Y. Gu, B. L. Hodous, X. Huang, J. L. Kim, J. Lin, A. M. Long, H. Nguyen, P. R. Olivieri, V. F. Patel, L. Wang, Y. Zhou, P. Hughes and S. Geuns-Meyer, Pyridyl-pyrimidine benzimidazole derivatives as potent, selective, and orally bioavailable inhibitors of Tie-2 kinase, Bioorg. Med. Chem. Lett., 2009, 19, 424–427 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, A semiempirical free energy force field with charge-based desolvation, J. Comput. Chem., 2007, 28, 1145–1152 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera-A visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- Discovery Studio, available online: https://discover.3ds.com/discovery-studio-visualizer-download Search PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- I. M. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. Gad- Elkareem, S. S. Abd El-Karim and E. S. Nossier, Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitors, RSC Adv., 2022, 12, 561–577 RSC.
- A. Guerraoui, M. Goudjil, A. Direm, A. Guerraoui, I. Y. Şengün, C. Parlak, A. Djedouani, L. Chelazzi, F. Monti, E. Lunedei and A. A. Boumaza, Rhodanine derivative as a potential antibacterial and anticancer agent: crystal structure, spectral characterization, DFT calculations, Hirshfeld surface analysis, in silico molecular docking and ADMET studies, J. Mol. Struct., 2023, 1280, 135025 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc,
Ayat K. Alsolimanic,
Mohammed A. Assiric,
Ali A. Shatid,
Mohammad Y. Alfaifid and
Serag E. I. Elbehairid
*bc,
Ayat K. Alsolimanic,
Mohammed A. Assiric,
Ali A. Shatid,
Mohammad Y. Alfaifid and
Serag E. I. Elbehairid