DOI:
10.1039/D5RA00216H
(Paper)
RSC Adv., 2025,
15, 14126-14135
Thermo-responsive methylcellulose/hyaluronic acid–mesalamine hydrogel in targeted drug delivery for ulcerative colitis
Received
13th January 2025
, Accepted 17th April 2025
First published on 1st May 2025
Abstract
Current treatments for ulcerative colitis (UC), including mesalamine (Me) enemas, face limitations such as poor colonic retention, systemic side effects, and suboptimal patient compliance. To address these challenges, this study developed a thermo-responsive hydrogel combining hyaluronic acid–mesalamine (HA–Me) conjugates with methylcellulose (MC), providing a targeted and sustained drug delivery platform for UC treatment. HA–Me conjugates were synthesized via a nucleophilic addition–elimination reaction, with FT-IR and 1H-NMR confirming successful conjugation and a grafting ratio of 12.45%. Rheological analysis revealed a lower critical solution temperature (LCST) of 36.7–37.7 °C, ensuring gelation at body temperature when the MC concentration was 5–7 wt%. The optimized hydrogel exhibits intestinal retention properties, thereby improving drug bioavailability. The results confirmed that this hydrogel not only improved drug release time but also provided a protective barrier for inflamed wounds, facilitating wound healing, reducing the risk of reinfection, and improving medical compliance. Its mucoadhesive properties further supported effective drug delivery and localized therapeutic effects. This study highlights the potential of the MC/HA–Me hydrogel as a platform for overcoming the limitations of conventional UC treatments, offering opportunities for tailored therapeutic applications and future clinical development.
1. Introduction
Inflammatory bowel disease (IBD) is a chronic autoimmune disorder of the gastrointestinal tract, with its global incidence having surged nearly 30-fold over the past few decades.1–3 IBD comprises two primary subtypes: ulcerative colitis (UC) and Crohn's disease (CD). The etiology and pathogenesis of IBD remain incompletely understood, with genetic predispositions, imbalances in gut microbiota, intestinal barrier dysfunction, and immune dysregulation being recognized as contributing factors.4,5 UC predominantly affects the rectum and colon, presenting as continuous inflammation extending proximally,6,7 whereas CD features discontinuous lesions that can involve the entire gastrointestinal tract, complicating localized treatment.8,9 Current therapies for UC primarily rely on 5-aminosalicylate (5-ASA, mesalamine) and corticosteroids, particularly for mild to moderate cases.10–13 Although mesalamine is widely used, its clinical benefits in severe cases are limited. Furthermore, patient intolerance to mesalamine is a significant issue, with symptoms ranging from mild headaches and rashes to severe reactions such as Stevens–Johnson syndrome and toxic epidermal necrolysis.14,15 These adverse effects often necessitate alternative formulations or therapies, including sulfasalazine (SASP), time-dependent mesalamine micro-granules, and pH-dependent mesalamine, which aim to improve colonic drug delivery.16–20 However, these approaches have shown mixed efficacy, particularly in patients with refractory UC. In addition to small-molecule drugs, the recent development of biologics has provided new options for UC treatment. Anti-TNF-α agents, such as infliximab, and IL-12/23 inhibitors have demonstrated success in modulating immune responses in severe UC cases.21–27 Despite their efficacy, biologics are associated with high costs, potential immunogenicity, and increased risk of infections, underscoring the need for alternative, cost-effective treatments.
Orally administered mesalamine is rapidly absorbed in the upper gastrointestinal tract, leading to suboptimal delivery to the colon. Topical preparations, such as suppositories, foams, or enemas, target the lower third of the colon more effectively but face retention challenges due to gastrointestinal motility.28–31 For example, after enema administration, the drug is often prematurely excreted due to mechanical peristalsis, significantly reducing its retention time and therapeutic efficacy. Additionally, patient compliance is hindered by the need to maintain fixed positions during administration, making these approaches less practical for long-term use.32,33
Hydrogels have emerged as promising drug delivery systems due to their unique properties, including biocompatibility, mucoadhesion, and customizable gelation behaviors.34 Among these, methylcellulose (MC) hydrogels are particularly notable for their thermo-responsive behavior, forming gels at physiological temperatures. Their ability to transition between sol and gel phases enables localized drug retention, potentially overcoming the limitations of traditional enemas.35,36 Additionally, the lower critical solution temperature (LCST) of MC hydrogels can be tuned to match human body temperature, further enhancing their applicability in rectal drug delivery systems. Hyaluronic acid (HA), a glycosaminoglycan widely distributed in the extracellular matrix, is recognized for its roles in wound healing and tissue repair. HA stimulates cellular migration and growth factor release, making it an attractive candidate for therapeutic applications.37–39 It also plays a crucial role in enhancing mucoadhesion and forming a protective barrier over inflamed mucosal tissues, characteristics that complement the drug delivery capabilities of MC hydrogels.40 Recent studies have demonstrated the potential of HA-drug conjugates in targeted therapies, such as hepatocyte-targeted HA–polyethylenimine (HA–PEI) conjugates, which synergistically enhance anti-inflammatory effects.41 Similarly, polyethyleneimine-conjugated organosilica nanoparticles (MON-PEI) have been developed for IBD treatment, leveraging reactive oxygen species (ROS) degradation to reduce inflammation.42
In our previous research, it was first revealed that combined treatment with HA and mesalamine (IBD98-M) protects rats from IBD disease induced by intracolonic administration of trinitrobenzenesulfonic acid (TNBS).43 The results showed that IBD98-M therapy strongly promoted wound healing in colonic injuries and significantly inhibited myeloperoxidase (MPO) activity in the inflamed colon tissue of rats. These findings underscore the synergistic potential of HA and mesalamine in treating IBD. Subsequently, other researchers have used HA to improve anti-inflammatory efficacy of rectal mesalamine administration in a murine colitis model, further validating its clinical potential.44
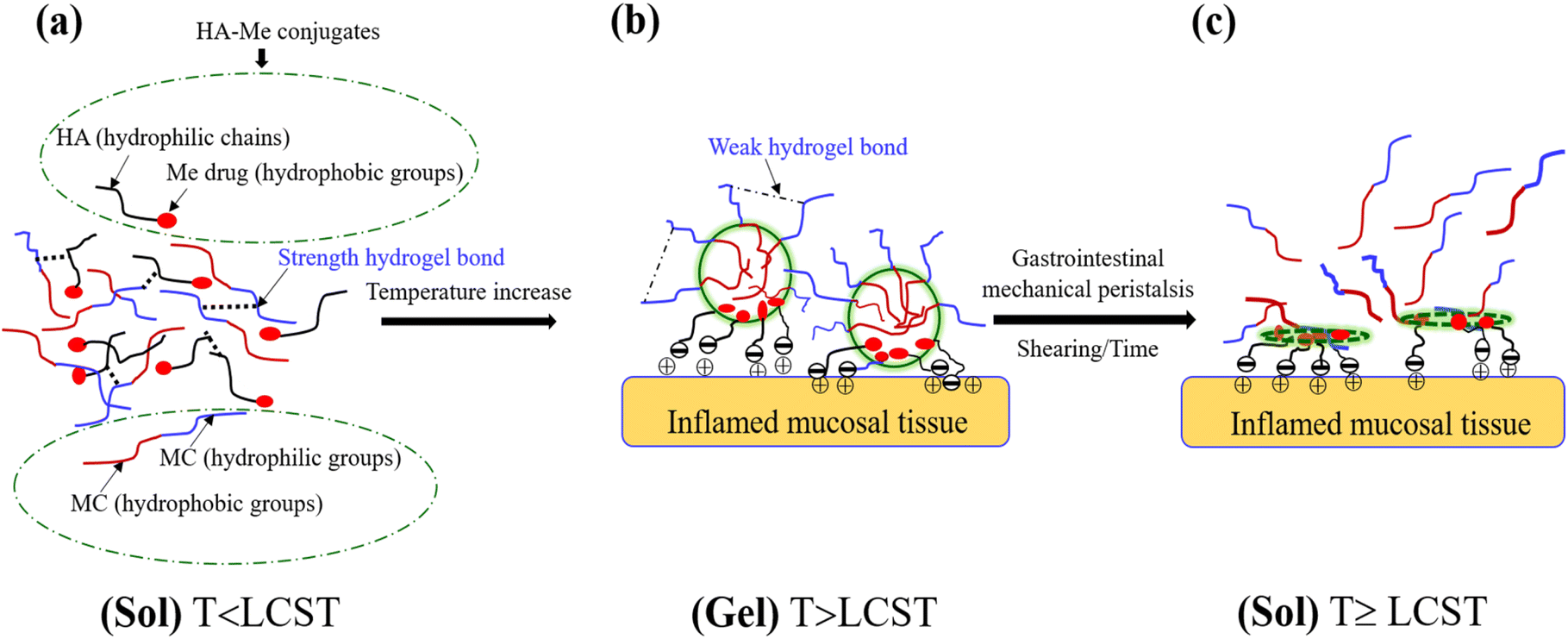
Building on these findings, this study pioneers a new approach by synthesizing a hyaluronic acid–mesalamine (HA–Me) conjugate to address key challenges such as hydrophobicity and phase separation observed in previous mesalamine formulations. This conjugate is incorporated into a MC hydrogel, forming a rectal enema designed for targeted UC treatment. The unique combination of HA and MC allows for a dual-action mechanism: HA provides anti-inflammatory benefits and enhances mucoadhesion, while MC ensures sustained drug release and improved retention at the inflamed site. The MC/HA–Me hydrogel adheres to inflamed mucosal tissues, forming a protective barrier that sustains drug release while minimizing systemic side effects. Its gelation properties are optimized for physiological temperatures, leveraging the electrostatic interactions between negatively charged HA–Me and positively charged inflamed tissues. The combination of MC and HA–Me is anticipated to provide synergistic therapeutic effects. Scheme 1 illustrates the proposed mechanisms of in situ gelation, de-gelation, and mucoadhesion.
 |
| | Scheme 1 Proposed mechanisms underlying the in situ physical gelation and de-gelation processes, along with the electrostatic interactions between inflamed mucosal tissues in the gastrointestinal tract and HA–Me conjugate drugs. (a) The mixing solution (sol) of MC/HA–Me conjugates are stabilized by hydrophilic chains forming strong hydrogen bonds at temperatures below the lower critical solution temperature (T < LCST). (b) In situ gelation of MC/HA–Me hydrogels occur through hydrophobic interactions as the temperature increases, with the disruption of weak hydrogen bonds at T > LCST. (c) A soft granular yield-stress sol is generated due to gastrointestinal mechanical peristalsis over time at temperatures reaching or exceeding the LCST (T ≥ LCST). | |
2. Materials and methods
2.1 Materials
Hyaluronic acid (HA) was purchased from Shandong Focusfreda Biotech Co., Ltd (Shandong, China). Methylcellulose (MC) was obtained from Shin-Etsu Chemical Co., Ltd (Niigata, Japan). Mesalamine (Me) was sourced from Chemi SPA (Patricia, Italy). Griess reagent was purchased from Abcam Co., Ltd (Cambridge, UK). Mouse TNF-α ELISA MAX™ Kit (cat. no. 430901) and Mouse IL-6 ELISA MAX™ Kit (cat. no. 431301) were procured from BioLegend Co., Ltd (San Diego, USA). Cell culture reagents were obtained from Corning Co., Inc. (New York, USA). All other experimental reagents were purchased from Sigma-Aldrich Co., LLC. (St. Louis, USA).
2.2 Synthesis and structural analysis of N-acyl HA–Me conjugates
2.2.1 Synthesis of HA–Me conjugates. A 30 mg mL−1 solution of hydroxybenzotriazole (HOBt) in DMSO was added to a 1 wt% aqueous solution of HA (molecular weight 100–500 kDa) and allowed to react for 6 min. Subsequently, a 5 mg mL−1 solution of Me in DMSO and a 5 mg mL−1 solution of N,N′-diisopropylcarbodiimide (DIC) were sequentially added, and the reaction mixture was stirred for over 18 h at 30 °C. Upon completion, the reaction product was filtered and transferred onto a dialysis membrane, which was immersed in 2 L of deionized water for dialysis to remove impurities. The resulting product, characterized by its light purple, cotton-like appearance, consisted of N-acyl HA–Me conjugates, which were then dried, sealed, and stored in a dry cabinet for later use.
2.2.2 Structural analysis of functional groups for HA–Me conjugates. The functional groups of HA–Me conjugates were analyzed using an attenuated total reflection Fourier transform infrared (ATR-FTIR) spectrometer (Thermo Nicolet™ iS™ 10, USA) over a wavenumber range of 600–4000 cm−1, with a scan rate of 0.7 scans per second.1H NMR spectra were acquired using a 600 MHz NMR spectrometer (VNMRS-600, VARIAN, USA), employing the D2O solvent peak as the internal standard. The structure and degree of substitution (DS)—defined as the number of conjugated Me groups per repeating unit of HA—were quantified by analyzing the integral ratios of the characteristic peaks corresponding to the HA backbone and the conjugated moiety.45–47
2.3 Evaluation of biocompatibility and anti-inflammatory efficacy for HA–Me conjugates
2.3.1 Cell culture. RAW 264.7 murine macrophage-like cells were obtained from the Bioresource Collection and Research Center (BCRC, Taiwan). The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 4 mM L-glutamine, 1.5 g per L sodium bicarbonate, and 4.5 g per L glucose, as well as 10% heat-inactivated fetal bovine serum (FBS), 100 IU per mL penicillin, and 100 μg per mL streptomycin. The cultures were maintained in a humidified incubator at 37 °C with 5% CO2.
2.3.2 Cytotoxicity. RAW 264.7 macrophages were seeded at a density of 1 × 104 cells per mL into 96-well plates and incubated for 24 h prior to treatment. The cells were then exposed to Me (3.06–12.24 mM), HA–Me (equivalent to 3.06–12.24 mM Me), HA (0.047–0.188%), or left untreated as a control group. Following an additional 24-hour incubation period, MTT reagent was added to each well and incubated for 4 h. The resulting formazan crystals were dissolved in dimethyl sulfoxide (DMSO), and absorbance was measured at 570 nm using a microplate reader (Bio-Tek, USA). All experiments were performed in triplicate.
2.3.3 Macrophage stimulation. RAW 264.7 macrophages were seeded at a density of 1 × 106 cells per mL into 6-well plates and incubated for 24 h prior to stimulation. The cells were treated with 2 μg per mL lipopolysaccharide (LPS), either alone or in combination with varying concentrations of Me or HA–Me (equivalent to 3.06 and 6.12 mM mesalamine), and incubated for 24 h at 37 °C with 5% of CO2. The culture supernatants were collected for subsequent ELISA and nitrite assays.
2.3.4 Determination of nitric oxide (NO). Nitrite concentration in the culture medium, indicative of NO secretion by macrophages, was quantified using a commercial Griess reagent according to the manufacturer's protocol. In brief, 100 μL of culture supernatant was transferred to a 96-well ELISA plate in triplicate and incubated with an equal volume of freshly prepared Griess reagent at room temperature for 15 min. Absorbance was measured at 490 nm using a microplate reader. Nitrite levels were calculated using a standard nitrite reference curve.
2.3.5 Determination of TNF-α and IL-6. The concentrations of TNF-α and IL-6 in the culture supernatants were measured using the Mouse TNF-α ELISA MAX™ Kit and Mouse IL-6 ELISA MAX™ Kit, following the manufacturer's instructions. All experiments were conducted in triplicate.
2.4 Preparation of MC/HA–Me conjugates hydrogels
2.4.1 Optimized formulation of hydrogels. The methylcellulose (MC)/hyaluronic acid methacrylate (HA–Me) hydrogel is prepared using the thermal dispersion method. First, 5–7 wt% MC is added to a 1% NaCl solution and magnetically stirred at room temperature until uniformly dispersed. The solution is then heated to 75 °C and continuously stirred for 1 h to ensure complete dissolution and hydration of MC, forming a stable polymer solution. Once MC is fully dissolved, stirring continues at room temperature, and after the temperature drops below 40 °C, 4–6 wt% HA–Me is gradually introduced. The mixture is then stirred at 4 °C overnight to achieve uniform dispersion. To further enhance homogeneity, ultrasonic treatment is applied, promoting interactions between MC and HA–Me and facilitating the formation of a stable interpenetrating network. The final MC/HA–Me hydrogel is stored at 4 °C for future use. This study investigates eight distinct formulations, with MC and HA–Me concentrations ranging from 5–7 wt% and 4–6 wt%, respectively, as detailed in Table 1.
Table 1 Average LCST, flowing distance and gelling time with different formulas
| No. |
Hydrogel formulationa |
LCST (°C) |
Flowing distance (cm) |
Gelling time (s) |
| All hydrogel formulations contain 1% NaCl. |
| H1 |
6.5% MC/4.0% HA–Me |
37.0 |
14.8 |
243 |
| H2 |
6.5% MC/4.5% HA–Me |
37.0 |
14.4 |
258 |
| H3 |
6.5% MC/5.0% HA–Me |
37.0 |
13.9 |
281 |
| H4 |
6.5% MC/6.0% HA–Me |
37.0 |
12.1 |
300 |
| H5 |
5.0% MC/4.5% HA–Me |
37.7 |
20.9 |
457 |
| H6 |
5.5% MC/4.5% HA–Me |
37.5 |
17.8 |
433 |
| H7 |
6.0% MC/4.5% HA–Me |
37.2 |
16.1 |
296 |
| H8 |
7.0% MC/4.5% HA–Me |
36.7 |
11.0 |
224 |
2.4.2 Rheological analysis. The rheological properties of MC/HA–Me hydrogels were evaluated using a rotational rheometer (Discovery HR 10, TA Instruments, USA) operating in dynamic mode. An oscillating temperature ramp program was employed, increasing the temperature from 25 °C to 50 °C at a rate of 5 °C min−1. Measurements were conducted at a constant frequency of 1 Hz and a strain of 1%. A 20 mm parallel-plate solvent trap on a Peltier surface was used as the testing geometry, with the gap set at 1000 μm. Hydrogel samples (0.3 mL) were tested, and changes in storage modulus (G′) and loss modulus (G′′) were recorded across the temperature range.
2.4.3 In vitro simulation of the gelling time and flowing distance. The gelling time and flowing distance of MC/HA–Me hydrogels were evaluated using excised porcine intestines. Colorectal segments (25 cm in length) were obtained from the Agricultural Technology Research Institute (ATRI, Taiwan) and prepared by thoroughly cleaning to remove residual excreta while preserving the intestinal mucosal tissue. The segments were stored at 4 °C until use. During the experiment, the intestinal segments were placed in a sample holder within an ultrasonic oscillation bath maintained at 37 °C and 20 Hz. A 2 mL hydrogel sample was injected into the intestinal segment, and the gelling time and flowing distance were observed and recorded.
2.4.4 In vitro drug release analysis of MC/HA–Me using Folin–Ciocalteu reagent. The in vitro release profile of mesalamine was assessed using the Folin–Ciocalteu reagent as a colorimetric detection method.48 The hydrogel formulation (6.5% MC, 4.5% HA–Me, 1.0% NaCl, H2) was subjected to shaking in an ultrasonic oscillator set at 37 °C and 20 Hz to simulate intestinal peristalsis. At predetermined time intervals, 1 mL of the release medium was withdrawn and replaced with an equal volume of fresh PBS to maintain sink conditions. The collected samples were then reacted with 0.1 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 diluted Folin–Ciocalteu reagent, followed by the addition of 0.2 mL of 0.5 M sodium hydroxide solution to stabilize the chromogenic complex. After incubating the samples in the dark for 15 min, the absorbance was measured at 655 nm using a UV-vis spectrophotometer. The concentration of released mesalamine was determined from a standard calibration curve, and the results were expressed as the cumulative percentage of drug released over time.
:3 diluted Folin–Ciocalteu reagent, followed by the addition of 0.2 mL of 0.5 M sodium hydroxide solution to stabilize the chromogenic complex. After incubating the samples in the dark for 15 min, the absorbance was measured at 655 nm using a UV-vis spectrophotometer. The concentration of released mesalamine was determined from a standard calibration curve, and the results were expressed as the cumulative percentage of drug released over time.
2.5 Statistical analysis
All experimental data were expressed as mean ± standard deviation (SD). Statistical analyses were performed using one-way analysis of variance (ANOVA) to determine significant differences between groups. Post hoc pairwise comparisons were conducted using Tukey's test, and a p-value of less than 0.05 was considered statistically significant. All statistical analyses were performed using Minitab statistical software (Minitab Inc., USA).
3. Results and discussion
3.1 Synthesis and structural characterization of HA–Me conjugates
3.1.1 Synthesis of HA–Me conjugates. Fig. 1a illustrates the reaction mechanism for the synthesis of HA–Me conjugates. The primary amine (–RNH2) group on the benzene ring of Me reacts with the carboxylic acid (–COOH) group of HA through nucleophilic addition–elimination reaction, forming a stable N-acyl HA–Me conjugate with secondary amide (–CONHPh) bond. Visually, pure HA appears white and transparent, while HA–Me conjugates exhibit a light purple hue due to the formation of secondary aromatic amide bonds (Fig. 1b). These observations confirm the successful conjugation of Me to HA.
 |
| | Fig. 1 Synthesis reaction process of HA–Me conjugates. | |
3.1.2 Structural analysis by ATR-FT/IR. The functional groups of Me, HA, and HA–Me conjugates were analyzed using ATR-FTIR spectroscopy. The spectrum of Me exhibits a prominent aromatic hydrogen absorption peak at 2980 cm−1, indicative of its benzene ring structure (Fig. 2a). In contrast, the spectrum of HA shows aliphatic hydrogen peaks within the range of 2950–2850 cm−1, corresponding to its polysaccharide backbone (Fig. 2b).42,43 The spectrum of HA–Me conjugates reveals absorption peaks corresponding to both aromatic and aliphatic hydrogen atoms, confirming the successful incorporation of Me into the HA structure (Fig. 2c). Notably, the characteristic amide (I) peaks of HA, located at 1646 cm−1 and 1560 cm−1, undergo a shift to 1654 cm−1 and 1561 cm−1, respectively, in HA–Me conjugates.49,50 These shifts are attributed to the formation of new amide (II) bonds during the conjugation process, which require higher energy for absorption due to their increased bond strength and structural rigidity. Additionally, the disappearance of the scissoring vibration peak of the –NH2 group from Me further supports the formation of N-acyl HA–Me conjugates. This indicates that the primary amine group on the benzene ring of Me has reacted with the carboxylic acid groups of HA, resulting in a stable amide bond. The combined observations demonstrate that HA–Me conjugates retain the functional characteristics of both HA and Me while forming new structural features unique to the conjugated compound.
 |
| | Fig. 2 ATR-FT/IR spectra of (a) Me, (b) HA, (c) HA–Me. | |
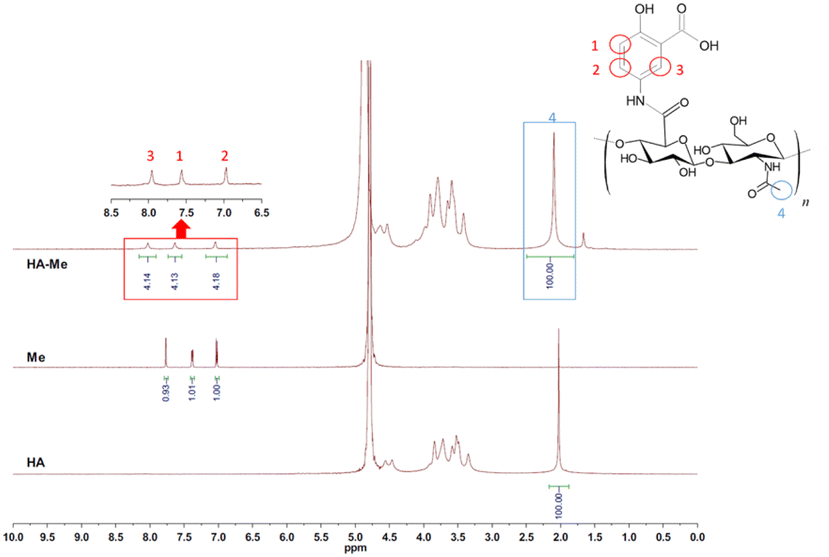
3.1.3 Structural analysis by 1H-NMR. Fig. 3 illustrates the 1H-NMR spectra of Me, HA, and HA–Me conjugates, highlighting their distinct chemical shift patterns. The spectrum of Me shows characteristic aromatic hydrogen peaks between δ 7 and δ 8 ppm, corresponding to the protons on the benzene ring.51 In contrast, HA lacks any peaks in this region due to the absence of aromatic hydrogen atoms, reflecting its polysaccharide structure. The spectrum of HA–Me conjugates reveals peaks in the δ 7–8 ppm region, similar to those observed for Me, indicating the retention of aromatic protons after conjugation. This observation confirms that the primary amine group (–NH2) of Me has been successfully grafted onto the carboxylic acid group of HA through a nucleophilic addition–elimination reaction, forming a stable secondary amide bond (amide II), as depicted in Scheme 1. The degree of substitution (DS) was quantified using the integrated peak areas in the 1H-NMR spectrum.47 The methyl groups on the original amide functional group (–CONHCH3) of HA exhibited a reference peak at δ 2 ppm, which was used as the baseline for normalization. For HA–Me conjugates, newly formed secondary aromatic amide (–CONHPh) peaks were observed in the δ 7–8 ppm region. The integrated area of these peaks (4.14 + 1.13 + 4.18) corresponded to a DS of approximately 12.45%, indicating that 12.45% of the repeating units in HA were successfully conjugated with Me. These findings provide clear evidence of successful chemical modification, with HA–Me conjugates retaining key functional characteristics of both HA and Me while forming new structural features specific to the conjugated compound.
 |
| | Fig. 3 1H-NMR spectra of HA–Me, Me and HA. | |
3.2 Anti-inflammatory efficacy of Me and HA–Me conjugated drugs
3.2.1 Cytotoxicity assay. Me has been extensively used in clinical settings for the treatment of mild to moderate UC due to its effective anti-inflammatory properties. However, higher concentrations of Me may induce cytotoxic effects, which could limit its therapeutic applications. Fig. 4 illustrates the cytotoxicity profiles of Me and HA–Me conjugates in RAW 264.7 macrophages. At a concentration of 12.24 mM, cell viability decreased to 72% for Me and 80% for HA–Me conjugates, indicating a dose-dependent cytotoxic response. In contrast, when the concentration was ≤6.12 mM, the cell viability remained above 90% for both Me and HA–Me conjugates, demonstrating their safety at lower concentrations. These results suggest that HA–Me conjugates exhibit slightly reduced cytotoxicity compared to Me alone, likely due to the controlled release properties imparted by the HA conjugation. Based on these findings, a concentration threshold of 6.12 mM or lower was selected for subsequent experiments to evaluate the anti-inflammatory efficacy of Me and HA–Me in the LPS-induced RAW 264.7 macrophage inflammation model. This ensures that the selected concentrations are both therapeutically effective and non-toxic, supporting their potential application in clinical treatment strategies.
 |
| | Fig. 4 The cytotoxic effects of Me and HA–Me conjugated drugs were evaluated in RAW264.7 macrophages, with untreated cells (N) serving as the negative control. Statistical significance is denoted as *p < 0.05 compared to the negative control (N). | |
3.2.2 Effect on NO production. Nitric oxide (NO) is a crucial mediator released by macrophages in response to infections or inflammatory stimuli. Excessive production of NO, primarily through the inducible nitric oxide synthase (iNOS) pathway, can result in oxidative damage to cells, thereby exacerbating inflammatory responses. RAW 264.7 cells, a well-established murine macrophage model, are commonly used in experimental studies to evaluate inflammation and immune responses. In these studies, lipopolysaccharide (LPS) is frequently employed as a stimulant to induce high levels of NO production.52–55 The inhibitory effects of Me and HA–Me conjugates on NO production were assessed in LPS-stimulated RAW 264.7 macrophages. As shown in Fig. 5, both Me and HA–Me significantly reduced NO levels in a dose-dependent manner. At a concentration of 3.06 mM, the inhibition efficiencies were 36.8 ± 4.8% and 31.1 ± 3.7% for Me and HA–Me, respectively. At a higher concentration of 6.12 mM, the inhibition efficiencies increased to 46.1 ± 3.4% for Me and 42.6 ± 4.7% for HA–Me. These reductions were statistically significant (p < 0.05). Importantly, the results indicate that the covalent bonding of Me to HA (forming HA–Me conjugates) does not compromise the anti-inflammatory efficacy of Me. Instead, the conjugation may offer additional benefits, such as improved drug delivery and retention, without diminishing the ability to inhibit NO production. These findings highlight the potential of HA–Me conjugates as a therapeutic option for managing inflammation associated with diseases such as UC. Moreover, the covalent conjugation of HA with Me may enhance drug retention at the inflamed site, thereby improving localized therapeutic efficacy and reducing systemic side effects—a crucial advantage over free Me administration.
 |
| | Fig. 5 Effects of Me and HA–Me conjugates at varying concentrations on NO production in LPS-stimulated RAW 264.7 macrophages. N denotes the negative control (untreated cells), and P represents the positive control (cells treated with LPS only). Both Me and HA–Me significantly inhibited NO production in a dose-dependent manner, with statistical significance indicated by *p < 0.05 compared to the LPS-stimulated positive control. | |
3.2.3 Effects on proinflammatory cytokines TNF-α and IL-6. Tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) are pivotal proinflammatory cytokines secreted by immune cells such as macrophages. TNF-α plays a central role in promoting inflammatory responses, and its overproduction is implicated in chronic inflammation and diseases such as rheumatoid arthritis, cancer, and IBD.53,56,57 Similarly, IL-6 contributes to both local and systemic inflammatory responses, with excessive IL-6 levels being associated with numerous inflammatory and autoimmune disorders, including cardiovascular disease and intestinal inflammation.58,59 The effects of Me and HA–Me conjugates on TNF-α and IL-6 production were evaluated in LPS-stimulated RAW 264.7 macrophages. Fig. 6a demonstrates that both Me and HA–Me significantly inhibited TNF-α expression in a dose-dependent manner. At a concentration of 3.06 mM, TNF-α levels were reduced to 197.5 ± 1.9 pg mL−1 and 230.6 ± 1.6 pg mL−1 for Me and HA–Me, respectively. Increasing the concentration to 6.12 mM further suppressed TNF-α expression to 162.1 ± 2.1 pg mL−1 for Me and 196.8 ± 41.3 pg mL−1 for HA–Me, with both reductions being statistically significant (*p < 0.05). Similarly, as shown in Fig. 6b, Me and HA–Me exhibited a significant inhibitory effect on IL-6 production. At 3.06 mM, IL-6 levels were reduced to 229.4 ± 5.0 pg mL−1 and 255.1 ± 7.2 pg mL−1 for Me and HA–Me, respectively. At 6.12 mM, the expression of IL-6 was further decreased to 164.5 ± 5.1 pg mL−1 and 212.1 ± 10.2 pg mL−1 for Me and HA–Me, respectively (*p < 0.05). These results highlight the comparable efficacy of HA–Me conjugates to free Me in suppressing key inflammatory mediators. The slightly reduced inhibitory effect observed with HA–Me may reflect a controlled release mechanism that moderates cytokine suppression over time. The ability of HA–Me conjugates to significantly inhibit both TNF-α and IL-6 reinforces their potential as therapeutic agents for managing inflammatory diseases.
 |
| | Fig. 6 Effects of Me and HA–Me conjugates at varying concentrations on proinflammatory cytokine production in LPS-stimulated RAW 264.7 macrophages. (a) TNF-α levels were significantly reduced in response to both Me and HA–Me treatments compared to the positive control (P, cells treated with LPS only). (b) IL-6 levels were similarly suppressed by Me and HA–Me in a dose-dependent manner. N represents the negative control (untreated cells), and P represents the positive control (cells treated with LPS only). Statistical significance is denoted by *p < 0.05 compared to the LPS-stimulated positive control. | |
3.3 Characterization of MC/HA–Me hydrogels
3.3.1 Rheological properties. The hydrogel formulations were prepared with varying concentrations of HA–Me (4–6 wt%) and methylcellulose (MC, 5–7 wt%) in distilled water (100 wt%), with a constant addition of 1 wt% NaCl to simulate physiological osmotic conditions. Eight distinct formulations were assembled, as summarized in Table 1. The thermo-responsive properties of MC/HA–Me hydrogels were analyzed through rheological measurements of the storage modulus (G′) and loss modulus (G′′), using an oscillatory shear rheometer. The temperature-dependent changes in G′ and G′′ were recorded to investigate the transition behavior of the hydrogels (Fig. 7). During the heating process, G′ represents the stored energy recoverable as elastic deformation, while G′′ quantifies the dissipated energy, such as heat generation or molecular reorganization. The intersection points of G′ and G′′ on the heating curve denotes the transition of the hydrogel from a sol state to a gel state, defining the LCST.60 Below the LCST, G′′ exceeds G′, indicating the predominance of viscous behavior. Conversely, above the LCST, G′ becomes greater than G′′, signifying the formation of a gel network with elastic properties. The results revealed that increasing the MC content from 5.0 wt% to 7.0 wt% at a constant HA–Me concentration (4.5 wt%) resulted in a gradual decrease in the LCST, from 37.7 °C to 36.7 °C. This behavior suggests that higher MC concentrations enhance the hydrophobic interactions within the hydrogel, promoting gelation at slightly lower temperatures. Notably, the HA–Me content did not significantly affect the LCST, indicating that MC concentration is the dominant factor governing thermo-responsiveness. These findings demonstrate that the gelling temperature of MC/HA–Me hydrogels can be fine-tuned to align with human body temperature when the MC concentration is maintained between 5.0 wt% and 7.0 wt%. This tunable property is critical for applications in rectal drug delivery systems, ensuring optimal gelation under physiological conditions for targeted and sustained therapeutic effects.
 |
| | Fig. 7 Dependence of G′ and G′′ on the temperature increase for various MC/HA–Me conjugates hydrogels with the content of MC from 5.0 wt% to 7.0 wt% increase at constant of 4.5%HA–Me/1.0% NaCl: (a) 5.0 wt%-MC, (b) 5.5 wt%-MC (c) 6.0 wt%-MC (d) 6.5 wt%-MC (e) 7.0 wt%-MC. | |
3.3.2 Gelling time and flowing distance. As shown in Fig. 8 H1–H8, increasing the HA–Me content from 4.0 wt% to 6.0 wt% at a constant MC concentration of 6.5 wt% resulted in a notable increase in the viscosity of the MC/HA–Me hydrogel. This viscosity increase reduced the hydrogel's fluidity and correspondingly prolonged its gelation time. Conversely, when the HA–Me content was held constant at 4.5 wt%, increasing the MC concentration reduced the flowing distance of the hydrogel. However, this adjustment exhibited the opposite effect on gelation time, which decreased as MC content increased, indicating that MC concentration strongly influences the gelation dynamics of the hydrogel system. Importantly, as shown in Fig. 8 No added MC, hydrogels lacking MC failed to form a stable gel after being injected into a simulated pig intestine. Without MC, the HA–Me conjugates were rapidly excreted, emphasizing the critical role of MC in forming a functional hydrogel matrix. The detailed data corresponding to these observations are presented in Table 1.
 |
| | Fig. 8 The gelation time and flow distance of various hydrogel formulations were evaluated using an ultrasonic oscillator in an in vitro simulation through porcine intestine at a constant temperature and frequency (37 °C and 20 Hz). H1–H4 hydrogel formulations: 6.5% MC mixed with 4.0%, 4.5%, 5.0% and 6.0% HAMe, respectively; H5–H8 hydrogel formulations: 4.5% HAMe mixed with 5.0%, 5.5%, 6.0% and 7.0% MC, respectively; no added MC: 4.5% HAMe. | |
The results from the pig intestine simulation provide valuable insights into optimizing hydrogel formulations. HA–Me content can be adjusted to balance drug efficacy and cytotoxicity, while MC content can be fine-tuned to optimize the hydrogel's flowing distance, ensuring it adequately covers the inflamed rectal segment. The rectum, located between the sigmoid colon and the anus, is approximately 15 cm in length, making flowing distance and gelling time crucial parameters for effective drug delivery. Based on these findings, H2 (6.5% MC, 4.5% HA–Me) and H7 (6.0% MC, 4.5% HA–Me) formulations were identified as optimal candidates. These hydrogels exhibit a favorable balance of short gelling time and sufficient flowing distance, ensuring high drug efficacy and targeted therapeutic delivery to inflamed rectal tissues.
3.3.3 In vitro drug release analysis of MC/HA–Me. The thermo-sensitive hydrogel exhibits a gel state at 37 °C (Fig. 9a). The hydrogel sample, H2, was subjected to ultrasonic oscillation at 37 °C to simulate intestinal peristalsis, and the drug release kinetics were measured. As shown in Fig. 9b, approximately 50% of the drug is released within 3 hours, followed by sustained release over a 12-hour period. These results suggest that the MC/HA–Me hydrogel can effectively prolong drug release, which is advantageous for maintaining therapeutic drug concentrations in the colon over an extended period. Importantly, this in vitro release behavior complements the findings from the pig intestine simulation experiments described in Section 3.3.2. In those tests, the optimized H2 formulation demonstrated both sufficient flowing distance and rapid gelation upon administration, enabling uniform coating of the rectal mucosa. The hydrogel's ability to form a stable gel in situ significantly enhanced its retention within the simulated pig colorectal tract, in contrast to HA–Me formulations without MC, which were rapidly expelled.
 |
| | Fig. 9 (a) The MC/HA–Me hydrogel in its gel form at 37 °C. (b) Cumulative drug release of the MC/HA–Me hydrogel at 37 °C. | |
4. Conclusions
This study successfully developed an intelligent MC/HA–Me hydrogel using a HA–Me conjugate and MC. The HA–Me conjugate retained the anti-inflammatory properties of mesalamine, significantly inhibiting NO, TNF-α, and IL-6 production in LPS- stimulated RAW 264.7 macrophages. Additionally, HA–Me improved the solubility of Me, facilitating its incorporation into enema preparations. Rheological analysis and in vitro pig intestine simulation demonstrated the hydrogel's tunable properties, with optimized formulations (H2 and H7) exhibiting ideal gelling temperatures (36.7–37.7 °C), short gelation times, and appropriate flowing distances for rectal application. The combination of MC and HA–Me ensures effective drug delivery, tailored to the rectal anatomy, while minimizing cytotoxicity. These findings establish the MC/HA–Me hydrogel as a promising candidate for targeted UC treatment, offering dual-action benefits: anti-inflammatory effects from HA–Me and prolonged retention through MC-mediated gelation. This work not only highlights the potential of hydrogel-based therapies in IBD management but also provides a foundation for future clinical studies, including in vivo evaluation, long-term safety assessments, and therapeutic efficacy testing in human trials.
Data availability
The datasets generated and analyzed in this current study are available from the corresponding author upon reasonable request.
Author contributions
Conceptualization and supervision: S. N. Kuo, S. L. Huang and J. H. Huang; methodology and investigation; S. N. Kuo, P. X. Wu and Y. C. Hsu; resources: P. X. Wu, Y. C. Hsu and S. N. Kuo; literature review and writing—original draft preparation: P. X. Wu, S. N. Kuo, S. N. Kuo and S. L. Huang; data curation: S. N. Kuo, P. X. Wu and Y. C. Hsu; writing—review and editing: S. N. Kuo, S. L. Huang and J. H. Huang. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
The authors are grateful for the financial support by National Science and Technology Council, Taiwan, ROC (NSTC 111-2221-E-239-001 and NSTC 112-2221-E-239 -018).
References
- N. A. Molodecky, I. S. Soon, D. M. Rabi, W. A. Ghali, M. Ferris, G. Chernoff, E. I. Benchimol, R. Panaccione, S. Ghosh, H. W. Barkema and G. G. Kaplan, Gastroenterology, 2012, 142, 46–54 CrossRef PubMed.
- H. S. de Souza and C. Fiocchi, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 13–27 CrossRef CAS PubMed.
- G. G. Kaplan and J. W. Windsor, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 56–66 CrossRef PubMed.
- B. Khor, A. Gardet and R. J. Xavier, Nature, 2011, 474, 307–317 CrossRef CAS PubMed.
- J. T. Chang, N. Engl. J. Med., 2020, 383, 2652–2664 CrossRef CAS PubMed.
- R. Ungaro, S. Mehandru, P. B. Allen, L. Peyrin-Biroulet and J. F. Colombel, Lancet, 2017, 389, 1756–1770 CrossRef PubMed.
- T. Kobayashi, B. Siegmund, C. Le Berre, S. C. Wei, M. Ferrante, B. Shen, C. N. Bernstein, S. Danese, L. Peyrin-Biroulet and T. Hibi, Nat. Rev. Dis. Primers, 2020, 6, 74 CrossRef PubMed.
- J. Torres, S. Mehandru, J. F. Colombel and L. Peyrin-Biroulet, Lancet, 2017, 389, 1741–1755 CrossRef PubMed.
- R. Voelker, JAMA, 2024, 331, 716 CrossRef PubMed.
- M. Harbord, R. Eliakim, D. Bettenworth, K. Karmiris, K. Katsanos, U. Kopylov, T. Kucharzik, T. Molnar, T. Raine, S. Sebastian, H. T. de Sousa, A. Dignass, F. Carbonnel, C. s. European and O. Colitis, J. Crohns Colitis, 2017, 11, 769–784 CrossRef PubMed.
- D. T. Rubin, A. N. Ananthakrishnan, C. A. Siegel, B. G. Sauer and M. D. Long, Am. J. Gastroenterol., 2019, 114, 384–413 CrossRef PubMed.
- J. D. Feuerstein, K. L. Isaacs, Y. Schneider, S. M. Siddique, Y. Falck-Ytter, S. Singh and A. G. A. I. C. G. Committee, Gastroenterology, 2020, 158, 1450–1461 CrossRef PubMed.
- P. Wangchuk, K. Yeshi and A. Loukas, Trends Pharmacol. Sci., 2024, 45, 892–903 CrossRef CAS PubMed.
- M. Naganuma, Y. Iwao, H. Ogata, N. Inoue, S. Funakoshi, S. Yamamoto, Y. Nakamura, H. Ishii and T. Hibi, Inflammatory Bowel Dis., 2001, 7, 221–225 CrossRef CAS PubMed.
- S. Hiraoka, A. Fujiwara, T. Toyokawa, R. Higashi, Y. Moritou, S. Takagi, K. Matsueda, S. Suzuki, J. Miyaike, T. Inokuchi, M. Takahara, J. Kato and H. Okada, J. Gastroenterol. Hepatol., 2021, 36, 137–143 CrossRef CAS PubMed.
- M. A. Kamm, G. R. Lichtenstein, W. J. Sandborn, S. Schreiber, K. Lees, K. Barrett and R. Joseph, Gut, 2008, 57, 893–902 CrossRef CAS PubMed.
- G. R. Lichtenstein and M. A. Kamm, Aliment. Pharmacol. Ther., 2008, 28, 663–673 CrossRef CAS PubMed.
- Y. Yan, J. Sun, X. Xie, P. Wang, Y. Sun, Y. Dong and J. Xing, RSC Adv., 2018, 8, 2561–2574 RSC.
- C. Le Berre, G. Roda, M. Nedeljkovic Protic, S. Danese and L. Peyrin-Biroulet, Expert Opin. Biol. Ther., 2020, 20, 363–378 CrossRef CAS PubMed.
- H. Deissler, H. Krammer and A. Gillessen, Biomed. Rep., 2021, 15, 96 CrossRef CAS PubMed.
- S. Ghosh and R. Panaccione, Ther. Adv. Gastroenterol., 2010, 3, 239–258 CrossRef CAS PubMed.
- H. Nakase, T. Yoshino and M. Matsuura, Inflammatory Bowel Dis., 2014, 20, 2151–2156 CrossRef PubMed.
- M. W. Teng, E. P. Bowman, J. J. McElwee, M. J. Smyth, J. L. Casanova, A. M. Cooper and D. J. Cua, Nat. Med., 2015, 21, 719–729 CrossRef CAS PubMed.
- S. J. Li, L. M. Perez-Chada and J. F. Merola, J. Psoriasis Psoriatic Arthritis, 2019, 4, 70–80 CrossRef PubMed.
- J. Guo, H. Zhang, W. Lin, L. Lu, J. Su and X. Chen, Signal Transduction Targeted Ther., 2023, 8, 437 CrossRef CAS PubMed.
- S. Honap, P. M. Irving and M. A. Samaan, Eur. J. Gastroenterol. Hepatol., 2023, 35, 1270–1277 CrossRef CAS PubMed.
- J. F. Merola, R. Landewe, I. B. McInnes, P. J. Mease, C. T. Ritchlin, Y. Tanaka, A. Asahina, F. Behrens, D. D. Gladman, L. Gossec, A. B. Gottlieb, D. Thaci, R. B. Warren, B. Ink, D. Assudani, R. Bajracharya, V. Shende, J. Coarse and L. C. Coates, Lancet, 2023, 401, 38–48 CrossRef CAS PubMed.
- M. S. Harris and G. R. Lichtenstein, Aliment. Pharmacol. Ther., 2011, 33, 996–1009 CrossRef CAS PubMed.
- R. Chibbar and A. C. Moss, Gastroenterol. Clin. North Am., 2020, 49, 689–704 CrossRef PubMed.
- R. D. Cohen and R. Weisshof, Gastroenterol. Hepatol., 2020, 16, 21–27 Search PubMed.
- H. Nakase, M. Uchino, S. Shinzaki, M. Matsuura, K. Matsuoka, T. Kobayashi, M. Saruta, F. Hirai, K. Hata, S. Hiraoka, M. Esaki, K. Sugimoto, T. Fuji, K. Watanabe, S. Nakamura, N. Inoue, T. Itoh, M. Naganuma, T. Hisamatsu, M. Watanabe, H. Miwa, N. Enomoto, T. Shimosegawa and K. Koike, J. Gastroenterol., 2021, 56, 489–526 CrossRef PubMed.
- P. Gionchetti, S. Ardizzone, M. E. Benvenuti, G. Bianchi Porro, G. Biasco, P. Cesari, G. D'Albasio, R. De Franchis, G. Monteleone, F. Pallone, T. Ranzi, G. Trallori, D. Valpiani, M. Vecchi and M. Campieri, Aliment. Pharmacol. Ther., 1999, 13, 381–388 CrossRef CAS PubMed.
- S. V. Kane, Aliment. Pharmacol. Ther., 2006, 23, 577–585 CrossRef CAS PubMed.
- S. Parveen, F. Arjmand and S. Tabassum, RSC Adv., 2019, 9, 24699–24721 RSC.
- F. P. P. L. Nasatto, J. L. M. Silveira, M. E. R. Duarte, M. D. Noseda and M. Rinaudo, Polymers, 2015, 7, 777–803 CrossRef.
- C. W. Qilu Zhang, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- M. Litwiniuk, A. Krejner, M. S. Speyrer, A. R. Gauto and T. Grzela, Wounds, 2016, 28, 78–88 Search PubMed.
- F. Shu, D. Lv, X. L. Song, B. Huang, C. Wang, Y. Yu and S. C. Zhao, RSC Adv., 2018, 8, 6581–6589 RSC.
- E. M. S. Magdalena Antoszewska and W. Barańska-Rybak, Sci. Pharm., 2024, 92, 23 CrossRef.
- A. Aprodu, J. Mantaj, B. Raimi-Abraham and D. Vllasaliu, Pharmaceutics, 2019, 11, 127 CrossRef CAS PubMed.
- H. X. Mingqiao Li, N. Zhao, L. Zhang, H. Xia, X. Zhang, Q. Li, M. Liao, Q. Pan, Z. Yi and C. Jin, Mater. Des., 2023, 233, 112212 CrossRef.
- C. Shi, J. Dawulieti, F. Shi, C. Yang, Q. Qin, T. Shi, L. Wang, H. Hu, M. Sun, L. Ren, F. Chen, Y. Zhao, F. Liu, M. Li, L. Mu, D. Liu, D. Shao, K. W. Leong and J. She, Sci. Adv., 2022, 8, eabj2372 CrossRef CAS PubMed.
- C. T. Chiu, S. N. Kuo, S. W. Hung and C. Y. Yang, Molecules, 2017, 22, 904 CrossRef PubMed.
- H. D. Jhundoo, T. Siefen, A. Liang, C. Schmidt, J. Lokhnauth, B. Moulari, A. Beduneau, Y. Pellequer, C. C. Larsen and A. Lamprecht, Biomol. Ther., 2021, 29, 536–544 CrossRef CAS PubMed.
- Y. Luo, M. R. Ziebell and G. D. Prestwich, Biomacromolecules, 2000, 1, 208–218 CrossRef CAS PubMed.
- E. K. Lim, H. O. Kim, E. Jang, J. Park, K. Lee, J. S. Suh, Y. M. Huh and S. Haam, Biomaterials, 2011, 32, 7941–7950 CrossRef CAS PubMed.
- Y. Song, H. Cai, T. Yin, M. Huo, P. Ma, J. Zhou and W. Lai, Int. J. Nanomed., 2018, 13, 1585–1600 CrossRef CAS PubMed.
- B. S. Chandra, S. S. Bhogela, M. Shaik, C. S. Vadlamudi, M. Chappa and N. S. Maddirala, Quim. Nova, 2011, 34, 1068–1073 CrossRef CAS.
- P. T. Michele Di Foggia, A. Torreggiani, M. Dettin and A. Tinti, Proteomics Res. J., 2011, 2, 231–272 Search PubMed.
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS PubMed.
- P. Ispas-Szabo, M. M. Friciu, P. Nguyen, Y. Dumoulin and M. A. Mateescu, Drug Dev. Ind. Pharm., 2016, 42, 1183–1193 CrossRef CAS PubMed.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed.
- J. Zhu, Y. Zhang, G. Wu, Z. Xiao, H. Zhou and X. Yu, Mol. Med. Rep., 2015, 11, 729–733 CrossRef CAS PubMed.
- R. P. Kumar and A. Abraham, Pharmacol. Rep., 2017, 69, 908–915 CrossRef PubMed.
- I. P. S. Fernando, W. W. Lee, T. U. Jayawardena, M. C. Kang, Y. S. Ann, C. I. Ko, Y. J. Park and Y. J. Jeon, RSC Adv., 2018, 8, 18626–18634 RSC.
- R. M. Locksley, N. Killeen and M. J. Lenardo, Cell, 2001, 104, 487–501 CrossRef CAS PubMed.
- M. Nourian, V. Chaleshi, L. Pishkar, P. Azimzadeh, S. Baradaran Ghavami, H. Balaii, S. Alinaghi, S. Shahrokh, H. Asadzadeh Aghdaei and M. R. Zali, Biomed. Rep., 2017, 6, 698–702 CrossRef CAS PubMed.
- E. Grebenciucova and S. VanHaerents, Front. Immunol., 2023, 14, 1255533 CrossRef CAS PubMed.
- A. Shahini and A. Shahini, J. Cell Commun. Signaling, 2023, 17, 55–74 CrossRef CAS PubMed.
- S. K. Faezeh Khoobbakht, F. Bahmanyar, S. Marzieh Hosseini, N. Aminikhah, M. Farhoodi and L. Mirmoghtadaie, Food Hydrocolloids, 2024, 147, 109411 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Pei-Xhan Wub,
Shu-Ling Huang
a,
Pei-Xhan Wub,
Shu-Ling Huang