
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an electrochemical sensor based on Ni-Bio-MOF and a molecular imprinted polymer for determination of diclofenac: electrochemical and DFT investigations†
Samaneh Ebadia,
Khadijeh Ghanbari *a and
Mansoureh Zahedi-Tabrizib
*a and
Mansoureh Zahedi-Tabrizib
aDepartment of Analytical Chemistry, Faculty of Chemistry, Alzahra University, P. O. Box 1993893973, Tehran, Iran. E-mail: Kh.ghanbari@alzahra.ac.ir; Kh_ghanb@yahoo.com
bDepartment of Physical Chemistry and Nanochemistry, Faculty of Chemistry, Alzahra University, P.O. Box 1993893973, Tehran, Iran
First published on 20th May 2025
Abstract
In this work, a metal–organic nickel framework (Ni-MOF) modified with a biological ligand (asparagine) (Ni-Bio-MOF) was synthesized by a hydrothermal method. Asparagine is believed to create defects on the surface of the MOF, thereby increasing its electrocatalytic activity. Then, a Diclofenac (DCF) polymer imprinted with L-methionine (PL-Met) was electrodeposited on a carbon paste electrode (CPE)/Ni-Bio-MOF and used as a new electrochemical sensor for highly selective and sensitive detection of DCF in biological and pharmaceutical samples. The Ni-Bio-MOF/MIP-PL-Met nanocomposite was characterized using the following techniques: FT-IR, FE-SEM, TEM, HR-TEM, XRD, XPS, and EDX. The electrochemical properties and performance of the sensor for the electrooxidation of DCF were assessed using CV, DPV, and EIS techniques. The electrochemical behavior of CPE/Ni-Bio-MOF/MIP-PL-Met and non-imprinted polymer (NIP) with an imprinting factor of 6.64 was investigated, and the influencing parameters in DCF measurement were optimized by cyclic voltammetry (CV). This modified sensor showed three dynamic ranges at 1.0–500.0 pM, 1.0–1000.0 nM, and 1.0–1000.0 μM of DCF with a limited detection (LOD) of 0.17 pM, sensitivity of 2015.5 μA μM−1 cm−2, relative standard deviation (RSD) of 3.3%, and reproducibility of 96.2%. Real samples of healthy human blood serum and DCF tablets were used to evaluate the practical application of the CPE/Ni-Bio-MOF/MIP-PL-Met electrochemical sensor. This method is simple, low-cost, with good limited detection, high sensitivity, and selectivity. The interactions of PL-Met with DCF were studied at the B3LYP/6-311++G(d,p) level of theory in both gaseous and aqueous phases. Additionally, the computational methodology investigated the thermodynamic stability of the proposed configurations and the role of hydrogen bonds in this system.
1 Introduction
Diclofenac, 2(2-((2,6-dichlophenyle) amino) phenyl) acetic acid, is a nonsteroidal anti-inflammatory medication (NSAID) with analgesic and antipyretic effects.1–3 It is often used to treat glaucoma, non-articular rheumatism, osteoarthritis, rheumatoid arthritis, and diseases involving muscular discomfort in sports injuries.4–6 In all patients, including those who take MD medications for blood pressure,4 DCF is often acknowledged as the most widely prescribed painkiller globally.1 The use of a normal therapeutic dose (50–150 mg) of DCF does not cause toxicity to humans,2 but the wide medicinal use of DCF as the first choice in the treatment leads to the incidence of hepatic injury and then causes toxicity.5 Prolonged and excessive usage of DCF may lead to myocardial infarction,3 aplastic anemia, gastrointestinal problems, agranulocytosis, and impaired renal function.2 In addition to the mentioned cases, it creates negative effects on the ecosystem's health and sustainability.7 As a result, it is crucial to determine DCF in medicines3 and biological samples.6 Therefore, it is crucial to develop a procedure that is uncomplicated, fast, specific, cost-effective, and dependable.4,5Several analytical methods, including gas chromatography-mass spectrometry (GC-MS),8 gas chromatography coupled with tandem mass spectrometry (GC-QqQ-MS/MS),9 high-performance liquid chromatography (HPLC),10 high-performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS/MS),11 alkaline-induced salting-out homogeneous liquid–liquid extraction (AI-SHLLE),12 mass spectrometry,13 automatic flow,14 calorimetry,15 and electrochemical,1–6,16 have been utilized for the determination of DCF. Although the mentioned methods have a high detection limit, electrochemical methods are notable for their unique features, including attractive analytical features, portability, simplicity,1 inexpensive,1,3,6 and the possibility of reducing the size.16
The selection of an appropriate working electrode is significantly important to increase selectivity and optimal sensitivity in electrochemical methods.16 One of the types of working electrodes used in electrochemical biosensors is CPE, which is prepared by mixing graphite powder and oil in an optimal weight ratio.16 These electrodes possess characteristics such as excellent conductivity, minimal background current within a broad potential range, ease of surface reproducibility, the ability to incorporate different modifiers, low potential,16 chemical stability, consistent analytical response, low resistance to electrical flow, and easy preparation.16 Despite these advantages, a lack of sufficient selectivity is their main weakness, which prevents their practical applications.16 Therefore, modifying the electrode surface can compensate for this weakness. Modifying the electrode surface may reduce overvoltage, increase the analyte's surface affinity, increase the analyte's effective preconcentration rate by mass transfer, and/or reduce interference.16 To enhance sensing performance in CPE, the most common modifiers include metal oxides,17–19 metal nanoparticles,20 polymeric materials,21,22 Metal–organic framework (MOF),23 biomolecules,24 and carbon-based materials25 in recent studies.
Among the mentioned modifiers, molecular imprinting polymers (MIPs) are a suitable choice for pharmaceutical sensing, drug analysis,22,26 and drug delivery.27 MIPs are a special class of polymeric materials that incorporate pre-engineered molecular recognition properties.28 In other words, those are formed by monomers and cross-linkers of organic polymeric materials, together with template molecules, to create a copolymer. The template molecules are extracted from the polymer using a solvent appropriate for elution. Consequently, the MIP retained distinct and easily identifiable three-dimensional (3D) cavity structures, known as binding sites, that were well-matched in size, shape, and functional groups.23,29 MIPs are stable synthetic polymers with particular sites30 that provide greater access to the sensing target molecules, even a macromolecule.21 MIPs, which are known as plastic antibodies and/or plastic enzymes, have emerged as efficient substances for creating biomimetic electrochemical sensors.31 In the past two decades, many research groups with different interests have been attracted to the application of MIPs32 and outstanding selective properties.33 The advantages of MIPs include cost-effective, low energy consumption and environmental effects,34 high selectivity, as signal converters,35 high affinity for the target molecule,23 more reusability and stability compared to biological receptors,35 easy preparation,26 high physical robustness,28 and long shelf life.31
The limitation of sensitivity, non-specific binding and indirect signal generation, synthesis and integration challenges, cost, and scalability barriers, and stability of MIPs in electrochemical biosensors is a challenge caused by the biological analytes' denaturation and dissociative affinity. In addition to these limitations, MIPs suffer from the following disadvantages: poor capacity, poor performance regarding site accessibility, removal of incomplete templates, and reorganization. As a result, it reduces the performance of the sensors/biosensors.36 Electrochemical biosensors can compete with other materials such as metal–organic frameworks (MOFs). Researchers have suggested using MOFs to produce materials with high sensitivity and selectivity for detection and binding.31 MOFs have a lot of imprinted sites that are spread out evenly, which can help MIPs work better because they have a lot of specific surface area.33 It would be good to use MOFs to help make MIPs that can hold monomers and templates in place.37 The addition of a MOF in the MIP structure is a novel and rapidly growing area of study. This advancement has yielded encouraging outcomes and has paved the way for the creation of a new breed of sensors.38,39 MOFs can play a role as a core in MIP patterns or be involved in molecular patterns.40 In recent years, there have been several investigations undertaken in the area of MOF-MIP sensors/biosensors.37,40
MOFs are coordination compounds that can be used for many different things. They are made by the combination of metal ions with organic linkers.41 MOFs are being used in chemical sensing applications because of their distinctive characteristics, including extremely large specific surface area, organized porosity, the ability to control the arrangement of active sites, and highly adjustable structures. Additionally, MOFs exhibit excellent thermal and chemical stability, as well as exposed active sites.40 Due to the mentioned properties, MOFs have become competitors for conventional porous materials, like activated carbons and zeolites.41 Despite all these significant advances, practical efforts are still required, particularly in developing MOF materials for electrochemical technologies. MOF materials alone have poor conductivity39 and low mechanical strength,42 not enough for electrochemical applications.39 The use of MOFs as electrochemical sensors is now in its early stages and poses a challenge in the domains of biological and chemical detection.43 The great performance of MOF precursors, including their large surface area, well-defined configuration, and adjustable pore size, is retained by the produced MOF derivatives. However, they also contribute other qualities, such as high electrical conductivity and stability.44 Hence, MOFs can be combined with specific materials such as metal oxides, quantum dots, carbon materials, polymers, polyoxometalates, biomolecules, and enzymes, and new MOFs are prepared with functional diversity in electrochemical sensors.
Since the first discovery of MOFs over two decades ago, more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 have been identified. Biological metal–organic frameworks (Bio-MOFs) are a recently discovered subgroup of MOFs that integrates the fields of MOF chemistry and bioscience. The definition of Bio-MOFs is currently ambiguous. For Bio-MOFs, which are MOFs utilized in biological or medical applications and have high porosity, it is necessary to have at least one biomolecule acting as an organic ligand. Using Bio-MOFs for biological applications must comprehensively characterize toxicity, stability, efficiency, particle size, and morphology. Bio-MOFs have many characteristics that distinguish them from traditional MOF materials. These include chirality, molecular recognition, low toxicity, biomolecular catalytic performance, and strong biocompatibility, in addition to their structural variety and intrinsic porosity. Despite being a relatively new area of study, Bio-MOFs have garnered significant interest due to their elegant structure, diverse supramolecular chemistry, and distinctive biomimetic features.45
000 have been identified. Biological metal–organic frameworks (Bio-MOFs) are a recently discovered subgroup of MOFs that integrates the fields of MOF chemistry and bioscience. The definition of Bio-MOFs is currently ambiguous. For Bio-MOFs, which are MOFs utilized in biological or medical applications and have high porosity, it is necessary to have at least one biomolecule acting as an organic ligand. Using Bio-MOFs for biological applications must comprehensively characterize toxicity, stability, efficiency, particle size, and morphology. Bio-MOFs have many characteristics that distinguish them from traditional MOF materials. These include chirality, molecular recognition, low toxicity, biomolecular catalytic performance, and strong biocompatibility, in addition to their structural variety and intrinsic porosity. Despite being a relatively new area of study, Bio-MOFs have garnered significant interest due to their elegant structure, diverse supramolecular chemistry, and distinctive biomimetic features.45
This work included the preparation of Ni-Bio-MOF by a hydrothermal method, utilizing BDC as a linker and Asparagine (Asn) as a modifier. Asn was selected for its ability to act as a modulator, stabilizer, or capping agent that can modify the surface of Ni-MOF.46 Furthermore, Asn may partly substitute ligands via its interaction with nickel ions, resulting in surface defects characterized by unsaturated metal centers. These defects significantly enhance the electrocatalytic activity of the materials. For example, Li et al. documented the modification of MIL-88A(Fe) with citric acid, leading to the formation of uniform particles and an improvement in the catalytic activity of MIL-88A(Fe).47 Dheyab et al. also demonstrated an increase in the catalytic activity of Fe3O4 after citric acid treatment.48
In this work, for the first time, the amino acid asparagine was used as a modifier in the synthesis of Ni-Bio-MOFs, and its effect on the chemical composition and crystal structure of the synthesized MOF, as well as its electrocatalytic activity, was investigated using XRD patterns and electrochemical methods. Then, the CPE was modified with a nanocomposite of Ni-Bio-MOF and MIP-PL-Met and used to detect DCF at very low concentrations in biological samples. The fabricated modified electrode was identified by FE-SEM, HR-TEM, XRD, XPS, FT-IR, electrochemical impedance spectroscopy (EIS), and CV. The CPE/Ni-Bio-MOF/MIP-PL-Met sensor has excellent sensitivity and specificity in detecting DCF and has been successfully employed for detecting DCF in biological samples, including blood serum and tablets. The steps for preparing the Ni-Bio-MOF/MIP-PL-Met and its electrochemical response to DCF are summarized in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the preparation of CPE/Ni-Bio-MOF/MIP-PL-Met for selective determination of DCF. | ||
2 Experimental
2.1 The supporting information provides the names of the materials and equipment used
2.2 Preparation of Ni-Bio-MOF
The synthesis of Ni-Bio-MOF is similar to MOF, except that in addition to BDC, the linker Asparagine (Asn) (with different Asn amounts 1.0, 3.0, 5.0, 6.0, 7.0 mg) has been used as a biomolecule modifier in the fabrication of MOF.2.3 Preparation of the CPE/Ni-Bio-MOF/MIP-PL-Met and CPE/Ni-Bio-MOF/NIP-PL-Met
The CPE unmodified was manufactured by mixing the fine graphite powder and paraffin oil (70:30% w/w) to form a homogeneous paste. The paste was poured and packed in a polyethylene syringe (diameter 3.5 mm) with a copper wire. The surface of the electrode becomes smooth by dragging it on clean paper.50 The CPE modified with Ni-Bio-MOF was prepared like the unmodified CPE method. The difference is that in addition to the fine graphite powder and paraffin oil, solid green Ni-Bio-MOF powder (3.0 mg) is also added.
For fabrication of the CPE/Ni-Bio-MOF/NIP-PL-Met sensor, firstly CPE/Ni-Bio-MOF electrode was cleaned by polishing on a piece of clean paper. Then, the electropolymerization of L-Met (0.25 mM) in 0.1 M PBS (pH 7.0) was performed by using the cyclic voltammetry (CV) method in the potential range of −0.6 to 2.0 V on CPE/Ni-Bio-MOF with 10 successive cycles. To form imprinted PL-Met film on CPE/Ni-Bio-MOF, the electropolymerization of L-Met was performed in the presence of 0.25 mM of L-Met and 0.083 mM DCF in 0.1 M PBS (pH 7).49 The CPE/Ni-Bio-MOF/MIP-PL-Met was placed on ethanol/acetonitrile/NaOH 0.2 M (1:1:1 V/V) solution for 5 min with a stirrer speed to DCF entrapped molecules were removed.
Nitrogen (3.04) in the L-methionine structure is more electronegative than sulfur (2.58). So, this suggests that nitrogen is more reactive. During the electropolymerization of L-Met within the voltage range of −0.6 to 2.0 V, the free radical of L-Met is first formed by removing a hydrogen radical. Then this formed free radical was attached to the surface of the Ni-Bio-MOF/CPE. After that, the resultant product combines with another L-Met molecule, and by the elimination of a water molecule, a conductive polymer layer is generated on the electrode surface.51 During the electropolymerization process, the current intensity increased with an increasing number of cycles of CV. Consequently, this led to the development of a durable and compact polymer film on the surface of the electrode (Fig. S1a†). The electropolymerization mechanism of L-Met is shown in Scheme S1†.52
To form imprinted PL-Met film on Ni-Bio-MOF/CPE by the electropolymerization of L-Met in the presence of DCF (Fig. S1b†), one anodic irreversible peak appeared around 0.7 V. As the number of cycles increases, the current intensity of this peak rises, indicating the production of DCF-imprinted PL-Met layers.52–54 During electropolymerization, DCF template molecules migrated toward the surface of Ni-Bio-MOF/CPE and were embedded in the polymer matrix as a consequence of non-covalent interactions such as hydrogen bonding and electrostatic interactions. Furthermore, because of the electrostatic interactions between the negatively charged DCF molecules and the positively charged PL-Met backbone, these molecules could become trapped in the PL-Met film.55 Next, DFT calculations show that hydrogen bonding plays an important role in the interactions between DCF molecules and PL-Met. Removal of template molecules results in the formation of many imprinting sites that specifically recognize DCF.
2.4 Real sample preparation
Tablet: DCF tablets (100 mg), Alborz Pharmaceutical Company, were purchased from a pharmacy. One tablet, with a weight of 284 mg, has been pounded and rubbed in a mortar. The amount of the powder dissolved in PBS of 0.1 M (pH 7.0) was sonicated for 5 min and filtered with a no. 41 Whatman filter paper. In the end, this solution reached 100 ml volume in the volumetric flask.Human serum: The healthy human blood serum sample was obtained from Bahar Medical Diagnostic Laboratory in Tehran (the pathology laboratory Bahar in Tehran) and kept in the refrigerator before use. 100 μl of sample in the volumetric flask of 10 ml was diluted with PBS of 0.1 M (pH 7.0).
Detection of DCF in complex matrices of tablet and human blood serum samples was made possible by using DPV and UV-Vis methods in conjunction with the conventional addition method for quantitative analysis under ideal conditions.
The nature of hydrogen bonds was studied by the quantum theory of atoms in molecules (QTAIM) and the AIM2000 program.60 The hydrogen bond energy has been estimated by the EHB = 0.5V(r) formula,61 which V(r) represents the interatomic interaction potential. The self-consistent reaction field (SCRF) and polarizable continuum model (PCM)62 were employed for the calculations in water with ε = 78.39. The Mercury63 and Avogadro64 programs were used to draw the molecular graphics and molecular electrostatic potential (MESP) plots. The interaction energies (Eint)65 were evaluated by eqn (1).
| Eint = Ecomplex − (EL-Met + EDCF) | (1) |
3 Results and discussion
3.1 Characterization
FT-IR spectra illustrate the bonding of organic ligands (BDC and Asn) and metal ions, as shown in Fig. 1a. A single band at 1678 cm−1 showed adsorption of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of COOH in BDC. In Asn, vibrations of symmetric stretching and asymmetric stretching occur between 3500-3300 cm−1 and 3420-3340 cm−1, respectively. In two groups of NH2, symmetric and asymmetric stretching vibration of –NH2 is 3420-3340 cm−1 and 3500-3420 cm−1, respectively. The scissoring mode lies in NH2 is between 1650-1529 cm−1.67 In the current study, stretching vibration is in 3269 cm−1, and scissoring mode lies in 1550 cm−1. The vibration analysis of CO in carboxylic acids is expected in the region 1740-1660 cm−1.67 In our study, CO stretching vibrations are assigned at 1643 cm−1. During the formation of Ni-MOF, the BDC will dissolve, so the COOH converts to COO− for forming Ni–O. A double band of asymmetric (1573 cm−1) and symmetric (1379 cm−1) stretching peaks of -COO− is shown. The intensity of the bands has increased due to conjugation with nickel metal or creating a hydrogenated bond in Ni-MOF and Ni-Bio-MOF. The peak of the Ni–O band was observed at 661 cm−1. Fig. 1b illustrates NIP and MIP composed of L-Met (monomer) and DCF (analyte). The spectrum of L-Met shows a broad peak at 2900-3300 cm−1 due to C–H, NH, and OH stretching vibration and C–S stretching vibration at 553 cm−1. These peaks are dedicated to the characteristic absorption of L-Met. During the polymerization process of L-Met, the peaks in the fingerprint area exhibited increased width, whereas the peaks associated with N–H bonds showed decreased intensity. As a result, the peak at 2096 cm−1 disappeared completely, which confirms the production of PL-Met.67 After the template molecules were taken out, the FT-IR spectra of NIP-PL-Met and MIP-PL-Met were the same. Stretching bands of the C–N, C–O, and CO were seen at wavenumbers of 1251 cm−1, 1380 cm−1, and 1741 cm−1 for NIP-PL-Met and MIP-PL-Met.68 The MIP-PL-Met spectrum does not provide clear information about the binding sites of DCF and L-Met. Therefore, hydrogen bonds can be proposed as the main interactions in this MIP pattern.
O bond of COOH in BDC. In Asn, vibrations of symmetric stretching and asymmetric stretching occur between 3500-3300 cm−1 and 3420-3340 cm−1, respectively. In two groups of NH2, symmetric and asymmetric stretching vibration of –NH2 is 3420-3340 cm−1 and 3500-3420 cm−1, respectively. The scissoring mode lies in NH2 is between 1650-1529 cm−1.67 In the current study, stretching vibration is in 3269 cm−1, and scissoring mode lies in 1550 cm−1. The vibration analysis of CO in carboxylic acids is expected in the region 1740-1660 cm−1.67 In our study, CO stretching vibrations are assigned at 1643 cm−1. During the formation of Ni-MOF, the BDC will dissolve, so the COOH converts to COO− for forming Ni–O. A double band of asymmetric (1573 cm−1) and symmetric (1379 cm−1) stretching peaks of -COO− is shown. The intensity of the bands has increased due to conjugation with nickel metal or creating a hydrogenated bond in Ni-MOF and Ni-Bio-MOF. The peak of the Ni–O band was observed at 661 cm−1. Fig. 1b illustrates NIP and MIP composed of L-Met (monomer) and DCF (analyte). The spectrum of L-Met shows a broad peak at 2900-3300 cm−1 due to C–H, NH, and OH stretching vibration and C–S stretching vibration at 553 cm−1. These peaks are dedicated to the characteristic absorption of L-Met. During the polymerization process of L-Met, the peaks in the fingerprint area exhibited increased width, whereas the peaks associated with N–H bonds showed decreased intensity. As a result, the peak at 2096 cm−1 disappeared completely, which confirms the production of PL-Met.67 After the template molecules were taken out, the FT-IR spectra of NIP-PL-Met and MIP-PL-Met were the same. Stretching bands of the C–N, C–O, and CO were seen at wavenumbers of 1251 cm−1, 1380 cm−1, and 1741 cm−1 for NIP-PL-Met and MIP-PL-Met.68 The MIP-PL-Met spectrum does not provide clear information about the binding sites of DCF and L-Met. Therefore, hydrogen bonds can be proposed as the main interactions in this MIP pattern.
 | ||
| Fig. 1 FT-IR of (a) Asn, BDC, Ni-MOF, Ni-Bio-MOF (b) L-Met, DCF, Ni-Bio-MOF/NIP-PL-Met, Ni-Bio-MOF/MIP-PL-Met (before and after removal of DCF). | ||
The field emission scanning electron microscope (FE-SEM) images illustrate the morphology of Ni-MOF and Ni-Bio-MOF in two magnifications. According to Fig. 2a and b, the Ni-MOF indicates a layer-cuboid structure with 16–21 nm in width and 4.12 to 4.99 μm in length. On the contrary, for Ni-Bio-MOF, Fig. 2c and d show a regular lamellar structure after the use of Asn, with a significant increase in thickness and length in the range of 94-61 nm and 9.35–9.73 μm, respectively. In the following, the FE-SEM images in Fig. 2e–h investigated the morphology of the PL-Met in the absence of template (NIP-PL-Met) (Fig. 2e), presence of DCF template (MIP-PL-Met before) (Fig. 2f), and leaving of DCF template (MIP-PL-Met after) (Fig. 2g and h). By adding the DCF template along L-Met, the surface of MIP gets wider and it shows NIP-PL-Met to MIP-PL-Met surface changes. After leaving the template, holes have been created in the polymer.
 | ||
| Fig. 2 FE-SEM images with different magnifications of (a and b) Ni-MOF, (c and d) Ni-Bio-MOF, (e) Ni-Bio-MOF/NIP-PL-Met, (f) Ni-Bio-MOF/MIP-PL-Met (before removal of DCF), (g and h) Ni-Bio-MOF/MIP-PL-Met (after removal of DCF). | ||
Fig. 3a and b represent the TEM images of Ni-Bio-MOF with different magnifications. According to this, the lamellar structure can be observed and confirms several thin sheets, and this is in good agreement with the FE-SEM images. The HR-TEM of Ni-Bio-MOF (Fig. 3c) demonstrated the presence of lattice fringes, indicating a crystalline structure. Also, this was confirmed by the corresponding selected area electron diffraction (SAED) pattern, which shows the expected structure and the absence of additional phases and impurities (Fig. 3d).
 | ||
| Fig. 3 (a and b) TEM images with different magnifications of Ni-Bio-MOF, (c) HR-TEM image of Ni-Bio-MOF, and (d) SAED pattern of Ni-Bio-MOF. | ||
Fig. 4a shows the XRD pattern of Ni-MOF and Ni-Bio-MOF. The pattern of major diffraction peaks of Ni-MOF was compared with synthesized Ni-MOF in the study of S. Gao, et al.49 and confirmed. The main diffraction peaks of the Ni-MOF XRD pattern were according to that Ni-Bio-MOF with a slight shift in position (due to lattice parameter variations and residual strain), intensity (due to the atomic arrangement, preferred orientation, and phase composition), and width (due to crystallite size, microstrain, defects, and disorder).68,69 The diffractogram peaks of Ni-MOF were appeared at 2θ = 11.75°, 12.19°, 15.50°, 18.39°, 23.74°, 28.09°, 29.09° and diffractogram peaks of Ni-Bio-MOF were observed at 2θ = 11.69°, 12.04°, 15.50°, 18.29°, 23.74°, 28.07°, 29.08°. According to Bragg's law,69 a little decrease in the angle of the Ni-Bio-MOF compared to the Ni-MOF suggests that the interlayer distance has increased. The microstrain values, calculated using the Williamson–Hall (W–H) method (Fig. S2†), for Ni-MOF and Ni-Bio-MOF were 0.002 and 0.004, respectively. The 2.0-fold increase in microstrain for Ni-Bio-MOF compared to Ni-MOF indicates a more defective structure in Ni-Bio-MOF. This can be attributed to the structural defects induced by the addition of Asn into the Ni-MOF.
 | ||
| Fig. 4 (a) XRD patterns of the Ni-MOF and Ni-Bio-MOF; (b) nitrogen adsorption–desorption isotherm and corresponding pore size distribution (inset) of the Ni-MOF and Ni-Bio-MOF. | ||
Nitrogen adsorption–desorption isotherms and Barrett–Joyner–Halenda (BJH) pore diameter distribution were utilized to evaluate the surface area and the porosity of Ni-MOF and Ni-Bio-MOF (Fig. 4b). The isotherms of Ni-MOF and Ni-Bio-MOF exhibit the characteristics typically associated with mesoporous materials as they display a type IV curve with H3-type hysteresis loops according to the IUPAC classification. The Brunauer–Emmett–Teller (BET) surface area of Ni-MOF and Ni-Bio-MOF were 8.603 and 16.487 m2 g−1, respectively. The calculated pore volume of Ni-MOF and Ni-Bio-MOF was 0.0247 and 0.0302 cm3 g−1, whereas the pore diameter was 11.493 and 7.322 nm, respectively. The BJH pore size distribution plots (insets of Fig. 4b) also confirm large amounts of mesopores with sizes smaller than 10 nm for the Ni-MOF and Ni-Bio-MOF. The properties of large specific surface area and high porosity of Ni-Bio-MOF increase the effective mass transfer and maximize the exposure of active sites.70
XPS was conducted to determine the elemental composition and oxidation states of the Ni-Bio-MOF nanocomposite. As shown in Fig. 5a, characteristic peaks for Ni, C, O, and N elements were found in the survey spectrum. HR-XPS of Ni 2p spectra both show 2p1/2 and 2p3/2 components due to spin–orbit coupling. In the high-resolution XPS spectrum of Ni 2p (Fig. 5b), peaks at 856.54 eV and 873.8 eV can be assigned to the Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively. The corresponding satellite peak is located at 862.62 eV. The presence of Ni2+ is derived from the connection between Ni2+ and BDC ligands.71 Furthermore, the presence of Ni–OH is indicated by the peaks seen at 859.86 eV (corresponding to Ni3+ 2p3/2) and 875.66 eV (corresponding to Ni3+ 2p1/2), as well as a satellite peak at 880.15 eV.72 Fig. 5c depicts the high-resolution XPS (HR-XPS) spectrum of C 1s of Ni-Bio-MOF, which can be fitted into four characteristic peaks at 284.59 eV, 285.38 eV, 286.39 eV, and 286.83 eV, belonging to the CC/C–C bonds, C–N/C–O bond, CN/CO, and the carboxyl (O–CO) groups in BDC ligands, respectively.60,73 The high-resolution XPS (HR-XPS) spectrum of O 1s (Fig. 5d) is deconvoluted into three peaks at binding energies 531.27 eV, 532.42 eV, and 533.65 eV, which can be attributed to the Ni–O bond, C–O, and the carboxylate moiety of the organic ligand, respectively. Furthermore, the HR-XPS of the N 1s spectrum shows two peaks at 400.23 eV and 402.64 eV, which can be assigned to the C–H/N–H and N–(CO), respectively (Fig. 5e).71
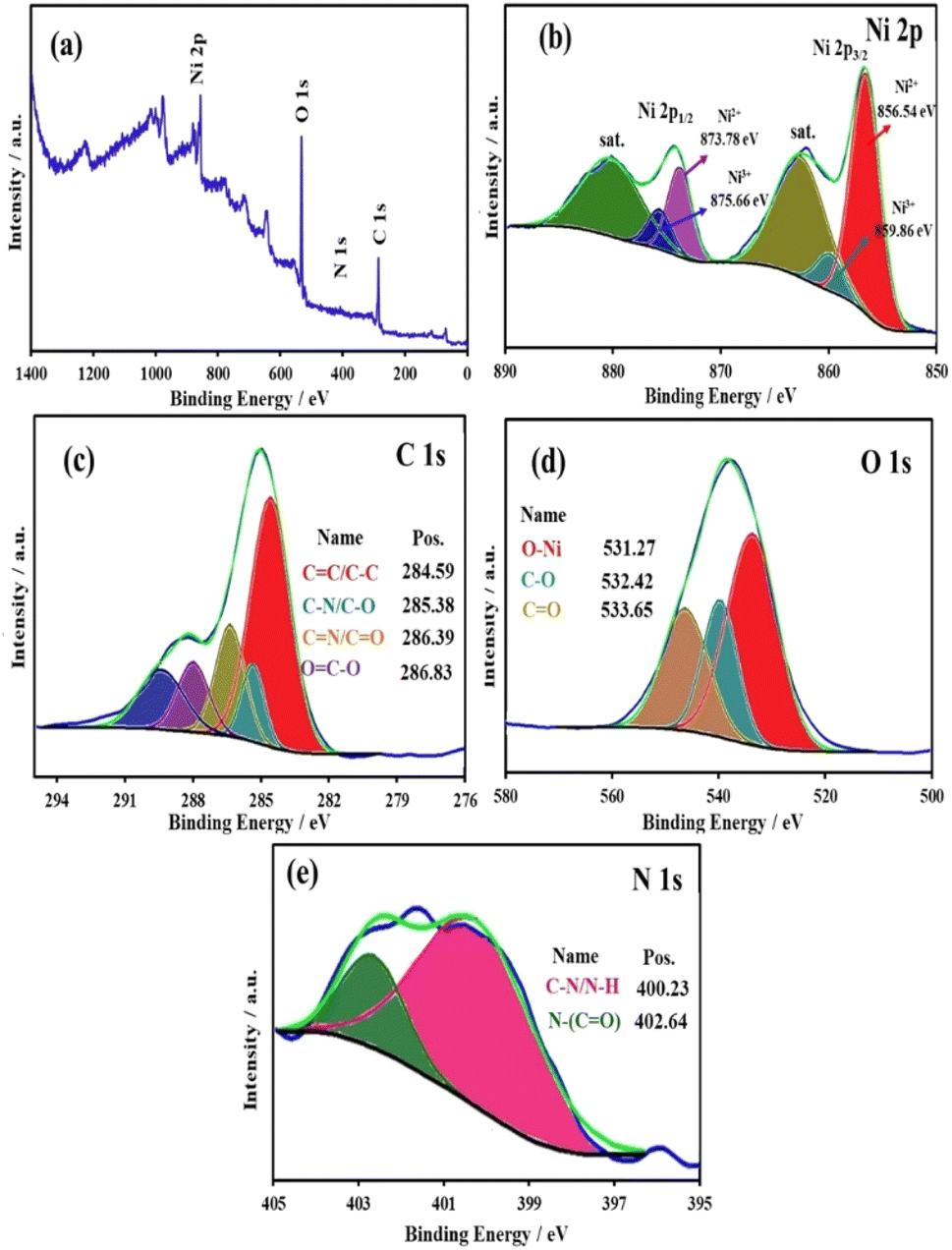 | ||
| Fig. 5 (a) XPS survey spectrum of the Ni-Bio-MOF, (b–e) high-resolution spectra of Ni 2p, C 1s, O 1s, and N 1s, respectively. | ||
3.2 Electrochemical characterization of different modified electrodes
By comparing the electrochemical behavior of modified electrodes with these materials using CV and EIS methods in a 0.1 M KCl solution containing 5.0 mM [Fe(CN)6]3−/4−, the produced materials' performance was assessed. The CV curves of CPE, CPE/Ni-MOF, CPE/Ni-Bio-MOF, CPE/Ni-Bio-MOF/NIP-PL-Met, and CPE/Ni-Bio-MOF/MIP-PL-Met (before and after removal of the template) are shown in Fig. 6a. The CV curve of bare CPE shows a pair of redox peaks. The peak current has increased significantly when the Ni-MOF and Ni-Bio-MOF are used as a modifier. These compositions with crystallite structures are specific patterns of organic ligands and metal, and pore compounds. | ||
| Fig. 6 (a) CVs at a scan rate of 50 mV s−1, (b) Nyquist plots from 105 Hz to 10−1 Hz frequencies in 0.1 M KCl solution containing 1.0 mM K3Fe(CN)6, and (c) CVs of the 1 mM solutions of the DCF in 0.1 M PBS (pH 7.0) on bare CPE, CPE/Ni-MOF, CPE/Ni-Bio-MOF, CPE/Ni-Bio-MOF/NIP-PL-Met, CPE/Ni-Bio-MOF/MIP-PL-Met (before and after removal of DCF). | ||
The two-dimensional structure of Ni-MOF and Ni-Bio-MOF has promoted diffusion speed and accumulation of ions across the electrode, shortening the diffusion distance of ions during electrochemical reactions.74 Besides that, the porosity of these compositions can facilitate the transfer of ions and electrons at the electrode/electrolyte interface.75 As previously explained, Asn can interact with nickel ions, partially substituting the ligand by Asn.47 This partial substitution creates defects on the MOF surface, which increases the metal center terminals and subsequently enhances its electrocatalytic activity. Furthermore, Asn can function as a capping agent that optimizes the crystallite structure of Ni-MOF, thereby enhancing the specific surface area and porosity, or, in other words, the availability of active sites.46
As seen in Fig. 6a, the modified electrode with Ni-Bio-MOF shows a higher peak current compared to CPE/Ni-MOF. When L-Met is electropolymerized on CPE/Ni-Bio-MOF (CPE/Ni-Bio-MOF/NIP-PL-Met), the peak current increased due to the high electrical conductivity and large surface area of the PL-Met. The oxidation of Fe (CN)63−/4− on the CPE/Ni-Bio-MOF/NIP-PL-Met electrode occurred at a lower potential (0.24 V) than that observed with the CPE/Ni-Bio-MOF electrode (0.26 V). After the inclusion of DCF at CPE/Ni-Bio-MOF/MIP-PL-Met, hindered electron conduction75 caused the peak current has decrease significantly. After removing the DCF from CPE/Ni-Bio-MOF/MIP-PL-Met, the peak current increases clearly, which is due to the selective recognition and capture of the DCF in the imprinted cavities of the DCF pattern in the MIP-PL-Met film. Furthermore, the peak potential difference (ΔEp) of bare CPE and MIP-PL-Met electrodes is shown as 0.154 V and 0.120 V, respectively. Such results were obtained due to the high conductivity, the larger specific surface area, and the catalytic effect of PL-Met and Ni-Bio-MOF.32
Nyquist diagrams of different modified electrodes are shown in Fig. 6b. The frequency of EIS analysis was set from 10 kHz to 0.1 Hz, with an amplitude of 0.2 V. The Nyquist diagram typically has a semicircular segment at high frequencies and a linear segment at low frequencies, corresponding to the electron transfer-limited process and the diffusion-controlled process, respectively. The obtained experimental EIS curves were fitted with the standard Randle's equivalent circuit, as shown in Fig. S3.† The equivalent circuit consists of solution resistance, charge transfer resistance, Warburg constant, and double-layer capacitance are expressed with Rs, Rct, Zw, and C, respectively.75 Rct in interaction electrolyte and electrode surface was calculated for CPE, CPE/Ni-MOF, CPE/Ni-Bio-MOF, CPE/Ni-Bio-MOF/NIP-PL-Met, CPE/Ni-Bio-MOF/MIP-PL-Met before remove of DCF, and CPE/Ni-Bio-MOF/MIP-PL-Met after remove of DCF, that were 8960, 6560, 4980, 4070, 8220, 861 Ω, respectively (Fig. S3(a–f)†). The Rct value of CPE/Ni-Bio-MOF/MIP-PL-Met was lower than that of CPE, CPE/Ni-MOF, CPE/Ni-Bio-MOF, and CPE/Ni-Bio-MOF/NIP, which suggested CPE/Ni-Bio-MOF/MIP-PL-Met has high electrocatalytic activity and fast charge transport kinetics. The results of EIS correspond with the CV results. Therefore, this modified electrode was applied as an electrochemical sensor for the determination of DCF.
The issue was examined by calculating the active surface area of the CP and MIP electrodes using the Randles–Sevcik equation.75
| Ip = 2.69 × 105n3/2AeffD1/2ν1/2C | (2) |
To assess the active surface area of the modified electrode, the cyclic voltammograms at different scan rates (10–100 mV s−1) were recorded in 0.1 M KC l solution containing 1.0 mM [Fe (CN)6]4−/[Fe (CN)6]3−, (Fig. S4(a–e)†). The active surface area of the electrodes was determined by plotting the change of “Ip” as a function of “υ1/2” (insets in Fig. S4†). The electrochemically active surface areas of CPE, CPE/Ni-MOF, CPE/Ni-Bio-MOF, CPE/Ni-Bio-MOF/NIP, and Ni-Bio-MOF/MIP-PL-Met after removal of DCF were found to be 0.0264, 0.0325, 0.0476, 0.0491, and 0.0511 cm−2, respectively. The active surface of the final modified electrode (Ni-Bio-MOF/MIP-PL-Met after) has increased by 1.94 times compared to the unmodified electrode (CPE). The findings revealed that the MIP has a function in increasing the active surface area and the Ip values, and it can also have a catalytic impact that may reduce the values of ΔEp.75
3.3 Electrochemical behavior of DCF at modified electrodes
DCF is an electrochemically active substance, and the electrochemical behavior of DCF (1.0 mM in 0.1 PBS at pH 7.0) was investigated on different modified electrodes in the −0.1 to 1.0 V potential window by CV in Fig. 6c. DCF is irreversibly oxidized at the CPE/Ni-Bio-MOF/MIP-PL-Met electrode and shows an anodic peak current at a potential of 0.6 V. A proposed mechanism for forming the irreversible anodic peak of DCF is shown in Scheme S2.† According to the study of Goyal,5 this peak is related to the electrochemical hydroxylation of DCF. In the first stage, the electron is released, and DCF becomes a nitrogen radical cation. In the second step, this cation is rearranged and loses a proton, leading to a carbon radical in the axis of the amine group. Then this radical loses an electron and is transformed into a carbocation that reacts with water in the last step and forms a DCF hydroxide.5The peak current of DCF on the bare CPE was lower than that of CPE/Ni-MOF. Following surface modification of Ni-Bio-MOF on CPE, the peak current further rises. This result may be explained by the electrocatalytic activity of the Ni-Bio-MOF for the electrochemical oxidation of DCF. Additionally, Ni-Bio-MOF has several attachment places because of the porous Ni-Bio-MOF structure. The oxidation peak current was significantly increased after modification of CPE with Ni-Bio-MOF/NIP-PL-Met, due to the higher electrical conductivity of the polymeric layer. The CPE/Ni-Bio-MOF/MIP-PL-MET exhibited a small signal, which may be attributed to physical and chemical obstruction caused by template molecules occupying the recognition sites and limiting the diffusion of DCF molecules and electron transfer. After removing template molecules, specific binding cavities are exposed, diffusion pathways are cleared, charge transfer resistance decreases, and functional groups become accessible for interactions. These changes collectively enhance electron transfer efficiency and increase peak current (CPE/Ni-Bio-MOF/MIP-PL-Met after).
3.4 Effect of scan rate
An investigation was conducted to examine the adsorption or diffusion of the electrode process using the cyclic voltammetry method. The study focused on the range of 10 to 100 mV s−1 and used CPE/Ni-Bio-MOF/MIP-PL-Met in a solution of 0.1 mM PBS (pH 7.0) containing 1.0 mM DCF. According to Fig. 7a, with increasing scan rate, Ipa of the DCF increases, and Epa shifts towards more positive values. The relationship between the oxidation peak currents (anodic peak current, Ipa) and the scan rate (υ), square root of scan rate (υ1/2), and log scanning rate (logυ) were investigated in Fig. 7. The plots of Ipa versus υ (Fig. 7b) and υ1/2 (Fig. 7c) is provided a prediction of the mechanism of redox reaction in the electrode. The linear relationships were obtained as follows:| Ipa = 0.59υ + 11.75 (R2 = 0.9873) | (3) |
| Ipa = 8.05υ1/2 − 12.85 (R2 = 0.9981) | (4) |
|
logIpa = 0.69logυ + 0.46 (R2 = 0.9995)
| (5) |
 | ||
| Fig. 7 (a) CVs of 1 mM solutions of the DCF at CPE/Ni-Bio-MOF/MIP-Met surface in 0.1 M PBS solution (pH 7.0) at various scan rates. Insets: (b) plot of Ip vs. υ, (c) plot of Ip vs. υ1/2, and (d) plot of logI vs. logυ obtained from the cyclic voltammograms; (e) cyclic voltammograms of 1 mM solutions of the DCF at the surface in 0.1 M PBS solution at different pH values (6.0 to 8.5) at a scan rate of 50 mV s−1 (Inset: dependence of peak potential and peak current on pH). Error bars indicate the standard deviations of three repeated measurements. | ||
Eqn (2) and (3) show a linear dependence of the anodic peak current with scan rate (υ) and the square root of scan rate (υ1/2), which considers control of redox reaction processes at the electrode surface by an adsorption and/or diffusion-controlled process, respectively.77 The values of R2 have suggested a diffusion-controlled process for the sensor. To confirm this proposal, the plot of logIpc versus logν has been investigated. The slope theoretical value of this plot is 0.5 for the pure diffusion-controlled process.78 This plot (Fig. 7d) was associated with a slope of 0.68 for the obtained data, indicating a mixed diffusion/adsorption process.79 It seems that the role of diffusion is more prominent than that of absorption.
3.5 Effect of pH
The proton is always involved in the electrochemical reaction, so the relationship between the pH of the electrolyte solution and the redox peak current was investigated. Fig. 7e illustrates the solution pH dependency of the DCF oxidation in the pH range of 6.0–8.5 at the CPE/Ni-Bio-MOF/MIP-PL-Met sensor by the voltammograms of CV. The anodic peak current increases to pH 7 and then decreases. No clear trend was observed between increasing pH and current.Scheme S2† shows the ionic position of DCF at acidic and basic pH in pKa 4.35. With a decrease in pH, the nitrogen atom is proteinization and the molecule is faced with a decrease in electrons, so the oxidation will be more difficult. The DCF is deprotonated at high pH, and the molecule is faced with a negative charge, which creates an electrostatic repulsion in the system.80 The highest peak current was determined by adjusting the solution to a pH of 7.0 in 0.1 M PBS at a voltage of 0.6 V. Fig. 7e shows the peak potential of DCF as a function of pH. The anodic peak potential moved towards a more negative potential, so the proton participation is affirmed in the CPE/Ni-Bio-MOF/MIP-PL-Met sensor.81 The linear regression equation for the peak potential vs. pH value is Ep (V) = − 0.0583 pH + 1.0126 (R2 = 0.9909). This slope is close to the theoretical value of 0.59 V pH−1 given by the Nernst equation, an equal number of protons and electrons in a half-reaction, Ox + ne− + mH+ → Red.50 The equation |Ep – Ep/2| = 0.477 V/αn has been used to determine the number of electrons involved in DCF oxidation for irreversible systems.82 Where Ep/2, α, and n are half-wave peak potential, transfer coefficient, and number of electrons, respectively. The value of Ep/2 was obtained by voltammogram for DCF oxidation in 0.1 M PBS (pH 7.0), which means the Ep – Ep/2 value was 0.495 V.
The value of αn is 0.97, and for an α value of 0.5, the number of electrons was found to be 2.02 (∼2). By replacing the linear regression slope (0.583 V) in the equation of DE/DpH = (0.591 V/n)NHb, the number of protons in the system is also calculated,81 where n and NHb are the number of electrons and the number of protons in DCF oxidation. The number of protons (NHb) was obtained by calculating the number of 1.97 (∼2). So, the electrochemical oxidation of DCF is suggested according to a two-electron/two-proton reaction.
3.6 DFT calculations
Fig. 8a shows the fully optimized structures of L-Methionine (PubChem CID: 6137), L-Met(I),83 and L-Met(II) (CCDC: 276855)84 at the B3LYP computational level with 6-311++G (d,p) basis set in the gas phase. The optimization process of L-Met(II) was initiated from the X-ray crystallographic data, in which the proton (H5) has been transferred from the –COOH group to the NH2 group, leading to the formation of Met zwitterion. The formation of an acid-base group affects the molecule's interaction behavior in aqueous environments. The natural charge of the H5 atom was evaluated at about +0.5e by NBO results. Comparing the natural charges on the N4 atoms in L-Met(I) and L-Met(II) shows an increase in the negative charge on the N4 atom in L-Met(II). MESP maps in Fig. 8a display a 3D viewpoint of electron density in molecules. The red regions show high electron density with an abundance of electrons. These regions could be appropriate sites for electrophilic attacks of adjacent molecules. Subsequently, the blue regions with low electron densities show suitable sites for nucleophilic attacks. Considering the experimental section of this research, we have studied all the calculations in the aqueous phase at a pH ≈ 7. Concerning the calculated net charge of amino acids based on the pK values in a nearly neutral solution, we have considered the net charge of Met to be about zero.85
 | ||
| Fig. 8 (a) The optimized structures of L-Met(I) and L-Met(II) at B3LYP/6-311++G(d,p) in the gas phase, the natural charges (e) over the atoms, and MESP maps; (b) the optimized structures of L-Met(I) and L-Met(II) at B3LYP/6-311++G(d,p) along with some bond lengths (in Å) in the gas and aqueous phases. ΔEelec., the total electronic energy difference, ΔG°, the standard Gibbs free energy change, and SE, the solvation energy, all in kcal mol−1; (c) the optimized structures of DCF at B3LYP/6-311++G(d,p) in two phases, selected bond lengths in Å, and its MESP map. ΔEelec, the total electronic energy difference, and SE, the solvation energy, both in kcal mol−1. | ||
The stability of L-Met(I) and L-Met(II) in both phases was compared in Fig. 8b. Comparing the total electronic energy differences (ΔEelec) and the standard Gibbs free energy change (ΔG°) of L-Met(I) to L-Met(II) exchange indicates a higher stability of L-Met(II), especially in an aqueous environment. Additionally, the SE of L-Met(II) was obtained about 2.73 kcal mol−1 more than the SE of Met(I), which can be due to the formation of a zwitterion.
The optimized structure of DCF (CCDC: 128771)86 in both phases, along with its MESP map, is shown in Fig. 8c. The SE of DCF (−7.13 kcal mol−1) validates its stability in an aqueous phase. The MESP map shows that the carboxyl group of the DCF could be a suitable site for intermolecular interactions. Also, based on the bond distances in Fig. 8c, a weak intramolecular hydrogen bond between the carbonyl and NH groups is expected.
In the next section of theoretical studies, we have considered the interactions between Met and DCF molecules in both phases. The possible configurations of L-Met(I) and L-Met(II) with DCF have been proposed by scanning the distance between two molecules in different directions, considering the MESP maps and natural charge distribution (Fig. 8) in both molecules. The most stable configurations of L-Met(I)- DCF and L-Met(II)-DCF in water are shown in Fig. 9a.
 | ||
| Fig. 9 (a) The optimized structures of under-studied complexes in an aqueous phase; (b) the molecular graph of PL-Met-DCF at B3LYP/6-311++G(d,p) in the gas phase. The small red and yellow spheres show BCPs and RCPs, respectively. The numbers demonstrate the relative HB strength. | ||
The interaction energies (Eint), and the binding thermodynamic parameters of L-Met(I)- DCF and L-Met(II)-DCF are collected in Table 1. Comparing these parameters shows that L-Met(I)-DCF complex formation is thermodynamically more favorable than L-Met(II)-DCF complex in both phases. To study the effect of H2O molecules in the interaction energies, we have considered L-Met(II)-DCF(2H2O) complex (see Fig. 9a). Although the Eint was increased in this complex, ΔG° was shifted to the positive values, which can be due to the significant negative value of ΔS° about 0.10 kcal mol−1 K−1 in both phases. The formation of this complex is exothermic and nonspontaneous in both phases, because of the decreasing entropy due to the accumulation of four separate molecules.
| Gaseous phase | Eint | ΔG° | ΔH° | ΔS° | SE |
|---|---|---|---|---|---|
| L-Met(I)-DCF | −16.18 | −2.96 | −14.84 | −39.85 | −10.25 |
| L-Met(II)-DCF | −9.65 | 2.06 | −8.12 | −34.14 | −14.81 |
| L-Met(II)-DCF(2H2O) | −26.26 | 8.83 | −21.71 | −102.41 | −16.20 |
| PL-Met-DCF | −16.23 | −3.53 | −14.85 | −37.97 | −17.68 |
| Aqueous phase | |||||
| L-Met(I)-DCF | −11.17 | 2.55 | −9.92 | −41.80 | — |
| L-Met(II)-DCF | −4.46 | 6.10 | −3.19 | −31.15 | — |
| L-Met(II)-DCF(2H2O) | −14.05 | 20.50 | −9.79 | −101.58 | — |
| PL-Met-DCF | −11.14 | 1.46 | −9.85 | −37.93 | — |
To study the interactions between L-Met polymer (PL-Met) and DCF, we have modeled a dimer molecule of L-Met based on the crystallographic data and the proposed electropolymerization mechanism of L-Met.84,87 The optimized structure of the PL-Met-DCF complex in water (Fig. 9a) confirms the important role of intermolecular hydrogen bonds in the stability of this configuration. The calculated thermodynamic parameters in Table 1 indicate that PL-Met-DCF's formation is spontaneous and exothermic in the gas phase. Additionally, the same parameters in the aqueous phase predict near-equilibrium conditions for PL-Met-DCF formation. It is worth mentioning that although the choice of computational level can influence the exact values of thermodynamic parameters, the overall trend in their variation remains almost identical.67,88 The SE values in Table 1 demonstrate that the stability of all complexes has increased in water, with PL-Met-DCF exhibiting the highest SE value.
In the last section of calculations, we compared the strengths of four HB bridges in the PL-Met-DCF complex by QTAIM analysis. According to this theory, by forming a bond, a bond critical point (BCP) forms between two neighboring atoms, which identifies the high electron density, and a ring critical point (RCP) shows delocalization of electrons across the ring. BCPs and RCPs can better describe the HB nature and the resonance-assisted hydrogen bonds (RAHB) within the molecules.
Fig. 9b shows the BCPs, RCPs, and the relative HB strength of inter/intramolecular HBs of PL-Met-DCF in the gas phase. According to these values, the intramolecular HB in PM is negligible. The intramolecular HB in the DCF is very weak, and intermolecular HBs play a significant role in PL-Met-DCF interactions. Fig. 9b shows that resonance effects can assist the RCP's creation, suggesting the formation of RAHB in the E1 and E2 intermolecular HBs. It is worth mentioning that the interactions in ionic liquids are very complicated. Our theoretical studies have been focused on hydrogen bond interactions (in the most stable L-Met-DCF configuration) with a polarizable continuum solvent model.
3.7 Optimization of the experimental conditions
In this step, the effects of key factors for preparing the fabricated sensor of CPE/Ni-Bio-MOF/MIP-PL-Met been investigated that contain (1) the amount of Asn in Ni-Bio-MOF, (2) the amount of Ni-Bio-MOF in bare CPE, (3) the typical of template extraction solvent, (4) the concentration of NaOH, (5) the cycle number of MIP polymerization, (6) the template to monomer ratio (T:M), and (7) elution time.
Fig. S6a† displays the cyclic voltammograms of DCF in the sensor with the amount of Ni-Bio-MOF. For this purpose, carbon paste electrodes were prepared with different compositions, including 70 mg of graphite powder, 30 mg of paraffin, and various amounts of Ni-Bio-MOF in the range of 1.0–7.0 mg. The examined sensors have seen a current decrease with Ni-Bio-MOF value above 1.0 mg. The optimal modified mass is chosen in such a way that it has the highest current compared to the background current,32 so the value of 1.0 mg was chosen.
:1, which has partially helped the removal of the template. In the second step, 0.2 M sodium hydroxide solution was used instead of water, which was removed better than in the previous stage. In the third step, a mixture of organic solvents was used together with a sodium hydroxide solution of 0.2 M, as shown in Fig. S6b and c.† So, the template extraction solvent of Ethanol/Acetonitrile/NaOH was chosen with a ratio (1:1:1). Also, the concentration of NaOH was optimized in the range of 0.01 M to 0.5 M in Fig. S6d.† The presence of the optimal amount of NaOH in the mixture of acetonitrile and ethanol solvents leads to the reduction of the hydrogen bond of nitrogen groups88 in DCF and PL-Met film, and the removal of DCF is facilitated from the cavities. The elution time was also examined, and 5 min was chosen as optimal Fig. S6e.†:5 to 3:1. As shown in Fig. S7a,† the anodic peak current of DCF decreased by increasing the amount of template. And with the increase of the amount of monomer up to the ratio of 1:3, the anodic peak current increased, and with the continuation of this increase to the ratio of 1:5, it decreased. So, the template-to-monomer ratio was chosen to be 1:3.The cycle number expresses the thickness of the MIP on the surface of the modified electrode, and then it affects the sensitivity of the designed sensor.90 As shown in Fig. S7b,† the anodic current gradually increased with the increase of cycle numbers from 2 to 5 and then decreased with a further increase in the number of cycles to 15 and remained constant from 15 to 25. By increasing the imprinted polymer membranes excessively, the MIP film thickens and prevents easy access of the template to the printed sites. So, the cycle number of MIP polymerization is chosen as the number of cycles 5.
3.8 Analytical performance
The electrochemical response of the CPE/Ni-Bio-MOF/MIP-PL-Met sensor and the CPE/Ni-Bio-MOF/NIP sensor to DCF in various concentrations was determined under the optimum experimental conditions by DPV. Fig. 10a demonstrates that the peak current in the modified electrode of CPE/Ni-Bio-MOF/MIP-PL-Met increases proportionally with the concentration of DCF. Linear relationships of the independent for CPE/Ni-Bio-MOF/MIP-PL-Met and CPE/Ni-Bio-MOF/NIP have been investigated in the concentration range of 1–500 pM, 1–1000 nM, and 1–1000 μM (Fig. 10b–d). Three concentration ranges are displayed for CPE/Ni-Bio-MOF/MIP-PL-Met and CPE/Ni-Bio-MOF/NIP. The linear regression equations for CPE/Ni-Bio-MOF/MIP-PL-Met are I (μA) = 0.016C (μM) + 8.9944 (R2 = 0.9985), I (μA) = 5.9581C (μM) + 2.94 (R2 = 0.9954) and I (μA) = 2015.5C (μM) + 0.5943 (R2 = 0.9999) and for CPE/Ni-Bio-MOF/NIP are as follows: I (μA) = 0.002C (μM) + 1.0189 (R2 = 0.997), and I (μA) = 0.897C (μM) + 0.089 (R2 = 0.9986).The limit of detection was calculated to be 0.17 pM by 3σ/s (where σ is the standard deviation of the blank (noise) and S is the slope of the calibration curve) (n = 3).91 where σ represents the standard deviation of the blank current and S represents the slope of the first calibration plot. | ||
| Fig. 10 (a) DPVs by the increasing concentration of DCF from 1.0 pM to 500 pM, 1 nM to 1000 nM, and 1.0 μM to 1000 μM in 0.1 M PBS solution (pH 7.0) at scan rate 50 mV s−1 on the CPE/Ni-Bio-MOF/MIP-PL-Met, and (b–d) the calibration curves. Error bars indicate the standard deviations of three repeated measurements. | ||
In Table 2, the comparison of this method with other reported methods is summarized for the analysis of DCF. The CPE-Ni-Bio-MOF/MIP-PL-Met has a wide linear range and a very low detection limit. The MIP sensor used in this study can directly adsorb and identify DCF molecules, which is better than previously reported work.
| Modified electrode | Technique | Detection limit (nM) | Linear range (μM) | Ref. |
|---|---|---|---|---|
| a Vanadium pentoxide nanosheet/screen-printed electrode.b Carboxyl-functionalized graphene oxide/glassy carbon electrode.c Copper nanoparticles/reduced graphene oxide/glassy carbon electrode.d Tyrosine-modified carbon paste electrode.e Edge plane pyrolytic graphite sensor.f Gold–platinum bimetallic nanoparticles/functionalized multi-walled carbon nanotubes/gold electrode.g Multiwalled carbon nanotube-ionic liquid/carbon ceramic electrode.h Poly(3,4-ethylene dioxythiophene)/polystyrene/glassy carbon electrode.i Zirconium–metal organic framework/molecularly imprinted polymer/glassy carbon electrode. | ||||
| V2O5/SPE a | DPV | 3.1 | 20–200 | 1 |
| GO-COOH/GCEb | LSV | 90.0 | 1.2–400.0 | 2 |
| Cu NPs/rGO/GCEc | Amperometry | 8.0 | 20–400 | 3 |
| TCPEd | DPV | 3280 | 10.0–140.0 | 4 |
| EPPGe | SWV | 6.2 | 0.01–1.0 | 5 |
| Au–PtNPs/f-MWCNTs/AuEf | DPV | 300.0 | 0.5–1000 | 6 |
| MWCNT–IL/CCEg | DPV | 18.0 | 0.05–50.0 | 7 |
| PEDOT/PS/GCE h | DPV | 77.5 | 0.1–0.5 | 55 |
| Zr-MOF/MIP/GCEi | DPV | 100.0 | 6.5–1.5 × 103 | 90 |
| CPE/Ni-Bio-MOF/MIP-PL-Met | DPV | 1.7 × 10−4 | 1.0 × 10−6–1.0 × 103 | This work |
3.9 Selectivity, reproducibility, and stability of CPE/Ni-Bio-MOF/MIP-PL-Met
The selectivity of the CPE/Ni-Bio-MOF/MIP-PL-Met sensor was studied by DPV response of 1.0 μM DCF in the presence of interfering species of glucose, glycine, uric acid, urea, ascorbic acid, and ions of chlorine, magnesium, ammonium, sulfate, sodium, nitrate, calcium, bromine, and potassium (Fig. 11a), and the current measured showed an error of less than or more than 5% (Fig. S8†). Table S1† exhibited that 1000-fold of glucose and glycine, 10-fold of uric acid and urea, 1000-fold of ascorbic acid, and 100-fold of chlorine, ammonium, magnesium, sulfate, sodium, nitrate, calcium, bromine, and potassium can interfere with 1 μM of DCF concentration. | ||
| Fig. 11 (a) Effect of various interferences on DCF sensing response of CPE/Ni-Bio-MOF/MIP-PL-Met; (b) CVs of CPE/Ni-Bio-MOF/MIP-PL-Met in 1.0 μM DCF and 0.1 M PBS solution (pH = 7.0) to investigate reproducibility; inset: corresponding bar diagram of current vs. number of the electrode. Error bars indicate the standard deviations of three repeated measurements. | ||
The reproducibility of the CPE/Ni-Bio-MOF/MIP-PL-Met sensor was assessed by studying 5 electrodes in a solution containing 1.0 μM of DCF, using the identical experimental conditions (Fig. 11b). The peak current observed from multiple sensors had a relative standard deviation (RSD) of 3.3%, indicating excellent reproducibility of the sensor. To investigate the stability of the CPE/Ni-Bio-MOF/MIP-PL-Met sensor, the peak current of the modified electrode was measured after it was kept in the refrigerator for 10 days. The peak current showed 96.2% of its initial value, indicating good stability.
3.10 Determination of DCF in biological fluids and pharmaceutical samples
The CPE/Ni-Bio-MOF/MIP-PL-Met sensor was used to determine DCF to evaluate operational applications. For this purpose, a pharmaceutical sample of a DCF tablet and a medical sample of blood serum of a healthy person were measured by the standard addition method. The precision of the suggested approach was evaluated with the reference UV-vis spectrophotometric method. The relative standard deviation and recovery values were computed and are shown in Table 3. As shown in Table 3, the recovery percent in the tablet sample ranged from 104.29% to 106.65%, indicating that the modified sensor is appropriate for the determination of DCF in complex compounds as a low-cost, easy, and fast method (Fig. S9a†).| Sample | Added (μM) | DPV | UV-vis | ||
|---|---|---|---|---|---|
| Found (μM) (%RSD) | Recovery (%) | Found (μM) (%RSD) | Recovery (%) | ||
| a ND: Not Detected. | |||||
| Human serum | 0 | ND | — | ND | — |
| 0.5 | 0.51 (2.43) | 102.00 | 0.51 (1.55) | 102.00 | |
| 0.7 | 0.70 (2.41) | 100.00 | 0.71 (2.78) | 101.43 | |
| 45 | 42.84 (0.34) | 95.20 | 44.10 (0.99) | 98.00 | |
| 80 | 83.19 (1.00) | 103.99 | 78.48 (0.95) | 98.10 | |
| DCF tablets | 0 | 5.62 (1.24) | — | 5.87 (0.60) | — |
| 40 | 42.66 (1.35) | 106.65 | 40.89 (0.34) | 102.22 | |
| 70 | 73.00 (1.48) | 104.29 | 75.79 (0.56) | 108.27 | |
A medical sample of blood serum was selected from a healthy person and was measured using the standard addition method. The achieved recoveries varied from 95.20% to 103.99%. Furthermore, the suggested approach exhibits an RSD of less than 2.43%, indicating the satisfactory precision of the voltammetric determination of DCF employing the CPE/Ni-Bio-MOF/MIP-PL-Met (Fig. S9b†). Therefore, the proposed method can be used to analyze real samples with different matrices.
In addition, we confirmed the precision of detecting Ni-Bio-MOF/MIP-PL-Met using UV-Visible spectrophotometry (Fig. S9c†). As shown in Fig. S9d,† standard solutions of DCF were prepared, and a DCF standard curve was established using UV-Visible spectroscopy, and it was obvious that a strong linear correlation was observed between the DCF concentration and the absorption peak. The linear regression equations were y = 0.1084 CDCF + 0.0188 (R2 = 0.9988) and y = 0.0106 CDCF + 0.1723 (R2 = 0.9883). To determine the DCF content in DCF tablet and human serum samples, they were diluted with water, and their absorption peaks at specific wavelengths were determined using UV-Visible spectroscopy (Fig. S9† (e and f)). Finally, we compared the electrochemical test results with the UV-Vis spectroscopy results, and the results indicate that the values obtained from the suggested modified electrode align well with those derived from the spectrophotometric approach.
4 Conclusion
In this study, a selective electrochemical sensor was designed and fabricated for the determination of DCF. The modification of Ni-BDC-based MOF using Asn was successfully carried out. We found that the amount of Asn influences the electrocatalytic activity of Ni-Bio MOF, where 3.0 mg of Asn resulted in the highest peak of the anodic current of DCF. The surface of the CPE was modified using a Ni-Bio-MOF (Ni-BDC-Asn), and then L-Met polymer was used for the solution of MIP. The calibration curve for determining the DCF was obtained using the designed sensor in differential pulse voltammetry. The suggested sensor has many benefits, including a broad linear range spanning from 1.0 pM to 1.0 mM, a limit of detection (LOD) as low as 0.17 pM, rapid DCF analysis, cost-effectiveness, high selectivity and accuracy, and low cost. This sensor can be successfully used in the identification of pharmaceutical samples and biological systems for the determination of DCF. The theoretical analysis at the B3LYP/6-311++G** computational level has confirmed the stability of L-Met-DCF complexes in both gaseous and aqueous phases. The calculated thermodynamic parameters of the binding show an exothermic process in the aqueous phase, with the binding standard Gibbs free energy change of about 1.46 kcal mol−1. The computed solvation energies of all complexes have confirmed their stability in water. The formation of intermolecular HBs between the carboxylic groups of PL-Met and DCF was approved by QTAIM analysis, confirming the significant role of HBs in this interaction.Data availability
Data will be made available on request.Author contributions
Samaneh Ebadi: conceptualization, methodology, writing – original draft, Khadijeh Ghanbari: writing – review & editing, supervision, project administration, validation, Mansoureh Zahedi-Tabrizi: writing – review & editing, software, visualization, investigation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge partial financial support from the Research Council of Alzahra University.References
- A. C. Lazanas and M. I. Prodromidis, Electrochim. Acta, 2022, 428, 140919 CrossRef CAS.
- C. Karuppiah, S. Cheemalapati, S.-M. Chen and S. Palanisamy, Ionics, 2015, 21, 231–238 CrossRef CAS.
- H. Yu, J. Jiao, Q. Li and Y. Li, Int. J. Electrochem. Sci., 2021, 16, 211024 CrossRef CAS.
- B. K. Chethana, S. Basavanna and Y. Arthoba Naik, Ind. Eng. Chem. Res., 2012, 51, 10287–10295 CrossRef CAS.
- R. N. Goyal, S. Chatterjee and B. Agrawal, Sens. Actuators, B, 2010, 145, 743–748 CrossRef CAS.
- M. M. Eteya, G. H. Rounaghi and B. Deiminiat, Microchem. J., 2019, 144, 254–260 CrossRef CAS.
- K. Sarhangzadeh, A. A. Khatami, M. Jabbari and S. Bahari, J. Appl. Electrochem., 2013, 43, 1217–1224 Search PubMed.
- A. Deng, M. Himmelsbach, Q. Z. Zhu, S. Frey, M. Sengl, W. Buchberger, R. Niessner and D. Knopp, Environ. Sci. Technol., 2003, 37, 3422–3429 CrossRef CAS PubMed.
- P. Szpot, O. Wachełko and M. Zawadzki, J. Chromatogr. B, 2021, 1179, 122860 CrossRef CAS PubMed.
- C. Coman, N. Hădade, S. Pesek, R. Silaghi-Dumitrescu and A. C. Mot, J. Inorg. Biochem., 2023, 249, 112400 Search PubMed.
- A. Shishov, A. Gerasimov, D. Nechaeva, N. Volodina, E. Bessonova and A. Bulatov, Microchem. J., 2020, 156, 104837 CrossRef CAS.
- A. Pochivalov, C. Vakh, V. Andruch, L. Moskvin and A. Bulatov, Talanta, 2017, 169, 156–162 CrossRef CAS PubMed.
- Y. M. Correa-Navarro, G. D. López, C. Carazzone, L. G. Moreno-Piraján and J. Carlos, ACS Omega, 2023, 8, 38905–38915 Search PubMed.
- N. Kaewwonglom, M. Oliver, D. J. Cocovi-Solberg, K. Zirngibl, D. Knopp, J. Jakmunee and M. Miró, Anal. Chem., 2019, 91, 13260–13267 Search PubMed.
- Y. M. Correa-Navarro, J. C. Moreno-Piraján and L. Giraldo, ACS Omega, 2023, 8, 1967–1978 Search PubMed.
- H. Afsharara, E. Asadian, B. Mostafiz, K. Banan, S. Arjomand Bigdeli, D. Hatamabadi, A. Keshavarz, C. M. Hussain, R. Keçili and F. Ghorbani-Bidkorpeh, TrAC, Trends Anal. Chem., 2023, 160, 116949 CrossRef CAS.
- P. Xu, S. Xie, X. Liu, L. Wang, X. Jia and C. Yang, Chem. Eng. J., 2022, 446, 136941 CrossRef CAS.
- B. Chandran, S. Ramasamy, S. K. Ponnaiah, E. Arumugam, S. Chandrasekaran, S. Karuppaiah and A. Ganesan, ACS Appl. Nano Mater., 2024, 7, 6839–6850 CrossRef CAS.
- B. Shobana, P. S. Kumar, K. Renugadevi and P. Prakash, Food Chem., 2024, 439, 138073 CrossRef CAS PubMed.
- M. Aliyari and Kh. Ghanbari, Anal. Lett., 2023, 56, 1081–1101 CrossRef CAS.
- S. Zwane, T. Ingwani, D. S. Dlamini, B. B. Mamba and A. T. Kuvarega, J. Photochem. Photobiol., A, 2021, 446, 115123 CrossRef.
- S. Bonyadi and Kh. Ghanbari, Mater. Chem. Phys., 2021, 367, 124683 CrossRef.
- Y. Liu, W. Li, Y. Gao, J. Wang, G. Cheng, J. Chen, X. Li and G. Zhu, Chem. Eng. J., 2022, 443, 136396 CrossRef CAS.
- G. S. Kassahun, S. Griveau, S. Guillard, A. Ringuedé, B. Bresson, Y. Tran, F. Bedioui and C. Slim, Langmuir, 2020, 36, 827–836 CrossRef CAS PubMed.
- B. Shobana, L. Gayathri, P. S. Kumar and P. Prakash, Microchem. J., 2023, 193, 109116 CrossRef CAS.
- Z. Guo, W. Xu, G. Xu and Q. Jia, TrAC, Trends Anal. Chem., 2023, 167, 117275 CrossRef CAS.
- S. Zamani, Kh. Ghanbari and S. Bonyadi, Anal. Methods, 2023, 15, 4606–4614 RSC.
- A. A. Lahcen, S. G. Surya, T. Beduk, M. T. Vijjapu, A. Lamaoui, C. Durmus, S. Timur, O. Shakhah, V. Mani, A. Amine, M. Eddaoudi and K. N. Salama, ACS Appl. Mater. Interfaces, 2022, 14, 49399–49424 CrossRef CAS PubMed.
- A. F. T. Waffo, C. Yesildag, G. Caserta, S. Katz, I. Zebger, M. C. Lensen, U. Wollenberger, F. W. Scheller and Z. Altintas, Sens. Actuators, B, 2018, 275, 163–173 CrossRef CAS.
- S. Bonyadi and Kh. Ghanbari, Microchem. J., 2023, 187, 108398 CrossRef CAS.
- M. E. Çorman, G. Ozcelikay, A. Cetinkaya, S. I. kaya, C. Armutcu, E. Özgür, L. Uzun and S. A. Ozkan, TrAC, Trends Anal. Chem., 2022, 150, 116573 CrossRef.
- M. Mostafavi, M. R. Yaftian, F. Piri and H. Shayani-Jam, Biosens. Bioelectron., 2018, 122, 160–167 CrossRef CAS PubMed.
- N. A. Samah, M. J. Sánchez-Martín, R. M. Sebastián, M. Valiente and M. López-Mesas, Sci. Total Environ., 2018, 631–632, 1534–1543 CrossRef PubMed.
- S. Bonyadi and Kh. Ghanbari, Microchem. J., 2021, 167, 106322 CrossRef CAS.
- L. Qian, S. Durairaj, S. Prins and A. Chen, Biosens. Bioelectron., 2021, 175, 112836 CrossRef CAS PubMed.
- D. Duan, X. Si, Y. Ding, L. Li, G. Ma, L. Zhang and B. Jian, Bioelectrochemistry, 2019, 129, 211–217 CrossRef CAS PubMed.
- Y. Wang, Q. Xiuzhen, L. Yangyang, G. Huishi, L. Wenguan and N. Libo, J. Nanosci. Nanotechnol., 2020, 20, 1807–1813 CrossRef CAS PubMed.
- T. Liang, S. Wang, L. Chen and N. Niu, Food Anal. Methods, 2019, 12, 217–228 CrossRef.
- Y. Xue, Y. Wang, S. Feng, M. Yan, J. Huang and X. Yang, Biosens. Bioelectron., 2022, 202, 113992 CrossRef CAS PubMed.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. M. Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- X. Xie, G. Mo and B. Hu, Sens. Actuators, B, 2023, 393, 134263 CrossRef CAS.
- P. Tong, J. Liang, X. Jiang and J. Li, Crit. Rev. Anal. Chem., 2020, 50, 376–392 CrossRef CAS PubMed.
- A. Portorreal-Bottier, S. Gutiérrez-Tarriño, J. J. Calvente, R. Andreu, E. Roldán, P. Oña-Burgos and J. L. Olloqui-Sariego, Sens. Actuators, B, 2022, 368, 132129 CrossRef CAS.
- L. Geng, S. Hu, F. Liu, L. Liu, X. He, Y. Wang, Y. Zhu, H. Xu, S. Zhao and F. Ye, Sens. Actuators, B, 2023, 390, 134010 CrossRef CAS.
- H. Cai, Y.-L. Huang and D. Li, Coord. Chem. Rev., 2019, 378, 207–221 CrossRef CAS.
- M. S. Annas, M. T. Ulhakim, N. L. W. Septiani, S. A. Abrori, A. Hermawan, O. Floweri, G. Gumilar, Nugraha and B. Yuliarto, Appl. Phys. A, 2024, 130, 956 CrossRef CAS.
- M. Li, C. Li, Z. Chunrui, T. Li, J. Jiang, Z. Han, C. Zhang, H. Sun and S. Dong, Sep. Purif. Technol., 2023, 308, 122945 CrossRef CAS.
- M. A. Dheyab, A. A. Aziz, M. S. Jameel, O. A. Noqta, P. M. Khaniabadi and B. Mehrdel, Sci. Rep., 2020, 10, 10793 CrossRef CAS PubMed.
- S. Gao, Y. Sui, F. Wei, J. Qi, Q. Meng and Y. He, J. Mater. Sci., 2018, 53, 6807–6818 CrossRef CAS.
- H. Razmi and Y. Bahadori, Microchem. J., 2021, 168, 106442 CrossRef CAS.
- H. R. Akbari Hasanjani and K. Zarei, Biosens. Bioelectron., 2019, 128, 1–8 CrossRef CAS PubMed.
- R. Ojani, A. Alinezhad and Z. Abedi, Sens. Actuators, B, 2013, 188, 621–630 CrossRef CAS.
- I. Seguro, J. G. Pacheco and C. Delerue-Matos, Sensors, 2021, 21, 1975 CrossRef CAS PubMed.
- D. E. Bayraktepe, C. Yıldız and Z. Yazan, Talanta, 2023, 257, 124361 CrossRef PubMed.
- D. H. N. Nguyen, Q. H. Le, T. L. Nguyen, V. T. Dinh, H. N. Nguyen, H. N. Pham, T. A. Nguyen, L. L. Nguyen, T. M. T. Dinh and V. Q. Nguyen, J. Electroanal. Chem., 2022, 921, 116709 CrossRef CAS.
- Z. Hadi and Kh. Ghanbari, New J. Chem., 2022, 46, 12941–12951 RSC.
- M. Zahedi-Tabrizi, B. Gerivani and S. F. Tayyari, Spectrochim. Acta, Part A, 2015, 136, 731–742 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Jr. Montgomery, R. E. Stratmann, J. C. Burant, et al., Gaussian 03, Revision A.7, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- E. D. Glendening, A. E. Reed, F. Weinhold, NBO, Version 3.1, Gaussian Inc., Pittsburgh, PA, 1992 Search PubMed.
- F. B. König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- J. Tomasi, R. Cammi, B. Mennucci, C. Cappelli and S. Corni, Phys. Chem. Chem. Phys., 2002, 4, 5697–5712 RSC.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. V. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–7 Search PubMed.
- E. Zarie-Moghaddam and M. Zahedi-Tabrizi, QTAIM, Monatsh. Chem., 2019, 150, 1267–1274 CrossRef CAS.
- S. F. S. Mazloum Hosseini, M. Zahedi-Tabrizi and Z. Moosavi-Tekyeh, Organomet. Chem. Res., 2022, 8, 48–56 Search PubMed.
- S. Sylvestre, S. Sebastian, S. Edwin, M. Amalanathan, S. Ayyapan, T. Jayavarthanan, K. Oudayakumar and S. Solomon, Spectrochim. Acta, Part A, 2014, 133, 190–200 CrossRef CAS PubMed.
- Y. Wang, X. Ouyang, Y. Ding, B. Liu, D. Xu and L. Liao, RSC Adv., 2016, 6, 10662–10669 RSC.
- M. Challa, S. Chinnam, A. M. Rajanna, A. Nandagudi, B. C. Yallur and V. Adimule, Heliyon, 2023, 9, 13223 CrossRef PubMed.
- Y. Yang, W. Ji, Y. Yin and N. Wang, Biosensors, 2023, 13, 508 CrossRef CAS PubMed.
- J. Li, J. Song, B.-Y. Huang, G. Liang, W. Liang, G. Huang, Y. Q. Jin, H. Zhang, F. Xie, J. Chen, N. Wang, Y. Jin, X.-B. Li and H. Meng, J. Catal., 2020, 389, 375–381 CrossRef CAS.
- Y. Tang, Q. Liu, L. Dong, H. B. Wu and X.-Y. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef CAS.
- D. Wang, F. Le, J. Lv, X. Chen, H. Yao and W. Jia, Molecules, 2023, 28, 4366 CrossRef CAS PubMed.
- G.-C. Li, P.-F. Liu, R. Liu, M. Liu, K. Tao, S.-R. Zhu, M.-K. Wu, F.-Y. Yi and L. Han, Dalton Trans., 2016, 45, 13311–13316 RSC.
- D. Duan, J. Ye, X. Cai and K. Li, Microchim. Acta, 2021, 188, 111 CrossRef CAS PubMed.
- M. Asif, H. Liu, A. Aziz, H. Wang, Z. Wang, M. Ajmal, F. Xiao and H. Liu, Biosens. Bioelectron., 2017, 97, 352–359 CrossRef CAS PubMed.
- C. M. A. Brett, A. M. O. Brett, Electrochemistry: Principles, Methods, and Applications. 1st edn, Oxford University, 1993 Search PubMed.
- A. J. Bard, L. R. Faulkner, H. S. White, Electrochemical Methods: Fundamentals and Applications. John Wiley & Sons, 2022 Search PubMed.
- L. M. Gonçalves, C. Batchelor-McAuley, A. A. Barros and R. G. Compton, J. Phys. Chem. C, 2010, 114, 14213–14219 CrossRef.
- M. M. Shanbhag, D. Ilager, S. Mahapatra, N. P. Shetti and P. Chandra, Mater. Chem. Phys., 2021, 273, 12044 CrossRef.
- W. Si, W. Lei, Z. Han, Y. Zhang, Q. Hao and M. Xia, Sens. Actuators, B, 2014, 193, 823–829 CrossRef CAS.
- E. S. Gomes, F. R. F. Leite, B. R. L. Ferraz, H. A. J. L. Mourão and A. R. Malagutti, J. Pharm. Anal., 2019, 9, 347–357 CrossRef PubMed.
- H. Lioe, A. J. Richard, S. Gronert, A. Austin and G. E. Reid, Int. J. Mass Spectrom., 2007, 267, 220–232 CrossRef CAS.
- B. Dalhus and C. H. Görbitz, Acta Chem. Scand., 1996, 50, 544–548 CrossRef CAS.
- J. C. Cameselle, J. M. Ribeiro and A. Sillero, Biochem. Educ., 1986, 4, 131–136 CrossRef.
- N. Muangsin, M. Prajuabsook, P. Chimsook, N. Chantarasiri, K. Siraleartmukul, N. Chaichit and S. Hannongbua, J. Appl. Crystallogr., 2004, 37, 288–294 CrossRef CAS.
- J. Thangphatthanarungruang, C. Chotsuwan, O. Chailapakul and W. Siangproh, Analyst, 2023, 148, 3107–3116 RSC.
- R. F. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- M. Shamsipur, N. Moradi and A. Pashabadi, J. Solid State Electrochem., 2018, 22, 169–180 CrossRef CAS.
- M. Malekzadeh, A. Mohadesi, M. A. Karimi and M. Ranjbar, Anal. Bioanal. Electrochem., 2020, 12, 402–414 CAS.
- M. C. Blanco-López, L. Fernández-Llano, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, Anal. Lett., 2004, 37, 915–927 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00194c |
| This journal is © The Royal Society of Chemistry 2025 |