DOI:
10.1039/D5RA00108K
(Paper)
RSC Adv., 2025,
15, 11675-11687
Self-assembled nanoplatform-mediated co-delivery of brusatol to sensitize sorafenib for hepatocellular carcinoma treatment†
Received
6th January 2025
, Accepted 18th March 2025
First published on 14th April 2025
Abstract
Sorafenib (Sor), recognized as a frontline multi-kinase inhibitor, constitutes the primary targeted therapy for hepatocellular carcinoma (HCC). Despite its potential, many HCC patients exhibit reduced responsiveness to Sor, thereby undermining its therapeutic efficacy. Recent studies highlight the importance of nuclear factor erythroid-2-related factor 2 (Nrf2) activation in HCC, which contributes to Sor resistance. Brusatol (Bru), a plant-derived Nrf2 inhibitor, counteracts this resistance but faces challenges due to its poor solubility in aqueous media. In this study, we developed a glutathione (GSH)-responsive nanoplatform that effectively dispersed in water for the co-delivery of Bru and Sor (B/S NP). This approach enhanced Bru's therapeutic efficacy and increased Sor sensitivity in HCC. Our nanoplatform significantly reduced Nrf2 expression, thereby increasing Sor sensitivity both in vitro and in vivo, while presenting a favorable biosafety profile. These findings suggest that the nanoplatform-mediated co-delivery of Bru and Sor offers an innovative approach to enhance Sor's effectiveness in HCC treatment.
Introduction
Hepatocellular carcinoma (HCC) ranks as the sixth most prevalent form of cancer and the second leading cause of cancer-related fatalities globally.1–3 Current clinical approaches to HCC encompass surgical resection, chemotherapy, and molecular targeted therapy.4–6 However, the insidious onset and rapid progression of HCC result in approximately 80% of patients being diagnosed when the cancer is no longer operable or has metastasized, thereby bypassing the optimal window for surgical intervention. Consequently, the five-year survival rate is a mere 12.1%.7 In advanced HCC, which often fails to respond to cytotoxic agents, such as chemotherapy, molecular targeted therapy, becomes the predominant treatment option. Sor, a type of tyrosine kinase inhibitor (TKI), was the first drug approved by the Food and Drug Administration (FDA) for specifically targeting HCC.8–10 Sor impedes HCC cell proliferation by disrupting the Raf protein kinase (Raf)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway and also prevents the development of new tumor vasculature by suppressing the vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor β (PDGFR-β).10 Despite these mechanisms, clinical observations indicate that approximately 70% of HCC patients do not respond to Sor therapy.11,12 Moreover, resistance to treatment often emerges within six months among those who initially respond.12 Addressing the pivotal factors that dictate Sor sensitivity is therefore essential for bolstering the drug's efficacy and improving survival rates among HCC patients, which has substantial medical and societal implications.13,14
Nuclear factor erythroid 2-related factor 2 (Nrf2) primarily regulates gene transcription through the Kelch-like ECH-associated protein 1 (Keap1)-Nrf2 signaling pathway.15,16 By regulating the expression of genes involved in antioxidant defense, Nrf2 prevents cellular damage caused by reactive oxygen species (ROS) and electrophiles, thereby maintaining redox homeostasis.17,18 Continuous activation of Nrf2 can significantly reduce ROS levels associated with Sor, contributing to Sor resistance and promoting tumor cell drug resistance.19–21 Thus, inhibiting the Nrf2 pathway could be a promising strategy to increase tumor sensitivity to chemotherapeutic drugs. Bru, a natural product extracted from Brucea javanica, is part of the quassinoid family and exhibits anti-tumor, anti-inflammatory, and antioxidant properties.22,23 Bru remains a potent Nrf2 pathway inhibitor that enhances chemotherapy efficacy by inhibiting Nrf2-mediated defense mechanism.24,25 Studies have demonstrated that Bru can make different types of cancer cells more responsive to chemotherapy drugs and enhance tumor cell apoptosis.26,27 However, free Bru suffers from poor aqueous solubility,28–30 which limits its pharmaceutical potential in the aqueous environment of the human body.31–35
Tumor sites typically exhibit significantly high levels of GSH compared to normal tissues, which can reach concentrations of 10 mM or higher.36 In addition, considering the poor aqueous solubility of Bru as a major challenge, we developed a GSH-responsive nanoplatform (Scheme 1) aimed at enhancing therapeutic outcomes. These nanoparticles were designed to release their payload in response to the high GSH levels in tumors, ensuring that the drug is released precisely at the target site. This not only enhances targeted therapeutic efficacy but also minimizes systemic side effects. This platform, which facilitates the co-delivery of Bru and Sor (B/S NP), was synthetized through a self-assembly process, utilizing a methyl-polyethyleneglycol-SS-poly(L-lactide-co-glycolide) copolymer (Meo-PEG-S-S-PLGA). The Meo-PEG segment of this construct markedly improves the aqueous solubility of the nanoplatform, while the disulfide bonds are susceptible to cleavage by the elevated GSH concentrations within tumor cells, thereby initiating a substantial release of the entrapped drugs. Furthermore, the hydrophobic nature of PLGA renders it an efficient vehicle for the delivery of hydrophobic medications, promoting their encapsulation and subsequent release. In this study, we conducted an array of in vitro assays, encompassing drug release studies, assessments of gene and protein expression, evaluations of cellular proliferation, and analyses of apoptotic responses. These experiments confirmed that Bru/Sor NP was capable of releasing encapsulated drugs in response to high GSH levels and effectively downregulating the expression of Nrf2, thereby sensitizing HCC cells to Sor treatment. In addition, using nude mouse subcutaneous tumor models, we observed that the B/S NP not only demonstrated significant accumulation at the tumor site but also exerted a potent inhibitory effect on HCC tumor growth. These findings indicated that the co-delivery of Bru and Sor based on this nanoplatform holds promise as a novel therapeutic approach to enhance the therapeutic efficacy of HCC treatment.
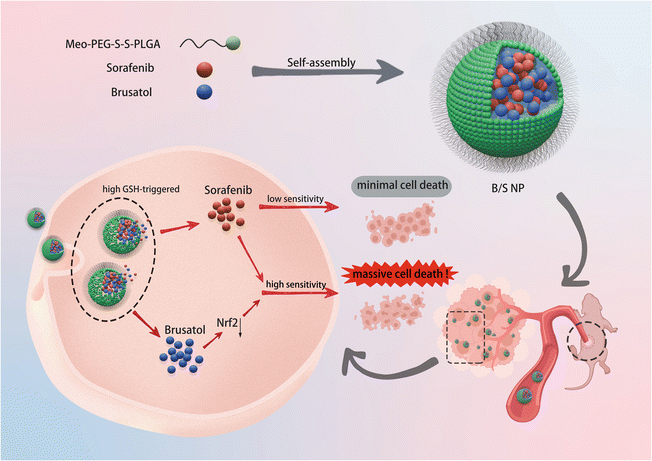 |
| | Scheme 1 Schematic of the self-assembled co-delivery nanoplatform for sorafenib and brusatol (B/S NP). The B/S NP, characterized by tumor site aggregation and high GSH-responsive drug release properties, reaches HCC cells (depicted as pink cells) via blood flow and downregulates Nrf2 expression by releasing brusatol, thereby enhancing sorafenib sensitivity in the treatment of HCC in vivo. | |
Experimental
Materials
Sorafenib (Sor), brusatol (Bru), and dimethyl sulfoxide (DMSO) were purchased from MCE (MedChemExpress, HY-Y0345, USA). Cy5-Bru and methyl-polyethyleneglycol-SS-poly(L-lactide-co-glycolide) copolymer (Meo-PEG-S-S-PLGA) was purchased from Suzhou Juling Polymer Technology Co., Ltd. Cy5-Sor was purchased from Xi'an QiYue Biology Co., Ltd. Unless specifically mentioned, all the reagents were used without any further purification or modification.
Preparation of the B/S NP
The B/S NP was synthesized using a nanoprecipitation method. To prepare the B/S NP, we combined 60 μL of Bru solution (100 μM in DMSO) with 40 μL of Sor solution (10 mM in DMSO), along with 400 μL of Meo-PEG-S-S-PLGA copolymer solution (25 g L−1 in DMSO). This blend was then introduced drop by drop into 10 mL of deionized water while stirring vigorously at 1500 rpm. The resulting B/S NP was purified using an ultrafiltration device with a molecular weight cut-off of 100 K via centrifugation. The NP loading Bru (denoted Bru NP) or Sor (denoted Sor NP) was also synthesized using the same procedure outlined previously.
Characterization
A transmission electron microscope (TEM, HT7800, Hitachi, Ltd, Japan) was used to analyze the morphology of the nanoparticles. Additionally, a Dynamic Light Scattering instrument (DLS, Nano ZS90 Zetasizer, Malvern Instruments Co. Ltd, UK) was used to track the size distribution and zeta potential of the particles. For determining the encapsulation efficiency (EE%) of Bru, nanoplatform loaded with Cy5-Bru was fabricated, and the fluorescence intensity of the encapsulated Cy5-Bru was quantified using a versatile microplate reader (SpectraMax® iD3, Molecular Devices, USA). Before the measurement, the structure of the B/S NP was destroyed by blending the nanoparticle suspension with DMSO in a volume ratio of 1/20. The EE% of Bru was ascertained by correlating the fluorescence intensity of Cy5-Bru with its corresponding standard curve. By fine-tuning the amount of feed, we achieved Bru-loaded NP with an EE% of ≈50%, and the EE% of Sor was also calculated using the above-mentioned method to be ≈70%.
Drug release in vitro
To evaluate the in vitro drug release from our nanoplatform, suspensions of Cy5-Bru-loaded nanoplatform (NP) and Cy5-Sor-loaded NP were prepared, each in 1 mL of phosphate-buffered saline (PBS). These were individually placed in dialysis bags with a 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular weight cut-off. The bags were then submerged in 10 mL of PBS containing 0.05% (w/v) Tween 80 and GSH concentrations of 0, 1, 5 mM, and 10 mM. This arrangement was kept at a temperature of 37 ± 0.5 °C with gentle agitation at 100 rpm to mimic the physiological conditions. At predetermined time intervals, the medium was collected and replaced with a pre-warmed PBS solution containing different concentrations of GSH. The residual Cy5-Bru and Cy5-Sor within the nanoparticles were quantified via fluorescence intensity, indicating the drug release amount over time.
000 molecular weight cut-off. The bags were then submerged in 10 mL of PBS containing 0.05% (w/v) Tween 80 and GSH concentrations of 0, 1, 5 mM, and 10 mM. This arrangement was kept at a temperature of 37 ± 0.5 °C with gentle agitation at 100 rpm to mimic the physiological conditions. At predetermined time intervals, the medium was collected and replaced with a pre-warmed PBS solution containing different concentrations of GSH. The residual Cy5-Bru and Cy5-Sor within the nanoparticles were quantified via fluorescence intensity, indicating the drug release amount over time.
Collection of HCC patient samples
HCC samples of patients, confirmed through post-operative pathological diagnosis, were collected from Tongliang District People's Hospital. The study was conducted after ethical review of Tongliang District People's Hospital (Approval number: TDPH-2024-137).
Cell culture
Both the normal human liver cell line (WRL68) and the human HCC cell lines (HepG2, MHCC-97H, Huh-7, SNU-449, and MHCC-LM3) were cultivated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Vazyme Biotech Co., Ltd, China), penicillin (100 U mL−1) and streptomycin (100 mg mL−1). These cultures were maintained in an incubator with a 5% CO2 environment at 37 °C, and the growth medium was refreshed every 48 hours. All cell experiments, unless specified otherwise, were conducted once the cells had grown to a confluence of approximately 70–80%.
Colony formation assay
Initially, six-well plates were seeded with MHCC-97H cells at a density of 5 × 103 cells per well. Cultivation continued for 7 days in DMEM supplemented with 10% FBS to facilitate colony formation. After the culture period, the growth medium was replaced with fresh DMEM. PBS, Bru NP, Sor NP, or B/S NP was sequentially introduced into each well to assess their effects. Incubation continued for an additional 24 hours. Following incubation, the medium was taken off and the cells were rinsed three times with PBS. Fresh DMEM was then added to each well, allowing cell growth until visible colonies indicated mature cell clusters. Once the colonies became apparent, cells were fixed with 4% paraformaldehyde, stained with crystal violet, and counted to assess the treatment's impact on proliferation and colony formation.
Flow apoptosis detection
MHCC-97H cells were initially seeded in six-well plates at a density of 5 × 103 cells per well, adhering and growing in 10% FBS-supplemented medium for 24 hours. Following this, the medium was replaced with a fresh one containing either PBS, Bru NP, Sor NP, or B/S NP, and the cells were incubated further for 24 hours to evaluate the treatment effects. Post-incubation, the medium from each well was aspirated, and the cells were washed three times with cold PBS. Trypsin treatment detached cells from the well surface. Once detached, the cells were collected via centrifugation and resuspended in 1 mL of cold binding buffer to achieve 1 × 106 cells per mL suspension. For the flow cytometry analysis, 100 μL cell suspension was pipetted into individual 5 mL flow tubes. Each tube received 5 μL Annexin V and 5 μL propidium iodide (PI) staining solution (BD Pharmingen, catalog number #556547, USA), as per the manufacturer's instructions. The tubes were then gently shaken and incubated at room temperature, in the darkness, for 15 minutes. Post-staining, 400 μL cold binding buffer was introduced to each tube to dilute the cell suspension. The stained cells were analyzed using a flow cytometer within one hour.
Cell viability assay
The cytotoxicity of Bru NP, Sor NP, and B/S NP was assessed using a cell counting kit-8 assay (CCK-8, Vazyme Biotech Co., Ltd, China). In a typical process, WRL68 cells were seeded into ninety-six-well plates at a density of 5 ×103 cells per well for 24 hours. A fresh medium with varying concentrations of nanoparticles was used to replace the initial medium, and the cells were incubated for an additional 24 hours. After that, the CCK-8 reagent was added and the cells were incubated for 2 hours. The absorbance was then measured using a microplate reader (Bio-Rad 680, USA) at 450 nm.
To evaluate the therapeutic effect of the nanoplatform on MHCC-97H cells, we also utilized the CCK-8 assay. The process began by plating cells into ninety-six-well plates and subsequently incubating them with different treatments: PBS, Bru NP, Sor NP, and B/S NP. Each well received a unique treatment, with the incubation period lasting 24 hours. After incubation, the treatment-containing medium was discarded, and the cells were gently rinsed with PBS. After rinsing, the CCK-8 assay was performed to assess the cell viability, as detailed previously.
NP-mediated Nrf2 silencing
MHCC-97H cells, plated in six-well plates at a density of 5 × 104 cells per well, were allowed to settle overnight in a medium supplemented with 10% FBS for 24 hours. After the initial step, cells were subjected to exposure with various concentrations of B/S NP. The plates underwent incubation for an additional 24 hours. Upon completion of this period, the incubation medium was discarded, and the cells were rinsed with PBS to remove any residual nanoparticles. A fresh medium was then introduced, and the cells were further incubated for another 48 hours before harvesting using trypsin to detach them. RNA was isolated with TRIzol for subsequent qRT-PCR analysis, while proteins were extracted in parallel using a lysis buffer supplemented with a cocktail of protease inhibitors and PMSF, in preparation for western blotting (WB).
EdU assay
MHCC-97H cells were plated in twenty-four-well plates at a density of 5 × 104 cells per well and cultured in a medium supplemented with 10% FBS for a period of 24 hours. After that, PBS, Bru NP, Sor NP, and B/S NP were added separately. After 24 hours incubation, 100 μL of EdU (Beyotime, C0071S, China) working solution was added to each well and incubation was continued at 37 °C for 2 hours. Then the culture medium was discarded and washed three times with PBS. Following that, 50 μL of 4% paraformaldehyde was added to each well and the cells were fixed for 30 minutes to maintain their morphology and structure. Afterwards, 100 μL of permeabilizing solution (0.5% Triton X-100 in PBS) was added to each well and incubated at room temperature for 15 minutes. To each well, 0.2 mL of the Click reaction mixture was added. The culture plate was agitated to ensure that the mixture evenly coated the samples, followed by incubation in the darkness for one hour. Finally, 500 μL of 1× Hoechst (1:1000) solution was added to each well and incubated at room temperature in the darkness for about 10 minutes. Then, the EdU-labeled and -unlabeled cells were observed using an inverted fluorescence microscope (OLYMPUS, IX73, Japan).
Establishment of HCC tumor-bearing mouse models
All in vivo studies were carried out in the designated animal research facility, following ethical guidelines and protocols set by the Institutional Animal Care and Use Committee at Army Medical University (No. AMUWEC20245253). Male BALB/c (nude) mice (6 weeks old) were purchased from the Army Medical University Experimental Animal Center. The MHCC-97H xenograft tumor models in mice were developed through the injection of 200 μL of a cell suspension, consisting of MHCC-97H cells mixed with the medium and Matrigel at equal volumes (1:1), at a concentration of 5 × 106 cells per mL, into the dorsal area of healthy nude mice. These mice, bearing tumors, were then utilized for subsequent in vivo studies once the tumors had grown to a size between 100 and 150 mm3. The tumor volume was determined using the formula: V = W2 × L/2, with W representing the smallest diameter and L representing the largest diameter.
Evaluation of tumor treatment efficacy
The MHCC-97H xenograft mice were allocated into four distinct groups and received intravenous injections of either PBS, Bru NP, Sor NP, or B/S NP every two days. The NP dosage was set at 2 mg kg−1 for Bru and/or 5 mg kg−1 for Sor per mouse. Each mouse underwent this treatment regimen three times in total. Tumor expansion was tracked every two days by measuring the orthogonal diameters using a caliper. After the systemic treatment was finished, the mice were euthanized in a humane manner, and the tumor tissues were harvested and weighed. Finally, the tumor samples were prepared for staining of Nrf2, TUNEL, and Ki67, following the manufacturer's prescribed protocol.
Hemolysis assay
First, 500 μL of whole blood was collected from a mouse's orbital sinus. Following this, this sample was diluted by adding 5 mL PBS. The assay was continued by centrifuging the mixture at a speed of 10000 rpm for 5 minutes to sediment the cells. The supernatant was removed and the centrifugation and supernatant removal steps were repeated until the supernatant was clear. Once clear, the remaining pellet, mainly composed of red blood cells, was resuspended in 10 mL of PBS to achieve a homogeneous red suspension. For the control samples, 200 μL of the red blood cell suspension was combined with 800 μL of deionized water to serve as the positive control, and with 800 μL of PBS for the negative control. For the experimental groups, 200 μL of the suspension was combined with 800 μL of the test material and previously dissolved in PBS containing different doses of B/S NP. All the prepared samples were incubated at a temperature of 37 °C for a period of 4 hours to allow for interaction between the red blood cells and the B/S NP. Post-incubation, the samples were centrifuged once more at 10000 rpm for 5 minutes to pellet unlysed red blood cells. Then, 100 μL of the supernatant was transferred from each group to a ninety-six-well plate in preparation for absorbance measurements at a wavelength of 540 nm, which will indicate the extent of hemolysis under each condition. Hemolysis rate (%) = [(abs. sample − abs. negative)/(abs. positive − abs. negative)] × 100%.
Statistical analysis
Data were expressed as mean ± standard deviation. GraphPad Prism software, version 10.1.2, was utilized for creating illustrations, data visualization, and performing statistical analyses. Differences were considered statistically significant at levels of *P < 0.05, **P < 0.01, and ***P < 0.001.
Results and discussion
Detection of Nrf2 expression in Sor-insensitive HCC patients
To explore the expression of Nrf2 in Sor-insensitive HCC patients, tumor samples from these patients (n = 8) were collected and subjected to immunohistochemical analysis (IHC) (Fig. 1). Compared to Sor-sensitive patients (n = 8), a higher expression level of Nrf2 protein was obviously found in tissues from Sor-insensitive patients.37 The results suggest that down-regulating Nrf2 expression could potentially restore sensitivity to Sor treatment in HCC.38
 |
| | Fig. 1 IHC analysis revealed that the Nrf2 expression level in HCC tissues from sorafenib insensitive patients (n = 8) is significantly higher than that in sorafenib-sensitive patients (n = 8). Scale bar = 100 μm; *P < 0.05, **P < 0.01, and ***P < 0.001. | |
Preparation and characterization of the B/S NP
Bru, as an alkaloid, contains multiple ring structures and non-polar groups within its hydrophobic molecular structure.39 Consequently, Bru exhibits very low solubility in an aqueous environment (Fig. 2A).31 The B/S NP was prepared via self-assembly,40–42 using a GSH-responsive Meo-PEG-S-S-PLGA copolymer as an encapsulating shell to co-encapsulate Bru and Sor (Fig. 2B).43 Following ultrafiltration purification, the B/S NP appeared as a white turbid liquid (Fig. 2C). Subsequent characterization of the B/S NP was carried out using TEM, DLS, and zeta potential measurements. As shown in Fig. 2D and F, the B/S NP appeared spherical in shape with an average diameter of around 145 nanometers and was homogeneously dispersed in an aqueous medium.44 Furthermore, the zeta potential was measured to be −10.9 mV (Fig. 2E). As demarcated in Fig. S1,† both the B NP and the S NP exhibit spherical shapes, with particle sizes of approximately 198 nm and 101 nm, respectively. In addition, their zeta potentials are −13.3 mV and −12.9 mV, respectively. As shown in Fig. S2,† the average particle sizes of the B/S NP, B NP, and S NP were basically stable. These results indicated that the primary B/S NP, B NP, and S NP were successfully synthesized. The presence of disulfide bonds on the surface of the Meo-PEG-S-S-PLGA copolymer imparted reduction-responsive properties to the NP.45–49 Drug release experiments were conducted at low GSH concentrations of 1 mM and 5 mM and a high GSH concentration of 10 mM (approximating tumor cellular levels). As shown in Fig. 2G and H, at low GSH concentrations, only minimal drug release occurred, whereas at high GSH concentrations, significant amounts of both Bru and Sor were released from the B/S NP.50,51 These drug release results demonstrated the favorable responsive release characteristics of B/S NP in a high GSH environment.
 |
| | Fig. 2 Preparation and characterization of the B/S NP. (A) Chemical structure of brusatol. (B) Schematic of the co-delivery nanoplatform (B/S NP) synthesized with sorafenib, brusatol and Meo-PEG-S-S-PLGA via self-assembly. (C) Appearance of the B/S NP at room temperature. (D) Particle size and distribution of the B/S NP. (E) Zeta potential of B/S NP. (F) TEM image of the B/S NP. (G) Sorafenib and (H) brusatol release from the B/S NP incubated in a PBS solution containing varying concentrations of GSH for different durations (n = 3). Scale bar = 50 nm. ns, no significance, *P < 0.05, **P < 0.01, and ***P < 0.001. | |
Combination therapy enhances the sorafenib sensitivity of HCC cells
To investigate the effect of combined treatment on enhancing HCC cells' responsiveness to Sor, we initially identified human HCC cell lines with high Nrf2 expression using WB and qRT-PCR experiments. As illustrated in Fig. 3A, the WB data unveiled that SNU-449 and MHCC-97H human HCC cell lines harbored elevated Nrf2 protein expression levels relative to the normal liver cell line, WRL68. Additionally, qRT-PCR results indicated that higher mRNA levels of Nrf2 across all human HCC cell lines versus the normal liver cell line suggest potential post-transcriptional Nrf2 regulation. Based on these findings, SNU-449 and MHCC-97H cell lines were chosen for subsequent experiments. We then determined the optimal concentrations to quell Nrf2 expression via a dose-escalation method (Fig. 3B). It was ascertained that 140 nM of Bru effectively downregulated Nrf2 in both SNU-449 and MHCC-97H cell lines. The half-maximal inhibitory concentrations (IC 50) of Bru in these cell lines amounted to 412.7 nM for SNU-449 and 598.3 nM for MHCC-97H (Fig. 3C). Fig. 3D displays that reducing Nrf2 expression with Bru augmented Sor sensitivity in both SNU-449 and MHCC-97H cells.52 In SNU-449, the IC50 value was reduced from 62.99 μM to 32.50 μM, while in MHCC-97H, the IC50 value decreased from 59.56 μM to 35.85 μM. Due to the similar IC50 values in SNU-449 (62.99 μM) and MHCC-97H (59.56 μM), we used one-fourth of these IC50 values (≈15 μM) to carry out subsequent experiments. Besides, the Loewe additivity principle (x/DA + y/DB = 1) was used to calculate the synergy value in SNU-449 and MHCC-97H, with the synergy values being 0.859 and 0.836 (<1), respectively. These results indicated that Bru and Sor have a synergistic therapeutic effect on HCC. Furthermore, colony formation inhibition assays conducted in the Sor and Bru combination groups revealed that the combined treatment resulted in the least colony formation compared to single-drug treatments (Fig. 3E). Flow cytometry apoptosis results similarly demonstrated that Bru-induced Nrf2 silencing enhanced Sor-induced apoptosis in the SNU-449 and MHCC-97H cell lines (Fig. 3F). These experimental results collectively demonstrate that the combination therapy can enhance the sensitivity to Sor treatment and amplify its therapeutic efficacy.32
 |
| | Fig. 3 Combination therapy enhances the responsiveness of HCC cells to sorafenib. (A) Results of WB and qRT-PCR analysis of Nrf2 expression levels in the human normal liver cell line and various human HCC cell lines. (B) Effective concentration range of Bru for inhibiting Nrf2. (C) IC50 of brusatol measured in two HCC cell lines through CCK-8 assay. (n = 3). (D) IC50 values of sorafenib, with or without brusatol, measured through the CCK-8 assay in two HCC cell lines (n = 3). (E and F) Combined application of brusatol and sorafenib in the colony formation assay and flow cytometry apoptosis assay (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. | |
NP-mediated Nrf2 downregulating enhances sorafenib sensitivity in vitro
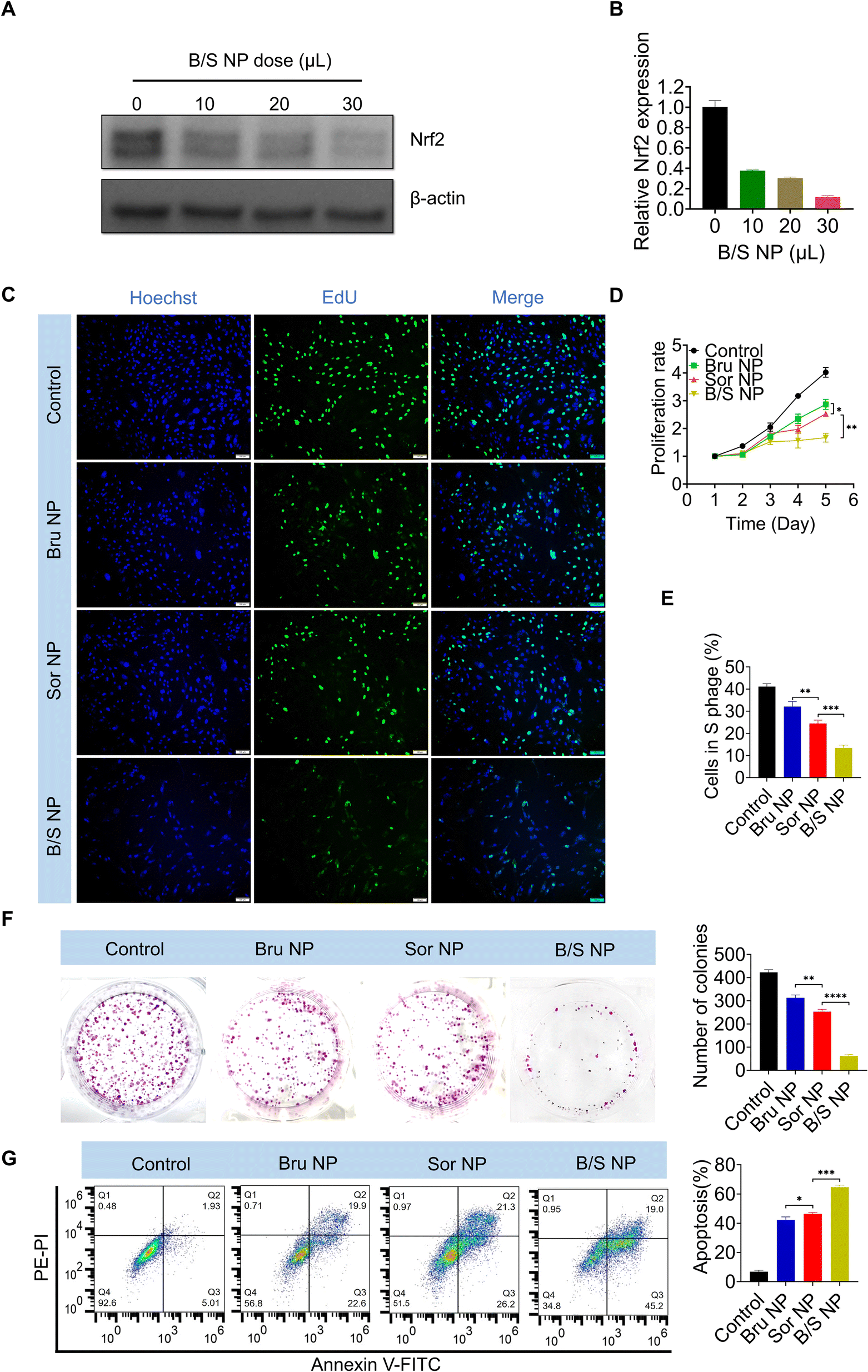
Given that Bru-induced downregulation of Nrf2 exhibited a strong capacity to modulate Sor sensitivity, and considering the poor water solubility of Bru, using a nanoplatform to co-deliver Bru and Sor (B/S NP) could effectively address the solubility issue and enhance their accumulation at the HCC site.53–57 Notably, western blot and qRT-PCR analysis demonstrated that the B/S NP efficiently downregulated Nrf2 expression in MHCC-97H cells in a dose-dependent manner (Fig. 4A and B). As shown in Fig. S3,† the B/S NP also showed advantageous performance over free B/S. As shown in the EdU assay, the presence of Bru significantly sensitized the inhibition of MHCC-97H cell growth by Sor, compared to the nanoplatform loaded with either Bru or Sor alone (i.e., Bru NP or Sor NP) (Fig. 4C and E). Besides, the sensitizing effect of Bru on Sor's inhibition of HCC cell proliferation was observed via the CCK-8 assay (Fig. 4D). The enhanced sensitivity to Sor treatment was further evident in the colony formation (Fig. 4F) and flow cytometry apoptosis assays (Fig. 4G), where the B/S NP demonstrated a stronger capability to inhibit colony formation and induce HCC apoptosis and in MHCC-97H cells than that of Bru NP or Sor NP. The phenomena of the assays above indicated that the B/S NP could significantly improve the sensitivity of Sor on HCC in vitro.
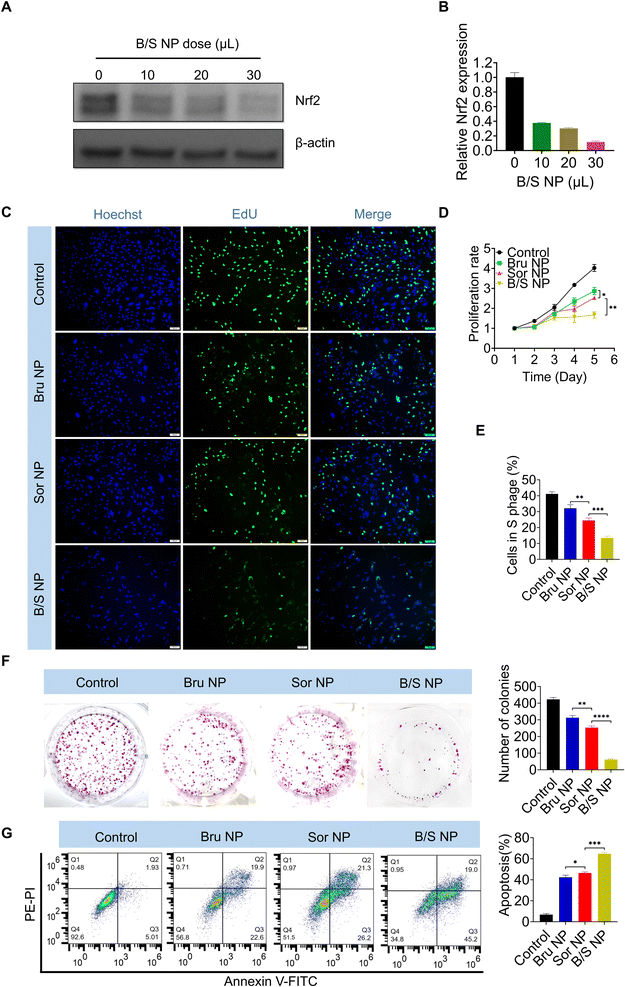 |
| | Fig. 4 NP-Mediated Nrf2 downregulation enhances sorafenib sensitivity in vitro. (A and B) WB and qRT-PCR results revealed the varying levels of Nrf2 expression in MHCC-97H cells following treatment with escalating doses of B/S NP. (C and E) EdU assay, (D) proliferation profile, (F) colony formation, and (G) flow cytometric analysis of MHCC-97H cells treated with PBS, Bru NP, Sor NP, and B/S NP (n = 3). Scale bar = 100 μm; *P < 0.05, **P < 0.01, and ***P < 0.001. | |
NP-mediated Nrf2 downregulation enhances sorafenib sensitivity in vivo
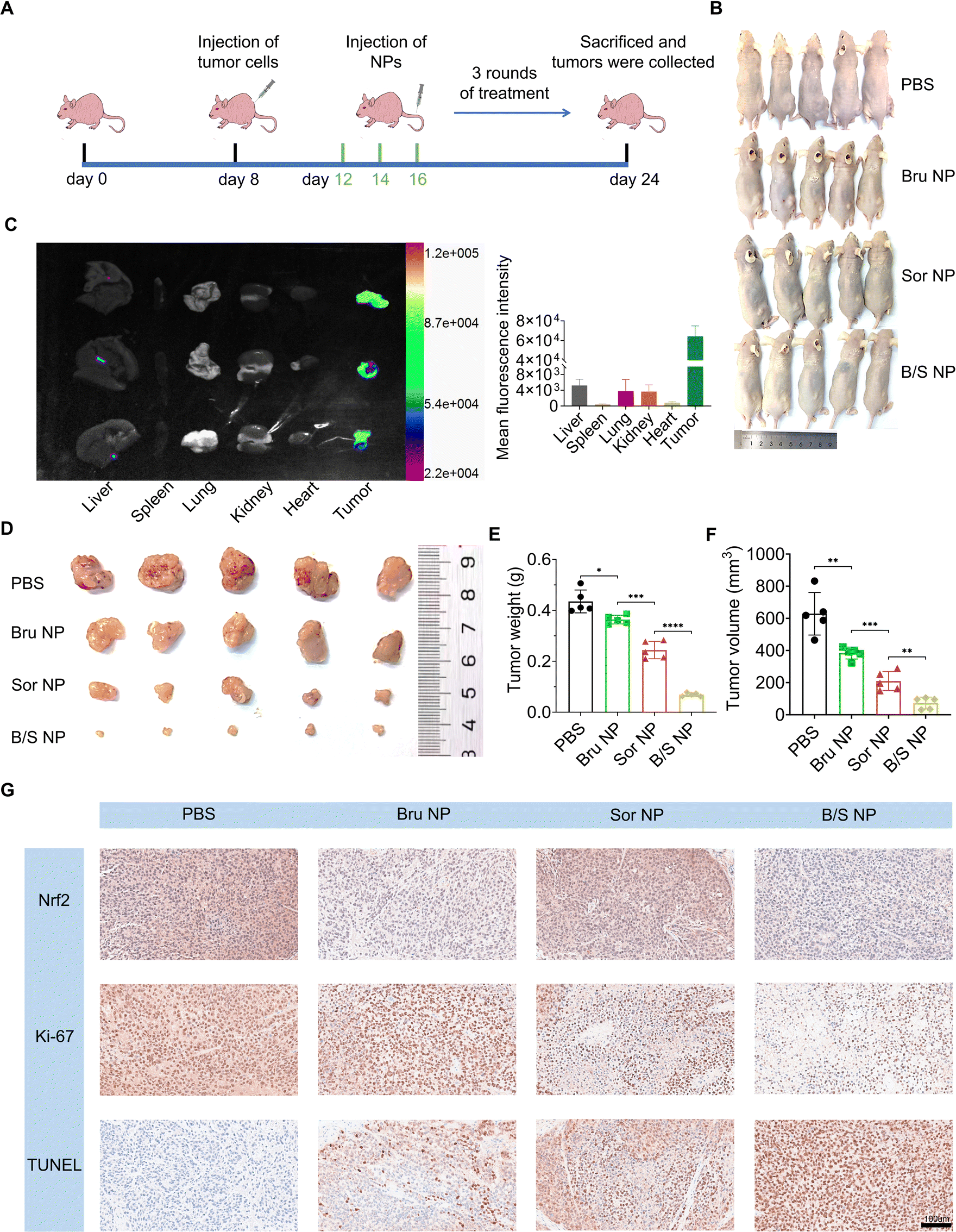
Having demonstrated that Nrf2 downregulation mediated and enhanced the HCC killing effect by the B/S NP in vitro, we proceeded to evaluate whether these nanoparticles could inhibit HCC tumor growth in vivo. We first established a subcutaneous tumor model in nude mice using MHCC-97H cells.58 PBS, Bru NP, Sor NP, and B/S NP were given through tail vein injections on alternate days, totaling three administrations (Fig. 5A). After 16 days of observation, all mice were photographed at the end of the treatment, and their tumors and major organs were collected for following experiments (Fig. 5B). First, we intravenously injected the B/S NP into three additional MHCC-97H xenograft tumor-bearing mice. After 24 hours, the mice were euthanized and the main organs and tumors were collected. We confirmed the significant enrichment of B/S NP at the tumor site compared to other major organs through small animal in vivo imaging.59–62 Due to the high tumor aggregation and GSH controlled release of B/S NP, coupled with the aggregation-caused quenching mechanism in the main organs,63 the overall fluorescence signal in the tumor region is significantly higher (Fig. 5C). As shown in Fig. 5E, the antitumor effects ranked from strongest to weakest were B/S NP, Sor NP, and Bru NP, compared to the PBS group. The B/S NP exhibited excellent tumor inhibition effects, effectively suppressing tumor growth in nude mice. Besides, the measurement results of tumor weight and volume also confirmed this conclusion (Fig. 5D and F). Finally, IHC findings from the tumor tissues also indicated that B/S NP had the strongest inhibitory effect on tumor growth, marked by decreased Nrf2 expression, reduced Ki67 staining (indicating less proliferation), and increased TUNEL staining (indicating more apoptosis) compared to mice treated with other regimens (Fig. 5G). The above-mentioned results indicated that the B/S NP also exhibited outstanding therapeutic efficacy against HCC in vivo.
 |
| | Fig. 5 NP-Mediated Nrf2 downregulating enhances sorafenib sensitivity in vivo. (A) Schematic illustration of MHCC-97H subcutaneous tumor models and the treatment process. (B) Pictures of tumor-carrying mice. (C) Fluorescent imaging of tumors and main organs. (D) Image of collected tumors, (E) tumor weight, and (F) tumor volume of tumor-carrying mice treated with PBS, Bru NP, Sor NP, and B/S NP (n = 5). (G) The levels of Nrf2, Ki-67, and TUNEL in mice’s tumor tissues were assessed following systemic treatment in each group through IHC staining analysis. Scale bar = 100 μm; *P < 0.05, **P < 0.01, and ***P < 0.001. | |
NP biosafety experiment
To determine the biosafety of this co-delivery nanoplatform, we carried out a comprehensive evaluation, encompassing both in vitro and in vivo studies.64–66 Initially, we conducted a hemolysis assay to assess the potential hemolytic effects of B/S NP. We observed that even with escalating concentrations, the B/S NP induced no significant hemolysis, indicating sufficient safety of these nanoparticles for red blood cells (Fig. 6A).67 Following this, the B/S NP was incubated with the normal human liver cell line WRL68 for 24 hours. Utilizing the CCK-8 assay, we determined that the B/S NP did not significantly inhibit the viability of WRL68 cells (Fig. 6B). These in vitro findings suggested that our nanoparticles exhibit no noticeable toxicity. Then in the in vivo experiment, we measured a spectrum of blood biochemical parameters to further probe the safety profile of the nanoparticles. These included alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), creatinine (CREA), urea, and total protein (TP) levels. Mice were treated with PBS, Bru NP, Sor NP, and B/S NP, and these parameters were analyzed 24 h post-treatment. The data revealed that all values fell within the normal range, indicating no adverse effects on renal or liver function (Fig. 6C). Besides, we tracked the body weight of the mice throughout the study and found no significant variations among the treatment groups (Fig. 6D). After three cycles of treatment, histological examination of major organs using hematoxylin-eosin (H&E) staining showed no discernible differences across the treatment groups, indicating the absence of treatment-induced organ damage (Fig. 6E). These observations collectively pointed to the good biocompatibility and tolerability of the B/S NP.44,68
 |
| | Fig. 6 Biosafety experimental results of the co-delivery nanoplatform in vitro and in vivo. (A) Hemolysis experiment results at different B/S NP doses (n = 3). (B) Viability of WRL68 cells incubated with different B/S NP doses, determined by CCK-8 (n = 3). (C) Serum levels of ALT, AST, ALP, CREA, urea and TP in healthy mice that received three consecutive intravenous injections of PBS, Bru NP, Sor NP, and B/S NP (n = 3) (the gray area represents the normal range of the corresponding serum biomarkers). (D) Nude mouse weight in different groups (n = 5). (E) H&E staining analysis of major organs from healthy mice that received three consecutive intravenous injections of PBS, Bru NP, Sor NP, and B/S NP (n = 3). Scale bar = 100 μm. | |
Conclusion and discussion
In summary, we have successfully developed a co-delivery nanoplatform, designated as B/S NP, aimed at improving HCC therapy. This platform is characterized by its high tumor targeting, outstanding GSH sensitivity, and excellent aqueous dispersibility, enabling effective co-encapsulation of Bru and Sor. Both in vitro and in vivo studies have demonstrated that the B/S NP effectively downregulated Nrf2 expression, the Sor resistance-associated gene in HCC cells, significantly inhibiting HCC tumor growth. This overcomes the issue of Bru's poor aqueous solubility and enhances the low sensitivity of Sor in treating HCC. Furthermore, the biosafety of the B/S NP has been validated. Although previous studies have revealed the potential of Bru to sensitize chemotherapy,69–72 our study, for the first time, demonstrated the previously unreported ability of Bru to synergistically sensitize the targeted drug Sor in the treatment of HCC both in vitro and in vivo through the construction of B/S NP. These features highlight B/S NP's potential in HCC combination therapy and drug delivery, presenting a promising future therapeutic strategy for sensitizing HCC treatment.
Our study has certain limitations. First, although our B/S NP ensures sufficient safety, it does not fully address the sensitivity and completeness of drug release. Second, the construction of B/S NP is currently in the proof-of-concept stage, and the specific arrangement of Sor and Bru molecules within the nanoplatform has not been thoroughly investigated. Future studies should focus on enhancing the safety and release efficiency of the nanoplatform, as well as exploring the optimized Sor and Bru molecule arrangement within the nanoplatform.
Data availability
The data are available upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by research grants from Chongqing Graduate Research Innovation Project (CYS240818), Key Project of Tongliang District People's Hospital (Y2024-3), the National Science Foundation of China (82403679 and 82203676), Young Doctoral Innovation Project of the Second Affiliated Hospital of Army Medical University (2024YQB011), and Sichuan Provincial Natural Science Foundation for Young Scholars (2024NSFSC1748).
References
- S. Qin, F. Bi, S. Gu, Y. Bai, Z. Chen, Z. Wang, J. Ying, Y. Lu, Z. Meng, H. Pan, P. Yang, H. Zhang, X. Chen, A. Xu, C. Cui, B. Zhu, J. Wu, X. Xin, J. Wang, J. Shan, J. Chen, Z. Zheng, L. Xu, X. Wen, Z. You, Z. Ren, X. Liu, M. Qiu, L. Wu and F. Chen, J. Clin. Oncol., 2021, 39, 3002–3011 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- J. Long, K. Cui, D. Wang, S. Qin and Z. Li, Cancer Control, 2024, 31, 10732748241310573 CrossRef PubMed.
- D. Anwanwan, S. K. Singh, S. Singh, V. Saikam and R. Singh, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1873, 188314 CrossRef CAS PubMed.
- C. Yang, H. Zhang, L. Zhang, A. X. Zhu, R. Bernards, W. Qin and C. Wang, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 203–222 CrossRef PubMed.
- C. R. de Lope, S. Tremosini, A. Forner, M. Reig and J. Bruix, J. Hepatol., 2012, 56(Suppl 1), S75–S87 CrossRef PubMed.
- F. H. Kong, Q. F. Ye, X. Y. Miao, X. Liu, S. Q. Huang, L. Xiong, Y. Wen and Z. J. Zhang, Theranostics, 2021, 11, 5464–5490 CrossRef CAS PubMed.
- J. M. Llovet, R. K. Kelley, A. Villanueva, A. G. Singal, E. Pikarsky, S. Roayaie, R. Lencioni, K. Koike, J. Zucman-Rossi and R. S. Finn, Nat. Rev. Dis. Primers, 2021, 7, 6 CrossRef PubMed.
- S. Qin, S. L. Chan, S. Gu, Y. Bai, Z. Ren, X. Lin, Z. Chen, W. Jia, Y. Jin, Y. Guo, X. Hu, Z. Meng, J. Liang, Y. Cheng, J. Xiong, H. Ren, F. Yang, W. Li, Y. Chen, Y. Zeng, A. Sultanbaev, M. Pazgan-Simon, M. Pisetska, D. Melisi, D. Ponomarenko, Y. Osypchuk, I. Sinielnikov, T. S. Yang, X. Liang, C. Chen, L. Wang, A. L. Cheng, A. Kaseb and A. Vogel, Lancet, 2023, 402, 1133–1146 CrossRef CAS PubMed.
- S. Xia, Y. Pan, Y. Liang, J. Xu and X. Cai, EBioMedicine, 2020, 51, 102610 CrossRef PubMed.
- J. K. Byun, S. Lee, G. W. Kang, Y. R. Lee, S. Y. Park, I. S. Song, J. W. Yun, J. Lee, Y. K. Choi and K. G. Park, J. Exp. Clin. Cancer Res., 2022, 41, 98 CrossRef CAS PubMed.
- J. M. Llovet, S. Ricci, V. Mazzaferro, P. Hilgard, E. Gane, J. F. Blanc, A. C. de Oliveira, A. Santoro, J. L. Raoul, A. Forner, M. Schwartz, C. Porta, S. Zeuzem, L. Bolondi, T. F. Greten, P. R. Galle, J. F. Seitz, I. Borbath, D. Häussinger, T. Giannaris, M. Shan, M. Moscovici, D. Voliotis and J. Bruix, N. Engl. J. Med., 2008, 359, 378–390 CrossRef CAS PubMed.
- A. D. Ladd, S. Duarte, I. Sahin and A. Zarrinpar, Hepatology, 2024, 79, 926–940 Search PubMed.
- Y. J. Zhu, B. Zheng, H. Y. Wang and L. Chen, Acta Pharmacol. Sin., 2017, 38, 614–622 CrossRef CAS PubMed.
- M. Yamamoto, T. W. Kensler and H. Motohashi, Physiol. Rev., 2018, 98, 1169–1203 CrossRef CAS PubMed.
- S. Adinolfi, T. Patinen, A. Jawahar Deen, S. Pitkänen, J. Härkönen, E. Kansanen, J. Küblbeck and A. L. Levonen, Redox Biol., 2023, 63, 102726 CrossRef CAS PubMed.
- D. Xue, X. Zhou and J. Qiu, Biomed. Pharmacother., 2020, 131, 110676 CrossRef CAS PubMed.
- I. S. Harris and G. M. DeNicola, Trends Cell Biol., 2020, 30, 440–451 CrossRef CAS PubMed.
- K. Chang, Y. Chen, X. Zhang, W. Zhang, N. Xu, B. Zeng, Y. Wang, T. Feng, B. Dai, F. Xu, D. Ye and C. Wang, Cancer Res., 2023, 83, 3940–3955 CrossRef CAS PubMed.
- Q. Wang, C. Bin, Q. Xue, Q. Gao, A. Huang, K. Wang and N. Tang, Cell Death Dis., 2021, 12, 426 CrossRef CAS PubMed.
- F. L. Xu, X. H. Wu, C. Chen, K. Wang, L. Y. Huang, J. Xia, Y. Liu, X. F. Shan and N. Tang, Cell Death Dis., 2023, 14, 22 CrossRef CAS PubMed.
- L. Avila-Carrasco, P. Majano, J. A. Sánchez-Toméro, R. Selgas, M. López-Cabrera, A. Aguilera and G. González Mateo, Front. Pharmacol., 2019, 10, 715 CrossRef CAS PubMed.
- Y. Zhang, P. Ye, H. Zhu, L. Gu, Y. Li, S. Feng, Z. Zeng, Q. Chen, B. Zhou and X. Xiong, CNS Neurosci. Ther., 2024, 30, e14456 Search PubMed.
- J. Yan, Z. Li, Y. Liang, C. Yang, W. Ou, H. Mo, M. Tang, D. Chen, C. Zhong, D. Que, L. Feng, H. Xiao, X. Song and P. Yang, Food Funct., 2023, 14, 10052–10068 Search PubMed.
- S. J. Cai, Y. Liu, S. Han and C. Yang, Cell Biosci., 2019, 9, 45 Search PubMed.
- D. D. Zhang and E. Chapman, Nat. Prod. Rep., 2020, 37, 797–826 RSC.
- J. Zhang, H. X. Xu, J. Q. Zhu, Y. X. Dou, Y. F. Xian and Z. X. Lin, Int. J. Biol. Sci., 2023, 19, 3029–3041 Search PubMed.
- L. E. Armstrong and E. C. Johnson, Nutrients, 2018, 10(12), 1928 Search PubMed.
- L. Petraccia, G. Liberati, S. G. Masciullo, M. Grassi and A. Fraioli, Clin. Nutr., 2006, 25, 377–385 Search PubMed.
- Y. Yamada, X. Zhang, M. E. T. Henderson, H. Sagayama, H. Pontzer, D. Watanabe, T. Yoshida, M. Kimura, P. N. Ainslie, L. F. Andersen, L. J. Anderson, L. Arab, I. Baddou, K. Bedu-Addo, E. E. Blaak, S. Blanc, A. G. Bonomi, C. V. C. Bouten, P. Bovet, M. S. Buchowski, N. F. Butte, S. G. Camps, G. L. Close, J. A. Cooper, R. Cooper, S. K. Das, L. R. Dugas, S. Eaton, U. Ekelund, S. Entringer, T. Forrester, B. W. Fudge, A. H. Goris, M. Gurven, L. G. Halsey, C. Hambly, A. El Hamdouchi, M. B. Hoos, S. Hu, N. Joonas, A. M. Joosen, P. Katzmarzyk, K. P. Kempen, W. E. Kraus, W. Kriengsinyos, R. F. Kushner, E. V. Lambert, W. R. Leonard, N. Lessan, C. K. Martin, A. C. Medin, E. P. Meijer, J. C. Morehen, J. P. Morton, M. L. Neuhouser, T. A. Nicklas, R. M. Ojiambo, K. H. Pietiläinen, Y. P. Pitsiladis, J. Plange-Rhule, G. Plasqui, R. L. Prentice, R. A. Rabinovich, S. B. Racette, D. A. Raichlen, E. Ravussin, L. M. Redman, J. J. Reilly, R. M. Reynolds, S. B. Roberts, A. J. Schuit, L. B. Sardinha, A. M. Silva, A. M. Sjödin, E. Stice, S. S. Urlacher, G. Valenti, L. M. Van Etten, E. A. Van Mil, J. C. K. Wells, G. Wilson, B. M. Wood, J. A. Yanovski, A. J. Murphy-Alford, C. U. Loechl, A. H. Luke, J. Rood, K. R. Westerterp, W. W. Wong, M. Miyachi, D. A. Schoeller and J. R. Speakman, Science, 2022, 378, 909–915 Search PubMed.
- J. Zhou, L. Tan, J. Xie, Z. Lai, Y. Huang, C. Qu, D. Luo, Z. Lin, P. Huang, Z. Su and Y. Xie, Drug Delivery, 2017, 24, 1667–1679 Search PubMed.
- T. He, F. Zhou, A. Su, Y. Zhang, Z. Xing, L. Mi, Z. Li and W. Wu, Biomed. Pharmacother., 2023, 158, 114134 Search PubMed.
- X. Ma, S. J. Li, Y. Liu, T. Zhang, P. Xue, Y. Kang, Z. J. Sun and Z. Xu, Chem. Soc. Rev., 2022, 51, 5136–5174 Search PubMed.
- Y. Zhu, Y. He, T. Su, C. Li, S. Cai, Z. Wu, D. Huang, X. Zhang, J. Cao and B. He, J. Mater. Chem. B, 2020, 8, 5109–5116 Search PubMed.
- S. Kiran, P. Dwivedi, R. Khatik, S. Hameed, M. Dwivedi, F. Huang and R. X. Xu, Chem. Commun., 2019, 56, 285–288 Search PubMed.
- Y. Hao, Y. Chen, X. He, R. Han, C. Yang, T. Liu, Y. Yang, Q. Liu and Z. Qian, Biomaterials, 2023, 293, 121975 Search PubMed.
- M. C. Jaramillo and D. D. Zhang, Genes Dev., 2013, 27, 2179–2191 Search PubMed.
- H. Kitamura and H. Motohashi, Cancer Sci., 2018, 109, 900–911 CrossRef CAS PubMed.
- J. Zhang, X. Fang, Z. Li, H. F. Chan, Z. Lin, Y. Wang and M. Chen, Int. J. Nanomed., 2018, 13, 939–956 CrossRef CAS PubMed.
- S. Li, L. Xu, G. Wu, Z. Huang, L. Huang, F. Zhang, C. Wei, Q. Shen, R. Li, L. Zhang and X. Xu, Adv. Sci., 2023, 10, e2207118 CrossRef PubMed.
- H. Yi, P. Liu, N. Sheng, P. Gong, Y. Ma and L. Cai, Nanoscale, 2016, 8, 5985–5995 RSC.
- Y. Tang, Y. Li, R. Xu, S. Li, H. Hu, C. Xiao, H. Wu, L. Zhu, J. Ming, Z. Chu, H. Xu, X. Yang and Z. Li, Nanoscale, 2018, 10, 17265–17274 Search PubMed.
- D. Zhou, Z. Fei, L. Jin, P. Zhou, C. Li, X. Liu and C. Zhao, J. Mater. Chem. B, 2021, 9, 801–808 Search PubMed.
- C. Hong, T. Chen, M. Wu, J. Lin, C. Gao, X. Ma, Z. Liu, X. Yang and A. Wu, J. Mater. Chem. B, 2023, 11, 8866–8882 RSC.
- W. Ma, X. Wang, D. Zhang and X. Mu, Int. J. Nanomed., 2024, 19, 7547–7566 CrossRef PubMed.
- S. Huang, Z. Xu, W. Zhi, Y. Li, Y. Hu, F. Zhao, X. Zhu, M. Miao and Y. Jia, J. Nanobiotechnol., 2024, 22, 324 CrossRef CAS PubMed.
- Q. Li, R. Wang, S. Han, N. Shi, J. Yang, C. Ping, L. Chai, R. Wang, B. Zheng, G. Ren and S. Zhang, Mol. Pharm., 2024, 21, 5551–5564 CrossRef CAS PubMed.
- Y. Pan, J. Cheng, Y. Zhu, J. Zhang, W. Fan and X. Chen, Chem. Soc. Rev., 2024, 53, 6399–6444 RSC.
- D. Zhou, S. Liu, Y. Hu, S. Yang, B. Zhao, K. Zheng, Y. Zhang, P. He, G. Mo and Y. Li, J. Mater. Chem. B, 2020, 8, 3801–3813 RSC.
- J. Qu, R. Wang, S. Peng, M. Shi, S. T. Yang, J. B. Luo, J. Lin and Q. H. Zhou, J. Mater. Chem. B, 2019, 7, 7129–7140 RSC.
- Z. Su, Y. Xu, Y. Wang, W. Shi, S. Han and X. Shuai, Biomater. Sci., 2019, 7, 3821–3831 RSC.
- Y. Murakami, K. Sugiyama, H. Ebinuma, N. Nakamoto, K. Ojiro, P. S. Chu, N. Taniki, Y. Saito, T. Teratani, Y. Koda, T. Suzuki, K. Saito, M. Fukasawa, M. Ikeda, N. Kato, T. Kanai and H. Saito, BMC Cancer, 2018, 18, 680 CrossRef PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
- V. Verma, K. M. Ryan and L. Padrela, Int. J. Pharm., 2021, 603, 120708 CrossRef CAS PubMed.
- K. U. Khan, M. U. Minhas, S. F. Badshah, M. Suhail, A. Ahmad and S. Ijaz, Life Sci., 2022, 291, 120301 CrossRef CAS PubMed.
- H. A. Santos and I. N. Savina, RSC Adv., 2023, 13, 1933–1934 RSC.
- W. M. Kedir, L. Li, Y. S. Tan, N. Bajalovic and D. K. Loke, J. Mater. Chem. B, 2024, 12, 12141–12173 RSC.
- Y. Zhou, F. Lin, T. Wan, A. Chen, H. Wang, B. Jiang, W. Zhao, S. Liao, S. Wang, G. Li, Z. Xu, J. Wang, J. Zhang, H. Ma, D. Lin and Q. Li, Theranostics, 2021, 11, 5926–5938 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- M. Ikeda-Imafuku, L. L. Wang, D. Rodrigues, S. Shaha, Z. Zhao and S. Mitragotri, J. Controlled Release, 2022, 345, 512–536 CrossRef CAS PubMed.
- Q. Guo, S. Wang, R. Xu, Y. Tang and X. Xia, RSC Adv., 2024, 14, 10608–10637 RSC.
- C. Wang, Y. Tang, C. Li, W. Wu and X. Jiang, Biomater. Sci., 2025, 13(3), 617–626 RSC.
- A. T. Turley, P. K. Saha, A. Danos, A. N. Bismillah, A. P. Monkman, D. S. Yufit, B. F. E. Curchod, M. K. Etherington and P. R. McGonigal, Angew Chem. Int. Ed. Engl., 2022, 61, e202202193 CrossRef CAS PubMed.
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS PubMed.
- F. Gao, L. Li, T. Liu, N. Hao, H. Liu, L. Tan, H. Li, X. Huang, B. Peng, C. Yan, L. Yang, X. Wu, D. Chen and F. Tang, Nanoscale, 2012, 4, 3365–3372 RSC.
- S. Duan, Y. Hu, Y. Zhao, K. Tang, Z. Zhang, Z. Liu, Y. Wang, H. Guo, Y. Miao, H. Du, D. Yang, S. Li and J. Zhang, RSC Adv., 2023, 13, 14443–14460 RSC.
- C. T. Pham, D. G. Thomas, J. Beiser, L. M. Mitchell, J. L. Huang, A. Senpan, G. Hu, M. Gordon, N. A. Baker, D. Pan, G. M. Lanza and D. E. Hourcade, Nanomedicine, 2014, 10, 651–660 CrossRef CAS PubMed.
- Y. Pan, W. Tang, W. Fan, J. Zhang and X. Chen, Chem. Soc. Rev., 2022, 51, 9759–9830 RSC.
- H. M. Chen, Z. Q. Lai, H. J. Liao, J. H. Xie, Y. F. Xian, Y. L. Chen, S. P. Ip, Z. X. Lin and Z. R. Su, Int. J. Mol. Med., 2018, 41, 1447–1454 CAS.
- D. Ren, N. F. Villeneuve, T. Jiang, T. Wu, A. Lau, H. A. Toppin and D. D. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1433–1438 CrossRef CAS PubMed.
- Y. Xiang, W. Ye, C. Huang, D. Yu, H. Chen, T. Deng, F. Zhang, B. Lou, J. Zhang, K. Shi, B. Chen and M. Zhou, Oxid. Med. Cell. Longevity, 2018, 2018, 2360427 CrossRef PubMed.
- J. Xie, Z. Lai, X. Zheng, H. Liao, Y. Xian, Q. Li, J. Wu, S. Ip, Y. Xie, J. Chen, Z. Su, Z. Lin and X. Yang, Toxicology, 2021, 451, 152680 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Lu Zheng*a
*a and
Lu Zheng*a