
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in cellulose-based hydrogels: tunable swelling dynamics and their versatile real-time applications
Md. Mahamudul Hasan Rumon
 *
*
Department of Mathematics and Natural Sciences, Brac University, 66 Mohakhali, Dhaka 1212, Bangladesh. E-mail: mhrumon.ku@gmail.com
First published on 14th April 2025
Abstract
Cellulose-derived hydrogels have emerged as game-changing materials in biomedical research, offering an exceptional combination of water absorption capacity, mechanical resilience, and innate biocompatibility. This review explores the intricate mechanisms that drive their swelling behaviour, unravelling how molecular interactions and network architectures work synergistically to enable efficient water retention and adaptability. Their mechanical properties are explored in depth, with a focus on innovative chemical modifications and cross-linking techniques that enhance strength, elasticity, and functional versatility. The versatility of cellulose-based hydrogels shines in applications such as wound healing, precision drug delivery, and tissue engineering, where their biodegradability, biocompatibility, and adaptability meet the demands of cutting-edge healthcare solutions. By weaving together recent breakthroughs in their development and application, this review highlights their transformative potential to redefine regenerative medicine and other biomedical fields. Ultimately, it emphasizes the urgent need for continued research to unlock the untapped capabilities of these extraordinary biomaterials, paving the way for new frontiers in healthcare innovation.
 Md. Mahamudul Hasan Rumon | Md. Mahamudul Hasan Rumon is a dedicated researcher in materials science, with expertise in supramolecular chemistry, hydrogels, nanocomposites, and biomaterials. He earned his BSc in Chemistry from Khulna University (KU), Bangladesh, and later completed his MSc in Chemistry at Bangladesh University of Engineering and Technology (BUET), Dhaka. Following his graduate studies, he joined BRAC University as a faculty member in the Mathematics and Natural Sciences (MNS) Department, where he contributed to teaching and research in advanced materials and their applications in drug delivery, nano crosslinkers, and wound healing. Currently, Rumon is pursuing a PhD in Materials Science at Indiana University Bloomington, USA, focusing on supramolecular polymers. His research aims to develop innovative, high-performance biomaterials with real-world applications in healthcare, pharmaceuticals, and sustainability. With a strong background in interdisciplinary research, Rumon has contributed to numerous scientific publications and collaborative projects. His passion for bridging fundamental science with practical applications drives his work in developing next-generation materials that address critical global challenges. |
1. Introduction
The effectiveness of hydrogels in biomedical applications is fundamentally influenced by their compatibility with natural tissues, mechanical properties, and swelling behaviours.1,2 These characteristics are predominantly governed by dynamic bonding interactions, including dynamic covalent bond, hydrogen bonding, hydrophobic interactions, Schiff-base interactions, π–π stacking, ionic bonding, and electrostatic forces.3–5 Additionally, external stimuli – such as temperature, pH, salt concentration, light, and electric fields – significantly impact the mechanical and swelling behaviour of gels.6 Despite considerable advancements in hydrogel research, conventional materials still encounter limitations, particularly regarding their responsiveness to stimuli like pH-triggered swelling and their other properties.7,8 Traditional hydrogels often lack adequate swelling ability and fail to deliver controlled release profiles, which can impede wound healing and other applications.9 Such deficiencies may lead to complications, such as adhesion and scab formation at the dressing site, thereby increasing the risk of exogenous infections.10–12 Furthermore, their effectiveness in managing infected wounds remains insufficient, highlighting the emergency for the development of bioactive dressings that can promptly address wound infections and promote the overall healing process.13Designing hydrogels for precise bioactive molecule delivery presents significant challenges, particularly in managing antibiotic release.14 An excessive release can lead to systemic toxicity, while insufficient release could promote the development of antibiotic-resistant bacteria.15,16 As bacterial proliferation occurs, the microenvironment of bio-tissues typically becomes acidic, emphasizing the necessity for hydrogels able to adjust antibiotic release in response to pH variations.17 An ideal pH-sensitive hydrogel would degrade in the presence of proliferating bacteria, facilitating the localized release of antibiotics to combat infections. Importantly, this degradation should cease once the wound area returns to a neutral pH.18 However, conventional hydrogels often exhibit uncontrolled swelling behaviour, which can compromise their efficacy.19 A promising strategy to address this issue involves the incorporation of polymers into hydrogel systems to develop interpenetrating or semi-interpenetrating networks, thus enhancing the mechanical strength of the hydrogels.20,21
Cellulose, the most abundant natural polymer, presents substantial potential for hydrogel fabrication due to its inherent hydrophilicity and biodegradability.22 Research has successfully demonstrated the development of macroporous hydrogels using cellulose as a support material, with acidic cellulose serving as a pore expander, resulting in hydrogels with high swelling ratios (SR) in aqueous environments.23,24 Recent studies have also reported the synthesis of carboxymethyl cellulose (CMC) through the carboxymethylation of cellulose using sodium monochloroacetate in a single solvent system.25 These advancements illustrate the growing interest in novel cellulose-based smart hydrogels that exhibit pH and salt sensitivity, underscoring cellulose 's versatility as a candidate for intelligent materials owing to its abundant hydroxyl groups.26 Furthermore, using an NaOH/urea aqueous solution as a homogeneous derivation system through environmentally friendly processes has emerged as a favourable method for constructing hydrogel networks via both chemical and physical cross-linking.27
This comprehensive review aims to provide a thorough overview for the research community, focusing on the mechanistic study of highly swellable gel materials. It will cover fundamental concepts, general classifications, and hydrogel structures, elucidating how the swelling behaviour of cellulose-based hydrogels is influenced by various physicochemical properties and their relevance to biomaterials. A deeper understanding of these mechanisms will enhance knowledge in polymer physics and chemistry, guiding the rational design of custom-tailored cellulose hydrogels for specific applications. From controlled drug delivery systems to responsive materials in smart devices, a nuanced comprehension of swelling behaviour mechanisms will foster innovation. Additionally, the review will highlight recent advancements in integrating highly swellable hydrogel-based materials into biomedical and environmental applications to improve performance and stability. In conclusion, this review will outline existing challenges and potential opportunities, emphasizing the promising future of intelligently designed hydrogels through gelation chemistry in biomedical applications.
2. Cellulose derivatives
Cellulose derivatives can be derived by the etherification process, wherein the OH groups of the glucose unit of cellulose are subjected to various reactions alongside organic compounds like methyl, amine, amide, and ethyl groups, shown in Scheme 1.28–30 However, most of the cellulose derivatives, e.g., NaCMC, methyl cellulose (MC), ethyl cellulose (EC), hydroxyethyl methyl cellulose (HEMC), and hydroxypropyl cellulose (HPC), are mainly ether-functionalized compounds, where OH functional groups present at C-2, C-3, and/or C-6 undergo dehydration to form ethers under various reaction conditions,28,31 shown in Scheme 2. | ||
| Scheme 1 Schematic presentation of cellulose and its various derivatise. The idea of this figure is taken from the ref. 28. Copyright © 2018, Springer Nature. | ||
 | ||
| Scheme 2 Representation of the various approached for the transformations of cellulose to its derivatives. | ||
The obtained derivative products are more reactive and have shown improved solubility in various polar and nonpolar solvents, allowing them more applicability than the mother cellulose structure.32 The controllable substitution of different reactive groups within the sugar subunit allows cellulose and its derivatives to possess sufficient water solubility and viscosity in aqueous solutions. Additionally, from the overall analysis, the average number of hydroxyl groups in a single glucose moiety that is etherified makes it possible to calculate the degree of substitution.33
3. Dynamic bonding mechanisms in cellulose-based hydrogels
Cellulose, composed of linear and fibrous chains of glucose units linked by β-glycosidic bonds (C1 → C4), exhibits numerous biocompatible properties, including biocompatibility, biodegradability, and mechanical toughness.34,35 The presence of polar and hydrophilic functional groups, such as –OH, –COOH, and –NH2, facilitates the application of cellulose and its derivatives in developing hydrogels for biomedical purposes.26 These hydrogels are synthesized through various dynamic and reversible physical and chemical bonding mechanisms. The inherent hydrophilicity and significant intramolecular crosslinking complicate their disintegration under specific conditions.36,37Recent advances in hydrogel synthesis have led to the development of physically crosslinked (PC) hydrogels that do not rely on chemical crosslinked (CC) hydrogels. Fig. 1 illustrating various physically crosslinking mechanism for the various cellulose based hydrogels.38 It is advisable to avoid chemical crosslinkers before clinical trials to minimize potential toxicity risks, as they may compromise the integrity of embedded compounds.39–41 PC hydrogels exhibit dynamic and reversible bond formation through non-covalent interactions, such as surface adsorption between adjacent polymer chains.41,42 Conformational optimization in PC hydrogels can be achieved through various interactions, including hydrophobic interactions, van der Waals forces, π–π stacking, hydrogen bonding, ionic interactions, and complexation.43 Various mechanisms of physically crosslinked hydrogels are represented in Fig. 1.
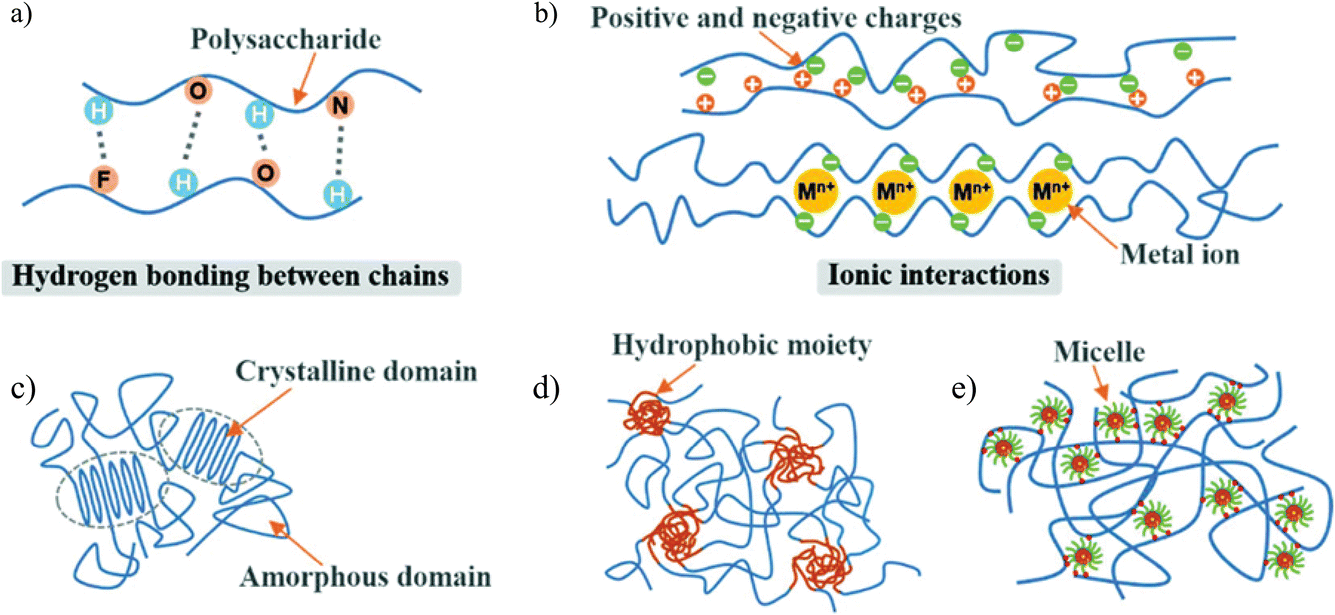 | ||
| Fig. 1 Illustrations of mechanism of various physically crosslinked cellulose-based hydrogels; (a) hydrogen bonding, (b) ionic bonding, (c) phase separation, or adsorption interactions, (d) hydrophobic interaction, and (e) micellar crosslinking. The figure is adopted with permission from ref. 38. Copyright (2020), Royal Society of Chemistry. | ||
Hydrogen bonding interactions (HBI) are a widely utilized approach in the physical crosslinking of supramolecular hydrogels.44,45 Due to its unique orientation and adaptability, HBI significantly influences essential biological processes, including DNA recombination, molecular recognition, and protein folding.46 HBI occurs when a hydrogen atom interacts with a highly electronegative heteroatom, such as oxygen, nitrogen, or halogens, which possess lone-pair electrons.47 This interaction is particularly advantageous in supramolecular hydrogel systems, as it enhances mechanical strength, promotes flexible chain orientation, and allows for reversible crosslinking, often outperforming other non-covalent interactions.48,49
The mechanical strength of HBI-based gels is primarily influenced by solvent polarity, the percentage of HBI active sites, the total number of hydrogen bond donors and acceptors, and the arrangement of these functional groups.50–52 Consequently, the strength of hydrogen bonds can vary from highly dynamic to nearly covalent, providing extensive opportunities for the design of HBI-based hydrogels.53 Researchers worldwide have leveraged the potential of HBI to develop diverse polymers, exploring novel concepts related to cellulose-based hydrogels.54 The numerous polar functional groups in cellulose, particularly the abundant hydroxyl groups on its surface, contribute to its supramolecular structure involving HBI.55,56 Inspired by the complex chemistry of mussels, Wu et al.,57 introduced a method for developing durable and responsive hydrogels using microcrystalline cellulose (MCC) and konjac glucomannan. This hydrogel exhibited significant intermolecular HBI, exceptional mechanical toughness, rapid self-healing abilities, improved pH properties, and reduced initial burst release compared to KGM hydrogels, highlighting its potential as a carrier for controlled biomolecule and drug delivery.57
Biyani et al.,58 explored the synthesis of a supramolecular polymer composite by modifying cellulose nanocrystals (CNCs) with 2-ureido-4-pyrimidone (UPy). The resulting nanocomposite displayed robust mechanical toughness and efficient optical healing, attributed to the increased HBI crosslinking density. Innovative research has combined hydrogen bonds with various bonding techniques, such as incorporating cellulose nanofibrils (CNFs) into a polyacrylic acid (PAA) gel matrix, which significantly enhanced the hydrogel's mechanical toughness and ionic conductivity due to the physical entanglements and HBI formed between PAA and the polar functional groups of CNFs.58
In another approach, Gong et al.,59 developed a hydrogel with a chain-like structure composed of aligned fibrous frameworks. This design led to significant enhancements in mechanical strength through dehydration, as the alignment of polymer chains contributed to the formation of intricate fibrous structures. The natural toughness and rigidity of these cellulose hydrogels enabled them to respond to mechanical signals, aided by structural HBI, which facilitated the formation of anisotropic structures.59 Mao et al.,60 engineered a gel network incorporating non-covalent bonds, demonstrating chain alignment, a crystalline network structure, reversible covalent bonding, and dynamic HBI. Physical crosslinking was induced using a CO2 medium to synthesize double-network gels from cellulose and silk fibroin. By adjusting the pH levels, the affinities between cellulose and other polymers led to the formation of hydrogels with significantly enhanced toughness, underscoring the practical implications of HBI in developing cellulose-based responsive biomimetic polymers.60 To improve the mechanical robustness and hemostatic performance of hydrogel dressings, we developed a novel cellulose/silk fibroin hydrogel (CSH) by combining two natural polymers-cellulose and SF-using an innovative CO2-incubation method (Fig. 2). This environmentally friendly CO2-assisted crosslinking approach enhances the mechanical integrity of the CSH while eliminating the toxicity associated with conventional crosslinking agents. Furthermore, the resulting hydrogel exhibits superior biocompatibility, making it a promising candidate for biomedical applications.60
 | ||
| Fig. 2 Development and analysis of highly flexible hydrogels. (a) Synthesis of cellulose/silk fibroin hydrogels (CSHs) using a two-step cross-linking approach. The initial step (i) involved the covalent bonding of cellulose and silk fibroin mediated. The subsequent cross-linking step (ii) leveraged hydrogen bonding among cellulose chains, facilitated by a CO2 exposure method. In this process, NaOH and urea were present in the hydrogel during the primary cross-linking phase (i), (b) images of various hydrogel samples captured at different fabrication stages, including precursor solution, after primary cross-linking, and following secondary cross-linking, (c) visual representation of mechanical performance tests on hydrogels, demonstrating superior mechanical properties in CO2-incubated samples (purple: hydrogels with primary cross-linking only; yellow: double cross-linked cellulose hydrogel; pink: double cross-linked cellulose and silk fibroin mediated hydrogel), (d and e) mechanical test results indicating that the strength of CSHs reached the megapascals range but decreased with increasing silk fibroin content. In contrast, SHs without CO2 treatment exhibited significantly lower mechanical strength compared to CO2-incubated CSHs. CSH-0, CSH-0.5, CSH-1, and CSH-1.5 corresponded to SF concentrations of 0, 0.5, 1, and 1.5%, respectively, while SH-0, SH-0.5, SH-1, and SH-1.5 represented equivalent SF contents in SHs. Data were presented as mean ± SD, n = 5, and signifies P < 0.001. The figure is adopted with permission from the ref. 60. Copyright © 2020 American Chemical Society. | ||
Electrostatic interactions (EIs), arising from the presence of opposite charges within molecules, can be integrated into supramolecular hydrogels through the strategic design and synthesis of charged monomers or polymer chains.61,62 Several studies have examined cellulosic hydrogels derived from EIs and their practical applications. Langer et al.,63 designed a self-assembled hydrogel utilizing EIs between negatively charged CMC and positively charged cetyltrimethylammonium bromide, demonstrating adjustable characteristics, reduced viscosity under shear stress, and self-repairing capabilities. MacLachlan et al.,64 synthesized a CO2-sensitive hydrogel by incorporating a combination of imidazole and monomers into CNCs, enabling control of the gelation process without chemical initiators or additional functionalities. This discovery holds potential for carbon dioxide extraction and sensing applications. Fig. 3 illustrates the mechanisms governing the CO2-responsive aggregation and re-dispersion of CNCs suspensions functionalized with imidazole groups. CNCs possess a net negative surface charge due to the presence of sulfate half-ester groups, which induce electrostatic repulsion and allow them to remain stably dispersed in aqueous media. Introducing CO2 into the suspension prompts its interaction with water, generating carbonic acid and consequently lowering the solution's pH. This pH reduction enhances the protonation of imidazole groups (pKaH 6.95), forming hydrogen carbonate ions and imidazolium salts. The emergence of these ionic species raises the ionic strength of the medium, neutralizes the CNC surface charges, and diminishes the electrostatic repulsion between CNC particles, facilitating aggregation (Fig. 3).64
 | ||
| Fig. 3 Cyclic aggregation and re-dispersion of CO2-responsive CNC suspensions incorporating imidazole, regulated by the addition and removal of CO2. The figure is adopted from ref. 64. Copyright © 2018 American Chemical Society. | ||
Huang et al.,32 engineered a biocompatible gel by integrating positively charged CNCs with negatively charged alginate (AG) in a double-network structure. The internal architecture, formed through ionic interactions between CNCs and AG, provided an efficient alternative without requiring chemical modifications.32 Yang et al.,65 developed a conductive hydrogel capable of self-healing and surface adhesion, where the combined effects of hydrogen bonding and EIs were critical. Tannic acid-coated CNCs served as reinforcing fillers within a doubly crosslinked network. Chang et al.,66 introduced quaternized tunicate CNCs into a crosslinked hydrogel matrix, significantly enhancing mechanical strength due to EIs between positively charged TCNCs and negatively charged PAA chains. Additionally, composite hydrogels with TCNCs exhibited pH-dependent swelling properties.66 Yang and Wang,67 developed a double-network structure utilizing quaternary ammonium group-modified β-cyclodextrin as the positively charged site and TCMC as the negatively charged site. These drug-loaded cellulosic gels, which interact through electrostatic and host–guest mechanisms, show promise for pharmaceutical applications. Liu et al. recently synthesized pH-responsive gels from CMC and bovine serum albumin (BSA) using an environmentally friendly method involving EIs, suggesting potential for combined chemo-radioisotope treatments for cancer.67
Within the category of thermo-reversible hydrogels, cellulose emerges as a prominent constituent, especially when engineered with hydrophobic functional groups.68 The incorporation of methyl or hydroxypropyl groups partially hinders hydrogen bond formation, enhancing the water solubility of the resulting derivatives.69 Gelation of cellulose derivatives occurs due to the exclusion of water from strongly methoxylated regions of the polymer. Aqueous solutions of MC exhibit unique properties, allowing reversible bond formation through hydrophobic interactions under thermal conditions.70 Compared to hydroxypropyl methylcellulose (HPMC), MC has a lower gelation temperature and exhibits fewer mechanical characteristics, such as mechanical stress and strain.71 Sekiguchi et al.,72 developed a near-infrared and small-angle X-ray scattering-induced MC-based thermos-responsive gelation system, where hydrophobic and HBI interactions contributed to gel formation. The results highlighted significant differences in gelation properties among regio-selectively methyl-substituted cellulose derivatives, indicating that gel formation results from the synergistic effects of hydrophobic interactions involving methyl groups and hydrogen bonds among hydroxyl groups, with the specific arrangement of methyl groups playing a crucial role. Subjecting the solution to repeated heating and cooling cycles ac cellulose rates the gelation of MC in cold water, particularly at higher concentrations.72 The gelation rate increases during the second heating–cooling cycle compared to the initial one. The practical application of MC hydrogels includes coating polystyrene dish surfaces to cultivate human embryonic stem cell (hES) clusters, facilitating the growth of embryoid bodies in suspended liquid culture. This process enhances the ionic strength and shielding effect of negatively charged CNCs, promoting gelation. The introduction of nitrogen gas into the solution reverses this process, reducing ionic strength and enabling CNC dispersion, resulting in a stable suspension.72 Notably, hES cells within embryoid bodies express molecular markers indicative of samples from all three embryonic germ layers, suggesting that the MC-coated medium can generate numerous hES cells clones. During HPMC synthesis, MC undergoes modification involving the addition of glycol ether groups to the glucose units of the cellulose structure, yielding a derivative with a molecular weight surpassing that of MC. Weiss et al.,73 successfully synthesized diverse biological materials using HPMC as a fundamental building block. Conversely, an electrostatic interaction-based gel system exhibits potential due to various remarkable properties, including controlled swelling, conductivity, and self-healing behaviours. The introduction of counterions as plasticizing agents into the hydrogel system fosters ionic interactions among polymers, facilitating hydrogel formation. The metal-coordinated approach involves the polymerization of a polyelectrolyte system in the presence of positively charged metal ions. Percec et al.,74 designed a neutral hydrogel with an equal number of positive and negative charges within the polymer network. Upon forming dynamic metal–ligand interactions, these hydrogels achieved a highly reversible and self-healing network that maintained stability under physiological conditions. The resultant gel exhibited a homogeneous, micro-sized porous structure characterized by well-organized chain networks. The hydrogels demonstrated dynamic properties, exhibiting dual-sensitive sol–gel transformations under varying pH and redox conditions, with hydrochloric acid (HCl) and trimethylamine (TEA) employed to modulate the solution pH. Notably, the hydrogel fractured upon the addition of 1,4-dithiol-dl-threitol (DTT) but was capable of self-healing through hydrogen peroxide (H2O2) oxidation. This phenomenon was prolonged due to the dynamic, reversible disruption and repair of acyl hydrazone and disulfide bonds within the hydrogels. Additionally, citric acid (CA) was incorporated with cysteamine dihydrochloride (CYS), which possesses disulfide bonding sites, resulting in a pH- and redox-responsive gel system.
The Diels–Alder reaction, a well-established chemical process, was utilized for dynamic bond formation, enabling these bonds to undergo reversible cleavage and reformation upon thermal stimulation.75 Nanocomposite gels were synthesized using maleimide-functionalized CNCs and furan-modified gelatine through a Diels–Alder process.76,77 Several studies reported that maleimide-terminated polyethylene glycol (PEG) and furan-modified CNCs were effective in developing strong, self-repairable polymeric gels.
Changyou Shao et al.,78 introduced a novel self-healing and self-recovering hydrogel by utilizing a furyl/maleimide pair and CNCs. In this system, CNCs function both as a reinforcing agent and a chemical crosslinker through a reversible Diels–Alder reaction. The study involved modifying CNCs with furyl groups to serve as multifunctional crosslinking agents, while PEG with maleimide terminal groups formed the polymer matrix. These components were combined to fabricate nanocomposite hydrogels capable of self-healing via the dynamic Diels–Alder reaction (Fig. 4).78 The gelation time and swelling behaviour of the hydrogels was analyzed under varying compositions. Results indicated that mechanical strength and self-healing efficiency could be fine-tuned by adjusting the substitution degree of furyl groups and the molar ratio between furyl and maleimide groups. To develop self-healing nanocomposite hydrogels, furyl-modified CNCs serve as multifunctional cross-linkers, while PEG with maleimide terminal groups functions as the polymer matrix. These components interact through a reversible DA reaction, enabling self-healing properties that can be fine-tuned by adjusting the substitution degree of furyl groups and the molar ratio of furyl to maleimide. The healing mechanism is driven by the dynamic cleavage and reformation of reversible cross-links, facilitated by thermally responsive DA bonds. This process promotes the formation of new phases at fractured surfaces, enhancing the hydrogel's self-repair ability. Moreover, a higher degree of furyl substitution combined with a lower furyl-to-maleimide molar ratio leads to an overall increase in healing efficiency over time. Since self-healing is primarily governed by DA bonds formed via furyl groups on CNCs and maleimide groups in PEG, a greater concentration of these bonds within the hydrogel network accelerates the repair process, ultimately improving healing efficiency.
 | ||
Fig. 4 (a) Self-healing performance of hydrogel DS1 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by direct visual inspection. (b) Stress–strain curves of the original and self-healed hydrogel DS1 1:1 at various healing times. (c and d) Self-healing efficiency of the hydrogel measured from tensile tests at room temperature. The figure is adopted from the ref. 78. Copyright © 2017 American Chemical Society. :1 by direct visual inspection. (b) Stress–strain curves of the original and self-healed hydrogel DS1 1:1 at various healing times. (c and d) Self-healing efficiency of the hydrogel measured from tensile tests at room temperature. The figure is adopted from the ref. 78. Copyright © 2017 American Chemical Society. | ||
To demonstrate self-healing, macroscopic tests were conducted, including visual observation and tensile analysis. In one test, a hydrogel sample (DS1 1:1) was split into two sections, with the fractured surfaces pressed together and incubated at 90 °C in an inert atmosphere. After cooling to room temperature, the merged hydrogel was subjected to tensile testing, showing no separation and confirming that the interface was robust enough to withstand applied stress. This self-healing process was attributed to the reversible cleavage and reformation of Diels–Alder bonds, which facilitated the reconstruction of the crosslinked network across the fractured regions. To evaluate the self-repairing performance, tensile properties were measured at various contact durations. Fig. 4 illustrates the stress–strain profiles of the repaired and pristine hydrogels. The data indicate that both the maximum elongation and tensile strength improved with extended healing time. For example, after 1 hour, the hydrogel achieved a fracture stress of 52 kPa, corresponding to a healing efficiency (HE) of 35%. With a 24-hour healing period, the recovered stress increased to 116 kPa, reflecting a remarkable HE of 78% compared to the original hydrogel.78
Yang et al.,79 developed cellulose-based self-healing hydrogels utilizing CMC-thermoplastic hydrogel (TPH) and PEG-diacrylate (PEG-DA) via dynamic covalent acylhydrazone linkages under the influence of the catalysis. The self-healing abilities of the CEC-TPH/PEG-DA hydrogels are illustrated in Fig. 5.79 Two stained hydrogel disks were cut in half; after keeping the semicircles in intimate contact along the cut line at room temperature for 6 hours, they completely merged into an integral hydrogel disk without any external intervention. The resultant hydrogel was sufficiently strong to withstand a tensile force applied perpendicularly to the cut surface without splitting.79 The self-healing process was documented using optical microscopy, revealing that the two dye molecules spread across the cut surfaces and ultimately interpenetrated, resulting in a purple coloration at the boundary. The strain compression test indicated that the fracture strength of the self-healed hydrogels increased with prolonged healing time, approaching values near the original strength. This finding suggests that the self-healed hydrogels effectively restored their original mechanical properties. Additionally, healing efficiency (HE) was found to depend on both healing time and 4a-Phe content, achieving a high HE of approximately 96% at 12 hours. Consequently, upon crack formation, CEC-TPH and PEG-DA macromolecules migrated towards the damaged interface, allowing for the reformation of reversible acylhydrazone linkages and recovery of the hydrogel networks.79
 | ||
| Fig. 5 Demonstration of the self-healing behaviour of CECT-ADH/PEG-DA hydrogels at 37 °C, (a) two Gel5 disks, colored with MeO and MeB dyes respectively, were each divided into halves, (b) semi-circular halves of Gel5 with different colors were combined into a single unified Gel5 disk after 6 hours of healing, (c) the self-repaired Gel5 disk being stretched to illustrate its mechanical integrity, (d) typical compression stress–strain profiles of Gel5 before healing and after healing for 3 and 6 hours, (e) compression stress–strain profiles of hydrogels with varying Phe-NH2 concentrations following 6 hours of healing, (f) influence of healing time and Phe-NH2 content on the healing efficiency (HE) of the hydrogels after 6 hours of contact. The figure is reprinted from ref. 79. Copyright © 2020 Published by Elsevier Ltd. | ||
Ding et al.,80 employed a telechelic cross-linking approach to impart pH-responsive self-healing properties to a composite hydrogel composed of chitosan and difunctional poly(ethylene glycol) (DF-PEG), as illustrated in Fig. 6. When utilized during the recovery process of rat-liver laceration, a thrombin-loaded hydrogel (CPT) applied to the liver capsule exhibited a smooth surface and vivid coloration, highlighting its potential as a highly efficient drug delivery system for treating wounds in vivo.
 | ||
| Fig. 6 (A) Visual comparison showcasing the pristine and repaired gel samples of COL-CS hydrogels: (a) COL-CS (1:1), (b) COL-CS (1:2), and (c) CS. (B) Schematic illustrating the hypothesized self-healing mechanism. Adapted with authorization from ref. 80. Copyright © 2020, American Chemical Society. | ||
In another study, Shao et al.,78 developed self-healing cellulose-based hydrogels characterized by high toughness and resilience, utilizing furyl-modified CNCs and maleimide end-functionalized PEG through a thermally reversible covalent Diels–Alder click reaction (Fig. 7).78 The hydrogels demonstrated remarkable mechanical properties, exhibiting a fracture elongation of up to 690% and a fracture strength of 0.3 MPa at a strain of 90%. The self-healing capability of these composite hydrogels was assessed via tension tests, revealing an efficiency of 78%. The researchers also constructed a tough and self-healing gel by establishing synergistic multiple coordination bonds among tannic acid (TA)-coated CNCs, PAA chains, and Al3+ ions within a covalent polymer network. The incision of the PAA-TA@CNC-Al3+ gel exhibited automatic self-healing, disappearing almost entirely within 30 minutes. The self-healed gel maintained sufficient strength to support itself and stretch without failure at the interface (Fig. 7A). The fracture stress of the healed gel significantly increased with extended healing time (Fig. 7B), reaching an equilibrium state with an HE of up to 92% after 30 minutes of healing. The HE was related to the TA@CNC content, with an increase in TA@CNC concentration enhancing the HE of the gel after 30 minutes (Fig. 7C), underscoring the importance of dynamic TA@CNC motifs for reversible rearrangement during the self-healing process. Microscopic self-healing behaviour of the gel was further validated through rheological experiments, which examined the breakup and reformation of the gel network (Fig. 7D). As anticipated, switching the strain from 100% to 1% at a fixed frequency allowed for immediate recovery of the gel-like character (G′ > G′′) without significant decrease across multiple recovery cycles. This sol–gel transition, accompanied by complete recovery of the gel network following disruption, was attributed to the reconstruction of reversible ionic coordination complexation within the gel network system.
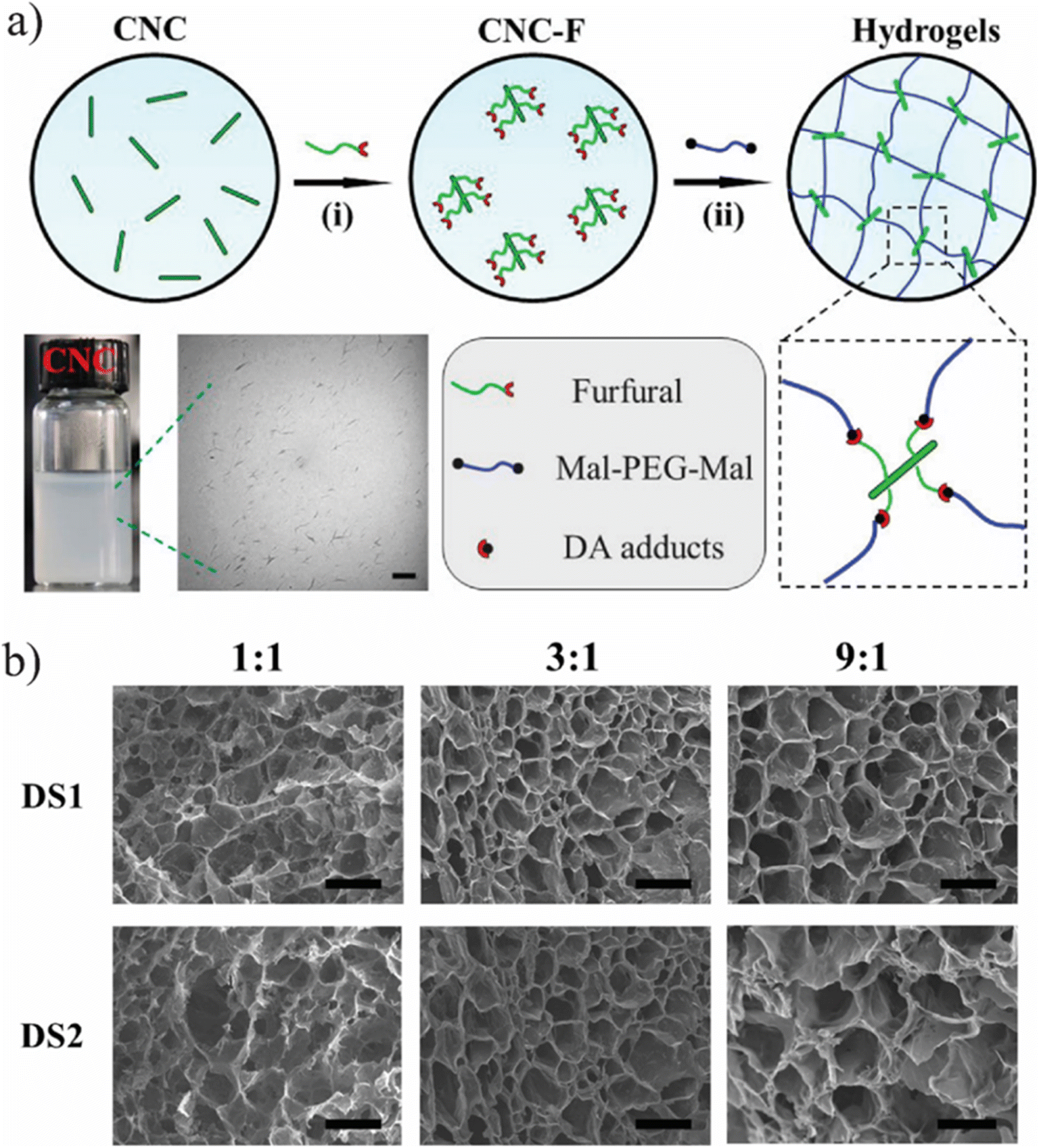 | ||
| Fig. 7 (a) Synthetic pathway for fabricating self-repairing CNC-PEG nanocomposite hydrogels, and (b) SEM images of cross-sectional views of CNC-PEG hydrogels. The figure is reprinted from ref. 78. Copyright © 2017 American Chemical Society. | ||
Chemical crosslinking is commonly employed to enhance the structural stability and desirable swelling properties of hydrogels derived from cellulose.81 Bi-functional molecules act as crosslinking agents, facilitating the formation of covalent bonds between cellulose or its derivatives and other polymer chains, thereby establishing a hydrophilic network.22 Sannino et al.,82 developed superabsorbent hydrogels based on cellulose by crosslinking CMC and hydroxyethyl cellulose (HEC) with divinyl sulfone (DVS). These hydrogels exhibited exceptional water absorption capacity, particularly under varying pH and ionic strength conditions. Moreover, these cellulose superabsorbent hydrogels were designed to address swelling by effectively extracting excess water from tissues. HPC hydrogels, synthesized under specific thermal conditions within an integrated phase system, demonstrated a nonporous structure throughout the biphasic system; however, alterations in conditions triggered the formation of a microporous structure within the hydrogels.
Hirsch and Spontak83 investigated the swelling capacities and dynamic mechanical properties of these pre-formed gels, analysing how they varied with changes in crosslinking temperature. The correlation between temperature and HPC gel swelling properties was limited. Conversely, the modulus of microporous HPC gels could be enhanced by extending the crosslinking duration. Thermally responsive hydrogels were synthesized through the crosslinking of HPC with PEG ether. Marsano et al.,84 developed hydrogels exhibiting swelling behaviour at lower temperatures (20 °C) and contraction activity at higher temperatures (60 °C). HPC hydrogels, fabricated through crosslinking with epichlorohydrin (ECH) and ammonia, demonstrated a remarkable capacity for absorbing anionic dyes, with a maximum adsorption capacity of 2478 g kg−1 at ambient temperature and a pH of 3.96.84
One crucial property of hydrogels is the SR, which can be manipulated through various means, including modifications in crosslinking length, alterations in the molecular weight of the gel systems, or the incorporation of filler materials to occupy the spaces between macromolecular chains,85,86 when DVS is used as a crosslinker. For gel structures to be suitable for applications in medicine, food, and biomaterials, the crosslinkers employed in their synthesis must be non-toxic. This is an essential area of research that scientists actively pursue, focusing on developing new biocompatible crosslinkers to create hydrogels based on cellulose.
Since carbodiimide molecules do not promote crosslinking, water-soluble carbodiimide is often utilized to enhance the biocompatibility of cellulose hydrogels through the crosslinking process.87 Furthermore, it is feasible to convert them into urea derivatives, which are less harmful to cellulose and can be easily washed out of the polymeric network. The SR of hydrogels is influenced by the chemical composition of CMC and HEC as well as the dehydration process.27 Dehydrated hydrogel pellets are designed for oral administration. According to Sannino et al.,88 in the acidic pH of the stomach, these pellets will swell, inducing a sensation of fullness before being excreted in the faces. Demitri et al.,89 successfully demonstrated the development of hydrogels composed of HEC and CMC, synthesized using CA as a crosslinking agent. This method offers several advantages, including reduced toxicity and cost-effectiveness compared to previous crosslinking agents. The proposed esterification process involves the synthesis of an anhydride intermediate, which is hypothesized to play a critical role in the interaction between cellulose and CA. The swelling degree is influenced by the duration of the active reaction and the concentration of CA; at a CA concentration of 3.75% (w/v), the hydrogel exhibits an SR of 90.
Various bonding mechanisms can influence the physical and chemical properties of cellulose-based hydrogels.70 The choice of suitable hydrophilic or hydrophobic groups, as well as the selection of compatible polymer matrices, plays a pivotal role in achieving desirable properties for specific biomedical applications. Continued exploration of cellulose-based hydrogels is essential to unlock their full potential, providing novel insights for advancing biocompatible materials and enhancing their performance across diverse biomedical fields.
4. Swelling properties of cellulose-based hydrogels
The swelling mechanism of polymeric hydrogels reveals the underlying processes responsible for water absorption and retention within the three-dimensional network when these materials are exposed to a solvent.90 The chain network of hydrogels contains charged polar functional groups, which primarily contribute to the hygroscopic properties of this advanced polymer. Several studies reveal that charged particles within the gel matrix generate repulsive forces and osmotic pressure (OP), which drive the swelling phenomenon.91 Specifically, negative charges within the network exert repulsive forces that facilitate the expansion of the gel structure.92 However, the presence of crosslinked polymers in hydrogels mitigates disintegration, thereby limiting excessive swelling, as illustrated in Fig. 8(a).93 | ||
| Fig. 8 Swelling mechanism of hydrogels under (a) changing the cross-linking length to adjust the gel mass size. The idea of this figure is taken from the ref. 93. Copyright © 2006 Royal Society of Chemistry. And (b) ionic conditions. The idea of this figure is taken from the ref. 94. Copyright © 2014 Royal Society of Chemistry. | ||
Hydrogels can absorb and retain significant quantities of water, contingent upon the nature and extent of cross-linking as well as the charge densities of their networks.95,96 The swelling degree (SD) quantifies the amount of water absorbed by a hydrogel. This property is critical for the in vivo application of hydrogels, as excessively swollen materials may disintegrate prematurely during wound healing or cause tissue damage in confined anatomical spaces.
The degree of swelling notably influences the diffusion of bioactive molecules or drugs from the swollen gel matrix. Given that hydrogels can absorb and retain water, they exhibit high swelling degrees and molecular diffusion rates.97 To assess the SR, hydrogels undergo various drying processes. Subsequently, the specimens are immersed in an adequate volume of water at room temperature until they reach an equilibrium state. During this period, the hydrogels undergo filtration, and their weights are measured. Equilibrium is achieved when the weight of the swollen hydrogels stabilizes. Thus, the equilibrium swelling degree (ESD) can be calculated using eqn (1).98,99
| ESD = (Ws − Wd)/Wd | (1) |
At equilibrium, the weights of dry and swollen gels are represented as Wd and Ws, respectively. Various factors, such as crosslinking density, type of crosslinking, solvent properties, and the interaction between the polymer and solvent, can influence swelling behaviours and the extent of swelling.100,101 Water acts as a softener or plasticizer in a hydrophilic polymer network structure. The swelling degree of gels can be explained using the Flory–Rehner equilibrium hypothesis (eqn (2)),101,102 which is mainly based on the Gibbs free energy concept, to analyze the swelling behaviour of the obtained hydrogels.103,104
| ΔGtotal = ΔGmix + ΔGel | (2) |
The variables denoted as ΔGtotal, ΔGmix, and ΔGel represent the free energy contributions from the mixing enthalpy and elastic retractile stresses within the polymer network. Initially, the change in Gibbs free energy for mixing, denoted as ΔGmix, has a substantially negative value. In contrast, the change in Gibbs free energy for an electrochemical process denoted as ΔGel is positive but with a magnitude smaller than that of ΔGmix. Therefore, the combined impact of these factors has a negative outcome (ΔGmix + ΔGel < 0). During this stage, swelling starts, leading to the migration of solvent molecules into the polymer network. Both are raised until they reach zero magnitudes during swelling, as shown by the formula |ΔGmix| = |ΔGel|. This results in zero total free energy, or ΔGtotal = ΔGmix. The development of hydrogel adhesives for wound closure involves essential elements, including the density of cross-linking (ρx), the polymer volume fraction when swollen (ν2,s), the molecular weight of polymer chains between neighbouring cross-links (Mc), and the mesh size (ξ). These characteristics play a vital role in determining the mechanical and diffusive properties of the material upon application to a wound site. As a result, there is no longer any driving force for the swelling, which causes it to cease and reach equilibrium.
However, the quantitative analysis of swelling behaviour can be obtained by using following equations,96
 | (3) |
By using this mathematical relationship, for example, Rahman et al.95 synthesized dicarboxylate nanocrystalline cellulose (DCNC) from microcrystalline cellulose (MCC) through acid hydrolysis and selective periodate oxidation. The polyelectrolyte (PE) nature of DCNC was analysed by measuring water uptake using a customized ion exchange system. Two DCNC samples with carboxylate contents of 9.65% and 33.33% exhibited increasing water absorption over time, reaching equilibrium. The higher carboxylate content (33.33%) resulted in the highest water uptake (≈60 times its weight), while the lower content (9.65%) absorbed ≈30 times its weight. In contrast, TEMPO-oxidized NCC (9.72% carboxylate content) absorbed only five times its weight, indicating that oxidation position significantly influences water uptake. DCNC exhibited nearly ten times greater equilibrium water absorption than TEMPO-oxidized NCC, confirming that C-2 and C-3 functionalization enhances water uptake performance.95 Another important example, Nguyen et al.,105 reported “dip-catalyst” hydrogels for methylene blue (MB) dye removal, where the catalyst is immobilized and uniformly distributed within the hydrogel matrix. TiO2 served both as a crosslinker and photocatalyst, enabling structural integrity and catalytic activity. The study examined crosslinking density by analyzing the swelling behaviour of cellulose-based hydrogels with different TiO2 crosslinkers. When immersed in distilled water at room temperature, all hydrogel samples exhibited a rapid increase in swelling capacity within the first 60 minutes, driven by OP and electrostatic repulsion between negatively charged groups. Beyond this period, the swelling rate gradually declined, reaching equilibrium after approximately 150 minutes. The cross-linked polymer network restricted excessive solvent absorption, thereby limiting the hydrogel's maximum swelling capacity.105
4.1. Structural properties
Several critical factors influence the SR properties of cellulose-based hydrogels, particularly in their application as structural biomaterials, such as wound dressings.106 The primary objective is to evaluate their effectiveness for these applications. The parameters include the optimal density of the polymer chain networks, denoted as ρx, which represents the proportion of polymer volume when fully swollen. These factors significantly affect the mechanical and diffusion properties of the hydrogel once it interfaces with the wound. Understanding and controlling the density of chain networks is essential, as it directly influences the gel's mechanical strength and its ability to adhere to the wound surface.107 Furthermore, the fully swollen polymer volume proportion affects the gel's capacity to absorb and release therapeutic agents, thereby promoting wound healing.108 Consequently, optimizing these parameters is vital for ensuring the successful application of cellulose hydrogels as effective wound dressings in biomedical contexts.The equilibrium-swelling (ES) theory and rubber-elasticity (RE) theory are key approaches for understanding the bonding and crosslinking characteristics of polymer network systems.109,110 The ES theory primarily focuses on the gel network's ability to integrate and describes how these networks interact within an aqueous environment. This analysis provides valuable insights into the structural parameters, particularly regarding the mechanical and crosslinking behaviour of the chain network. Conversely, the RE theory examines the elastic properties of the gel matrix and its ability to maintain structural integrity under applied stress.111,112 Upon the removal of stress, the theory elucidates how the matrix reverts to its original state, which is fundamentally dependent on the nature of crosslinking within the gel matrix.
Analysing material properties through RE is significant for investigating correlations and structural parameters. Numerous equations have been developed in the scientific literature to compute these parameters, which vary based on the gel structure and the methodologies employed for their determination. For example, in a study examining the deformation of a crosslinked polymer gel under stress, researchers applied varying stress levels and utilized rheological techniques to assess changes in the network structure.107,113 By observing how the gel's structure altered under stress and subsequently returned to its initial form, they gleaned insights into the type and strength of crosslinking within the gel matrix. This information was instrumental in understanding the material's response to external forces and enabled the determination of critical structural parameters, such as elasticity and stability.
Moreover, this knowledge served as a foundation for further optimization and development of the gel material. The researchers successfully modified the gel formulation and crosslinking process to enhance both elasticity and stability, resulting in a more robust gel capable of withstanding higher stress levels without significant structural changes. These advancements opened avenues for diverse applications, including biomedical engineering, where the gel could serve as a scaffold for tissue regeneration or as a drug delivery system. The researchers remained optimistic that ongoing optimization efforts would yield even more versatile and efficient gel materials in the future.
4.2. Water vapor transmission rate
Hydrogels possess a unique ability to absorb and retain water molecules within their matrix while also desorbing them under varying atmospheric conditions.114,115 Due to their properties closely resembling those of soft natural tissues, hydrogels are widely utilized in the biomedical field, particularly in wound treatment.1,116–118 In an ideal hydrogel application for wound care, the hydrogel encapsulates the wound surface, effectively absorbing wound exudates and preventing dehydration. This mechanism significantly improves the wound healing process compared to conventional treatment methods.When applied to a wound, cellulose-based hydrogels absorb exudates such as pus and fluid, thereby maintaining a moist environment conducive to faster healing.119 Additionally, cellulose-based hydrogels can be easily removed from the wound, minimizing pain and discomfort during dressing changes and enhancing patient comfort. Furthermore, these hydrogels exhibit antimicrobial properties, reducing the risk of infection and associated complications. Overall, cellulose-based hydrogels represent a promising approach in wound treatment, not only accelerating the healing process but also improving patient comfort and minimizing infection risks.120
The rate of water vapor transmission (RWVT) is a widely employed technique for quantifying the amount of water vapor released from the hydrogel network. This measurement is obtained by analysing the vapor process across a unit surface area of hydrogels (cm2) under controlled conditions, including a fixed time, specific temperature, and constant humidity level. The RWVT is expressed in grams per square meter per hour (g m−2 h−1) using the following formula:121
| RWVT = t·A/G | (4) |
The weight change (G) is measured in grams, the time (t) is expressed in hours, and the tested surface area (A) is denoted in m2. For instance, wound treatments exhibiting an RWVT ranging from 80 to 100 g m−2 h−1 are effective in maintaining moisture within the wound, thereby facilitating swift recovery.
In a standard procedure, a hydrogel is applied to the opening of a sealed cylindrical plastic container filled with purified water to prevent vapor escape. This setup is maintained at 37 °C with controlled humidity levels and monitored periodically by weighing. A graph representing weight loss over time is generated, and the RWVT is calculated by dividing the slope of this curve by the tested surface area. In a wound healing study, a hydrogel with a known RWVT is applied to the surface of a wound of a defined area.122–124 The wound is then covered with a transparent film to prevent moisture loss and placed in a controlled environment. By periodically weighing the wound and monitoring weight loss over time, the RWVT can be determined using the slope of the weight loss curve divided by the wound's surface area (A). The RWVT is a critical parameter for assessing the rate at which the hydrogel releases moisture or other substances into the wound.124 This information is essential for understanding the healing process and evaluating the hydrogel's efficacy in promoting wound healing. Additionally, the RWVT can be utilized to compare various hydrogel formulations, aiding in the selection of the most appropriate option for wound care. Ultimately, determining the RWVT offers valuable insights into the performance and therapeutic potential of hydrogels in wound healing applications.
4.3. Total charge balance stoichiometry
Chunyu Chang et al.,125 broadly discuss the total charge balance stoichiometry (CBS) of as-prepared hydrogels underwent examination through theoretical and experimental approaches. When reaching the equilibrium of the charge balance, it is expected to be fulfilled.125| Q+ = Q− | (5) |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) q = SqM1 + (1 − Sq)M0 q = SqM1 + (1 − Sq)M0
| (6) |
|
c = ScM2 + (1 − Sqc)M0
| (7) |
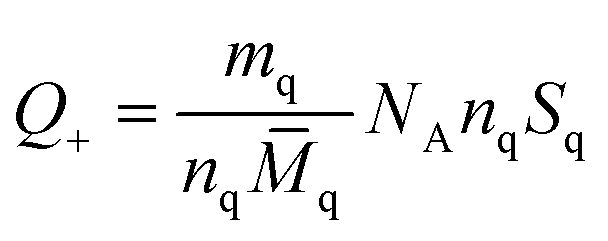 | (8) |
 | (9) |
The masses of QC (mq) and CMC (mc) used in hydrogel preparation, along with their respective glucose units (nq and nc), were considered alongside Avogadro's number (NA). The determined weight ratio of QC to CMC (mq/mc) was 1.5 upon achieving charge equilibrium (Q+/Q−).125
4.4. Effect of crosslinking materials for cellulose-based hydrogels
The synthesis of hydrogels is heavily influenced by the selection and role of crosslinking materials (CLMs).126,127 The primary challenge in this process lies in constructing a three-dimensional network through various synthetic methodologies, including physical crosslinking (PC), chemical crosslinking (CC), and other interactions.128 Chemical bonding facilitates the aggregation of adjacent chains into a continuous, interconnected network.129,130 CLMs can be derived from both natural and synthetic sources. Depending on the cellulose derivative employed, a variety of CLMs, such as ECH, aldehyde-functionalized materials, carboxylic acids (COOH), and urea derivatives, may be utilized in hydrogel fabrication.131,132 It is crucial to acknowledge that certain reagents, particularly aldehydes, pose risks in their unreacted forms. Despite thorough washing with distilled water to remove unreacted chemicals following crosslinking, it is vital to select non-toxic CLMs to ensure the biocompatibility and environmentally friendly production of the final hydrogel.133Numerous studies have explored the advancement of polymeric gels utilizing CMCNa and incorporating natural CLMs.134,135 The production of these hydrogels often involves genipin (GP) and CA as natural crosslinking agents.136 GP, derived from the gardenia fruit, is a favourable alternative to dialdehyde crosslinkers due to its excellent biocompatibility.136 This inherent catalyst fosters the formation of covalent bonds between the gel and biological or natural tissues, such as chitosan (CS) and gelatin. Notably, GP exhibits a toxicity threshold that is 10000 times lower than that of the commonly used glutaraldehyde (GA), which minimizes hazardous effects during hydrogel crosslinking. Muhammad et al.,137 reported the successful synthesis of a gel by combining κ-carrageenan (κC) with CMCNa using GP as a CLM. The highest SR was observed in hydrogel beads composed of a κC ratio of 90:10, with decreasing swelling efficiencies noted for 80:20, 70:30, and 60:40 ratios. This phenomenon can be attributed to the increased weight of carrageenan, which enhances the number of counterions (SO3−) in the solution, thereby strengthening the electrostatic attraction between these groups. The elevated OP resulting from the higher concentration of SO3− ions facilitate polymer expansion.137
The swelling behaviour of the beads was assessed in both an acidic solution at pH 1.2 and a neutral medium at pH 7.4.138 Interestingly, most bead compositions exhibited greater swelling at pH 7.4 compared to pH 1.2, with SRs of 109% and 100% for the 70:30 bead ratio, respectively.139 The diminished ionization of carboxymethyl groups leads to the conversion of COONa to COOH at lower pH levels. Consequently, the ionization of carboxylic groups intensifies with increasing pH, promoting bead swelling and network repulsion. The 70:30 bead ratio was selected for further investigation, as the 80:20 and 90:10 ratios, despite exhibiting greater swelling, proved unsuitable for bead development due to practical synthesis challenges.139 Recent studies have demonstrated the effectiveness of CA as a highly compatible crosslinker in hydrogel research. Demitri et al.89 successfully synthesized hydrogels using CA, with SR analysis showing that CMCNa crosslinked with 10% CA exhibited superior swelling behaviour compared to HEC at the same CA concentration. However, at 20% CA, the swelling behaviour of HEC-based hydrogels became comparable to that of CMCNa-based hydrogels. This suggests that CA reacts more rapidly with HEC than with CMCNa, likely due to reduced steric hindrance, allowing for faster crosslinking. Gorgieva and Kokol discovered that the swelling capacity of CA-crosslinked CMCNa/HEC gels increased with higher concentrations of CMCNa.140 Hydrogels with a CMCNa/HEC ratio of 3:1 exhibited a notable swelling increase of approximately 10–20% compared to those with a 1:1 ratio. Conversely, hydrogels with elevated HEC concentrations showed decreased stability due to a reduction in crosslinking points, influenced by a high degree of substitution leading to fewer OH groups compared to CMCNa. At pH levels ranging from 6.25 to 6.5, the ionization of carboxylic acid groups (COO−) was noted, corresponding to the cellulose pKa value of 4.6. Changes in pH instigated a breakdown of hydrogen bonding interactions (HBIs), attributed to the repulsion among macromolecules and water absorption. It is noteworthy that the hydrogel with a molar ratio of 1:1 exhibited fewer HBIs than the hydrogel with a 3:1 ratio. Additionally, the CMCNa/HEC hydrogel displayed a reduction in HB content with a higher CA crosslinker concentration (5.75% w/w) compared to 3.75% (w/w) CA. The response to pH changes was immediate; a 3.75% (w/w) CA concentration elicited greater swelling in alkaline conditions than in acidic environments. Gels with a higher pH and abundant COO− groups experienced increased ionic repulsion, resulting in the expansion of the gel network and enhanced water absorption.
Durpekova et al.,141 investigated a hydrogel composed of CMCNa and HEC, with CA acting as the crosslinker. The resultant gel exhibited superior swelling behaviour when exposed to water compared to individual CMCNa or HEC hydrogels at equivalent CA concentrations. However, the incorporation of CA diminished the stability of the HEC-based hydrogel, reducing its absorption capacity. The limited crosslinking efficiency of HEC can be attributed to its higher degree of substitution compared to CMCNa. As a polyelectrolyte, CMCNa exhibits pH sensitivity and ionic strength, enhancing swelling through the Gibbs–Donnan effect and increasing OP. This elevated OP mitigates the ionic strength of the external solution, facilitating water ingress into the hydrogel.141 Nonetheless, CMCNa's poor crosslinking efficiency results from ionic repulsion among charged polyelectrolyte chains, favouring the formation of intermolecular over intramolecular crosslinks.
Numerous investigations have indicated that the monomer ratio and the manipulation of CLM concentration significantly affect the degree of swelling.142 An elevated concentration of CA in the solution mixture, resulting from enhanced crosslinking density, corresponded to a decrease in water uptake. Conversely, hydrogels with inadequate crosslinking percentages exhibited poor formation due to insufficient crosslinking.143,144 CMCNa/HEC (3:1) demonstrated an increased absorption capacity with a CA weight percentage of 5.75% in water-prepared samples, with comparable results observed in whey-prepared samples at a CA concentration of 5%.89 The gel synthesized at pH 4.5 displayed the most favourable SR. The introduction of whey acid (WC), a low-protein acid, as an alternative to distilled water in gel synthesis, effectively repurposed waste from the dairy industry. The water uptake capability of WC/cellulose-based hydrogels was remarkably high (1000–1700%), comparable to hydrogels synthesized with distilled water. Various pH environments were examined to assess their influence on the swelling behaviour of WC/cellulose gels. The gel reached maximum swelling at pH 7.2 in distilled water and at pH 10.0 (994%) in saline solution. A significant reduction in swelling was observed when pH decreased to 2.5 due to the protonation of COOH groups. Above the pKa value of 4–5 for COOH groups, deprotonation occurred, leading to ionic repulsion among the negatively charged groups within the hydrogels and enhancing swelling performance. Heat application to the mechanism of CA action facilitates the removal of water from the carboxylic acid group, resulting in the formation of a cyclic anhydride.145 This cyclic anhydride of CA then esterifies by linking with hydroxyl groups on cellulose. According to Demitri et al.,89 dehydration of carboxylic acid to cyclic anhydride initiates at 60 °C, while degradation occurs at 160 °C. CMCNa demonstrates thermal stability below 100 °C but degrades at elevated temperatures. Therefore, the optimal temperature for crosslinking CMCNa with CA is identified as 80 °C.
Zhang and Qiao146 developed a polymeric gel using CMCNa as a monomer and ECH as a crosslinker. The addition of superabsorbent polymers (SAPs) to soil has been shown to reduce water evaporation and percolation. However, the as-prepared SAPs failed to meet necessary requirements due to challenges with consistent water uptake and salt tolerance. This study explored the effects of valence cations, specifically sodium, calcium, and Al3+ ions, on the structural modifications of ECH-crosslinked hydrogels based on CMCNa. Results indicated that the presence of additional COOH groups enhanced water absorption capacity, with a hydrogel containing 5% CMCNa and 3% NaOH demonstrating an exceptional water absorption capacity of 969.0 g g−1 in deionized water.146 However, increased cation valence in solution reduced hydrophilicity and salt resistance. The interaction between the COOH group and the Na cation resulted in proton replacement, while Ca-ion interactions involved bidentate bridging chelation, with Al3+ ions exhibiting tridentate chelation.
Although the inclusion of polyvalent cations stabilized the COOH group, it concurrently hindered water uptake due to reduced swelling capacity.147 Following this study, Peptu et al.,148 developed a hydrogel using ECH crosslinking, where the stabilization of the COOH group by polyvalent cations also led to diminished swelling capacity. They produced an ECH-crosslinked hydrogel exhibiting excellent superabsorbent characteristics with a supreme SR of 1273% at a 1:1 AG molar ratio and a 6.6% monomer mixture with 0.75 mL ECH. This high SR was attributed to the porous structure observed in scanning electron microscopy (SEM) images (Fig. 9).148 In contrast, a sample with the same molar ratio but a higher ECH concentration (3 mL) exhibited a lower SR of 362%, likely due to an enlarged network structure resulting from excessive ECH crosslinking. This highlights the importance of the concentrations of monomers and CLMs in influencing the SR.
 | ||
| Fig. 9 SEM images of the AG/CMC films with different resolutions. The figure is adopted from the ref. 148. Copyright 2021, MDPI. | ||
The swelling behaviour in a polyelectrolyte system is driven by the combined effects of Donnan OP and the elasticity of the polymeric chain network. The OP increases steadily with the number of active molecules and charges but decreases at higher ionic strengths. This relationship between OP and ionic strength is contingent upon the number of ions present in the system. At lower ionic strengths, repulsion among charged molecules prevails, with Donnan OP dominating the swelling process.149,150 Conversely, an increased ionic presence at higher ionic strengths screens charged molecules, reducing repulsion and consequently lowering Donnan OP. Understanding and managing the swelling behaviour of polyelectrolyte systems hinges on this interaction between Donnan OP and ionic strength.149,150 Table 1 represents various crosslinking mechanism for various cellulose based hydrogels.
| Hydrogels | Synthetic method | Crosslinker | Crosslinking mechanism | Ref. |
|---|---|---|---|---|
| a ECH: epichlorohydrin, CMC: carboxymethyl cellulose, AM: acrylamide, GO: graphene oxide, BC: bacterial cellulose, SA: sodium alginate, HAP: hydroxyapatite. | ||||
| Cellulose–bentonite hydrogel | Crosslinking | ECH | Electrostatic interaction | 151 |
| CMC-based hydrogel | Inverse suspension crosslinking | ECH | Electrostatic interaction | 152 |
| Chitosan and cellulose-based hydrogels | Freeze-dried | Electrostatic and hydrogen bonding | 153 | |
| CMC-based hydrogels | γ-irradiation | Cu2+ | Chelation | 154 |
| CMC–AM–GO-based hydrogels | Radical polymerization | GO | Covalent and electrostatic | 155 |
| BC | SA | Hydrophobic interaction | 156 | |
| Cellulose | Freeze thawing | HAP | Hydrogen-bonding | 157 |
4.5. Effect of ionic strength on swelling behaviour
Three main components of a salt-responsive hydrogel are typically its three-dimensional structure, interstitial fluid, and many ionic groups (Fig. 10).158,159 However, the pH-sensitive gel networks, including the primary and acidic groups BOH and AH. A−/B+ and the related conjugate base/acid H+/−OH are formed when some acidic or basic groups dissociate when the solvent penetrates the gel.159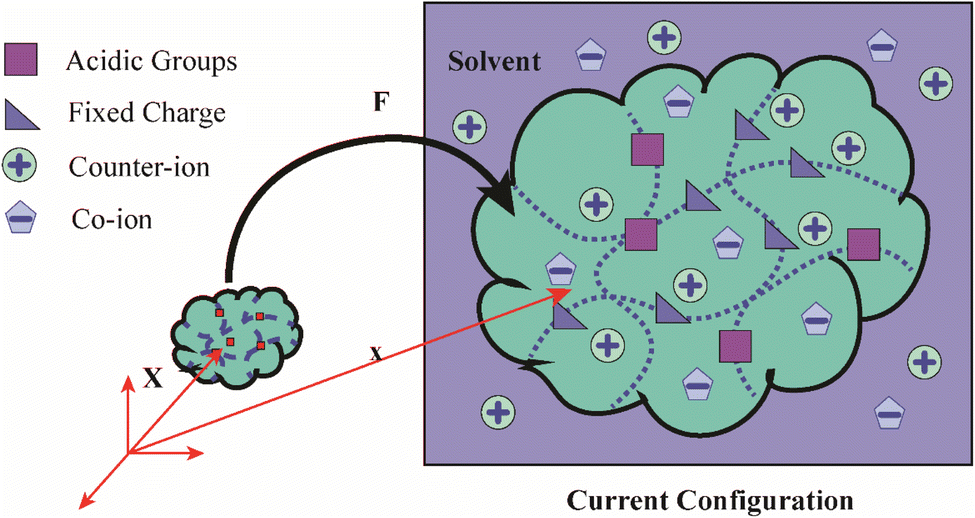 | ||
| Fig. 10 Schematic of the reference and current configurations of a pH-sensitive hydrogel with an arbitrary shape undergoing deformation due to external stimuli. The idea of the diagram is taken from ref. 158. Copyright© 2020, Elsevier B.V. | ||
The fixed charge, denoted as the conjugate base/acid, arises from this process, contributing to a network-bound charge. Importantly, these reactions are reversible.
| AH ↔ A− + H+ | (10) |
| BOH ↔ B+ + OH− | (11) |
Chemical equilibrium is achieved when the concentrations of the reactive chemicals AH/BOH, A−/B+, and H+/OH− remain constant over time. The dissociation constant is,
 | (12) |
 | (13) |
 | (14) |
 | (15) |
The concentration of constant charges can be obtained by combining eqn (12) and (13) for anionic hydrogels and eqn (14) and (15) for cationic hydrogels,160 as follows:
 | (16) |
 | (17) |
For simplicity, we write anionic hydrogel equations, which are also pH sensitive. However, the movement of ionic species typically arises from diffusion, convection, and electrical migration. The flux is mathematically described by the Nernst–Planck equation, as outlined by Kirby.161
| jm = ϕ(−Dm∇Cm − zmμmFCm∇ψ) + cmv | (18) |
The Debye length is a measure of the EIs between charged particles in the gel. The Debye length describes how far the double layer goes in pH-responsive gels when the pH level changes and there are different ionic atmospheres (higher or lower salt concentrations) in the solution. The variable f expresses the hydrogel's porosity, and Dm stands for the mth species' diffusion coefficient. The number of charges on ionic species is shown by zm, the mobility of the mth species' ions is shown by μm, F stands for Faraday's constant, c stands for electric potential, and v stands for fluid velocity in relation to the polymer network. The Einstein equation governs the connection between diffusivity and ionic mobility. The relationship between diffusivity and ionic mobility is given by the Einstein equation.
Hydrogels responsive to pH changes, the Nernst–Planck equation often emphasizes the concentration gradient of species when there is no external electric field. Simply put, diffusion takes precedence over convection and electrical migration since OP primarily drives swelling.162,163 However, to ascertain the spatial electric field in eqn (18), one can employ Poisson's equation, as elucidated by Jeans.163
 | (19) |
This equation is specifically applicable to anionic gels. It's important to note that in this expression, denotes the permittivity of the vacuum and the gel, respectively. The normalized representation of Poisson's equation takes the following form:
 | (20) |
4.6. Effect of structural properties on swelling behaviour
The SR of ionic polymers is influenced by several factors, including the inherent mechanical strength of the polymer, the interactions between the polymer and solvent, and the OP resulting from ions present on the interior and exterior surfaces of the gel.166 These elements collectively affect the hydrogel's behaviour, which ultimately influences its stability and performance. For instance, in drug delivery applications, the stability and effectiveness of ionic hydrogels can be significantly affected by variations in temperature and pH.18,167 When the temperature deviates from a specific range, the gel may undergo phase transitions, leading to alterations in its structure and properties.Similarly, fluctuations in pH can cause the gel to either expand or contract, impacting the kinetics of drug release.168 Thus, designing hydrogels with specific drug delivery characteristics necessitates a thorough understanding and control of these factors.169 For example, changes in pH can be utilized to trigger drug release in pH-responsive hydrogels, enhancing the controlled delivery of therapeutics.170,171 The differential diffusion of drug molecules across gels that swell or shrink in response to pH variations allows for improved therapeutic effectiveness and enables the development of customized treatment strategies.
OP and electrostatic repulsion within polyelectrolyte gels are key factors influencing hydrogel swelling behaviour, particularly in quaternized chitosan/carrageenan hydrogels. The swelling capacity depends on the gel's chemical composition. In cationic polyelectrolyte gels, repulsive forces between fixed positive charges push water molecules and polymer chains apart, leading to expansion. Conversely, in anionic polyelectrolyte gels, fixed negative charges attract water molecules, promoting swelling and water absorption. These distinct swelling mechanisms highlight the importance of optimizing hydrogel properties through a deeper understanding of their chemical composition and interactions. Interestingly, cationic polyelectrolyte gels can exhibit water absorption without the expected repulsion between water molecules and polyelectrolyte chains. In some cases, favorable interactions between fixed positive charges and water molecules promote hydration, challenging the assumption that positive charges always induce repulsion. This highlights the need for a detailed understanding of chemical composition and interaction mechanisms in material design. Researchers have successfully engineered positively charged hydrogels with high water absorption capacity, making them ideal for applications requiring high water content, such as contact lenses and wound dressings.
Furthermore, researchers have developed nanomaterials with unique surfaces that exhibit both hydrophilic and hydrophobic characteristics.172,173 These materials have proven beneficial in oil spill cleanup applications, selectively attracting and absorbing organic molecules while repelling water. This advancement refutes the idea that materials can possess only a singular characteristic and emphasizes the need for a comprehensive understanding of chemical compositions and interactions to create multifunctional materials. For instance, this nanomaterial can effectively separate oil droplets from water due to its hydrophobic surface, which prevents water from mixing with the oil.174 At the same time, the hydrophilic surface draws in and absorbs organic contaminants, facilitating efficient spill remediation. This innovative approach demonstrates how materials with dual functionalities can revolutionize environmental cleanup efforts.
Another notable example is the development of self-healing materials that leverage adaptable components with specific functions. These materials can autonomously repair themselves when damaged, reducing the need for regular maintenance and replacement. For instance, a self-healing polymer coating on a vehicle's exterior can repair minor dings and scratches over time, thereby preserving the vehicle's integrity and aesthetic appeal.175 This breakthrough in material science has the potential to significantly enhance durability and reduce waste across various industries. Additionally, advances in regenerative medicine may lead to the creation of medically functional, self-healing tissues and organs. Researchers are exploring methods, such as utilizing stem cells, to repair damaged heart tissue following myocardial infarction. Such innovations could potentially eliminate the need for heart transplants in patients with heart disease, significantly improving their quality of life.
4.7. Effect of stimuli on swelling behaviour
Stimuli-sensitive hydrogels can undergo significant changes in shape or volume in response to specific environmental triggers.176 These stimuli can include physical factors such as light, pressure, temperature, electric and magnetic fields, and ultrasound. Additionally, they can respond to chemical stimuli, including glucose, CO2, ionic strength, pH, and redox conditions.177,178 Biological signals, such as DNA, glutathione, antigens, and enzymes, may also activate these hydrogels.179,180 Stimuli influencing hydrogels can be classified as internal or external, based on their in vivo origin. Chemical and biological factors are internal stimuli, while physical factors (excluding temperature) are external stimuli. Often termed “smart” or “intelligent” hydrogels, these materials can detect stimuli and respond by modifying their physical or chemical properties, enabling the controlled release of encapsulated drugs.Among stimuli-responsive polymeric gels, pH-sensitive hydrogels have attracted significant research interest due to their ability to undergo abrupt volume phase transitions, resulting in swelling or collapse under specific conditions. Their responsiveness depends on factors such as size, shape, cross-linking density, ionic group content, and overall composition, all of which can be tailored accordingly. The response rate varies based on pore size, ionic group concentration, and cross-linking density. Another intriguing class of intelligent materials is shape-memory polymeric hydrogels, which can retain a fixed shape and recover it upon exposure to a specific trigger. Achieving shape memory requires (i) a polymer that undergoes phase transitions in response to stimuli and (ii) a memory code (chemical or physical) that enables shape recovery when triggered. Guo et al.,181 designed pH-sensitive hydrogels capable of shape memory using acrylamide and deoxyribonucleic acid (DNA) as cross-linked polymers. At pH 5, the hydrogel maintains its shape due to cross-linked polyacrylamide and duplex bridges within the DNA. Conversely, at pH 8, the hydrogel deforms and enters a quasi-liquid state, where the DNA duplex bridges do not contribute to restoring the hydrogel to its original shape upon returning to pH 5.
Redox-responsive hydrogels are a unique class of intelligent materials that react to reducing conditions, particularly elevated levels of glutathione (GSH) tripeptide. These hydrogels incorporate strategically placed disulfide linkages within their main or side chains or as cross-linkers. In the circulatory system and extracellular matrix (ECM), where glutathione levels are low (2.0–20 μM), disulfide bonds remain stable. However, inside cells, where glutathione concentrations rise to 0.5–10 mM, these bonds undergo thiol-disulfide exchange, leading to rapid cleavage. Notably, tumour tissues have glutathione levels four times higher than normal tissues, causing disulfide bond disruption and facilitating drug or bioactive compound release from the intracellular matrix. Yu et al.,182 synthesized hydrogels that can be reduced by incorporating poly(ethylene glycol) monomethyl ether (mPEG). This compound, mPEG-g-SS-PAA, consists of disulfide-linked poly(amido-amine) grafted with α-cyclodextrin. The researchers integrated BSA into the hydrogels and investigated how reductions in the intracellular matrix affected its release.
Ionic hydrogels exhibit pH-responsive swelling due to the presence of pendant groups with charges.183 Several variables influence this swelling behaviour, including ionic charge, the pKa or pKb values of ionizable groups, degree of ionization, hydrophilicity, polymer content, and the pH of the swelling medium. The composition of pendant groups and the pH of the environment are critical determinants of the properties of pH-sensitive hydrogels. For instance, cationic hydrogels such as poly(ethylene imine) and chitosan expand in acidic environments as amino/imine groups become protonated, leading to the formation of positively charged species that induce repulsion and swelling as shown in Fig. 11.184 These hydrogels are advantageous for drug delivery in the stomach within injectable drug delivery systems. In contrast, anionic hydrogels such as carboxymethyl chitosan swell in alkaline solutions due to the ionization of their acidic groups. The presence of negatively charged pendant groups induces repulsion and swelling, making these hydrogels suitable for drug administration in the gut at a physiological pH of 7.4. Another effective drug delivery strategy involves using polyelectrolyte complex (PEC) hydrogels, which eliminate the need for hazardous covalent cross-linkers. PEC hydrogels typically consist of a positively charged polymer, like chitosan, and a negatively charged polymer, such as carboxymethyl chitosan, held together by Eis. Zaino et al.,185 studied the drug delivery characteristics of dexamethasone using pH-sensitive PEC hydrogels composed of N-trimethyl chitosan (positively charged) and N-carboxymethyl chitosan (negatively charged).
 | ||
| Fig. 11 (a) pH-dependent ionization of specific acidic or basic functional groups on hydrogel chains responsible for swelling, (b) pH-dependent swelling and drug release mechanism. The figure is adapted from ref. 184 Copyright 2017, MDPI. | ||
The swelling behaviour of hydrogels upon exposure to water or physiological fluids is primarily driven by OP, influenced by the hydrophilic nature of polymers, static charges within the polymer network, and counterions present in the hydrogel matrix. This process occurs in three sequential stages: (i) initial water infiltration into the hydrogel network, (ii) polymer chain relaxation and hydration, and (iii) expansion of the network as polymer chains continue to relax. Swelling is primarily induced by primary-bound water, which forms through interactions between hydrophilic and polar groups and water molecules. Simultaneously, hydrophobic regions interact with water, leading to the formation of secondary-bound water. The overall increase in water content is regulated by osmotic forces, counteracted by the elastic retractive forces of cross-linked polymer chains. As the swelling progresses, excess water (free water) accumulates, guiding the hydrogel toward equilibrium swelling. At this point, swelling is stabilized by a delicate balance between elastic retractive forces and OP. This phenomenon is well explained by the Flory–Rehner theory, which attributes hydrogel swelling to the elastic properties of polymer chains and the thermodynamic compatibility between water molecules and the polymer matrix.186,187 A volume-phase transition occurs when stimulus-responsive hydrogels interact with specific triggers. The swelling behaviour of ionic hydrogels is primarily governed by two factors: (i) the intrinsic properties of the hydrogel-forming polymer, including cross-linking density, hydrophilicity, ionic charge, pKa, and pKb values; and (ii) the characteristics of the swelling medium, including ionic concentration, acidity, and counterion types.
The rate of water uptake in hydrogels is contingent upon the surrounding medium's pH in relation to the pKa and pKb values of the pendant groups in the polymer chains.188 For an anionic network (e.g., with carboxylic acid, COOH pendant groups), when the surrounding medium's pH exceeds the pKa of the acidic pendant groups, ionization occurs, resulting in fixed negative charges on the polymer chains and mobile positive charges in the solution. This change affects (i) the hydrophilicity of the hydrogels, (ii) the quantity of fixed negative charges, and (iii) the electrostatic repulsion between the chains, leading to an expansion of the hydrogel network; the reverse occurs when pH is lower than the pKa. Conversely, in a cationic network (e.g., containing amino or NH2 pendant groups), if the pH of the surrounding medium is below the pKb of the pendant basic groups, protonation occurs, increasing the number of fixed positive charges and inducing swelling due to enhanced hydrophilicity and electrostatic repulsion. Conversely, swelling contracts when the pH exceeds the pKb.
Zare-Akbari et al.,189 synthesized ionic cross-linked hydrogel beads based on CMC and ZnO nanoparticles (NPs) using Fe3+ ions as a physical cross-linking agent. The prepared hydrogel beads exhibited pH-sensitive swelling behaviour when immersed in phosphate-buffered saline (PBS) at pH levels of 1.2, 6.8, and 7.4 at room temperature, simulating various physiological environments. The swelling degree at pH 7.4 and 6.8 was found to be approximately three times greater than that at pH 1.2. At pH 6.8 and 7.4, the carboxyl groups in CMC dissociated in aqueous media, converting to negatively charged carboxylate ions, which led to increased electrostatic repulsion and enhanced water penetration into the hydrogel matrix. The pH-sensitive drug release behaviour was demonstrated using propranolol hydrochloride as a model drug, suggesting that the hydrogel beads could be potential candidates for controlled drug delivery applications.
Additionally, various polymers containing weakly acidic or basic groups that readily undergo hydrolysis or protonation have been developed for pH-sensitive hydrogels with cellulose, including gelatine, PAA, hyaluronic acid (HA), and poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA). The swelling behaviour of these hydrogels can be modulated by altering the pH, enabling controlled release of therapeutic agents.190 Furthermore, hydrogels with anionic and cationic polymeric networks can be synthesized through polyelectrolyte complexation, where cationic and anionic polymers form stable complexes, leading to enhanced swelling capacity in response to changes in pH.
5. Applications of cellulose-based hydrogels
In the biomedical field, hydrogels are prominent among polymer types due to their unique properties that render them suitable for various applications. These materials exhibit characteristics such as compatibility with living tissue, which opens numerous possibilities for use in biomedical settings. Their resemblance to biological tissues creates significant opportunities in areas such as drug delivery, wound care, and the rapidly advancing field of tissue engineering. The diverse attributes of hydrogels make them invaluable, offering versatile solutions to complex biomedical challenges. However, while different polymer types may possess unique properties for biomedical applications, their similarity to living tissue does not necessarily guarantee efficacy in drug delivery, wound care, or tissue engineering.5.1. Drug delivery
Hydrogel-based drug delivery systems demonstrate efficacy in precisely targeting and delivering drugs to specific sites within the body.191–193 These hydrogels can respond to various environmental stimuli, including light, temperature, pH fluctuations, and chemical interactions, as well as electric and magnetic fields.22 Their intriguing ability to expand and contract facilitates the clustering of polymers, leading to changes in transparency, size, and the formation of hydrogen bonds between molecules. Upon disruption of the hydrogel structure, drugs encapsulated within are released, with swelling occurring in response to nearby stimuli. Understanding the swelling process is crucial for studying the composition of hydrogels and its impact on response rates. Moreover, the degree of swelling influences the mechanical properties of the hydrogel, including flexibility and durability.194 A deep understanding of swelling behaviour is crucial for optimizing hydrogels in drug delivery and tissue engineering. Their stimuli-responsive nature enables precise and controlled drug release, improving therapeutic outcomes. The incorporation of cellulose and its derivatives significantly modifies hydrogel structures, creating larger pores due to intra-carboxyl group repulsion, which enhances the SR. Additionally, cellulose's responsive properties make it an excellent candidate for regulated drug release. Its proven biocompatibility, as confirmed by viability assessments, further supports its potential for biomedical applications. Numerous studies have focused on the role of hydrogels in regulating drug release. For instance, Villalba-Rodríguez et al.,195 conducted a comprehensive review on drug delivery using biodegradable hydrogels made from various materials, including chitosan, poly (lactic-co-glycolic acid), and bacterial cellulose. Hydrogels that adapt to changes in temperature, pH, and redox conditions exhibit considerable potential for drug delivery, aligning well with the body's natural mechanisms. Researchers have developed membranes from pH-responsive CMC-based hydrogels for applications in drug release and wound care. Distinct volumetric characteristics of HPC and poly(N-isopropyl acrylamide) (PNIPA) hydrogels have also been highlighted, indicating their promise in diverse biomedical applications.196 Additionally, thiolated HPC has shown potential as a hydrogel responsive to both redox reactions and temperature variations, making it suitable for controlled drug release.Zuwu Tang et al.197 developed a biocompatible, injectable, and self-healing PVP/CMC hydrogel using N,N′-methylenebis(acrylamide) (MBA) as a crosslinker and potassium persulfate (KPS) as an initiator. This hydrogel was designed for drug encapsulation and controlled release of 4-ASA, a drug commonly used to treat tuberculosis and inflammatory bowel diseases. Given that 4-ASA is sensitive to heat and moisture, its amphoteric nature-which depends on the surrounding pH-made it a suitable water-soluble model drug for this study. The drug loading and release properties of PVP/CMC hydrogels were tested in PBS buffer solutions at pH 2.0 and 7.4 at 37 °C (Fig. 12A). Increasing CMC concentration led to higher drug loading due to the greater availability of carboxyl and hydroxyl groups, which interacted with 4-ASA via hydrogen bonding. At 1.0 g CMC content, drug loading was 2.5 in pH 2.0 and 3.5 in pH 7.4, with higher loading at pH 7.4 due to reduced hydrogen bond interference in a less acidic environment. Drug release profiles (Fig. 12D and E) showed that hydrogels at pH 7.4 released more drug than those at pH 2.0. For instance, the PVP/CMC1.5 hydrogel released 70% of the drug in pH 7.4, compared to only 50% in pH 2.0. This difference was due to protonation of CMC's carboxyl groups at low pH, which led to hydrophobic region formation, causing the hydrogel network to contract and restrict drug diffusion. Overall, this study demonstrated that CMC concentration and pH significantly influence the drug loading and release behaviour of PVP/CMC hydrogels, making them promising for controlled drug delivery applications. In contrast, at pH = 7.4, the electrostatic repulsion among ions weakens the contraction, facilitating drug release.197 Additionally, as the CMC content increased, the drug release rate also increased. These findings suggest that the PVP/CMC hydrogel is highly efficient for drug release, and both the loading and release of 4-ASA are pH-dependent and can be optimized by adjusting the composition of the PVP/CMC hydrogel.197
 | ||
| Fig. 12 (A) A diagram illustrating the drug loading and release process of the PVP/CMC hydrogel. The loading behaviour of the hydrogel at varying CMC concentrations in PBS is shown in (B) at pH 2.0 and (C) at pH 7.4. The release profile of 4-ASA from different hydrogel formulations over time in PBS is depicted in (D) at pH 2.0 and (E) at pH 7.4. The figure is reprinted from ref. 197. Copyright © 2024 The Authors. Published by American Chemical Society. This publication is licensed under CC-BY-NC-ND 4.0. | ||
Many studies provide insights into the release profiles of various proteins using a range of hydrogel types, including dextran, ketoprofen, BSA, and alaptide.198 Various hydrogels utilized in these studies encompass interpenetrating networks (IPN), semi-IPN HPC-poly(acrylic acid), polyacrylamide grafted with xanthan gum (PAAm-g-XG), and different cellulose variations. Dutta et al.,199 reported on the development of triple stimuli-responsive hydrogels demonstrating the versatility of CMC and PNIPA. Research continues to explore various types of cellulose hydrogels that selectively release drugs in response to stimuli, including pH, magnetic fields, and temperature.200 While cellulose hydrogels function optimally in neutral environments, chitosan-based hydrogels are particularly effective in acidic conditions, which corresponds with the diverse pH levels found in different organs.
Fekete et al.,201 discussed how CMC/starch superabsorbent hydrogels synthesized via gamma irradiation exhibit superior water absorption compared to pure cellulose hydrogels, especially at high electrolyte concentrations. Conversely, cellulose/pectin hydrogels have been designed for targeted delivery to the colon, relying on their adhesive properties and enzymatic degradation.202 pH-sensitive hydrogels are emerging as prominent carriers for oral drug delivery to the intestine or colon, demonstrating their potential in non-invasive drug delivery strategies. Ahmadi et al.,203 highlighted the versatility of chitosan-based hydrogels for drug delivery systems via multiple routes, including oral, ocular, and nasal administration. In a different context, Vlaia et al.,204 emphasized the role of cellulose-derived hydrogels in dermal and transdermal drug delivery systems, showcasing the flexibility of cellulose hydrogels for diverse drug delivery needs. While these examples illustrate the potential of hydrogels in drug delivery, they do not confirm widespread clinical application or effectiveness.
Beyond drug delivery, cellulose-based hydrogels present substantial opportunities for advancements in disease detection and treatment.205 Various NPs, including lipid-based celluloses, liposomes, polymeric celluloses, dendrimers, carbon nanomaterials, and both inorganic and metallic NPs, are integrated with cellulose derivatives.206 This incorporation enhances the solubility and bioavailability of active pharmaceutical compounds, which is vital for developing dosage forms and achieving accurate release profiles in pharmaceutical formulations. However, concerns about the potential risks and long-term effects of NPs in drug delivery systems must be addressed, as enhanced drug absorption and solubility may not justify these hazards. Additionally, the use of NPs may increase production costs and limit the accessibility of certain medications.207,208
In cancer treatment, NPs can be employed to reduce the adverse effects on healthy tissues by selectively delivering chemotherapeutic agents to tumour cells.209 This targeted drug delivery approach enhances therapeutic efficacy while minimizing toxicity. However, concerns about the potential long-term effects and health risks associated with nanoparticle accumulation in organs and tissues persist.210 Moreover, the complex manufacturing processes and specialized equipment required for integrating NPs into drug formulations can significantly escalate production costs, restricting access to these therapies. Researchers are actively developing nanoparticle-based drug delivery systems in oncology to enhance targeting and reduce harm to healthy organs while effectively treating cancer cellulose.
Farnoush Ahmadpour et al.,211 conducted research exploring the distinctive characteristics and multifunctionality of hydrogel structures used in biomedicine. In their study, they synthesized a cross-linked Pec-Cel hydrogel using CaCl2 as a cross-linker. The synthesis of Fe3O4 magnetic NPs (MNPs) was then performed in the presence of the cross-linked hydrogel, resulting in the formation of a novel magnetic cross-linked Pec-Cel hydrogel nanobiocomposite. To assess the efficacy of this magnetic nanobiocomposite for in vitro hyperthermia applications, varying concentrations of the nanostructure were tested under alternating magnetic fields (AMF) with different frequencies. As anticipated, the highest specific absorption rate (SAR) was observed in the sample with the lowest concentration of magnetic nanobiocomposite, as the dipole–dipole interactions between NPs can disrupt their heat generation. Moreover, a significant reduction in SAR was noted as the sample concentration increased from 0.5 mg mL−1 to 10.0 mg mL−1, with SAR dropping from 126.0 W g−1 at 0.5 mg mL−1 to approximately 5.0 W g−1 at 10.0 mg mL−1. While the sample concentration had a substantial impact on the SAR, the influence of AMF frequency was comparatively less pronounced.211 The SAR decreased by over 95.0% as the concentration increased from the lowest to the highest, while the variation in SAR due to changes in AMF frequency was only 6, 7, 14, 30, and 2 percent for the 0.5, 1.0, 2.0, 5.0, and 10.0 mg mL−1 samples, respectively. These results indicate that the effect of AMF frequency differs based on sample concentration. Although the data shows that lowering the concentration of the magnetic Nanobiocomposite increases the thermal power per unit mass, it is crucial to also compare the temperature differences generated by each sample for a more comprehensive evaluation of their performance.211
5.2. Wound healing
The natural healing of wounds involves a complex interplay of biological systems that work together to restore the integrity of damaged skin.212 During this intricate process, tissue layers and cellulose Lular structures are replaced. For example, Hao et al. developed an injectable gel that aids in the healing of diabetic wounds by preventing infections and promoting angiogenesis.213 Hydrogels have proven effective for treating wounds in both diabetic and healthy individuals.11,214 Cellulose hydrogels and their derivatives, available in various forms such as fibres, membranes, and sponges, are widely utilized in wound care products.99,215 Due to the natural polycationic and hemostatic properties of deacetylated chitin or chitosan, these materials are particularly beneficial for wound healing applications. It is common practice to incorporate zinc oxide or silver NPs into cellulose gel systems to impart antimicrobial effects, as cellulose lacks inherent antibacterial activity.Initially, hydrogel wound dressings aimed to maintain a moist environment and provide physical protection.216 However, as clinical demands for improved performance in wound healing have evolved, hydrogel dressings with enhanced single or multiple biological activities have emerged. In addition to facilitating in situ creation and rapid hemostasis, hydrogels serve as essential physical barriers, preventing external bacterial infections.217 Nevertheless, conventional hydrogels are susceptible to rupture and damage upon exposure to tissue movement or external strain, compromising their integrity and function while increasing the risk of bacterial invasion and wound infection. Consequently, hydrogel dressings must maintain structural integrity throughout the healing process. Self-healing hydrogels have emerged as an innovative approach characterized by materials that can autonomously repair structural and functional damage.218 This technique employs constitutional dynamic chemistry to create a cross-linked hydrogel network through dynamic and reversible chemical interactions. Self-healing hydrogels can be categorized into two types based on their healing mechanisms: physical and chemical.219 Physical self-healing hydrogels reconstruct networks through non-covalent interactions, such as hydrophobic interactions, host–guest interactions, hydrogen bonding, crystallization, and various intermolecular forces among molecules or polymer chains. For instance, Liu et al.,220 developed a composite double-network hydrogel using tannic acid and gelatin methacrylate, with the hydrogel's efficient self-healing properties attributed to the dynamic hydrogen bonds of tannic acid. Another strategy involved modifying bacterial cellulose with both positive and negative fragments to create a self-healing hydrogel in a pH 7.4 buffer solution via an ionic interlocking mechanism.
Hao Chen et al.,221 introduced an injectable, self-repairing hydrogel with antibacterial and angiogenesis-promoting features for the treatment of diabetic wounds. This hydrogel, named Ag-SH-PEG, was synthesized through a straightforward method involving the coordination-based crosslinking of multi-arm thiolated polyethylene glycol (SH-PEG) with silver nitrate. Thanks to the reversible and dynamic nature of the Ag–S coordination bonds, the hydrogel demonstrated both self-healing behaviour after mechanical disruption and the ability to be administered via a medical syringe. The researchers proposed that this multifunctional hydrogel could effectively combat infections, promote blood vessel formation, and expedite tissue recovery in diabetic skin wounds.221
Additionally, the hydrogel matrix gradually released antimicrobial silver ions, making it particularly suitable for managing vulnerable open wounds in diabetic patients. By incorporating desferrioxamine (DFO), an angiogenic agent, into the hydrogel, the team developed a material that combined stress resistance, antibacterial efficacy, and enhanced angiogenic potential. In their experimental model using full-thickness dorsal skin wounds in SD rats, three groups were treated: a control group, a group receiving the plain hydrogel, and a group treated with the DFO-loaded hydrogel.221 Wound healing was assessed on days 0, 4, 7, 10, and 14 through digital imaging and quantitative analysis of wound size using ImageJ software.221 By day 7, wounds treated with the hydrogel showed significantly smaller diameters, with those in the DFO-hydrogel group shrinking by 50% of their original size. This reduction correlated with better tissue repair and a dry appearance, suggesting prevention of bacterial contamination and early tissue remodeling. In contrast, wounds in the control and plain hydrogel groups showed only a 30% size reduction by the same time. By day 14, nearly complete closure was observed in the DFO-hydrogel-treated wounds.221 The comparative analysis of wound areas revealed minimal differences between the groups during the initial inflammatory phase (first 2–3 days post-injection), likely due to limited vessel formation and epithelialization during this period. However, a noticeable acceleration in wound closure occurred after day 7 in the DFO-hydrogel group, persisting until day 14. The marked difference in wound size between this group and the others highlighted the biocompatibility of the DFO-loaded hydrogel and its ability to support tissue regeneration effectively.221
Esteban Guamba et al.,222 extracted cellulose from four distinct plants native to Ecuador and utilized it as a polymer to synthesize cellulose-based hydrogels. These hydrogels were subsequently compared to a reference hydrogel derived from commercially available cellulose. Comprehensive characterization was performed on both the cellulose powder isolates and the resulting hydrogels. Notably, cellulose from pear mesocarp (F1) failed to produce a hydrogel, leaving hydrogels prepared from pear epicarp (F4), tomato (F12), pitahaya (F53), and commercial cellulose for further investigation. These selected hydrogels underwent in vitro antimicrobial assays and were evaluated for wound dressing applications using a pigskin model as a conceptual demonstration.222
The findings revealed that hydrogels fabricated using pitahaya cellulose demonstrated the highest potential in inhibiting bacterial proliferation, making them favourable for wound healing applications.222 This efficacy was attributed to their antifouling properties, which appeared to impede bacterial adhesion and growth. However, all hydrogels-except for F53-exhibited enhanced bacterial colonization, with the commercial wound dressing showing the most significant colony formation under identical conditions. Interestingly, F53 effectively controlled bacterial growth within the initial 48 hours, after which normal bacterial proliferation resumed. These observations highlight F53 as the most promising candidate for developing wound dressings, surpassing both commercial cellulose-based hydrogels and standard wound bandages in bacterial growth resistance. While hydrogels from F12, CMC, and F53 displayed no colony formation in the in vitro tests, only F53 demonstrated reduced bacterial growth in the ex vivo pigskin model. This discrepancy is likely due to the variation in bacterial strains present in pigskin samples compared to the single-strain inoculum used in the in vitro assessments.222
Xiaotong Yi et al.,223 designed and developed an innovative, adhesive, antibacterial, self-healing hydrogel wound dressing by incorporating a PVA-borax hydrogel matrix with bacterial cellulose (BC), dopamine (DA), and doxycycline (Doxy). To improve the adhesion and mechanical strength of the PB hydrogel, a reinforcement material, PDA@BC, was synthesized via dopamine-induced self-polymerization. This material was then coated onto the BC surface, allowing for uniform dispersion within the hydrogel matrix. The resulting PB-PDA@BC hydrogels, loaded with doxycycline, exhibited outstanding antibacterial properties. These effects were primarily due to the local release of the antibiotic, alongside the hydrogels' favourable cytocompatibility and blood compatibility.223 The wound healing efficiency of the PB-PDA@BC/Doxy hydrogel was compared to the commercial Tegaderm film dressing (control group) across three time points-day 3, day 7, and day 14-as shown in Fig. 13(a–c). All groups demonstrated a reduction in wound area, but the PB-PDA@BC/Doxy group exhibited the most significant wound closure, with a 20.1% reduction on day 3 (p < 0.05). By day 7, the PB-PDA@BC/Doxy hydrogel group showed a 5.7% and 11.9% improvement in wound closure compared to the PB-PDA@BC hydrogel and control groups, respectively. On day 14, the PB-PDA@BC/Doxy group achieved near-complete healing, with a wound closure of 97.1%, which was approximately 10% higher than the control group, as confirmed by quantitative analysis of the wound area (p < 0.05). These results suggest that doxycycline-loaded PB-PDA@BC hydrogels offer superior performance in wound healing compared to Tegaderm, thanks to the combined effects of antibiotic properties, hemostasis, and the moist environment provided by the hydrogel matrix. However, the antibiotic-coated PB-PDA@BC hydrogel efficiently eliminates bacteria at the wound site, promotes a conducive microenvironment for cellular proliferation and epidermal regeneration, and accelerates the overall healing process. To further assess the wound healing process, tissue samples from the wound area were subjected to histological analysis.223 Hematoxylin and eosin (H&E) staining results (Fig. 13(d)) revealed mild inflammatory responses across all groups after 3 days of treatment. However, the PB-PDA@BC/Doxy hydrogel group showed significantly fewer inflammatory cells compared to the control group, likely due to the antibacterial effects of doxycycline. After 7 days, all groups exhibited the formation of a new epidermal layer. Notably, the PB-PDA@BC/Doxy hydrogel group displayed a thicker epidermis than both the control and PB-PDA@BC hydrogel groups. By day 14, the PB-PDA@BC/Doxy group also showed the emergence of hair follicles and blood vessels, signalling the hydrogel's ability to support skin regeneration. These findings demonstrate that the drug-loaded hydrogels not only enhance wound healing but also promote skin regeneration, making them a promising option for accelerated tissue repair. However, challenges remain in developing hydrogels that provide the optimal balance of structural integrity, mechanical flexibility, and controlled drug release for effective wound healing. Hydrogels with superior mechanical properties can withstand various stresses while providing sufficient softness and pliability for enhanced comfort in wearability. Developing next-generation cellulose hydrogels that address these factors will significantly improve wound care solutions.223
 | ||
| Fig. 13 Highlights the application and effectiveness of hydrogels in wound healing. Panel (a) demonstrates the use of the hydrogel as a therapeutic agent to enhance the wound recovery process. Panel (b) compares the wound closure rates achieved through various treatments, showcasing the superior efficacy of the PB-PDA@BC/Doxy hydrogel. Panel (c) provides histological evidence of wound regeneration on the 3rd, 7th, and 14th days, comparing commercial Tegaderm film dressing, PB-PDA@BC, and PB-PDA@BC/Doxy hydrogels. Notably, the PB-PDA@BC/Doxy hydrogel exhibits enhanced regenerative capabilities, as evidenced by improved tissue organization and closure over time. Finally, panel (d) offers a detailed depiction of the PB-PDA@BC/Doxy hydrogel actively promoting wound healing, underlining its potential as an advanced material in tissue repair applications. The figure is adopted from ref. 223. Copyright © 2023, The Author(s), under exclusive licence to Springer Nature B.V. | ||
5.3. Tissue engineering
Tissue engineering requires hydrogels that fulfil specific criteria, balancing physical properties such as degradation rates and mechanical strength with biological performance indicators like cell adhesion.224,225 Biocompatibility is of paramount importance, ensuring that these hydrogels can exist within the body without causing harm or eliciting adverse responses.226,227 This aspect is particularly crucial, as any inflammatory reaction to the hydrogel could influence the immune response towards transplanted cells and vice versa. Naturally derived polymers generally exhibit suitable biocompatibility; however, synthetic polymers may trigger negative bodily reactions, which limits their application in hydrogel preparation for tissue engineering purposes.228A promising strategy for treating patients in need of new organs or tissues involves the engineering of artificial tissues or organs.229 Various techniques have been developed for this purpose, with one particularly appealing approach involving the use of a combination of patient-derived cells and polymer scaffolds.230,231 Cells isolated from a patient's tissue biopsy are cultured in vitro and subsequently integrated into three-dimensional polymer scaffolds that mimic the natural extracellular matrices found in tissues. These scaffolds facilitate cell delivery to the targeted site, create space for new tissue formation, and may regulate the structure and function of the engineered tissue. This approach has been employed in engineering different types of tissues, including arteries, bladders, skin, cartilage, bone, ligaments, and tendons, some of which are nearing clinical use. Additionally, techniques have been developed to guide cells towards the desired phenotype, a critical aspect of successful tissue engineering.
CNCs, typically isolated from cellulose, have gained popularity as reinforcing fillers in polymeric hydrogels due to their favourable mechanical properties and intrinsic biocompatibility.232,233 The unique aspect ratio and abundant active –OH groups on their surface make CNCs an excellent precursor for preparing robust stabilizers. Yang et al.,234 reported injectable hydrogels based on adipic acid dihydrazide-modified CMC and aldehyde-modified dextran, reinforced with CNCs and aldehyde-functionalized CNCs. Gelation occurred within seconds when the hydrogel components were extruded from a double-barrel syringe, and CNCs were observed to be evenly distributed throughout the composites. Swelling tests indicated that all CNC-reinforced hydrogels maintained their structural integrity for over 60 days in both water and 10 mM PBS, suggesting their potential for long-term applications. The CHO-CNCs acted as both a filler and a chemical cross-linker, enhancing the elasticity and stability of the CHO-CNC-reinforced hydrogels, allowing for higher nanoparticle loadings without compromising mechanical strength compared to hydrogels containing unmodified CNCs. Cytotoxicity tests demonstrated that both the initial components and the hydrogels exhibited good cytocompatibility with NIH 3T3 fibroblast Cells. Together with their syringe ability, these CNC-reinforced injectable hydrogels show promise for filling irregular cavities and shapes without requiring pre-shaped forming processes, making them ideal for bone tissue engineering.234
The introduction of inorganic NPs, such as LAPONITE®, hydroxyapatite (HAp), and titanium dioxide, can further enhance cellulose-based hydrogels for bone tissue engineering applications.235 Boyer and colleagues,236 developed a LAPONITE® NP-reinforced Si-HPMC hydrogel, in which LAPONITE® NPs self-assembled within the Si-HPMC gel structure, resulting in a hybrid interpenetrating network (IPN). This IPN structure significantly improved the mechanical properties of the hydrogel while maintaining oxygen diffusion and cell viability after gelation. The ability of the hybrid scaffold, composed of the Si-HPMC/LAPONITE® hydrogel and chondrogenic cells, to form cartilaginous tissue in vivo was investigated over six weeks of implantation in subcutaneous pockets of nude mice. Histological analysis of the composite constructs revealed the formation of cartilage-like tissue with an extracellular matrix rich in glycosaminoglycans and collagen. These findings indicate that the prepared Si-HPMC/LAPONITE® hydrogels possess considerable potential for repairing cartilage defects. Furthermore, Tohamy et al.,237 constructed composite scaffolds of sodium alginate (SA)/HEC/HAp using a lyophilization technique followed by cross-linking in the presence of Ca2+ ions. The study found that a higher HAp concentration (40 wt%) effectively enhanced the mechanical properties (23.9 MPa), bioactivity, and protein adsorption. Cell experiments confirmed the non-toxicity and robust proliferation capability of the SA/HEC/HAp scaffold, likely attributed to the biocompatibility, bioactivity, strong cell adhesiveness, and excellent mechanical strength of HAp.237
The polymer scaffold is a critical component in nearly all tissue engineering approaches, aiming to replicate various functions of natural extracellular matrices.238 These matrices, composed of amino acids and sugar-based macromolecules, regulate tissue structure, cell function, and nutrient diffusion. Various polymers, such as aliphatic polyesters like poly(glycolic acid) (PGA), poly(lactic acid) (PLA), and copolymers (PLGA), have been extensively studied and utilized in tissue engineering.239 These polymers, considered safe in numerous medical contexts by the FDA, require incisions for the implantation of polymer/cell constructs. An intriguing approach for cell delivery in tissue engineering involves the use of injectable polymers, specifically hydrogels.240 This method allows clinicians to minimally invasively transplant cell–polymer combinations. Hydrogels, akin to the macromolecular components found in the body, are regarded as biocompatible and serve a variety of applications, including tissue engineering and drug delivery. Their recent application in tissue engineering includes functioning as scaffolds for the development of new tissues. Tissue engineering leverages innovative hydrogels based on cellulose technology, as highlighted by Radhakrishnan et al.,241 which act as frameworks resembling extracellular matrices to support the growth of new tissues. These frameworks create favourable conditions, providing essential space and nutrients crucial for targeted tissue development. Marler et al.,242 demonstrated their ability to engineer tissues such as cartilage, bone, muscle, skin, fat, arteries, ligaments, tendons, liver, bladder, and neurons. Furthermore, hydrogel frameworks contribute to surface modifications that facilitate tissue growth in biomedical implants. This modification can inhibit specific cell attachment while promoting the binding of others or securing biological components under specific conditions. Tallawi et al.,243 explained that these approaches control cellular interactions by reducing unwanted cell adhesion or improving connections between implanted biomaterials and bone or skin.
To address challenges associated with mesenchymal stem cell (MSC) culture, thermo-responsive hydrogels, particularly chitosan-g-PNIPAAm, present a promising solution. Lihui Peng et al.244 discussed the difficulties in MSC culture, especially the challenges in detaching MSCs from their culture carriers due to their strong adhesive properties, which often require harsh conditions. Therefore, further exploration of cell culture carrier materials and gentle methods for harvesting cultured cells is necessary. One potential strategy involves utilizing thermo-responsive hydrogels as cell culture carriers, which allow for easy detachment of cultured cells by simply lowering the temperature below their lower critical solution temperature (LCST). Chitosan-g-PNIPAAm demonstrates favourable characteristics for cellular attachment, proliferation, viability, and chondrocyte differentiation. This advancement led to the development of an injectable gel material based on chitosan/PNIPAAm for the chondrogenic differentiation of human MSCs. Subsequent assessments evaluated cartilage formation in vivo following the injection of a cell-thermo-sensitive gel complex. Animal experiments aimed to assess cartilage formation in the submucosal layer of rabbit bladders. Although the thermo-sensitive gel system may not be suitable for in vivo applications due to its high LCST of 32 °C, the innovative combination of chondrogenic ally differentiated MSCs with a thermo-sensitive polymer is proposed for use as an injectable cell–polymer complex. Observations of chondrogenic differentiation, both in vitro and in vivo, indicate the potential of this gel for a simplified treatment approach for vesicoureteral reflux using an endoscopic single injection technique, potentially eliminating the need for a dual injection system.
5.4. Smart materials
Cellulose-based hydrogels have emerged as crucial components in the development of advanced devices, particularly in the field of biochemical sensors. Their exceptional biocompatibility, substantial capacity for cell and small molecule storage, and low interfacial tension with aqueous solutions render them suitable for various applications.184 These sophisticated materials are essential for interpreting chemical information and translating specific analyte concentrations into comprehensive compositional analyses.Typically, chemical sensors consist of two primary components: a chemical recognition system (receptor) and a physico-chemical transducer. Biosensors, a specialized subclass of chemical sensors, leverage biochemical mechanisms within their recognition systems.183 Recent studies have extensively explored the mechanical strength and biocompatibility of cellulose-based smart materials, which have applications ranging from electro-responsive electro-rheological (ER) suspensions to innovative composites with CNTs for conducting materials, wearable electronics, smart textiles, and various sensor types.186–189
Stimuli-responsive polymer systems are vital for effective transduction mechanisms, making hydrogels particularly well-suited for sensor applications.245 For instance, hydrogel films demonstrate exceptional sensitivity as pH-responsive nanosensors, characterized by rapid response times, highlighting their potential in diverse sensing applications.190 The rising demand for reliable, microfabricated chemical biosensors for real-time monitoring in biotechnology, food production, pharmaceuticals, and environmental sectors underscores the necessity for efficient and cost-effective analytical solutions. Functionalized hydrogel-coated biosensors can detect, transmit, and record variations in analyte concentration or the presence of specific functional groups, generating signals that correspond to the target analyte's concentration.191 The sensitivity of these hydrogels is influenced by various factors, including temperature, applied electrical voltage, pH, and the concentrations of organic compounds and salts in aqueous solutions. Stimuli-responsive hydrogels that can convert chemical energy into reversible mechanical work are becoming increasingly valuable in sensor technology. The swelling behaviour of pH-sensitive hydrogels, which is determined by the presence of acidic or basic groups within the polymer backbone, dynamically changes in response to pH shifts and charge interactions.192,193
The development of smart wearable devices has become a prominent research focus due to their potential in health monitoring.246 Self-healing wearable devices can restore their structure and functionality after damage, enhancing durability and safety.247 Self-healing hydrogels, as soft and flexible materials, are particularly appealing for the development of wearable pressure sensors that detect human motion.248 To meet the required mechanical toughness and mobility properties, two seemingly contradictory characteristics, self-healing hydrogels must effectively balance dynamic cross-links for healing and stable cross-links for mechanical strength. The integration of self-healing abilities with robust mechanical properties into a single conductive hydrogel presents a significant challenge. CNCs are commonly utilized as fillers to reinforce hydrogels due to their unique properties, including mechanical strength, biodegradability, biocompatibility, and modifiability. Recent advancements have successfully employed CNCs to create conductive hydrogels that exhibit both self-healing and mechanical performance.29,30
Inspired by the hierarchical structures of biological soft tissues, Liu et al. developed a conductive, elastic, self-healing, and strain-sensitive functional network hydrogel (F-hydrogel) formed from a “soft” homogeneous polymer network via covalent cross-linking of polyvinyl alcohol (PVA) and polyvinylpyrrolidone (PVP).249 This network incorporated a “hard” Fe3+-cross-linked CNC network with dynamic CNC-Fe3+ coordination bonds as reinforcing domains.250 Under stress, these dynamic coordination bonds functioned as sacrificial bonds to efficiently dissipate energy, while the PVA-PVP network facilitated smooth stress transfer. As a result, the F-hydrogels exhibited exceptional mechanical properties, characterized by a tensile strength of 2.1 MPa, toughness of approximately 9.0 MJ m−3, and stretchability of 830%.249 These properties allow the hydrogels to withstand significant deformations, such as compression and knotting, without visible damage. Furthermore, the F-hydrogels demonstrated autonomous self-healing capabilities within just 5 minutes, without external stimuli or healing agents, while maintaining their sensing performance before and after restoration. They exhibited ultrasensitive, stable, and repeatable resistance variations in response to mechanical deformations, making them promising candidates for wearable devices.
Based on the characteristics of the F-hydrogels, a wearable soft strain sensor was assembled by Liu et al.,249 to monitor finger joint motions, breathing patterns, and slight blood pulses (Fig. 14). As the bending angle of the finger increased from 0° to 120° during a controlled bending process, the relative resistance of the F-hydrogel sensor increased correspondingly (Fig. 14(a)). The sensor displayed repeatable responses at a bending angle of 90° with a working frequency of 0.3 Hz (Fig. 14(a)). When attached to the skin of a volunteer's lower left rib cage, the sensor effectively monitored and distinguished various breathing modes-including regular breathing, rapid deep breathing, and breath-holding-based on changes in relative resistance (Fig. 14(b)). Additionally, when placed on the wrist of a volunteer (Fig. 14(c)), the sensor accurately reflected changes in blood pulse before and after exercise, as shown on the relative resistance curve (Fig. 14(d)), confirming its high strain sensitivity and rapid response-ideal for human health monitoring.249
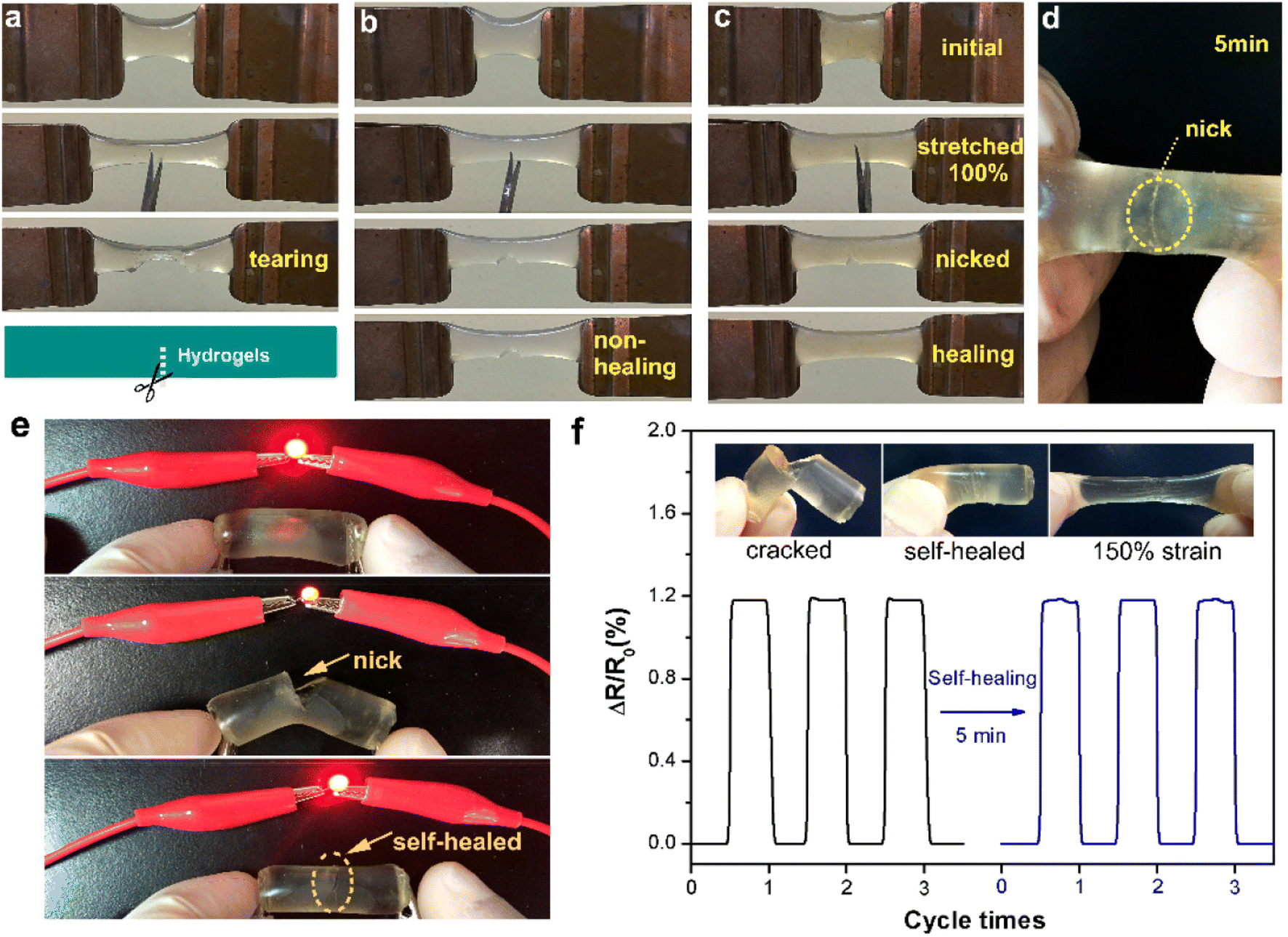 | ||
| Fig. 14 Real images of notch sensitivity testing. This series of photographs illustrates the notch sensitivity experiments conducted on hydrogels. A notch (one-third of the width) was created in the edge of the hydrogels, which were initially stretched to 100% using scissors: (a) the notch in the tearing H-hydrogels, (b) the undamaged notch in C-hydrogels-15, and (c) the intact notch demonstrating self-healing in F-hydrogels-15/2.5. (d) Evidence of self-healing in F-hydrogels-15/2.5 is shown after only 5 minutes post-unloading, occurring without any external stimuli or healing agents. (e) Changes in the LED light within the electric circuit are depicted before and after the self-healing process. (f) The sensor performance of the F-hydrogels during a loading–unloading cycle at 150% strain is shown, along with an inset featuring photographs of the F-hydrogels in cracked, self-healed, and 150% strain conditions. This image is adopted from the ref. 249. Copyright © 2017 American Chemical Society. | ||
Similarly, Shao et al.,251 developed a tough, self-healing, and self-adhesive gel by constructing synergistic multiple coordination bonds among tannic acid (TA)-coated CNCs, PAA chains, and Al3+ ions within a covalent polymer network. The TA@CNCs acted as dynamic bridges in a hierarchically porous network, mediated by multiple reversible coordination bonds. This architecture endowed the PAA-TA@CNCs-Al3+ gels with superior mechanical properties, including ultra stretchability (fracture strain of 2952%), high compression performance (95% strain without fracture), and toughness (5.60 MJ m−3). The dynamic features of the reversible coordination bonds also provided the gels with excellent recoverability and electrical self-healing properties. Incisions in the gels self-healed automatically, disappearing nearly completely within 30 minutes, achieving an equilibrium state with a healing efficiency of 92%. Additionally, due to the presence of catechol groups from the incorporated TA, these gels displayed durable and repeatable adhesion to various substrates, including human skin, rubber, aluminium, glass, and polytetrafluoroethylene, without inducing inflammatory responses or leaving residues. This makes them suitable as flexible strain sensors directly adhered to the skin.
Owing to their excellent self-adhesiveness, high strain sensitivity, remarkable electrical stability, and fast self-healing capabilities, these gels can detect substantial motions, such as joint bending and stretching during various human activities, including opisthenar, elbow, and shoulder joint bending in standard shooting actions.251 Furthermore, subtle motions like pulse and breath, crucial for real-time healthcare monitoring during sports training, can be accurately detected and promptly recognized by the gel strain sensors.252 This research paves the way for developing biocompatible cellulose-based conductive hydrogels for applications in wearable electronic sensors, healthcare monitoring, and even soft intelligent robots.
Pressure sensors have become indispensable in the realm of wearable devices due to their outstanding sensing capabilities.253 These sensors offer a wide range of applications, including pulse detection, voice recognition, and motion sensing. A notable example is the work by Li et al.,254 who utilized a simple pyrolysis technique to produce carbon cottons (CCs) with remarkable properties such as low density and an electrical conductivity of approximately 11 S m−1, utilizing a cotton substrate. The composite material was synthesized by infusing polydimethylsiloxane (PDMS) resin into the CC structure under vacuum conditions. The resulting CC/PDMS composite pressure sensor exhibited a high sensitivity of 6.04 kPa−1, a large operational pressure ranges up to 700 kPa, a broad frequency response from 0.01 Hz to 5 Hz, and outstanding durability, maintaining performance over 1000 cycles. The practical application of this sensor was demonstrated by embedding it in a sports shoe and a waist belt, where it was used to monitor health metrics and track athletic performance. The sensor's manufacturing process is cost-effective and scalable, primarily due to the use of cotton, a readily available and inexpensive material. These attributes make this sensor highly promising for future applications in health monitoring and the development of wearable electronics, such as prosthetic skins.
In a different approach, cellulose fibres were coated with multi-walled carbon nanotubes (MWCNTs) to create flexible and pressure-responsive sensors.255 The team employed cotton cellulose in combination with MWCNTs to fabricate these sensors and analysed their morphology using SEM (Fig. 15).255 The mechanical performance of the sensors, in terms of stress–strain behaviour, was assessed for varying MWCNT compositions. These sensors exhibited key advantages such as flexibility, porosity, cost-effectiveness, and significant potential as textiles for various applications. Further innovation was seen in the work of Li et al.,256 who developed conductive fibres to fabricate a highly stable and sensitive textile sensor with excellent electrical properties. In this process, silver nanocomposite particles and elastic rubber were combined to fabricate the conductive fibres via a coating method, achieving an electrical conductivity of 0.15 Ω cm−1. These fibres formed a well-organized electrical network, demonstrating both durability and resistance to external forces for about 3000 bending cycles. The inclusion of elastic rubber enhanced the fibres' stretchability. Additionally, the researchers employed PDMS-coated conductive fibres to build a textile-based capacitive pressure sensor. This sensor exhibited remarkable features, including ultra-high sensitivity of around 0.21 kPa−1 in the low-pressure region, a rapid response time of under 10 ms, exceptional durability for over 10000 cycles, and minimal hysteresis. The textile sensor was then woven into a fabric, effectively creating a skin-like interface for human–machine interactions. With the potential to be seamlessly integrated into clothing and gloves, these sensors offer exciting possibilities for wireless human motion tracking, making them strong candidates for smart textile applications.
 | ||
| Fig. 15 (a, i, and ii) Depicts the chemical structure of cotton cellulose and illustrates its swelling behaviour. (b, i, and ii) Shows the chemical structure of multi-walled carbon nanotubes (MWCNTs) and their dispersion characteristics. (c) Presents a SEM image of the pristine cellulose network in its substrate form. (d and e) Demonstrates pressure sensors incorporating varying mass fractions of MWCNTs, with concentrations of (d) 1 wt% and (e) 3 wt%. (f) Provides a planar view of the strain sensor, while (g) displays its axial view. (h) Depicts the stress–strain curves for the pressure sensor, as shown in the referenced work. Reproduced with permission from ref. 255. Copyright © 2019 American Chemical Society. | ||
Capacitive sensors, typically composed of two parallel plates separated by a dielectric material such as air, are well-known for their superior linearity and minimal hysteresis, characteristics that make them particularly effective for a wide range of practical applications. In recent research, integrating single-walled carbon nanotube (SWCNT) paper into a PDMS matrix has enabled the development of flexible, highly sensitive strain sensors. For example, Zhou et al.,257 demonstrated a novel approach where SWCNT paper-based sensors achieved an impressive gauge factor (GF) of 107 when subjected to a 50% applied strain. This fabrication technique allows for the incorporation of SWCNT paper of varying thicknesses within PDMS substrates, resulting in a range of smart materials with distinct properties, such as enhanced sensitivity and moderate flexibility. To optimize the performance of these sensors, the introduction of inter- or intra-laminar cracks has proven effective, with each method influencing the sensor's response in unique ways. This innovative strategy not only holds promise for strain detection but also presents an opportunity for expanding the use of other cellulose derivatives or nanomaterials, paving the way for the development of highly effective strain sensors.257
In another significant breakthrough, researchers have designed a flexible, ultra-sensitive pressure sensor utilizing a simple, cost-effective, and scalable approach based on silver nanoparticle (AgNP)-poly(3,4-ethylenedioxythiophene) (PEDOT)-paper composites.258 This sensor consists of intertwined fibres coated with a conductive PEDOT layer, supported by a permeable and stretchable paper substrate. The PEDOT coating enhances the electrical conductivity of the cellulose fibres, while the addition of AgNPs to the PEDOT matrix increases the surface roughness of the polymer, significantly improving its conductive properties. These modifications-lowered resistance and increased surface roughness-lead to notable improvements in sensor performance, particularly in terms of sensitivity and detection range. The sensor demonstrates an impressive sensitivity of 0.119 kPa−1 within a pressure range of 0–12 kPa, along with excellent stability over 2000 cycles. These outstanding characteristics make the sensor highly suitable for applications in human movement detection, including monitoring of breathing, phonation, pulse, heartbeat, and voice recognition. Furthermore, the sensor has been successfully integrated into a human–machine interaction system, enabling voice recognition and even musical translation from a piano keyboard. This advancement not only creates new opportunities for bi-directional communication channels but also facilitates the integration of real-time human behaviour monitoring, advanced digital signal processing, and artificial intelligence technologies.258
6. Conclusions and future aspects
Cellulose-based hydrogels hold immense promise for biomedical applications due to their biocompatibility, biodegradability, and versatile mechanical properties.259,260 As we look ahead, further advancements in cellulose-based hydrogel technology will open up new avenues for their use in regenerative medicine, drug delivery, wound healing, and tissue engineering. One key area of focus will be enhancing the mechanical toughness and swelling behaviour of these hydrogels simultaneously to meet the demanding requirements of biomedical applications.1,261,262By incorporating dynamic noncovalent interactions, the hydrogel can exhibit reversible responses to external stimuli, enabling dynamic changes in structure and properties.263,264 This adaptability allows the hydrogel to withstand mechanical stresses and undergo controlled swelling without compromising its integrity. Simultaneously, the introduction of non-reversible covalent bonds provides stability and permanence to the hydrogel network, ensuring long-term structural integrity and resistance to degradation. These covalent bonds act as anchors within the hydrogel matrix, reinforcing its mechanical strength and preventing excessive swelling.
The synergistic combination of dynamic noncovalent interactions, such as HB and host–guest interactions, with non-reversible covalent bonds offers a promising approach to overcome the limitations associated with mechanical toughness and swelling behaviour in cellulose-based hydrogels. Through the synergistic interplay between dynamic noncovalent and non-reversible covalent bonds, cellulose-based hydrogels can achieve a balance between toughness and swelling behaviour, making them ideal candidates for biomedical applications. The dynamic noncovalent interactions allow for reversible deformation and toughening, while the non-reversible covalent interactions provide structural stability and control over swelling behaviour. This synergistic approach can lead to the development of cellulose-based hydrogels with enhanced properties for biomedical applications, paving the way for new advances in regenerative medicine, drug delivery, and tissue engineering. Here Fig. 16 represent the network structure of crosslinked and uncross linked hydrogels at swollen condition. Herein, the uncross linked gel network absorbed a larger amount of water in an uncontrolled manner compared to the crosslinked network. However, after certain time, the uncross linked hydrogels get disintegrated while the crosslinked hydrogel remained same. However, the proper combination of those techniques would be offering a combination of mechanical toughness and control swelling properties.
 | ||
| Fig. 16 Swollen network structure of uncross-linked and cross-linked hydrogels. | ||
There is limited understanding regarding the establishment of theoretical models that can quantitatively predict the mechanical properties of cellulose-based hydrogels based on their composition. Factors such as sacrificed physical and chemical bonds, degree of homogeneity, cross-linking density, concentration, entanglement, and network structure all play crucial roles.
Molecular simulations are essential for advancing the study of cellulose-based hydrogels. These simulations can (i) expedite the modelling of large and complex hydrogel structures; (ii) develop precise force fields to analyse the contributions of various factors; (iii) assess the mechanical properties of specific compositions before experimental testing; and (iv) design cellulose-based hydrogels tailored to specific applications, such as ion and electron transport in energy storage and conversion. Additionally, it is important to elucidate the mechanical behaviour of these hydrogels around their yield point.
Cellulose-based smart materials offer immense potential for advancing wearable sensors, yet significant challenges persist in improving their biocompatibility, multi-functionality, and seamless integration with human health monitoring systems. Future research should prioritize the creation of innovative microstructures, the development of skin-like electronics with optimized mechanical and electrical properties, and the application of advanced processing techniques to produce intelligent, multi-functional devices. Furthermore, overcoming limitations in sensitivity, stretchability, and wireless functionality is crucial for transitioning these materials into commercially viable, scalable, and durable technologies. Such advancements promise not only to revolutionize health monitoring but also to expand their utility into areas like drug delivery and broader biological research, heralding groundbreaking progress in biomedical engineering.
Research on cellulose-based hydrogels for various innovative applications is still in its early stages. Potential applications include injectability and 3D printing, optical, electric, and magnetic tough materials, actuators, ionotropic devices, flexible electronic skins, and impact-resistant materials. Despite being in the nascent phase, the inherent mechanical properties and cytocompatibility of cellulose-based hydrogels offer promising pathways for their use across multiple fields.
Abbreviations
| CMC | Carboxymethyl cellulose |
| EC | Ethyl cellulose |
| MC | Methylcellulose |
| HEMC | Hydroxyethyl methyl cellulose |
| MCC | Microcrystalline cellulose |
| HBI | Hydrogen bonding interactions |
| EIS | Electrostatic interactions |
| PAA | Polyacrylic acid |
| HPC | Hydroxypropyl cellulose |
| CNCs | Cellulose nanocrystals |
| CC | Chemical crosslinked |
| Upy | 2-Ureido-4-pyrimidone |
| PC | Physical crosslinked |
| CNFs | Cellulose nanofibrils |
| CSH | Cellulose/silk fibroin hydrogel |
| DTT | 1,4-Dithiol-dl-threitol |
| CA | Citric acid |
| CYS | Cysteamine dihydrochloride |
| SD | Swelling degree |
| SR | Swelling ratio |
| ES | Equilibrium-swelling theory |
| RE | Rubber-elasticity |
| PGA | Poly(glycolic acid) |
| PLGA | Copolymers |
| IPN | Interpenetrating network |
| PLA | Poly(lactic acid) |
| HEC | Hydroxyethyl cellulose |
| SA | Sodium alginate |
| DVS | Divinyl sulfone |
| PNIPAAm | (N-isopropylacrylamide) |
| MSC | Mesenchymal stem cell |
| PDMS | Polydimethylsiloxane |
| CYS | Cysteamine dihydrochloride |
| AG | Alginate |
| ECH | Epichlorohydrin |
| OP | Osmotic pressure |
Data availability
There is no supporting document.Author contributions
Conceptualization: M. M. H. R.; original manuscript writing, M. M. H. R., figure drawing: M. M. H. R.; revision and editing, M. M. H. R., The author has read and agreed to the published version of the manuscript.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
The authors are thankful to the School of Data and Sciences Course Waiver for Research Policy of Brac University, Bangladesh.References
- H. Cao, L. Duan, Y. Zhang, J. Cao and K. Zhang, Current hydrogel advances in physicochemical and biological response-driven biomedical application diversity, Signal Transduction Targeted Ther., 2021, 6(1), 426, DOI:10.1038/s41392-021-00830-x.
- M. M. H. Rumon, S. D. Sarkar, M. M. Uddin, M. M. Alam, S. N. Karobi, A. Ayfar, M. S. Azam and C. K. Roy, Graphene oxide based crosslinker for simultaneous enhancement of mechanical toughness and self-healing capability of conventional hydrogels, RSC Adv., 2022, 12(12), 7453–7463, 10.1039/D2RA00122E.
- M. M. H. Rumon, S. D. Sarkar, M. M. Alam and C. K. Roy, Nanomaterials for Self-Healing Hydrogels, ACS Appl. Nano Mater., 2023, 141, 270–293 CAS.
- K. Sahajpal, S. Shekhar, A. Kumar, B. Sharma, M. K. Meena, A. K. Bhagi and S. Sharma, Dynamic protein and polypeptide hydrogels based on Schiff base co-assembly for biomedicine, J. Mater. Chem. B, 2022, 10(17), 3173–3198, 10.1039/D2TB00077F.
- S.-W. Zhou, C. Yu, M. Chen, C.-Y. Shi, R. Gu and D.-H. Qu, Self-healing and shape-shifting polymers controlled by dynamic bonds, Smart Mol., 2023, 1(2), e20220009, DOI:10.1002/smo.20220009.
- M. M. Rumon, A. A. Akib, F. Sultana, M. Moniruzzaman, M. S. Niloy, M. S. Shakil and C. K. Roy, Self-Healing Hydrogels: Development, Biomedical Applications, and Challenges, Polymers, 2022, 14, 4539 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review, Biomaterials, 2016, 85, 152–167, DOI:10.1016/j.biomaterials.2016.01.061.
- J. Singh and P. Nayak, pH-responsive polymers for drug delivery: Trends and opportunities, J. Polym. Sci., 2023, 61(22), 2828–2850, DOI:10.1002/pol.20230403.
- Z. Bei and J. Zheng, Recent advances in the application of functional hydrogels in skin wound healing, MedComm: Biomater. Appl., 2024, 3(4), e101, DOI:10.1002/mba2.101.
- M. M. Rahman Khan and M. M. Rumon, Synthesis of PVA-Based Hydrogels for Biomedical Applications: Recent Trends and Advances, Gels, 2025, 11, 88 CrossRef CAS PubMed.
- M. M. Rahman Khan, M. M. H. Rumon and M. Islam, Synthesis, Rheology, Morphology, and Mechanical Properties of Biodegradable PVA-Based Composite Films: A Review on Recent Progress, Processes, 2024, 12(12), 2880 CrossRef CAS.
- H.-S. Song, M. M. Rumon, M. M. Rahman Khan and J.-H. Jeong, Toward Intelligent Materials with the Promise of Self-Healing Hydrogels in Flexible Devices, Polymers, 2025, 17, 542 CrossRef CAS PubMed.
- R. Yadav, R. Kumar, M. Kathpalia, B. Ahmed, K. Dua, M. Gulati, S. Singh, P. J. Singh, S. Kumar and R. M. Shah, et al., Innovative approaches to wound healing: insights into interactive dressings and future directions, J. Mater. Chem. B, 2024, 12(33), 7977–8006, 10.1039/D3TB02912C.
- N. Ahmad, S. N. A. Bukhari, M. A. Hussain, H. Ejaz, M. U. Munir and M. W. Amjad, Nanoparticles incorporated hydrogels for delivery of antimicrobial agents: developments and trends, RSC Adv., 2024, 14(19), 13535–13564, 10.1039/D4RA00631C.
- Z. Hu, J. Shan, X. Jin, W. Sun, L. Cheng, X.-L. Chen and X. Wang, Nanoarchitectonics of in Situ Antibiotic-Releasing Acicular Nanozymes for Targeting and Inducing Cuproptosis-like Death to Eliminate Drug-Resistant Bacteria, ACS Nano, 2024, 18(35), 24327–24349, DOI:10.1021/acsnano.4c06565.
- X. Li, W. Wang, Q. Gao, S. Lai, Y. Liu, S. Zhou, Y. Yan, J. Zhang, H. Wang and J. Wang, et al., Intelligent bacteria-targeting ZIF-8 composite for fluorescence imaging-guided photodynamic therapy of drug-resistant superbug infections and burn wound healing, Exploration, 2024, 20230113, DOI:10.1002/EXP.20230113.
- A. Dsouza, C. Constantinidou, T. N. Arvanitis, D. M. Haddleton, J. Charmet and R. A. Hand, Multifunctional Composite Hydrogels for Bacterial Capture, Growth/Elimination, and Sensing Applications, ACS Appl. Mater. Interfaces, 2022, 14(42), 47323–47344, DOI:10.1021/acsami.2c08582.
- P. Lu, D. Ruan, M. Huang, M. Tian, K. Zhu, Z. Gan and Z. Xiao, Harnessing the potential of hydrogels for advanced therapeutic applications: current achievements and future directions, Signal Transduction Targeted Ther., 2024, 9(1), 166, DOI:10.1038/s41392-024-01852-x.
- I. J. Das and T. Bal, pH factors in chronic wound and pH-responsive polysaccharide-based hydrogel dressings, Int. J. Biol. Macromol., 2024, 279, 135118, DOI:10.1016/j.ijbiomac.2024.135118.
- X. Ding, M. Wang, L. Wei, L. Wang, M. Yuan, F. Yu and H. Yang, Mechanical properties of three N-isopropylacrylamide-based semi-interpenetrating network bilayer hydrogels and their effects on multi-stimuli responsiveness, Iran. Polym. J., 2024, 33(3), 273–287, DOI:10.1007/s13726-023-01247-9.
- Q.-J. Xu, Z.-Y. Yuan, C.-C. Wang, H. Liang, Y. Shi, H.-T. Wu, H. Xu, J. Zheng and J.-R. Wu, Tough Semi-interpenetrating Polyvinylpyrrolidone/Polyacrylamide Hydrogels Enabled by Bioinspired Hydrogen-bonding Induced Phase Separation, Chin. J. Polym. Sci., 2024, 42(5), 591–603, DOI:10.1007/s10118-024-3066-z.
- M. M. H. Rumon, A. A. Akib, S. D. Sarkar, M. A. R. Khan, M. M. Uddin, D. Nasrin and C. K. Roy, Polysaccharide-Based Hydrogels for Advanced Biomedical Engineering Applications, ACS Polym. Au, 2024, 4(5), 463–486, DOI:10.1021/acspolymersau.4c00028.
- K. Li, H. Wang, J. Yan, Z. Shi, S. Zhu and Z. Cui, Emulsion-Templated Gelatin/Amino Acids/Chitosan Macroporous Hydrogels with Adjustable Internal Dimensions for Three-Dimensional Stem Cell Culture, ACS Biomater. Sci. Eng., 2024, 10(8), 4878–4890, DOI:10.1021/acsbiomaterials.4c00501.
- B. Teng, Y. Zhong, J. Wu, J. Zhu, L. Cai, P. Qi and Z. Luo, Transforming watermelon (Citrullus lanatus) rind into durable superabsorbent hydrogels for enhanced soil water retention properties and adsorbs dye in water, Heliyon, 2024, 10(19) DOI:10.1016/j.heliyon.2024.e38656.
- X. Dong, Z. Mei, W. Pan, X. Sheng, N. Li, J. Zhang and Q. Ping, One-path preparation of carboxymethylcellulose from bamboo and its application in all-biodegradable films, Ind. Crops Prod., 2024, 222, 119925, DOI:10.1016/j.indcrop.2024.119925.
- X. Li, C. Wan, T. Tao, H. Chai, Q. Huang, Y. Chai and Y. Wu, An overview of the development status and applications of cellulose-based functional materials, Cellulose, 2024, 31(1), 61–99, DOI:10.1007/s10570-023-05616-8.
- F. Persano, C. Malitesta and E. Mazzotta, Cellulose-Based Hydrogels for Wastewater Treatment: A Focus on Metal Ions Removal, Polymers, 2024, 16(9), 1292, DOI:10.3390/polym16091292.
- S. M. F. Kabir, P. P. Sikdar, B. Haque, M. A. R. Bhuiyan, A. Ali and M. N. Islam, Cellulose-based hydrogel materials: chemistry, properties and their prospective applications, Prog. Biomater., 2018, 7(3), 153–174, DOI:10.1007/s40204-018-0095-0.
- R. Das, N. Mallik, A. Adhikari and A. Bhattarai, A Comprehensive Review on the Creation, Description, and Utilization of Surfactants Containing Multiple Hydroxyl Groups, Int. J. Polym. Sci., 2024, 2024(1), 6120535, DOI:10.1155/2024/6120535.
- M. M. Mahmoud, K. Chawraba and S. A. El Mogy, Nanocellulose: A Comprehensive Review of Structure, Pretreatment, Extraction, and Chemical Modification, Polym. Rev., 2024, 64(4), 1414–1475, DOI:10.1080/15583724.2024.2374929.
- M. Niaounakis, 10 – Building and Construction Applications, in Biopolymers: Applications and Trends, ed. M. Niaounakis, William Andrew Publishing, 2015, pp. 445–505 Search PubMed.
- J. Huang, M. Frauenlob, Y. Shibata, L. Wang, T. Nakajima, T. Nonoyama, M. Tsuda, S. Tanaka, T. Kurokawa and J. P. Gong, Chitin-Based Double-Network Hydrogel as Potential Superficial Soft-Tissue-Repairing Materials, Biomacromolecules, 2020, 21(10), 4220–4230, DOI:10.1021/acs.biomac.0c01003.
- A. Puri, D. M. Syukri, E. Silvia, F. Ladyani, P. Mohite, N. Ade, S. Munde, V. R. Chidrawar, S. Singh and S. Shafi, Waste-to-Value-Added Customized Cationic Banana Starch for Potential Flocculant Application, J. Polym. Environ., 2024, 32(11), 6096–6113, DOI:10.1007/s10924-024-03349-8.
- Y. Lu, M. Mehling, S. Huan, L. Bai and O. J. Rojas, Biofabrication with microbial cellulose: from bioadaptive designs to living materials, Chem. Soc. Rev., 2024, 53(14), 7363–7391, 10.1039/D3CS00641G.
- T. Yang, S.-H. Lu, H. Zhu, A. Patetsos, E. McDonald, M. D. Mellor, Y. Luo, J. F. Rusling, X. Wang and J. He, Tough and Elastic Cellulose Composite Hydrogels/Films for Flexible Wearable Sensors, ACS Appl. Mater. Interfaces, 2024, 16(30), 40018–40029, DOI:10.1021/acsami.4c08592.
- W. H. Binder and J. F. F. Thümmler, Compartmentalised Single-Chain Nanoparticles and their Function, Chem. Commun., 2024, 14332–14345, 10.1039/D4CC04387A.
- T. P. Le, L. Cavalcanti, J. P. Tellam and P. Malo de Molina, Effect of the Protein Chain Conformation on the Collapse into Nanoparticles, Biomacromolecules, 2024, 25(10), 6602–6610, DOI:10.1021/acs.biomac.4c00754.
- H. Hu and F.-J. Xu, Rational design and latest advances of polysaccharide-based hydrogels for wound healing, Biomater. Sci., 2020, 8(8), 2084–2101, 10.1039/D0BM00055H.
- H. Cao, M. Wang, J. Ding and Y. Lin, Hydrogels: a promising therapeutic platform for inflammatory skin diseases treatment, J. Mater. Chem. B, 2024, 12(33), 8007–8032, 10.1039/D4TB00887A.
- S. Mantry, A. Behera, S. Pradhan, L. Mohanty, R. Kumari, A. Singh and M. K. Yadav, Polysaccharide-based chondroitin sulfate macromolecule loaded hydrogel/scaffolds in wound healing- A comprehensive review on possibilities, research gaps, and safety assessment, Int. J. Biol. Macromol., 2024, 279, 135410, DOI:10.1016/j.ijbiomac.2024.135410.
- I. Condò, S. M. Giannitelli, D. Lo Presti, B. Cortese and O. Ursini, Overview of Dynamic Bond Based Hydrogels for Reversible Adhesion Processes, Gels, 2024, 10, 442 CrossRef PubMed.
- F. Gao, X. Yang and W. Song, Bioinspired Supramolecular Hydrogel from Design to Applications, Small Methods, 2024, 8(4), 2300753, DOI:10.1002/smtd.202300753.
- Y. Hao, H. Ji, L. Gao, Z. Qu, Y. Zhao, J. Chen, X. Wang, X. Ma, G. Zhang and T. Zhang, Self-assembled carrier-free formulations based on medicinal and food active ingredients, Biomater. Sci., 2024, 6253–6273, 10.1039/D4BM00893F.
- N. Anghel, I. Spiridon, M.-V. Dinu, S. Vlad and M. Pertea, Xanthan–Polyurethane Conjugates: An Efficient Approach for Drug Delivery, Polymers, 2024, 16, 1734 CrossRef CAS PubMed.
- Z. Bai, X. Li, T. Wu, H. Jiang, X. Zhang, L. Zhang, Y. Yang, S. Liu, L. Lei and N. Song, et al., A facile and on-demand optimizing strategy for polyurethane elastomers via programmable hydrogen bonding, Chem. Eng. J., 2024, 492, 152110, DOI:10.1016/j.cej.2024.152110.
- T. Ozawa, H. Yoshimura and S. B. Kim, Advances in Fluorescence and Bioluminescence Imaging, Anal. Chem., 2013, 85(2), 590–609, DOI:10.1021/ac3031724.
- K. Fu, X. Yang, Z. Yu, L. Song and L. Shi, Revealing the nature of covalently tethered distonic radical anions in the generation of heteroatom-centered radicals: evidence for the polarity-matching PCET pathway, Chem. Sci., 2024, 15(31), 12398–12409, 10.1039/D4SC02602K.
- L. Chen, J. Xu, M. Zhu, Z. Zeng, Y. Song, Y. Zhang, X. Zhang, Y. Deng, R. Xiong and C. Huang, Self-healing polymers through hydrogen-bond cross-linking: synthesis and electronic applications, Mater. Horiz., 2023, 10(10), 4000–4032, 10.1039/D3MH00236E.
- Y. Ren and X. Dong, Dynamic polymeric materials via hydrogen-bond cross-linking: Effect of multiple network topologies, Prog. Polym. Sci., 2024, 158, 101890, DOI:10.1016/j.progpolymsci.2024.101890.
- P. A. G. Cormack and A. Z. Elorza, Molecularly imprinted polymers: synthesis and characterisation, J. Chromatogr. B, 2004, 804(1), 173–182, DOI:10.1016/j.jchromb.2004.02.013.
- A. Suvitha, N. S. Venkataramanan, R. Sahara and Y. Kawazoe, A theoretical exploration of the intermolecular interactions between resveratrol and water: a DFT and AIM analysis, J. Mol. Model., 2019, 25(3), 56, DOI:10.1007/s00894-019-3941-7.
- K. Tanabe, Chapter 5 Solid Acid and Base Catalysts, De Gruyter, 1982, vol. 2, pp 231–274 Search PubMed.
- Y. Deng, Q. Zhang and D.-H. Qu, Emerging Hydrogen-Bond Design for High-Performance Dynamic Polymeric Materials, ACS Mater. Lett., 2023, 5(2), 480–490, DOI:10.1021/acsmaterialslett.2c00865.
- C. Yadav, J.-M. Lee, P. Mohanty, X. Li and W.-D. Jang, Graft onto approaches for nanocellulose-based advanced functional materials, Nanoscale, 2023, 15(37), 15108–15145, 10.1039/D3NR03087C.
- B. Medronho and B. Lindman, Brief overview on cellulose dissolution/regeneration interactions and mechanisms, Adv. Colloid Interface Sci., 2015, 222, 502–508, DOI:10.1016/j.cis.2014.05.004.
- M. Wohlert, T. Benselfelt, L. Wågberg, I. Furó, L. A. Berglund and J. Wohlert, Cellulose and the role of hydrogen bonds: not in charge of everything, Cellulose, 2022, 29(1), 1–23, DOI:10.1007/s10570-021-04325-4.
- L. Wang, Y. Du, Y. Yuan, R.-J. Mu, J. Gong, Y. Ni, J. Pang and C. Wu, Mussel-inspired fabrication of konjac glucomannan/microcrystalline cellulose intelligent hydrogel with pH-responsive sustained release behavior, Int. J. Biol. Macromol., 2018, 113, 285–293, DOI:10.1016/j.ijbiomac.2018.02.083.
- M. V. Biyani, E. J. Foster and C. Weder, Light-Healable Supramolecular Nanocomposites Based on Modified Cellulose Nanocrystals, ACS Macro Lett., 2013, 2(3), 236–240, DOI:10.1021/mz400059w.
- M. T. I. Mredha, Y. Z. Guo, T. Nonoyama, T. Nakajima, T. Kurokawa and J. P. Gong, A Facile Method to Fabricate Anisotropic Hydrogels with Perfectly Aligned Hierarchical Fibrous Structures, Adv. Mater., 2018, 30(9), 1704937, DOI:10.1002/adma.201704937.
- Z. Wang, W. Hu, Y. Du, Y. Xiao, X. Wang, S. Zhang, J. Wang and C. Mao, Green Gas-Mediated Cross-Linking Generates Biomolecular Hydrogels with Enhanced Strength and Excellent Hemostasis for Wound Healing, ACS Appl. Mater. Interfaces, 2020, 12(12), 13622–13633, DOI:10.1021/acsami.9b21325.
- S. J. K. O’Neill, Z. Huang, X. Chen, R. L. Sala, J. A. McCune, G. G. Malliaras and O. A. Scherman, Highly stretchable dynamic hydrogels for soft multilayer electronics, Sci. Adv., 2024, 10(29), eadn5142, DOI:10.1126/sciadv.adn5142.
- W. Xu and A. Chen, Application of supramolecular hydrogel in supercapacitors: Opportunities and challenges, Aggregate, 2024, 5(5), e581, DOI:10.1002/agt2.581.
- E. A. Appel, M. W. Tibbitt, J. M. Greer, O. S. Fenton, K. Kreuels, D. G. Anderson and L. Robert, Exploiting Electrostatic Interactions in Polymer-Nanoparticle Hydrogels, ACS Macro Lett., 2015, 848–852, DOI:10.1021/acsmacrolett.5b00416.
- A.-L. Oechsle, L. Lewis, W. Y. Hamad, S. G. Hatzikiriakos and M. J. MacLachlan, CO2-Switchable Cellulose Nanocrystal Hydrogels, Chem. Mater., 2018, 30(2), 376–385, DOI:10.1021/acs.chemmater.7b03939.
- C. Shao, L. Meng, C. Cui and J. Yang, An integrated self-healable and robust conductive hydrogel for dynamically self-adhesive and highly conformable electronic skin, J. Mater. Chem. C, 2019, 7(48), 15208–15218, 10.1039/C9TC05467G.
- T. Zhang, Q. Cheng, D. Ye and C. Chang, Tunicate cellulose nanocrystals reinforced nanocomposite hydrogels comprised by hybrid cross-linked networks, Carbohydr. Polym., 2017, 169, 139–148, DOI:10.1016/j.carbpol.2017.04.007.
- L. Yang and T. Wang, Preparation of cellulosic drug-loaded hydrogel beads through electrostatic and host–guest interactions, J. Appl. Polym. Sci., 2018, 135(31), 46593, DOI:10.1002/app.46593.
- S. Pardeshi, F. Damiri, M. Zehravi, R. Joshi, H. Kapare, M. K. Prajapati, N. Munot, M. Berrada, P. S. Giram and S. Rojekar, et al., Functional Thermoresponsive Hydrogel Molecule to Material Design for Biomedical Applications, Polymers, 2022, 14, 3126 CrossRef CAS PubMed.
- Y. Yang, Q. Zhang, Y. Weng, A. Jiao and Z. Jin, Effects of hydrocolloids on the structure and physicochemical properties of triticale starch during fermentation, Int. J. Biol. Macromol., 2024, 282, 137327, DOI:10.1016/j.ijbiomac.2024.137327.
- L. Yang, H. Wang, Y. Yang and Y. Li, Self-healing cellulose-based hydrogels: From molecular design to multifarious applications, Carbohydr. Polym., 2025, 347, 122738, DOI:10.1016/j.carbpol.2024.122738.
- J. D. dos Santos Carvalho, R. S. Rabelo and M. D. Hubinger, Thermo-rheological properties of chitosan hydrogels with hydroxypropyl methylcellulose and methylcellulose, Int. J. Biol. Macromol., 2022, 209, 367–375, DOI:10.1016/j.ijbiomac.2022.04.035.
- Y. Sekiguchi, C. Sawatari and T. Kondo, A gelation mechanism depending on hydrogen bond formation in regioselectively substituted O-methylcelluloses, Carbohydr. Polym., 2003, 53(2), 145–153, DOI:10.1016/S0144-8617(03)00050-X.
- P. Weiss, O. Gauthier, J. M. Bouler, G. Grimandi and G. Daculsi, Injectable bone substitute using a hydrophilic polymer, Bone, 1999, 25(2), 67S–70S, DOI:10.1016/S8756-3282(99)00146-5.
- V. Percec, T. K. Bera and R. J. Butera, A New Strategy for the Preparation of Supramolecular Neutral Hydrogels, Biomacromolecules, 2002, 3(2), 272–279, DOI:10.1021/bm015614q.
- Z. Lei, H. Chen, S. Huang, L. J. Wayment, Q. Xu and W. Zhang, New Advances in Covalent Network Polymers via Dynamic Covalent Chemistry, Chem. Rev., 2024, 124(12), 7829–7906, DOI:10.1021/acs.chemrev.3c00926.
- L. Gautam, A. Jain, P. Shrivastava, S. Vyas and S. P. Vyas, Natural Polymers-Based Biocomposites: State of Art, New Challenges, and Opportunities, in Polymeric and Natural Composites: Materials, Manufacturing and Biomedical Applications, ed. M. S. Hasnain, A. K. Nayak and S. Alkahtani, Springer International Publishing, 2022, pp. 1–24 Search PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels–Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46(16), 4895–4950, 10.1039/C7CS00184C.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, A Self-Healing Cellulose Nanocrystal-Poly(ethylene glycol) Nanocomposite Hydrogel via Diels–Alder Click Reaction, ACS Sustain. Chem. Eng., 2017, 5(7), 6167–6174, DOI:10.1021/acssuschemeng.7b01060.
- X. Yang, H. Yang, X. Jiang, B. Yang, K. Zhu, N. C.-H. Lai, C. Huang, C. Chang, L. Bian and L. Zhang, Injectable chitin hydrogels with self-healing property and biodegradability as stem cell carriers, Carbohydr. Polym., 2021, 256, 117574, DOI:10.1016/j.carbpol.2020.117574.
- C. Ding, M. Tian, R. Feng, Y. Dang and M. Zhang, Novel Self-Healing Hydrogel with Injectable, pH-Responsive, Strain-Sensitive, Promoting Wound-Healing, and Hemostatic Properties Based on Collagen and Chitosan, ACS Biomater. Sci. Eng., 2020, 6(7), 3855–3867, DOI:10.1021/acsbiomaterials.0c00588.
- Q. Mo, L. Huang, Y. Sheng, Z. Wei, S. Zhang, Y. Li, X. Wang, Y. Wang, X. Lu and C. Huang, et al., Crosslinking strategy and promotion role of cellulose as a composite hydrogel component for three-dimensional printing – A review, Food Hydrocolloids, 2024, 154, 110079, DOI:10.1016/j.foodhyd.2024.110079.
- A. Sannino, M. Madaghiele, M. G. Lionetto, T. Schettino and A. Maffezzoli, A cellulose-based hydrogel as a potential bulking agent for hypocaloric diets: An in vitro biocompatibility study on rat intestine, J. Appl. Polym. Sci., 2006, 102(2), 1524–1530, DOI:10.1002/app.24468.
- S. G. Hirsch and R. J. Spontak, Temperature-dependent property development in hydrogels derived from hydroxypropylcellulose, Polymer, 2002, 43(1), 123–129, DOI:10.1016/S0032-3861(01)00608-5.
- E. Marsano, E. Bianchi and A. Viscardi, Stimuli responsive gels based on interpenetrating network of hydroxy propylcellulose and poly(N-isopropylacrylamide), Polymer, 2004, 45(1), 157–163, DOI:10.1016/j.polymer.2003.10.088.
- F. Ganji, S. Vasheghani Farahani and E. Vasheghani-Farahani, Theoretical Description of Hydrogel Swelling: A Review, Iran. Polym. J., 2010, 19, 375–398 CAS.
- Z. Xu, P. Yue and J. J. Feng, A theory of hydrogel mechanics that couples swelling and external flow, Soft Matter, 2024, 20(27), 5389–5406, 10.1039/D4SM00424H.
- M. T. P. Paiva, J. O. F. Kishima, J. B. M. D. Silva, J. Mantovan, F. G. Colodi and S. Mali, Crosslinking Methods in Polysaccharide-Based Hydrogels for Drug Delivery Systems, Biomed. Mater. & Devices, 2024, 2(1), 288–306, DOI:10.1007/s44174-023-00118-4.
- A. Sannino, A. Maffezzoli and L. Nicolais, Introduction of molecular spacers between the crosslinks of a cellulose-based superabsorbent hydrogel: Effects on the equilibrium sorption properties, J. Appl. Polym. Sci., 2003, 90(1), 168–174, DOI:10.1002/app.12625.
- C. Demitri, R. Del Sole, F. Scalera, A. Sannino, G. Vasapollo, A. Maffezzoli, L. Ambrosio and L. Nicolais, Novel superabsorbent cellulose-based hydrogels crosslinked with citric acid, J. Appl. Polym. Sci., 2008, 110(4), 2453–2460, DOI:10.1002/app.28660.
- S. Ma, B. Yu, X. Pei and F. Zhou, Structural hydrogels, Polymer, 2016, 98, 516–535, DOI:10.1016/j.polymer.2016.06.053.
- C. A. Barbero, M. V. Martínez, D. F. Acevedo, M. A. Molina and C. R. Rivarola, Cross-Linked Polymeric Gels and Nanocomposites: New Materials and Phenomena Enabling Technological Applications, Macromol, 2022, 2, 440–475 CAS.
- I. Adroher-Benítez, A. Martín-Molina, S. Ahualli, M. Quesada-Pérez, G. Odriozola and A. Moncho-Jordá, Competition between excluded-volume and electrostatic interactions for nanogel swelling: effects of the counterion valence and nanogel charge, Phys. Chem. Chem. Phys., 2017, 19(9), 6838–6848, 10.1039/C6CP08683G.
- G. Marcì, G. Mele, L. Palmisano, P. Pulito and A. Sannino, Environmentally sustainable production of cellulose-based superabsorbent hydrogels, Green Chem., 2006, 8(5), 439–444, 10.1039/B515247J.
- X. Shi, W. Wang, Y. Zheng and A. Wang, Utilization of hollow kapok fiber for the fabrication of a pH-sensitive superabsorbent composite with improved gel strength and swelling properties, RSC Adv., 2014, 4(92), 50478–50485, 10.1039/C4RA10866C.
- M. Motiur Rahman, A. Hasan Howlader Hasan, E. Rahman, H. Roy, M. Mozammal Hosen, M. M. Rahman, S. D. Sarkar, C. K. Roy, A. Bin Imran and S. H. Firoz, Development of functionalized nanocrystalline cellulose-based polyelectrolytes with high water uptake, Polym. J., 2021, 53(8), 913–921, DOI:10.1038/s41428-021-00483-1.
- S. D. Sarkar, M. M. Uddin, C. K. Roy, M. J. Hossen, M. I. Sujan and M. S. Azam, Mechanically tough and highly stretchable poly(acrylic acid) hydrogel cross-linked by 2D graphene oxide, RSC Adv., 2020, 10(18), 10949–10958, 10.1039/D0RA00678E.
- B. S. Neves, R. C. Gonçalves, J. F. Mano and M. B. Oliveira, Controlling the diffusion of small molecules from matrices processed by all-aqueous methodologies: towards the development of green pharmaceutical products, Green Chem., 2024, 26(8), 4417–4431, 10.1039/D3GC04183B.
- M. J. Hossen, S. D. Sarkar, M. M. Uddin, C. K. Roy and M. S. Azam, Mussel-Inspired Adhesive Nano-Filler for Strengthening Polyacrylamide Hydrogel, ChemistrySelect, 2020, 5(29), 8906–8914, DOI:10.1002/slct.202001632.
- M. Motiur Rahman, A. Hasan Howlader, E. Rahman, H. Roy, M. Mozammal Hosen, M. M. Rahman, S. D. Sarkar, C. K. Roy, A. Bin Imran and S. H. Firoz, Development of functionalized nanocrystalline cellulose-based polyelectrolytes with high water uptake, Polym. J., 2021, 53(8), 913–921 Search PubMed.
- X. Li, X. Deng, J. Liu and X. Zhang, Phase behavior of rubber and supercritical CO2: Operating parameters and swelling characteristics, Polym. Eng. Sci., 2024, 64(8), 3473–3487, DOI:10.1002/pen.26763.
- C. W. H. Rajawasam, O. J. Dodo, M. A. S. N. Weerasinghe, I. O. Raji, S. V. Wanasinghe, D. Konkolewicz and N. De Alwis Watuthanthrige, Educational series: characterizing crosslinked polymer networks, Polym. Chem., 2024, 15(4), 219–247, 10.1039/D3PY00914A.
- C. Cecone, G. Hoti, F. Caldera, M. Ginepro, A. Matencio and F. Trotta, Evaluation of the Swelling Properties and Sorption Capacity of Maltodextrin-Based Cross-Linked Polymers, Gels, 2024, 10, 232 Search PubMed.
- P. J. Flory and J. Rehner Jr, Statistical Mechanics of Cross-Linked Polymer Networks I. Rubberlike Elasticity, J. Chem. Phys., 1943, 11(11), 512–520, DOI:10.1063/1.1723791.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Stimulus-responsive hydrogels: Theory, modern advances, and applications, Mater. Sci. Eng., R, 2015, 93, 1–49, DOI:10.1016/j.mser.2015.04.001.
- B. C. Nguyen, T. M. Truong, N. T. Nguyen, D. N. Dinh, D. Hollmann and M. N. Nguyen, Advanced cellulose-based hydrogel TiO2 catalyst composites for efficient photocatalytic degradation of organic dye methylene blue, Sci. Rep., 2024, 14(1), 10935, DOI:10.1038/s41598-024-61724-w.
- M. Momin, V. Mishra, S. Gharat and A. Omri, Recent advancements in cellulose-based biomaterials for management of infected wounds, Expert Opin. Drug Delivery, 2021, 18(11), 1741–1760, DOI:10.1080/17425247.2021.1989407.
- C. Xu, Y. Chen, S. Zhao, D. Li, X. Tang, H. Zhang, J. Huang, Z. Guo and W. Liu, Mechanical Regulation of Polymer Gels, Chem. Rev., 2024, 124(18), 10435–10508, DOI:10.1021/acs.chemrev.3c00498.
- A. G. F. Shoair, H. A. Sahyon, M. A. E. Shishtawy, A. S. A. Almalki, A. A. Alqarni, F. Althobaiti, M. M. A. H. Shanab and H. A. Khalaf, Improved Hemostatic Efficacy of Graviola-Modified PVA–HA Hydrogels for Wound Healing: In Vitro and In Vivo Analysis, ChemistrySelect, 2024, 9(43), e202404438, DOI:10.1002/slct.202404438.
- B. Liu and K. Chen, Advances in Hydrogel-Based Drug Delivery Systems, Gels, 2024, 10, 262 CrossRef CAS PubMed.
- R. Wang, C. Cheng, H. Wang and D. Wang, Swollen hydrogel nanotechnology: Advanced applications of the rudimentary swelling properties of hydrogels, ChemPhysMater, 2024, 3(4), 357–375, DOI:10.1016/j.chphma.2024.07.006.
- X. Li and J. P. Gong, Design principles for strong and tough hydrogels, Nat. Rev. Mater., 2024, 9(6), 380–398, DOI:10.1038/s41578-024-00672-3.
- W. Xian, Y.-S. Zhan, A. Maiti, A. P. Saab and Y. Li, Filled Elastomers: Mechanistic and Physics-Driven Modeling and Applications as Smart Materials, Polymers, 2024, 16, 1387 CrossRef CAS PubMed.
- R. G. Ricarte and S. Shanbhag, A tutorial review of linear rheology for polymer chemists: basics and best practices for covalent adaptable networks, Polym. Chem., 2024, 15(9), 815–846, 10.1039/D3PY01367G.
- T. Lyu, Y. Han, Z. Chen, X. Fan and Y. Tian, Hydrogels and hydrogel derivatives for atmospheric water harvesting, Mater. Today Sustainability, 2024, 25, 100693, DOI:10.1016/j.mtsust.2024.100693.
- H. Mittal, A. Al Alili and S. M. Alhassan, Development of high efficacy super-porous hydrogel composites-based polymer desiccants to capture water vapors from moist air, Adsorption, 2024, 30(6), 841–857, DOI:10.1007/s10450-024-00463-w.
- S. Khan, L. Maryam, A. Gulzar, M. A. Mansoor and M. Iqbal, Review: smart and active hydrogels in biotechnology—synthetic techniques and applications, J. Mater. Sci., 2024, 59(35), 16449–16471, DOI:10.1007/s10853-024-10187-5.
- B. Sim, J. J. Chang, Q. Lin, J. H. M. Wong, V. Ow, Y. Leow, Y. J. Wong, Y. J. Boo, R. Goh and X. J. Loh, Hydrogels Based on Polyelectrolyte Complexes: Underlying Principles and Biomedical Applications, Biomacromolecules, 2024, 25(12), 7563–7580, DOI:10.1021/acs.biomac.4c01240.
- K. Zöller, D. To and A. Bernkop-Schnürch, Biomedical applications of functional hydrogels: Innovative developments, relevant clinical trials and advanced products, Biomaterials, 2025, 312, 122718, DOI:10.1016/j.biomaterials.2024.122718.
- M. Ribeiro, M. Simões, C. Vitorino and F. Mascarenhas-Melo, Hydrogels in Cutaneous Wound Healing: Insights into Characterization, Properties, Formulation and Therapeutic Potential, Gels, 2024, 10, 188 CrossRef CAS PubMed.
- D. M. Alshangiti, T. K. El-damhougy, A. Zaher, M. Madani and M. Mohamady ghobashy, Revolutionizing biomedicine: advancements, applications, and prospects of nanocomposite macromolecular carbohydrate-based hydrogel biomaterials: a review, RSC Adv., 2023, 13(50), 35251–35291, 10.1039/D3RA07391B.
- G. S. Banker, A. Y. Gore and J. Swarbrick, Water vapour transmission properties of free polymer filmsFn fn1, J. Pharm. Pharmacol., 1966, 18(7), 457–466, DOI:10.1111/j.2042-7158.1966.tb07906.x.
- D. Queen, J. D. S. Gaylor, J. H. Evans, J. M. Courtney and W. H. Reid, The preclinical evaluation of the water vapour transmission rate through burn wound dressings, Biomaterials, 1987, 8(5), 367–371, DOI:10.1016/0142-9612(87)90007-X.
- M. Sirousazar and M. Yari, Dehydration kinetics of polyvinyl alcohol hydrogel wound dressings during wound healing process, Chin. J. Polym. Sci., 2010, 28(4), 573–580, DOI:10.1007/s10118-010-9099-5.
- R. Xu, H. Xia, W. He, Z. Li, J. Zhao, B. Liu, Y. Wang, Q. Lei, Y. Kong and Y. Bai, et al., Controlled water vapor transmission rate promotes wound-healing via wound re-epithelialization and contraction enhancement, Sci. Rep., 2016, 6(1), 24596, DOI:10.1038/srep24596.
- C. Chang, M. He, J. Zhou and L. Zhang, Swelling Behaviors of pH- and Salt-Responsive Cellulose-Based Hydrogels, Macromolecules, 2011, 44(6), 1642–1648, DOI:10.1021/ma102801f.
- X. Gan, C. Li, J. Sun, X. Zhang, M. Zhou, Y. Deng and A. Xiao, GelMA/κ-carrageenan double-network hydrogels with superior mechanics and biocompatibility, RSC Adv., 2023, 13(3), 1558–1566, 10.1039/D2RA06101E 10.1039/D2RA06101E.
- V. M. Gun’ko, I. N. Savina and S. V. Mikhalovsky, Properties of Water Bound in Hydrogels, Gels, 2017, 3, 37 CrossRef PubMed.
- B.-X. Wang, W. Xu, Z. Yang, Y. Wu and F. Pi, An Overview on Recent Progress of the Hydrogels: From Material Resources, Properties, to Functional Applications, Macromol. Rapid Commun., 2022, 43(6), 2100785, DOI:10.1002/marc.202100785.
- Y. Liu, L. Wang, L. Zhao, Y. Zhang, Z.-T. Li and F. Huang, Multiple hydrogen bonding driven supramolecular architectures and their biomedical applications, Chem. Soc. Rev., 2024, 53(3), 1592–1623, 10.1039/D3CS00705G.
- S. Seiffert and J. Sprakel, Physical chemistry of supramolecular polymer networks, Chem. Soc. Rev., 2012, 41(2), 909–930, 10.1039/C1CS15191F.
- A. Etale, A. J. Onyianta, S. R. Turner and S. J. Eichhorn, Cellulose: A Review of Water Interactions, Applications in Composites, and Water Treatment, Chem. Rev., 2023, 123(5), 2016–2048, DOI:10.1021/acs.chemrev.2c00477.
- C. Wu, J. Li, Y.-q. Zhang, X. Li, S.-y. Wang and D.-q. Li, Cellulose Dissolution, Modification, and the Derived Hydrogel: A Review, ChemSusChem, 2023, 16(21), e202300518, DOI:10.1002/cssc.202300518.
- A. P. C. Almeida, J. N. Saraiva, G. Cavaco, R. P. Portela, C. R. Leal, R. G. Sobral and P. L. Almeida, Crosslinked bacterial cellulose hydrogels for biomedical applications, Eur. Polym. J., 2022, 177, 111438, DOI:10.1016/j.eurpolymj.2022.111438.
- J. Jiang, M. Liu, D. Xu, T. Jiang and J. Zhang, Quantitative detection of microcystin-LR in Bellamya aeruginosa by thin-layer chromatography coupled with surface-enhanced Raman spectroscopy based on in-situ ZIF-67/Ag NPs/Au NWs composite substrate, Food Chem., 2024, 452, 139481, DOI:10.1016/j.foodchem.2024.139481.
- G. P. Udayakumar, S. Muthusamy, B. Selvaganesh, N. Sivarajasekar, K. Rambabu, S. Sivamani, N. Sivakumar, J. P. Maran and A. Hosseini-Bandegharaei, Ecofriendly biopolymers and composites: Preparation and their applications in water-treatment, Biotechnol. Adv., 2021, 52, 107815, DOI:10.1016/j.biotechadv.2021.107815.
- D.-J. Lim, Cross-Linking Agents for Electrospinning-Based Bone Tissue Engineering, Int. J. Mol. Sci., 2022, 23(10), 5444, DOI:10.3390/ijms23105444.
- M. A. U. R. Qureshi, N. Arshad, A. Rasool, N. K. Janjua, M. S. Butt, M. N. U. R. Qureshi and H. Ismail, Kappa-carrageenan and sodium alginate-based pH-responsive hydrogels for controlled release of methotrexate, R. Soc. Open Sci., 2024, 11(4) DOI:10.1098/rsos.231952.
- S. Hua, H. Ma, X. Li, H. Yang and A. Wang, pH-sensitive sodium alginate/poly(vinyl alcohol) hydrogel beads prepared by combined Ca2+ crosslinking and freeze-thawing cycles for controlled release of diclofenac sodium, Int. J. Biol. Macromol., 2010, 46(5), 517–523, DOI:10.1016/j.ijbiomac.2010.03.004.
- K. S. Postolović, M. D. Antonijević, B. Ljujić, M. Miletić Kovačević, M. Gazdić Janković and Z. D. Stanić, pH-Responsive Hydrogel Beads Based on Alginate, κ-Carrageenan and Poloxamer for Enhanced Curcumin, Natural Bioactive Compound, Encapsulation and Controlled Release Efficiency, Molecules, 2022, 27(13), 4045, DOI:10.3390/molecules27134045.
- S. Gorgieva and V. Kokol, Synthesis and application of new temperature-responsive hydrogels based on carboxymethyl and hydroxyethyl cellulose derivatives for the functional finishing of cotton knitwear, Carbohydr. Polym., 2011, 85(3), 664–673, DOI:10.1016/j.carbpol.2011.03.037.
- S. Durpekova, K. Filatova, J. Cisar, A. Ronzova, E. Kutalkova and V. Sedlarik, A Novel Hydrogel Based on Renewable Materials for Agricultural Application, Int. J. Polym. Sci., 2020, 2020(1), 8363418, DOI:10.1155/2020/8363418.
- X. Hu and C. Gao, Photoinitiating polymerization to prepare biocompatible chitosan hydrogels, J. Appl. Polym. Sci., 2008, 110(2), 1059–1067, DOI:10.1002/app.28704.
- L. T. Mashabela, M. M. Maboa, N. F. Miya, T. O. Ajayi, R. S. Chasara, M. Milne, S. Mokhele, P. H. Demana, B. A. Witika and X. Siwe-Noundou, et al., A Comprehensive Review of Cross-Linked Gels as Vehicles for Drug Delivery to Treat Central Nervous System Disorders, Gels, 2022, 8(9), 563, DOI:10.3390/gels8090563.
- X. Xue, Y. Hu, S. Wang, X. Chen, Y. Jiang and J. Su, Fabrication of physical and chemical crosslinked hydrogels for bone tissue engineering, Bioact. Mater., 2022, 12, 327–339, DOI:10.1016/j.bioactmat.2021.10.029.
- Y. He, Y. Zheng, C. Liu, H. Zhang and J. Shen, Citric acid cross-linked β-cyclodextrins: A review of preparation and environmental/biomedical application, Carbohydr. Polym., 2024, 323, 121438, DOI:10.1016/j.carbpol.2023.121438.
- Z. Zhang and X. Qiao, Influences of cation valence on water absorbency of crosslinked carboxymethyl cellulose, Int. J. Biol. Macromol., 2021, 177, 149–156, DOI:10.1016/j.ijbiomac.2021.02.080.
- H. Nasution, H. Harahap, N. F. Dalimunthe, M. H. S. Ginting, M. Jaafar, O. O. H. Tan, H. K. Aruan and A. L. Herfananda, Hydrogel and Effects of Crosslinking Agent on Cellulose-Based Hydrogels: A Review, Gels, 2022, 8(9), 568, DOI:10.3390/gels8090568.
- C. A. Peptu, E. S. Băcăiţă, C.-L. Savin, M. Luţcanu and M. Agop, Hydrogels Based on Alginates and Carboxymethyl Cellulose with Modulated Drug Release—An Experimental and Theoretical Study, Polymers, 2021, 13(24), 4461, DOI:10.3390/polym13244461.
- M. Muthukumar, 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions, Macromolecules, 2017, 50(24), 9528–9560, DOI:10.1021/acs.macromol.7b01929.
- J. Travas-Sejdic and A. Easteal, Swelling equilibria and volume phase transition of polyelectrolyte gel with strongly dissociated groups, Polym. Gels Networks, 1998, 5(6), 481–502, DOI:10.1016/S0966-7822(97)00019-1.
- S. P. Santoso, A. Kurniawan, F. E. Soetaredjo, K.-C. Cheng, J. N. Putro, S. Ismadji and Y.-H. Ju, Eco-friendly cellulose–bentonite porous composite hydrogels for adsorptive removal of azo dye and soilless culture, Cellulose, 2019, 26(5), 3339–3358, DOI:10.1007/s10570-019-02314-2.
- S. Yang, S. Fu, H. Liu, Y. Zhou and X. Li, Hydrogel beads based on carboxymethyl cellulose for removal heavy metal ions, J. Appl. Polym. Sci., 2011, 119(2), 1204–1210, DOI:10.1002/app.32822.
- H. Tu, Y. Yu, J. Chen, X. Shi, J. Zhou, H. Deng and Y. Du, Highly cost-effective and high-strength hydrogels as dye adsorbents from natural polymers: chitosan and cellulose, Polym. Chem., 2017, 8(19), 2913–2921, 10.1039/C7PY00223H.
- K. Hara, M. Iida, K. Yano and T. Nishida, Metal ion absorption of carboxymethylcellulose gel formed by γ-ray irradiation: For the environmental purification, Colloids Surf., B, 2004, 38(3), 227–230, DOI:10.1016/j.colsurfb.2004.02.024.
- K. Varaprasad, T. Jayaramudu and E. R. Sadiku, Removal of dye by carboxymethyl cellulose, acrylamide and graphene oxide via a free radical polymerization process, Carbohydr. Polym., 2017, 164, 186–194, DOI:10.1016/j.carbpol.2017.01.094.
- A. B. Tomé Constantino and E. E. Garcia-Rojas, Microencapsulation of betanin by complex coacervation of carboxymethylcellulose and amaranth protein isolate for application in edible gelatin films, Food Hydrocolloids, 2022, 133, 107956, DOI:10.1016/j.foodhyd.2022.107956.
- G. Wang, T. Lu, X. Zhang, M. Feng, C. Wang, W. Yao, S. Zhou, Z. Zhu, W. Ding and M. He, Structure and properties of cellulose/HAP nanocomposite hydrogels, Int. J. Biol. Macromol., 2021, 186, 377–384, DOI:10.1016/j.ijbiomac.2021.07.060.
- M. R. Bayat, R. Dolatabadi and M. Baghani, Transient swelling response of pH-sensitive hydrogels: A monophasic constitutive model and numerical implementation, Int. J. Pharm., 2020, 577, 119030, DOI:10.1016/j.ijpharm.2020.119030.
- M. R. Bayat and M. Baghani, A review on swelling theories of pH-sensitive hydrogels, J. Intell. Mater. Syst. Struct., 2021, 32(18–19), 2349–2365, DOI:10.1177/1045389X21995880.
- P. E. Grimshaw, J. H. Nussbaum, A. J. Grodzinsky and M. L. Yarmush, Kinetics of electrically and chemically induced swelling in polyelectrolyte gels, J. Chem. Phys., 1990, 93(6), 4462–4472, DOI:10.1063/1.458729.
- B. J. Kirby, Micro-and Nanoscale Fluid Mechanics: Transport in Microfluidic Devices, Cambridge university press, 2010 Search PubMed.
- S. K. De, N. R. Aluru, B. Johnson, W. C. Crone, D. J. Beebe and J. Moore, Equilibrium swelling and kinetics of pH-responsive hydrogels: models, experiments, and simulations, J. Microelectromech. Syst., 2002, 11(5), 544–555, DOI:10.1109/JMEMS.2002.803281.
- S. K. De and N. R. Aluru, A chemo-electro-mechanical mathematical model for simulation of pH sensitive hydrogels, Mech. Mater., 2004, 36(5), 395–410, DOI:10.1016/S0167-6636(03)00067-X.
- M. Dehghany, H. Zhang, R. Naghdabadi and Y. Hu, A thermodynamically-consistent large deformation theory coupling photochemical reaction and electrochemistry for light-responsive gels, J. Mech. Phys. Solids, 2018, 116, 239–266, DOI:10.1016/j.jmps.2018.03.018.
- N. G. Hoogeveen, M. A. Cohen Stuart, G. J. Fleer and M. R. Böhmer, Formation and Stability of Multilayers of Polyelectrolytes, Langmuir, 1996, 12(15), 3675–3681, DOI:10.1021/la951574y.
- J. M. Korde and B. Kandasubramanian, Fundamentals and Effects of Biomimicking Stimuli-Responsive Polymers for Engineering Functions, Ind. Eng. Chem. Res., 2019, 58(23), 9709–9757, DOI:10.1021/acs.iecr.9b00683.
- A. Revete, A. Aparicio, B. A. Cisterna, J. Revete, L. Luis, E. Ibarra, E. A. Segura González, J. Molino and D. Reginensi, Advancements in the Use of Hydrogels for Regenerative Medicine: Properties and Biomedical Applications, Int. J. Biomater., 2022, 2022(1), 3606765, DOI:10.1155/2022/3606765.
- M. V. S. Varma, A. M. Kaushal and S. Garg, Influence of micro-environmental pH on the gel layer behavior and release of a basic drug from various hydrophilic matrices, J. Controlled Release, 2005, 103(2), 499–510, DOI:10.1016/j.jconrel.2004.12.015.
- J. Li and D. J. Mooney, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater., 2016, 1(12), 16071, DOI:10.1038/natrevmats.2016.71.
- H. Ding, P. Tan, S. Fu, X. Tian, H. Zhang, X. Ma, Z. Gu and K. Luo, Preparation and application of pH-responsive drug delivery systems, J. Controlled Release, 2022, 348, 206–238, DOI:10.1016/j.jconrel.2022.05.056.
- Y.-J. Zhu and F. Chen, pH-Responsive Drug-Delivery Systems, Chem.–Asian J., 2015, 10(2), 284–305, DOI:10.1002/asia.201402715.
- D. Ahmad, I. van den Boogaert, J. Miller, R. Presswell and H. Jouhara, Hydrophilic and hydrophobic materials and their applications, Energy Sources, Part A, 2018, 40(22), 2686–2725, DOI:10.1080/15567036.2018.1511642.
- Y.-L. Zhang, H. Xia, E. Kim and H.-B. Sun, Recent developments in superhydrophobic surfaces with unique structural and functional properties, Soft Matter, 2012, 8(44), 11217–11231, 10.1039/C2SM26517F.
- R. Shakil, Y. A. Tarek, M. H. Rumon, C. K. Roy, A.-N. Chowdhury and R. Das, Polymeric Membranes for O2/N2 Separation, Polymeric Membranes for Water Purification and Gas Separation, 2021, vol. 113, pp. 171–202 Search PubMed.
- H. Vafaeenezhad and R. Eslami-Farsani, Self-Healing and Self-Lubricating Nano-Hybrid Smart Coatings, in Nano-Hybrid Smart Coatings: Advancements in Industrial Efficiency and Corrosion Resistance, ACS Symposium Series, American Chemical Society, 2024, vol. 1469, pp 303–352 Search PubMed.
- Y. Qiu and K. Park, Environment-sensitive hydrogels for drug delivery, Adv. Drug Delivery Rev., 2001, 53(3), 321–339 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani and M. Bozorgomid, et al., Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems, Chem. Soc. Rev., 2016, 45(5), 1457–1501, 10.1039/C5CS00798D.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Bioresponsive materials, Nat. Rev. Mater., 2016, 2(1), 16075, DOI:10.1038/natrevmats.2016.75.
- D. Buenger, F. Topuz and J. Groll, Hydrogels in sensing applications, Prog. Polym. Sci., 2012, 37(12), 1678–1719, DOI:10.1016/j.progpolymsci.2012.09.001.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Disulfide-Cleavage-Triggered Chemosensors and Their Biological Applications, Chem. Rev., 2013, 113(7), 5071–5109, DOI:10.1021/cr300358b.
- W. Guo, C.-H. Lu, R. Orbach, F. Wang, X.-J. Qi, A. Cecconello, D. Seliktar and I. Willner, pH-stimulated DNA hydrogels exhibiting shape-memory properties, Adv. Mater., 2014, 27(1), 73–78 CrossRef PubMed.
- C.-C. Yu, Y.-C. Su and Y.-C. Yeh, Fabrication of multi-responsive dynamic poly(ethylene glycol)-iron oxide nanoparticle nanocomposite hydrogels through interfacial boronate-catechol crosslinking, Eur. Polym. J., 2024, 220, 113469, DOI:10.1016/j.eurpolymj.2024.113469.
- G. Kocak, C. Tuncer and V. Bütün, pH-Responsive polymers, Polym. Chem., 2017, 8(1), 144–176 Search PubMed.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, pH Sensitive Hydrogels in Drug Delivery: Brief History, Properties, Swelling, and Release Mechanism, Material Selection and Applications, Polymers, 2017, 9(4), 137, DOI:10.3390/polym9040137.
- C. Zaino, Ylenia Zambito, G. Mollica, M. Geppi, M. F. Serafini, C. Vera and D. C. Giacomo, A Novel Polyelectrolyte Complex (PEC) Hydrogel for Controlled Drug Delivery to the Distal Intestine, Open Drug Delivery J., 2007, 1, 68–75, DOI:10.2174/1874126600701010068.
- H. Fan, J. He, M. Heiranian, W. Pan, Y. Li and M. Elimelech, The physical basis for solvent flow in organic solvent nanofiltration, Sci. Adv., 2024, 10(24), eado4332, DOI:10.1126/sciadv.ado4332.
- S. J. Pai, E. J. Kang, W. M. Ahn, J. S. Kim, Y. C. Bae, J. W. Kwon and J. S. Oh, Swelling Behaviors of Natural Rubber/Solvent Systems Based on the Extended Modified Double Lattice Model, Macromol. Theory Simul., 2024, 33(4), 2400015, DOI:10.1002/mats.202400015.
- S. Ghorbani, H. Eyni, S. R. Bazaz, H. Nazari, L. S. Asl, H. Zaferani, V. Kiani, A. A. Mehrizi and M. Soleimani, Hydrogels based on cellulose and its derivatives: applications, synthesis, and characteristics, Polym. Sci., Ser. A, 2018, 60, 707–722 CrossRef CAS.
- Z. Zare-Akbari, H. Farhadnejad, B. Furughi-Nia, S. Abedin, M. Yadollahi and M. Khorsand-Ghayeni, PH-sensitive bionanocomposite hydrogel beads based on carboxymethyl cellulose/ZnO nanoparticle as drug carrier, Int. J. Biol. Macromol., 2016, 93, 1317–1327, DOI:10.1016/j.ijbiomac.2016.09.110.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Injectable and self-healing nanocomposite hydrogels with ultrasensitive pH-responsiveness and tunable mechanical properties: implications for controlled drug delivery, Biomacromolecules, 2020, 21(6), 2409–2420 CrossRef CAS PubMed.
- A. A. Akib, M. M. H. Rumon, M. Moniruzzaman, C. K. Roy and A.-N. Chowdury, PLA-PEG Diblock Copolymer Micelle as Nanocarrier for Anti-Obesity Drug Delivery System, ECS Trans., 2022, 107(1), 19031 CrossRef CAS.
- A. A. Akib, R. Shakil, M. M. H. Rumon, C. K. Roy, E. H. Chowdhury and A.-N. Chowdhury, Natural and synthetic micelles for the delivery of small molecule drugs, imaging agents and nucleic acids, Curr. Pharm. Des., 2022, 28(17), 1389–1405 CrossRef CAS PubMed.
- M. M. H. Rumon, M. S. Rahman, A. A. Akib, M. S. Sohag, M. R. A. Rakib, M. A. R. Khan, F. Yesmin, M. S. Shakil and M. M. Rahman Khan, Progress in hydrogel toughening: addressing structural and crosslinking challenges for biomedical applications, Discover Mater., 2025, 5(1), 5, DOI:10.1007/s43939-025-00178-x.
- G. Jiang, C. Liu, X. Liu, Q. Chen, G. Zhang, M. Yang and F. Liu, Network structure and compositional effects on tensile mechanical properties of hydrophobic association hydrogels with high mechanical strength, Polymer, 2010, 51(6), 1507–1515 CrossRef CAS.
- A. M. Villalba-Rodriguez, R. Parra-Saldivar, I. Ahmed, K. Karthik, Y. S. Malik, K. Dhama and H. Iqbal, Bio-inspired biomaterials and their drug delivery perspectives-A review, Curr. Drug Metab., 2017, 18(10), 893–904 CAS.
- S. S. A. Sharma, S. Bashir, R. Kasi and R. T. Subramaniam, The significance of graphene based composite hydrogels as smart materials: A review on the fabrication, properties, and its applications, FlatChem, 2022, 33, 100352 CrossRef.
- Z. Tang, Y. Yang, Y. Pan, M. Yu, X. Lin and A. K. Mondal, Biocompatible, Injectable, and Self-Healing Poly(N-vinylpyrrolidone)/Carboxymethyl Cellulose Hydrogel for Drug Release, ACS Omega, 2024, 9(5), 5854–5861, DOI:10.1021/acsomega.3c09156.
- V. S. Sivasankarapillai, S. S. Das, F. Sabir, M. A. Sundaramahalingam, J. C. Colmenares, S. Prasannakumar, M. Rajan, A. Rahdar and G. Z. Kyzas, Progress in natural polymer engineered biomaterials for transdermal drug delivery systems, Mater. Today Chem., 2021, 19, 100382 CrossRef CAS.
- S. Dutta, R. S. Gupta, S. Pathan and S. Bose, Interpenetrating polymer networks for desalination and water remediation: A comprehensive review of research trends and prospects, RSC Adv., 2023, 13(9), 6087–6107 RSC.
- Y. Zhang, L. Dong, L. Liu, Z. Wu, D. Pan and L. Liu, Recent advances of stimuli-responsive polysaccharide hydrogels in delivery systems: A review, J. Agric. Food Chem., 2022, 70(21), 6300–6316 CrossRef CAS PubMed.
- T. Fekete, J. Borsa, E. Takács and L. Wojnárovits, Synthesis of carboxymethylcellulose/starch superabsorbent hydrogels by gamma-irradiation, Chem. Cent. J., 2017, 11, 1–10 CrossRef PubMed.
- N. Udaipuria and S. Bhattacharya, Novel Carbohydrate Polymer-Based Systems for Precise Drug Delivery in Colon Cancer: Improving Treatment Effectiveness With Intelligent Biodegradable Materials, Biopolymers, 2025, 116(1), e23632 CrossRef CAS PubMed.
- F. Ahmadi, Z. Oveisi, S. M. Samani and Z. Amoozgar, Chitosan based hydrogels: characteristics and pharmaceutical applications, Res. Pharm. Sci., 2015, 10(1), 1–16 CAS.
- L. Vlaia, G. Coneac, I. Olariu, V. Vlaia and D. Lupuleasa, Cellulose-derivatives-based hydrogels as vehicles for dermal and transdermal drug delivery, Emerging Concepts Anal. Appl. Hydrogels, 2016, 2, 64 Search PubMed.
- J. Gong, L. Hou, Y. C. Ching, K. Y. Ching, N. Dai Hai and C. H. Chuah, A review of recent advances of cellulose-based intelligent-responsive hydrogels as vehicles for controllable drug delivery system, Int. J. Biol. Macromol., 2024, 130525 CrossRef CAS PubMed.
- P. Taneja, S. Sharma, V. B. Sinha and A. K. Yadav, Advancement of nanoscience in development of conjugated drugs for enhanced disease prevention, Life Sci., 2021, 268, 118859 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20(2), 101–124 CrossRef CAS PubMed.
- R. H. Muller and C. M. Keck, Challenges and solutions for the delivery of biotech drugs–a review of drug nanocrystal technology and lipid nanoparticles, J. Biotechnol., 2004, 113(1–3), 151–170 CrossRef CAS PubMed.
- E. Pérez-Herrero and A. Fernández-Medarde, Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed.
- M. Sajid, M. Ilyas, C. Basheer, M. Tariq, M. Daud, N. Baig and F. Shehzad, Impact of nanoparticles on human and environment: review of toxicity factors, exposures, control strategies, and future prospects, Environ. Sci. Pollut. Res., 2015, 22, 4122–4143 CrossRef PubMed.
- F. Ahmadpour, F. Ganjali, F. Radinekiyan, R. Eivazzadeh-Keihan, M. Salimibani, H. Bahreinizad, M. Mahdavi and A. Maleki, Fabrication and characterization of a novel magnetic nanostructure based on pectin–cellulose hydrogel for in vitro hyperthermia during cancer therapy, RSC Adv., 2024, 14(19), 13676–13684, 10.1039/D3RA08067F.
- S. A. Eming, P. Martin and M. Tomic-Canic, Wound repair and regeneration: mechanisms, signaling, and translation, Sci. Transl. Med., 2014, 6(265), 265sr266 CrossRef PubMed.
- M. Hao, C. Ding, S. Sun, X. Peng and W. Liu, Chitosan/sodium alginate/velvet antler blood peptides hydrogel promotes diabetic wound healing via regulating angiogenesis, inflammatory response and skin flora, J. Inflammation Res., 2022, 4921–4938 CrossRef PubMed.
- M. M. Rahman Khan and M. M. H. Rumon, Recent progress on the synthesis, morphological topography, and battery applications of polypyrrole-based nanocomposites, Polymers, 2024, 16(23), 3277 CrossRef CAS PubMed.
- A. R. Shakil, M. L. Begum, M. A. A. Shaikh, S. Sultana, M. S. Rahman, M. M. H. Rumon, C. K. Roy and M. A. Haque, Jute Fiber Reinforced Hydrogel Composite for Removal of Methylene Blue Dye from Water, Dhaka Univ. J. Sci., 2022, 70(2), 59–64 CrossRef.
- V. Brumberg, T. Astrelina, T. Malivanova and A. Samoilov, Modern wound dressings: Hydrogel dressings, Biomedicines, 2021, 9(9), 1235 CrossRef CAS PubMed.
- S. Sarkhel and A. Jaiswal, Emerging Frontiers in In Situ Forming Hydrogels for Enhanced Hemostasis and Accelerated Wound Healing, ACS Appl. Mater. Interfaces, 2024, 16(45), 61503–61529 CrossRef CAS PubMed.
- D. L. Taylor and M. In Het Panhuis, Self-healing hydrogels, Adv. Mater., 2016, 28(41), 9060–9093 CrossRef CAS PubMed.
- B. Li, P.-F. Cao, T. Saito and A. P. Sokolov, Intrinsically self-healing polymers: from mechanistic insight to current challenges, Chem. Rev., 2022, 123(2), 701–735 CrossRef PubMed.
- B. Liu, Y. Wang, Y. Miao, X. Zhang, Z. Fan, G. Singh, X. Zhang, K. Xu, B. Li and Z. Hu, et al., Hydrogen bonds autonomously powered gelatin methacrylate hydrogels with super-elasticity, self-heal and underwater self-adhesion for sutureless skin and stomach surgery and E-skin, Biomaterials, 2018, 171, 83–96, DOI:10.1016/j.biomaterials.2018.04.023.
- H. Chen, R. Cheng, X. Zhao, Y. Zhang, A. Tam, Y. Yan, H. Shen, Y. S. Zhang, J. Qi and Y. Feng, et al., An injectable self-healing coordinative hydrogel with antibacterial and angiogenic properties for diabetic skin wound repair, NPG Asia Mater., 2019, 11(1), 3, DOI:10.1038/s41427-018-0103-9.
- E. Guamba, N. S. Vispo, D. C. Whitehead, A. K. Singh, R. Santos-Oliveira, D. Niebieskikwiat, C. Zamora-Ledezma and F. Alexis, Cellulose-based hydrogels towards an antibacterial wound dressing, Biomater. Sci., 2023, 11(10), 3461–3468, 10.1039/D2BM01369J.
- X. Yi, J. He, X. Wei, H. Li, X. Liu and F. Cheng, A mussel-inspired multifunctional hydrogel reinforced by bacterial cellulose for wound healing: sustained drug release, enhanced adhesion and self-healing property, Cellulose, 2023, 30(10), 6523–6538, DOI:10.1007/s10570-023-05297-3.
- K. Y. Lee and D. J. Mooney, Hydrogels for tissue engineering, Chem. Rev., 2001, 101(7), 1869–1880 CrossRef CAS PubMed.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Supramolecular Adhesive Hydrogels for Tissue Engineering Applications, Chem. Rev., 2022, 122(6), 5604–5640, DOI:10.1021/acs.chemrev.1c00815.
- Y. Li, J. Rodrigues and H. Tomás, Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications, Chem. Soc. Rev., 2012, 41(6), 2193–2221 RSC.
- D. F. Williams, Biocompatibility pathways: biomaterials-induced sterile inflammation, mechanotransduction, and principles of biocompatibility control, ACS Biomater. Sci. Eng., 2017, 3(1), 2–35 CrossRef CAS PubMed.
- Z. Terzopoulou, A. Zamboulis, I. Koumentakou, G. Michailidou, M. J. Noordam and D. N. Bikiaris, Biocompatible synthetic polymers for tissue engineering purposes, Biomacromolecules, 2022, 23(5), 1841–1863 Search PubMed.
- A. Atala, Engineering tissues, organs and cells, J. Tissue Eng. Regener. Med., 2007, 1(2), 83–96 CrossRef CAS PubMed.
- M. C. Barsotti, F. Felice, A. Balbarini and R. Di Stefano, Fibrin as a scaffold for cardiac tissue engineering, Biotechnol. Appl. Biochem., 2011, 58(5), 301–310 CrossRef CAS PubMed.
- G. Rubi-Sans, O. Castaño, I. Cano, M. A. Mateos-Timoneda, S. Perez-Amodio and E. Engel, Engineering Cell-Derived Matrices: From 3D Models to Advanced Personalized Therapies, Adv. Funct. Mater., 2020, 30(44), 2000496 Search PubMed.
- R. M. A. Domingues, M. E. Gomes and R. L. Reis, The potential of cellulose nanocrystals in tissue engineering strategies, Biomacromolecules, 2014, 15(7), 2327–2346 CrossRef CAS PubMed.
- L.-H. Fu, C. Qi, M.-G. Ma and P. Wan, Multifunctional cellulose-based hydrogels for biomedical applications, J. Mater. Chem. B, 2019, 7(10), 1541–1562 Search PubMed.
- X. Yang, E. Bakaic, T. Hoare and E. D. Cranston, Injectable Polysaccharide Hydrogels Reinforced with Cellulose Nanocrystals: Morphology, Rheology, Degradation, and Cytotoxicity, Biomacromolecules, 2013, 14(12), 4447–4455, DOI:10.1021/bm401364z.
- P. Shrivastav, S. Pramanik, G. Vaidya, M. A. Abdelgawad, M. M. Ghoneim, A. Singh, B. M. Abualsoud, L. S. Amaral and M. A. S. Abourehab, Bacterial cellulose as a potential biopolymer in biomedical applications: A state-of-the-art review, J. Mater. Chem. B, 2022, 10(17), 3199–3241 RSC.
- C. Boyer, L. Figueiredo, R. Pace, J. Lesoeur, T. Rouillon, C. L. Visage, J.-F. Tassin, P. Weiss, J. Guicheux and G. Rethore, Laponite nanoparticle-associated silated hydroxypropylmethyl cellulose as an injectable reinforced interpenetrating network hydrogel for cartilage tissue engineering, Acta Biomater., 2018, 65, 112–122, DOI:10.1016/j.actbio.2017.11.027.
- K. M. Tohamy, M. Mabrouk, I. E. Soliman, H. H. Beherei and M. A. Aboelnasr, Novel alginate/hydroxyethyl cellulose/hydroxyapatite composite scaffold for bone regeneration: In vitro cell viability and proliferation of human mesenchymal stem cells, Int. J. Biol. Macromol., 2018, 112, 448–460, DOI:10.1016/j.ijbiomac.2018.01.181.
- J. Leppiniemi, P. Lahtinen, A. Paajanen, R. Mahlberg, S. Metsä-Kortelainen, T. Pinomaa, H. Pajari, I. Vikholm-Lundin, P. Pursula and V. P. Hytönen, 3D-Printable Bioactivated Nanocellulose–Alginate Hydrogels, ACS Appl. Mater. Interfaces, 2017, 9(26), 21959–21970, DOI:10.1021/acsami.7b02756.
- A. C. B. Benatti, A. F. Pattaro, A. A. Rodrigues, M. V. Xavier, A. Kaasi, M. I. R. Barbosa, A. L. Jardini, R. Maciel Filho and P. Kharmandayan, Bioreabsorbable polymers for tissue engineering: PLA, PGA, and their copolymers, in Materials for Biomedical Engineering, Elsevier, 2019, pp 83–116 Search PubMed.
- A. Sivashanmugam, R. A. Kumar, M. V. Priya, S. V. Nair and R. Jayakumar, An overview of injectable polymeric hydrogels for tissue engineering, Eur. Polym. J., 2015, 72, 543–565 CrossRef CAS.
- J. Radhakrishnan, A. Subramanian, U. M. Krishnan and S. Sethuraman, Injectable and 3D bioprinted polysaccharide hydrogels: from cartilage to osteochondral tissue engineering, Biomacromolecules, 2017, 18(1), 1–26 CrossRef CAS PubMed.
- J. J. Marler, J. Upton, R. Langer and J. P. Vacanti, Transplantation of cells in matrices for tissue regeneration, Adv. Drug Delivery Rev., 1998, 33(1–2), 165–182 CrossRef CAS PubMed.
- M. Tallawi, E. Rosellini, N. Barbani, M. G. Cascone, R. Rai, G. Saint-Pierre and A. R. Boccaccini, Strategies for the chemical and biological functionalization of scaffolds for cardiac tissue engineering: a review, J. R. Soc. Interface, 2015, 12(108), 20150254 CrossRef PubMed.
- L. Peng and E. G. Julien, Long term expansion profile of mesenchymal stromal cells at protein nanosheet-stabilised bioemulsions for next generation cell culture microcarriers, Mater. Today Bio, 2021, 12, 100159, DOI:10.1016/j.mtbio.2021.100159.
- A. V. Goponenko and Y. A. Dzenis, Role of mechanical factors in applications of stimuli-responsive polymer gels–Status and prospects, Polymer, 2016, 101, 415–449 CrossRef CAS PubMed.
- M. Chan, D. Estève, J.-Y. Fourniols, C. Escriba and E. Campo, Smart wearable systems: Current status and future challenges, Artif. Intell. Med., 2012, 56(3), 137–156 Search PubMed.
- Y. Zhou, L. Li, Z. Han, Q. Li, J. He and Q. Wang, Self-healing polymers for electronics and energy devices, Chem. Rev., 2022, 123(2), 558–612 CrossRef PubMed.
- X. Jing, H. Li, H.-Y. Mi, Y.-J. Liu, P.-Y. Feng, Y.-M. Tan and L.-S. Turng, Highly transparent, stretchable, and rapid self-healing polyvinyl alcohol/cellulose nanofibril hydrogel sensors for sensitive pressure sensing and human motion detection, Sens. Actuators, B, 2019, 295, 159–167 Search PubMed.
- Y.-J. Liu, W.-T. Cao, M.-G. Ma and P. Wan, Ultrasensitive wearable soft strain sensors of conductive, self-healing, and elastic hydrogels with synergistic “soft and hard” hybrid networks, ACS Appl. Mater. Interfaces, 2017, 9(30), 25559–25570 CrossRef CAS PubMed.
- J. Yang, F. Xu and C.-R. Han, Metal Ion Mediated Cellulose Nanofibrils Transient Network in Covalently Cross-linked Hydrogels: Mechanistic Insight into Morphology and Dynamics, Biomacromolecules, 2017, 18(3), 1019–1028, DOI:10.1021/acs.biomac.6b01915.
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Mussel-inspired cellulose nanocomposite tough hydrogels with synergistic self-healing, adhesive, and strain-sensitive properties, Chem. Mater., 2018, 30(9), 3110–3121 Search PubMed.
- Y. Wang, Y. Yu, X. Wei and F. Narita, Self-powered wearable piezoelectric monitoring of human motion and physiological signals for the postpandemic era: a review, Adv. Mater. Technol., 2022, 7(12), 2200318 CrossRef.
- H. Yuan, Q. Zhang, T. Zhou, W. Wu, H. Li, Z. Yin, J. Ma and T. Jiao, Progress and challenges in flexible capacitive pressure sensors: Microstructure designs and applications, Chem. Eng. J., 2024, 149926 CrossRef CAS.
- G.-X. Li, P.-X. Hou, S.-Y. Zhao, C. Liu and H.-M. Cheng, A flexible cotton-derived carbon sponge for high-performance capacitive deionization, Carbon, 2016, 101, 1–8, DOI:10.1016/j.carbon.2015.12.095.
- H. Zhang, X. Sun, M. Hubbe and L. Pal, Flexible and Pressure-Responsive Sensors from Cellulose Fibers Coated with Multiwalled Carbon Nanotubes, ACS Appl. Electron. Mater., 2019, 1(7), 1179–1188, DOI:10.1021/acsaelm.9b00182.
- Y. Li, Y. A. Samad and K. Liao, From cotton to wearable pressure sensor, J. Mater. Chem. A, 2015, 3(5), 2181–2187, 10.1039/C4TA05810K.
- J. Zhou, H. Yu, X. Xu, F. Han and G. Lubineau, Ultrasensitive, Stretchable Strain Sensors Based on Fragmented Carbon Nanotube Papers, ACS Appl. Mater. Interfaces, 2017, 9(5), 4835–4842, DOI:10.1021/acsami.6b15195.
- Y.-J. Tsai, C.-M. Wang, T.-S. Chang, S. Sutradhar, C.-W. Chang, C.-Y. Chen, C.-H. Hsieh and W.-S. Liao, Multilayered Ag NP–PEDOT–Paper Composite Device for Human–Machine Interfacing, ACS Appl. Mater. Interfaces, 2019, 11(10), 10380–10388, DOI:10.1021/acsami.8b21390.
- K. Upadhyay, R. K. Tamrakar, S. Thomas and M. Kumar, Surface functionalized nanoparticles: A boon to biomedical science, Chem.-Biol. Interact., 2023, 380, 110537, DOI:10.1016/j.cbi.2023.110537.
- H. Lee, Y. Jung, N. Lee, I. Lee and J. H. Lee, Nature-Derived Polysaccharide-Based Composite Hydrogels for Promoting Wound Healing, Int. J. Mol. Sci., 2023, 24, 16714 Search PubMed.
- C. Xu, G. Dai and Y. Hong, Recent advances in high-strength and elastic hydrogels for 3D printing in biomedical applications, Acta Biomater., 2019, 95, 50–59, DOI:10.1016/j.actbio.2019.05.032.
- X. Kuang, M. O. Arıcan, T. Zhou, X. Zhao and Y. S. Zhang, Functional Tough Hydrogels: Design, Processing, and Biomedical Applications, Acc. Mater. Res., 2023, 4(2), 101–114, DOI:10.1021/accountsmr.2c00026.
- M. M. Perera and N. Ayres, Dynamic covalent bonds in self-healing, shape memory, and controllable stiffness hydrogels, Polym. Chem., 2020, 11(8), 1410–1423, 10.1039/C9PY01694E.
- Z. Tong, L. Jin, J. M. Oliveira, R. L. Reis, Q. Zhong, Z. Mao and C. Gao, Adaptable hydrogel with reversible linkages for regenerative medicine: Dynamic mechanical microenvironment for cells, Bioact. Mater., 2021, 6(5), 1375–1387, DOI:10.1016/j.bioactmat.2020.10.029.
| This journal is © The Royal Society of Chemistry 2025 |