DOI:
10.1039/D5RA00032G
(Paper)
RSC Adv., 2025,
15, 9408-9419
A hollow nanozyme-based multifunctional platform enhances sonodynamic–chemodynamic-induced ferroptosis for cancer therapy†
Received
2nd January 2025
, Accepted 13th March 2025
First published on 27th March 2025
Abstract
Ferroptosis, a novel form of cell death driven by lipid peroxides (LPO) accumulation, holds promise for personalized cancer therapy. However, its efficacy is constrained by the tumor microenvironment (TME), which is characterized by hypoxia, insufficient endogenous hydrogen peroxide (H2O2), and glutathione (GSH) overabundance. To address these limitations, we developed a multifunctional nanoplatform, HMnO2–VC@mPEG–Ce6 (HMVC), which integrates sono–chemodynamic strategies to induce synergistic ferroptosis in prostate cancer. The therapeutic superiority of HMVC stems from three coordinated mechanisms. Firstly, HMnO2 catalyze H2O2 decomposition to generate oxygen (O2), alleviating tumor hypoxia and amplifying the sonodynamic effect of chlorin e6 (Ce6). Secondly, vitamin C (VC) sustains H2O2 production via chemodynamic therapy (CDT), driving a burst of reactive oxygen species (ROS). Thirdly, GSH-triggered reduction of Mn4+ to Mn2+ depletes GSH reserves and suppresses glutathione peroxidase 4 (GPX4) activity. These cascading actions disrupt the ROS–GPX4 equilibrium, leading to irreversible LPO accumulation and subsequent ferroptosis. Our work establishes a generalizable nanotechnology paradigm to overcome TME barriers and achieve precise ferroptosis regulation, offering a transformative strategy for cancer treatment.
1. Introduction
Ferroptosis, an iron-dependent form of programmed cell death driven by lipid peroxidation, has emerged as a novel strategy for cancer treatment due to its ability to bypass traditional apoptosis resistance mechanisms.1–3 Its core regulatory axis lies in the dynamic balance between reactive oxygen species (ROS) and glutathione peroxidase 4 (GPX4): ROS promote the accumulation of lipid peroxides (LPO), while GPX4 relies on glutathione (GSH) to eliminate LPO and maintain cell survival.4,5 Therefore, ROS levels are critical, primarily including hydrogen peroxide (H2O2), singlet oxygen (1O2), superoxide anion (O2−), hypochlorous acid (HClO), and hydroxyl radical (˙OH), which can kill tumor cells by damaging cellular lipids, proteins, DNA, and other biomolecules.6–8 Based on this, sonodynamic therapy (SDT) and chemodynamic therapy (CDT) have become effective approaches for inducing ferroptosis by generating 1O2 and ˙OH to elevate ROS levels.9–12 However, the therapeutic efficacy of these approaches is severely constrained by three major barriers in the tumor microenvironment (TME).13–15 Firstly, hypoxia limits the activation efficiency of sonosensitizers in SDT;16,17 secondly, insufficient endogenous H2O2 weakens the Fenton reaction in CDT; and thirdly, excessive GSH neutralizes ROS and sustains GPX4 activity,18–21 thereby resisting ferroptosis. Consequently, developing synergistic strategies that simultaneously remodel the TME and achieve bidirectional regulation of ROS–GPX4 is key to overcoming the bottleneck in ferroptosis induction.
In recent years, manganese dioxide (MnO2) has garnered significant attention due to its unique properties in modulating the tumor microenvironment (TME).22,23 MnO2 exhibits multiple enzyme-mimicking activities, including oxidase, peroxidase, catalase, and superoxide dismutase, making it an important manganese-based nanomaterial. Although hypoxia significantly suppresses the immune activity of natural killer cells, MnO2 can effectively alleviate tumor hypoxia by converting endogenous H2O2 into O2 through its catalase and peroxidase activities.24–26 Additionally, the high concentration of GSH in the TME promotes the decomposition of MnO2, releasing Mn2+ and making it a promising GSH-depleting agent.27–30 The released Mn2+ can further enhance tumor-killing effects by catalyzing the generation of ˙OH from H2O2 via Fenton-like reactions.31,32 Notably, manganese, as an essential trace element, has well-regulated uptake and excretion mechanisms in biological systems, which ensures the low toxicity and excellent biocompatibility of MnO2-based nanomaterials. Compared to traditional MnO2 nanoparticles, hollow mesoporous manganese dioxide (HMnO2) nanoparticles not only inherit these advantages but also exhibit superior performance due to their unique structural characteristics. Specifically, HMnO2 demonstrates faster degradation rates, larger specific surface areas, excellent colloidal dispersibility, and enhanced drug-loading capacity.33 However, single-component HMnO2 systems face significant limitations: (1) the endogenous H2O2 levels in tumors (100 μM to 1 mM)34 are insufficient to sustain continuous O2 supply and ˙OH generation; (2) after GSH depletion, the lack of direct inhibition on GPX4 activity prevents an increase in lipid peroxidation levels. These challenges highlight the urgent need to design a multi-mechanism synergistic nanoplatform that integrates H2O2 self-supply, dual blockade of the GSH–GPX4 pathway, and controlled ROS generation.
Based on this, we constructed a multifunctional nanosystem, HMnO2–VC@mPEG–Ce6 (HMVC), and proposed a triple-interlocked ferroptosis induction mechanism of “TME remodeling-ROS storm-GPX4 silencing” (Fig. 1). The innovation of this system lies in the following aspects: firstly, vitamin C (VC) specifically generates H2O2 at the tumor site, which serves as both a substrate for the Fenton-like reaction in CDT and a source for O2 production catalyzed by HMnO2.35 This dual function synergistically alleviates hypoxia and amplifies the sonodynamic effect of the sonosensitizer Ce6. Secondly, HMnO2 continuously depletes GSH through the Mn4+/Mn2+ redox cycle while simultaneously suppressing GPX4 activity. Thirdly, The nanocarrier regulates the release kinetics of VC and Mn2+, preventing off-target toxicity caused by premature ROS bursts and ensuring the cascade amplification of ferroptosis signaling. This study not only demonstrates the potent antitumor efficacy of HMVC in prostate cancer models but also provides deeper insights into the molecular mechanisms by which TME-adaptive nanomaterials enhance ferroptosis through metabolic reprogramming. These findings highlight its significant potential for clinical translation.
 |
| | Fig. 1 Preparation process and therapeutic mechanism of the HMnO2–VC@mPEG–Ce6 nanoparticles (HMVC NPs). The acidic tumor microenvironment and US trigger the decomposition of HMVC NPs. HMnO2 catalyzes the conversion of H2O2 into O2 and releases Mn2+, synergizing with Ce6 to mediate SDT/CDT for ROS generation. Vit C releases H2O2 to enhance the Fenton-like reaction and SDT effects, while GSH depletion inhibits GPX4 activity, and the accumulation of ROS and LPO jointly promotes ferroptosis. | |
2. Experimental section
2.1. Materials and reagents
Tetraethyl orthosilicate (TEOS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydro-chloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich. N,N-Dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were purchased from Junsei Chemicals (Tokyo, Japan). Methoxy polyethylene glycol amine (mPEG-amine) with a molecular weight of 2000 Da was purchased from Sunbio (Orinda, CA, USA). Chlorin e6 (Ce6) was purchased from Frontier Scientific, Inc. (Salt Lake City, UT, USA). Potassium permanganate (KMnO4), sodium carbonate (Na2CO3), hydrogen peroxide (H2O2), sodium bicarbonate (NaHCO3), methylene blue (MB), manganese chloride (MnCl2), and glutathione (GSH) were purchased from Sinopharm Chemical Reagent Co. Ltd. 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) was purchased from Alad-din Chemical Co. Ltd. Singlet oxygen sensor green (SOSG), C11-BODIPY 581/591 and 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from Beyotime (Shanghai, China). Calcein-AM/PI was purchased from Solarbio Science and Technology Co. Ltd (Beijing, China). Cell Counting Kit-8 (CCK-8) was purchased from Proteintech (Wuhan, China). Cyanine-5.5 (Cy5.5) was purchased from MedChemExpress. All other chemical reagents used in this experiment were analytically pure without further purification.
2.2. Cell lines and cell culture
PC-3 cells were obtained from Procell Life Science and Technology Co., Ltd. These cell lines were cultured in F-12K medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin solution at 37 °C under 5% carbon dioxide.
2.3. Animals
BALB/c mice (male, 4 weeks old) were purchased from Beijing Viton Lihua Laboratory Animal Technology Co. and were reared in an SPF-grade animal center. A tumor model was created by subcutaneously injecting 0.1 mL of PC-3 cells (1 × 106 cells per mL) into the left side of the abdomen. When the tumors reached a magnitude of 100 mm3, the mice were randomly assigned to different treatment groups. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shandong Provincial Hospital and approved by the Animal Ethics Committee of Shandong Provincial Hospital (no. 2023-0012).
2.4. Synthesis of HMnO2
Firstly, synthesis of SiO2 core: in a mixed solution containing ethanol (25 mL), ultrapure water (0.5 mL), and ammonia (1.8 mL, 28%), 0.75 mL of TEOS was rapidly added. The mixture was placed in a constant-temperature water bath and reacted under magnetic stirring at 40 °C and 150 rpm for 12 hours. SiO2 nanoparticles with a particle size of 80 ± 5 nm were obtained. Secondly, preparation of SiO2@MnO2: the synthesized SiO2 nanoparticles were dispersed in 40 mL of ultrapure water. Under ultrasonic conditions (150 W, 40 kHz), 47 mL of KMnO4 solution (0.02 mmol L−1) was slowly added dropwise. After 6 hours of ultrasonic dispersion, the precipitate was collected by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rpm, 10 minutes, 4 °C) and washed three times with ultrapure water and ethanol, respectively, to obtain SiO2@MnO2 nanoparticles. The chemical reaction equations is: 2KMnO4 + 3SiO2 + 2H2O → 2MnO2 + 3SiO32− + 2KOH. Thirdly, synthesis of HMnO2: the SiO2@MnO2 nanoparticles were mixed with 2 mmol per L Na2CO3 solution and reacted under magnetic stirring at 60 °C and 150 rpm for 12 hours to remove the SiO2 template. After the reaction, the precipitate was collected by centrifugation (12000 rpm, 10 minutes, 4 °C) and washed three times with ultrapure water. The final product was dispersed in 40 mL of ultrapure water to obtain HMnO2 nanoparticles at a concentration of 2 mg mL−1.
800 rpm, 10 minutes, 4 °C) and washed three times with ultrapure water and ethanol, respectively, to obtain SiO2@MnO2 nanoparticles. The chemical reaction equations is: 2KMnO4 + 3SiO2 + 2H2O → 2MnO2 + 3SiO32− + 2KOH. Thirdly, synthesis of HMnO2: the SiO2@MnO2 nanoparticles were mixed with 2 mmol per L Na2CO3 solution and reacted under magnetic stirring at 60 °C and 150 rpm for 12 hours to remove the SiO2 template. After the reaction, the precipitate was collected by centrifugation (12000 rpm, 10 minutes, 4 °C) and washed three times with ultrapure water. The final product was dispersed in 40 mL of ultrapure water to obtain HMnO2 nanoparticles at a concentration of 2 mg mL−1.
2.5. Synthesis of mPEG–Ce6
60 mg of Ce6 was dissolved in 10 mL of anhydrous DMF under vigorous stirring until complete dissolution. To the Ce6 solution, 19 mg of EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) and 12 mg of NHS (N-hydroxysuccinimide) were added. The mixture was reacted at 25 °C under magnetic stirring at 500 rpm for 4 hours to activate the carboxyl groups of Ce6. 200 mg of mPEG-amine was dissolved in 10 mL of anhydrous DMF and set aside. The activated Ce6 solution was then slowly added dropwise to the mPEG-amine solution. The reaction was carried out under nitrogen protection (the reaction flask was purged with nitrogen for 10 minutes prior to the reaction, and a continuous nitrogen flow was maintained throughout the reaction) at 25 °C under magnetic stirring at 500 rpm for 24 hours. This allowed the terminal amino groups of mPEG-amine to react with the activated carboxyl groups of Ce6, forming the mPEG–Ce6 conjugate via an esterification reaction. After the reaction, the mixture was transferred into a dialysis bag and dialyzed against ultrapure water at 4 °C for 72 hours to remove unreacted Ce6, EDC, NHS, and other small-molecule byproducts. The purified product was then freeze-dried for 24 hours to obtain the mPEG–Ce6 conjugate as a solid powder.
2.6. Synthesis of HMnO2–VC@mPEG–Ce6
0.5 mg of HMnO2 nanoparticles was added to 5 mL of ultrapure water containing 10 mg of vitamin C (VC). The mixture was sonicated at 40 kHz and 100 W for 5 minutes, followed by magnetic stirring at 200 rpm in the dark for 2 hours to obtain the HMnO2–VC dispersion. The prepared mPEG–Ce6 solution (5 mg mL−1) was mixed with the HMnO2–VC dispersion. The mixture was sonicated at 40 kHz and 100 W for 5 minutes, followed by magnetic stirring at 200 rpm in the dark for 24 hours. After the reaction, the supernatant was removed by centrifugation (10000 rpm, 10 minutes, 4 °C), and the precipitate was collected. The final product was redispersed in 10 mL of ultrapure water to obtain the HMnO2–VC@mPEG–Ce6 nanoparticle dispersion, which was stored at 4 °C in the dark. The concentration of free Ce6 was measured using a multifunctional microplate reader (Varioskan LUX, Thermo Scientific, USA) at a wavelength of 402 nm. The encapsulation efficiency (EE) was calculated using the following formula: EE (%) = (total Ce6 − free Ce6)/total Ce6 × 100%.
2.7. Characterization of HMnO2–VC@mPEG–Ce6
Scanning electron microscopy (SEM, Zeiss Gemini 300, Carl Zeiss Co., Germany) was used to observe the dispersion of the nanoparticles. Transmission electron microscopy (TEM, FEI Talos F200X, FEI Co., USA), high-angle annular dark field scanning TEM (HAADF-STEM), and elemental mapping images were used to observe the morphology and elemental distribution of the nanoparticles. Fourier transform infrared spectroscopy (FTIR) were generated on a Thermo Scientific Nicolet iS 20 FTIR spectrum analyzer using a KBr disk in the range of 4000–400 cm−1. Ultraviolet-visible-near infra-red spectrophotometry (UV-vis-NIR) was tested on a PerkinElmer Lambda 950 instrument. The hydrodynamic particle size distribution and ζ-potential of the synthesized nanoparticles were tested on a Malvern Zetasizer Nano ZS90 nanoparticle size and zeta potential analyzer. Specific surface area and pore size of the nanoparticles were studied by N2 adsorption–desorption analysis and Brunauer–Emmett–Teller analysis.
2.8. Detection of O2
Prior to use, the portable dissolved oxygen meter (WLDO-300, Shanghai Seawater Instrumentation Technology Co., Ltd) was calibrated using air-saturated water and zero-oxygen solution in a two-point calibration procedure to ensure measurement accuracy. Calibration was performed before each experiment. To assess oxygen production, HMnO2–VC@mPEG–Ce6 solutions at different concentrations (100, 200, and 500 μg mL−1) were mixed with H2O2 solution ([H2O2] = 100 μM, pH 7.4). The dissolved oxygen concentration was measured at time points of 0, 5, 10, 15, 20, and 30 minutes using the portable dissolved oxygen meter. Similarly, the oxygen production under different pH conditions (6.5 and 7.4) was evaluated by mixing HMnO2–VC@mPEG–Ce6 solution (200 μg mL−1) with H2O2 solution ([H2O2] = 100 μM) at the specified pH values.
2.9. Detection of ˙OH
Methylene blue (MB) was used as an assay reagent to verify the ˙OH production capacity of HMnO2–VC@mPEG–Ce6. MB solution (10 mg L−1) containing 0.5 mM MnCl2 was mixed with different concentrations of H2O2 (1.25, 2.5, 5, and 10 mM) in NaHCO3/CO2 buffer and incubated for 30 minutes, and the change in absorbance of MB at 665 nm was observed to verify the production of ˙OH. In addition, the effect of GSH on ˙OH generation was verified by mixing an MB solution (10 mg L−1) containing H2O2 and 0.5 mM MnCl2 with a mixture of different concentrations of GSH (0, 2, 4, 6, 8, and 10 mM) in NaHCO3/CO2 buffer and by incubating for 30 minutes to measure the change in absorbance of MB at 665 nm.
2.10. Detection of 1O2
Singlet oxygen sensor green (SOSG) is a common 1O2 detection kit that is highly selective for 1O2 but not cell membrane permeable, and the change in the fluorescence density of SOSG endoperoxide (SOSG-EP) produced by the reaction of SOSG with 1O2 reflects the amount of 1O2 produced. The solutions of different experimental groups—(1) mPEG–Ce6; (2) mPEG–Ce6 + HMnO2; and (3) mPEG–Ce6 + HMnO2 + H2O2—were mixed with SOSG (2.5 μM) and exposed to low-frequency ultrasound (US) irradiation (1 W cm−2, 10 minutes) using an ultrasonic therapeutic instrument (WED-100, China). At time points of 0, 1, 3, 6, and 10 minutes, 100 μL aliquots were sampled and transferred to a 96-well plate. The fluorescence intensity of SOSG was measured using a fluorescence spectrophotometer (PerkinElmer Lambda 950, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
2.11. GSH consumption
5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) was used to quantify GSH. Briefly, mixtures of DTNB (400 μM) was mixed with different concentrations of GSH (0.2, 0.4, 0.6, 0.8, 1, and 1.2 mM) in PBS buffer (pH 7.4) with a reaction volume of 1 mL. The mixtures were incubated in a constant-temperature water bath at 37 °C for 10 minutes, and the absorbance at 412 nm was measured using a spectrophotometer. A calibration curve was constructed using linear fitting. Different concentrations of HMVC (0, 50, 100, 200, 300, 400, and 500 μg mL−1) were mixed with GSH (1 mM) in PBS buffer (pH 7.4) with a reaction volume of 1 mL. The mixtures were incubated in a constant-temperature water bath at 37 °C for 30 minutes, followed by the addition of DTNB (500 μM). The UV-vis-NIR absorption spectra were recorded. Similarly, different concentrations of GSH (0, 0.25, 0.5, 1, and 1.2 mM) were mixed with HMnO2–VC@mPEG–Ce6 (100 μg mL−1) in PBS buffer (pH 7.4) with a reaction volume of 1 mL. The mixtures were incubated in a constant-temperature water bath at 37 °C for 30 minutes, followed by the addition of DTNB (500 μM). The UV-vis-NIR absorption spectra were recorded using a spectrophotometer.
2.12. Cellular uptake of HMnO2–VC@mPEG–Ce6
The cellular uptake of HMnO2–VC@mPEG–Ce6 nanoparticles was observed using an inverted fluorescence microscope (Zeiss Axio Observer Z1, USA). HMnO2–VC@mPEG–Ce6 was incubated with the fluorophore FITC overnight. Subsequently, the used medium was replaced with fresh medium containing HMnO2–VC@mPEG–Ce6/FITC. The uptake of HMnO2–VC@mPEG–Ce6/FITC nanoparticles by cells was assessed by observing the FITC (green fluorescence) and DAPI (blue fluorescence) signals using the inverted fluorescence microscope.
2.13. Live/dead cell staining assay
To analyze the in vitro antitumor efficacy of HMnO2–VC@mPEG–Ce6 in combination with SDT/CDT, the survival and death of PC-3 cells were detected using a CAM/PI double-staining kit. PC-3 cells were seeded at a density of 1.5 × 105 cells per well in a 6-well plate containing 1 mL of culture medium, when the cells reached approximately 70% confluence, the used medium was removed and replaced with fresh culture medium. The cells were then divided into the following experimental groups according to the planned design: (1) control group; (2) HMnO2; (3) mPEG–Ce6 + US; (4) HMnO2–VC@mPEG–Ce6; (5) HMnO2–VC@mPEG–Ce6 + US. The cells were incubated in a 37 °C incubator for 4 hours, after which the groups requiring ultrasound irradiation were exposed to ultrasound at an intensity of 1 W cm−2 for 10 minutes. Finally, the cells were stained with the CAM/PI double-staining kit for 15 minutes, and images were observed and captured using an inverted fluorescence microscope.
2.14. Quantitative determination of cytotoxicity
PC-3 cells were seeded at a density of 5000 cells per well in a 96-well plate containing 100 μL of culture medium. When the cells reached approximately 70% confluence, the used medium was removed and replaced with fresh culture medium. Then, the cells were treated according to the grouping described above. The cells were incubated in a 37 °C incubator for 4 hours, after which the groups requiring ultrasound irradiation were exposed to ultrasound at an intensity of 1 W cm−2 for 10 minutes. Cell viability was measured using a CCK-8 assay kit, and the cell survival rate was calculated using the formula: cell survival rate (%) = (OD of the treatment group/OD of the control group) × 100%.
2.15. Cellular ROS generation assessment
PC-3 cells were seeded at a density of 1.5 × 105 cells per well in a 6-well plate containing 1 mL of culture medium, when the cells reached approximately 70% confluence, the used medium was removed and replaced with fresh culture medium. Then, the cells were treated according to the grouping described above, stained with 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) probe, and images were taken using an inverted fluorescence microscope.
2.16. Cellular LPO generation assessment
Lipid peroxides (LPO) were assessed using C11-BODIPY 581/591, an oxidation-sensitive LPO-specific fluorescent probe that accumulates in the cell membrane. PC-3 cells were seeded at a density of 1.5 × 105 cells per well in a 6-well plate containing 1 mL of culture medium, when the cells reached approximately 70% confluence, the used medium was removed and replaced with fresh culture medium. Then, the cells were treated according to the grouping described above. The treated cells were incubated with the C11-BODIPY 581/591 fluorescent probe in a 37 °C incubator for 30 minutes. Images were observed and captured using an inverted fluorescence microscope.
2.17. In vivo biodistribution of HMnO2–VC@mPEG–Ce6
HMnO2–VC@mPEG–Ce6 (5 mg kg−1, labeled with cyanine-5.5) was intravenously injected into tumor-bearing BALB/c mice via the tail vein. At 0.5, 4, 12, and 24 hours post-injection, the mice were anesthetized (by intraperitoneal injection of 1.5% pentobarbital sodium solution, 50 mg kg−1) and placed on the detection platform of a small animal in vivo imaging system. The imaging parameters were adjusted, and fluorescence signal distribution images within the mice were captured. At the 24 hour time point, the mice were euthanized (using carbon dioxide inhalation), and the heart, liver, spleen, lungs, kidneys, and tumor tissues were rapidly excised. The isolated tissues were placed in the small animal in vivo imaging system to capture fluorescence signal images.
2.18. In vivo antitumor therapeutic efficacy
PC-3 tumor-bearing mice with tumor volumes of approximately 100 mm3 were randomly divided into five groups (n ≥ 5/group): (1) control group; (2) HMnO2; (3) mPEG–Ce6 + US; (4) HMnO2–VC@mPEG–Ce6; (5) HMnO2–VC@mPEG–Ce6 + US. Different formulations were administered via tail vein injection (5 mg kg−1). For the groups receiving ultrasound, the tumors were irradiated with US (1 W cm−2, 10 minutes) 12 hours after administration. The treatment was performed three times. Body weight and tumor size were recorded every other day after treatment. Tumor volume was calculated using the following formula: tumor volume = (width2 × length)/2. All mice were euthanized and photographed 21 days post-injection, and the tumors were excised and photographed. The collected tumors were fixed in 4% paraformaldehyde solution for 24 hours. After fixation, the tissues were dehydrated, embedded in paraffin, and sectioned (5 μm thickness). Histological analyses, including ROS detection, H&E staining, and immunohistochemical staining for HIF-1α, PCNA, and GPX4, were performed on the tissue sections.
2.19. Data analysis
All quantitative data were analyzed using SPSS 25.0 software. All data were expressed as mean ± standard deviation (SD). Differences among groups were analyzed using one-way ANOVA. Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
3. Results and discussion
3.1. Characterization of HMnO2–VC@mPEG–Ce6
The HMnO2–VC@mPEG–Ce6 nanoparticles were observed using SEM and TEM, and Fig. 2A–C clearly show the spherical morphology, porous structure, and well-dispersed nature. HAADF-STEM further confirmed the hollow structure of the nanoparticle shells and visualized the distribution of the major elements (Fig. 2D). Analyzed using the BET method, the specific surface area of HMnO2 was 129.43 m2 g−1. An obvious hysteresis loop caused by capillary condensation could be observed in the desorption isotherm, indicating the presence of mesopores in the nanoparticles (Fig. S1†). By analyzing the desorption branching curve using the BJH method, the pore size was found to be concentrated around 3.9 nm (Fig. S2†). The HMnO2 nanoparticles exhibited a large specific surface area and mesopore size, suggesting their potential for effective drug loading. FTIR analysis was performed on HMnO2, VC, Ce6, and HMnO2–VC@mPEG–Ce6. The infrared spectra of HMnO2 changed after compounding with VC and Ce6. The relative intensities of the absorption peaks in the bands of 1711, 1434, and 1208 cm−1 increased, which mainly originated from the structures of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and C–O structures in Ce6 and VC. Notably, the typical characteristic peaks of the monosubstitution of the pyridinium ring in Ce6 appeared at 725 cm−1, confirming the successful loading of Ce6 into the HMnO2 matrix. Additionally, the significant increase in the relative intensity of the C–OH absorption peak at 1020 cm−1 and the change in the peak shape at 3441 cm−1 were attributed to the introduction of a large number of hydroxyl groups onto the surface of HMnO2, indicating that VC was successfully loaded on the surface of HMnO2 and affected intermolecular hydrogen bonding. These results confirm the successful loading of VC and Ce6 onto the surface of HMnO2, demonstrating the successful synthesis of HMnO2–VC@mPEG–Ce6 (Fig. 2E). Furthermore, the UV-vis-NIR absorption spectra were shown in Fig. 2F. In the spectrum of HMnO2–VC@mPEG–Ce6, the characteristic absorption peaks of mPEG–Ce6 were observed at 401 and 656 nm, confirming its successful coupling. As shown in Fig. S3,† the regression equation of the mPEG–Ce6 concentration–absorbance standard curve is y = 0.0089x + 0.0034, R2 = 0.9866. The linear relationship between mPEG–Ce6 concentration and absorbance is strong within the concentration range of 0–200 μg mL−1, allowing for quantitative analysis. The entrapment efficiency was calculated as follows: entrapment efficiency = (total Ce6 − free Ce6)/total Ce6 × 100% = 15.17%. Additionally, the zeta potential of HMnO2–VC@mPEG–Ce6 was −21.9 ± 0.68 mV (Fig. 2G), and the average hydrodynamic diameter of HMnO2–VC@mPEG–Ce6 was approximately 189.3 nm (Fig. 2H).
O, CC, and C–O structures in Ce6 and VC. Notably, the typical characteristic peaks of the monosubstitution of the pyridinium ring in Ce6 appeared at 725 cm−1, confirming the successful loading of Ce6 into the HMnO2 matrix. Additionally, the significant increase in the relative intensity of the C–OH absorption peak at 1020 cm−1 and the change in the peak shape at 3441 cm−1 were attributed to the introduction of a large number of hydroxyl groups onto the surface of HMnO2, indicating that VC was successfully loaded on the surface of HMnO2 and affected intermolecular hydrogen bonding. These results confirm the successful loading of VC and Ce6 onto the surface of HMnO2, demonstrating the successful synthesis of HMnO2–VC@mPEG–Ce6 (Fig. 2E). Furthermore, the UV-vis-NIR absorption spectra were shown in Fig. 2F. In the spectrum of HMnO2–VC@mPEG–Ce6, the characteristic absorption peaks of mPEG–Ce6 were observed at 401 and 656 nm, confirming its successful coupling. As shown in Fig. S3,† the regression equation of the mPEG–Ce6 concentration–absorbance standard curve is y = 0.0089x + 0.0034, R2 = 0.9866. The linear relationship between mPEG–Ce6 concentration and absorbance is strong within the concentration range of 0–200 μg mL−1, allowing for quantitative analysis. The entrapment efficiency was calculated as follows: entrapment efficiency = (total Ce6 − free Ce6)/total Ce6 × 100% = 15.17%. Additionally, the zeta potential of HMnO2–VC@mPEG–Ce6 was −21.9 ± 0.68 mV (Fig. 2G), and the average hydrodynamic diameter of HMnO2–VC@mPEG–Ce6 was approximately 189.3 nm (Fig. 2H).
 |
| | Fig. 2 Schematic of the characterization of HMnO2–VC@mPEG–Ce6 nanoparticles. (A) SEM image. (B and C) TEM images. (D) HAADF-STEM image and elemental mapping. (E) Fourier transform infrared spectra of the HMnO2, VC, Ce6, and HMnO2–VC@mPEG–Ce6 nanoparticles. (F) UV-vis-NIR absorption spectra of HMnO2, mPEG–Ce6, HMnO2–VC, and HMnO2–VC@mPEG–Ce6. (G) Zeta potential of the HMnO2, HMnO2–VC, and HMnO2–VC@mPEG–Ce6. (H) Particle size distribution map. | |
3.2. O2 production of HMnO2–VC@mPEG–Ce6
We evaluated the O2-generating capacity of HMnO2–VC@mPEG–Ce6 using a dissolved oxygen meter to assess its hypoxia-alleviating potential. Upon H2O2 addition, HMVC demonstrated concentration-dependent catalase-like activity, with dissolved O2 levels rising proportionally to HMVC concentration (Fig. 3A). This confirms its ability to catalyze H2O2 decomposition into water-soluble O2. Notably, at a fixed concentration (200 μg mL−1), HMVC exhibited pH-responsive catalytic behavior, showing enhanced O2 generation efficiency under acidic conditions (Fig. 3B). In the acidic and hypoxic environment within the tumor, the addition of HMnO2–VC@mPEG–Ce6 significantly increased the O2 yield. Compared with similar MnO2-based nanomaterials, HMnO2–VC@mPEG–Ce6 has an approximately 2-fold increase in O2 yield in the acidic tumor microenvironment.36 This advantage stems from the synergistic catalytic effect of its mesoporous structure and VC loading on H2O2 decomposition.
 |
| | Fig. 3 Performance testing of HMnO2–VC@mPEG–Ce6 nanoparticles. (A) The curves show the O2 production of HMnO2–VC@mPEG–Ce6 solutions at different concentrations (100, 200 and 500 μg mL−1). (B) O2 production of HMnO2–VC@mPEG–Ce6 solution (200 μg mL−1) at pH 6.5 and 7.4. (C) UV-vis-NIR absorption spectra of MB after degradation by Mn2+-mediated Fenton-like reaction in the presence of HCO3−. (D) MB degradation with the addition of different concentrations of H2O2. (E) MB degradation by Mn2+-mediated Fenton-like reaction in the presence or absence of GSH. (F) MB degradation by H2O2 plus different concentrations of GSH. (G) Typical ESR signals of DMPO incubated with different groups in 25 mM NaHCO3/CO2 buffer. (H) Typical ESR signals of TEMP incubated with different groups. (I) Detection of GSH (1 mM) consumption by different concentrations of HMnO2–VC@mPEG–Ce6 (0, 50, 100, 200, 300, 400 and 500 μg mL−1) after 30 min of reaction. | |
3.3. In vitro CDT effects
To systematically evaluate the ˙OH generation in Mn2+-mediated Fenton-like reaction within physiological bicarbonate buffer (HCO3−/CO2), we employed MB degradation assays and electron spin resonance (ESR) spectroscopy. Significant MB absorbance reduction (Fig. 3C) confirmed efficient ˙OH production via Mn2+/H2O2 interactions in NaHCO3/CO2 buffer, with ˙OH yields showing H2O2 concentration dependence (Fig. 3D). Strikingly, the introduction of 10 mM glutathione (GSH) caused a five-fold increase in residual MB levels (Fig. 3E and F), demonstrating GSH's dual role as both a ˙OH scavenger and CDT efficacy suppressor through redox equilibrium modulation. ESR spectra with characteristic 1:2:2:1 quartet signals provided direct evidence of ˙OH generation (Fig. 3G). Notably, signal attenuation in the Mn2+ + H2O2 + GSH group versus the Mn2+ + H2O2 control visually corroborated GSH's radical quenching capacity.
3.4. In vitro SDT effects
The integration of mPEG–Ce6 endowed HMnO2–VC@mPEG–Ce6 with US-responsive sonodynamic capability. Using SOSG fluorescence probes and ESR with TEMP trapping, we systematically evaluated 1O2 generation under US irradiation. Both assays revealed time-dependent 1O2 production across all test groups, with the H2O2-supplemented group showing the highest yield (Fig. S4–S6†). ESR spectra exhibited characteristic 1:1:1 triplet signals, confirming US-triggered 1O2 generation (Fig. 3H). Notably, H2O2 co-treatment amplified these signals, suggesting peroxide-enhanced sonodynamic activity. The H2O2-dependent 1O2 enhancement implies a self-reinforcing therapeutic mechanism. Tumor-associated H2O2 not only provides oxygen sources via HMVC's catalase-like activity but also amplifies SDT efficacy, creating a feedback loop for targeted ROS storm generation.
3.5. GSH consumption
We quantitatively characterized the GSH-depleting ability of HMnO2–VC@mPEG–Ce6 through DTNB, which has a typical absorption peak at 325 nm (Fig. S7†) and a new absorption peak at 412 nm after reacting with GSH to form TNB (Fig. S8†). As shown in Fig. 3I, the intensity of the absorption peak at 412 nm decreased with increasing HMVC concentration. In addition, in the degradation experiments of HMVC with the addition of various concentrations of various concentrations of GSH (0, 0.25, 0.5, 1, 1.2 mM), the absorbance at 412 nm increased with increasing GSH concentration (Fig. S9†). Since the concentration of GSH in cancer cells (1–10 mM) is at least four times higher than that in normal cells,37–39 HMnO2 can be completely degraded by GSH in cancer cells. These results suggest that HMVC is able to act as a GSH-depleting agent that regulates intracellular redox homeostasis and provides large amounts of Mn2+ to CDT. Compared to Fe3+ or Cu2+-based GSH depleting agents,40,41 HMnO2–VC@mPEG–Ce6 rapidly degrades and depletes GSH at GSH concentrations (1–10 mM) in tumor cells through Mn2+-mediated redox reactions, while releasing Mn2+-enhancing CDT, resulting in a “self-reinforcing” effect.
3.6. Cellular uptake of HMnO2–VC@mPEG–Ce6
The prerequisite for achieving a perfect therapeutic effect is that the nanoparticles can be effectively internalized by the tumor cells. As shown in Fig. 4A, green fluorescence was observed around the cells, and the fluorescence intensity was significantly enhanced with increasing incubation time, indicating the time-dependent internalization of HMVC nanoparticles by PC-3 cells.
 |
| | Fig. 4 Cellular uptake and in vitro therapeutic efficacy of HMnO2–VC@mPEG–Ce6 nanoparticles. (A) Images of PC-3 cells incubated with FITC-labeled HMnO2–VC@mPEG–Ce6 for 0, 1, 2, and 4 h. Scale bar = 20 μm. (B) The relative viability of PC-3 cells after different treatments was quantitatively analyzed using CCK-8. (C) Fluorescence images stained with calcein-AM (green) and PI (red) after different treatments. Scale bar = 200 μm. (D) Fluorescence images of PC-3 cells stained with intracellular ROS probe DCFH-DA after different treatments. Scale bar = 200 μm. (E) Fluorescence images of PC-3 cells stained with C11-BODIPY 581/591 after different treatments. Scale bar = 200 μm. | |
3.7. In vitro cytotoxicity assay
The combinatorial therapeutic effect of HMVC was systematically evaluated using CAM/PI double-staining kit and CCK-8 assays in PC-3 prostate cancer cells. Fluorescence quantification revealed maximal red fluorescence intensity in the HMVC + US group (Fig. 4C), demonstrating superior tumor eradication capacity. Correspondingly, CCK-8 assays showed 76.4% cell death under HMVC + US treatment – a 1.2-fold enhancement over SDT therapy and 2.2-fold improvement versus CDT alone (Fig. 4B). This suggests that HMVC + US treatment has the strongest cell-killing effect on PC-3 cells. Different from the nanosystems of CDT or SDT alone,42,43 HMnO2–VC@mPEG–Ce6 alleviates hypoxia, ROS outburst, and GSH depletion through O2, resulting in cell mortality of 76.4% in vitro, which is significantly higher than similar studies.
3.8. Intracellular ROS and LPO level
Intracellular ROS production and LPO accumulation were respectively monitored using DCFH-DA and C11-BODIPY 581/591 probes. As shown in Fig. 4D, PC-3 cells treated with HMnO2, mPEG–Ce6 + US, and HMVC groups all showed some degree of green fluorescence due to the SDT or CDT effects. Notably, the HMVC + US group showed the strongest green fluorescence, which was attributed to the synergistic effect of SDT/CDT. LPO analysis demonstrated parallel enhancement, with the HMVC + US group exhibiting maximal green fluorescence ratio (Fig. 4E), confirming US-potentiated ferroptosis initiation. These results confirm that HMVC + US treatment efficiently generated large amounts of ROS and promoted LPO accumulation.
3.9. In vivo anticancer studies
Building upon the demonstrated tumor-targeting capability and synergistic in vitro efficacy, we evaluated the combined SDT/CDT performance in PC-3 tumor-bearing mice (tumor volume ≈ 100 mm3). Animals were randomized into five treatment groups (n ≥ 5/group): (a) PBS (control); (b) HMnO2; (c) mPEG–Ce6 + US; (d) HMVC; (e) HMVC + US. Ultrasound irradiation (1 W cm−2, 10 minutes) was applied 24 h post-injection to groups of mPEG–Ce6 + US and HMVC + US, ensuring optimal nanoparticle accumulation at tumor sites (Fig. 5A).
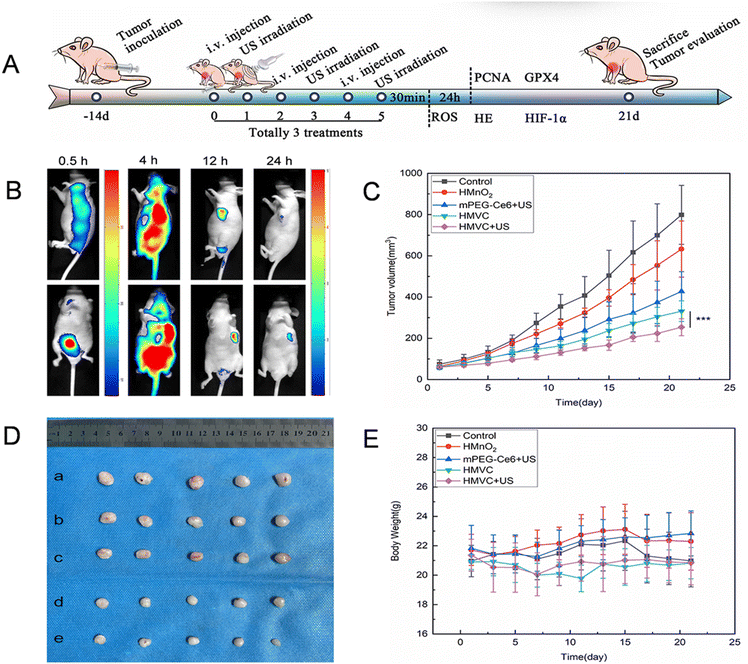 |
| | Fig. 5 In vivo therapeutic efficacy of HMnO2–VC@mPEG–Ce6 nanoparticles. (A) Overview of the in vivo treatment process. (B) Fluorescence images of mice 24 hours after injection of Cy5.5-HMnO2–VC@mPEG–Ce6 via tail vein. (C) Curves show the changes in tumor volume of mice in each group after different treatments. (D) Appearance of tumors isolated from mice: (a) the control, (b) HMnO2, (c) mPEG–Ce6 + US, (d) HMVC, and (e) HMVC + US. (E) Curves show the changes in the body weight of the mice in each group after different treatments. | |
3.10. Biodistribution of HMnO2–VC@mPEG–Ce6
To evaluate the in vivo biodistribution and tumor accumulation behavior of HMnO2–VC@mPEG–Ce6, we injected HMVC labeled with Cy5.5 into PC-3 tumor-bearing mice via the tail vein and observed their biodistribution. As shown by in vivo fluorescence imaging, the fluorescence intensity of the tumor gradually increased over time, peaked at the tumor site at 12 h postinjection, and then decreased because of the in vivo metabolism (Fig. 5B) (a separate scale was used because of the high fluorescence intensity at 0.5 hours). This suggests that the distribution of HMnO2–VC@mPEG–Ce6 is time-dependent, which may be due to the enhanced permeability and retention (EPR) effect of the tumor. We also excised tumors and major organs for in vitro imaging 24 h after injection. We found that tumors in mice showed the strongest Cy5.5 fluorescence signals, whereas resected organs no longer showed fluorescence, suggesting that HMnO2–VC@mPEG–Ce6 nanoparticles were still heavily enriched at the tumor site 24 hours after injection (Fig. S10†).
3.11. In vivo anticancer efficacy of HMnO2–VC@mPEG–Ce6
Mouse body weights and tumor volumes were monitored for 21 days after the administration of the different treatments. As shown in Fig. 5C, tumor growth was rapid in the control group and somewhat inhibited in the mPEG–Ce6 + US and HMVC treatment groups, with the best tumor suppression in the HMVC + US treatment group. As shown in Fig. 5D and S11,† tumor images from different treatment groups on day 21 further confirmed that HMVC + US treatment resulted in greater tumor growth inhibition. The favorable efficacy of the above treatments may be attributed to the SDT/CDT synergistic effect. At the end of the experiment, the body weights of the mice were relatively constant and showed no significant changes, and no statistical differences were observed between groups (Fig. 5E). This indicates that HMnO2–VC@mPEG–Ce6 is biologically safe and has no significant toxic effects on animals.
3.12. Tissue analysis
To further evaluate the effects of different treatments on tumor tissues, dissected tumor tissues from each group were stained with antibodies against hematoxylin eosin (H&E), proliferating cell nuclear antigen (PCNA), GPX4, and ROS. The hypoxia-relieving capacity of HMnO2–VC@mPEG–Ce6 was then explored by HIF-1α staining of the tumor site. Compared with the control group, HIF-1α expression was significantly decreased in the HMVC + US group, suggesting that HMnO2–VC@mPEG–Ce6 can effectively alleviate tumor hypoxia (Fig. 6A). Dihydroethidium (DHE) staining was used to detect intracellular ROS, which produce red fluorescence in the presence of ROS. The ROS staining showed clear red fluorescence in the HMVC + US group, which suggested that it can synergize with SDT/CDT to produce ROS to kill tumor cells (Fig. 6B). In addition, H&E and PCNA staining also demonstrated the effects of the above treatments (Fig. 6C and D). GPX4 staining was used to further detect the expression of the GPX4 protein in tumor tissues after different treatments. We found that the expression of the GPX4 protein was downregulated in the HMVC + US group compared with the control group (Fig. 6E). Finally, major organs were harvested from each group of mice to evaluate the biosafety of HMnO2–VC@mPEG–Ce6. No obvious inflammatory reaction was observed in the H&E sections of each major organ. And no abnormalities were detected in routine examinations and blood biochemical analyses in all groups of mice (Fig. S12 and S13†). Compared with some metal-based nanomaterials, the Mn2+ metabolites of HMnO2 can be excreted through the kidney, and no significant hepatorenal toxicity has been observed, which has better clinical translation potential.
 |
| | Fig. 6 Representative images of stained tumor tissue sections of nude mice after treatment in each experimental group. Each set of images was stained for HIF-1α (A), ROS (B), H&E (C), PCNA (D) and GPX4 (E). Scale bar = 50 μm. | |
4. Conclusion
We present a biodegradable HMnO2-based nanoplatform (HMnO2–VC@mPEG–Ce6) that synergizes SDT/CDT with ferroptosis induction, overcoming key limitations of conventional apoptosis-focused therapies. HMnO2–VC@mPEG–Ce6 not only synergized with SDT and CDT effects to generate sufficient ROS, thereby inducing the accumulation of LPO in tumor cells, but also achieves GSH depletion via the HMnO2–Mn2+ transition, leading to GPX4 inhibition to induce ferroptosis in cancer cells. Our HMnO2-based therapeutic nanoplatforms hold great promise for clinical translation because of inherent biodegradability. Future work will focus on active targeting ligand conjugation to further enhance tumor specificity.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Qi Shen: conceptualization, formal analysis, investigation, methodology, writing-original draft. Xi Zhu: conceptualization, methodology, investigation, data curation. Mengping Huo: conceptualization, methodology, supervision, resources. Yafei Lin: conceptualization, resources. Wenting Zhang: conceptualization, resources. Ming Yang: conceptualization, resources, supervision, writing-review and editing, funding acquisition, project administration. Yang Zhang: conceptualization, resources, supervision, writing-review and editing, funding acquisition, project administration. Long Zhang: conceptualization, resources, supervision, writing-review and editing, funding acquisition, project administration. Yonghao Gai: conceptualization, resources, supervision, writing-review and editing, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82102040), the Natural Science Foundation of Shandong Province (ZR2021QH228 and ZR2022MH251), the Taishan Scholars Program of Shandong Province (tsqnz20221171), Scientific Research Project of Weifang Health Commission (WFWSJK-2023-352).
Notes and references
- S. Fan, Q. Yang, Q. Song, M. Hong, X. Liu, H. Chen, J. Wang, C. Li and S. Cheng, Chem. Commun., 2022, 58, 6486–6489 CAS.
- W. S. Yang, R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell, Cell, 2014, 156, 317–331 CAS.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 Search PubMed.
- A. Knickle, W. Fernando, A. L. Greenshields, H. P. V. Rupasinghe and D. W. Hoskin, Food Chem. Toxicol., 2018, 118, 154–167 CAS.
- B. Wang, Y. Wang, J. Zhang, C. Hu, J. Jiang, Y. Li and Z. Peng, Arch. Toxicol., 2023, 97, 1439–1451 CAS.
- C. Qian, P. Feng, J. Yu, Y. Chen, Q. Hu, W. Sun, X. Xiao, X. Hu, A. Bellotti, Q.-D. Shen and Z. Gu, Angew Chem. Int. Ed. Engl., 2017, 56, 2588–2593 CAS.
- R. Stefanatos and A. Sanz, FEBS Lett., 2018, 592, 743–758 CAS.
- Y. Chen, X. Luo, Z. Zou and Y. Liang, Curr. Drug Targets, 2020, 21, 477–498 CrossRef CAS PubMed.
- H. Shibaguchi, H. Tsuru, M. Kuroki and M. Kuroki, Anticancer Res., 2011, 31, 2425–2429 Search PubMed.
- K. Wei, Y. Wu, X. Zheng, L. Ouyang, G. Ma, C. Ji and M. Yin, Angew Chem. Int. Ed. Engl., 2024, 63, e202404395 CrossRef CAS.
- H. Gao, Z. Cao, H. Liu, L. Chen, Y. Bai, Q. Wu, X. Yu, W. Wei and M. Wang, Theranostics, 2023, 13, 1974–2014 CAS.
- C. Jia, Y. Guo and F.-G. Wu, Small, 2022, 18, e2103868 CrossRef.
- I. A. Khawar, J. H. Kim and H.-J. Kuh, J. Controlled Release, 2015, 201, 78–89 CrossRef CAS PubMed.
- Y. Xiao and D. Yu, Pharmacol. Ther., 2021, 221, 107753 CrossRef CAS PubMed.
- J. E. Bader, K. Voss and J. C. Rathmell, Mol. Cell, 2020, 78, 1019–1033 CAS.
- S. Lu, W. Feng, C. Dong, X. Song, X. Gao, J. Guo, Y. Chen and Z. Hu, Adv. Healthcare Mater., 2022, 11, e2102135 Search PubMed.
- J. Li, Z. Yue, M. Tang, W. Wang, Y. Sun, T. Sun and C. Chen, Adv. Healthcare Mater., 2024, 13, e2302028 Search PubMed.
- Y. Liu, Y. Wan, Y. Jiang, L. Zhang and W. Cheng, Biochim. Biophys. Acta, Rev. Cancer, 2023, 1878, 188890 CAS.
- B. Niu, K. Liao, Y. Zhou, T. Wen, G. Quan, X. Pan and C. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed.
- A. C. González-Garzón, J. P. Ramón-Ugalde, D. A. Ambríz-García, J. R. Vazquez-Avendaño, J. E. Hernández-Pichardo, J. L. Rodríguez-Suastegui, C. Cortez-Romero and M. D. C. Navarro-Maldonado, Animals, 2023, 13, 3602 Search PubMed.
- H. Wu, X. Gao, Y. Luo, J. Yu, G. Long, Z. Jiang and J. Zhou, Adv. Sci., 2022, 9, e2203894 CrossRef.
- G. Yang, J. Ji and Z. Liu, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2021, 13, e1720 CAS.
- Y. Chen, H. Cong, Y. Shen and B. Yu, Nanotechnology, 2020, 31, 202001 CrossRef CAS.
- J. Wang, S. Zhao, J. Chen, X. Liu, H. Chen, T. Lu, M. Xu, X. Guo, X. Shen, C. Liu and C. Li, ACS Appl. Mater. Interfaces, 2023, 15, 21904–21916 CrossRef CAS.
- Y. Liu, Y. Huang, P. Lu, Y. Ma, L. Xiong, X. Zhang, Z. Yin, H. Xu, Y. Nie, J. Luo, Z. Xiong and X. Liang, ACS Appl. Mater. Interfaces, 2023, 15, 54207–54220 CrossRef CAS.
- Z. Chen, Z. Liu, Q. Zhang, S. Huang, Z. Zhang, X. Feng, L. Zeng, D. Lin, L. Wang and H. Song, Front. Pharmacol, 2023, 14, 1133011 CrossRef CAS PubMed.
- L.-S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H.-H. Yang and X. Chen, Angew Chem. Int. Ed. Engl., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- J. Ou, H. Tian, J. Wu, J. Gao, J. Jiang, K. Liu, S. Wang, F. Wang, F. Tong, Y. Ye, L. Liu, B. Chen, X. Ma, X. Chen, F. Peng and Y. Tu, ACS Appl. Mater. Interfaces, 2021, 13, 38050–38060 CrossRef CAS PubMed.
- S. Cheng, Y. Shi, C. Su, Y. Li and X. Zhang, Biosens. Bioelectron., 2022, 214, 114550 CAS.
- B. Zhou, C. Yin, Q. Feng, Y. Wu, X. Pan, C. Liu, J. Tian, S. Geng, K. Wang, J. Xing, Y. Cao, P. Shou, Z. Yu and A. Wu, Nanoscale, 2021, 13, 19085–19097 CAS.
- L. Feng, R. Xie, C. Wang, S. Gai, F. He, D. Yang, P. Yang and J. Lin, ACS Nano, 2018, 12, 11000–11012 CAS.
- Y. Liu, W. Zhen, Y. Wang, J. Liu, L. Jin, T. Zhang, S. Zhang, Y. Zhao, S. Song, C. Li, J. Zhu, Y. Yang and H. Zhang, Angew Chem. Int. Ed. Engl., 2019, 58, 2407–2412 CAS.
- L. Liang, M. Jia, M. Zhao, Y. Deng, J. Tang, X. He, Y. Liu, K. Yan, X. Yu, H. Yang, C. Li, Y. Li and T. Li, Int. J. Nanomed., 2024, 19, 8883–8900 Search PubMed.
- Y. Zhang, Y. Cao, T. Gao, Y. Kuang, Z. An, Z. Mao, Y. He, J. Yan, Z. Lu and R. Pei, ACS Appl. Bio Mater., 2020, 3, 3880–3893 CAS.
- L. Deng, M. Liu, D. Sheng, Y. Luo, D. Wang, X. Yu, Z. Wang, H. Ran and P. Li, Biomaterials, 2021, 271, 120710 CAS.
- T. Lin, X. Zhao, S. Zhao, H. Yu, W. Cao, W. Chen, H. Wei and H. Guo, Theranostics, 2018, 8, 990–1004 CAS.
- C. Hwang, A. J. Sinskey and H. F. Lodish, Science, 1992, 257, 1496–1502 CAS.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671–691 Search PubMed.
- N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 2013, 972913 Search PubMed.
- J. Bonet-Aleta, M. Sancho-Albero, J. Calzada-Funes, S. Irusta, P. Martin-Duque, J. L. Hueso and J. Santamaria, J. Colloid Interface Sci., 2022, 617, 704–717 CAS.
- J. Bai, X. Zhang, Z. Zhao, S. Sun, W. Cheng, H. Yu, X. Chang and B. Wang, Small, 2024, 20, e2400326 Search PubMed.
- Q. Chen, N. Li, X. Wang, Y. Yang, Y. Xiang, X. Long, J. Zhang, J. Huang, L. Chen and Q. Huang, Front. Pharmacol, 2022, 13, 847048 CAS.
- F. He, W. Li, B. Liu, Y. Zhong, Q. Jin and X. Qin, ACS Biomater. Sci. Eng., 2024, 10, 298–312 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a





