
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a scalable and sustainable continuous-flow microreaction process for mononitration of aromatic compounds with high selectivity and yield†
Dong Xing a,
Xiangui Leia,
Yufeng Fua,
Zhijie Xua,
Dengyun Luoa,
Jiujun Chenb,
Yong Xiangb,
Zhouyu Wang*a and
Qiao Song*a
a,
Xiangui Leia,
Yufeng Fua,
Zhijie Xua,
Dengyun Luoa,
Jiujun Chenb,
Yong Xiangb,
Zhouyu Wang*a and
Qiao Song*a
aSichuan Engineering Research Center for Molecular Targeted Diagnostic & Therapeutic Drugs, Asymmetric Synthesis and Chiral Technology Key Laboratory of Sichuan Province, Research and Application of Small Organic Chiral Molecules Key Laboratory of Yibin City, Department of Chemistry, Xihua University, Chengdu 610039, China. E-mail: songqiao@xhu.edu.cn; zhouyuwang@mail.xhu.edu.cn
bSichuan North Hongguang Special Chemical Co., Ltd, Yinguang Group, Yibin 644000, China
First published on 4th February 2025
Abstract
Nitroaromatic compounds are extensively used in industries such as pharmaceuticals, pesticides, and dyes. However, traditional synthesis methods often face challenges, including high safety risks, significant environmental pollution, and poor selectivity in mononitration reactions. In this study, we developed an efficient and safe continuous-flow microreaction process for mononitration, which achieves high yield and excellent selectivity. This process is applicable for the continuous synthesis of various mononitro compounds, including nitro-p-xylene, nitro-o-xylene, nitro-chlorobenzene, and nitro-toluene. Furthermore, the process was successfully applied to the synthesis of a key intermediate in the anticancer drug erlotinib, achieving a yield of 99.3%. The process has also been scaled up for the continuous production of nitro-p-xylene and nitro-o-xylene, with a product output of 800 g h−1. Under the same reaction conditions, the yield and selectivity were consistent with, or even improved over, those obtained in small-scale experiments, demonstrating the scalability and industrial potential of the process. Additionally, the process incorporates a waste acid recycling strategy, which has no significant impact on product yield, thus enhancing economic benefits and reducing environmental pollution. This continuous nitration process not only shows broad application potential but also offers a safe and efficient solution for nitration in the pharmaceutical and chemical industries.
Introduction
Nitroaromatic compounds represent a vital class of chemicals that serve as key precursors for aromatic amines, azo compounds, and nitrosoarenes.1,2 Their derivatives are extensively utilized in industries such as explosives,3 dyes,4 pesticides,5,6 and pharmaceuticals,7–9 highlighting their significant role in the chemical industry and related sectors. The nitration of aromatic compounds, one of the most fundamental aromatic substitution reactions, has seen notable advancements in synthesis methods in recent years.10 Common nitrating agents include mixed acid of nitric acid (HNO3) and sulfuric acid (H2SO4),11 HNO3,12,13 solid acid,14 nitrogen oxides,15–17 N-nitro reagents,18,19 and nitrates.20–23 Although some of these methods are considered more environmentally sustainable, their adoption in industrial production is often constrained by high costs or difficulties in achieving large-scale implementation.The nitration process using a mixed acid of HNO3 and H2SO4 remains the dominant method for synthesizing nitroaromatic compounds, but it is fraught with significant challenges. Among these, safety concerns are particularly critical.24 Nitro compounds are highly explosive, and their production involves flammable raw materials. Additionally, the highly exothermic nature of the electrophilic nitration reaction exacerbates the difficulty of maintaining stable reaction conditions in traditional batch reactors, thereby heightening safety risks.25 Furthermore, this process generates substantial amounts of waste acid, which, if not properly treated, can lead to severe environmental contamination, such as acidification of water bodies and soil degradation. Lastly, traditional nitration methods suffer from limitations in reaction selectivity and yield. The inability to precisely control reaction parameters often results in inconsistent selectivity and yield, posing significant challenges for achieving reliable and efficient production.
In recent years, continuous-flow microreaction technology has demonstrated remarkable advantages in nitration processes, such as excellent mixing efficiency, high heat and mass transfer efficiency, minimal reactant volume, short residence times, and rapid responsiveness.24,26 Building on these benefits, numerous successful applications of microreactor technology in nitration have emerged, offering an efficient, safe, and sustainable direction for the development of industrial nitration processes.27–34 In 2015, Kulkarni and his team investigated a continuous nitration process for o-xylene using a tubular reactor.35 The process employed fuming HNO3 (FNA) as the nitrating agent, achieving a 99% conversion rate of o-xylene with 7.2% dinitro impurity content. However, the required FNA amount was six times the molar quantity of o-xylene, leading to substantial reagent waste, while the maximum yield of nitro-o-xylene reached only 91.8%, leaving room for optimization.
In 2023, Shū Kobayashi and colleagues introduced a continuous-flow nitration approach using solid acid catalysts in the presence of HNO3.36 While this method demonstrated effectiveness, its industrial scalability was hindered by the high cost of solid acid catalysts and the use of organic solvents. In 2022, our team developed an efficient continuous-flow nitration process for o-xylene and its analogues (Fig. 1). This approach utilized 10 microreaction modules arranged in series, where the double-stage nitration reaction overcame the reaction equilibrium, resulting in a mononitro-o-xylene yield of 94.1%. Despite improving substrate selectivity and yield, implementing multiple microreactor modules prolonged reaction times, reduced production efficiency, and increased fixed capital costs. Additionally, the double-stage nitration step introduced procedural complexity and elevated waste acid generation, which together limited its industrial scalability.37
 | ||
| Fig. 1 Our previous work and this work. | ||
In this study, we propose an innovative nitration strategy for aromatic compounds that delivers high yields and enhanced mononitration selectivity. By optimizing the molar ratio of acid to substrate, the number of microreactor modules was reduced to five, shortening the reaction time from 90 seconds to 29 seconds. This adjustment significantly lowered fixed costs associated with industrial implementation while enhancing process feasibility. Furthermore, integrating a waste acid recycling strategy effectively mitigated the issue of acid disposal. Compared to our previous methodology, this new process achieved notable improvements in product yield and mononitro compound selectivity.
Results and discussion
Optimization of nitration in continuous-flow
p-Xylene, an important chemical feedstock,8,38 is widely utilized in the synthesis of dyes, plastics, pharmaceuticals, and pesticides. In this study, p-xylene was chosen as a model substrate for optimizing reaction conditions. First, 98% concentrated H2SO4 was diluted with water to the desired concentration and mixed with 97% concentrated HNO3 at a specific molar ratio to prepare the mixed acid. The mixed acid and p-xylene were then fed into a continuous-flow microreactor (Corning AFR Lab reactor equipped with a 2.7 mL microreactor module) through separate inlet streams (Fig. 2). After separating the organic phase from the reaction mixture, the products were analyzed via gas chromatography. | ||
| Fig. 2 Continuous-flow reactor setup for condition optimization. | ||
The effect of temperature on the reaction was first investigated under initial conditions: p-xylene flow rate at 2 g min−1, H2SO4 concentration at 70%, H2SO4/HNO3 molar ratio at 1.2, and HNO3/p-xylene molar ratio at 1.1. Within the temperature range of 30 °C to 100 °C, substrate conversion increased with rising temperature, while mononitration selectivity initially increased and then declined. The highest selectivity was observed at 60 °C (Fig. 3a), with a conversion rate of 46.1% and selectivity of 83.3%. The impact of the H2SO4/HNO3 molar ratio was then examined. Conversion and selectivity improved as the H2SO4/HNO3 ratio increased but declined after exceeding 2.0. While slightly higher ratios offered marginal improvements, 1.6 was chosen as the optimal ratio for its ability to achieve a good balance between high conversion and selectivity, while minimizing the environmental burden associated with excessive acid usage (Fig. 3b). H2SO4 concentration played a critical role in the nitration reaction, functioning as both a catalyst and a dehydrating agent. Insufficient H2SO4 concentration hindered the reaction, while excessive concentrations led to an overabundance of nitronium ions (NO2+), promoting the formation of unwanted polynitro by-products. Optimization experiments showed that a H2SO4 concentration of 70% provided the best selectivity (Fig. 3c).
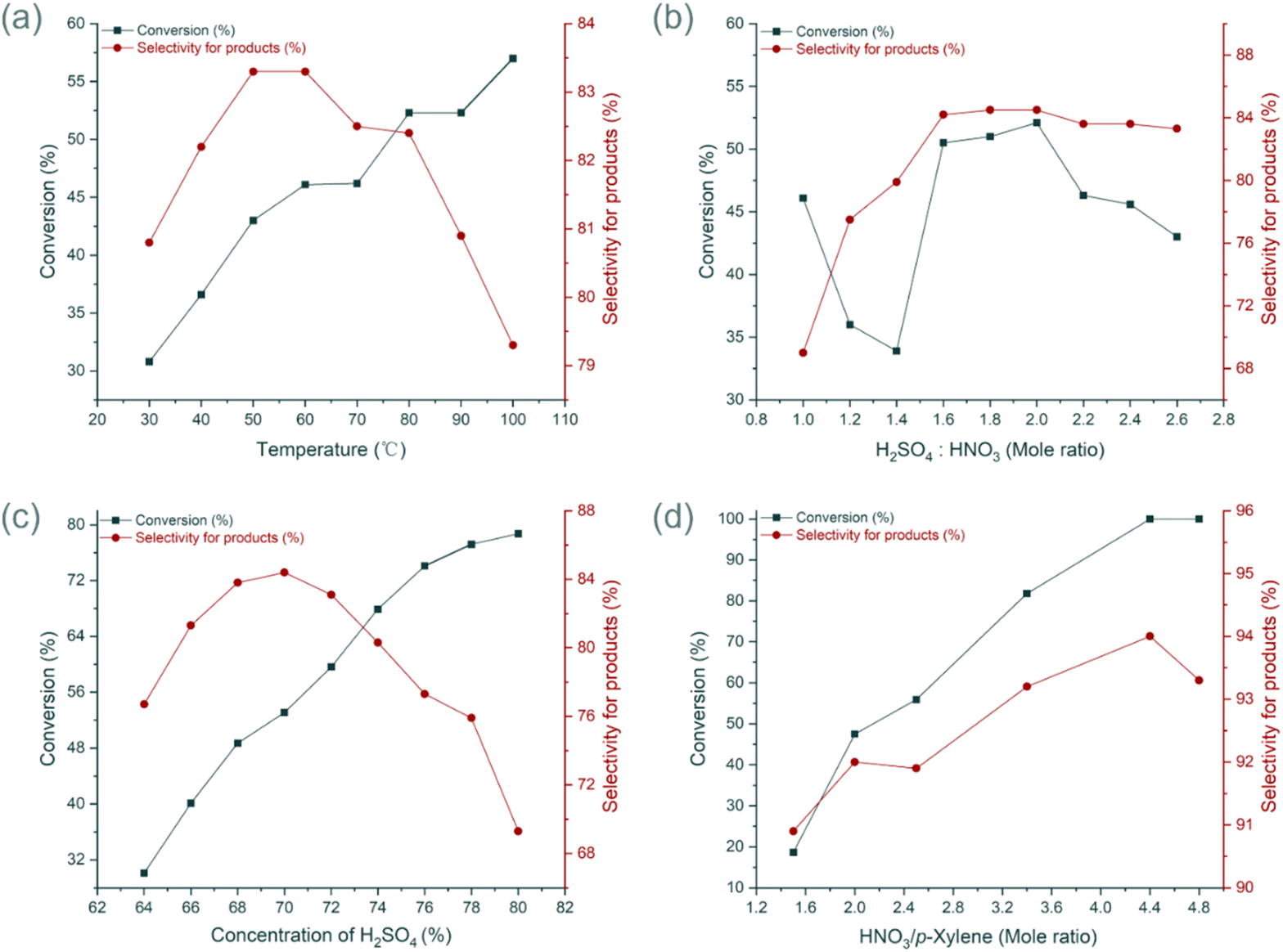 | ||
| Fig. 3 Optimization of nitration in continuous-flow. (a) The influence of temperature. (b) The influence of mole ratio of H2SO4 and HNO3. (c) The influence of H2SO4 concentration. (d) The influence of mole ratio of HNO3 and p-xylene. | ||
Despite these optimizations, substrate conversion remained suboptimal, primarily due to the short residence time in the continuous-flow reactor, which limited reaction completion. Increasing the number of microreactor modules to extend the residence time could enhance conversion but would significantly raise the fixed investment costs for industrial-scale equipment. To maintain a short reaction time, the reaction was intensified by increasing the molar ratio of HNO3 to substrate. Further studies revealed that as the HNO3/p-xylene ratio increased, both conversion and selectivity improved. Complete substrate conversion was achieved when the molar ratio reached 4.4 (Fig. 3d). Ultimately, under optimized conditions—a substrate flow rate of 1 g min−1 and a mixed acid flow rate of 12 g min−1—the yield of mononitro-p-xylene reached 94.0%, with a residence time of only 19 seconds.
Extension to analogues
To assess the general applicability of this method, we expanded the reaction conditions to include the synthesis of structurally related compounds (Table 1). Nitro-o-xylene serves as a precursor for the synthesis of important compounds, including riboflavin (vitamin B2) and the herbicide dymron, was synthesized using this approach with a yield of 96.1% and a reaction residence time of 19 seconds (Table 1, entry 1). Nitro-chlorobenzene, a key intermediate for azo dyes and herbicides like alachlor, was obtained with a yield of 99.4% within a residence time of 21 seconds (Table 1, entry 2). Similarly, nitrotoluene, a widely used chemical intermediate in the synthesis of dyes and explosives, was produced with a high yield of 98.1% within a residence time of 17 seconds (Table 1, entry 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Nitro-product(s) | Selectivity of productsb (%) | Reaction temperature (°C) | Yieldc (%) | Residence time (s) |
| a Reaction conditions: H2SO4 concentration = 70%, H2SO4/HNO3 mole ratio = 4.4, HNO3/substrate mole ratio = 1.6 and flow rate of substrates = 1 g min−1.b Selectivity of products = (total yield of mononitro-products/conversion of substrates) × 100%.c Yields and selectivities were determined by GC.d Numbers in parentheses represent regioselectivity. | ||||||
| 1 | 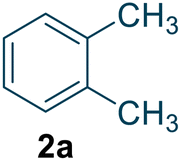 |
 |
96.7 | 80 | 96.1 (o = 54.6) d (p = 45.4) d | 19 |
| 2 | 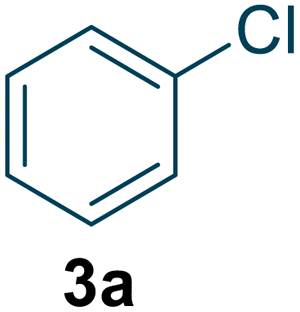 |
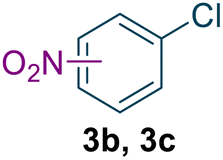 |
99.5 | 100 | 99.4 (o = 65.1) d (p = 34.9) d | 21 |
| 3 |  |
 |
98.2 | 70 | 98.1 (o = 61.6) d (p = 39.4) d | 17 |

Compound 5b, a crucial intermediate in the synthesis of Erlotinib, a targeted therapy drug for non-small cell lung cancer (NSCLC) and pancreatic cancer, was also synthesized to further evaluate the versatility of this method (Fig. 4). Under optimized conditions of 30 °C and using dichloromethane (DCM) as the solvent, the substrate achieved complete conversion, resulting in the successful synthesis of 5b. The reaction had a residence time of 65 seconds and an impressive yield of 99.3%, with a product output rate of 34 g h−1.
 | ||
| Fig. 4 Continuous-flow synthesis of Erlotinib key intermediate 5b. | ||
Scale-up to pilot plant scale
We then performed scale-up experiments for the continuous nitration of o-xylene and p-xylene (using the Corning AFR G1 reactor equipped with five standard flow control modules, each with a volume of 8.3 mL) (Fig. 5). In these experiments, the feed rate of the substrates was scaled up to 600 g h−1 and the reaction residence time was 29 seconds. By fine-tuning the reaction temperature, p-nitroxylene was successfully synthesized at 60 °C with a yield of 94.6% and a product throughput of 809 g h−1. Similarly, the nitration of o-xylene was carried out at 78 °C, achieving a total yield of 97.6% and a throughput of 835 g h−1. Compared to the small-scale process, only minor temperature adjustments were required for the scaled-up throughput, while all other parameters remained unchanged. The yields of the final products remained stable and even showed slight improvement, highlighting the excellent scalability of the process and its strong potential for industrial applications. | ||
| Fig. 5 Continuous-flow scale-up experimental setup. | ||
Recycling and reuse of waste acid
A major drawback of the mixed acid nitration process is the generation of large quantities of waste acid, which, if not properly managed, leads to resource loss and significant environmental burdens. Recycling and reusing waste acid, therefore, offer substantial practical and industrial value. Theoretically, nitration of 1 mol of o-xylene consumes 1 mol of HNO3 and produces an equivalent molar amount of H2O. Based on the initial composition of the mixed acid, the required amounts of supplemental H2SO4 and HNO3 can be calculated. Under optimal reaction conditions, the waste acid is separated and treated, after which the theoretical amount of H2SO4 is replenished. To account for HNO3 decomposition, 1.2 times the theoretical amount of HNO3 is added. Experimental results confirm that, compared to the standard conditions (Table 2, entry 1), the use of recycled waste acid (Table 2, entry 2) has a negligible impact on reaction outcomes, demonstrating the feasibility and effectiveness of the waste acid recycling strategy.| Entry | 3-Nitro product (%) | 4-Nitro product (%) | Conversion (%) | Yield b (%) | Impurities b (%) | Selectivity of products c (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: T = 80 °C, H2SO4 concentration = 70%, H2SO4/HNO3 mole ratio = 1.6, HNO3/o-xylene mole ratio = 4.4.b yield and impurities were determined by GC.c Selectivity of products = (total yield of mononitro-products/conversion) × 100%.d Use freshly prepared mixed acid.e Using recycled mixed acid. | ||||||
| 1d | 53.5 | 44.1 | 99.9 | 97.6 | 2.4 | 97.7 |
| 2e | 52.5 | 44.8 | 99.9 | 97.3 | 2.6 | 97.4 |
Conclusions
In conclusion, this study developed an efficient and safe continuous-flow microreaction mononitration process, achieving high yields and exceptional mononitration selectivity for a variety of mononitro compounds, including nitro-p-xylene, nitro-o-xylene, nitro-chlorobenzene, nitro-toluene, and a key intermediate in the synthesis of the anticancer drug erlotinib. The process was successfully scaled up to a production capacity of 800 g h−1 for the continuous synthesis of nitro-o-xylene and nitro-p-xylene, confirming its scalability and feasibility for industrial implementation. Additionally, the integration of a waste acid recovery and recycling strategy significantly enhanced economic efficiency while mitigating environmental pollution. These advancements highlight the potential of this process for widespread application in the pharmaceutical and chemical industries.Data availability
The data presented in this study are available in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22208268), Sichuan Science and Technology Program (2023NSFSC1977) and Technology Department of Yibin Program (2024GY008 and 2023SF009).References
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2018, 119, 2611–2680 CrossRef PubMed.
- J. Gui, C. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts and T. E. La Cruz, Science, 2015, 348, 886–891 CrossRef CAS PubMed.
- O. T. O'Sullivan and M. J. Zdilla, Chem. Rev., 2020, 120, 5682–5744 CrossRef PubMed.
- K. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- M. M. L. Nigro and M. A. Carballo, Toxicol. Lett., 2008, 180, 46–52 CrossRef PubMed.
- H. B. Fung and T. L. Doan, Clin. Ther., 2005, 27, 1859–1884 CrossRef CAS PubMed.
- U. A. Boelsterli, H. K. Ho, S. Zhou and K. Yeow Leow, Curr. Drug Metab., 2006, 7, 715–727 CrossRef CAS PubMed.
- K. Nepali, H. Y. Lee and J. P. Liou, J. Med. Chem., 2018, 62, 2851–2893 CrossRef PubMed.
- M. J. Strauss, Ind. Eng. Chem. Prod. Res. Dev., 1979, 18, 158–166 CrossRef CAS.
- G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554–2566, 10.1039/C3OB27354G.
- Z. Yu, P. Zhou, J. Liu, W. Wang, C. Yu and W. Su, Org. Process Res. Dev., 2016, 20, 199–203 CrossRef CAS.
- Y. Wu, W. Lu, Y. Ma, F. Chen, W. Ren and X. Chen, J. Org. Chem., 2023, 88, 11322–11327 CrossRef CAS PubMed.
- L. Lu, H. Liu and R. Hua, Org. Lett., 2018, 20, 3197–3201 CrossRef CAS PubMed.
- X. M. Ma, B. D. Li, L. Chen, M. Lu and C. X. Lv, Chin. Chem. Lett., 2012, 23, 809–812 CrossRef CAS.
- J. M. Bakke and E. Ranes, Synthesis, 1997, 1997, 281–283 CrossRef.
- B. Tang, S. Wei and X. Peng, Synth. Commun., 2014, 44, 2057–2065 CrossRef CAS.
- H. Liu, C. Ji, X. Dong, X. Peng and C. Shi, Chem. Lett., 2014, 43, 817–819 CrossRef CAS.
- F. Jia, A. Li and X. Hu, Org. Lett., 2023, 25, 4605–4609 CrossRef CAS PubMed.
- T. Yang, X. Li, S. Deng, X. Qi, H. Cong, H. Cheng, L. Shi, Q. Zhou and L. Zhuang, JACS Au, 2022, 2, 2152–2161 CrossRef CAS PubMed.
- H. Sun, R. Hua and Y. Yin, J. Org. Chem., 2005, 70, 9071–9073 CrossRef CAS PubMed.
- Y. Zheng, Q. Hu, Q. Huang and Y. Xie, Org. Lett., 2024, 26, 3316–3320 CrossRef CAS PubMed.
- A. K. Bose, S. N. Ganguly, M. S. Manhas, S. Rao, J. Speck, U. Pekelny and E. Pombo-Villars, Tetrahedron Lett., 2006, 47, 1885–1888 CrossRef CAS.
- S. Liu, Z. Gan, M. Jiang, Q. Liao, Y. Lu, H. Wang, Z. Xue, Z. Chen, Y. Zhang and X. Yang, JACS Au, 2024, 4, 4899–4909 CrossRef CAS PubMed.
- A. A. Kulkarni, Beilstein J. Org. Chem., 2014, 10, 405–424 CrossRef PubMed.
- G. A. Olah, S. C. Narang, J. A. Olah and K. Lammertsma, Proc. Natl. Acad. Sci. U.S.A., 1982, 4487–4494, DOI:10.1073/pnas.79.14.4487.
- S. Guo, L. Zhan and B. Li, Chem. Eng. J., 2023, 468, 143468 CrossRef CAS.
- A. A. Kulkarni, V. S. Kalyani, R. A. Joshi and R. R. Joshi, Org. Process Res. Dev., 2009, 13, 999–1002 CrossRef CAS.
- A. A. Kulkarni, N. T. Nivangune, V. S. Kalyani, R. A. Joshi and R. R. Joshi, Org. Process Res. Dev., 2008, 12, 995–1000 CrossRef CAS.
- B. Cardinal-David, K. C. Harper, A. Verma, D. Hanna, D. D. Caspi, C. Vitale, J. T. Bien, Z. Wang and M. Diwan, Org. Process Res. Dev., 2021, 25, 2473–2481 CrossRef CAS.
- C. E. Brocklehurst, H. Lehmann and L. La Vecchia, Org. Process Res. Dev., 2011, 15, 1447–1453 CrossRef CAS.
- Z. Wen, F. Jiao, M. Yang, S. Zhao, F. Zhou and G. Chen, Org. Process Res. Dev., 2017, 21, 1843–1850 CrossRef CAS.
- Y. Chen, Y. Zhao, M. Han, C. Ye, M. Dang and G. Chen, Green Chem., 2013, 15, 91–94 RSC.
- F. Xu, Z. Chen, L. Ni, G. Fu, J. Liu and J. Jiang, Org. Process Res. Dev., 2023, 27, 2134–2145 CrossRef CAS.
- D. Kyprianou, M. Berglund, G. Emma, G. Rarata, D. Anderson, G. Diaconu and V. Exarchou, Molecules, 2020, 25, 3586 CrossRef CAS PubMed.
- Y. Sharma, R. A. Joshi and A. A. Kulkarni, Org. Process Res. Dev., 2015, 19, 1138–1147 CrossRef CAS.
- H. Ishitani, M. Sasaya and S. Kobayashi, ACS Sustainable Chem. Eng., 2023, 11, 5826–5833 CrossRef CAS.
- Q. Song, X. Lei, S. Yang, S. Wang, J. Wang, J. Chen, Y. Xiang, Q. Huang and Z. Wang, Molecules, 2022, 27, 5139 CrossRef CAS PubMed.
- M. B. Sagandira, C. R. Sagandira and P. Watts, J. Flow Chem., 2021, 11, 193–208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, NMR data and spectrum. See DOI: https://doi.org/10.1039/d4ra09115a |
| This journal is © The Royal Society of Chemistry 2025 |