
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTough and self-healing waterborne polyurethane elastomers via hydrogen bonds and oxime–carbamate design for wearable flexible strain sensors†
Wei Zhang‡
b,
Mengqing Ren‡a,
Ming Chena and
Lili Wu *a
*a
aSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: polym_wl@whut.edu.cn
bWuhan Star Waterproofing Co., Ltd, P. R. China
First published on 25th February 2025
Abstract
Self-healing waterborne polyurethane elastomers, as functional materials, have been widely applied in flexible wearable devices. However, inevitable crack damage during use significantly limits their service life. Achieving an optimal balance between mechanical strength and self-healing ability remains a major challenge in the development of high-performance self-healing polyurethane materials. In this study, a rigid-soft phase-separated structure with multilayered rigid and flexible supramolecular segments was designed using isophorone isocyanate (IPDI), dimethylglyoxime (DMG), and 6-methyl-1,3,5-triazine-2,4-diamine (AGM) as hard segments, and polytetramethylene ether glycol (PTMEG) as soft segments. AGM establishes a multi-hydrogen bonding network with urethane groups, and DMG introduces dynamically reversible oxime–carbamate bonds, enabling the PU elastomers to achieve a self-healing efficiency of up to 95.5%. Benefiting from the gradient rigidity of the polyurethane, the strong hydrogen bond formed by the urea–carbamate bond and the triazine ring, the elastomer exhibited excellent mechanical properties, with a tensile strength of 33.04 MPa, an elongation at break of 954.79%, and a toughness of 90.66 MJ m−3. Moreover, when the composite conductor was used as an electronic skin, it possessed good self-healing properties with electrical response properties.
1 Introduction
With the gradual electronic station of human society, flexible strain sensors play an important role in wearable electronic devices,1,2 medical devices,3,4 electronic skins (e-skins)5,6 and soft robots.7–9 However, during use, sensors may be damaged by physical damage such as friction, abrasion, or impact, leading to a significant reduction in their reliability, durability, and service life, which triggers high economic losses.10 Ideal flexible sensors should have high elasticity to ensure recovery and repeatability, high healing efficiency to repair damage, excellent sensitivity for fast response, and reasonable durability for long-term use.11–15The introduction of self-repair function in stretchable conductive materials can effectively enhance the safety, reliability and sustainability of electronic devices, which has become a research hotspot.14,16–18 Waterborne emulsions (WPUs) are widely used as substrates in flexible electronic devices due to their environmentally friendly production process, flexible structural design, and adjustable performance.19,20 Polymer elastomers are often subjected to large dynamic loads in practical applications, so high strength, high toughness and good elasticity are key to their usefulness. In addition, elastomers have excellent self-healing capabilities, which is important for extending service life and enabling reuse.
Self-healing strategies are classified into two categories, exogenous and endogenous, according to the energy supply method.21–23 Exogenous self-healing relies on doping the repair medium, and although it can restore the damage, there are limitations such as poor reproducibility, restricted repair area, and possible degradation of polymer properties.24,25 Endogenous self-healing methods, on the other hand, self-repair through the introduction of dynamic reversible structures, including dynamic covalent bonds (e.g., disulfide, Diels–Alder, imine, and borate ester bonds) and dynamic non-covalent interactions (e.g., hydrogen bonds, metal–ligand bonds, and host–guest interactions).26–28 These bonds can be dynamically exchanged or dissociated/reorganised in response to external stimuli (e.g. heat, light or catalysts) when the material is damaged, conferring self-healing, reprocessing and recycling capabilities.29–32 Introducing dynamic covalent bonds and non-covalent interactions into the elastic matrix is an effective way to achieve self-healing and recyclability, but such materials are often challenged with insufficient mechanical properties.33–35 Therefore, how to balance the mechanical strength and self-healing properties of PU remains a key challenge for the application of self-healing PU materials in flexible electronics.
Shi et al. prepared environmentally friendly WPU systems containing multiple hydrogen and boron ester bonds in the polymer backbone by introducing 2,6 diaminopyridine and boric acid as a dynamic chain extender and reversible cross-linking agent, respectively. The chain structure of polymers was adjusted by controlling the DAP/BA ratio of the hard segments, and the requirements of mechanical robustness and self-healing efficiency can be effectively met.14 Zeng fabricated self-healing waterborne polyurethane (DTe-WPU-Zn-x) with double dynamic bonds and double crosslinked network by introducing dynamic telluride (DiTe-DiOH), trimethylolpropane, and zinc ions into waterborne polyurethane, and the temperature sensors prepared from the obtained polyurethane elastomers and LiTFSI were very sensitive to monitoring the changes in ambient temperature.36 Li used borate and BN coordination bonds as composite matrix to synthesise PU elastomers with high mechanical and self-healing properties. Highly conductive flexible strain sensors were fabricated using multi-walled carbon nanotubes (CNTs) and graphene nanosheets (GNSs) as fillers.37 Song et al. designed a self-repairing WPU elastomer by incorporating 2′-deoxythymidine (2′-dT) and isophorone diamine (IPDA) into the polymer chain, which when used as a flexible conductive substrate can quickly restore conductivity after surface damage after surface damage and increase the lifetime of the material.38 However, currently, WPUs synthesized with isophorone isocyanate (IPDI) as the hard segment and polytetramethylene ether glycol (PTMEG) as the soft segment have excellent performance in self-healing in flexible electronic devices, but often have difficulty in meeting the requirements of mechanical strength. The introduction of rigid fillers, trifunctional cross-linkers or rigid small molecules can improve the mechanical properties, but usually at the expense of reducing the self-healing efficiency, it is difficult to flexibly and effectively balance the self-healing performance and mechanical strength.39–41
In this paper, a soft-rigid phase-separated material with multilayered rigid and flexible supramolecular structures was designed using IPDI, dimethylglyoxime (DMG) and 6-methyl-1,3,5-triazine-2,4-diamine (AGM) as the hard segments and PTMEG as the soft segments, and excellent mechanical and self-healing properties were successfully achieved. The formation of the soft-rigid phase-separated structure promotes the free movement of the flexible PTMEG chain segments, which endows the material with high elasticity and provides space for the reorganisation of the molecular chains after breakage, thus enhancing the self-healing properties. The multiple hydrogen bonds, high-temperature dissociable oxime–carbamate bonds, and rigid triazine rings in the system then constitute a multilayered rigidity-regulating mechanism, which enables the materials to maintain high strength at low temperatures and enhance molecular mobility and self-healing ability at high temperatures. The emulsion and elastomer properties were characterised by infrared spectroscopy, particle size and zeta potential, differential scanning calorimetry (DSC) and tensile tests, and the ratio of DMG to AGM was adjusted to achieve a dynamic equilibrium, resulting in the preparation of a waterborne polyurethane elastomer, DxAyWPU, which combines good mechanical and self-healing properties. Subsequently, the elastomer was used as a matrix, and CNTs were doped as a conductive filler, to produce a self-repairable composite conductor. Self-repairable composite conductor and further explored its conductivity and electro-responsiveness.
2 Experimental section
2.1 Materials
Polytetramethylene ether glycol (PTMEG, Mn = 1000 g mol−1) was provided by BASF Co., Ltd. Isophorone isocyanate (IPDI, 99%), dimethylglyoxime (DMG, AR), dibutyltin dilaurate (DBTDL, 95%), 6-methyl-1,3,5-triazine-2,4-diamine (AGM, 98%), triethylamine (TEA) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Dimethylolpropionic acid (DMPA, 98%), acetone (AR) and ethanol absolute (AR) were kindly supplied by Shanghai Bide Pharmatech Ltd (China). Carbon nanotubes (CNT, 99%) was provided by Shenzhen Suheng Technology Co., Ltd. Deionized water was self-made in the laboratory.2.2 Synthesis of self-healing polyurethane elastomers (DxAyWPU)
The Scheme 1 illustrates the synthetic route for waterborne polyurethane emulsions. DMG and AGM were used as dynamic chain extenders to prepare self-healing waterborne polyurethane emulsions labeled DxAyWPU, where x and y denote the molar ratios of DMG to AGM, and the specific compositions of which are shown in Table S1.† The dehydrated PTMEG (30.0 g, 30 mmol) was placed in a glass flask equipped with N2. IPDI (17.2 g, 77.4 mmol) and DMG (2.75 g, 23.7 mmol) were added at 70 °C. Dibutyltin dilaurate (DBTDL, 0.14 g) was added dropwise when the system was cooled to 60 °C and kept at 80 °C for 3 h. Next, dihydroxymethylpropionic acid (DMPA, 4.03 g) was added to extend the chain for 3 h. Acetone can be added to reduce viscosity. Then, triethylamine (TEA, 3.0 g) was neutralised dropwise into the flask for 0.5 h at 30 °C to obtain polyurethane prepolymer. Finally, it was emulsified with deionised water dissolved in AGM for 1 h at 1500 rpm under vigorous stirring to obtain DxAyWPU emulsion. The required amounts of emulsions were cast into PTFE molds, left to dry at room temperature for 7 days. Testing was only conducted once the membrane reached a constant weight. | ||
| Scheme 1 Synthetic procedure of DxAyWPU. | ||
2.3 Composite conductor and flexible sensor preparation
CNT dissolved in acetone were mixed with DxAyWPU emulsion and added to 30 mL of deionized water, which was homogeneously dispersed under high speed stirring (1000 rpm) for 1 hour. The dispersion was spread evenly on a PTFE mould and dried at 80 °C for 6 h to obtain the composite conductor (DxAyWPU-CNT).The obtained composite conductor DxAyWPU-CNT (3 mm × 1 mm × 0.3 mm) was tightly connected to the finger joints or elbow joints of the volunteers by commercial medical tape to detect the movements of the finger and elbow joints, respectively, and the output metal wires were connected to the two ends of the composite conductor to obtain the desired flexible strain sensors.
2.4 Characterizations
The prepared DxAyWPU emulsion was diluted 10 times by adding deionised water, and then the particle size and the zeta potential of the DxAyWPU emulsion were tested by LS-909E laser particle size analyser.The chemical structure of the films was probed with a Nicolet ATR-FTIR spectrometer in the range of 600–4000 cm−1. Thermogravimetric analysis (TGA) was carried out on a STA449F3 integrated thermal analyser, and 3–5 mg of the samples were heated from 25 °C to 600 °C under N2 atmosphere with a ramp rate of 10 °C min−1.
Glass transition temperature (Tg) was obtained by using DSC-Q2000 (TA meter) from −80 °C to 100 °C under N2 atmosphere with a temperature increase rate of 10 °C min−1.
A WDW-L02 universal testing machine was used for mechanical testing. The films were made into dumbbell-shaped specimens of 50 mm × 4 mm with a thickness of 0.5–0.1 mm. All the specimens were set at a tensile speed of 50 mm min−1 according to the standard of GB/T13022-91, and at least three tests were carried out.
The dumbbell-shaped specimens were cut from the middle with a sharp blade and were quickly spliced at room temperature, then repaired at 40 °C, 60 °C and 80 °C for 6 h or 12 h.
The mechanical properties before and after repair were tested, and the formula for calculating the healing efficiency (η) was as follows, and the self-repairing of the specimens was observed with an optical microscope at a constant temperature heating at 80 °C.
A Tektronix DMM7510 digital multimeter was used to record the resistance of the flexible strain transducer in real time. The square resistance of the composite conductor DxAyWPU-CNT was tested using a four-probe resistivity tester FT-341 manufactured by Rico Instruments, taking three different positions on the conductor, and the test results are averages.
3 Results and discussion
3.1 Properties of DxAyWPU emulsions
The particle size and long term stability are very important characters for the application of DxAyWPU emulsions. The particle size and appearance of DxAyWPU emulsions were showed in Fig. 1(a) and S1.† It can be seen that transparent, bluish and no precipitation appeared in all samples. And it is found that the particle size of DxAyWPU emulsions were enlarged with the addition of DMG and AGM and the polydispersity become wider. When the mole ratio of DMG/AGM increased from 10/0 to 0/10, the latex particle size of emulsions rose from 6.24 nm to 11.34 nm. This could be explained by the multiple hydrogen bonding network resulting in increased intermolecular forces. Zeta potential is another important index to evaluate the stability of WPU emulsion. As shown in Fig. 1(b), the zeta potential values of the emulsions prepared with different molar ratios of DMG/AGM are about −38.6 to −40.3 mV, which are in the range of stable emulsions. Generally, an absolute value of zeta potential higher than 25 mV represents good dispersion of latex particles. The results show that DxAyWPU emulsions have good stability. | ||
| Fig. 1 (a) Particle size and distribution of DxAyWPU emulsions. (b) Zeta potential of DxAyWPU emulsions. | ||
3.2 Structural characterizations of DxAyWPU
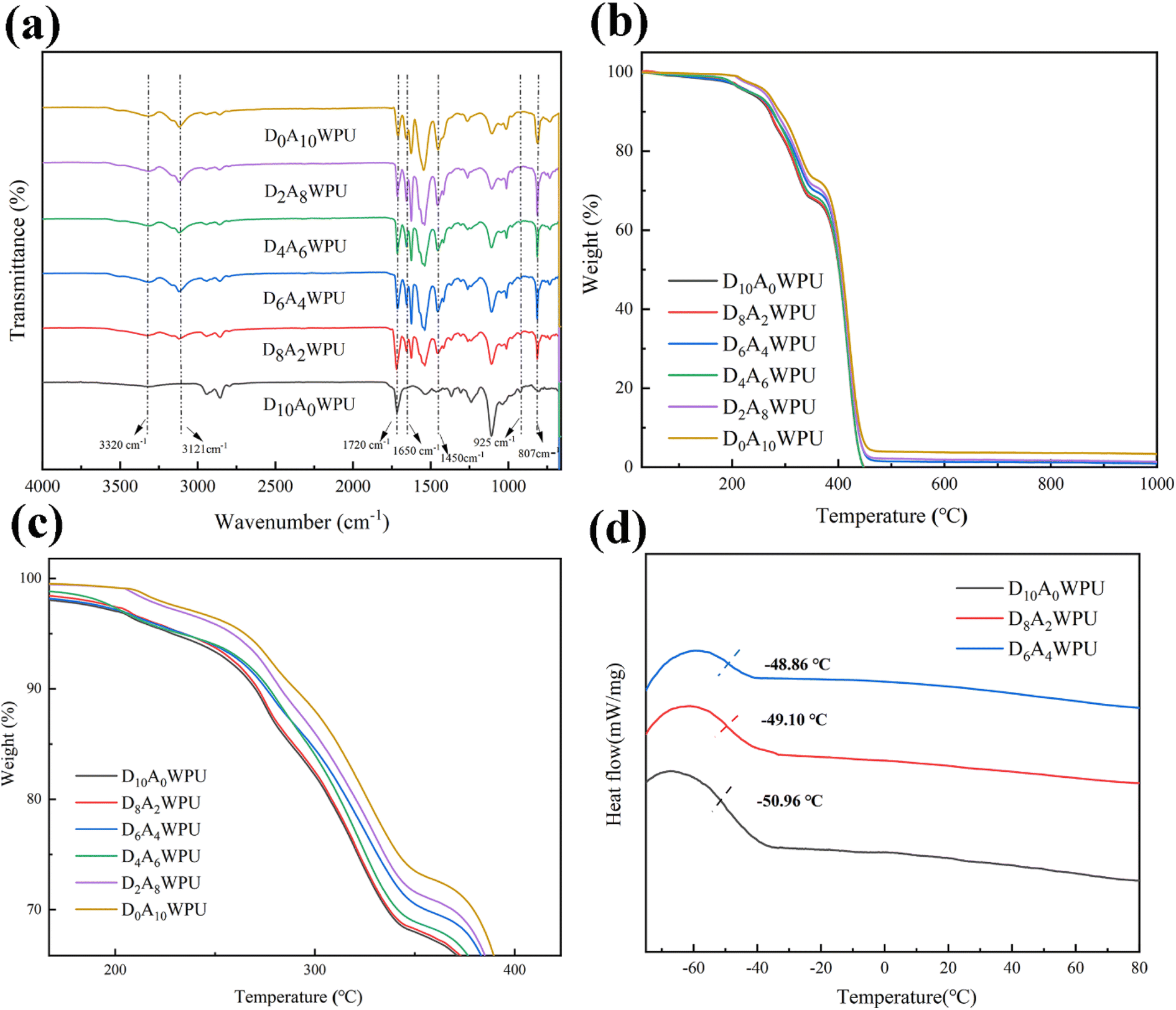
The synthesis of the self-healing and tough DxAyWPU was carried out through a two-step procedure in Scheme 1. The chemical structure of DxAyWPU was confirmed via FT-IR analysis, as shown in Fig. 2(a). The peaks at 3320 cm−1 and 1525 cm−1 were attributed to the N–H stretching and bending vibrations in urethanes, and the characteristic peak of isocyanate at 2270 cm−1 was not observed, suggesting a successful synthesis of the polyurethane prepolymer. A weak![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O– stretching vibration peak was observed at 925 cm−1, and the peak strength became more and more obvious as the content of DMG increased, indicating that the oxime was present in the polyurethane backbone. The peaks at 2950 cm−1, 2853 cm−1 and 1050 cm−1 corresponded to the characteristic absorption peaks of PTMG. The peaks at 807 cm−1, 1450 cm−1 and 1650 cm−1 were extra-planar and intra-planar bending vibrations of the triazine ring. Notably, all samples showed the characteristic peaks of the triazine ring except D10A0WPU, which indicated that AGM was successfully introduced into the polyurethane backbone. In conclusion, FT-IR analysis showed that DMG and AGM were successfully introduced into the waterborne polyurethane system and DxAyWPU was synthesised.
N–O– stretching vibration peak was observed at 925 cm−1, and the peak strength became more and more obvious as the content of DMG increased, indicating that the oxime was present in the polyurethane backbone. The peaks at 2950 cm−1, 2853 cm−1 and 1050 cm−1 corresponded to the characteristic absorption peaks of PTMG. The peaks at 807 cm−1, 1450 cm−1 and 1650 cm−1 were extra-planar and intra-planar bending vibrations of the triazine ring. Notably, all samples showed the characteristic peaks of the triazine ring except D10A0WPU, which indicated that AGM was successfully introduced into the polyurethane backbone. In conclusion, FT-IR analysis showed that DMG and AGM were successfully introduced into the waterborne polyurethane system and DxAyWPU was synthesised.
 | ||
| Fig. 2 (a) FTIR spectra of DxAyWPU elastomers. (b) TGA and (c) enlarged diagram of DxAyWPU elastomers. (d) DSC of D10A0WPU, D8A2WPU and D6A4WPU. | ||
3.3 Thermal performance of DxAyWPU
TGA is the main method for evaluating the thermal stability and decomposition behavior of materials. As shown in Fig. 2(b) and (c), the weight loss of all samples was less than 3% under 200 °C, which was attributed to the presence of traces of water in the samples. In general, the thermal decomposition of polyurethanes consists of two main steps: the first step of thermal decomposition is related to the decomposition of oxime and urea bonds from 200 to 370 °C. The second step occurs at 350–410 °C and is mainly attributed to the decomposition of soft segment polyols. It can be seen that the initial decomposition temperatures corresponding to 5% weight loss are 245 °C, 239 °C and 233 °C, respectively, decreasing sequentially with increasing AGM content. The result was attributed to the triazine ring and/or urea–carbamate bonds providing abundant hydrogen bonding sites between the polymer chains to form a dynamic crosslinked network, which improves the thermal stability of the waterborne polyurethanes. The Tg of self-healing elastomers was analyzed by DSC. As can be seen from the DSC curves in Fig. 2(d), the Tg was about −50 °C, indicating good mobility of the chain segments.3.4 Mechanical properties
A series of elastomers with different characteristic mechanical properties were synthesized by adjusting the molar ratios between DMG and AGM (Table S1†). As shown in Fig. 3(a) and (b), the D10A0WPU with containing only DMG has an elongation at break of 1119%, but a tensile strength of only 11.65 MPa, showing soft and weak characteristics, mainly due to the lack of sufficient physical or chemical cross-linking points or rigid components. When the molar ratios of AGM increased, the tensile strength and toughness of the elastomers increased significantly. Among them, the D8A2WPU showed excellent mechanical properties, with tensile strength at 33.04 MPa, toughness at 90.66 MJ m−3 and elongation at break at 954.79%. The introduction of AGM and DMG gives polyurethane hard segments multilevel stiffness. Moreover, the mechanical properties of PU elastomers are improved through the construction of stable covalent crosslinking networks and the formation of abundant multilevel hydrogen bonds. | ||
| Fig. 3 (a) Stress–strain curves of DxAyWPU. (b) Tensile strength and toughness of DxAyWPU. (c) Hysteresis curves of DxAyWPU specimen at 400% fixed strain with different loading-unloading times. (d) Residual strain and hysteresis energy of DxAyWPU. (e) Cyclic tensile curve of D8A2WPU at 100–600% strain. | ||
3.5 Cyclic tensile tests
To evaluate the elastic recovery ability and to investigate the effect of multiple hydrogen bonds and reversible oxime–carbamate bonds on elastomers, cyclic tensile tests with different number of cycles and strains were carried out. DxAyWPU were successively loaded-unloaded for one cycles at 400% strain (Fig. 3(c) and (d)). All elastomers formed a hysteresis line during stretching, and the area of the ring indicates the energy dissipated per unit volume. With the introduction of AGM component in the system, the residual strain was adjusted. Specifically, the residual strains of D8A2WPU and D6A4WPU were reduced by 18.02% and 4.07%, respectively, compared to D10A0WPU. The energy dissipation mechanism of the system is initially caused by reversible hydrogen bonding. The formation of multiple hydrogen bonds in the system with an appropriate ratio of DMG/AGM resists large deformations at high strains (400%) for a short period of time. It is noted that with continued addition of the AGM component, the high content of hard segments and the excessive density of hydrogen bonding resulted in restricted movement of the chain segments.Fig. 3(e) depicts cyclic tensile tests with progressively increasing strain and no waiting time (100–600%). At low strains, the hydrogen bond breaks first as a sacrificial bond and recovers quickly as the external force was released, so the residual strain remains low. However, under large strains, once the reversible covalent bonds were broken, complete recovery was impossible within a short time, resulting in large residual strains. Nevertheless, D8A2WPU retains a better deformation recovery ability with a residual strain of only 88.02% after several cycles of stretching. The results indicate that the crosslinked networks at different levels in DxAyWPUs, consisting of reversible hydrogen and oxime–carbamate bonds, can effectively dissipate strain energy and exhibit good elastic recovery.
3.6 Self-healing properties and mechanisms
Scratch and tensile tests were conducted to evaluate the impact of reversible hydrogen and oxime–carbamate bonds on the self-repairing properties of DxAyWPUs. Representative D10A0WPU, D8A2WPU and D6A4WPU elastomers were selected as examples, and a crack was cut on the surface of the elastomers with a razor blade and placed on a heating table at 80 °C for different times (0, 0.5 h, 1 h). The optical microscopic images of the cut elastomers before and after healing were compared, as shown in Fig. 4(a). The scratches of D10A0WPU and D8A2WPU almost disappeared after 1 h of thermal repair, whereas the scratch of D6A4WPU was still relatively obvious, which was consistent with the self-repairing efficiency described in the previous section. This was due to the fact that the decrease of DMG/AGM ratio represented a sharp increase in the rigid triazine ring and the hydrogen bonding density, which was unfavourable for chain segment migration. | ||
| Fig. 4 (a) Scratch healing of D10A0WPU, D8A2WPU and D6A4WPU at 80 °C for different time. D8A2WPU after 6 h healing at 80 °C (b) tensile test and (c) loaded with 2.5 kg weights. Stress–strain curves of (d) D6A4WPU, (e) D8A2WPU, (f) D10A0WPU healed for 24 hours at 40 °C, 60 °C, 80 °C. | ||
Subsequently, the dumbbell-shaped film D8A2WPU was cut in half, quickly gentle contact, and placed in an oven at 80 °C for 6 h for self-healing. The repaired specimen can be stretched to four times its original length without cracking, and the healed D8A2WPU elastomer specimen can withstand a 2.5 kg weight (Fig. 4(b) and (c)). In summary, D8A2WPU exhibited satisfactory mechanical properties after self-healing.
In addition, we also evaluated the self-healing properties of elastomers with different molar ratios of DMG/AGM prepared by tensile testing (Fig. 4(d)–(f) and S2–S4†). This indicated that the healing effect of DxAyWPUs had a significant dependence on the healing temperature, and the higher temperature could significantly improve the self-healing efficiency. Furthermore, the self-healing efficiency of DxAyWPUs elastomers was compared (Fig. 5(a)). The tensile strength and elongation at break of D8A2WPU and D6A4WPU were 90.1%, 86.6% and 76.5%, 95.5%, respectively, after repairing for 24 h at 80 °C. A particular comparison of tensile strength and healing efficiency were provided in Fig. 5(b). In contrast to D10A0WPU with DMG only, the introduction of AGM provided a hydrogen bonding network with different crosslink density with dynamic covalent bonding (oxime–carbamate bonding) synergistically promoting rapid self-healing of the polyurethane. However, as the molar ratio of DMG/AGM continues to decrease from 6/4, the rigidity of the triazine ring and the high strength spatial site resistance restrict the molecular chain movement, resulting in a relatively slow rate of dynamic bond exchange and leading to reduced healing efficiency.
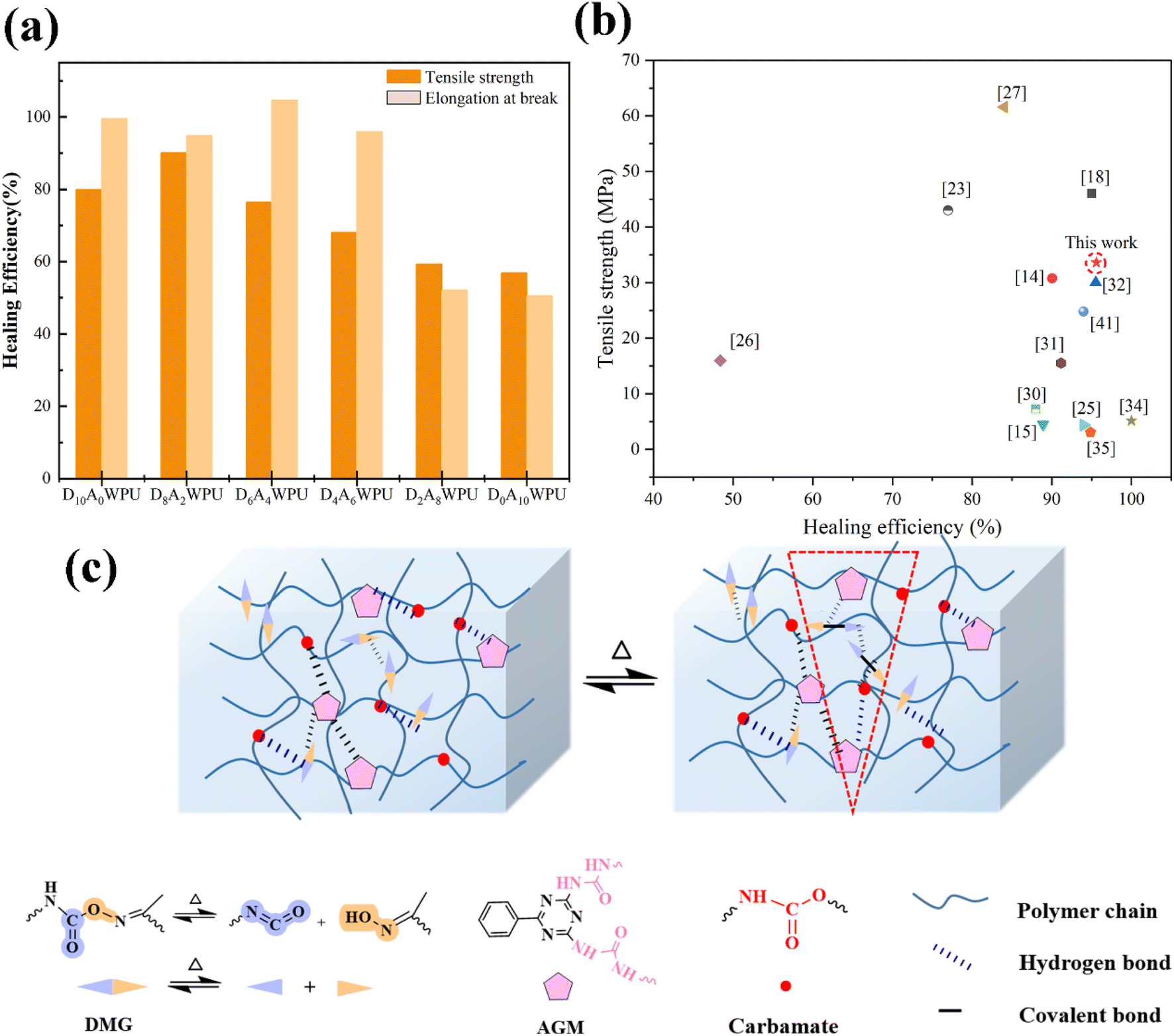 | ||
| Fig. 5 (a) Self-repair efficiency of DxAyWPU repaired at 80 °C for 24 h. (b) Comparison of tensile strength and healing efficiency for the representative D8A2WPU films in this work and other self-healable polymers reported in literature. (c) Schematic diagram of the self-healing mechanism of DxAyWPU films. | ||
The excellent self-healing properties of DxAyWPU elastomers are attributed to the synergistic effect of reversible oxime–carbamate covalent bonds and multiple reversible hydrogen bonds in its system (Fig. 5(c)). Employing IPDI, DMG and AGM as hard segments and PTMEG as soft segments, a soft- and hard-phase separation structure containing multilayered rigid and flexible supramolecular segments was designed. PTMEG is used as a soft segment because of its chain flexibility, which is conducive to the movement of polymer chains, and DMG provides dynamic oxime–carbamate bonds that can be reversibly dissociated and bonded under thermal drive, which endows waterborne polyurethanes with the self-healing ability. AGM endows the system with a multiple hydrogen-bonded physically-crosslinked network, which drives the accelerated diffusion and rearrangement of the molecular chains. Meanwhile, the strong hydrogen bonding in the urea–carbamate bond generated by the reaction between –NH2 and isocyanate in AGM, and the triazine ring in AGM itself, successfully balance the self-healing and mechanical properties.
3.7 Flexible strain sensors application
Due to the excellent mechanical properties and good self-healing ability, a stretchable and self-healing wearable sensor was fabricated by D8A2WPU with the addition of CNT. The preparation process of flexible composite conductor DxAyWPU-CNT is shown in Fig. 6(a). The composite conductor is not conductive when the CNT content is 10%, and the square resistance of the composite conductor is 2462.2 Ω, and 379.4 Ω when the content is 15% and 20%, respectively, so the following test, D8A2WPU-CNT-20%, was taken as a sample. Connect the D8A2WPU-CNT composite conductor in series with a 3 V power supply and an LED as shown in Fig. 6(b). When power is applied, the LED lights up, indicating that the composite conductor has the ability to conduct electricity. When the conductor is cut, the circuit is interrupted and the LED goes off. Subsequently, the broken D8A2WPU-CNT specimen was spliced and reconnected to the circuit after healing for 4 hours at 100 °C, and the LED lit up again, verifying its self-repairing performance.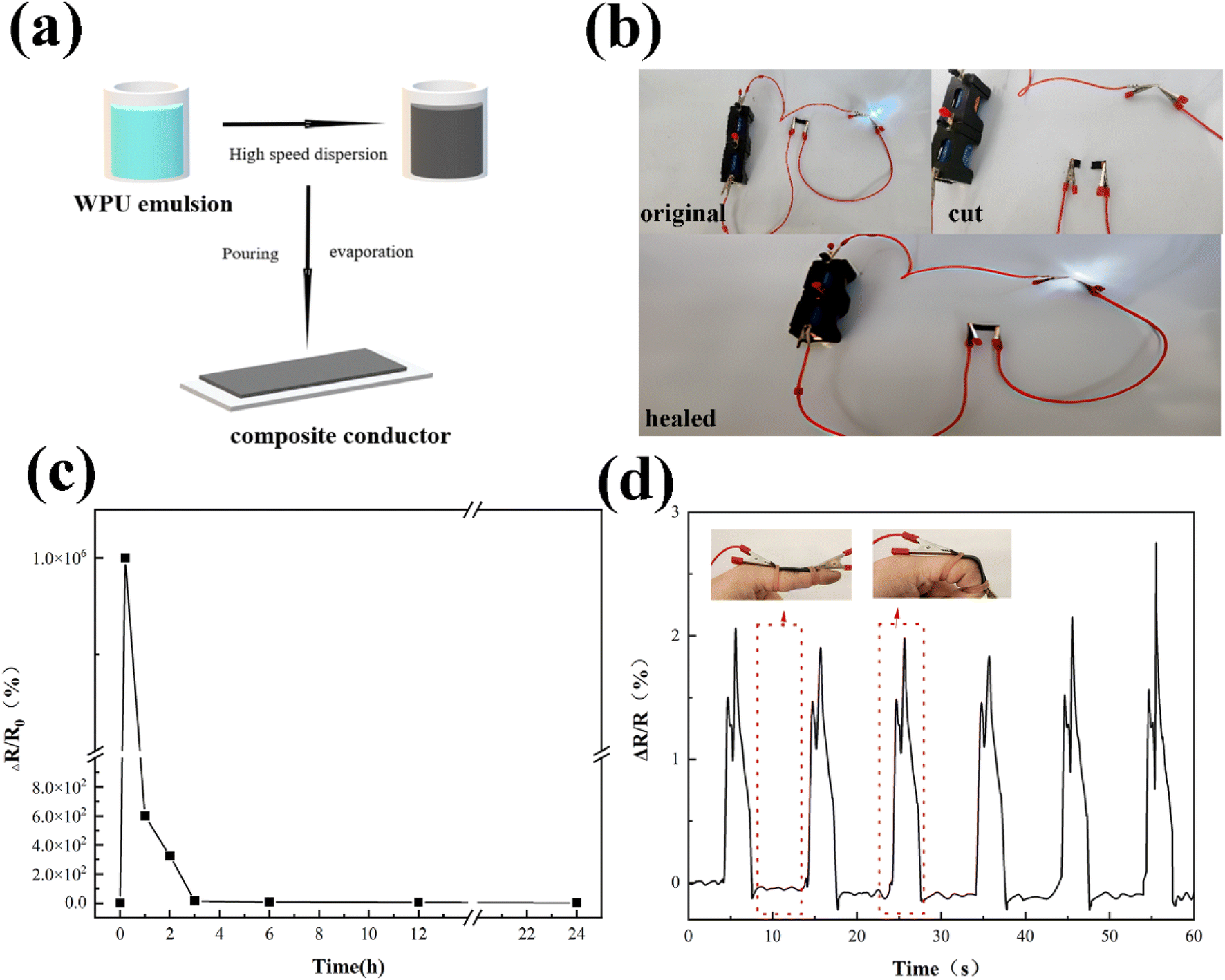 | ||
| Fig. 6 (a) Preparation of composite conductors. (b) Schematic diagram of the healing of the D8A2WPU-CNT composite conductor with LEDs in series. (c) ΔR/R0 for different times of healing at 100 °C. (d) D8A2WPU-CNT flexible strain sensor detects ΔR/R0 vs. finger movement time. | ||
To investigate the change of relative resistance of the composite conductor during healing, the sheared D8A2WPU-CNT composite conductor was contacted and healed by heating at 100 °C for 24 h, and the change of relative resistance ΔR/R0 (ΔR = R − R0) was recorded in time. As the healing time increased from 3 to 24 h, the relative resistance decreased from 15 to 1.5 in Fig. 6(c). The dissociation and recombination of reversible covalent and hydrogen bonds in the composite conductor allowed the efficient reconstruction of the conductive network, which paved the way for the reuse of elastomers in flexible electronic devices. Based on the flexibility and conductivity of D8A2WPU-CNT, it was assembled into a wearable flexible strain sensor for monitoring daily human movement. As shown in Fig. 6(d), a tensile strain sensor was attached at the knuckle, and the sensing resistance increased when the 90° bending behaviour was executed. Then, the resistance also recovers quickly as the finger was straightened. Repeated actions were tested repeatedly with essentially the same change in ΔR/R0, indicating that the electrical response behaviour of the flexible sensor was stable, sensitive and repeatable. It proved that the self-healable conductive composite D8A2WPU-CNT had the prospect that it can be used as a wearable flexible device application.
4 Conclusion
In this study, a self-healing waterborne polyurethane elastomer (DxAyWPU) based on hydrogen bonding with oxime–carbamate bonding was successfully synthesised, and the conductive composite was further prepared by introducing CNTs. Studies have shown that DxAyWPU exhibits excellent performance in terms of mechanical properties, self-healing ability and thermal stability. By modulating the molar ratio of DMG and AGM, not only the mechanical strength and toughness of the material were significantly enhanced, but also a crosslinked network with reversible covalent bonds and multiple hydrogen bonds was constructed, which endowed the material with excellent self-healing ability. D8A2WPU has the optimal comprehensive performance with a tensile strength of 33.04 MPa, an elongation at break of 954.79%, and a self-healing efficiency of up to 90.1%. After the innovative introduction of CNT, the prepared D8A2WPU-CNT composite conductor can recover the electrical conductivity by heating and healing after fracture, showing good self-healing ability. Tests show that the flexible strain sensors based on D8A2WPU-CNT are able to sensitively monitor human body movements with stable and repeatable resistance response. This study provides new ideas for the design of high-performance self-healing conductive composites and promotes their application development in flexible electronics, smart sensors and wearable devices.Data availability
Direct request to the corresponding author.Author contributions
Wei Zhang: conceptualization, methodology, software, investigation, writing original draft. Mengqing Ren: writing – review & editing, supervision, data curation. Ming Chen: writing – review & editing. Lili Wu: resources, writing – review & editing, supervision, data curation. All authors reviewed and approved the final version of the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was supported by Guangdong Pustar Adhesives & Sealants Co. Ltd (2020AG028).References
- X. Xiao, Z. Zheng, X. Zhong, R. Gao, Z. Piao, M. Jiao and G. Zhou, ACS Nano, 2023, 17, 1764–1802 CrossRef CAS PubMed.
- S. Yu, W. Zhang, J. An, T. Wang, H. Ling, T. Zhang and L. Chen, Electrochim. Acta, 2023, 441, 141802 CrossRef CAS.
- Y. Khan, A. E. Ostfeld, C. M. Lochner, A. Pierre and A. C. Arias, Adv. Mater., 2016, 28, 4373–4395 CrossRef CAS PubMed.
- L. Wang, K. Jiang and G. Shen, Adv. Mater. Technol., 2021, 6, 2100107 CrossRef.
- H. Yuan, T. Lei, Y. Qin and R. Yang, Nano Energy, 2019, 59, 84–90 CrossRef CAS.
- R. Dahiya, D. Akinwande and J. S. Chang, Proc. IEEE., 2019, 107, 2011–2015 Search PubMed.
- Z. Shen, F. Liu, S. Huang, H. Wang, C. Yang, T. Hang, J. Tao, W. Xia and X. Xie, Biosens. Bioelectron., 2022, 211, 114298 CrossRef CAS PubMed.
- J. Qu, G. Cui, Z. Li, S. Fang, X. Zhang, A. Liu, M. Han, H. Liu, X. Wang and X. Wang, Adv. Funct. Mater., 2024, 34, 2401311 CrossRef CAS.
- H.-L. Park, Y. Lee, N. Kim, D.-G. Seo, G.-T. Go and T.-W. Lee, Adv. Mater., 2020, 32, 1903558 CrossRef CAS PubMed.
- Y. Chen, M. Gao, K. Chen, H. Sun, H. Xing, X. Liu, W. Liu and H. Guo, Small, 2024, 20, 2400593 CrossRef CAS PubMed.
- Z. Tang, Y. Yuan, C. Zhang, Z. Yang and X. Wang, ACS Appl. Polym. Mater., 2023, 5, 9297–9306 CrossRef CAS.
- N. Zhang, G. Zhao, F. Gao, Y. Wang, W. Wang, L. Bai, H. Chen, H. Yang and L. Yang, ACS Appl. Polym. Mater., 2022, 4, 3394–3407 CrossRef CAS.
- S. A. Chowdhury, M. C. Saha, S. Patterson, T. Robison and Y. Liu, Adv. Mater. Technol., 2019, 4, 1800398 CrossRef.
- Q. Shi, W. Wu, B. Yu, M. Ren, L. Wu and C. Zhang, RSC Adv., 2022, 12, 35396–35408 RSC.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- Y. Sheng, Z. Li, D. Gao, P. Niu, X. Gao, Y. Huang, C. Li, J. Qiu, R. Zhang and Y. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 63840–63850 CrossRef CAS PubMed.
- S.-Y. Zhang, D.-W. Pan, H.-D. Fan, L. Shi, Y.-M. Yang, Z. Liu, X.-J. Ju, R. Xie, W. Wang and L.-Y. Chu, J. Polym. Sci., 2024, 62, 3236–3246 CrossRef CAS.
- M. Ren, Z. Wu, L. Wu, B. Yu, W. Wu, B. Liu and S. Xiao, J. Appl. Polym. Sci., 2024, 141, e54911 CrossRef CAS.
- Y. Wang, S. Wang, P. Li, S. Cao, Y. Liu and C. Gao, React. Funct. Polym., 2023, 192, 105736 CrossRef CAS.
- Y. Guo, X. An and X. Qian, Ind. Crops Prod., 2024, 208, 117800 CrossRef CAS.
- K. Han, J. Wang, C. Li, C. Zhou, J. Yuan, Z. Pan and M. Pan, Chem. Eng. J., 2023, 468, 143573 CrossRef CAS.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- Y. Eom, S.-M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 CrossRef CAS PubMed.
- Y. Peng, S. Gu, Q. Wu, Z. Xie and J. Wu, Acc. Mater. Res., 2023, 4, 323–333 CrossRef CAS.
- X. Zhou, H. Wang, S. Li and M. Liu, Eur. Polym. J., 2021, 159, 110769 CrossRef CAS.
- J. Xu, J. Chen, Y. Zhang, T. Liu and J. Fu, Angew. Chem., Int. Ed., 2021, 60, 7947–7955 CrossRef CAS PubMed.
- X. Feng and G. Li, ACS Appl. Mater. Interfaces, 2021, 13, 53099–53110 CrossRef CAS PubMed.
- Y. Li, Y. Jin, W. Zeng, R. Zhou, X. Shang, L. Shi, L. Bai and C. Lai, Prog. Org. Coat., 2023, 174, 107256 CrossRef CAS.
- Y. Yang, D. Davydovich, C. C. Hornat, X. Liu and M. W. Urban, Chem, 2018, 4, 1928–1936 CAS.
- Y. Li, M. Zhou, R. Wang, H. Han, Z. Huang and J. Wang, Eur. Polym. J., 2024, 214, 113159 CrossRef CAS.
- Z. Cui, X. Yue, Y. Wang, Y. Zhang, Z.-H. Ren and Z.-H. Guan, Chem. Eng. J., 2024, 479, 147538 CrossRef CAS.
- H. Wang, J. Xu, H. Wang, X. Cheng, S. Wang and Z. Du, Prog. Org. Coat., 2022, 167, 106837 CrossRef CAS.
- J. Liu, X. Ma, Y. Tong and M. Lang, Appl. Surf. Sci., 2018, 455, 318–325 CrossRef CAS.
- G.-F. Pan, Z. Wang, X.-B. Gong, Y.-F. Wang, X. Ge and R.-G. Xing, Chem. Eng. J., 2022, 446, 137228 CrossRef CAS.
- Y. Fang, X. Du, Z. Du, H. Wang and X. Cheng, J. Mater. Chem. A, 2017, 5, 8010–8017 RSC.
- W. Zeng, Y. Jin, R. Zhou, Y. Li and H. Chen, Chem. Eng. J., 2024, 482, 148994 CrossRef CAS.
- Z. Li, C. Ge, X. Li, L. Zhou, S. Liu and X. Zhang, Mater. Today Commun., 2024, 38, 108195 CrossRef CAS.
- Y. Song, J. Li, G. Song and X. Li, ACS Appl. Mater. Interfaces, 2024, 16, 2683–2691 CrossRef CAS PubMed.
- J. Yao, Z. Fu, H. Yang, L. Gao, X. Jiang, W. Nie, Z. Sun, H. Lu, M. Lin and J. Xu, Mol. Syst. Des. Eng., 2024, 9, 1167–1178 RSC.
- S. Wang, Y. Hu, T. B. Kouznetsova, L. Sapir, D. Chen, A. Herzog-Arbeitman, J. A. Johnson, M. Rubinstein and S. L. Craig, Science, 2023, 380, 1248–1252 CrossRef CAS PubMed.
- F. Dong, X. Yang, L. Guo, Y. Wang, H. Shaghaleh, Z. Huang, X. Xu, S. Wang and H. Liu, J. Mater. Chem. A, 2022, 10, 10139–10149 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra09084e |
| ‡ Wei Zhang and Mengqing Ren contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |