
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis, in silico and in vitro evaluation of pyrrole–indole hybrids as dual tubulin and aromatase inhibitors with potent anticancer activities†
Rungroj Saruengkhanphasit *ab,
Jaruwan Chatwichienac,
Lukana Ngiwsarad,
Kriengsak Lirdprapamongkolbd,
Worawat Niwetmarina,
Chatchakorn Eurtivong*e,
Prasat Kittakoopabf,
Jisnuson Svastid and
Somsak Ruchirawatabg
*ab,
Jaruwan Chatwichienac,
Lukana Ngiwsarad,
Kriengsak Lirdprapamongkolbd,
Worawat Niwetmarina,
Chatchakorn Eurtivong*e,
Prasat Kittakoopabf,
Jisnuson Svastid and
Somsak Ruchirawatabg
aChulabhorn Graduate Institute, Program in Chemical Sciences, 54 Kamphaeng Phet 6, Talat Bang Khen, Lak Si, Bangkok 10210, Thailand. E-mail: rungrojs@cgi.ac.th; Tel: +66 25541900 ext. 2629
bCenter of Excellence on Environmental Health and Toxicology (EHT), OPS, Ministry of Higher Education, Science, Research and Innovation, Bangkok, Thailand
cChulabhorn Royal Academy, Bangkok 10210, Thailand
dLaboratory of Biochemistry, Chulabhorn Research Institute, Bangkok, 10210, Thailand
eDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Mahidol University, 447 Si Ayutthaya Road, Ratchathewi, Bangkok 10400, Thailand. E-mail: chatchakorn.eur@mahidol.edu; Tel: +66 26448677-91 ext. 5402
fLaboratory of Natural Products, Chulabhorn Research Institute, Bangkok 10210, Thailand
gLaboratory of Medicinal Chemistry, Chulabhorn Research Institute, Bangkok 10210, Thailand
First published on 27th June 2025
Abstract
Twenty-four new pyrrolyl-3-phenyl-1H-indole-2-carbohydrazide derivatives were designed, synthesized and evaluated for their anticancer activities and dual inhibition properties against tubulin and aromatase. Their anticancer activities were highly potent against the NCI60 human cancer cell line panel. Amongst them, single chloro-substituted derivative 3h was the strongest tubulin inhibitor, disrupting the microtubule structure by inhibiting the colchicine site, while potently inhibiting aromatase (IC50 = 1.8 µM) with strong activity against the estrogen receptor-positive T47D breast cancer cell line (IC50 = 2.4 µM). Ester derivative 3k showed the best aromatase inhibitory activity (IC50 = 18 nM) with moderate anti-T47D activity (IC50 = 10.6 µM). Molecular docking predicted the derivatives inhibited the colchicine site of tubulin by forming mainly hydrophobic interactions with the surrounding amino acid residues. Moreover, heme chelation with the pyrrole ring was predicted as a key interaction, and the formation of intermolecular bonds with adjacent amino acid residues was predicted as important for inhibitory activity.
1. Introduction
Cancer has been reported by the World Health Organization (WHO) as a leading cause of death, with almost 10 million deaths in 2020.1 Furthermore, the WHO estimated that there would be around 20 million new cancer cases and 9.7 million deaths globally in 2022, and also projected that by 2050, there would be a 77% increase in new cases.2 Statistics revealed that lung (12.6%), breast (11.6%), and colon (9.6%) cancers were the first, second and third most common types of new cases in 2022, respectively.2 In the same report, lung cancer was the most common cause of deaths at 18.7% followed by colon cancer at 9.3%, and liver cancer and breast cancer at 7.8% and 6.9%, respectively. These statistics clearly indicate that cancer is a major global health burden.2Estrogen receptor-positive (ER+) breast cancer is the most prevalent type of breast cancer, and is characterized by the predominant expression of estrogen and progesterone receptors, which drives cell proliferation in the breast.3 The chemotherapeutic approach to treat ER+ breast cancer is mainly by administration of hormonal drugs, i.e., selective estrogen receptor modulators (SERMs), selective estrogen receptor degrader (SERD) and aromatase inhibitors to suppress estrogenic signals.4 Aromatase is a cytochrome P450 enzyme that contains a heme cofactor that is central to the catalytic conversion of androgens into estrogens.3 Aromatase activity is often upregulated in ER+ breast cancer tissues and has been designated as a key drug target for many clinically approved aromatase inhibitors: exemestane, letrozole and anastrozole (Fig. 1).3 Despite their clinical effectiveness, they are currently being challenged by drug resistance and undesirable side effects in patients, such as musculoskeletal pain, osteoporosis and cardiovascular problems.5–8 Other anticancer agents used to treat breast cancers include drugs that modulate microtubule activities such as vinorelbine and docetaxel (Fig. 1), which belong to the Vinca alkaloid and taxane classes of drugs, respectively.9 Microtubule-modulating agents exert their mechanism of action by binding to tubulin monomers that interfere with microtubule dynamics during mitosis, subsequently leads to mitotic arrest.9 Although clinically effective, they are limited by their large size, and are administered via injection and non-oral routes leading to inconvenience and compliancy issues for patients. Furthermore, cases of multidrug resistance were observed through various mechanisms, such as overexpression of drug efflux transporter MDR-1 (P-glycoprotein) resulting in reduced drug presence in cancer cells, and alteration of signal transduction pathways.10 Given these drawbacks, there is a need for the discovery and development of new anti-breast cancer agents with orally acceptable pharmacokinetic profiles.
 | ||
| Fig. 1 The chemical structures of clinically approved aromatase inhibitors (exemestane, anastrozole and letrozole), and microtubule-modulating agents (vinorelbine and docetaxel) for breast cancer treatment. | ||
It has been observed that co-administration of aromatase inhibitor and microtubule-modulating drugs can improve clinical effectiveness in breast cancer patients.11,12 Despite of these positive outcomes, such combinations can increase the risk of adverse effects, drug interactions, and detrimental physiological change. An alternative approach is to use dual-targeting strategies by employing dual-targeting agents, i.e., agents that simultaneously interact with two different targets or pathways. This strategy has been applied broadly in medicinal chemistry with proven enhanced pharmacological potencies, clinical effectiveness and to overcome multidrug resistance mechanisms, e.g. antitubercular agents,13,14 anti-Alzheimer's agents,15,16 antimalarials,17 anti-rheumatics18 and anticancer agents.19–23 Regardless of the potential benefits, marginal development of agents that modulate aromatase and tubulin activities were reported such as the 1-(diarylmethyl)-1H-azoles series,24 whereas, dual modulators of tubulin and estrogen receptor activities have shown slightly more traction, e.g., modulation of tubulin and antagonism of estrogen receptors.25,26
In our previous work, we have identified furanyl derivatives 1a, and thiophenyl derivatives 2a as promising anticancer agents, which subsequently led to the synthesis of 1b and 2b (Fig. 2) that exhibited potent anti-tubulin and anticancer activities.27,28 Typically, it was revealed that several derivatives from the thiophenyl subseries, including 2b, displayed potent antiproliferative activities and selectivity for certain cancer cell lines.28 The series is based on the 3-phenyl-1H-indole-2-carbohydrazide as the core scaffold. Consequently, this scaffold was used in this work as a platform for the design of new anticancer agents. Prior to this work, we were interested in attaching a pyrrole ring to our core scaffold, since pyrrole is a natural chelator of metals in many biological structures such as magnesium in chlorophyll, heme iron in hemoglobin and cytochrome P450 enzymes, including aromatase. Given this rationale, the pyrrole ring was attached to the side chain of the 3-phenyl-1H-indole-2-carbohydrazide core scaffold affording 3a (Fig. 2) with the expectation of a dual-targeting mechanism, i.e., tubulin and aromatase inhibition. This was followed by establishing a preliminary structure–activity relationship (SAR) by the synthesis of twenty-four new derivatives with various substituents installed at the pyrrole ring followed by experimental validation of their anticancer, anti-tubulin, and anti-aromatase activities. Furthermore, molecular modeling was employed to gain an understanding of the interactions between the compounds and the protein targets, and their physicochemical properties were evaluated. It should be noted that previously, aryl-substituted derivatives were synthesised by Cai and co-workers,29 however these derivatives displayed only weak anticancer effects, Moreover, cycloalkyl derivatives were previously prepared by our groups, but they exhibited poor anticancer activities.27 The present work focuses on anticancer pyrrole–indole hybrids with dual tubulin and aromatase inhibitory activities.
 | ||
| Fig. 2 The chemical structures of compounds 1a–b, 2a–b, and 3a. | ||
2. Results and discussion
2.1. Chemistry
The synthesis of pyrrolyl-3-phenyl-1H-indole-2-carbohydrazide 3a–x was achieved by the condensation of hydrazide 5 (ref. 28) with various pyrrole aldehyde (Scheme 1). The synthesis was initiated by replacing the ethoxy group of indole 4 (ref. 27) with hydrazine. Condensation of hydrazide 5 under acid condition with various substituted aldehydes afforded indole hydrazides. The indole hydrazide products from the condensation reaction could generate mixtures of E- and Z-isomers, but the E-isomer was isolated due to its high stability.27,28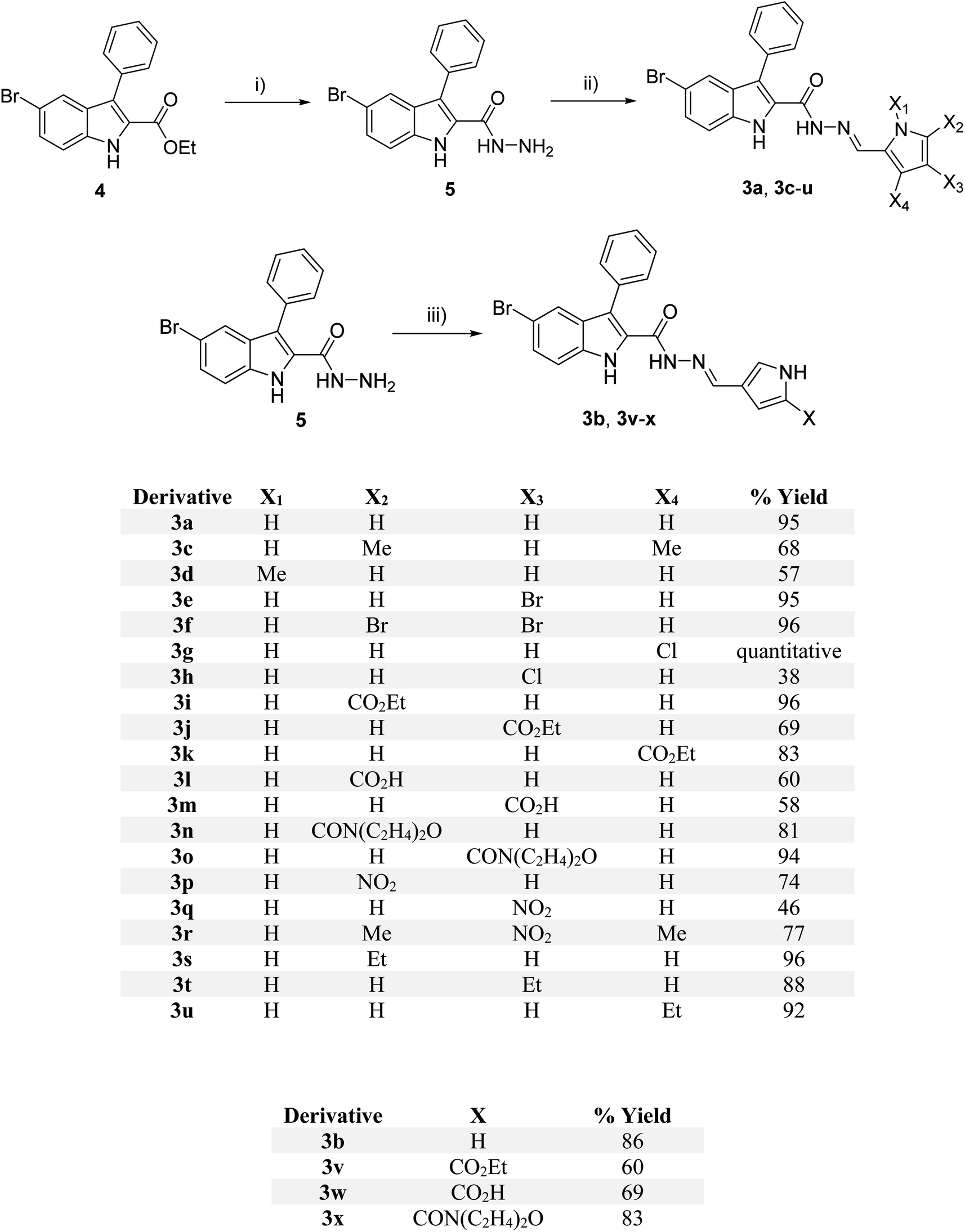 | ||
| Scheme 1 Synthesis of indole hydrazide derivatives; reagent and conditions: (i) N2H4, EtOH, 80 °C, 17 h, 70%; (ii) ArCHO, few drops AcOH, EtOH, 80 °C, 17 h, 46–100%; (iii) ArCHO, few drops AcOH, EtOH, 80 °C, 17 h, 67%. | ||
The dimethyl substitutions at positions X2 and X4 on the pyrrole ring gave 3c, whereas N-methyl afforded 3d. The halogen-containing derivatives include bromo at X3 (3e), dibromo (3f) at X2 and X3, single chloro substitution at X4 (3g) and X3 (3h). The electron-withdrawing groups were introduced for ester substitution at X2 (3i), X3 (3j), and X4 (3k), carboxylic acid at X2 (3l) and X3 (3m), morpholine-4-carbonyl at positions X2 (3n), and X3 (3o), and nitro at positions X2 (3p) and position X3 (3q). Nitro substitution at X3 and dimethyl at X2 and X4 positions generated 3r. Weak electron-donating ethyl group was introduced at positions X2 (3s), X3 (3t), and X4 (3u). Altering the connectivity from 2-pyrrolyl to 3-pyrrolyl afforded 3b, and varying the functionality at position X produced ester (3v), carboxylic acid (3w), and amide (3x) derivatives. The purity of the compounds was assessed by 1H NMR spectroscopy, which showed the purity of ca. 93–97% of the final products. The 1H NMR spectra of some compounds have additional peaks due to the existence of rotamers arising from the amide bond rotation. To prove the existence of rotamers of these compounds, compounds 3d, 3i, and 3p were selected for the 1H NMR experiment at 90 °C (see 1H and 13C spectra in ESI†). Indeed, 1H NMR spectra of compounds 3d, 3i, and 3p at 90 °C showed only one set of a single isomer, confirming the presence of rotamers.
2.2. Biological evaluation
The pyrrole derivatives are mostly strong anticancer agents (GI50 < 1 µM), with some exhibiting strong lethal effects (LC50 < 10 µM). The parent compound 3a displayed strong growth inhibition, and was most lethal against five cancer cell lines: HT29 colon adenocarcinoma (LC50 = 9.31 µM), UAAC-62 melanoma (LC50 = 8.03 µM), OVCAR-8 ovarian carcinoma (LC50 = 6.55 µM), SN12C renal cell carcinoma (LC50 = 3.97 µM), and BT-549 breast carcinoma (LC50 = 6.39). Derivative 3c exhibited moderate growth inhibition activities with GI50 values between 1.77 and 8.25 µM, and was most lethal against SK-MEL-5 melanoma cell line (LC50 = 19.3 µM). Derivative 3d displayed potent growth inhibition activities and was best at suppressing growth of MCF-7 breast cancer cells at double-digit nanomolar potency (GI50 = 72 nM), whilst exhibited selectivity for NCI/ADR-RES doxorubicin-resistant ovarian cancer cell line (LC50 = 40.9 µM) that overexpresses MDR1 drug efflux transporter.31 Single bromo-substituted derivative 3e displayed potent growth inhibition and exhibited most potent lethal effects against COLO 205 (LC50 = 0.89 µM), HT29 (LC50 = 8.24 µM), LOX IMVI (LC50 = 6.98 µM), SK-MEL-5 (LC50 = 1.22 µM), NCI/ADR-RES (LC50 = 0.97 µM), and SN12C (LC50 = 5.49 µM) cancer cell lines, whereas dibromo-substituted derivative 3f displayed moderate growth inhibition effects with GI50 values ranging between 0.53 to 6.8 µM, and was potent at killing SK-MEL-5 (LC50 = 8.65 µM) and NCI/ADR-RES (LC50 = 9.73 µM). Single chloro-substituted derivatives 3g and 3h were effective at suppressing growth inhibition, albeit 3g was less lethal. Derivative 3h displayed potent lethal effects against HL-60 (TB), K-562 and SR leukemia, NCI-H460 non-small cell lung carcinoma, COLO 205, HCC-2998 and HT29 colon carcinoma cell lines, SK-MEL-5 melanoma, and OVCAR-8 ovarian carcinoma with LC50 values in the range of 0.80 to 7.20 µM. Introduction of electron-withdrawing ester groups to derivatives 3i and 3j resulted in moderate growth inhibition activities. Nevertheless, 3i showed strong lethal effects against SK-MEL-5 (LC50 = 7.68 µM) and NCI/ADR-RES cell lines (LC50 = 9.2 µM), and 3j was potent against HL-60 (TB) (LC50 = 9.88 µM). Ester derivative 3k, and the carboxylic acid derivatives 3l and 3m were opted out from the five-dose experiment, i.e., 3k displayed moderate growth inhibition (NCI mean = 49%), whereas 3l and 3m exhibited mild effects. Morpholine-4-carbonyl derivative 3n was potent at growth suppression and exhibited potent lethality against HL-60 (TB) with LC50 = 8.56 µM, whereas 3o showed moderate growth inhibition (NCI mean = 45%), and was subsequently excluded from the five-dose experiment. Single electron-withdrawing nitro-substituted derivatives 3p and 3q showed potent growth inhibition effects; 3p was most lethal against COLO 205 (LC50 = 8.08 µM) and SK-MEL-5 (LC50 = 8.69 µM), and 3q was most selective for U251 glioblastoma (LC50 = 66.8 µM), as well as MDA-MB-435 melanoma (LC50 = 8.78 µM) and SK-MEL-5 melanoma (LC50 = 69.9 µM). Derivative 3r exhibited moderate growth inhibition activities, but displayed lethal effects against NCI-H460, COLO 205, HT29, LOX IMVI, SK-MEL-5, NCI/ADR-RES and SN12C cell lines. Ethyl-substituted derivatives 3s, 3t and 3u exhibited strong growth inhibition activities with moderate lethal effects for 3s and 3u, whilst 3t displayed potent lethal effects for LOX IMVI and SK-MEL-5 melanoma cell lines, and MDA-MB-468 breast carcinoma. Altering the connectivity of the pyrrole ring resulted in apparent reduction in growth inhibition activities (3a vs. 3b) with moderate lethal activities for most cell lines. The ester derivative 3v exhibited strong lethal effects for MDA-MB-435 melanoma with LC50 = 9.35 µM, and the substituted carboxylic acid (3w) and morpholine-4-carbonyl (3x) derivatives displayed moderate growth inhibition in the single dose experiment with NCI mean values ranging between 66–68%.
Six derivatives were the most potent: compound 3a, single electron-donating chloro- (3e) and bromo- (3h) substituted derivatives at X2 position, a single nitro-substituted derivative 3p at X1 position, and a dimethylated and nitro-substituted derivative 3r, and an ethylated derivative 3t at X2 position (Table 1). Small electron-donating and withdrawing substituents are favored for the anticancer effects, whereas derivatives with larger electron-withdrawing groups such as carbonyl-substituted derivatives are unfavorable. This can be attributed to an increase in molecular size and change in molecular shape which affects target binding. Additionally, an increase in molecular size and polarity perturbs drug-like behavior, e.g., decrease passive diffusion of drugs into cancer cells. Comparing the NCI60 screening results from this work with our previous studies on the furanyl and thiophenyl derivatives, all three subseries possess strong efficacies for COLO 205 colon carcinoma, SK-MEL-5 and MDA-MB-435 melanoma, and OVCAR-8 ovarian carcinoma suggesting part of the mechanism of action is influenced by the presence of the same core scaffold.28 Interestingly, 3d displayed selectivity for the NCI/ADR-RES cell line, suggesting it as a highly promising candidate to treat multidrug-resistant ovarian cancers. In addition, 3q was revealed to be the most selective at killing MDA-MB-435 and SK-MEL-5 melanoma cell lines implying it as an appealing candidate for invasive forms of melanoma treatment. Moreover, 3q was identified to be relatively potent against U251 glioblastoma, which is highly associated with acquired drug resistance CNS cancer.32
| 3a | 3e | 3h | 3p | 3r | 3t | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 | LC50 | GI50 | LC50 | GI50 | LC50 | GI50 | LC50 | GI50 | LC50 | GI50 | LC50 | |
| HL-60 (TB) | 0.242 | >100 | 0.327 | >100 | 0.185 | 0.886 | 0.374 | >100 | 2.31 | >100 | 0.237 | 91.9 |
| COLO 205 | 0.261 | 35.1 | 0.192 | 0.884 | 0.251 | 1.88 | 1.52 | 8.08 | 1.9 | 7.51 | 0.207 | — |
| LOX IMVI | 0.244 | 19 | 0.36 | 6.98 | 0.37 | 16.7 | 0.519 | 35.8 | 1.63 | 5.73 | 0.427 | 8.27 |
| MDA-MB-435 | 0.105 | — | 0.207 | 24.3 | 0.185 | — | 0.253 | 29.8 | 1.77 | 17.9 | 0.23 | 38.7 |
| SK-MEL-5 | 0.302 | 15.7 | 0.195 | 1.22 | 0.228 | 3.08 | 1.02 | 8.69 | 1.5 | 5.87 | 0.279 | 6.96 |
| OVCAR-8 | 0.323 | 6.55 | 0.304 | 18.2 | 0.214 | 0.988 | 1.02 | 39 | 3.08 | 36.5 | 0.316 | 26 |
| NCI/(ADR-RES) | 0.44 | 59.2 | 0.197 | 0.971 | 0.2 | — | 0.285 | 16.2 | 1.89 | 9.15 | 0.247 | 22 |
| SN12C | 0.371 | 3.97 | 0.310 | 5.49 | 0.391 | 15.3 | 1.24 | 36.7 | 1.74 | 6.73 | 0.372 | 30 |
 | ||
| Fig. 3 Effects of derivatives 3a–b, 3d–e, 3g–h, 3p–q, and 3s–u at a concentration of 10 µM on tubulin polymerization assay at 37 °C with colchicine (10 µM) as appositive control and paclitaxel (3 µM) as negative control. The curve is the average from two experiment runs. | ||
 | ||
| Fig. 4 Effects of derivative 3h on cell morphology and cell cycle progression of A549 human non-small cell lung cancer cells. The cells were treated with 3h at indicated concentrations for 24 h. The cells were subjected to cell cycle analysis by flow cytometric method. (A) Cell morphology after treatment, images of a representative experiment are shown with original magnification of ×200. (B) DNA content histograms of a representative experiment. (C) Percentages of cell populations in G0/G1, S, and G2/M phases. Data are shown as average with SD from three independent experiments, *p < 0.05, **p < 0.01, and ***p < 0.001 versus control group. | ||
| Derivative | Aromatase (µM) | T47Da (µM) | L929a (µM) | Selectivity indexd (SI) |
|---|---|---|---|---|
| a IC50 values determined by protocol in Methodology section 4.6.b Not determined.c No toxicity observed at 50 µM single concentration (>80% cell survival).d = IC50 (L929)/IC50 (T47D). | ||||
| 1a | 11.3 ± 1.7 | —b | —b | — |
| 2a | >12.5 | —b | —b | — |
| 3a | 3.4 ± 0.6 | 1.6 ± 1.0 | 26.3 ± 14.5 | 16.4 |
| 3b | 11.0 ± 1.3 | —b | —b | — |
| 3c | 11.1 ± 2.0 | 11.6 ± 0.3 | 29.5 ± 12.6 | 2.54 |
| 3d | >12.5 | 1.0 ± 0.4 | —c | — |
| 3e | 4.0 ± 1.0 | 2.4 ± 1.4 | 22.3 ± 11.9 | 9.29 |
| 3f | 1.5 ± 0.3 | 19.1 ± 5.4 | 47.9 ± 0.5 | 2.51 |
| 3g | 2.6 ± 0.5 | 8.2 ± 1.8 | 20.1 ± 10.3 | 2.45 |
| 3h | 1.8 ± 0.4 | 2.4 ± 1.8 | 53.7 ± 30.4 | 22.4 |
| 3i | 2.6 ± 0.8 | 11.8 ± 1.4 | —c | — |
| 3j | 1.3 ± 0.4 | 10.4 ± 2.9 | 12.2 ± 1.0 | 1.17 |
| 3k | 0.018 ± 0.002 | 10.6 ± 1.1 | 15.0 ± 8.9 | 1.42 |
| 3l | >12.5 | —b | —b | — |
| 3m | >12.5 | —b | —b | — |
| 3n | 3.3 ± 0.6 | 5.4 ± 4.8 | 21.8 ± 11.5 | 4.04 |
| 3o | 1.6 ± 0.4 | 14.9 ± 1.0 | 43.2 ± 9.9 | 3.57 |
| 3p | 1.4 ± 0.3 | —b | —b | — |
| 3q | >12.5 | —b | —b | — |
| 3r | 0.2 ± 0.1 | 12.3 ± 0.3 | —c | — |
| 3s | 4.6 ± 0.5 | 3.8 ± 1.0 | 19.5 ± 11.8 | 5.13 |
| 3t | 4.7 ± 0.5 | 4.6 ± 2.9 | 7.1 ± 3.6 | 1.54 |
| 3u | 9.6 ± 1.0 | 6.2 ± 3.6 | 12.2 ± 0.7 | 1.97 |
| 3v | 2.5 ± 0.6 | 24.9 ± 8.4 | 90.1 ± 40.3 | 3.62 |
| 3w | >12.5 | —b | —b | — |
| 3x | 3.7 ± 0.6 | —b | —b | — |
| Letrozole | 0.0014 ± 0.0003 | —b | —b | — |
| Cisplatin | —b | >100 | 23.7 ± 8.9 | — |
2.3. Molecular modelling
 | ||
| Fig. 5 The predicted binding orientation of derivative 3h at the colchicine binding site. The dipole–dipole interactions between Asn349 and the ligands are shown. The surface of tubulin was rendered orange for the β-subunit and light pink for the α-subunit. Amino acid residues Ala316, Ile318, Leu255, Ala250, Leu248 and Asn349 are depicted as sticks and colored as red, blue, black, brown, lime green and turquoise, respectively. | ||
 | ||
| Fig. 6 The predicted binding orientation of derivative 3k in the aromatase binding site. Potential chelation between 3k and heme (blue spherical shape) is depicted as red dotted lines. The surface of the aromatase was rendered in green. Amino acid residues Ile70, Phe134, Tyr220, Phe221, Trp224, Val369, Ile305, Leu372, and Met374 are depicted in sticks and colored as red, turquoise, orange, yellow, black, green, blue, and olive green, respectively. | ||
The toxicity profiles of the most active compounds were predicted using ADMETlab 3.0.39 The results of the mainstream toxicity types for the compounds are shown in Table S9.† The compounds were predicted to have low cardiotoxic and immunotoxic effects with moderate chances of being mutagens, albeit they are generally well-tolerated. Nonetheless, strong heptotoxic effects were predicted, which indicate further need for hepatotoxicity studies.
3. Conclusion
In this study, twenty-four new pyrrolyl-3-phenyl-1H-indole-2-carbohydrazides were successfully designed, synthesized and evaluated for their dual inhibitory effects against two targets: tubulin and aromatase. Derivative 3h which has a single chloro substitution at X3 was the strongest tubulin inhibitor with strong aromatase inhibitory activity. Furthermore, strong lethal effects and growth inhibition activities were observed for 3h, which was confirmed to cause mitotic arrest at the G2/M phase. Ester derivative 3k was the best aromatase inhibitor but exhibited moderate antiproliferative effects. In general, electron-withdrawing substituents tended to favor aromatase inhibition. However, the series lacked correlation between aromatase inhibition and anti-T47D breast cancer activities. All derivatives displayed lower cytotoxic effects against the non-tumoral L929 cell line compared to the T47D cancer cell line. Molecular docking predicted the subseries interact with and inhibit tubulin at the colchicine site and revealed that in addition to the pyrrole as a chelating group, the hydrazone functionality is predicted to contribute to heme chelation for some derivatives resulting in the subsequent inhibition of aromatase. Eight cancer cell lines were particularly sensitive to the subseries: HL-60 (TB), COLO 205, LOX IMVI, MDA-MB-435, SK-MEL-5, OVCAR-8, NCI/(ADR-RES) and SN12C. Strong antiproliferative effects against COLO 205, MDA-MB-435, SK-MEL-5 and OVCAR-8 cancer cell lines are shared with precedent furanyl and thiophenyl subseries. The derivatives were mostly predicted to be drug-like with acceptable oral pharmacokinetics. The most active compounds were generally predicted to have adequate toxicity profiles, aside from speculation of strong hepatotoxic effects.4. Experimental section
4.1. Synthesis
All chemicals for synthesis were purchased from commercial suppliers and used directly unless specifically stated. Thin layer chromatography was performed on Merck silica gel 60 F254 plates and visualized by UV irradiation at 254 nm. Flash column chromatography was performed using SilicaFlash® F60 (40–63 µm) or (63–200 µm) to obtain purified products. Automated flash chromatography was performed on Ultra Performance Flash Purification Chromatography (Model: Interchim puriflash 5.050) to obtain purified products. Bruker AVANCE-400 and 600 MHz were used to collect 1H and 13C NMR spectra. Chemical shifts (δ) are reported in parts-per-million (ppm) with respect to the solvent peaks and coupling constants (J) are in Hertz. Multiplicity of NMR peaks are given the abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Thermo Scientific Nicolet iS 5 Fourier Transform IR spectrometer was used to record Infrared (IR) spectra. Only selected peaks are reported and absorption maxima are given in cm−1. Thermo Scientific Orbitrap Q Exactive Focus mass spectrometer was used to record High Resolution mass spectra (HR-MS). Melting points were measured by using a Stuart SMP30 capillary melting point apparatus.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 to 1:1), gave indole hydrazide 3a (0.085 g, 0.21 mmol, 95%) as a yellow solid; Rf 0.5 [EtOAc–hexane (1:1)]; mp 206 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.14 (s, 1H), 11.51 (s, 1H), 11.07 (s, 1H), 7.87 (s, 1H), 7.71 (s, 1H), 7.50–7.44 (m, 5H), 7.38 (dd, J = 8.6, 1.9 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 6.89 (s, 1H), 6.42 (s, 1H), 6.10 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.66, 140.54, 134.29, 133.10, 129.53, 129.28, 128.58, 127.88, 126.85, 126.75, 126.34, 122.78, 121.83, 116.76, 114.47, 113.62, 112.91, 109.40; νmax/cm−1 3254, 1629, 1599, 1290; HRMS (ESI) m/z [M + H]+ calcd for C20H16ON479Br 407.0502; found 407.0504. Compounds 3e, 3g, 3f and 3h were synthesized following the procedure described here.:1)]; mp 231–233 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.05 (s, 1H), 11.76 (s, 1H), 11.15 (s, 1H), 7.76 (s, 1H), 7.62 (s, 1H), 7.41–7.35 (m, 5H), 7.30–7.23 (m, 2H), 6.91 (s, 1H), 6.44 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.98, 139.39, 134.40, 133.07, 129.62, 129.01, 128.66, 127.94, 127.87, 126.98, 126.55, 122.31, 121.95, 117.19, 114.58, 114.35, 113.04, 96.05; νmax/cm−1 3305, 1645, 1615, 1538, 1288; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br2 484.9607; found 484.9604.:3)]; mp 164 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.64 (s, 1H), 12.08 (s, 1H), 11.23 (s, 1H), 7.73 (s, 1H), 7.64 (s, 1H), 7.42–7.26 (m, 7H), 6.56 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 158.03, 138.69, 134.39, 133.03, 129.60, 129.13, 128.89, 128.63, 127.91, 126.97, 126.57, 121.95, 117.28, 115.11, 114.57, 113.04, 105.49, 99.00; νmax/cm−1 3259, 3066, 1646, 1536, 1244; HRMS (ESI) m/z [M + H]+ calcd for C20H14ON479Br3 562.8712; found 562.8714.:1)]; mp 170 °C decomposed; 1H NMR (400 MHz (CD3)2SO) δ 12.34 (s, 1H), 12.17 (s, 1H), 11.18 (s, 1H), 7.84 (s, 1H), 7.74 (s, 1H), 7.54–7.36 (m, 7H), 6.47 (s, 1H), 6.11 (s, 1H); 13C NMR (100 MHz (CD3)2SO) δ 157.83, 139.78, 134.34, 133.07, 129.56, 129.11, 128.60, 127.91, 126.91, 126.77, 126.44, 121.88, 118.52, 116.99, 114.53, 114.18, 112.96, 107.96; νmax/cm−1 3241, 3080, 1645, 1538, 1246; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br35Cl 441.0112; found 441.0112.:1)]; mp 190 °C decomposed; 1H NMR (400 MHz (CD3)2SO) δ 12.14 (s, 1H), 11.78 (s, 1H), 11.22 (s, 1H), 7.84 (s, 1H), 7.71 (s, 1H), 7.51–7.33 (m, 7H), 6.97 (s, 1H), 6.48 (s, 1H); 13C NMR (100 MHz (CD3)2SO) δ 157.81, 139.51, 134.30, 133.02, 129.52, 129.02, 128.55, 127.84, 126.86, 126.41, 121.85, 119.76, 117.01, 114.49, 112.92, 111.73, 111.57; νmax/cm−1 3254, 1613, 1537, 1237; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br35Cl 441.0112; found 441.0113.:100 to 100:0), gave indole hydrazide 3b (0.096 g, 0.24 mmol, 86%) as a brown solid; Rf 0.5 [EtOAc–hexane (100:0)]; mp 177–180 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.09 (s, 1H), 11.15 (s, 1H), 10.88 (s, 1H), 7.90 (s, 1H), 7.69 (s, 1H), 7.50–7.44 (m, 5H), 7.37–7.33 (m, 2H), 7.15 (s, 1H), 6.80 (s, 1H), 6.36 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.48, 144.53, 134.22, 133.15, 129.57, 129.42, 128.57, 127.96, 126.83, 126.25, 122.01, 121.78, 120.10, 119.01, 116.61, 114.44, 112.85, 105.30; νmax/cm−1 3239, 3049, 1629, 1557, 1288; HRMS (ESI) m/z [M + H]+ calcd for C20H16ON479Br 407.0502; found 407.0495. Compounds 3k, 3o (eluted with MeOH–CH2Cl2 (0:100 to 15:85)), and 3s–u were synthesized following the procedure described here.:1)]; mp 259–261 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.16 (s, 1H), 12.04 (s, 1H), 11.54 (s, 1H), 8.59 (s, 1H), 7.72 (s, 1H), 7.51–7.33 (m, 7H), 6.86 (s, 1H), 6.47 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 163.79, 158.06, 139.50, 134.35, 133.07, 129.94, 129.56, 128.86, 128.56, 127.87, 126.88, 126.51, 122.15, 121.91, 117.33, 116.43, 114.53, 112.97, 111.09, 59.51, 14.35; νmax/cm−1 3262, 3061, 2853, 1698, 1640, 1538, 1268; HRMS (ESI) m/z [M + H]+ calcd for C23H20O3N479Br 479.0713; found 479.0712.:93)]; mp 198–200 °C; 1H NMR (600 MHz (CD3)2SO) δ 12.15 (s, 1H), 11.94 (s, 1H), 11.23 (s, 1H), 7.90 (s, 1H), 7.71 (s, 1H), 7.51–7.44 (m, 5H), 7.38 (dd, J = 8.6, 1.9 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.20 (s, 1H), 6.64 (s, 1H), 3.57 (s, 8H); 13C NMR (150 MHz (CD3)2SO) δ 164.66, 157.82, 139.89, 134.33, 133.04, 129.53, 129.07, 128.58, 127.86, 127.00, 126.88, 126.43, 125.09, 121.87, 119.01, 116.98, 114.50, 113.61, 112.95, 66.27; νmax/cm−1 3213, 2856, 1601, 1544, 1225; HRMS (ESI) m/z [M + H]+ calcd for C25H23O3N579Br 520.0979; found 520.0977.:1)]; mp 136 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.08 (s, 1H), 11.23 (s, 1H), 10.93 (s, 1H), 7.72 (s, 1H), 7.66 (s, 1H), 7.45–7.39 (m, 5H), 7.33 (dd, J = 8.7, 1.9 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 6.25 (s, 1H), 5.79 (s, 1H), 2.52 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.59, 140.63, 139.96, 134.32, 133.16, 129.55, 129.42, 128.62, 127.94, 126.89, 126.33, 125.49, 121.84, 116.67, 114.66, 114.49, 112.94, 106.43, 20.41, 13.96; νmax/cm−1 3313, 2925, 1612, 1544, 1247; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0812.:1)]; mp 141 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.08 (s, 1H), 11.13 (s, 1H), 10.98 (s, 1H), 7.75 (s, 1H), 7.66 (s, 1H), 7.45–7.38 (m, 5H), 7.32 (dd, J = 8.6, 1.9 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 6.63 (s, 1H), 6.24 (s, 1H), 2.34 (q, J = 7.6 Hz, 2H), 1.05 (t, J = 7.5 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.65, 140.57, 134.33, 133.14, 129.58, 129.32, 128.62, 127.94, 126.89, 126.57, 126.51, 126.37, 121.87, 119.93, 116.81, 114.50, 113.15, 112.95, 19.49, 15.36; νmax/cm−1 3313, 2928, 1613, 1544, 1247; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0812.:1)]; mp 154 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.09 (s, 1H), 11.17 (s, 1H), 10.96 (s, 1H), 7.95 (s, 1H), 7.68 (s, 1H), 7.48–7.41 (m, 5H), 7.34 (dd, J = 8.6, 1.9 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 6.76 (s, 1H), 5.95 (s, 1H), 2.43 (q, J = 7.5 Hz, 2H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.47, 139.53, 134.30, 133.17, 130.74, 129.59, 129.39, 128.59, 127.93, 126.82, 126.34, 122.29, 122.25, 121.85, 116.76, 114.47, 112.93, 109.21, 18.54, 16.03; νmax/cm−1 3312, 3058, 2925, 1606, 1538, 1248; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0811.
:9 to 1:1), gave indole hydrazide 3a (0.085 g, 0.21 mmol, 95%) as a yellow solid; Rf 0.5 [EtOAc–hexane (1:1)]; mp 206 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.14 (s, 1H), 11.51 (s, 1H), 11.07 (s, 1H), 7.87 (s, 1H), 7.71 (s, 1H), 7.50–7.44 (m, 5H), 7.38 (dd, J = 8.6, 1.9 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 6.89 (s, 1H), 6.42 (s, 1H), 6.10 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.66, 140.54, 134.29, 133.10, 129.53, 129.28, 128.58, 127.88, 126.85, 126.75, 126.34, 122.78, 121.83, 116.76, 114.47, 113.62, 112.91, 109.40; νmax/cm−1 3254, 1629, 1599, 1290; HRMS (ESI) m/z [M + H]+ calcd for C20H16ON479Br 407.0502; found 407.0504. Compounds 3e, 3g, 3f and 3h were synthesized following the procedure described here.:1)]; mp 231–233 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.05 (s, 1H), 11.76 (s, 1H), 11.15 (s, 1H), 7.76 (s, 1H), 7.62 (s, 1H), 7.41–7.35 (m, 5H), 7.30–7.23 (m, 2H), 6.91 (s, 1H), 6.44 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.98, 139.39, 134.40, 133.07, 129.62, 129.01, 128.66, 127.94, 127.87, 126.98, 126.55, 122.31, 121.95, 117.19, 114.58, 114.35, 113.04, 96.05; νmax/cm−1 3305, 1645, 1615, 1538, 1288; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br2 484.9607; found 484.9604.:3)]; mp 164 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.64 (s, 1H), 12.08 (s, 1H), 11.23 (s, 1H), 7.73 (s, 1H), 7.64 (s, 1H), 7.42–7.26 (m, 7H), 6.56 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 158.03, 138.69, 134.39, 133.03, 129.60, 129.13, 128.89, 128.63, 127.91, 126.97, 126.57, 121.95, 117.28, 115.11, 114.57, 113.04, 105.49, 99.00; νmax/cm−1 3259, 3066, 1646, 1536, 1244; HRMS (ESI) m/z [M + H]+ calcd for C20H14ON479Br3 562.8712; found 562.8714.:1)]; mp 170 °C decomposed; 1H NMR (400 MHz (CD3)2SO) δ 12.34 (s, 1H), 12.17 (s, 1H), 11.18 (s, 1H), 7.84 (s, 1H), 7.74 (s, 1H), 7.54–7.36 (m, 7H), 6.47 (s, 1H), 6.11 (s, 1H); 13C NMR (100 MHz (CD3)2SO) δ 157.83, 139.78, 134.34, 133.07, 129.56, 129.11, 128.60, 127.91, 126.91, 126.77, 126.44, 121.88, 118.52, 116.99, 114.53, 114.18, 112.96, 107.96; νmax/cm−1 3241, 3080, 1645, 1538, 1246; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br35Cl 441.0112; found 441.0112.:1)]; mp 190 °C decomposed; 1H NMR (400 MHz (CD3)2SO) δ 12.14 (s, 1H), 11.78 (s, 1H), 11.22 (s, 1H), 7.84 (s, 1H), 7.71 (s, 1H), 7.51–7.33 (m, 7H), 6.97 (s, 1H), 6.48 (s, 1H); 13C NMR (100 MHz (CD3)2SO) δ 157.81, 139.51, 134.30, 133.02, 129.52, 129.02, 128.55, 127.84, 126.86, 126.41, 121.85, 119.76, 117.01, 114.49, 112.92, 111.73, 111.57; νmax/cm−1 3254, 1613, 1537, 1237; HRMS (ESI) m/z [M + H]+ calcd for C20H15ON479Br35Cl 441.0112; found 441.0113.:100 to 100:0), gave indole hydrazide 3b (0.096 g, 0.24 mmol, 86%) as a brown solid; Rf 0.5 [EtOAc–hexane (100:0)]; mp 177–180 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.09 (s, 1H), 11.15 (s, 1H), 10.88 (s, 1H), 7.90 (s, 1H), 7.69 (s, 1H), 7.50–7.44 (m, 5H), 7.37–7.33 (m, 2H), 7.15 (s, 1H), 6.80 (s, 1H), 6.36 (s, 1H); 13C NMR (150 MHz (CD3)2SO) δ 157.48, 144.53, 134.22, 133.15, 129.57, 129.42, 128.57, 127.96, 126.83, 126.25, 122.01, 121.78, 120.10, 119.01, 116.61, 114.44, 112.85, 105.30; νmax/cm−1 3239, 3049, 1629, 1557, 1288; HRMS (ESI) m/z [M + H]+ calcd for C20H16ON479Br 407.0502; found 407.0495. Compounds 3k, 3o (eluted with MeOH–CH2Cl2 (0:100 to 15:85)), and 3s–u were synthesized following the procedure described here.:1)]; mp 259–261 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.16 (s, 1H), 12.04 (s, 1H), 11.54 (s, 1H), 8.59 (s, 1H), 7.72 (s, 1H), 7.51–7.33 (m, 7H), 6.86 (s, 1H), 6.47 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 163.79, 158.06, 139.50, 134.35, 133.07, 129.94, 129.56, 128.86, 128.56, 127.87, 126.88, 126.51, 122.15, 121.91, 117.33, 116.43, 114.53, 112.97, 111.09, 59.51, 14.35; νmax/cm−1 3262, 3061, 2853, 1698, 1640, 1538, 1268; HRMS (ESI) m/z [M + H]+ calcd for C23H20O3N479Br 479.0713; found 479.0712.:93)]; mp 198–200 °C; 1H NMR (600 MHz (CD3)2SO) δ 12.15 (s, 1H), 11.94 (s, 1H), 11.23 (s, 1H), 7.90 (s, 1H), 7.71 (s, 1H), 7.51–7.44 (m, 5H), 7.38 (dd, J = 8.6, 1.9 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.20 (s, 1H), 6.64 (s, 1H), 3.57 (s, 8H); 13C NMR (150 MHz (CD3)2SO) δ 164.66, 157.82, 139.89, 134.33, 133.04, 129.53, 129.07, 128.58, 127.86, 127.00, 126.88, 126.43, 125.09, 121.87, 119.01, 116.98, 114.50, 113.61, 112.95, 66.27; νmax/cm−1 3213, 2856, 1601, 1544, 1225; HRMS (ESI) m/z [M + H]+ calcd for C25H23O3N579Br 520.0979; found 520.0977.:1)]; mp 136 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.08 (s, 1H), 11.23 (s, 1H), 10.93 (s, 1H), 7.72 (s, 1H), 7.66 (s, 1H), 7.45–7.39 (m, 5H), 7.33 (dd, J = 8.7, 1.9 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 6.25 (s, 1H), 5.79 (s, 1H), 2.52 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.59, 140.63, 139.96, 134.32, 133.16, 129.55, 129.42, 128.62, 127.94, 126.89, 126.33, 125.49, 121.84, 116.67, 114.66, 114.49, 112.94, 106.43, 20.41, 13.96; νmax/cm−1 3313, 2925, 1612, 1544, 1247; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0812.:1)]; mp 141 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.08 (s, 1H), 11.13 (s, 1H), 10.98 (s, 1H), 7.75 (s, 1H), 7.66 (s, 1H), 7.45–7.38 (m, 5H), 7.32 (dd, J = 8.6, 1.9 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 6.63 (s, 1H), 6.24 (s, 1H), 2.34 (q, J = 7.6 Hz, 2H), 1.05 (t, J = 7.5 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.65, 140.57, 134.33, 133.14, 129.58, 129.32, 128.62, 127.94, 126.89, 126.57, 126.51, 126.37, 121.87, 119.93, 116.81, 114.50, 113.15, 112.95, 19.49, 15.36; νmax/cm−1 3313, 2928, 1613, 1544, 1247; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0812.:1)]; mp 154 °C decomposed; 1H NMR (600 MHz (CD3)2SO) δ 12.09 (s, 1H), 11.17 (s, 1H), 10.96 (s, 1H), 7.95 (s, 1H), 7.68 (s, 1H), 7.48–7.41 (m, 5H), 7.34 (dd, J = 8.6, 1.9 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 6.76 (s, 1H), 5.95 (s, 1H), 2.43 (q, J = 7.5 Hz, 2H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (150 MHz (CD3)2SO) δ 157.47, 139.53, 134.30, 133.17, 130.74, 129.59, 129.39, 128.59, 127.93, 126.82, 126.34, 122.29, 122.25, 121.85, 116.76, 114.47, 112.93, 109.21, 18.54, 16.03; νmax/cm−1 3312, 3058, 2925, 1606, 1538, 1248; HRMS (ESI) m/z [M + H]+ calcd for C22H20ON479Br 435.0815; found 435.0811.* Rotamer.
1H NMR (600 MHz (CD3)2SO, 90 °C) δ 7.99 (s, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.54–7.53 (m, 2H), 7.48–7.46 (m, 3H), 7.37–7.34 (m, 2H), 6.89 (s, 1H), 6.41 (s, 1H), 6.07 (s, 1H), 3.74 (s, 3H).
* Rotamer.
1H NMR (600 MHz (CD3)2SO, 90 °C) δ 11.84 (s, 2H), 11.08 (s, 1H), 8.03 (s, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.53–7.50 (m, 3H), 7.6 (t, J = 7.6 Hz, 2H), 7.40 (dd, J = 8.7, 1.9 Hz, 1H), 7.35 (t, J = 6.8 Hz, 1H), 6.81 (d, J = 3.9 Hz, 1H), 6.54 (s, 1H), 4.29 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H).
* Rotamer.
1H NMR (600 MHz (CD3)2SO, 90 °C) δ 13.23 (s, 1H), 11.88 (s, 1H), 11.36 (s, 1H), 8.03 (s, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.52–7.50 (m, 3H), 7.45 (t, J = 7.6 Hz, 2H), 7.40 (dd, J = 8.7, 1.9 Hz, 1H), 7.35 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 4.3 Hz), 6.62 (s, 1H).
4.2. NCI60 screening
The synthesized compounds were evaluated their anticancer properties by submitted to National Cancer Institute 60 human tumour cell lines (NCI60) screening program in the USA.000 cells/well and incubated at 37 °C for 24 hours. After incubation, some plates were treated with trichloroacetic acid (TCA) to determine the time of drug addition (Tz) for each cell line. The plates containing the test compounds at five different concentrations were incubated for 48 hours, fixed, stained with sulforhodamine B (SRB), and incubated at room temperature for 10 minutes. An automate plate was used to measure the absorbance at a wavelength of 515 nm. The percentage of growth inhibition was calculated by use the absorbance from time zero (Tz), control growth (C), and test growth in the presence of drug at the five concentration levels (Ti) using this equation.| [(Ti − Tz)/(C − Tz)] × 100 for concentrations which Ti ≥ Tz |
| [(Ti − Tz)/Tz] × 100 for concentration which Ti < Tz |
The results are reported as Growth Inhibition at 50% (GI50), Total Growth Inhibition (TGI), and Median Lethal Concentration (LC50).
4.3. Tubulin polymerisation assay
The tubulin polymerisation experimental protocol followed the method described in this literature.28 The experimental was using fluorescence-based assay kit (Cytoskeleton, catalog no. BK011P). In Brief, 5 µL of test compounds was added to a 96-well plate, then mix with 50 µL of a solution containing porcine brain tubulin (2 mg mL−1) in buffer with 20% glycerol and 1 mM GTP. A microplate reader (Varioskan™ LUX multimode microplate reader, Thermo Scientific™) was used to measure fluorescence intensity every 60 seconds for 60 minutes, with excitation at 360 nm and emission at 450 nm, maintained at 37 °C throughout the experiment.4.4. Cell cycle
A549 human non-small cell lung cancer cell line (ATCC CCL-185) was purchased from American Type Culture Collection (Rockville, MD, USA). Analysis of cell cycle phase distribution was performed as previously described.40 Briefly, A549 cells in 6-well plate (1 × 105 cells per well) were treated with test compound (or 0.2% DMSO for control). After 24 h treatment, the treated cells were harvested and subjected to cell cycle analysis using Muse Cell Cycle Assay kit (Luminex, Austin, TX, USA) and analyzed with Muse Cell Analyzer™ with (Luminex). The number of cells in each cell cycle phase was expressed as percentage of total cells.4.5. Aromatase inhibition assay
Aromatase enzyme was purchased from GENTEST, Woburn, MA01801, USA (Supersom human CYP19+oxidoreductase, baculovirus/insect cell-expressed; Product No. 456260). The aromatase inhibitory activity was performed using Human CYP19 enzymes and DBF as a fluorometric substrate as described by Stresser, et al.41 with minor modifications. The mixture, containing 100 µL of cofactor/serial dilution buffer was pre-incubated in 37 °C for 10 min. The reaction was initiated by addition of 100 µL of enzyme/substrate and 10 µL of test compounds (in 10% DMSO) or 10% DMSO as solvent control or 5 µM letrozole (6.3 nM final concentration) as positive control. After incubation at 37 °C for 30 min, the reaction was stopped by addition of 50 µL of 2.2 M NaOH. Fluorescence signal was monitored using an excitation wavelength of 490 nm and emission wavelength of 530 nm. The test compounds showing inhibition >50% were expressed as the half-maximal inhibitory concentration (IC50).4.6. Cytotoxicity assay
T47D (ATCC HTB-133) was purchased from Biomedia (Thailand) Co., LTD. L929 (ATCC CCL-1) was received as a gift from Prof. Tanapat Palaga, Department of Microbiology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand. The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone), 100 U mL−1 penicillin and 100 µg mL−1 streptomycin (Gibco) at 37 °C with 5% CO2.The antiproliferative activity was determined by MTT assay. Briefly, the cells were plated into a 96-well microplate at the density of 10000 cells per well. The cells were allowed to grow overnight before treated with the solutions of compounds in culture medium at the designated concentrations for 48 hours. The medium was then replaced with MTT solution (0.5 mg mL−1 MTT in culture medium) and incubated for 3 hours. A 100 µL of DMSO was used to dissolve the produced formazan. Absorbance at 570 nm was then measured using a microplate reader (Varioskan™ LUX multimode microplate reader, Thermo Scientific™). Percent viability was calculated relative to the vehicle control (DMSO).
4.7. Molecular docking calculations
The ligands were constructed using Chem3D Pro 12.0 and energy minimized using the MM2 (ref. 42) force field. The tubulin structure was downloaded from the Protein Data Bank (PDB):43 tubulin (PDB ID: 4O2B, resolution 2.3 Å),44 as aromatase (PDB ID: 3S7S, resolution 3.2 Å).45 The protein structure were prepared for the docking simulations, i.e., co-crystallised colchicine and spectator ligands were removed. The centre of the colchicine binding pocket of tubulin, and aromatase binding site were defined at coordinates (x = 13.222, y = 8.371, z = −23.331) and (x = 85.219, y = 49.732, z = 42.202), respectively, with 10 Å radius. The docking runs were set at fifty docking runs per ligand with default search efficiency (100%) to permit thorough searches for the best bioactive conformations. Basic amino acids including lysine and arginine were defined as protonated, whilst acidic amino acids including aspartic and glutamic acids were deprotonated. The GoldScore (GS) scoring function was implemented to calculate the binding score and predict the binding modes, which was included in the GOLD version 2023.2.0 software suite.46 Discovery Studio 4.5 visualizer was used as a visualizer to examine and analyse the predicted binding modes and intermolecular interactions.4.8. Calculations of physicochemical properties
The Dragon 7.0 software package was automated to calculate the molecular descriptors of the derivatives: MWs, number of HAs and HDs, number of RBs, PSA and MlogP. The ADMETlab 3.0 web server was automated to predict for the toxicities of the compounds.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Rungroj Saruengkhanphasit: conceptualization, project administration, methodology, data curation, writing – original draft, writing – review & editing, funding acquisition. Jaruwan Chatwichien: methodology. Lukana Ngiwsara: methodology. Kriengsak Lirdprapamongkol: methodology, writing – review & editing, funding acquisition. Worawat Niwetmarin: methodology, writing – review & editing. Chatchakorn Eurtivong: conceptualization, methodology, writing – review & editing. Prasat Kittakoop: conceptualization, data curation, supervision, writing – review & editing. Jisnuson Svasti: data curation, writing – review & editing. Somsak Ruchirawat: data curation, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work (Grant No. RGNS 65-207) was supported by Office of the Permanent Secretary, Ministry of Higher Education, Science, Research and Innovation (OPS MHESI), Thailand Science Research and Innovation (TSRI) and Chulabhorn Graduate Institute. Additional supports in part were from the Center of Excellence on Environmental Health and Toxicology (EHT), OPS, Ministry of Higher Education, Science, Research and Innovation; and Thailand Science Research and Innovation (TSRI), Chulabhorn Research Institute (Grant No. 49890/4759797 and 49893/4759807).We thank S. Sitthimonchai from Biological Activity Testing Service, CRI, Thailand, for aromatase assays.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef.
- R. W. Brueggemeier, J. C. Hackett and E. S. Diaz-Cruz, Endocr. Rev., 2005, 26, 331–345 CrossRef CAS PubMed.
- T. Reinert, B. D. Paula, M. N. Shafaee, P. H. Souza, M. J. Ellis and J. Bines, Chin. J. Clin. Oncol., 2018, 7, 25 CrossRef.
- E. Amir, B. Seruga, S. Niraula, L. Carlsson and A. Ocaña, J. Natl. Cancer Inst., 2011, 103, 1299–1309 CrossRef CAS PubMed.
- A. S. Coates, A. Keshaviah, B. Thürlimann, H. Mouridsen, L. Mauriac, J. F. Forbes, R. Paridaens, M. Castiglione-Gertsch, R. D. Gelber, M. Colleoni, I. Láng, L. D. Mastro, I. Smith, J. Chirgwin, J.-M. Nogaret, T. Pienkowski, A. Wardley, E. H. Jakobsen, K. N. Price and A. Goldhirsch, J. Clin. Oncol., 2007, 25, 486–492 CrossRef CAS PubMed.
- P. E. Lønning and H. P. Eikesdal, Endocr.-Relat. Cancer, 2013, 20, R183–R201 Search PubMed.
- A. F. Sobral, C. Amaral, G. Correia-da-Silva and N. Teixeira, J. Steroid Biochem. Mol. Biol., 2016, 163, 1–11 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS.
- T. Fojo and M. Menefee, Ann. Oncol., 2007, 18, v3–v8 CrossRef.
- N. Watanabe, Y. Ootawa, K. Kodama, A. Kaide, N. Ootsuka and J. Matsuoka, Breast Cancer, 2010, 17, 247–253 CrossRef.
- P. A. Young, D. C. Márquez-Garbán, Z. S. Noor, N. Moatamed, D. Elashoff, T. Grogan, T. Romero, H. Sasano, R. Saito, R. Rausch, N. Hamilton, S. M. Dubinett, E. B. Garon and R. J. Pietras, JTO Clin. Res. Rep., 2021, 2, 100150 Search PubMed.
- M. M. Shaaban, M. Teleb, H. M. Ragab, M. Singh, B. H. Elwakil, L. A. Heikal, D. Sriram and M. A. Mahran, Bioorg. Chem., 2024, 145, 107179 CrossRef CAS.
- A. B. Velappan, D. Kesamsetty, D. Datta, R. Ma, N. Hari, S. G. Franzblau and J. Debnath, Eur. J. Med. Chem., 2020, 208, 112835 CrossRef CAS.
- J. He and K. Y. Tam, Drug Discovery Today, 2024, 29, 103914 CrossRef CAS.
- J. P. S. Ferreira, H. M. T. Albuquerque, S. M. Cardoso, A. M. S. Silva and V. L. M. Silva, Eur. J. Med. Chem., 2021, 221, 113492 CrossRef CAS.
- N. S. Tibon, C. H. Ng and S. L. Cheong, Eur. J. Med. Chem., 2020, 188, 111983 CrossRef CAS.
- T. Liang, L. Cen, J. Wang, M. Cheng, W. Guo, W. Wang, C. Yu, H. Zhang, Y. Wang, Z. Hao, J. Jin, Y. Wu, T. Jiang, Q. Zhu and Y. Xu, Bioorg. Med. Chem., 2023, 96, 117354 CrossRef CAS.
- J. Zhang, X. Liu, N. Sa, J.-H. Zhang, Y.-S. Cai, K.-M. Wang, W. Xu, C.-S. Jiang and K.-K. Zhu, Eur. J. Med. Chem., 2024, 269, 116341 CrossRef CAS.
- D. Yevale, N. Teraiya, T. Lalwani, M. Dalasaniya, K. Kapadiya, R. K. Ameta, C. B. Sangani and Y.-T. Duan, Bioorg. Chem., 2024, 147, 107323 CrossRef CAS.
- S. Kurup, D. Gesinski, K. Assaad and A. Reynolds, Bioorg. Med. Chem. Lett., 2024, 100, 129612 CrossRef CAS PubMed.
- A. A. Mohamed, S. S. A. El-Hddad, A. K. B. Aljohani, F. Khedr, O. M. Alatawi, D. E. Keshek, S. Ahmed, M. Alsulaimany, S. A. Almadani, K. El-Adl and N. S. Hanafy, Bioorg. Chem., 2024, 143, 107062 CrossRef CAS PubMed.
- M. K. Verma, C. Samant, R. Kale, S. Patra, N. Mahajan, M. K. Gholve, A. Marisetti, B. Sunkara, A. Naik, M. Shingare, M. Reddy, A. M. Bokare, A. Akarte, S. Koul, P. B. Nigade, V. B. Patil, D. Modi, P. Ahirrao, S. Pawar, S. Kuldharan, L. Dinchhana, M. Mehta, J. Gundu, N. Jana, P. Vidhate, S. C. Mahangare, M. R. Shukla, R. N. Goel, M. Bhonde, R. K. Kamboj and V. P. Palle, Biochem. Biophys. Res. Commun., 2022, 637, 267–275 CrossRef CAS PubMed.
- G. Ana, P. M. Kelly, A. M. Malebari, S. Noorani, S. M. Nathwani, B. Twamley, D. Fayne, N. M. O'Boyle, D. M. Zisterer, E. F. Pimentel, D. C. Endringer and M. J. Meegan, Pharmaceuticals, 2021, 14, 169 CrossRef CAS.
- X. Deng, X. Deng, W. Ning, L. Xin, Q. Li, Z. Hu, B. Xie, K. Liang, C. Min, C. Dong, J. Huang and H.-B. Zhou, J. Med. Chem., 2023, 66, 11094–11117 CrossRef CAS PubMed.
- N. M. O'Boyle, J. K. Pollock, M. Carr, A. J. S. Knox, S. M. Nathwani, S. Wang, L. Caboni, D. M. Zisterer and M. J. Meegan, J. Med. Chem., 2014, 57, 9370–9382 CrossRef.
- R. Saruengkhanphasit, C. Butkinaree, N. Ornnork, K. Lirdprapamongkol, W. Niwetmarin, J. Svasti, S. Ruchirawat and C. Eurtivong, Bioorg. Chem., 2021, 110, 104795 CrossRef CAS PubMed.
- R. Saruengkhanphasit, L. Ngiwsara, K. Lirdprapamongkol, J. Chatwichien, W. Niwetmarin, C. Eurtivong, P. Kittakoop, J. Svasti and S. Ruchirawat, RSC Med. Chem., 2024, 15, 2483–2495 RSC.
- H.-Z. Zhang, J. Drewe, B. Tseng, S. Kasibhatla and S. X. Cai, Bioorg. Med. Chem., 2004, 12, 3649–3655 CrossRef CAS PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- M. Alvarez, K. Paull, A. Monks, C. Hose, J. S. Lee, J. Weinstein, M. Grever, S. Bates and T. Fojo, J. Clin. Invest., 1995, 95, 2205–2214 CrossRef CAS.
- Q. Pan, X. J. Yang, H.-M. Wang, X.-T. Dong, W. Wang, Y. Li and J.-M. Li, Cell Biochem. Biophys., 2012, 62, 185–191 CrossRef CAS PubMed.
- A. V. Taubenberger, B. Baum and H. K. Matthews, Front. Cell Dev. Biol., 2020, 8, 687 CrossRef.
- E. W. Bell and Y. Zhang, J. Cheminf., 2019, 11, 40 Search PubMed.
- D. Ramírez and J. Caballero, Molecules, 2018, 23, 1038 CrossRef.
- I. Moriguchi, S. Hirono, Q. Liu, I. Nakagome and Y. Matsushita, Chem. Pharm. Bull., 1992, 40, 127–130 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- L. Fu, S. Shi, J. Yi, N. Wang, Y. He, Z. Wu, J. Peng, Y. Deng, W. Wang, C. Wu, A. Lyu, X. Zeng, W. Zhao, T. Hou and D. Cao, Nucleic Acids Res., 2024, 52, W422–W431 CrossRef PubMed.
- W. Rodphon, P. Laohapaisan, N. Supantanapong, O. Reamtong, L. Ngiwsara, K. Lirdprapamongkol, C. Thongsornkleeb, N. Khunnawutmanothan, J. Tummatorn, J. Svasti and S. Ruchirawat, ChemMedChem, 2021, 16, 3750–3762 CrossRef CAS PubMed.
- D. M. Stresser, S. D. Turner, J. McNamara, P. Stocker, V. P. Miller, C. L. Crespi and C. J. Patten, Anal. Biochem., 2000, 284, 427–430 CrossRef CAS PubMed.
- N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- A. E. Prota, F. Danel, F. Bachmann, K. Bargsten, R. M. Buey, J. Pohlmann, S. Reinelt, H. Lane and M. O. Steinmetz, J. Mol. Biol., 2014, 426, 1848–1860 CrossRef CAS.
- D. Ghosh, J. Lo, D. Morton, D. Valette, J. Xi, J. Griswold, S. Hubbell, C. Egbuta, J. Wenhua, J. An and H. M. L. Davies, J. Med. Chem., 2012, 55, 8464–8476 CrossRef CAS.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra09000d |
| This journal is © The Royal Society of Chemistry 2025 |