
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of Mo2C nanoparticles on N-doped carbon as an electrocatalyst for efficient electrocatalytic hydrogen evolution†
Zheng Chen ,
Shuting Wang,
Hongyan Zhou and
Suyuan Zeng*
,
Shuting Wang,
Hongyan Zhou and
Suyuan Zeng*
School of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng, 252059, PR China. E-mail: drzengsy@163.com; Fax: +86-635-8230196; Tel: +86-635-8230614
First published on 12th March 2025
Abstract
Molybdenum carbide (Mo2C) has emerged as a subject of considerable research interest as an intrinsically Pt-like electronic and cost-effective alternative to platinum-based catalysts for the sustainable hydrogen evolution reaction (HER). However, a high carbonization temperature can easily cause the agglomeration of nanoparticles. Therefore, we employed a zeolitic imidazolate framework (ZIF)-assisted synthesis strategy, obtaining Mo2C/CN-T nanoparticles by calcining the precursor Mo-ZIF at high temperature and low pressure. Utilizing the carbon defects, nitrogen doping, and nanosized active sites provided by ZIFs, the as-prepared electrocatalyst enables swift catalytic reaction kinetics and superior activity stability. The as-prepared Mo2C/CN-850 manifests a high HER catalytic activity in acid and alkaline media with a small overpotential of 97 and 117 mV at 10 mA cm−2, respectively and good stability at the industrial current density above 200 mA cm−2.
1 Introduction
Hydrogen energy, characterized by its high energy density and zero carbon emissions, holds tremendous potential as an ideal candidate to solve the challenges of energy scarcity and environmental pollution.1–3 However, the majority of hydrogen is currently produced from fossils fuels rather than water electrolysis, which is apparently unsustainable in the long term.4–6 The electrocatalytic hydrogen evolution reaction (HER) represents the most promising approach for industrial hydrogen production because of the extensive variety of feedstocks, the minimal level of product impurities, and the exceptional purity of the resulting hydrogen.7,8 It is well-known that Pt-based materials are currently the most efficient catalysts for hydrogen evolution, however, their high cost and limited availability pose significant constraints on their large-scale application.9,10 Consequently, in HER research, a critical challenge is to develop cost-efficient and accessible electrocatalysts, prioritizing the creation of non-precious metal catalysts with Pt-like performance to promote sustainable and economical hydrogen production.11–13Among various alternatives, transition metal carbides (TMCs), which are primarily composed of earth-abundant transition metal elements and carbon, have attracted substantial attention in the scientific community. Incorporating carbon into the molybdenum lattice will result in an expanded d-band in the catalyst, which exhibits an electronic configuration akin to platinum, thus showing Pt-like catalytic activity in HER.14,15 Consequently, Mo2C has emerged as a highly promising noble metal-free catalyst with the potential to replace Pt.16,17
The conventional synthetic methods for nanosized carbides can be categorized into two types: solid–solid thermal reduction and gas–solid reaction. The former uses carbon-containing solid compounds such as, carbon nanotubes, active carbon, or organics as the carbon source, while the later involves gaseous carbon sources such as CH4, C4H10, C2H6, C3H8, and aromatic compounds14,18,19 as the starting materials. Despite the tremendous progress using the two methods mentioned above, the HER properties of the materials are still greatly influenced by the excessive carbon during the synthetic process. This phenomena may be associated with the scattered carbide phase nestled within a carbon matrix exhibits which limited the accessibility of reactants to the catalytic sites.20 Meanwhile, the excess carbon derived from the gaseous carbon source tends to accumulate on the surface of the carbide, envelope the active sites and consequently diminishes the HER electrocatalytic activity.21 Recently, numerous approaches to the synthesis of carbides to further improve the HER performance have been consistently put forward. Nguyen et al.22 utilized a solution process and electrospinning technique to achieve a liquid–solid–solid reaction, resulting in the production of nano-coral W2C with a uniform distribution of active sites. Qiang et al.19 have reported a different gas–solid–solid–solid reaction for the synthesis of Mo2C nanowires, wherein the carbon cloth acts as the solid carbon source, and NaCl-assisted sublimable MoO3 serves as the vapor phase molybdenum precursor, promoting the formation of active sites and preventing the aggregation of gaseous carbon. However, the methodologies reported for nanosized carbide synthesis tend to be intricate and expensive, which restricted their large-scale application. Therefore, identifying a scalable and straightforward method to enhance the hydrogen evolution performance and mitigate the agglomeration of MoxC-based electrocatalysts is crucial.
In this study, we propose a solution to address this challenge by employing the calcination of non-volatile, crystalline, pre-formed Mo-ZIF nanoparticles under high-temperature, low-pressure conditions. During the synthetic process, ZIFs are chosen as the solid carbon precursor due to their unique structural properties, abundant organic ligands, and controllable frameworks, which will slow the diffusion rate of carbon atoms from ZIFs to the molybdenum lattice, thereby not only suppressing the sintering of carbide nanoparticles but also preventing excessive carbon deposition on their surfaces. In this manner, we can accomplish the synthesis of nanosized and phase-pure Mo2C particles, exhibiting excellent HER catalytic activity together with long-term HER stability in both acid and alkaline electrolyte. Our research introduces a scalable and straightforward approach for the synthesis of high-quality nanostructured Mo2C using ZIFs, providing a valuable strategy for the design and fabrication of other high-performance TMCs catalysts for energy conversion applications beyond water electrocatalysis.
2 Experimental methods
2.1 Materials
2-Methylimidazole (C4H6N2, 98%), Nafion solution (5 wt%) and molybdenum(V) chloride (MoCl5, 99.6%) were purchased from Shanghai Aladdin Reagent Co., Ltd., methanol anhydrous (CH3OH, ≥99.8%), ethanol anhydrous (C2H6O, ≥99.7%) were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd., 20% Pt/C were purchased from MACKLIN Reagent Co., Ltd. All reagents were used directly without further purification.2.2 Preparation of electrocatalysts
2.3 Electrochemical measurements
For the electrochemical tests, 8.5 mg of the as-fabricated catalyst powder was dispersed in 1 mL of Milli-Q water, at room temperature under slightly ultrasonicated for 40 min to obtain a homogenous catalyst ink. Then, 20 μL of the ink was applied to a mirror-polished glass carbon electrode with a diameter of 5 mm. After air-drying, 5 μL of 0.5 wt% Nafion aqueous solution was dripped to the electrode and allowed to dry at room temperature, serving as a binder.In the HER test, the working electrode was placed into a three-electrode cell setup. In this setup, either an Ag/AgCl/KCl (3.3 M) electrode or a Hg/HgO electrode was selected as the reference electrode, while a platinum sheet served as the counter electrode in a cell filled with electrolyte (0.5 M H2SO4 or 1 M KOH). All measured potentials were converted to the reversible hydrogen electrode (RHE) scale using the equations: ERHE = EAg/AgCl + (0.2046 + 0.0592pH) for the Ag/AgCl reference electrode, and ERHE = EHg/HgO + 0.098 + 0.0592pH for the Hg/HgO reference electrode The polarization curves were corrected with 100% iR compensation.
The data were recorded using an electrochemical analysis station, (References 3000, Gamry Instruments, USA). Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) test were conducted using a glassy carbon rotating disk electrode (RDE). The LSV was employed in 1 M KOH and 0.5 M H2SO4 solutions at a scan rate of 5 mV s−1 to obtain the polarization curves, respectively. The double-layer capacitances (Cdl) using CV measurements were performed in the potential ranges of 0.075 to 0.125 V (vs. RHE, 1 M KOH) and 0.185 to 0.235 V (vs. RHE, 0.5 M H2SO4) utilizing scan rates varying from 10 to 100 mV s−1. Electrochemical impedance spectroscopy (EIS) was performed with the frequency from 105 Hz to 0.1 Hz to assess the resistance of the synthesized materials. The long-term stability was assessed through amperometric i–t curves recorded at overpotentials corresponding to current densities of 10 and 240 mA cm−2.
3 Results and discussion
3.1 Synthesis and characterizations of Mo2C/CN-T
As shown in Fig. 1, the preparation of Mo2C/CN-T begins with the self-assembly of 2-methylimidazole and molybdenum chloride to form Mo-ZIF at room temperature, followed by a simple carbonization reaction. During the synthetic process, the XRD peaks of MoO2 becomes weaker and weaker until it disappears at 850 °C. Meanwhile, the XRD peaks of Mo2C became stronger and stronger (Fig. S1a†). This indicates that during the carbonizing process, the precursor firstly transformed into MoO2 because of the residual oxygen in the quartz tube. Meanwhile, the 2-methylimidazole molecules will gradually carbonize to form the N doped carbon. As the temperature increased further, the as-formed MoO2 will be reduced by carbon and further react with carbon to form Mo2C. When the temperature continues to rise after the complete formation of molybdenum carbide, the plane of (002) will transform into (101) because the peak intensity ratio of (101) to (002) becomes larger and larger (Fig. S1b†), and the weak and broad peak of graphitic carbon (at approximately 25°) will become smaller and smaller.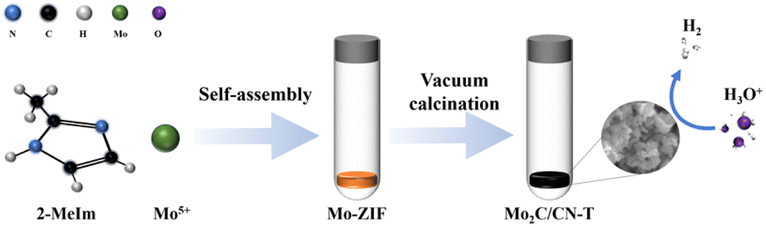 | ||
| Fig. 1 Schematic illustration for the synthesis of Mo2C/CN-T. | ||
The X-ray diffraction (XRD) pattern of the as-synthesized Mo-ZIF is depicted in Fig. S2,† showing consistency with the results reported previously.23 As illustrated in Fig. 2a, the XRD patterns of Mo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950 display typical peaks centering at 34.3, 37.9, 39.3, 52.1, 61.5, 69.5, 72.3, 74.6, and 75.5°, which can be designated to be the (100), (002), (101), (102), (110), (103), (200), (112), and (201) planes of Mo2C (JCPDS 35-0787), respectively. This outcome indicates that all the three samples have the same crystalline phase, and simultaneously confirms the successful synthesis of β-Mo2C,24 whose Fermi level energy aligns more closely to the corresponding value of Pt. Meanwhile, it can be inferred from the peak intensity of the (101) planes in the XRD pattern that the crystallinity of Mo2C/CN-T increases with the rise of temperature. Additionally, the sample of Mo2C/CN-850 has a better ratio of Mo2C (002) and (101) crystal planes, thereby contributing to excellent HER performance due to their synergistic effects.15,25
 | ||
| Fig. 2 (a) XRD pattern, (b) Raman spectrum of Mo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950 (c) N2 adsorption/desorption isotherms curve, the inset is pore size distribution, (d) TGA curve, (e) XPS survey spectrum, and (f) Mo 3d, (g) C 1s, (h) N 1s XPS high-resolution spectra of Mo2C/CN-850. | ||
Raman spectroscopy was employed to further prove the existence of carbon in Mo2C/CN-T. As shown in Fig. 2b, two distinct carbon bands can be observed: the D band at 1343 cm−1 and the G band at 1600 cm−1, which is related to the defective carbon and ordered graphitic carbon, respectively. With a peak intensity ratio (ID/IG) of approximately 0.98, Mo2C/CN-850 exhibits a significant degree of graphitic carbon amidst its structure, indicative of a carbonization process that has taken place under vacuum conditions. The ID/IG ratio for Mo2C/CN-900 is 0.91, while Mo2C/CN-950 exhibits almost no peak intensity, suggesting that higher temperatures are not favorable for the formation of defective carbon or the carbonization process. Such partial graphitization is beneficial to the enhancements in catalytic conductivity and can bring more electrons transport channels, which is consistent with the XRD analysis.26,27
The N2 adsorption/desorption isotherm reveals the specific surface and porous characteristics of samples Mo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950 (Fig. 2c and S3a, S3b†). Sample Mo2C/CN-850 exhibits a type IV isotherm with an H3-type hysteresis loop, which is typical for mesoporous materials. Samples Mo2C/CN-900 and Mo2C/CN-950 have type III isotherms. The Brunnauer–Emmett–Teller (BET) specific surface areas of the three samples are determined to be 20.2 m2 g−1, 22.3 m2 g−1 and 26.2 m2 g−1, respectively. Further analysis of the pore size distribution, as illustrated in the inset of Fig. 2c and S3a, S3b,† confirms the mesoporous properties of the three as-synthesized samples. The smaller pore size of Mo2C/CN-850, which offers the advantages of higher internal space utilization and shorter ion diffusion paths, facilitates the rapid diffusion of ions and the efficient transport of substances, benefiting from ZIFs having controllable frameworks and can suppress the sintering of carbide nanoparticles. According to the thermogravimetric analysis (TGA) in oxygen (Fig. 2e), in the sample of Mo2C/CN-850 is approximately 87 wt%. (More detailed TGA analysis are shown in Fig. S4†). This finding is crucial for enhancing HER performance, as it represents about 6.7 times the content of the carbon matrix, which is beneficial with an increased exposure of active sites.
The surface chemical information of chemical state and valence of the prepared catalysts was studied by X-ray photoelectron spectroscopy (XPS). As revealed in Fig. 2f, the typical survey spectrum indicates that sample Mo2C/CN-850 is primarily comprised of elements Mo, C, O, and N. The high resolution Mo 3d spectrum of Mo2C/CN-850 could be deconvoluted into three double peaks (Fig. 2g). The binding energy peaks centering at 228.6/231.7 eV are associated with Mo2+, indicating the existence of Mo2C species, which are highly active for the electrocatalytic HER process.28 The double peaks centering at 229.1/232.1 eV and 232.1/235.8 eV rare related to Mo4+ (MoO2) and Mo6+ (MoO3), respectively, originate from the Mo2C species.29 The presence of the Mo–Ox peaks is attributed to the inevitable surface oxidation of Mo2C upon exposure to the air.30 The high-resolution C 1s XPS spectrum (Fig. 2h) shows four fitted peaks. The binding energy peak centering at 283.6 eV is attributed to the Mo–C bonds,31 indicating the formation of Mo2C along with the ZIF-aided carbonization of Mo2C/CN-850. The primary peak at 284.8 eV and two weaker peaks at 286.3 and 286.7 eV are assigned to the C–C, C–O/C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CN bonds, respectively. The deconvolution of the N 1s XPS spectrum (Fig. 2i) reveals the characteristic peak of pyridinic-N at 397.8 eV, indicating the N doping into Mo2C/CN-850. The N element in Mo2C/CN-850 material may originate from 2-methylimidazole which can promote electrochemical performance of carbon-based molybdenum carbide by increasing the material's conductivity and the stability of the carbon matrix.26,32
O/CN bonds, respectively. The deconvolution of the N 1s XPS spectrum (Fig. 2i) reveals the characteristic peak of pyridinic-N at 397.8 eV, indicating the N doping into Mo2C/CN-850. The N element in Mo2C/CN-850 material may originate from 2-methylimidazole which can promote electrochemical performance of carbon-based molybdenum carbide by increasing the material's conductivity and the stability of the carbon matrix.26,32
Field emission scanning electron microscopy (FESEM) was also employed for the characterization of the fabricated catalysts. As presented Fig. S5,† the Mo-ZIF intermediate is consisted of nanoparticles ranging from approximately 200 to 400 nm in diameter, which could mitigate the agglomeration after carbonization. The series of Mo2C/CN-T samples were prepared by changing the temperature of carbonization. After carbonization, Mo2C/CN-850 still retains its 3D architecture but becomes smaller and loosely stacked (Fig. 3a and b), indicating that the original polyhedral morphology of the precursor was well preserved after the annealing process. The FESEM images show that Mo2C/CN-900 and Mo2C/CN-950 have a certain degree of agglomeration in contrast to Mo2C/CN-850, which is not conducive to the exposure of active sites (Fig. S6†).
 | ||
| Fig. 3 (a) and (b) FESEM images, (c–e) TEM and HRTEM images and (f) EDS color mapping images of Mo2C/CN-850. | ||
The microstructure of Mo2C/CN-850 catalyst was further characterized by the Transmission electron microscopy (TEM). As demonstrated in Fig. 3c, a rough surface can be observed, which originated from the phase generation of the newly formed Mo2C and generated amorphous carbon wrapped around the bulk surface. From Fig. 3d, the enlarged image exhibited a structure which was consistent with the SEM results, showing large number of active sites embedded in carbon matrix. The carbon matrix may not only help limit the coalescence and agglomeration of heterogeneous structure but also protect the active site from leaching in the harsh corrosive environments. Fig. 3e shows high-resolution TEM (HRTEM) image with clear lattice fringes of 0.26 nm, corresponding to the (100) crystal planes of Mo2C, consistent with other samples (Fig. S7†), indicating the excellent crystallinity of the as-prepared catalyst. Moreover, the Energy Dispersive Spectroscopy (EDS) mapping images in Fig. 3f suggest that Mo2C/CN-850 is composed of Mo, C and trace N elements, which is consistent with the XPS analysis.
3.2 Electrochemical measurements
To demonstrate the electrocatalytic performance, the sample powders were loaded onto the glassy carbon electrode and the HER performance was measured in 0.5 M H2SO4 using a standard three-electrode configuration. As shown in Fig. 4a, the commercial Pt/C catalyst exhibits the excellent electrocatalytic properties, consistent with ref. 4 and 19, with an onset overpotential of nearly zero. The LSV curves revealed that Mo2C/CN-850 has a low overpotential of 97 mV at a current density of 10 mA cm−2 (η10), in the Fig. 4b, which is lower than the corresponding values of samples Mo2C/CN-900 (139 mV) and Mo2C/CN-950 (158 mV), indicating that the degree of agglomeration of Mo2C/CN-T can affect catalytic activity. The outstanding HER properties is comparable to those of other reported molybdenum carbide-based catalysts such as Mo2C–Mo3C2 HWNs (η10 = 134 mV)33 MoC/Mo2C (η10 = 152 mV),34 Mo2C-900 (η10 = 187 mV),19 β-Mo2C (η10 = 220 mV),35 Mo2C@BNC(η10 = 184 mV),36 and many others37–42 (Fig. 4c). The Tafel slope is crucial for evaluating the performance of HER, identifying the rate-limiting step, and serves as a valuable tool in studying the mechanism of HER. The three reaction equations in acid medium as an example are shown as follows,43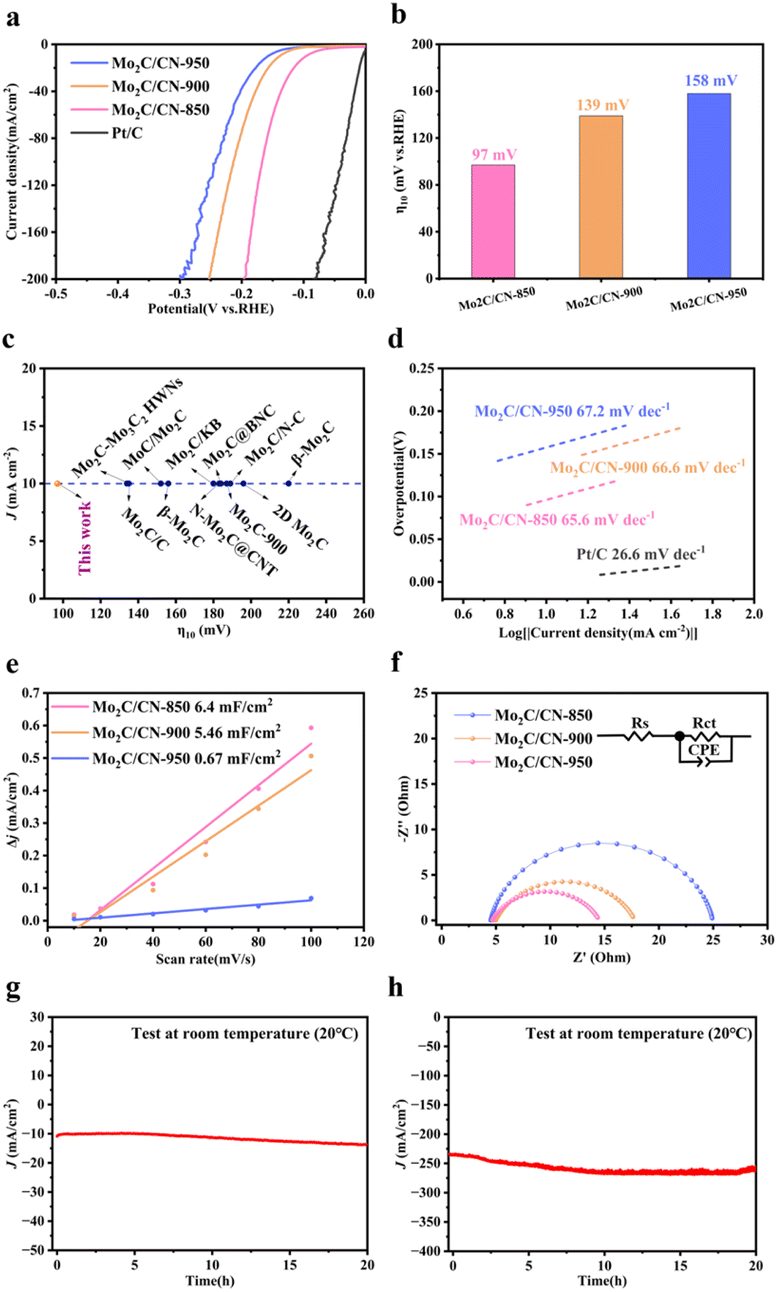 | ||
| Fig. 4 (a) Polarization curves of Mo2C/CN-850, Mo2C/CN-900, Mo2C/CN-950 and Pt/C in 0.5 M H2SO4 solution. (b) HER activity (overpotentials at 10 mA cm−2). (c) Comparison of overpotential values of the HER with previous reports. (d) Tafel plots of Mo2C/CN-850, Mo2C/CN-900, Mo2C/CN-950 and Pt/C. (e) Estimated Cdl, and (f) Nyquist plots of Mo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950, the inset is the equivalent circuit model. (g) and (h) Chronopotentiometry test of Mo2C/CN-850 at current densities 10 and 240 mA cm−2. | ||
Volmer step (discharge step):
| H3O+ + M + e− → M−Had + H2O | (1) |
Heyrovsky step (electrochemical atom + ion reaction step):
| M−Had + H3O+ + e− → H2 + H2O + M | (2) |
Tafel step (atomic combination step):
| 2M−Had → H2 + 2M | (3) |
The Tafel slopes for the Volmer reaction, Heyrovsky reaction, and Tafel reaction are 120, 40, and 30 mV dec−1, respectively. In the Fig. 4d, the Tafel slope of Mo2C/CN-850 is 65.6 mV decade−1, smaller than those of Mo2C/CN-900 (66.6 mV decade−1) and Mo2C/CN-950 (67.2 mV decade−1). This exhibits that the hydrogen evolution on Mo2C/CN-850 follows the Volmer–Heyrovsky reaction mechanism, with the rate-limiting step being determined to be Heyrovsky reaction. Additionally, the exchange current density (j0) is another important parameter obtained by extrapolating of the Tafel plot to zero overpotential, which can be calculated to evaluate the intrinsic catalytic activity and reflects the inherent rate of electron transfer between the catalyst and the electrolyte. The j0 values, as shown in Fig. S8,† indicate that Mo2C/CN-850 (0.3406 mA cm−2) is larger than those for Mo2C/CN-900 (0.087 mA cm−2) and Mo2C/CN-950 (0.0454 mA cm−2). Obviously, the results indicate that Mo2C/CN-850 exhibits superior inherent activity for HER performance. The electrochemical double-layer capacitance (Cdl), which is equivalent to the electrochemical active area (ECSA), reflects the active sites of the catalysts. To estimate the Cdl values of the as-prepared electrocatalysts from the linear relationship between the current density and scan rate, CV curves were collected with scan rates ranging from 10 to 100 mV s−1 (Fig. S9†). As shown in Fig. 4e, the Cdl values for samples Mo2C/CN-900 and Mo2C/CN-950 are determined to be 5.46 and 0.67 mF cm−2. Notably, Mo2C/CN-850 shows the higher Cdl value (6.4 mF cm−2), indicating there are more exposed electroactive sites and larger ECSA.
To further analyzed the intrinsic property of Mo2C/CN-850 and investigate the electron transfer kinetics, the charge transfer resistance (Rct) of the HER process was analyzed using the electrochemical impedance spectroscopy (EIS). A smaller value indicates a faster electron transfer rate for HER. As shown in Fig. 4f, compared to the Rct values of samples Mo2C/CN-900 (12.84 Ω) and Mo2C/CN-950 (20.44 Ω), the corresponding value of sample Mo2C/CN-850's R (9.852 Ω) is much smaller, revealing a faster charge-transfer process. More intuitive EIS fitting parameters are shown in Table S2.† This result is consistent with the trend observed in their HER activities. For the purpose of practical use, stability is a crucial parameter for the evaluation of a newly designed catalyst. The chronopotentiometric curves were conducted at constant current densities of 10 and 240 mA cm−2 in 0.5 M H2SO4 (Fig. 4h and i). The continuous i–t test for 20 hours showed no obvious change at the industrial current density (larger than 200 mA cm−2), indicating the high electrochemical stability.
The hydrogen evolution performances of the as-prepared samples were also investigated in 1 M KOH electrolyte (Fig. 5a). Fig. 5b illustrates that sample Mo2C/CN-850 exhibits a lower overpotential of 117 mV at a current density of 10 mA cm−2, which is much lower than the corresponding values of samples Mo2C/CN-900 (171 mV) and Mo2C/CN-950 (228 mV). As shown in Fig. 5c, Mo2C/CN-850 displays an excellent HER activity and outperforms most previously reported values for carbide-based electrocatalysts, such as Mo2C@BNC (145 mV),36 2D Mo2C (152 mV)38 Mo2C@N-CNFs (168 mV),44 Mo2C/NC-2 (180 mV),45 and many others.46–50
 | ||
| Fig. 5 (a) Polarization curves of Mo2C/CN-850, Mo2C/CN-900, Mo2C/CN-950 and Pt/C in 1 M KOH solution. (b) HER activity (overpotentials at 10 mA cm−2). (c) Comparison of overpotential values of the HER with previous reports. (d) Tafel plots of Mo2C/CN-850, Mo2C/CN-900, Mo2C/CN-950 and Pt/C. (e) Estimated Cdl, and (f) Nyquist plotsMo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950, the inset is the equivalent circuit model. (g) and (h) Chronopotentiometry test of Mo2C/CN-850 at current densities 10 and 240 mA cm−2. | ||
Tafel slopes for the as-prepared samples were also calculated, with Mo2C/CN-850 displaying the smallest value of 77.8 mV dec−1 compared to Mo2C/CN-900 (83.8 mV dec−1) and Mo2C/CN-950 (139.7 mV dec−1), indicating the Volmer–Heyrovsky kinetic mechanism (Fig. 5d). Fig. S10† indicates that the j0 of sample Mo2C/CN-900 and Mo2C/CN-950 are 0.087 and 0.0454 mA cm−2, respectively, which are lower than the corresponding value of Mo2C/CN-850 (0.3406 mA cm−2). The electrochemical Cdl of Mo2C/CN-T materials was obtained by the CV (Fig. S11†). As shown in Fig. 5d, Mo2C/CN-850 (5.04 mF cm−2) shows the larger Cdl value than Mo2C/CN-900 (0.55 mF cm−2) and Mo2C/CN-950 (0.11 mF cm−2), indicating there are more exposed electroactive sites and larger ECSA. The Nyquist plots of the Mo2C/CN-T are shown in Fig. 5f. Clearly, the Rct of Mo2C/CN-850, Mo2C/CN-900 and Mo2C/CN-950 are 2.812, 6.563 and 22.68 Ω, respectively. These results indicated that Mo2C/CN-850 have obviously advantage in HER. Fig. 5g and h demonstrate the excellent stability of Mo2C/CN-850 at constant current densities of 10 and 240 mA cm−2 over a period of 20 hours.
We found that Mo2C/CN-850 is a better electrocatalyst in both acidic and basic solutions. The high activity can be attributed to the carbon structures with a superior ID/IG ratio, which facilitate a better conductive environment, coupled with a more complete morphology that effectively exposes more active sites. At the same time, the sample's suitable ratio of Mo2C facets, (101) and (002), synergistically contributes to its excellent HER performance. The results implies that the carbonization temperature affects the morphology and crystallinity of molybdenum carbide, and high temperatures are detrimental to the HER performance of molybdenum carbide, which is detrimental to the catalytic conductivity and the peak intensity ratio of (101) to (002).15,25
4 Conclusion
In this study, we introduced a straightforward and adjustable ZIFs-assisted strategy that employs the carburization of non-volatile, crystalline, pre-formed Mo-ZIF nanoparticles under high-temperature, low-pressure conditions. Those nanosized ZIFs prevent Mo2C nanoparticles agglomeration and enhance their electron-transfer ability. The characteristics of the N-doped β-Mo2C were analyzed using different techniques, such as XRD, XPS, TG, BET, Raman spectroscopy, SEM and TEM. All data indicated the successful preparation of the Mo2C/CN-850 material. Electrochemical HER results indicate that Mo2C/CN-850 exhibited superior catalytic performance and stability, and it was found that calcination temperatures significantly influence the electrocatalytic activities of the materials. Hence, the as-prepared Mo2C/CN-T demonstrates providing insights into the role of TMCs electrocatalysts synthesized from ZIF precursors.Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Author contributions
Zheng Chen: writing – original draft and investigation. Suyuan Zhen: supervision and writing – review and editing. Shuting Wang and Hongyan Zhou: discussing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science Foundation of Shandong Province (ZR2016BQ41).References
- B. Yang, C. G. Wei, X. H. Wang, H. C. Fu, X. H. Chen, Q. Zhang, Y. H. Luo, H. Q. Luo and N. B. Li, Mater. Today Nano, 2023, 23, 100350 CrossRef CAS.
- X. Zhang, T. Liu, T. Guo, Z. Mu, X. Hu, K. He, X. Chen, V. P. Dravid, Z. Wu and D. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 40705–40712 CrossRef CAS PubMed.
- J. Hao, K. Wu, C. Lyu, Y. Yang, H. Wu, J. Liu, N. Liu, W.-M. Lau and J. Zheng, Mater. Horiz., 2023, 10, 2312–2342 RSC.
- H.-Q. Chang, G.-H. Zhang and K.-C. Chou, Electrochim. Acta, 2021, 394, 139119 CrossRef CAS.
- K. Zhu, Y. Zhang, R. Jiao, Y. Zhai, D. Wei, S. Zeng and L. Wang, CrystEngComm, 2023, 25, 2196–2203 RSC.
- R. Jiao, Z. Chen, S. Zeng, D. Wang and J. Li, J. Environ. Chem. Eng., 2023, 11, 111275 CrossRef CAS.
- W. Han, L. Chen, B. Ma, J. Wang, W. Song, X. Fan, Y. Li, F. Zhang and W. Peng, J. Mater. Chem. A, 2019, 7, 4734–4743 RSC.
- M.-I. Jamesh, H. Tong, S. P. Santoso, W. Niu, J.-J. Kai, C.-W. Hsieh, K.-C. Cheng, F.-F. Li, B. Han, J. C. Colmenares and H.-Y. Hsu, Nanoscale, 2024, 16, 18213–18250 RSC.
- P. Zhang, Y. Liu, T. Liang, E. H. Ang, X. Zhang, F. Ma and Z. Dai, Appl. Catal. B Environ., 2021, 284, 119738 CrossRef CAS.
- Q. Yu, H. Li, R. Li, S. Zeng, R. Li, Q. Yao, H. Chen, K. Qu and Y. Zheng, Carbon, 2022, 186, 171–179 CrossRef CAS.
- C. Yang, R. Zhao, H. Xiang, J. Wu, W. Zhong, W. Li, Q. Zhang, N. Yang and X. Li, Adv. Energy Mater., 2020, 10, 2002260 CrossRef CAS.
- S. Sarwar, A. Ali, Z. Liu, J. Li, S. Uprety, H. Lee, R. Wang, M. Park, M. J. Bozack, A. J. Adamczyk and X. Zhang, J. Colloid Interface Sci., 2021, 581, 847–859 CrossRef CAS PubMed.
- P. S. Kumar and P. Prakash, J. Environ. Chem. Eng., 2023, 11, 109045 CrossRef CAS.
- Y. Ma, G. Guan, X. Hao, J. Cao and A. Abudula, Renew. Sustain. Energy Rev., 2017, 75, 1101–1129 CrossRef CAS.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun and J. Zhou, Chem. Eng. J., 2023, 451, 138977 CrossRef CAS.
- Z. Zhou, Z. Liu, Q. Wang, Y. Jia, L. Zhang, J. Xiao and L. Guo, ACS Appl. Energy Mater., 2023, 6, 12084–12094 Search PubMed.
- I. H. Sajid, M. Z. Iqbal and S. Rizwan, RSC Adv., 2024, 14, 6823–6847 RSC.
- Q. Gong, Y. Wang, Q. Hu, J. Zhou, R. Feng, P. N. Duchesne, P. Zhang, F. Chen, N. Han, Y. Li, C. Jin, Y. Li and S.-T. Lee, Nat. Commun., 2016, 7, 13216 CrossRef CAS PubMed.
- M. Qiang, X. Zhang, H. Song, C. Pi, X. Wang, B. Gao, Y. Zheng, X. Peng, P. K. Chu and K. Huo, Carbon, 2022, 197, 238–245 CrossRef CAS.
- Q. Du, R. Zhao, T. Guo, L. Liu, X. Chen, J. Zhang, J. Du, J. Li, L. Mai and T. Asefa, Small Methods, 2021, 5, 2100334 CrossRef CAS PubMed.
- D. Geng, X. Zhao, Z. Chen, W. Sun, W. Fu, J. Chen, W. Liu, W. Zhou and K. P. Loh, Adv. Mater., 2017, 29, 1700072 CrossRef PubMed.
- T. V. Nguyen, Q. V. Le, C. C. Nguyen, T. P. Nguyen, D. V. Dao, J. H. Cho, S. H. Ahn and S. Y. Kim, Int. J. Energy Res., 2022, 46, 13089–13098 CrossRef CAS.
- S. Gong, Y. Niu, X. Teng, X. Liu, M. Xu, C. Xu, T. J. Meyer and Z. Chen, Appl. Catal. B Environ., 2022, 310, 121333 CrossRef CAS.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS PubMed.
- X. Chen, A. Jiang, X. Cao, S. Tao, L. Chen, H. Liu, L. Liu, X. Li and J. Xiao, J. Colloid Interface Sci., 2025, 680, 53–65 CrossRef CAS PubMed.
- D. Wang, X. Bai, H. Yang, G. Du, Z. Wang, J. Man, F. Du and P. Zhang, J. Energy Storage, 2024, 99, 113302 CrossRef.
- Y. Xiang, M.-Q. Li, P. Li, Y.-T. Ren, H.-G. Hao, J.-M. Dou, H.-Y. Ma, S.-N. Wang and Y.-W. Li, ACS Appl. Nano Mater., 2024, 8, 613–621 CrossRef.
- C. Shi, X. Li, W. Yang, X. Liu, Y. An, L. Zhou and L. Mai, Chem. Commun., 2022, 58, 5269–5272 RSC.
- N. Malone, H. Fiedler, D. R. G. Mitchell, J. V. Kennedy, G. I. N. Waterhouse and P. Gupta, ACS Appl. Eng. Mater., 2023, 1, 2377–2385 CrossRef CAS.
- W. Liu, X. Wang, J. Qu, X. Liu, Z. Zhang, Y. Guo, H. Yin and D. Wang, Appl. Catal. B Environ., 2022, 307, 121201 CrossRef CAS.
- J. Dong, Q. Wu, C. Huang, W. Yao and Q. Xu, J. Mater. Chem. A, 2018, 6, 10028–10035 RSC.
- B. Yu, D. Yang, Y. Hu, J. He, Y. Chen and W. J. S. m. He, Small Methods, 2019, 3, 1800287 CrossRef.
- L. Jia, C. Li, Y. Zhao, B. Liu, S. Cao, D. Mou, T. Han, G. Chen and Y. Lin, Nanoscale, 2019, 11, 23318–23329 RSC.
- P. Chen, L. Ouyang, C. Lang, H. Zhong, J. Liu, H. Wang, Z. Huang and M. Zhu, ACS Sustain. Chem. Eng., 2023, 11, 3585–3593 CrossRef CAS.
- B. Deng, Z. Wang, W. Chen, J. T. Li, D. X. Luong, R. A. Carter, G. Gao, B. I. Yakobson, Y. Zhao and J. M. Tour, Nat. Commun., 2022, 13, 262 CrossRef CAS PubMed.
- S. Wu, M. Chen, W. Wang, J. Zhou, X. Tang, D. Zhou and C. Liu, Carbon, 2021, 171, 385–394 CrossRef CAS.
- J. Fan, X. Wu, A. Piñeiro-García, N. Boulanger, Y. Panecatl-Bernal, A. Ashok, S. Koroidov and E. Gracia-Espino, ACS Appl. Nano Mater., 2021, 4, 12270–12277 CrossRef CAS.
- R. Liu, Q. Du, R. Zhao, X. Nie, L. Liu, J. Li and J. Du, ChemCatChem, 2020, 12, 3195–3201 CrossRef CAS.
- Y. Wang, R. A. Senthil, J. Pan, Y. Sun, S. Osman, A. Khan and X. Liu, Ionics, 2019, 25, 4273–4283 CrossRef CAS.
- M. Ji, S. Niu, Y. Du, B. Song and P. Xu, ACS Sustain. Chem. Eng., 2018, 6, 11922–11929 CrossRef CAS.
- D. Wang, J. Wang, X. Luo, Z. Wu and L. Ye, ACS Sustain. Chem. Eng., 2017, 6, 983–990 CrossRef.
- J. Guo, J. Wang, Z. Wu, W. Lei, J. Zhu, K. Xia and D. Wang, J. Mater. Chem. A, 2017, 5, 4879–4885 RSC.
- J. Hu, C. Zhang, X. Meng, H. Lin, C. Hu, X. Long and S. Yang, J. Mater. Chem. A, 2017, 5, 5995–6012 RSC.
- Z.-Y. Wu, B.-C. Hu, P. Wu, H.-W. Liang, Z.-L. Yu, Y. Lin, Y.-R. Zheng, Z. Li and S.-H. Yu, NPG Asia Mater., 2016, 8, e288 CrossRef CAS.
- S. Li, B. Dong, Z. Yuanyuan and P. Xu, ChemistrySelect, 2020, 5, 14307–14311 CrossRef CAS.
- Y. Liu, B. Huang, X. Hu and Z. Xie, Int. J. Hydrogen Energy, 2019, 44, 3702–3710 CrossRef CAS.
- X. Nie, T. Guo, Q. Du, R. Liu, L. Liu, R. Zhao, J. Zhang, J. Du and J. Li, ChemistrySelect, 2020, 5, 5974–5980 CrossRef CAS.
- M. Liu, Y. Yang, X. Luan, X. Dai, X. Zhang, J. Yong, H. Qiao, H. Zhao, W. Song and X. Huang, ACS Sustain. Chem. Eng., 2018, 6, 14356–14364 CrossRef CAS.
- X. Zhu, X. Zhang, B. Huang, J. Li and E. Wang, J. Mater. Chem. A, 2019, 7, 18304–18310 RSC.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, J. Chem. Sci., 2016, 7, 3399–3405 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08969c |
| This journal is © The Royal Society of Chemistry 2025 |