
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 methanation over Ni/SiO2–Al2O3 catalysts: effect of Ba, La, and Ce addition†
Muhammad Usman,
Seetharamulu Podila *,
Abdulrahim A. Al-Zahrani and
Majed A. Alamoudi
*,
Abdulrahim A. Al-Zahrani and
Majed A. Alamoudi
Chemical and Materials Engineering Department, Faculty of Engineering, King Abdulaziz University, P. O. Box 80204, Jeddah 21589, Saudi Arabia. E-mail: srpodila@kau.edu.sa; Tel: +966 531596482
First published on 7th April 2025
Abstract
One of the most technologically and financially feasible methods for managing anthropogenic CO2 emissions is CO2 hydrogenation to methane. However, the high efficiency of mostly used nickel-based catalysts is still a challenge in the CO2 methanation process. Herein, 10% silica–90% alumina, commercially known as SIRAL-10, was used as a support for nanostructured Ni catalysts. Modified SIRAL-supported nickel catalysts (Ni/SA) with Ba, La, and Ce metals as promoters were prepared by a simple wet impregnation method. These catalysts were tested for atmospheric CO2 methanation reaction in a 250–500 °C temperature range in a tubular fixed bed reactor with a H2/CO2 molar ratio of 4. As prepared samples were characterized by X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) analysis, hydrogen temperature-programmed reduction (H2-TPR), carbon dioxide temperature-programmed desorption (CO2-TPD), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), and X-ray photoelectron spectroscopy (XPS). CO2 methanation was found to be highly dependent on surface basic sites and Ni dispersion. Ni active sites were mainly obtained from the reduction of strongly interacted NiO at temperatures >700 °C. All promoted catalysts showed better catalytic activity than unpromoted nickel catalysts. Maximum CO2 conversion of 85.6% was obtained on the Ba-promoted sample at 400 °C, while low-temperature catalyst activity was achieved in the case of Ce–Ni/SA. The catalysts exhibited CH4 selectivity in the following order: Ce–Ni/SA > Ba–Ni/SA > La–Ni/SA > Ni/SA. The Ce-containing sample showed exceptional catalytic performance with about 78.4% CO2 conversion and 98% CH4 selectivity at 350 °C. Both Ba and Ce-promoted catalysts exposed the best stability for 24 hours. Unique features of the SIRAL support and the addition of basic promoters facilitated the sequential hydrogenation of CO2 to produce almost CO-free CH4.
1 Introduction
CO2 methanation is one of the core strategies in “Power to Gas (PtG)” technology that plays a crucial role in achieving carbon neutrality. This pathway uses H2 and CO2 to produce methane, which is a major constituent of natural gas.1,2 The rapid increase in natural gas costs and the depletion of its reserves have increased interest in synthetic or substitute natural gas (SNG) as a clean energy carrier due to its many advantages, including its high combustion efficiency and the ability to utilize existing gas pipelines.3,4 Modern methanation technology has advanced to the point where it is considerably easier to transform the CO2 methanation process into industrial-scale.4,5 However, the extremely exothermic nature of the CO2 methanation reaction (eqn (1)) makes thermal sintering and stability formidable obstacles, which in turn can reduce the efficiency of the most used catalysts for this process.6| CO2 + 4H2 ⇆ CH4 + 2H2O ΔH0298K = −165.5 kJ mol−1; ΔG = −113 kJ mol−1 | (1) |
Various catalytic systems have been suggested throughout the years to circumvent the kinetic obstacles of the CO2 hydrogenation to methane.7–10 Among the catalysts tested, Ru and Ni showed the greatest activity in CO2 methanation.11–14 As a result of their inexpensive cost and high CH4 selectivity, nickel-based catalysts are widely used in hydrogenation of CO2 to CH4.15–19 However, unlike precious Ru-based catalysts, Ni is susceptible to deactivation caused by carbon deposition and nickel sintering.20
The type of support greatly affects the distribution of active metallic sites and the interaction between the metal and the support material.21,22 Recent research has shown that specific acid–base or redox properties of supports can aid in the activation and ensuing transformation of CO2 and H2.23 As a possible support, alumina is utilized because of its large surface area and excellent thermal stability.3 Ru/Al2O3 is commonly used for low-temperature applications, whereas Ni/Al2O3 catalysts, such as those produced by Clariant and BASF, are preferred for situations where temperature control is less crucial.24 Furthermore, a lot of research on CO2 methanation with catalysts based on Ni/SiO2 has been conducted with CO2 conversion rates ranging from 30 to 90% depending on the catalyst preparation method, Ni loading, and reaction conditions.25–31 According to the literature,24,32 incorporating trace amounts of silica into alumina does not alter its chemical surface characteristics, but it does enhance the stability of Al2O3 concerning surface area loss and phase transitions of Al2O3.33. However, at higher loadings, the bulk and surface chemical properties are altered substantially. Zhang et al.34 presented data on a Ni/SiO2–Al2O3 catalyst synthesized using the grinding–mixing process. The catalyst exhibited a 76% conversion of CO with a selectivity of 80% towards CH4 at 350 °C and a pressure of 2.0 MPa.34 It was observed that the silica-containing catalyst exhibits higher activity compared to the catalyst supported by bare alumina.
The silica–alumina family of compounds is extremely valuable for adsorption and catalytic applications. Their acid strength can be adjusted by changing Lewis and Brønsted sites. These materials are very stable even at high temperature (>700 °C). They are preferable for some applications due to their decreased inclination to coke.33 These catalytic supports are highly versatile and beneficial because of their tunable dispersion ability for metal and cationic centers. Due to all these characteristics, silica–alumina materials have been utilized in light olefin oligomerization,35,36 diesel oxidation catalysts,37 hydrogenation of conjugated olefins,38 and CO & CO2 hydrogenation.32,39–42 The SIRAL series, made of commercially available amorphous silica–alumina, have been used as possible silica–alumina compounds. By adjusting the silica content, its combined Lewis and Brønsted acidic features can be fine-tuned.35,43
According to the literature, the addition of alkaline and alkaline earth metals raised the catalyst basicity to improve CO2 chemisorption, and the number of oxygen vacancies on the metal surface.44 Research on Ni/Al2O3-based catalysts indicates that Ba is the most effective promoter for CO2 methanation among Mg, Ca, Ba, and Sr. This is thought to be due to the surface formation of activated *CO and H2CO* species and a decrease in Ni particle size.45 The use of lanthanides as promoters to boost catalytic activity has recently exploded in popularity. This is because lanthanides can be highly incorporated into catalyst supports, leading to an increase in nickel dispersion within catalyst frameworks and improved catalytic activity.45–48 Cerium dioxide exhibits fascinating structural, redox, and alkaline properties and has been extensively employed in diverse applications.49 It has been shown that catalysts supported by various materials, such as alumina, silica, and zeolites, can enhance their catalytic performance in CO2 methanation when small quantities of cerium are added.50–54
Therefore, in this study, we provide our investigation on the utilization of Ni/SIRAL-10 (SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3 = 10:90 wt%) as a heterogeneous catalyst for the methanation of CO2. This catalyst can be generated simply and inexpensively by impregnating a Ni precursor on commercially available SIRAL-10. The effect of a small addition of Ba, La, and Ce on SIRAL-supported Ni catalysts is investigated and their physicochemical characteristics are analyzed by using different characterization techniques.
:Al2O3 = 10:90 wt%) as a heterogeneous catalyst for the methanation of CO2. This catalyst can be generated simply and inexpensively by impregnating a Ni precursor on commercially available SIRAL-10. The effect of a small addition of Ba, La, and Ce on SIRAL-supported Ni catalysts is investigated and their physicochemical characteristics are analyzed by using different characterization techniques.
2 Experimental
2.1. Materials
All chemicals were purchased from different suppliers (Table 1) and used without further purification except SIRAL-10, which was calcined at 750 °C for 5 hours in a static air.24,41| Reagents | Composition/formula | Purity | Supplier |
|---|---|---|---|
| SIRAL-10 | 10% SiO2–90% Al2O3 | >99% | Sasol (Germany) |
| Nickel precursor | Ni(NO3)2·6H2O | 99.9% | Fisher chemicals (UK) |
| Barium precursor | Ba (NO3)2 | 99% | Panreac (Spain) |
| Lanthanum precursor | La (NO3)3·6H2O | 99.9% | Fluka-SIGMA ALDRICH (Austria) |
| Cerium precursor | Ce(NO3)3·6H2O | 99.9% | Thermo scientific (China) |
2.2. Catalyst preparation
All the catalysts were prepared by using the wet impregnation method. The nickel loading was kept constant as 10 wt% for all samples. Nickel precursor was dissolved in an appropriate volume of deionized water and added to the support in a round bottom flask. The impregnation was conducted in a rotary evaporator at 80 °C and 150 rpm for 2 hours. After impregnation the solid dried at 110 °C overnight then calcined the sample at 750 °C for 5 hours with a heating rate of 10 °C min−1.In a similar fashion, promoted nickel catalysts supported on SIRAL-10 were prepared. Typically, to produce 10 gm of 10% Ni and 4% Ba on SIRAL-10; a solution of 8.6 gm of calcined support and 20 ml of deionized water was taken in a round bottom flask. Before adding nickel salt solution to the support solution, 0.4 gm of barium salt solution was added dropwise so that Ba could mix with acidic support. After that, 4.95 gm of nickel precursor solution was added to it. The final mixture was loaded in a rotary evaporator and then followed the same steps as mentioned above. All catalysts maintained the same loading (4 wt%) of the promoter. Hereafter, SIRAL-10 is denoted as “SA” and promoted-nickel over SIRAL-10 catalysts are designated as “X–Ni/SA (X = Ba, La, Ce)”.
2.3. Catalyst characterization
The crystal structures of calcined catalysts were investigated using X-ray powder diffraction (XRD) on a Shimadzu X-ray Diffractometer-6100. The diffraction was recorded with Cu-Kα radiation (λ = 1.54056 Å) in the 2θ range of 20–80°, with a scan speed of 5° min−1. The instrument operated at 30 kV and 30 mA. The crystalline structure size was calculated using Scherrer's equation:
 | (2) |
The percentage of metal dispersion of each catalyst was investigated by volumetric CO chemisorption analysis on Autochem 2950 Micromeritics apparatus. The detailed procedure is described in ESI and ref. 55.† The following equation56 was used to determine Ni dispersion (%).
 | (3) |
The surface area and pore size distribution of calcined catalysts were determined using the NOVA 2200e (manufactured by Quantachrome Instruments). The measurements were conducted at −196 °C. Prior to measurement, the samples underwent degassing at 200 °C for 2 hours under vacuum.
The H2-TPR experiments were conducted using a Micromeritics Autochem II 2950 apparatus with a thermal conductivity detector (TCD) at atmospheric pressure. Firstly, 0.1 g of calcined catalyst was loaded in a U-shaped quartz tube. Subsequently, the catalyst temperature raised linearly from room temperature to 120 °C under helium (He) with a flow rate of 15 ml min−1 and kept on the same condition for 60 minutes. This pretreatment step aimed to eliminate physically adsorbed water on the catalyst. Next, the catalyst was cooled down to 40 °C in the He environment. Finally, the analysis was conducted by heating the sample to 850 °C at a rate of 10 °C min−1. This analysis was carried out in a gas mixture of 10% hydrogen and 90% argon, with a flow rate of 40 ml min−1.
Temperature-programmed desorption with carbon dioxide (CO2-TPD) analyses were performed to assess the basicity of synthesized catalysts. The experiments conducted on the same instrument were used for H2-TPR analysis. Before analysis, 0.1 g of catalyst was reduced with 10% H2 and 90% Ar gas mixture (50 ml min−1) and subsequently, the catalyst was flushed with He gas. Next, the sample temperature was cooled to 40 °C under He flow. After that, the 10% CO2/He mixture gas was used to adsorb on the surface of reduced samples for 1 h at 40 °C. After completion of adsorption, the sample was flushed with He gas for one hour to remove physisorbed CO2. Then the temperature of the sample raised progressively at a ramping rate of 10 °C per minute from 40 °C to 850 °C.
The catalysts' surface morphology was examined using a scanning electron microscope (JSM-IT500 integrated with EDX). Using energy dispersive X-ray spectroscopy, an elemental composition analysis was performed to estimate the weight percentages of Ni, Ba, La, and Ce on SiO2–Al2O3.
X-ray photoelectron spectroscopy (XPS) was used to investigate the surface elemental composition of the calcined catalysts. Finely crushed samples were evaluated using Mg-Kα X-ray source in Specs GmbH (Berlin, Germany) equipment. The C 1s peak at around 285.0 eV was used for charge referencing to mitigate charging effects. XPSPEAK41 software was used to deconvolute the spectra peaks. Following the elimination of the non-linear (Shirley) background, high-resolution core-level spectra was utilized for quantitative surface chemical studies.
2.4. Catalyst activity
The CO2 methanation reaction was carried out at atmospheric pressure using MA-Effi (PID Eng & Tech, Spain) vertical continuous-flow fixed-bed reactor with an inner diameter of 9.1 mm and a length of 300 mm. Mass flow controllers (Bronkhorst) were used to regulate the movement of the feed gases. A k-type thermocouple was utilized to monitor the precise reaction temperature of the catalyst, which was positioned at the center of the tubular reactor, as shown in Fig. 1. A quantity of 300 mg of catalyst was placed onto quartz wool. The reactor's temperature was brought up to 750 °C at a rate of 10 °C min−1 under an Ar flow of 20 ml min−1. The catalyst was subjected to an in situ reduction process at 750 °C and 1 atm followed by a half-hour exposure to a flow of 20 ml min−1 of 99.99% pure hydrogen gas, prior to the reaction. The reduction conditions were adopted by referring to previous works of Riani et al.24,41 about Ni/SiO2–Al2O3 for CO2 methanation. After reduction, the temperature was reduced to 250 °C (initial reaction temperature) and the mixture of H2 and CO2 (with a molar ratio of H2/CO2 = 4/1) was fed into the reactor at a gas hourly space velocity (GHSV) of 12000 ml g−1 h−1. The catalytic experiments were conducted at various temperatures spanning from 250 to 500 °C with an increment of each 50 °C. Products were analyzed after 0.5 hours at each temperature, and an average of three readings is reported.
 | ||
| Fig. 1 Experimental setup for the CO2 methanation reaction. | ||
An online gas chromatogram (Agilent 7890 B) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID) was used to evaluate gas products. CO2, CO, and CH4 were analyzed by using TCD with a HayeSep Q-packed column. An FID detector with an HP-Pona capillary column (19091S-001E) was also employed for CH4 detection. Following equations57,58 were used to determine the CO2 conversion (XCO2), CH4 selectivity (SCH4), CO selectivity (SCO), and yield (Y) of methane.
 | (4) |
 | (5) |
 | (6) |
 | (7) |
 | (8) |
3 Results and discussion
3.1. Physiochemical characteristics of calcined catalysts
The powder XRD patterns of all calcined samples and support SA (10% SiO2–90% Al2O3) are shown in Fig. 2. Support sample revealed two clear diffraction peaks at 2θ = 66.7°, 46.5° and 37.64° representing γ-Al2O3.35 These peaks correspond to the alumina crystal plane of (440), (400) and (311) respectively (PDF#10-0425).60 All Ni/SA samples showed peaks at 2θ = 37.5° corresponding to the (111) plane of the NiO phase, consistent with previous studies.28,61,62 The appearance of sharp peak at 37.5° for all samples excluding the support shows that the presence of NiO formed by calcination at higher temperature (750 °C). The γ-Al2O3 peaks remained the same at both positions, but a small shift towards the lower angle and peak broadening observed in Ba, La and Ce promoted Ni/SA samples.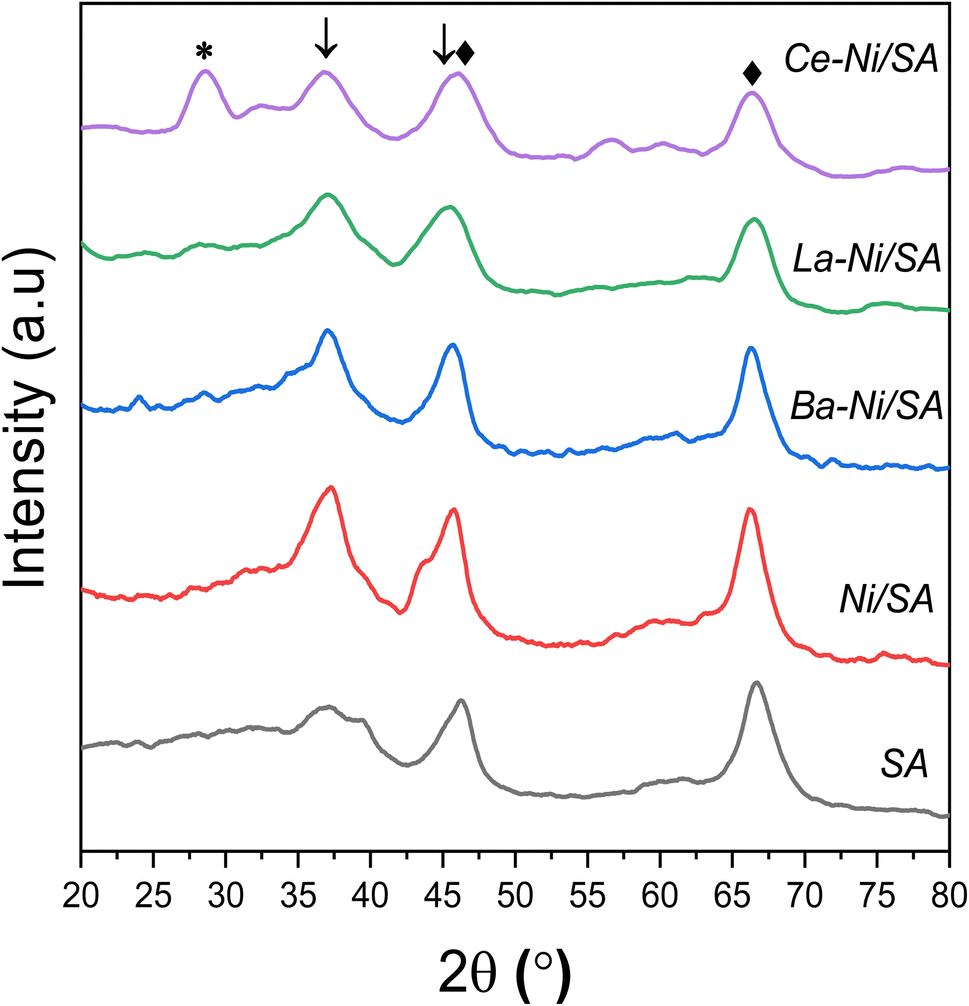 | ||
| Fig. 2 XRD patterns of calcined catalysts ♦-γ-Al2O3 [PDF#10-0425], *-CeO2 [PDF#034-0394], ↓-NiO [PDF#044-1159]. | ||
In the case of Ce promoted sample, CeO2 (111) plane at 28.5° and CeO2 (311) peak at 2θ = 56.3° were observed (PDF#034-0394).63–65 A minor shift toward lower angle from 2θ = 46.5° to 45° and from 67.4° to 66° in the case of Ce promoted sample is correlated with the gradual incorporation of Ni2+ into the Al–O crystal structure.66 No XRD signal at 28.2° observed corresponds to La2O3 (JCPDS 065-3185) or La(OH)3 (JCPDS 13-1481).67,68 This might be due to low crystal size or amorphous in nature. In the case of all promoted catalysis, broadening of diffraction peak of NiO at 37.5° was observed which might be due to the transformation of NiO to NiAl2O4 as described in previously reported Ni over silica doped alumina catalysts.32,41
The crystallite size and FWHM values of NiO are listed in Tables 2 and S2,† respectively. Lower Ni crystal size in promoted samples showed that addition of small amount of Ba, La and Ce is beneficial in increasing Ni dispersion on the silica–alumina support.
| Catalyst | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Pore sizeb (nm) | Total H2 consumptionc (μmol g−1) | Total CO2 desorptiond (μmol g−1) | NiO crystallinee size (nm) | Metal dispersionf (%) |
|---|---|---|---|---|---|---|---|
| a Calculated by BET method from N2 adsorption–desorption analysis.b Calculated by BJH method from N2 adsorption–desorption analysis.c Obtained from H2-TPR results from 100–850 °C range.d Obtained from CO2-TPD results from 100–850 °C range.e Calculated from XRD profiles of calcined sample at 2θ = 37.5°.f Estimated by CO pulse chemisorption analysis. | |||||||
| SA | 339 | 0.71 | 5.7 | — | 471.24 | — | — |
| Ni/SA | 276 | 0.61 | 5.7 | 761.27 | 975.61 | 3.84 | 5.33 |
| Ba–Ni/SA | 203 | 0.49 | 5.68 | 765.95 | 624.69 | 4.46 | 4.16 |
| La–Ni/SA | 234 | 0.53 | 5.68 | 817.85 | 585.39 | 3.30 | 5.62 |
| Ce–Ni/SA | 245 | 0.54 | 6.24 | 692.48 | 912.99 | 2.99 | 5.98 |
These results are in good correlation with CO chemisorption data displayed in Table S3† where we observed that the order of Ni average particle size in the catalyst samples is Ba–Ni/SA > Ni/SA > La–Ni/SA > Ce–Ni/SA.
Low temperature nitrogen adsorption and desorption experiments were performed to analyze textural properties of synthesized catalysts. A summary of obtained results is presented in Table 2. The addition of Ni on support (SA) results in decreased BET surface area and pore volume from 339 to 276 m2 g−1 and 0.71 to 0.61 cm3 g−1 respectively. This is due to blocking of support pores with Ni during preparation of Ni/SA catalyst.35 The introduction of promoters in Ni/SA the surface area and total pore volume further lowered. This is because the promoter loading causes a partial blockage of the support's pores. Maximum decline was experienced in the case of Ba promoted sample. It is interesting to observe that the pore size of Ce containing sample was more than all other samples including support. The interaction between Ni and CeO2 may be the reason for the larger pore size in the Ce-containing sample. This interaction alters the structure of support, facilitating the creation of oxygen vacancies.66 CeO2 has the capacity to produce oxygen vacancies and is well-known for its strong oxygen mobility; these properties can impact how Ni particles sinter during catalyst synthesis.69 These interactions may cause the support structure to reorganize, which in turn may alter the pore size and distribution. Increased mesopore size could be due to improved thermal stability and resistance to pore collapse on the Ce containing Ni/SA. The XRD study further indicated that the crystalline size of NiO in Ce–Ni/SA is smaller than that of other samples, which could lead to a bigger pore size.51 All samples exhibited type IV (a) isotherm and H1 hysteresis loop according to IUPAC isotherm classification (Fig. 3a) indicating mesoporous solids with open and some partially blocked pores.70
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of (a) fresh calcined catalysts (b) spent catalysts after reaction. | ||
Relative saturation point (p/po) detained at about 0.85 for all samples and for promoted catalysts a slight shift to 0.8 was observed. The occurrence of a hysteresis loop at high relative pressure (p/po > 0.7) indicates mesopores.71 There is no change observed in isotherm patterns in promoted catalysts comparison to that of Ni/SA catalyst. The pore size distribution of all samples is shown in Fig. 3b, representing a narrow distribution with pore diameter below 10 nm. Fig. 3B shows the outcomes of the physisorption isotherms for the spent catalysts. While the surface area values changed, the N2 adsorption–desorption isotherm patterns remained unchanged in the exhausted catalysts. The surface area values of all the spent catalysts decreased, except for Ba–Ni/SA, whose surface area rose from 203 to 220. The reduction of surface area might be due to the agglomeration of Ni particles on the surface72 and blockages of some mesopores.42 An increase in surface area after the reaction in the case of Ba containing sample might be due to the structural changes with enlargements of pores.73
Table S1† lists the BET surface area, pore volume, and pore size values of all the used catalysts.
Hydrogen temperature-programmed reduction (H2-TPR) was conducted to evaluate the reducibility and metal–support interactions of the synthesized catalysts. The H2-TPR profiles for both unpromoted and promoted nickel catalysts are presented in Fig. 4a. All catalysts exhibited a single reduction peak above 700 °C, indicating the presence of strongly interacting Ni2+ species with the support. Based on literature, high-temperature reduction peaks for Ni2+ can be attributed to two distinct species: (1) Ni2+ ions with strong direct interactions with the support and/or promoter, and (2) Ni2+ ions within the NiAl2O4 spinel phase.74 On other hand, some studies have reported low-temperature reduction peaks in nickel-based catalysts, corresponding to weakly interacting or bulk NiO, alongside the high-temperature peaks. This observation is typically associated with higher nickel loadings, which can lead to the formation of less dispersed NiO phases.42
 | ||
| Fig. 4 Pore size distribution for the catalysts (a) before reaction (b) after reaction. | ||
Table 2 presents hydrogen consumption for all catalysts. Notably, the total hydrogen consumption decreased upon the introduction of promoters. This reduction likely stems from increased NiO dispersion and enhanced interaction between NiO and the promoters & support. Among the promoted catalysts, La–Ni/SA exhibited the highest hydrogen consumption, potentially due to lanthanum facilitating the reduction of a greater proportion of Ni2+ species compared to barium and cerium promoted catalysts.75,76
Temperature Programmed Desorption (TPD) experiments were performed to analyze the total basicity of all prepared samples including the support. CO2 is a weak Lewis acidic molecule that can be absorbed on certain basic catalyst sites during the hydrogenation process. Adsorption of CO2 is used to recognize the strength and type of basic sites present in the catalyst. Reaction temperature is another factor that determines the adsorbed quantity.77,78 In the case of monometallic (unpromoted) sample, the Lewis basic sites of Ni and the Brønsted basic hydroxyl groups of the Al2O3 can adsorb CO2.79 The addition of promoter could form additional metal bicarbonates and shift the nature of catalysts80 Using TPD analysis up to 850 °C, we determined the amount of CO2 adsorbed at temperatures that are kinetically relevant for methanation. CO2-TPD profiles of all reduced samples are presented in Fig. 4b, while quantitative results are shown in Table 2. There are two clear peaks in the CO2 desorption profile: one at low temperatures (around 90–110 °C) and another at high temperatures (about 350 °C). The first peak represents physiosorbed CO2 (weak basicity) on SA surface. While the second peak suggested the chemisorption of CO2 on medium and strong basic sites.81 No more desorption peaks were observed for all samples until reaching 850 °C.
The amount of CO2 desorption was low on SA support, indicating mildly acidic nature of silica–alumina support. Impregnation of Ni on SA enhances the amount of CO2 desorption and peak maxima was shifted from around 360 °C to about 280 °C, showing the moderated basic nature of Ni supported catalyst. For all promoted samples, CO2 desorption value was increased, and maximum desorption peak was shifted toward lower temperature. The order of total basicity was found to be as: Ni/SA > Ce–Ni/SA > Ba–Ni/SA > La–Ni/SA > SA. According to the literature, the presence of strong basic sites does not favor methanation process, probably because CO2 sticks to the basic sites too tightly at reaction temperature, allowing it to be captured instead of converted.71,81,82
The surface morphology of the as prepared X–Ni/SA samples and pure SA support are presented in Fig. S4 and S5† respectively. Surface morphology of unpromoted Ni/SA is changed from irregular spherical form to spherical clusters with aggregates after the addition of La and Ba promoters. Plates like structures appeared at La and Ba containing samples. These agglomerations might be due to the basic nature of La and Ba, further discussed in XPS analysis. Ce–Ni/SA exhibits roughly spherical structure suggesting absence of any agglomeration of the particles on silica–alumina support.
The expected elemental composition in SA supported catalysts is indicated by the EDS peaks. With a uniform distribution of essential elements and effective dispersion, EDS analysis validates the theoretical weight% of Ni and promoters used during the synthesis of X–Ni/SA catalysts. In all samples, wt% of Ni was found above 8% while wt% of promoter was found above 3%, as shown in Fig. S4.† Overall, the SEM and EDS findings suggest that applied synthesis method for preparation of SiO2–Al2O3 supported Ni catalysts was appropriate.
X-ray photoelectron spectroscopy is a powerful surface analysis technique that provides information about the outermost few nanometers of heterogenous catalysts responsible for reaction. Species and their oxidation states present on the surface of X–Ni/SA catalysts were identified using XPS. Fig. 6 displays the Ni 2p spectra, which provides insights into the surface state of the active nickel species. The high-resolution XPS spectra of Ni 2p3/2 revealed three distinct peaks at binding energies of 853 eV, 856.5 eV, and 861 eV. Riani et al.41 investigated the interaction of Ni with SiO2–Al2O3 and reported that the presence of SiO2 in the support inhibits the formation of NiAl2O4, leading to the existence of two distinct Ni2+ species in the catalysts. Similarly, De Piano et al. examined the interaction of Ni with Al2O3 in the presence of Ce as a promoter. Their findings indicate the presence of two types of Ni2+ species: one exhibiting strong interaction with the support and the other forming Ni–Ox–Ce interfacial interactions.66,83 Numerous other studies have also reported the existence of various Ni2+ species in Ni/Al2O3 catalysts with different promoters.84–88 Based on these literature reports, the peaks observed in Fig. 6 can be attributed to different Ni2+ species, including direct interaction with the support, interface interactions with promoters (such as Ni–Ce and Ni–La), and Ni2+ species associated with NiAl2O4. In a barium-containing sample, the Ni 2p XPS signal observed at 860.9 eV could indicate the presence of NiO or a barium-interacted nickel species.89 This interaction may alter the sample's electronic environment, influencing the nickel binding energy. Notably, the binding energies of Ni2+ species are higher in Ba–Ni/SA catalysts compared to Ce–Ni/SA catalysts, suggesting a change in the nickel interaction strength due to the promoter, shifting from barium to cerium.
High-resolution XPS spectra of O 1s (Fig. 7) for all X–Ni/SA samples revealed two distinct peaks, corresponding to lattice oxygen (530 eV) and absorbed oxygen species (535 eV). Notably, the O 1s spectra of Ni/SA and Ba–Ni/SA exhibited no significant differences. Similar results were reported by Ayub et al.58 In contrast, La–Ni/SA and Ce–Ni/SA samples showed a significant increase in the intensity of the adsorbed oxygen peak. This adsorbed oxygen signal (OH−) is attributed to oxygen species trapped by electron-rich oxygen vacancies, with its intensity directly correlating to the surface oxygen vacancy concentration.90 The enhanced adsorbed oxygen peak in Ce–Ni/SA indicates stronger electronic interactions within this catalyst. To further understand the impact of the promoter on Ni/SA, surface atom quantification was performed, and the results are summarized in Table 3. The atomic-oxygen content followed the trend: Ce–Ni/SA > Ni/SA > La–Ni/SA > Ba–Ni/SA. This suggests that Ce–Ni/SA possesses a higher concentration of surface oxygen vacancies, facilitating the adsorption of gaseous oxygen.65
| Catalyst | Ni 2p3/2 | O 1s | Al 2p3/2 | Si 2p3/2 | C 1s | Ce 4d5/2 | Ba 3d5/2 | La 4d5/2 |
|---|---|---|---|---|---|---|---|---|
| Ni/SA | 19.08 | 23.22 | 22.28 | 12.39 | 23.03 | — | — | — |
| Ce–Ni/SA | 18.29 | 25.22 | 20.62 | 11.35 | 18.31 | 6.21 | — | — |
| Ba–Ni/SA | 16.88 | 18.98 | 20.80 | 15.68 | 20.39 | — | 7.27 | — |
| La–Ni/SA | 13.39 | 20.32 | 23.72 | 11.71 | 28.65 | — | — | 2.21 |
Surface active metal (Ni) percentages were high in Ce and Ba promoted, at 18% and 16%, respectively. Al 2p3/2 XPS spectra of all calcined samples are shown in Fig. S1.† As expected from the quantification results, the highest Al peak was observed at 78 eV in the case of Ni–La/SA.
3.2. Catalytic performance in CO2 methanation
The CO2 conversion and CH4 selectivity for each catalyst are presented in Fig. 8. The main products detected were CH4 and CO, confirming the occurrence of both CO2 methanation (eqn (1)) and the reverse water-gas shift (RWGS) reaction (eqn (9)). The CO2 conversion and CH4 selectivity increased with temperature, reaching their highest values at 400 °C, beyond which selectivity declined due to increased CO production via RWGS. This trend aligns with previous findings on Ni-based catalysts, where higher temperatures favor CO formation over CH4 due to thermodynamic limitations.27| CO2 + H2 ⇆ CO + H2O ΔH0298K = +41 kJ mol−1 | (9) |
The monometallic Ni/SA catalyst exhibited moderate CO2 conversion, reaching 61.2% at 400 °C, but suffered from lower CH4 selectivity (84.7%) due to significant CO formation. Maximum CO selectivity was observed in monometallic Ni/SA sample (Fig. S3†). After introducing promoters (Ba, La, and Ce), both CO2 conversion and CH4 selectivity improved significantly. The Ce–Ni/SA catalyst exhibited the highest CH4 selectivity (97.5% at 350 °C), while Ba–Ni/SA achieved the highest CO2 conversion (85.6% at 400 °C). The product yield was higher on Ce–Ni/SA at lower temperatures till 350 °C (Fig. S2†). The superior low-temperature activity of Ce–Ni/SA can be attributed to smaller NiO crystallite size (2.99 nm) (Table 2), leading to higher Ni dispersion and more active sites,91 formation of oxygen vacancies, and moderate basicity,84 ensuring effective CO2 adsorption while avoiding excessive carbonate formation that could hinder reaction kinetics (from CO2-TPD results). These findings match with studies reporting that Ce-doped Ni catalysts exhibit enhanced methanation activity at lower temperatures (<400 °C) due to oxygen vacancies and better Ni dispersion.6,51,66,91 In contrast, Ba–Ni/SA showed the highest CO2 conversion at 400 °C due to enhanced CO2 adsorption capacity (Fig. 5b), which facilitated surface hydrogenation. This behavior aligns with previous reports where alkaline earth metal promoters increase CO2 chemisorption, boosting conversion at higher temperatures.44,86 The La–Ni/SA catalyst, while improving over the unpromoted Ni/SA, showed relatively lower CO2 conversion (78.9% at 400 °C) and CH4 selectivity (93.4%). This could be due to its strong Ni–support interactions, leading to larger NiO crystallites (3.3 nm) and lower reducibility, as indicated by H2-TPR analysis (Fig. 5a). In summary, the best catalytic activity of Ce–Ni/SA sample can be attributed due to interaction of Ce with Ni2+ on SA support (from XPS results), large amount of medium basic sites and more oxygen vacancies that provide sites for hydrogen dissociation result in considerably higher CH4 selectivity.66
 | ||
| Fig. 5 (a) H2-TPR and (b) CO2-TPD profiles of X–Ni/SA catalysts. | ||
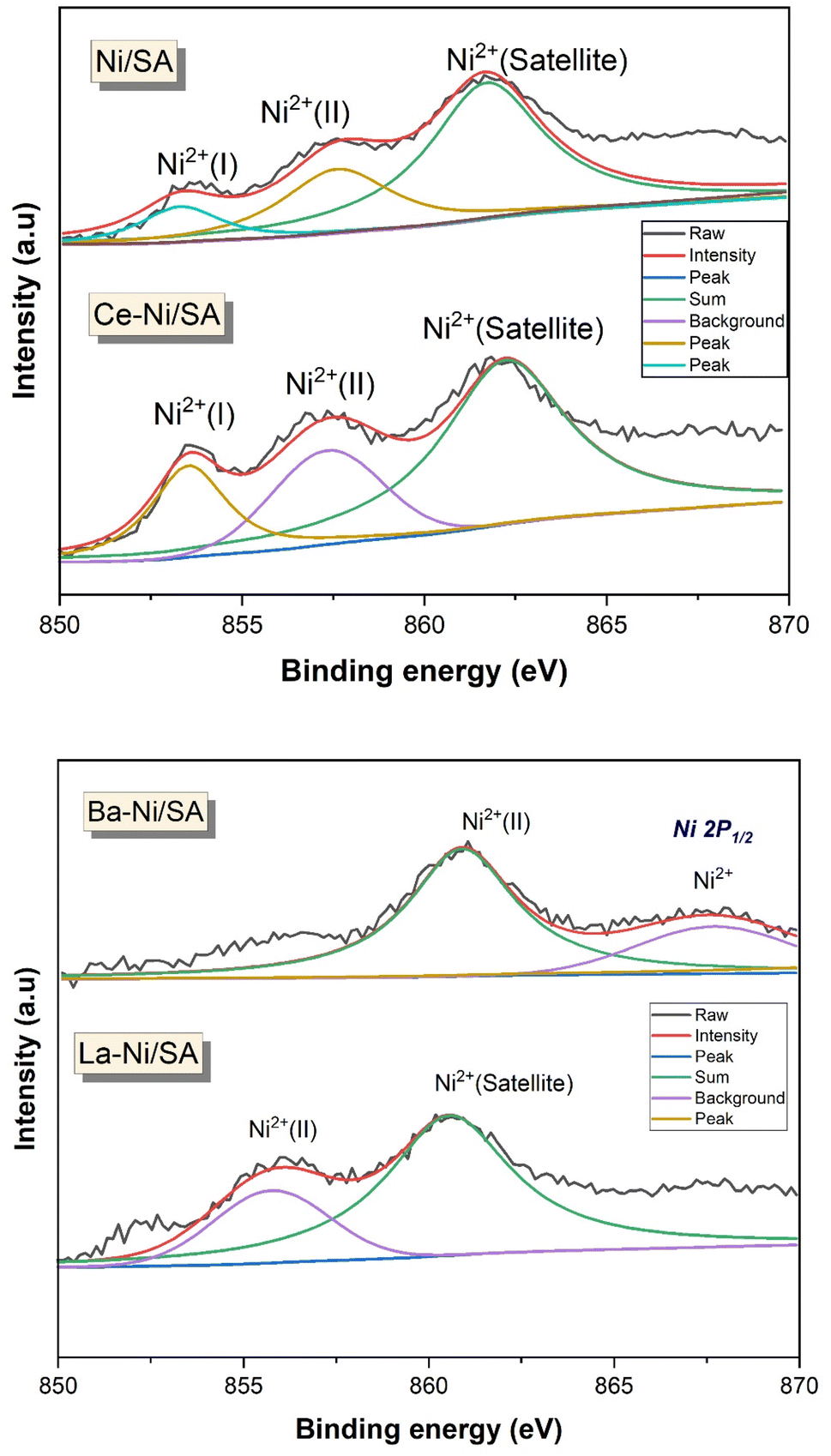 | ||
| Fig. 6 Ni 2p XPS spectra of calcined X–Ni/SA catalysts. | ||
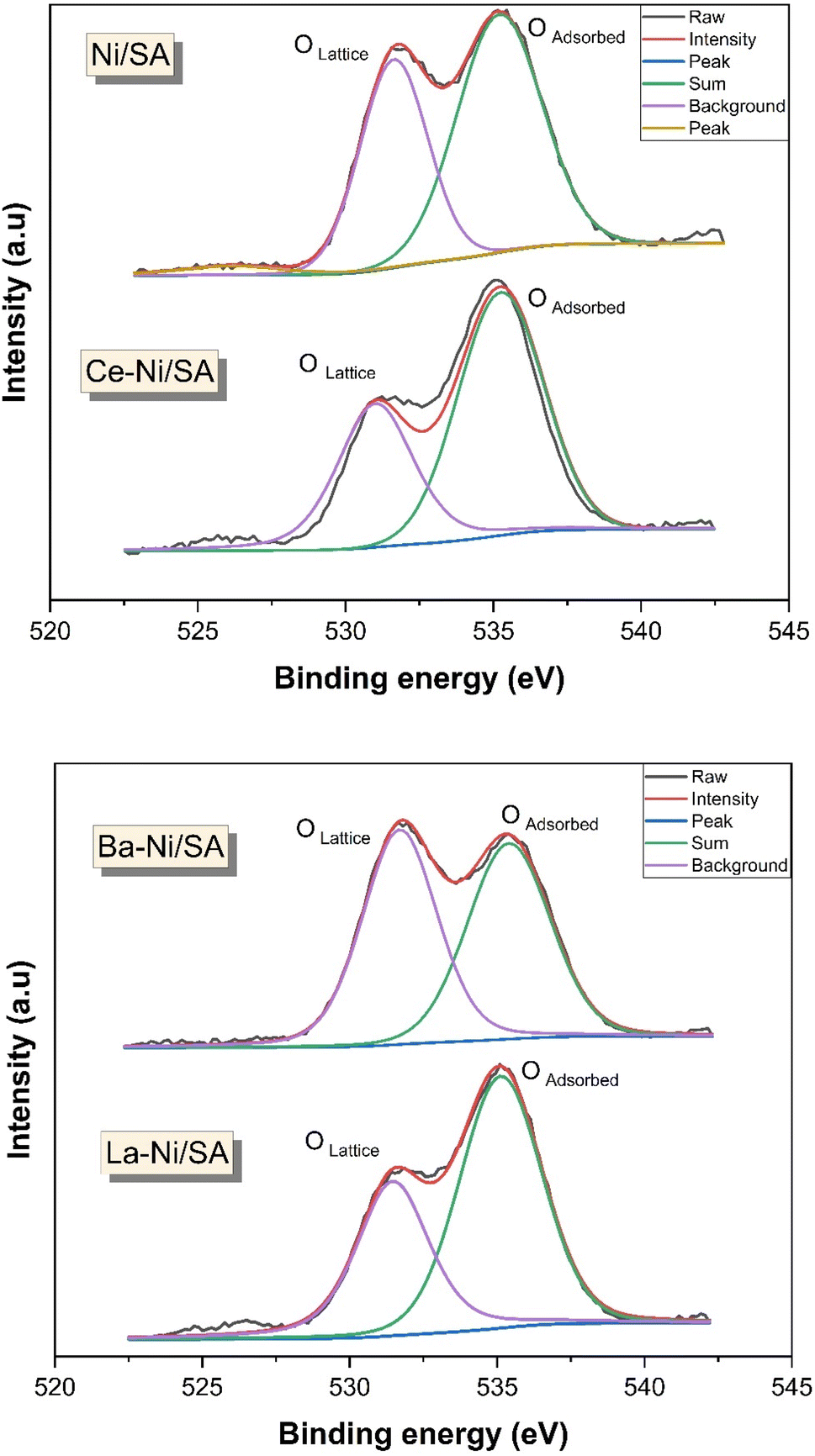 | ||
| Fig. 7 O 1s XPS spectra of calcined X–Ni/SA catalysts. | ||
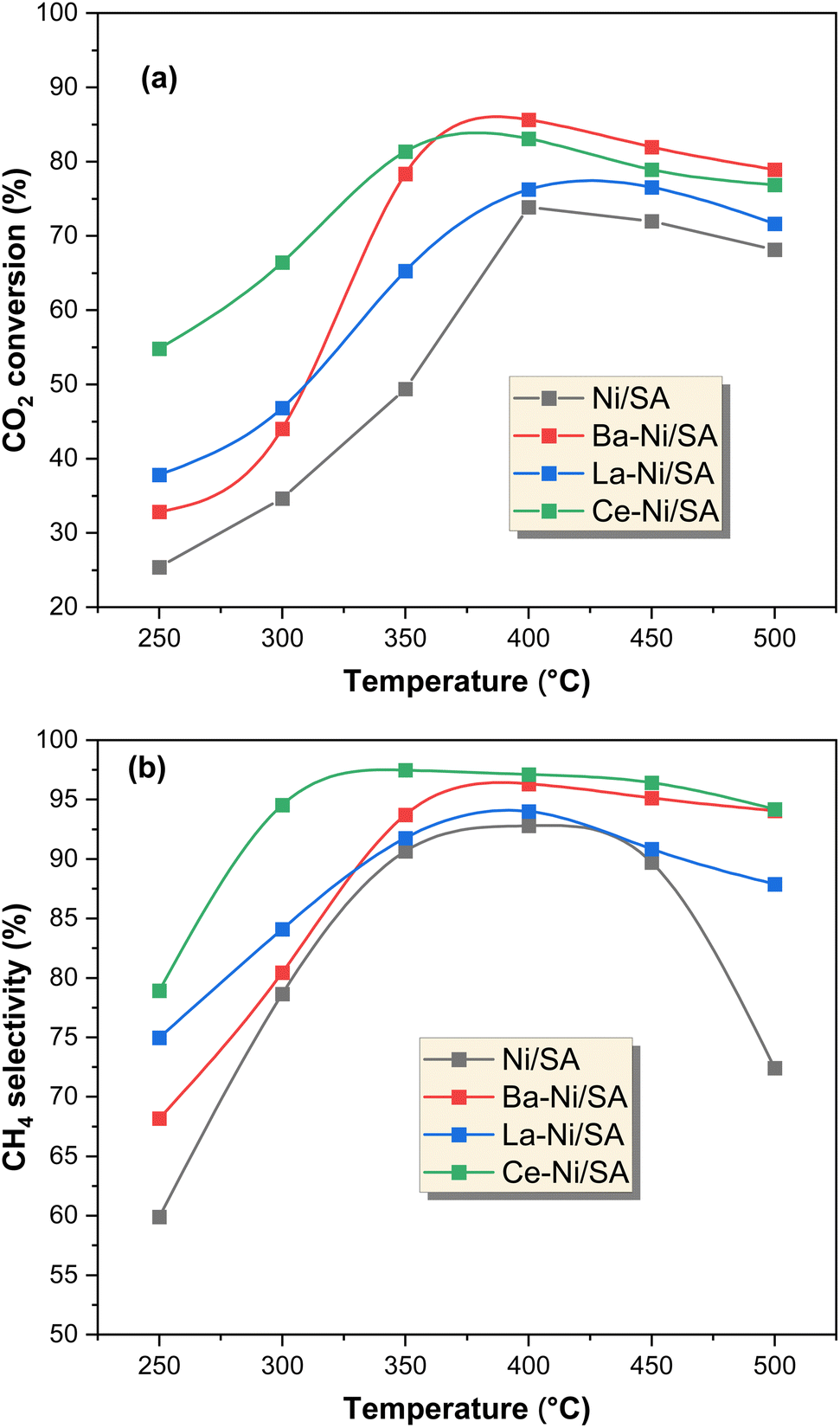 | ||
| Fig. 8 Catalytic activity results of X–Ni/SA catalysts at 1 atm, 250–500 °C, 12000 ml g−1 h−1, H2/CO2 = 4/1; (a) CO2 conversion, (b) CH4 selectivity. | ||
To evaluate the catalyst performance under long-term operation, Ce–Ni/SA and Ba–Ni/SA were tested for 24 hours at 400 °C (Fig. 10). Both catalysts exhibited stable CH4 selectivity, with Ba–Ni/SA maintaining 85.6% CO2 conversion, while Ce–Ni/SA showed a slight decline (2.5%) due to minor deactivation, possibly a lower Ni–Ce interactions at high temperatures. Furthermore, CO2 consumption rates at 250–350 °C (Fig. 9) confirmed that Ce–Ni/SA exhibited the highest reaction rate at lower temperatures, correlating well with its high Ni dispersion and oxygen vacancies. Table 4 compares the performance of our catalysts with previously reported Ni-based systems. The Ce–Ni/SA catalyst (97.5% CH4 selectivity at 350 °C) and Ba–Ni/SA (85.6% CO2 conversion at 400 °C) outperform many reported catalysts in terms of low-temperature CH4 selectivity and overall CO2 conversion, confirming the effectiveness of SIRAL-10 support and strategic promoter selection.
 | ||
| Fig. 9 CO2 consumption rates at three initial temperatures. | ||
 | ||
| Fig. 10 Catalyst stability of Ce and Ba promoted Ni/SA catalysts at 400 °C. | ||
| Catalyst | Reaction conditions | Performance | Ref. |
|---|---|---|---|
| 13.6% Ni/20% SiO2–80% Al2O3 | T = 250–500 °C; P = 1 atm | XCO2 = ∼66% | 24 |
| GHSV = 55000 h−1; H2/CO2 = 5 |
SCH4 = ∼100%; at 400 °C | ||
| 10% Ni/SiO2 | T = 200–450 °C; P = 1 atm | XCO2 = 68% | 92 |
| GHSV = 2400 h−1; H2/CO2 = 4 | SCH4 = 66%; at 400 °C | ||
| Ni-0.25% Mn/SIRAL-20 (20% SiO2–Al2O3) | T = 200–400 °C; P = 20 bar | XCO2 = 83% | 39 |
| GHSV = 60000 ml g−1 h−1; H2/CO2 = 4 |
SCH4 = >95% at 300 °C | ||
| 30% Ni/0.25% SiO2–99.75% Al2O3 | T = 200–500 °C; P = 1 atm | XCO2 = 82% | 93 |
| GHSV = 9000 ml g−1 h−1; H2/CO2 = 3.5 | SCH4 = 96% at 350 °C | ||
| 10% Ni/SiO2 | T = 200–400 °C; P = 1 atm | XCO2 = 67% | 94 |
| GHSV = 30 L g−1 h−1; H2/CO2 = 4 | SCH4 = 94% at 350 °C | ||
| 40% Ni/SiO2 | T = 290–470 °C; P = 1 atm | XCO2 = 30% | 42 |
| GHSV = 10000 ml g−1 h−1; H2/CO2 = 4 |
SCH4 = 83% at 330 °C | ||
| 13.6% Ni/1% SiO2–99% Al2O3 | T = 250–500 °C; P = 1 atm | XCO2 = ∼79% | 24 |
| GHSV = 55000 h−1; H2/CO2 = 5 |
SCH4 = ∼100%; at 400 °C | ||
| 5% Ni–5% Ba/Sm2O3 | T = 200–450 °C; P = 1 atm | XCO2 = 72.7% | 58 |
| GHSV = 2400 ml g−1 h−1; H2/CO2 = 4 | SCH4 = 100% at 450 °C | ||
| 10% Ni/(Al2O3–ZrO2) | T = 160–460 °C; P = 1 atm | XCO2 = 77% | 95 |
| GHSV = 6000 ml g−1 h−1; H2/CO2 = 4 | SCH4 = 100% at 340 °C | ||
| 15% Ni–1% Ru/Al2O3 | T = 250–550 °C; P = 1 atm | XCO2 = 88% | 96 |
| GHSV = 5835 h−1; H2/CO2 = 5 | SCH4 = 93% at 400 °C | ||
| 10% Ni–5% Ce/Al2O3 | T = 50–650 °C; P = 1 atm | XCO2 = 72% | 97 |
| GHSV = 7200 h−1; H2/CO2 = 4 | SCH4 = 98% at 400 °C | ||
| 10% NiO/(CaO–Al2O3) | T = 250–550 °C; P = 1 atm | XCO2 = 79.1% | 98 |
| GHSV = 18000 ml g−1 h−1; H2/CO2 = 4 |
SCH4 = 98.1% at 450 °C | ||
| 10% Ni–4% Ce/SIRAL-10 | T = 250–500 °C; P = 1 atm | XCO2 = 81.3% | This work |
| GHSV = 12000 ml g−1 h−1; H2/CO2 = 4 |
SCH4 = 97.5% at 350 °C | ||
| 10% Ni–4% Ba/SIRAL-10 | T = 250–500 °C; P = 1 atm | XCO2 = 85.6% | This work |
| GHSV = 12000 ml g−1 h−1; H2/CO2 = 4 |
SCH4 = 96% at 400 °C |
4 Conclusions
The catalytic activity was increased with a small addition of Ba, La and Ce on SiO2–Al2O3 supported Ni catalysts for CO2 methanation. The 10% Ni–4% Ce/SIRAL-10 catalyst displayed the highest CO2 methanation activity at 250–400 °C. Upon the addition of promoter, the incorporation of Ni into Al2O3 and oxygen vacancies increased. At high temperatures the Ba promoted catalyst is more active than the Ce promoted catalyst. The addition of Ce to the catalyst system plays a crucial role in optimizing Ni dispersion and preventing sintering. This dual function leads to catalysts with high activity and durability. The best catalytic activity of Ce–Ni/SA sample can be attributed due to better reducibility, high N2+ incorporation on SA support, large amount of medium basic sites and more oxygen vacancies that provide sites for hydrogen dissociation result in considerably higher CH4 selectivity. This study presents a simple preparation method to use silica–alumina class of compounds as a prospective support material for Ni-based catalysts for efficient CO2 conversion to methane.Data availability
All the data concerning this study added in the manuscript. There is no additional data available for the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by KAU Endowment (WAQF) at King Abdulaziz University, Jeddah, under grant no. WAQF:217-135-2024. The authors, therefore, acknowledge with thanks WAQF and the Deanship of Scientific Research (DSR) for technical and financial support.References
- R. Zhu, Q. Liu, Y. He and P. Liang, J. Colloid Interface Sci., 2024, 668, 352–365 CrossRef CAS PubMed.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10(1), 1–12 CrossRef CAS PubMed.
- J. Lin, C. Ma, J. Luo, X. Kong, Y. Xu, G. Ma, J. Wang, C. Zhang, Z. Li and M. Ding, RSC Adv., 2019, 9, 8684–8694 RSC.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- W. Gac, W. Zawadzki, M. Kuśmierz, G. Słowik and W. Grudziński, Appl. Surf. Sci., 2023, 157542, DOI:10.1016/j.apsusc.2023.157542.
- W. Gac, W. Zawadzki, G. Słowik, W. Grudziński and S. Dzwigaj, Catal. Today, 2024, 114728, DOI:10.1016/j.cattod.2024.114728.
- G. Yergaziyeva, Z. Kuspanov, M. Mambetova, N. Khudaibergenov, N. Makayeva and C. Daulbayev, J. CO2 Util., 2024, 80, 102682 CrossRef CAS.
- M. Younas, L. Loong Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- W. Gac, W. Zawadzki, M. Rotko, M. Greluk, G. Słowik and G. Kolb, Catal. Today, 2020, 357, 468–482 CrossRef CAS.
- M. A. Memon, Y. Jiang, M. A. Hassan, M. Ajmal, H. Wang and Y. Liu, Catalysts, 2023, 13, 1514 CrossRef CAS.
- K. Jalama, Catal. Rev., 2017, 59, 95–164 CrossRef CAS.
- L. P. Merkouri, J. L. Martín-Espejo, L. F. Bobadilla, J. A. Odriozola, M. S. Duyar and T. R. Reina, Nanomaterials, 2023, 13, 506 CrossRef CAS PubMed.
- S. Kumar, K. Singh Grewal and P. Kumar, A review of current developments in Sabatier reaction-based methanation of carbon dioxide: Thermocatalytic approach Search PubMed.
- M. Usman, A. G. Fareed and M. Amin, J. Iran. Chem. Soc., 2024, 21(5), 1185–1201 CrossRef.
- Z. Wang, L. Wang, Y. Cui, Y. Xing and W. Su, J. CO2 Util., 2022, 63, 102117 Search PubMed.
- L. Shen, J. Xu, M. Zhu and Y. F. Han, ACS Catal., 2020, 10, 14581–14591 CAS.
- L. Li, W. Zeng, M. Song, X. Wu, G. Li and C. Hu, Catalysts, 2022, 12, 244 CAS.
- G. Busca, E. Spennati, P. Riani and G. Garbarino, Energies, 2023, 16, 5304 CAS.
- N. D. M. Ridzuan, M. S. Shaharun, M. A. Anawar and I. Ud-Din, Catalysts, 2022, 12, 469 Search PubMed.
- M. Usman, S. Podila, M. A. Alamoudi and A. A. Al-Zahrani, Catalysts, 2025, 15, 203 CAS.
- Y. H. Lee, J. Y. Ahn, D. D. Nguyen, S. W. Chang, S. S. Kim and S. M. Lee, RSC Adv., 2021, 11, 17648–17657 CAS.
- M. A. Memon, W. Zhou, M. Ajmal, A. Afzal, Y. Jiang, C. Zhang, J. Zhang and Y. Liu, Int. J. Hydrogen Energy, 2024, 92, 1202–1213 CAS.
- S. Ewald, M. Kolbeck, T. Kratky, M. Wolf and O. Hinrichsen, Appl. Catal., A, 2019, 570, 376–386 CAS.
- P. Riani, E. Spennati, M. V. Garcia, V. S. Escribano, G. Busca and G. Garbarino, Int. J. Hydrogen Energy, 2023, 48, 24976–24995 CAS.
- M. Mihet, M. Dan, L. Barbu-Tudoran and M. D. Lazar, Catalysts, 2021, 443, DOI:10.3390/catal11040443.
- H. Ma, K. Ma, J. Ji, S. Tang, C. Liu, W. Jiang, H. Yue and B. Liang, Chem. Eng. Sci., 2019, 10–21 CAS.
- K. Wang, Y. Men, S. Liu, J. Wang, Y. Li, Y. Tang, Z. Li, W. An, X. Pan and L. Li, Fuel, 2021, 121388, DOI:10.1016/j.fuel.2021.121388.
- R. P. Ye, L. Liao, T. R. Reina, J. Liu, D. Chevella, Y. Jin, M. Fan and J. Liu, Fuel, 2021, 119151, DOI:10.1016/j.fuel.2020.119151.
- Y. R. Dias and O. W. Perez-Lopez, Energy Convers. Manage., 2020, 112214, DOI:10.1016/j.enconman.2019.112214.
- W. Gac, W. Zawadzki, G. Słowik, A. Sienkiewicz and A. Kierys, Microporous Mesoporous Mater., 2018, 272, 79–91 CAS.
- H. Liu, Y. Zhou, H. Cui, Z. Cheng and Z. Zhou, Ind. Eng. Chem. Res., 2024, 63, 10172–10183 CAS.
- G. Garbarino, S. Chitsazan, T. K. Phung, P. Riani and G. Busca, Appl. Catal., A, 2015, 505, 86–97 CAS.
- G. Busca, Catal. Today, 2020, 357, 621–629 CrossRef CAS.
- H. Zhang, Y. Dong, W. Fang and Y. Lian, Chin. J. Catal., 2013, 34, 330–335 CrossRef CAS.
- M. Lee, J. W. Yoon, Y. Kim, J. S. Yoon, H. J. Chae, Y. H. Han and D. W. Hwang, Appl. Catal., A, 2018, 562, 87–93 CrossRef CAS.
- S. Moussa, M. A. Arribas, P. Concepción and A. Martínez, Catal. Today, 2016, 277, 78–88 CrossRef CAS.
- J. Uchisawa, A. Obuchi, T. Tango and T. Murakami, J. Jpn. Pet. Inst., 2015, 58, 9–19 Search PubMed.
- M. A. Alamoudi, H. Almohamadi and K. J. Smith, Energy Fuels, 2021, 35, 10113–10121 Search PubMed.
- W. L. Vrijburg, G. Garbarino, W. Chen, A. Parastaev, A. Longo, E. A. Pidko and E. J. M. Hensen, J. Catal., 2020, 382, 358–371 Search PubMed.
- H. Zhang, Y. Dong, W. Fang and Y. Lian, Chin. J. Catal., 2013, 34, 330–335 CrossRef CAS.
- P. Riani, I. Valsamakis, T. Cavattoni, V. Sanchez Escribano, G. Busca and G. Garbarino, Appl. Catal., B, 2021, 119697, DOI:10.1016/j.apcatb.2020.119697.
- S. V. Moghaddam, M. Rezaei, F. Meshkani and R. Daroughegi, Int. J. Hydrogen Energy, 2018, 43, 19038–19046 Search PubMed.
- S. A. D'Ippolito, C. Especel, L. Vivier, F. Epron and C. L. Pieck, Appl. Catal., A, 2014, 469, 532–540 CrossRef.
- C. Liang, X. Hu, T. Wei, P. Jia, Z. Zhang, D. Dong, S. Zhang, Q. Liu and G. Hu, Int. J. Hydrogen Energy, 2019, 44, 8197–8213 CrossRef CAS.
- E. Spennati, P. Riani and G. Garbarino, Catal. Today, 2023, 114131, DOI:10.1016/j.cattod.2023.114131.
- J. B. Branco, P. E. Brito and A. C. Ferreira, Chem. Eng. J., 2020, 380, 122465 CrossRef CAS.
- M. Biset-Peiró, J. Guilera, T. Zhang, J. Arbiol and T. Andreu, Appl. Catal., A, 2019, 575, 223–229 Search PubMed.
- M. A. Salaev, L. F. Liotta and O. V. Vodyankina, Int. J. Hydrogen Energy, 2022, 47, 4489–4535 Search PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- M. C. Bacariza, I. Graça, J. M. Lopes and C. Henriques, ChemCatChem, 2018, 10, 2773–2781 CrossRef CAS.
- W. Gac, W. Zawadzki, M. Greluk, G. Słowik, M. Rotko and M. Kuśmierz, Catalysts, 2021, 13, DOI:10.3390/catal12010013.
- X. Wang, L. Zhu, Y. Liu and S. Wang, Sci. Total Environ., 2018, 625, 686–695 CrossRef CAS PubMed.
- W. Gac, W. Zawadzki, M. Rotko, G. Słowik and M. Greluk, Top. Catal., 2019, 62, 524–534 CrossRef CAS.
- Y. Xie, J. Wen, Z. Li, J. Chen, Q. Zhang, P. Ning, Y. Chen and J. Hao, Green Chem., 2023, 25, 130–152 RSC.
- S. Podila, A. A. Al-Zahrani, N. Pasupulety and M. A. Alamoudi, Arabian J. Chem., 2023, 105235, DOI:10.1016/j.arabjc.2023.105235.
- P. Yan, E. Kennedy and M. Stockenhuber, Green Chem., 2021, 23, 4673–4684 CAS.
- K. Wang, Y. Men, S. Liu, J. Wang, Y. Li, Y. Tang, Z. Li, W. An, X. Pan and L. Li, Fuel, 2021, 121388, DOI:10.1016/j.fuel.2021.121388.
- N. A. Ayub, H. Bahruji and A. H. Mahadi, RSC Adv., 2021, 11, 31807–31816 CAS.
- A. I. Tsiotsias, N. D. Charisiou, C. Italiano, G. D. Ferrante, L. Pino, A. Vita, V. Sebastian, S. J. Hinder, M. A. Baker, A. Sharan, N. Singh, K. Polychronopoulou and M. A. Goula, Appl. Surf. Sci., 2024, 158945, DOI:10.1016/j.apsusc.2023.158945.
- F. M. Segal, M. F. Correa, R. Bacani, B. Castanheira, M. J. Politi, S. Brochsztain and E. R. Triboni, Mater. Res., 2018, 21, e20170674 Search PubMed.
- P. Zhu, Q. Chen, Y. Yoneyama and N. Tsubaki, RSC Adv., 2014, 4, 64617–64624 CAS.
- X. Wang, S. Zhu, S. Wang, J. Wang, W. Fan and Y. Lv, Appl. Catal., A, 2018, 568, 231–241 CAS.
- M. Mihet, M. Dan, L. Barbu-Tudoran and M. D. Lazar, Catalysts, 2021, 443, DOI:10.3390/catal11040443.
- X. Yan, T. Hu, P. Liu, S. Li, B. Zhao, Q. Zhang, W. Jiao, S. Chen, P. Wang, J. Lu, L. Fan, X. Deng and Y. X. Pan, Appl. Catal., B, 2019, 246, 221–231 Search PubMed.
- L. Li, L. Jiang, D. Li, J. Yuan, G. Bao and K. Li, Appl. Catal. O: Open, 2024, 192, 206956 CAS.
- G. De Piano, J. J. Andrade Gamboa, A. M. Condó and F. C. Gennari, Int. J. Hydrogen Energy, 2024, 56, 1007–1019 CrossRef CAS.
- M. Dan, M. Mihet, Z. Tasnadi-Asztalos, A. Imre-Lucaci, G. Katona and M. D. Lazar, Fuel, 2015, 147, 260–268 CrossRef CAS.
- D. Méndez-Mateos, V. L. Barrio, J. M. Requies and M. Gil-Calvo, Environ. Sci. Pollut. Res., 2024, 31, 36093–36117 CrossRef PubMed.
- C. Tang, J. Li, X. Yao, J. Sun, Y. Cao, L. Zhang, F. Gao, Y. Deng and L. Dong, Appl. Catal., A, 2015, 494, 77–86 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- C. Italiano, J. Llorca, L. Pino, M. Ferraro, V. Antonucci and A. Vita, Appl. Catal., B, 2020, 264, 118494 CrossRef CAS.
- N. L. Visser, J. C. Verschoor, L. C. J. Smulders, F. Mattarozzi, D. J. Morgan, J. D. Meeldijk, J. E. S. van der Hoeven, J. A. Stewart, B. D. Vandegehuchte and P. E. de Jongh, Catal. Today, 2023, 418, 114071 CrossRef CAS.
- M. Usman, S. Podila and A. A. Al-Zahrani, Int. J. Hydrogen Energy, 2024, 95, 173–184 CrossRef CAS.
- A. J. Maia, B. Louis, Y. L. Lam and M. M. Pereira, J. Catal., 2010, 269, 103–109 CrossRef CAS.
- J. M. Rynkowski, T. Paryjczak and M. Lenik, Appl. Catal., A, 1993, 106, 73–82 CrossRef CAS.
- T. K. Phung and G. Garbarino, J. Ind. Eng. Chem., 2017, 47, 288–296 CrossRef CAS.
- A. Bustinza, M. Frías, Y. Liu and E. García-Bordejé, Catal. Sci. Technol., 2020, 10, 4061–4071 RSC.
- T. Burger, F. Koschany, A. Wenng, O. Thomys, K. Köhler and O. Hinrichsen, Catal. Sci. Technol., 2018, 8, 5920–5932 RSC.
- Y. Pan, C. J. Liu and Q. Ge, Langmuir, 2008, 24, 12410–12419 CrossRef CAS PubMed.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- E. García-Bordejé, A. B. Dongil, J. M. Conesa, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Nanomaterials, 1052, DOI:10.3390/nano12071052.
- X. Wang, Y. Hong, H. Shi and J. Szanyi, J. Catal., 2016, 343, 185–195 CrossRef CAS.
- J. M. Rynkowski, T. Paryjczak and M. Lenik, Appl. Catal., A, 1993, 106, 73–82 CrossRef CAS.
- C. Yang, J. Zhang, W. Liu, X. Yang, Y. Wang and W. Wang, J. Catal., 2024, 115811, DOI:10.1016/j.jcat.2024.115811.
- S. López-Rodríguez, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, I. J. Villar-Garcia, V. P. Dieste, J. A. O. Calvo, J. R. G. Velasco and A. Bueno-López, J. CO2 Util., 2022, 60, 101980 CrossRef.
- D. Méndez-mateos, V. Laura Barrio, J. M. Requies and J. F. Cambra, Catalysts, 2021, 11, 1–28 CrossRef.
- A. I. Tsiotsias, N. D. Charisiou, A. AlKhoori, S. Gaber, V. Stolojan, V. Sebastian, B. van der Linden, A. Bansode, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, J. Energy Chem., 2022, 71, 547–561 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Mater. Today, 2020, 19, 100591 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- Y. He, H. Shen, Y. Bai, X. Niu, Y. Zhao, C. Wu, S. Yang, Y. Cao, Q. Zhang and H. Zhang, Ind. Eng. Chem. Res., 2022, 61, 15948–15960 CAS.
- W. Gac, W. Zawadzki, M. Rotko, G. Słowik and M. Greluk, Top. Catal., 2019, 62, 524–534 Search PubMed.
- X. Guo, A. Traitangwong, M. Hu, C. Zuo, V. Meeyoo, Z. Peng and C. Li, Energy Fuels, 2018, 32, 3681–3689 CrossRef CAS.
- S. V. Moghaddam, M. Rezaei, F. Meshkani and R. Daroughegi, Int. J. Hydrogen Energy, 2018, 43, 19038–19046 Search PubMed.
- Y. Xu, Y. Wu, J. Li, S. Wei, X. Gao and P. Wang, Int. J. Hydrogen Energy, 2021, 46, 20919–20929 Search PubMed.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Appl. Catal., B, 2019, 243, 262–272 Search PubMed.
- R. Y. Chein and C. C. Wang, Catalysts, 2020, 10, 1112 CrossRef CAS.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612–619 CrossRef CAS.
- A. Rajabzadeh Nobakht, M. Rezaei, S. M. Alavi, E. Akbari, M. Varbar and J. Hafezi-Bakhtiari, Int. J. Hydrogen Energy, 2023, 48, 38664–38675 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08895f |
| This journal is © The Royal Society of Chemistry 2025 |