DOI:
10.1039/D4RA08875A
(Paper)
RSC Adv., 2025,
15, 8346-8353
Insight into the key factors of β-Mo2C catalyst for the reverse water gas shift reaction†
Received
19th December 2024
, Accepted 12th March 2025
First published on 18th March 2025
Abstract
In this study, we found that β-Mo2C prepared at different carbonization temperatures exhibited significantly different catalytic activities for the reverse water gas shift (RWGS) reaction. The β-Mo2C synthesized at 600 °C demonstrated notably higher activity compared to those prepared at 700 °C and 800 °C. This enhanced activity was likely attributed to its improved redox properties, which were primarily driven by a smaller crystallite size and the presence of MoxOCy species. Therefore, we proposed that the crystallite size and MoxOCy content were the key factors governing the RWGS activity of β-Mo2C. Clearly, both factors were strongly influenced by the carbonization temperature. Notably, the β-Mo2C prepared at 600 °C even outperformed Cu-doped β-Mo2C prepared at 700 °C under similar reaction conditions.
1 Introduction
The heightened levels of carbon dioxide (CO2) in the atmosphere are widely acknowledged as a key contributor to climate change and ocean acidification.1 Consequently, there has been considerable interest in capturing CO2 and converting it into fuels and vital chemicals. The reverse water gas shift (RWGS) reaction is a promising pathway for CO2 utilization, as the product CO can be directly utilized as a feedstock in the Fischer–Tropsch process, enabling further conversion into fuels and chemicals.2,3
Molybdenum carbide, in particular, has received much attention due to its capacity to facilitate both H2 dissociation and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage.4 It was reported that Mo2C exhibited even higher activity and selectivity for the RWGS reaction compared to noble metal-based bimetallic catalysts such as PtCo/CeO2 and PdNi/CeO2.5 Since then, the molybdenum carbides have been widely studied as catalysts in the RWGS reaction and these studies mainly focus on the following aspects.6 (i) The effect of crystal phase. To date, various phases of molybdenum carbide (e.g., α-MoC1−x, β-Mo2C and α-Mo2C) have been investigated for the RWGS reaction.7–18 It was found that β-Mo2C showed higher CO2 conversion compared to α-MoC1−x below 500 °C, but the latter exhibited higher CO selectivity.8 Very recently, α-Mo2C had been discovered to be highly efficient and selective for the RWGS reaction.7,11 This cubic molybdenum carbide can achieve equilibrium conversion and 100% CO selectivity at 600 °C for 500 h.11 Additionally, Sun et al. had recently identified a unique Mo oxycarbide (MoxOCy) structure that formed in situ on the surface of Mo oxide during the RWGS reaction. This structure was demonstrated to be more active than Mo carbide in the process.15 (ii) The effect of support. Various common supports, such as γ-Al2O3, SiO2, SBA-15, CN and CNTs, had been utilized for supporting Mo2C in the RWGS process.19–24 It was suggested that these supports generally offered surfaces for Mo2C dispersion and contributed to its stabilization against high-temperature sintering during the reaction.20 (iii) The effect of metal promoter. Promotion of molybdenum carbides with other metals (e.g. Co, Cu, Na, K and Cs) also significantly modified the activity, selectivity and stability in the RWGS reaction.5,19,25–31 Zhang et al. demonstrated that the strong interaction between Cu and β-Mo2C played a pivotal role in enhancing Cu dispersion and preventing agglomeration.25 This phenomenon contributed significantly to the remarkable activity and stability. Additionally, Cs-doped Mo2C catalysts can exhibit higher CO selectivity than non-doped one.27 The authors suggested that the electropositive nature of Cs facilitated electron transfer from Cs to Mo, resulting in a surface enriched with electrons, thereby favoring CO production.
O bond cleavage.4 It was reported that Mo2C exhibited even higher activity and selectivity for the RWGS reaction compared to noble metal-based bimetallic catalysts such as PtCo/CeO2 and PdNi/CeO2.5 Since then, the molybdenum carbides have been widely studied as catalysts in the RWGS reaction and these studies mainly focus on the following aspects.6 (i) The effect of crystal phase. To date, various phases of molybdenum carbide (e.g., α-MoC1−x, β-Mo2C and α-Mo2C) have been investigated for the RWGS reaction.7–18 It was found that β-Mo2C showed higher CO2 conversion compared to α-MoC1−x below 500 °C, but the latter exhibited higher CO selectivity.8 Very recently, α-Mo2C had been discovered to be highly efficient and selective for the RWGS reaction.7,11 This cubic molybdenum carbide can achieve equilibrium conversion and 100% CO selectivity at 600 °C for 500 h.11 Additionally, Sun et al. had recently identified a unique Mo oxycarbide (MoxOCy) structure that formed in situ on the surface of Mo oxide during the RWGS reaction. This structure was demonstrated to be more active than Mo carbide in the process.15 (ii) The effect of support. Various common supports, such as γ-Al2O3, SiO2, SBA-15, CN and CNTs, had been utilized for supporting Mo2C in the RWGS process.19–24 It was suggested that these supports generally offered surfaces for Mo2C dispersion and contributed to its stabilization against high-temperature sintering during the reaction.20 (iii) The effect of metal promoter. Promotion of molybdenum carbides with other metals (e.g. Co, Cu, Na, K and Cs) also significantly modified the activity, selectivity and stability in the RWGS reaction.5,19,25–31 Zhang et al. demonstrated that the strong interaction between Cu and β-Mo2C played a pivotal role in enhancing Cu dispersion and preventing agglomeration.25 This phenomenon contributed significantly to the remarkable activity and stability. Additionally, Cs-doped Mo2C catalysts can exhibit higher CO selectivity than non-doped one.27 The authors suggested that the electropositive nature of Cs facilitated electron transfer from Cs to Mo, resulting in a surface enriched with electrons, thereby favoring CO production.
Among these research strategies, modifying β-Mo2C with a metal, particularly Cu, has been regarded as one of the most effective methods to enhance catalyst's performance. However, the key factors influencing the RWGS activity of β-Mo2C remained unclear. In this study, for the first time, we found that β-Mo2C prepared at different carbonization temperatures exhibited significantly different RWGS activities. Interestingly, the carbonization temperature had a greater effect on RWGS activity than the Cu promoter. Herein, the key factors governing the RWGS activity of β-Mo2C were clarified.
2 Experimental
2.1 Sample preparation
β-Mo2C was synthesized using the traditional CH4/H2-temperature-programmed reduction method. Ammonium paramolybdate ((NH4)6Mo7O24·4H2O; purchased from Aladdin Biochemical Technology Co. Ltd) was heated to 500 °C at 10 °C min−1 and held at this temperature for 4 h to prepare precursor MoO3 in a muffle furnace. Initially, 1.0 g of MoO3 precursor was loaded into a micro-reactor. A flow of 20%CH4/H2 (300 ml min−1) was then introduced into the system. The temperature was raised from room temperature (RT) to 300 °C over 30 min, followed by a further increase to a specified temperature at a rate of 1 °C min−1. Subsequently, the temperature was maintained at this level for 2 h before rapid cooling to RT under a 20%CH4/H2 flow. Finally, the material underwent passivation in 1%O2/Ar for 2 h prior to exposure to air. Samples obtained from heating at 600, 700, and 800 °C were labeled as β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800, respectively. The precursor for the Cu/β-Mo2C catalyst was synthesized via a coprecipitation method. A mixed aqueous solution containing (NH4)6Mo7O24·4H2O and Cu(NO3)2·6H2O, with a Cu content of 1 wt% relative to MoO3, was stirred at RT for 4 h. Subsequently, it was dried using a water bath at 80 °C, followed by overnight drying at 110 °C, and then calcination at 500 °C for 4 h to obtain the precursor. The carbonization and passivation processes followed a similar approach to the temperature-programmed reaction method described above. The samples synthesized at 600 and 700 °C were designated as 1 wt%Cu/β-Mo2C-600 and 1 wt%Cu/β-Mo2C-700, respectively.
2.2 Sample characterizations
X-ray diffraction (XRD) analysis was conducted employing an X-ray diffractometer (X'Pert Pro MPD) featuring a Cu Kα source. The average crystallite size of β-Mo2C was calculated using the Scherrer equation d = 0.9λ/(B·cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ), where λ was the X-ray wavelength (0.15406 nm), B was the peak width at half peak height in radians, and θ was the angle between the incident and diffracted beams in degrees. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were obtained using Hitachi S-4800 instrument and Philips Tecnal 10 microscope, respectively. The BET surface area of the samples was determined using a Nova 4200e instrument from Quantachrome. X-ray photoelectron spectroscopy (XPS) was conducted utilizing a Kratos Axis ultra (DLD) instrument equipped with an Al Kα X-ray source. The binding energies (±0.2 eV) were calibrated to the C 1s peak at 284.8 eV, attributed to adventitious carbon. H2-TPR experiments were conducted in a quartz tube micro-reactor, with effluent gases monitored using a thermal conductivity detector. Prior to each H2-TPR run, the samples (0.1 g) were subjected to heating from RT to 300 °C in a He atmosphere and maintained at this temperature for 30 min. Subsequently, the sample was cooled back to 100 °C before undergoing further heating from 100 to 900 °C at a rate of 10 °C min−1 in a gas mixture of 5%H2/Ar (30 ml min−1). CO2-TPO was conducted using a flow of 6%CO2/Ar (50 ml min−1). Prior to the reaction, the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples were preheated in H2 at temperatures of 600, 700 and 800 °C, respectively, for a duration of 30 min. Subsequently, they were cooled to RT under Ar before being heated under the reactant gas from RT to 900 °C at a rate of 4 °C min−1. The gas-phase product evolution during the reaction was monitored using gas chromatography (GC). CO-TPD experiments were carried out in a quartz tube micro-reactor. Prior to the CO-TPD process, 0.1 g of catalyst underwent the same pretreatment procedures as those employed for the CO2-TPO.
θ), where λ was the X-ray wavelength (0.15406 nm), B was the peak width at half peak height in radians, and θ was the angle between the incident and diffracted beams in degrees. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were obtained using Hitachi S-4800 instrument and Philips Tecnal 10 microscope, respectively. The BET surface area of the samples was determined using a Nova 4200e instrument from Quantachrome. X-ray photoelectron spectroscopy (XPS) was conducted utilizing a Kratos Axis ultra (DLD) instrument equipped with an Al Kα X-ray source. The binding energies (±0.2 eV) were calibrated to the C 1s peak at 284.8 eV, attributed to adventitious carbon. H2-TPR experiments were conducted in a quartz tube micro-reactor, with effluent gases monitored using a thermal conductivity detector. Prior to each H2-TPR run, the samples (0.1 g) were subjected to heating from RT to 300 °C in a He atmosphere and maintained at this temperature for 30 min. Subsequently, the sample was cooled back to 100 °C before undergoing further heating from 100 to 900 °C at a rate of 10 °C min−1 in a gas mixture of 5%H2/Ar (30 ml min−1). CO2-TPO was conducted using a flow of 6%CO2/Ar (50 ml min−1). Prior to the reaction, the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples were preheated in H2 at temperatures of 600, 700 and 800 °C, respectively, for a duration of 30 min. Subsequently, they were cooled to RT under Ar before being heated under the reactant gas from RT to 900 °C at a rate of 4 °C min−1. The gas-phase product evolution during the reaction was monitored using gas chromatography (GC). CO-TPD experiments were carried out in a quartz tube micro-reactor. Prior to the CO-TPD process, 0.1 g of catalyst underwent the same pretreatment procedures as those employed for the CO2-TPO.
After being cooled to RT in He flow, a stream of 10%CO/He was introduced into the system for 20 min. After CO adsorption, the gas flow was switched to He (30 ml min−1) to remove physically adsorbed CO on catalyst surface 30 min. Finally, the sample was heated from RT to 900 °C at a rate of 10 °C min−1 in He (30 ml min−1), and then was held at 900 °C for 30 min. The effluent gases were monitored by means of a GC thermal conductivity detector.
2.3 Catalyst performance tests
The catalytic performance of the catalyst for the RWGS reaction was evaluated using a micro-reactor with an inner diameter of 8 mm. Prior to the reaction, the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples were preheated in H2 at temperatures of 600, 700 and 800 °C, respectively, for a duration of 30 min. Subsequently, a gas mixture consisting of H2 and CO2 in a ratio of 2:1 was introduced into the catalyst bed. The gas-phase products were continuously monitored using online GC. The conversion of CO2, the selectivities of CO and CH4 as well as carbon balance were determined as follows:
| CO2 conversion (%) = ([CO2]in − [CO2]out)/([CO2]in) × 100 |
| CO selectivity (%) = ([CO]out)/([CO]out + [CH4]out) × 100 |
| CH4 selectivity (%) = ([CH4]out)/([CO]out + [CH4]out) × 100 |
| Carbon balance (%) = ([CO2]out + [CO]out + [CH4]out)/([CO2]in) ×100 |
3 Results and discussion
The XRD patterns of the β-Mo2C catalysts obtained at different carbonization temperatures of 600, 700 and 800 °C are shown in Fig. 1. The diffraction peaks at 34.5, 38.0, 39.6, 52.3, 61.9, 69.8, 75.0 and 76.0° were corresponded to the (100), (002), (101), (102), (110), (103), (112) and (201) reflections of β-Mo2C (JCPDS 65-8766). As the carbonization temperature increased from 600 to 800 °C, the diffraction peaks became sharper and more intense, indicating the enhanced crystallinity of β-Mo2C. Therefore, it was reasonable to estimate the crystallite size of β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples using the Scherrer method as 20, 34 and 45 nm, respectively (see Table 1). The increase in crystallite size corresponded to a decrease in surface area. It was well known that the complete synthesis process involved reduction-carbonization of MoO3 precursor by 20%CH4/H2 mixture via the pathway of MoO3 → MoO2 → MoxOCy → β-Mo2C.32 This process was strongly influenced by temperature and contact time, with the formation of the MoxOCy phase typically occurring between 500 and 650 °C. Due to the broad XRD peaks (2θ = 37 and 44°) associated with MoxOCy,33 they can easily be masked by the β-Mo2C peaks, making it difficult to exclude the presence of this phase in the β-Mo2C-600 sample.
 |
| | Fig. 1 XRD patterns of the β-Mo2C catalysts obtained at different carbonization temperatures of 600, 700 and 800 °C. | |
Table 1 BET surface area and crystallite size of the β-Mo2C catalysts obtained at different carbonization temperatures of 600, 700 and 800 °C
| Sample |
BET surface area (m2 g−1) |
Crystallite size (nm) |
| β-Mo2C-600 |
34.0 |
20 |
| β-Mo2C-700 |
30.3 |
34 |
| β-Mo2C-800 |
17.9 |
45 |
The crystallite size of β-Mo2C was calculated by Scherrer equation according to (101) plane.
The morphologies of these β-Mo2C catalysts were observed using SEM and TEM (refer to Fig. 2). All three samples exhibited similar irregular particle aggregates. Detailed analysis of primary particle sizes using TEM was not easy due to significant agglomeration. Therefore, it must be taken into account the average size measured from XRD. The insets in Fig. 2a displayed EDX element mapping images, confirming the presence of Mo, C, and O elements in the β-Mo2C samples, and those in Fig. 2b clearly showed the β-Mo2C (002) crystal lattices, proving the existence of β-Mo2C phase in these samples. It was worth noting that the MoOC (111) crystal lattice, as reported by Sun et al.,15 was also observed in multiple regions (more than three) of the β-Mo2C-600 sample (Fig. 2 and S1†), providing strong evidence for the coexistence of β-Mo2C and MoxOCy phases. Moreover, the H2-TPR result (Fig. 3) demonstrated an obvious hydrogen consumption peak when non-passivated β-Mo2C-600 was subjected to further treatment in a 5% H2/Ar atmosphere, thereby confirming the presence of MoxOCy species. In contrast, the non-passivated β-Mo2C-700 and β-Mo2C-800 samples showed no H2-TPR signal, further confirming that they contained only the β-Mo2C phase.
 |
| | Fig. 2 (a) SEM images and (b) TEM images of β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. The insets in the SEM images show the Mo, O and C element mapping images and those in the TEM images show the β-Mo2C (002) and MoOC (111) crystal lattices. | |
 |
| | Fig. 3 H2-TPR profiles of non-passivated β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. The non-passivated samples were directly subjected to H2-TPR experiment, where they were heated from 100 to 700 °C at a rate of 5 °C min−1 in a gas mixture of 5%H2/Ar (60 ml min−1). | |
Subsequently, further information regarding these β-Mo2C surfaces was obtained by XPS. Fig. 4 shows the XPS spectra of Mo 3d for the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. Based on the curve fitting of Mo 3d levels, Table 2 summarizes the Mo 3d5/2 binding energy, distribution of Mo species and Mo2+ content. Because the as-prepared metal carbides were usually passivated before exposure to air, the surface regions of all the samples should be dominated by oxidized species and the underlying (oxy)carbide species. As shown in Fig. 4 and Table 2, four Mo 3d5/2 binding energy peaks were observed on the β-Mo2C-700 surface at 228.7, 229.1, 229.9 and 232.6 eV. These peaks were assigned to Mo2+ (carbide),34,35 Moδ+ (oxycarbide),36,37 Mo4+ (oxide)38 and Mo6+ (oxide),39 respectively. However, unlike β-Mo2C-700, the surfaces of β-Mo2C-800 and β-Mo2C-600 showed only two (Mo2+ carbide and Moδ+ oxycarbide) and three Mo species (Moδ+ oxycarbide, Mo4+ oxide and Mo6+ oxide), respectively. The Mo2+ contents of the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples estimated by quantitative XPS analysis (Table 2) were 0, 32.0 and 63.2%, respectively. This indicated that the oxophilicity ranked in the order of β-Mo2C-600 > β-Mo2C-700 > β-Mo2C-800.
 |
| | Fig. 4 XPS spectra of Mo 3d region of β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. | |
Table 2 Mo 3d5/2 binding energies and Mo2+ contents measured by XPS for β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts
| Sample |
Mo 3d5/2 (eV) |
Mo2+ content (%) |
| Mo2+ carbide |
Moδ+ oxycarbide |
Mo4+ oxide |
Mo6+ oxide |
| β-Mo2C-600 |
— |
229.2 |
229.9 |
232.7 |
0 |
| β-Mo2C-700 |
228.7 |
229.1 |
229.9 |
232.6 |
32.0 |
| β-Mo2C-800 |
228.6 |
229.3 |
— |
— |
63.2 |
The RWGS performance variation of the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts with temperature is shown in Fig. 5a, b and S2.† A clear correlation was observed between the carbonization synthesis temperature and the RWGS activity, with β-Mo2C-600 exhibiting the highest activity, followed by β-Mo2C-700 and β-Mo2C-800 in the entire temperature range. Arrhenius curve is presented in Fig. 5c. The Ea values followed the order of β-Mo2C-800 > β-Mo2C-700 > β-Mo2C-600, which was just the opposite of their ranks in RWGS activity. To compare the influence of carbonization synthesis temperature and Cu doping on the RWGS activity of β-Mo2C, a Cu-doped β-Mo2C catalyst was synthesized at 700 °C (XRD pattern shown in Fig. S3†). The crystallite size of β-Mo2C in this Cu doped catalyst was estimated to be 31 nm. Noticeably, the β-Mo2C-600 even displayed slightly higher RWGS activity compared to Cu-doped β-Mo2C-700 under similar reaction conditions. In terms of selectivity, although CO selectivities increased with carbonization synthesis temperature, they can remain above 95% throughout the entire temperature range. The main side product was CH4, resulting from the methanation of CO2. And the carbon balances ranged from 0.98 to 1.00 (see Fig. S2†). The errors in CO2 conversion, CO/CH4 selectivity as well as carbon balance were all within ±2%, which was consistent with previously reported results and deemed acceptable.40 Finally, the catalytic stability of the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts at 600 °C was depicted in Fig. S4.† It was evident that all three catalysts exhibited excellent stability throughout the 10 h test period. Given the superior performance of the β-Mo2C-600 catalyst, Cu/β-Mo2C-600 was subsequently synthesized (XRD pattern shown in Fig. S3†) and its RWGS performance shown in Fig. S5† was compared with those of other mainstream catalysts (Table S1† and ref. 41–45). It was clear that the Cu/β-Mo2C-600 catalyst exhibited superior activity to traditional Pt and Cu-based as well as Mo2C-based catalysts.
 |
| | Fig. 5 (a) CO2 conversion and (b) CO selectivity over β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts in the RWGS reaction as well as 1 wt%Cu/β-Mo2C-700 catalyst for comparison (reaction conditions: CO2/H2 = 1/2, WHSV = 150000 cm3 g−1 h−1, T = 300–600 °C) and (c) Arrhenius plots and Ea values below 15% of CO2 conversion. | |
The CO adsorption properties of β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts were estimated by CO-TPD, as presented in Fig. 6. The CO-TPD profiles revealed an obvious desorption peak at around 870 °C for all samples, consistent with previously reported data.22 The amount of CO desorption ranked in the order of β-Mo2C-600 (69.4 μmol g−1) > β-Mo2C-700 (35.5 μmol g−1) > β-Mo2C-800 (13.1 μmol g−1). The observed order showed an opposite trend to their crystallite size variation. The results suggested an inverse relationship between CO affinity and β-Mo2C crystallite size, with smaller crystallite size demonstrating stronger CO affinity. In the RWGS reaction, the higher the CO affinity was, the easier the CH4 formation via CO hydrogenation. This was probably the reason why the CO selectivity followed the order: β-Mo2C-800 > β-Mo2C-700 > β-Mo2C-600.
 |
| | Fig. 6 CO-TPD curves of β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. | |
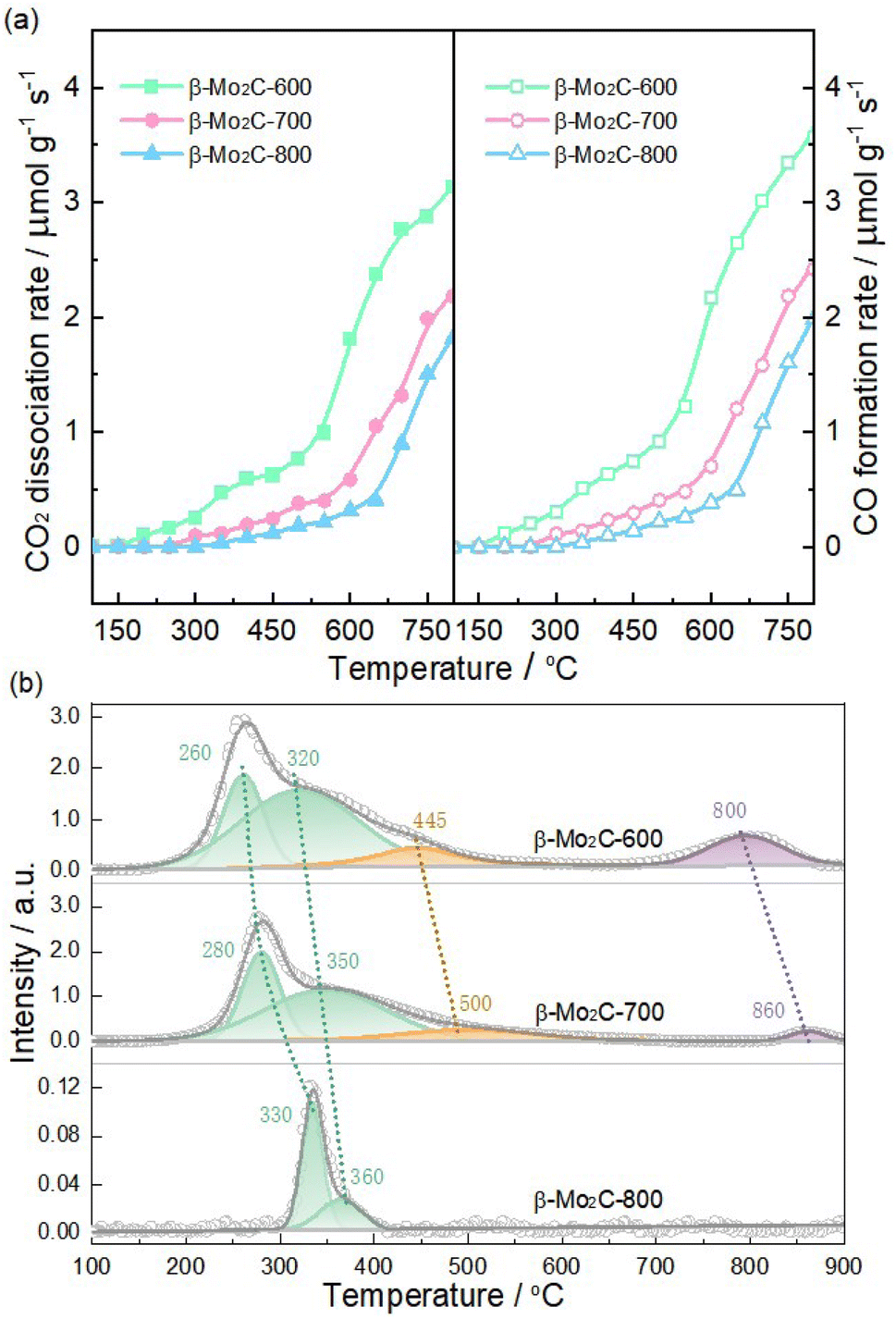
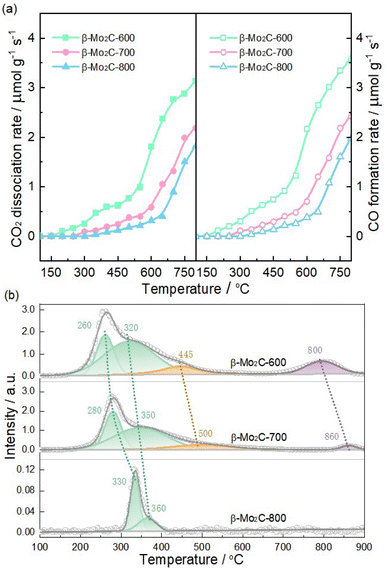
Compared to the associative routes that involved the formation of formate and carboxyl, the redox pathway, MoxCy + CO2 = MoxOCy + CO and MoxOCy + H2 = MoxCy + H2O, had been identified as the key steps for achieving high RWGS activity of Mo carbide.11,13 Therefore, it became evident that the redox ability of β-Mo2C predominantly influenced its RWGS activity. Subsequently, the oxidizability and reducibility of β-Mo2C were characterized using CO2-TPO and H2-TPR techniques, respectively. As depicted in Fig. 7a, all three samples exhibited a consistent increase in CO2 consumption accompanied by CO generation as the temperature was elevated. It was noteworthy that significant increases in CO2 dissociation and CO formation rates were observed above 550, 600, and 650 °C for β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800, respectively. These results suggest that severe bulk oxidation occurred in these samples under high-temperature conditions.33,37 This trend clearly indicated that the catalyst's oxidizability followed the sequence: β-Mo2C-600 > β-Mo2C-700 > β-Mo2C-800, which corresponded with the sequence of their oxophilicity estimated from XPS data. Fig. 7b shows the H2-TPR profiles for the passivated β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 samples and the H2-TPR data are summarized in Table 3. The H2 consumption peaks corresponding to the reduction of these Mo species were evident in the H2-TPR profiles. According to XPS results, the β-Mo2C-600 and β-Mo2C-700 exhibited three oxygen-containing Mo species (MoxOCy, MoO2 and MoO3), whereas the β-Mo2C-800 showed only one oxygen-containing Mo species (MoxOCy). Therefore, the peaks below 360 °C were assigned to the reduction of MoxOCy.7,46 Reduction of MoO3 to MoO2 could contribute to H2 consumption at temperatures above 400 °C,47 and the reduction of MoO2 to Mo was expected at temperatures above 800 °C.48 These reduction peaks associated with MoxOCy shifted to higher temperatures in the sequence β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800, indicating a trend of decreasing reducibility: β-Mo2C-600 > β-Mo2C-700 > β-Mo2C-800. Moreover, the total H2-consumption values in Table 3 provide additional confirmation of the sequence of oxophilicity determined by XPS. Correlating CO2-TPO and H2-TPR results with crystallite size analysis suggested that reducing crystallite size can enhance the redox ability of β-Mo2C, thereby improving the its RWGS activity through the redox pathway.
 |
| | Fig. 7 (a) CO2-TPO and (b) H2-TPR profiles of passivated β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts. | |
Table 3 The summarized data for the H2-TPR in Fig. 7b
| Sample |
Peak temperature (°C) |
Total H2 consumption (μmol g−1) |
| MoxOCy → MoxCy |
MoO3 → MoO2 |
MoO2 → Mo |
| β-Mo2C-600 |
260, 320 |
445 |
800 |
1344.1 |
| β-Mo2C-700 |
280, 350 |
500 |
860 |
934.7 |
| β-Mo2C-800 |
330, 360 |
— |
— |
13.1 |
It was obvious that both MoxCy and MoxOCy phases served as active sites in the redox pathway, with this unsaturated MoxOCy phase being reported to be more active than MoxCy.15 It was reasonable to deduce that the presence of incompletely carbonized MoxOCy in β-Mo2C would enhance its RWGS activity. The TEM and H2-TPR results (Fig. 2 and 3) revealed that only β-Mo2C-600 contained the incompletely carbonized MoxOCy species before the reaction, which might contribute to its superior activity compared to β-Mo2C-700 and β-Mo2C-800. Finally, the β-Mo2C-600, β-Mo2C-700, and β-Mo2C-800 catalysts after the reaction were characterized by XRD and H2-TPR (see Fig. S6 and S7†). It can be seen from Fig. S6† that the XRD patterns of used samples resembled those of fresh samples, indicating these β-Mo2C catalysts can keep their bulk structure well at 600 °C in RWGS. Note that the H2-TPR results indicated H2 consumption below 400 °C for all three used samples. The H2 consumption values followed the order of β-Mo2C-600 > β-Mo2C-700 > β-Mo2C-800, suggesting that the amount of MoxOCy decreased in the same sequence. This finding further confirmed the correlation between the MoxOCy content in β-Mo2C and its RWGS activity. Therefore, in addition to crystallite size, the MoxOCy content was proposed to be another key factor influencing the RWGS activity of β-Mo2C catalyst.
4 Conclusions
A clear correlation was observed between the carbonization synthesis temperature and the RWGS activity, with the activity following the order: β-Mo2C-600 > β-Mo2C-700 > β-Mo2C-800. Even β-Mo2C-600 showed slightly superior RWGS activity compared to Cu-doped β-Mo2C-700 under similar reaction conditions. It was proposed that crystallite size and MoxOCy content were key factors influencing the RWGS activity of β-Mo2C. These factors can effectively regulate the redox properties of β-Mo2C catalyst, thereby affecting its catalytic activity in the RWGS reaction. This study underscored a strategy to enhance the RWGS activity of β-Mo2C catalysts by reducing crystallite size or increasing the MoxOCy content.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 21978125), the Project of Guangdong Province Department of Education (No. 2023ZDZX3035) and the Project of Liaoning Province Department of Education (No. LJ212410148022).
Notes and references
- D. Zagoraios, N. Kokkinou, G. Kyriakou and A. Katsaounis, Catal. Sci. Technol., 2022, 12, 1869–1879 RSC.
- A. R. Querido, L. P. L. Gonçalves, Y. V. Kolen'ko, M. F. R. Pereira and O. S. G. P. Soares, Catal. Sci. Technol., 2023, 13, 3606–3613 RSC.
- C. F. Holder, J. R. Morse, P. M. Barboun, A. R. Shabaev, J. W. Baldwinc and H. D. Willauera, Catal. Sci. Technol., 2023, 13, 2685–2695 RSC.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- L. Wang, H. Wang, H. Huang, T. Yun, C. Song and C. Shi, Adv. Funct. Mater., 2024, 34, 2309850 CrossRef CAS.
- X. Liu, C. Kunkel, P. R. Piscina, N. Homs, F. Viñes and F. Illas, ACS Catal., 2017, 7, 4323–4335 CrossRef CAS.
- J. Gao, Y. Wu, C. Jia, Z. Zhong, F. Gao, Y. Yang and B. Liu, Catal. Commun., 2016, 84, 147–150 CrossRef CAS.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- M. Attwood, H. Akutsu, L. Martin, D. Cruickshank and S. S. Turner, Dalton Trans., 2019, 48, 90–98 RSC.
- M. A. Khoshooei, X. Wang, G. Vitale, F. Formalik, K. O. Kirlikovali, R. Q. Snurr, P. Pereira-Almao and O. K. Farha, Science, 2024, 384, 540–546 CrossRef PubMed.
- M. Dixit, X. Peng, M. D. Porosoff, H. D. Willauer and G. Mpourmpakis, Catal. Sci. Technol., 2017, 7, 5521–5529 RSC.
- A. Jurado, Á. Morales-García, F. Viñes and F. Illas, ACS Catal., 2022, 12, 15658–15667 CrossRef CAS.
- X. Du, R. Li, H. Xin, Y. Fan, C. Liu, X. Feng, J. Wang, C. Dong, C. Wang, D. Li, Q. Fu and X. Bao, Angew. Chem., Int. Ed., 2024, e202411761 CAS.
- X. Sun, J. Yu, H. Zada, Y. Han, L. Zhang, H. Chen, W. Yin and J. Sun, Nat. Chem., 2024, 16, 2044–2053 CrossRef CAS PubMed.
- F. G. Baddour, E. J. Roberts, A. T. To, L. Wang, S. E. Habas, D. A. Ruddy, N. M. Bedford, J. Wright, C. P. Nash, J. A. Schaidle, R. L. Brutchey and N. Malmstad, J. Am. Chem. Soc., 2020, 142, 1010–1019 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
- J. Lu, S. Zhang, H. Zhou, C. Huang, L. Xia, X. Liu, H. Luo and H. Wang, Acta Phys. Chim. Sin., 2023, 39, 2302021 Search PubMed.
- M. D. Porosoff, J. W. Baldwin, X. Peng, G. Mpourmpakis and H. D. Willauer, ChemSusChem, 2017, 10, 2408–2415 CrossRef CAS PubMed.
- Y. Ma, Z. Guo, Q. Jiang, K. H. Wu, H. Gong and Y. Liu, J. Energy Chem., 2020, 50, 37–43 CrossRef.
- W. Marquart, S. Raseale, G. Prieto, A. Zimina, B. B. Sarma, J. D. Grunwaldt, M. Claeys and N. Fischer, ACS Catal., 2021, 11, 1624–1639 CrossRef CAS.
- Q. Rong, W. Ding, G. Liu, Z. Zhang and Z. Yao, Catal. Sci. Technol., 2023, 13, 6360–6365 RSC.
- A. G. Galallah, M. K. Albolkany, A. E. Rashed, W. Sadik, A. G. El-Demerdash and A. A. El-Moneim, J. Environ. Chem. Eng., 2024, 12, 113380 CrossRef CAS.
- S. Cao, Z. Guan, Y. Ma, B. Xu, J. Ma, W. Chu, R. Zhang, G. Giambastiani and Y. Liu, ACS Catal., 2024, 14, 10939–10950 CrossRef CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y. W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS.
- E. Heracleous, V. Koidi and A. A. Lappas, Catal. Sci. Technol., 2021, 11, 1467–1480 RSC.
- Q. Zhang, L. Pastor-Pérez, W. Jin, S. Gu and T. R. Reina, Appl. Catal., B, 2019, 244, 889–898 CrossRef CAS.
- J. Xu, X. Gong, R. Hu, Z. Liu and Z. Liu, Mol. Catal., 2021, 516, 111954 CrossRef CAS.
- M. Figueras, R. A. Gutiérrez, F. Viñes, P. J. Ramírez, J. A. Rodriguez and F. Illas, ACS Catal., 2021, 11, 9679–9687 CrossRef CAS.
- W. Zhang, A. Vidal-López and A. Comas-Vives, Front. Chem., 2023, 11, 11444189 Search PubMed.
- R. Liu, C. Chen, W. Chu and W. Sun, Materials, 2022, 15, 3775 CrossRef CAS PubMed.
- A. Hanif, T. Xiao, A. P. E. York, J. Sloan and M. L. H. Green, Chem. Mater., 2002, 14, 1009–1015 CrossRef CAS.
- P. Liang, H. Gao, Z. Yao, R. Jia, Y. Shi, Y. Sun, Q. Fan and H. Wang, Catal. Sci. Technol., 2017, 7, 3312–3324 RSC.
- Z. Yao, J. Jiang, Y. Zhao, F. Luan, J. Zhu, Y. Shi, H. Gao and H. Wang, RSC Adv., 2016, 6, 19944–19951 RSC.
- H. Gao, Z. Yao, Y. Shi and S. Wang, Catal. Sci. Technol., 2018, 8, 697–701 RSC.
- H. Wang, S. Liu, B. Liu, V. Montes, J. M. Hill and K. J. Smith, J. Solid State Chem., 2018, 258, 818–824 CrossRef CAS.
- H. Gao, Z. Yao, Y. Shi, R. Jia, F. Liang, Y. Sun, W. Ma and H. Wang, Inorg. Chem. Front., 2018, 5, 90–99 RSC.
- A. Kurlov, E. B. Deeva, P. M. Abdala, D. Lebedev, A. Tsoukalou, A. Comas-Vives, A. Fedorov and C. R. Müller, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- S. Upadhyay and O. P. Pandey, Int. J. Hydrogen Energy, 2020, 45, 27114–27128 CrossRef CAS.
- L. Yang, L. Pastor-Pérez, J. J. Villora-Pico, A. Sepúlveda-Escribano, F. Tian, M. Zhu, Y. F. Han and T. R. Reina, ACS Sustainable Chem. Eng., 2021, 9, 12155–12166 CrossRef CAS.
- S. Kim, H. Lee and S. Hong, Appl. Catal., A, 2012, 423–424, 100–107 CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Catal. Lett., 2000, 68, 45–48 CrossRef CAS.
- L. Wang, S. Zhang and Y. Liu, J. Rare Earths, 2008, 26, 66–70 CrossRef.
- X. Pan, H. Sun, M. Ma, H. Liao, G. Zhan, K. Wang, M. Fan, J. Xu, L. Ding, K. Sun and J. Jiang, Biochar, 2024, 6, 93 CrossRef CAS.
- M. Zhang, Y. Zhu, J. Yan, J. Xie, T. Yu, M. Peng, J. Zhao, Y. Xu, A. Li, C. Jia, L. He, M. Wang, W. Zhou, R. Wang, H. Jiang, C. Shi, J. Rodriguez and D. Ma, Angew. Chem., Int. Ed., 2025, 64, e202418645 CrossRef CAS PubMed.
- G. Wang, J. A. Schaidle, M. B. Katz, Y. Li, X. Pan and L. T. Thompson, J. Catal., 2013, 304, 92–99 CrossRef CAS.
- P. Sun, F. Teng, Z. Yang, X. Yang, S. Zhai, S. Liang, W. Gu, W. Hao and S. Shi, J. Mater. Chem. C, 2020, 8, 2475–2482 RSC.
- P. Chen, Z. Xie, Z. Zhao, J. Li, B. Liu, B. Liu, X. Fan, L. Kong and X. Xiao, Catal. Sci. Technol., 2021, 11, 4083–4097 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Zhiwei Yao
*a and
Zhiwei Yao