
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilizing red mud from industrial waste as catalysts for the hydrogenation of CO2 into value added chemicals†
Mahbuba Aktary a,
Mohammed A. Sanhoobb,
Atif S. Alzharani*ac,
Huda S. Alghamdib,
Afnan M. Ajeebib,
Md. Abdul Azizb and
M. Nasiruzzaman Shaikh*ab
a,
Mohammed A. Sanhoobb,
Atif S. Alzharani*ac,
Huda S. Alghamdib,
Afnan M. Ajeebib,
Md. Abdul Azizb and
M. Nasiruzzaman Shaikh*ab
aDepartment of Materials Science and Engineering (MSE), King Fahd University of Petroleum and Minerals (KFUPM), Dhahran, Saudi Arabia
bInterdisciplinary Research Center for Hydrogen Technologies and Carbon Management (IRC-HTCM), King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
cInterdisciplinary Research Center for Sustainable Energy Systems (IRC-SES), King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia. E-mail: mnshaikh@kfupm.edu.sa; atif@kfupm.edu.sa
First published on 14th February 2025
Abstract
Finding environmentally acceptable and long-lasting catalysts that can convert carbon dioxide into compounds with additional value is of great interest. Using Red Mud (RM), a waste product from the aluminum industry, as a CO2 reduction agent is also a great idea, given the current environmental problems. In this research, we developed, characterized, and evaluated a series of metal-promoted (M = Na, K, Cs, Ba, Mg, and Sr) RM catalysts for CO2 hydrogenation to produce olefins and CO. In the beginning, we synthesized RM that had been treated with acid and base by employing hydrochloric acid (HCl) and potassium hydroxide (KOH), and then we examined the activity of these catalysts in CO2 hydrogenation. Surprisingly, when tested at 375 °C and 30 bar pressure (CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:3), pure RM converted 22% CO2 compared to acid-and base-treated RM, which converted 16% of CO2. Under identical reaction conditions, the 3%K-promoted RM (3%K@RM) catalyst achieved over 27% of CO2 conversion activity compared to the other 3%M@RM catalysts (where M = Na, Cs, Ba, Mg, and Sr) in terms of conversion and selectivity for light olefins (C2–4=).
:H2 = 1:3), pure RM converted 22% CO2 compared to acid-and base-treated RM, which converted 16% of CO2. Under identical reaction conditions, the 3%K-promoted RM (3%K@RM) catalyst achieved over 27% of CO2 conversion activity compared to the other 3%M@RM catalysts (where M = Na, Cs, Ba, Mg, and Sr) in terms of conversion and selectivity for light olefins (C2–4=).
1. Introduction
Environmental concerns that become apparent from the rising levels of CO2 are mostly caused by human activities.1 Numerous initiatives have been undertaken to prevent catastrophic environmental disasters due to greenhouse gas emissions, including the implementation of technologies and policies to mitigate the negative consequences of using fossil fuels.2–4 Carbon capture, utilization, and storage (CCUS) technologies have become vital mitigation strategies for the sustainable development of nations.1,5 Specifically, a workable solution to this urgent problem is utilizing captured CO2 and converting it through photoreduction,6–8 electroreduction,9–11 photo-chemical,12 thermo-chemical,13–15 electrochemical,13,16–20 or biological methods into value-added products. Among these, thermochemical processes are preferred for CO2 conversion in many industrial applications due to the process flexibility, scalability, and economic and environmental advantages.In this regard, catalytic hydrogenation is one well-established thermo-chemical strategy for converting CO2 to a variety of high-value-added products, including CO, CH4, methanol, dimethyl ether, olefins, etc. For instance, the bimetallic 40CuCe(rod) catalyst showed 36.5% to 41.1% conversion with 100% CO selectivity at 400 °C.21 Al-Fatesh et al., demonstrated, 2.5 wt% Ni and 2.5 wt% Co supported over ZrO2/Al2O3 catalysts for CO2 hydrogenation and produce 83.8% CO2 converted product with 73.7% CH4 selectivity.22 Chuan-Shu et al. reported a novel method using Ni–NP catalysts to hydrogenate CO2 into formic acid.23 Also, numerous catalytic systems for CO2 hydrogenation including Zr12-bpdc-CuCs,24 Pd2Cu NPs/P25,25 CoMoCx26, Cu@Na-Beta,27 Au/a-TiO2 (ref. 28) are developed for CO2 to ethanol. However, the production of olefins from CO2 is an intensive research area. Because olefins are the primary raw material used in the industrial manufacturing of plastics and polymers,29,30 lubricants,31 and detergents32 where lower-olefins (C2=–C4=), such as ethylene, propylene, and butylene are the major component.33 Specifically, ethylene (C2=) is widely employed as a precursor in the synthesis of thermoplastic oligomers or polymers.34 It is also employed in the synthesis of vinyl chloride, acetate, styrene, ethylene glycol, etc.35,36 Today, lower olefins production is the few hundreds of billions dollar of industry.37 However, very few studies are dedicated to CO2 to lower olefin formation.38
In this context, several low-cost metal-based catalysts, such as Fe2O3-CT600, Fe–Co(0.17)/K(1.0)/Al2O3, ZnZrO/SAPO-34, and Na–Fe3O4/HZSM-5, have demonstrated good CO2 hydrogenation performance for the production of C2+ hydrocarbons through the modified Fischer–Tropsch (FT) process.39 The reverse water-gas shift (RWGS) reaction (eqn (1)) transforms CO2 to more reactive CO by the endothermic reaction. Further hydrogenation of CO can produce C2+ hydrocarbons followed by the reactions of eqn (2)–(4)
| CO2 + H2 ⇌ CO + H2O; ΔH = +41.2 kJ mol−1 | (1) |
| nCO + (2n + 1)H2 ⇌ CnH2n+2 + nH2O | (2) |
| nCO + 2nH2 ⇌ CnH2n + nH2O | (3) |
| nCO + 2nH2 ⇌ CnH2n+1OH + (n − 1)H2O | (4) |
Also, Fe–Co–K/Al2O3-400 catalysts showed 49% CO2 conversion and 37% olefins selectivity at 340 °C.40 The addition of Zr with K–Fe catalyst enhanced conversion (>54%) as well as olefins selectivity (>53%) at 320 °C and 2 MPa.41 Researchers fabricated a bulk Fe catalyst with a large surface area that is promoted by K and activated during the hydrogenation of CO2 to C2–C4 olefins without the requirement for extra heteroatoms or structural stabilizers at 5 bar pressure and 300 °C. The catalysts K–FeX (X = 15, 30, 60) powdered catalyst have more activity than materials based on commercial composition, phase and structure of Fe3O4 and α-Fe2O3.38 The K–Fe15 catalyst achieved a CO2 conversion of approximately 38% at steady-state conditions whereas the selectivity to C2–C4 olefins reached about 40%.42 Additionally, a moderate chain growth probability and low secondary hydrogenation of olefins contribute to the high selectivity towards these olefins. A recent study found that the K–Fe–Al–O catalyst (prepared by electrospinning method) improved CO2 conversion by 48% and selectivity to C2–C5 light olefins by 52%.43
Transformation of CO2 focuses on direct hydrogenation of CO2 to lower olefins for a while. Some research on the mixed catalyst consisting of the ZnZrO solid solution and Zn-modified SAPO-34 zeolite mentioned this heterogeneous mixture allows for selective conversion of CO2 into short-chain olefins instead of alkanes and selectivity to form these compounds is close to 80–90%.44
The catalyst support has played a major role in the metal-support interaction and is essential in spreading the active metal.45 Catalytic activity varies, for example, whether the active metal is uniformly immobilized on its surface chemical states and exposed crystal facet.46 Metal oxides,45,47,48 clays,49,50 zeolites,51–54 activated carbon,55–58 porous silica,59–62 and MOFs,63–67 have all been used as supports for CO2 hydrogenations. Because of their chemical, mechanical, and thermal stabilities, inorganic metal oxides are more advantageous than the others.
Recently, RM has attracted a lot of attention due to its toxic nature and, most importantly, abundant byproducts, generating hundreds of thousands of tons every year from the aluminum industry. Eliminating it from the earth's surface is an urgent priority.68 It primarily consists of a diverse mixture of metal oxides and hydroxides of iron, aluminum, titanium, silicon, and calcium in the form of very fine particles <10 μm.69 Although the actual composition varies globally, it is generalized as a mixture of metal oxides. However, the potential of red mud (RM) is less explored as catalyst as well as support for CO2 hydrogenation reaction. All the active components, such as Fe2O3, Fe3O4 etc., are required to activate the CO2 molecules and then transform them into high-value chemicals, such as lower olefins (C2=–C4=). Therefore, a combination of RM and a suitable promoter could be the best choice as a catalyst for CO2 conversion in terms of economics with enhanced environmental credentials.
In this study, bifunctional catalysts were developed for CO2 hydrogenation to lower olefins by inhibiting the production of CH4, using a series of alkali and alkaline earth metal-promoted RM obtained from MA’ADEN, Saudi Arabia. We also intended to study the impact of monovalent (Na+ from NaCl, K+ from KNO3, and Cs+ from CsOH) and divalent metal cations (Mg2+ from MgO, Ba2+ from BaO, and Sr2+ from SrCl2) on the production of olefins through CO2 hydrogenation reactions. The methodology we have adopted involves only two key processes: mixing and calcination. Both of these are well-established, efficient, and cost-effective operations commonly used in industry, making them highly favorable for large-scale implementation. Since these steps do not require complex equipment or conditions, the use of Red Mud in this process is not only technically feasible but also economically viable.
2. Experimental
2.1 Reagents and chemicals
In this study, the following materials and chemicals are used without further purification:1 raw red mud (RM) from MA’ADEN Bauxite and Alumina Company (MBAC) of Saudi Arabia,2 deionized water (DI-H2O),3 different metal salts including sodium chloride (NaCl, Sigma Aldrich), potassium nitrate (KNO3), cesium hydroxide (CsOH), magnesium oxide (MgO), barium oxide (BaO), and strontium chloride (SrCl2),4 potassium hydroxide (KOH),5 hydrochloric acid (HCl).2.2 Catalyst preparation
A schematic diagram for the steps involved in preparing the RM and K@RM catalyst is shown in Scheme 1. The raw RM was dried in an oven at 80 °C, grinded using mortar and pestle, and then sieved to a size of <200 μm. The powdered material was calcined at 500 °C for 5 hours and then divided into two portions. One was produced via acid treatment and the second by base treatment. The first one was produced by adding HCl (10 mL) to a well-dispersed suspension of the calcined RM catalyst (3 g) in deionized water (DI-H2O) (50 mL) and continued stirring (250 rpm) overnight. The product of RM catalyst was then filtered, washed with DI-H2O, and dried in the oven at 80 °C overnight. The acid-tread sample was named HCl-treated RM. The second treated sample was modified using potassium hydroxide. The sample was prepared by solid mixing method using mortar and pestle which was grinded for 30 minutes. The base-treated sample was named KOH treated RM. | ||
| Scheme 1 Steps of RM-based catalyst preparation. | ||
2.3 Analytical techniques
Using Quantachrome Instruments (version 6.0), the N2 adsorption–desorption isotherm was used to calculate the Brunauer–Emmett–Teller (BET) to evaluate the catalytic surface area, and the pore volume. The Non-linear Density Functional Theory (NLDFT) equilibrium model is used to calculate the micropore volume. The degassing of the sample should be performed at a temperature of 150 °C for 12 hours. Raman spectroscopy was used to analyze and measure the catalyst's surface before the reaction. A 455 nm DXR laser and a Thermo Scientific DXR Raman spectroscope were used to record the Raman spectra. Temperature-programmed-reduction (H2-TPR, BELCAT II from MacrotracBEL) method was used to observe the reduction capability of catalysts by H2. H2-TPR was carried out by placing 50 mg of catalysts in a reactor tube and preheating it at 500 °C for 30 minutes with argon flow (50 mL min−1). After cooling down to 50 °C, a mixture of H2 and Ar gas (containing 10% of H2 with a flow rate 5 mL min−1 and 90% Ar with a flow rate 50 mL min−1) was introduced into the reactor tube. The sample was then heated to 900 °C at a rate of 10 °C min−1 while the TCD signal was captured concurrently.
2.4 Catalytic activity evaluation
The RM catalysts with different promoters were tested for CO2 hydrogenation inside a fixed bed reactor (PID Microactivity-Effi reactor). Each time, 0.20 g of the catalyst was placed inside SS316 tube with an inner diameter of 9.11 mm in a particular location of the 300 mm long tube. The catalyst sample was heated at 500 °C for 50 min with 20 mL per min N2 gas to eliminate moisture as moisture increases the reaction barrier for CO2. The catalysts were reduced in the presence of H2 gas (3 mL min−1) for 30 min. The sample was cooled to 150 °C. Then, the reactor tube was pressurized to 30 bar with a CO2/H2 molar ratio of 1:3 and then heated to the reaction temperature of 375 °C. The required activation energy for the catalyst, which has a strong drive for the creation of hydrocarbons, was guaranteed by this reaction temperature. Catalyst deterioration and unwanted side reactions can result from extremely high temperatures. The pressure of 30 bar that we chose was in line with the working limitations of our experimental apparatus and was based on best practices in CO2 hydrogenation investigations.70 To provide a sufficient supply of hydrogen for the process while preventing excessive H2, a CO2:H2 ratio of 1:3 was used. Additionally, this ratio shows a compromise between the stoichiometric needs for olefin synthesis and industrial viability. The catalytic evaluation was investigated at gas hourly space velocity (GHSV) of 4500 mL g−1 h−1. The evaluation of catalysts was conducted using an online gas chromatography system (Shimadzu, GC-2014) equipped with two flammable ionization detectors (FIDs) and a thermal conductivity detector (TCD). The first FID tracked anticipated hydrocarbons, and aromatics, while the second FID monitored oxygenates and acidic products. The TCD detector was responsible for monitoring permanent gases and catalytic conversions. Eqn (5)–(7) reported by Peng Gao et al. were used to determine CO2 conversion and hydrocarbon selectivity on a carbon atom basis.71
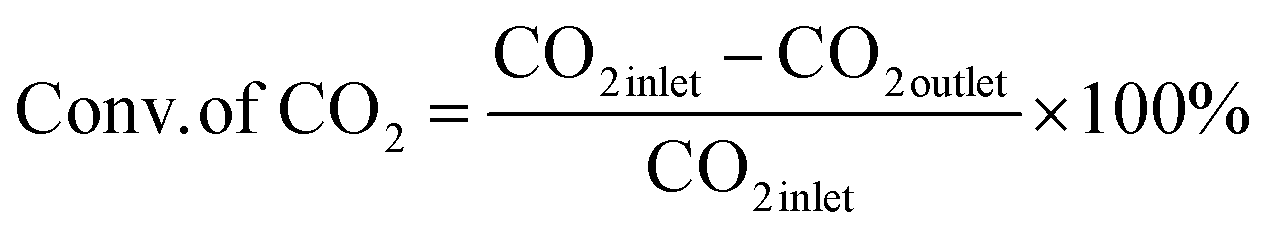 | (5) |
 | (6) |
 | (7) |
3. Results and discussion
3.1 Characterization of the RM-supported catalysts
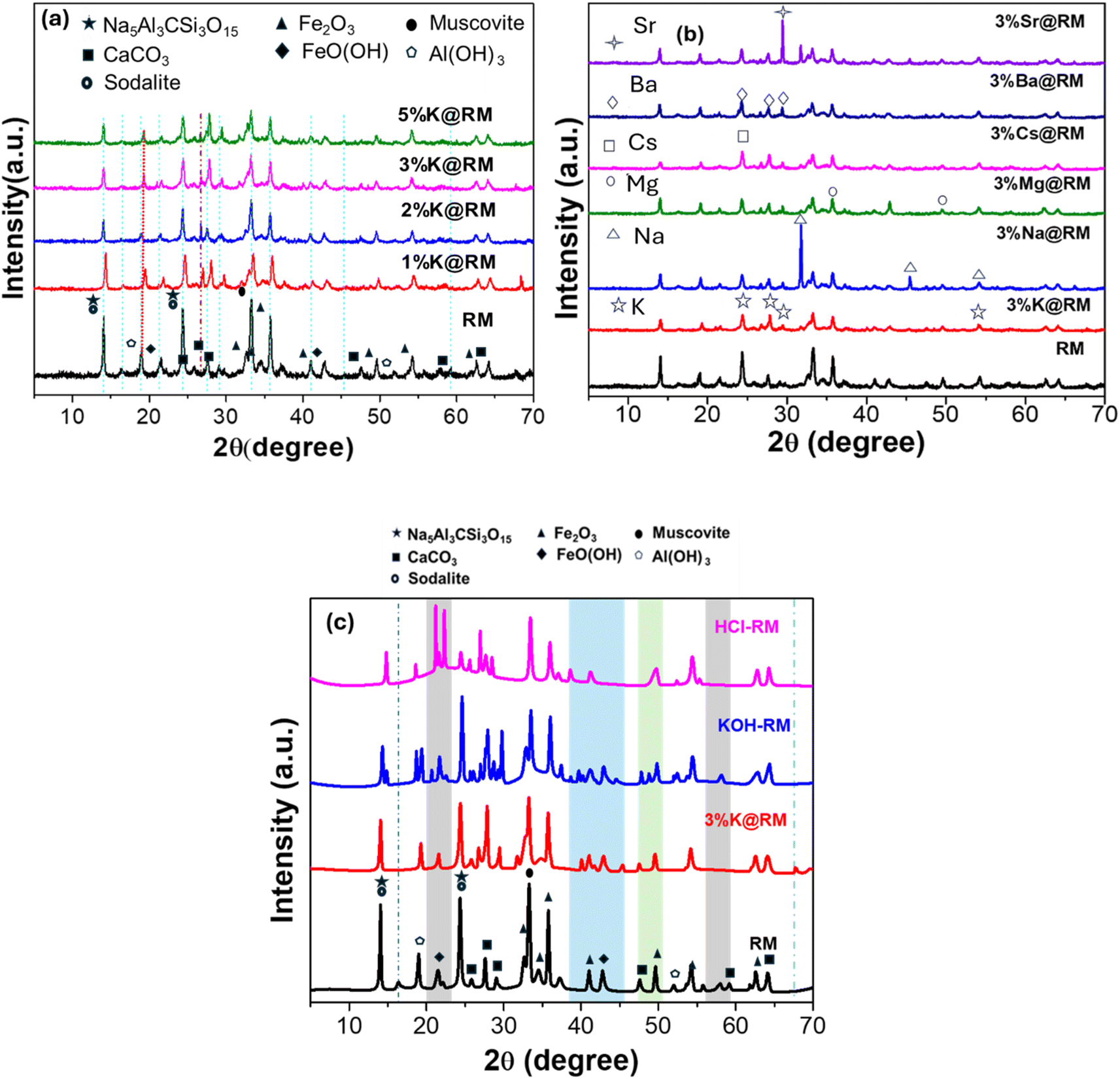 | ||
| Fig. 1 XRD of (a) pure RM and different percent K-promoted RM, (b) pure RM and 3 wt% different metal-promoted RM, (c) pure RM and 3%K@RM, KOH-treated RM (KOH-RM), and HCl-treated RM (HCl-RM). | ||
The peak at 19° for Al in RM has shown a little shift to the right with the increase of K loading which was observed as a maximum shift by 0.3° for the addition of 5%K in RM (Fig. 1a). This was due to the crystal lattice strain or lattice deformation resulting in smaller grain size, as evidenced in HRTEM the 5%K@RM (Fig. 5). However, the excess amount of K (5%K@RM) incorporates higher basicity, which breaks down larger crystals as supported by TEM data.76
Then, different metal ions (3%) (X= Na, Mg, Cs, Ba, and Sr) in addition to the K are loaded into the RM, and its XRD signatures were evaluated, and the results are shown in Fig. 1b. The diffraction peaks of each individual metal ions were detected in XRD as shown in Fig. 1b. In Fig. 1c, the addition of KOH by wet method has showed the presence of many more microcrystalline structures in the catalyst. The XRD data that appeared for KOH-treated RM has many new reflection peaks (from 2θ = 20 to 2θ = 46°, shaded regions). These peaks were generated from different planes of the corresponding metal compounds (that were absent in RM). HCl-treated RM has mainly bigger ions as smaller ions were washed away such as Na, Al, and other composite materials Na5Al3CSi3O15. From the XRD data of HCl-treated RM (Fig. 1c), the diminished intensity of Na5Al3CSi3O15, CaCO3, Fe2O3, FeO(OH), Al(OH)3 established the proof of this hypothesis.
FTIR spectra of pure RM and K-promoted RM with comparable transmission bands are presented in Fig. 2(a and b) and the dominant bands are listed in Table 1. No noticeable change in the absorption band is observed when the concentration of K is increased from 1 to 5% (Fig. 2a). However, a broad peak at 3433 cm−1 appeared in the stretching vibration of –OH groups due to adsorbed water molecules in the surface or the envelope formed by hydrogen-bonded surface –OH groups, when the data is compared with pure RM and 3%K@RM in Fig. 2b.77 The bands at 555-565 cm−1 were assigned to the Fe–O stretching vibration arising from the hematite present in the RM. Also, a prominent peak at 1435 cm−1 appears due to CaCO3 formed after calcination of RM at 500 °C.
 | ||
| Fig. 2 FT-IR data of (a) K-promoted RM, (b) pure RM vs. 3wt% K-promoted RM (c) Raman spectroscopy of K-promoted RM. | ||
| Wavenumbers (cm−1) | Phases |
|---|---|
| 3433 | OH group, H-bond |
| 1640 | H2O in cancrinite |
| 1435 | CaCO3 |
| 1113 | FeO(OH) |
| 986 | Ca2SiO4, Si–O–Si(Al) |
| 612–695 | Al–O |
| 563 | Fe2O3 |
Additionally, the peak at 1113 cm−1 suggests asymmetric stretching of Si–O–Al framework in cancrinite and hibschite, as well as O–Fe–O in goethite. Furthermore, the peak at 986 cm−1 signifies Si–O–Si(Al) bonds, corresponding to the anti-symmetric stretching vibrations (υ3) in SiO4 tetrahedra of Ca2SiO4. The peaks at 563 and below 500 cm−1 in RM are attributed to the stretching vibration of Fe–O, some minor peaks at the range of 612-695 cm−1 correspond to Al–O band, which are predominant in 3% and 5%K promoted RM (Fig. 2a).77 This may be due to the phase changes during the activation process.
The presence of metal-oxides is further supplemented by Raman spectroscopy and the results are shown in Fig. 2c. The distinct Raman peaks at specific wavelengths in the analysis represent various metal oxides in RM. For example, five prominent peaks at 145, 211, 275, and 620 cm−1, are associated with hematite (Fe2O3), which is one of the major components in RM. Additionally, the peaks at 145, 280, 400, and 508 cm−1 could be attributed to the presence of anatase (TiO2), calcium carbonate (CaCO3), alumina (Al2O3), and quartz (SiO2), respectively.78–81 Also, the presence of Fe2O3 is more noticeable when 3% K was added to RM.
A scanning electron microscope (SEM) imaging study is performed to investigate the surface morphology of the pure RM and alkali and alkaline earth metal-promoted RM catalysts. As depicted in Fig. 3, the heterogeneous internal structure of RM is confirmed by the SEM imaging of pure RM, which is constituted by different sizes of particles and crystals with various shapes and morphology ranging from nanometer to a few microns sizes formed by agglomeration (Fig. 3). The higher agglomeration of particles was observed with the KOH-treated samples, thereby decreasing the surface area and leading to the loss of active sites.
 | ||
| Fig. 3 SEM image of pure RM, KOH- and HCl-treated RM, and different metal-promoted and different wt% K-promoted RM. | ||
It is noticeable in SEM that the monovalent Na, K, and Cs metal-promoted RM contributed to an increase in the particle size than in di-valent Mg, Ba, and Sr metal-promoted RM with the fixed concentration of metal ions (3%) (Fig. 3a–i). Also, no significant effect in morphology is noted when the K content is increased from 1 to 3% (Fig. 3j–l). However, a higher amount of K (5%) in RM undoubtedly promotes further agglomeration, diminishing catalytic activity. X-ray dispersive spectroscopy (EDS) attached to SEM confirms the presence of constituent elements in the RM, its promoter metal elements, and their distribution. The detailed composition and contents of Si, Fe, Al, Na, Ti, and Ca of RM and other catalysts were found by EDS mapping data and from XRF normalized wt% (ESI, Fig. S1, S2, and Table S1†).
The transmission electron microscopy (TEM) images are depicted in Fig. 4. The particles present in the RM and RM-based catalysts exhibit diverse sizes and shapes with different crystal planner orientations due to the presence of different metal nanocrystals. KOH-treated RM showed irregularly shaped smaller particles, while HCl-treated RM exposed longer arrays of crystal planes, which resulted in TiO2 rod-like structures.82 The introduction of K and other metals had an impact on the grain size and crystallinity of the constituent materials (ESI†). Pure RM crystallinity was enhanced by a slight addition of K. This trend leveled off to 3 wt% of K. However, the higher amount of K (5%) in RM starts to break the long-range order of the lattice. This claim has been justified by the XRD peak analysis presented in Fig. 1c. It caused smaller grains and a large amount of grain boundary. However, it did not bring significant changes in the morphology of the particles. Upon examination, it was noted that K-promoted RM resulted in the formation of particles with pores and rod-like structures, attributed to the contribution of quartz and rutile82 in RM. The unidirectional crystal growth, which gives the tubular shape, comes from the combined effect of Al substitution on hematite.83 The effect of Na, Cs, Mg, and Sr on RM particle shape and size has shown a similar effect. However, 3%Ba@RM has produced bigger cubic particles due to the cubic nature of BaO.
 | ||
| Fig. 4 TEM image of pure RM and different metal-promoted RM. | ||
HRTEM picture has shown slip dislocation, or planar defect on the structure (Fig. 5). HRTEM of 3%K@RM revealed the clear and bright surface of the particle. As mentioned earlier, RM is a complex mixture of different metal oxides, HRTEM image clearly established the presence of Al2O3 with d(110) = spacing 0.24 nm,84 hematite Fe2O3 with d(311) = 0.25 nm.85 Also, the presence of hexagonal anatase TiO2 nanoparticles with d(101) = 0.35 nm (ref. 82) and rutile phase TiO2 nanorods with d(110) = 0.33 nm82 is confirmed by the matching of inter-planar distance (d).
 | ||
| Fig. 5 (a and b) TEM, (c) HRTEM and SAED image(inserted) of 3 wt%K-promoted red mud. | ||
The hydrogen consumption-ability of pure and modified RM samples was assessed using temperature-programmed reducibility studies (H2-TPR) and spectra, as shown in Fig. 6a. It demonstrated the well-defined reduction peak at 502 °C for the pure RM. Generally, hematite (α-Fe2O3) is reduced by H2 in the range of 220 to 680 °C,86 while magnetite reduces in the range of 600 to 800 °C.87 The other metals available in the RM sample such as sodium, potassium, magnesium, calcium, and titanium are also reduced in the range of 250 to 850 °C, which concludes that the broad peak is composed of overlapping of a series of peaks of each constituent element. However, modifying the RM sample with different concentrations of potassium led to a reduction in H2 consumption, as shown in Fig. 6a. It was observed that the total H2 consumption for the parent RM catalyst was 502 μmol g−1 at temperatures ranging between 200 to 850 °C. Moreover, as the concentration of the K increased to 5 wt%, a reduction in H2 consumption (502 to 423 μmol g−1) was observed. Additionally, it was observed that the first peak, centered around 600 °C, shifted to a lower temperature as the concentration of K increased in the sample. On the other hand, the second peak located after 800 °C was shifted toward higher temperatures, which confirms the contribution of K in changing the elemental phase of the RM.
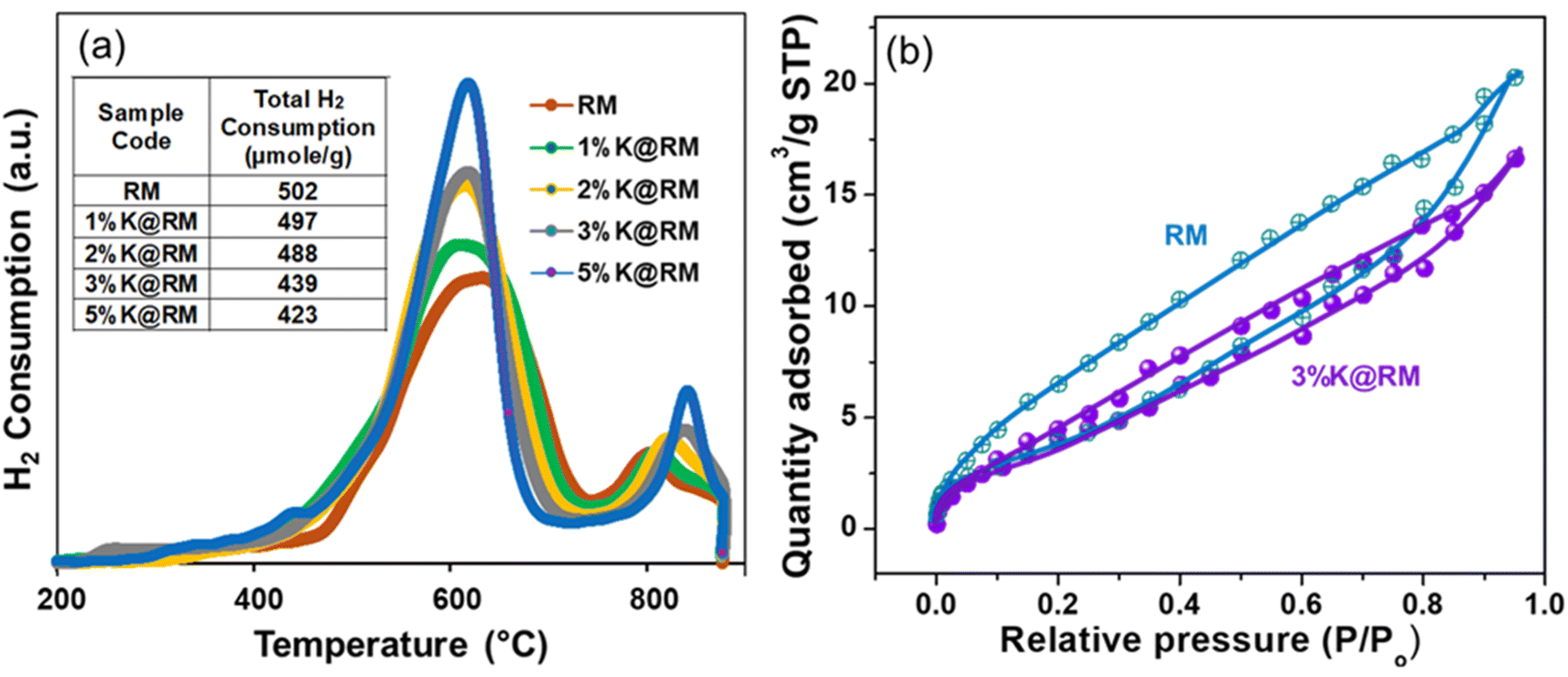 | ||
| Fig. 6 (a) H2-TPR profile, (b) N2 adsorption–desorption isotherms of RM and K-promoted RM samples. | ||
Fig. 6b illustrates the textural properties of the pure RM and 3% alkali or alkaline earth metal-loaded RM catalysts. In the curve of RM, it is evident that the adsorption and desorption curves diverged when the relative pressure was increased. A substantial hysteresis loop was observed between relative pressures of >0 and 1.0, indicating the presence of huge mesoporosity in the prepared catalysts. According to IUPAC isotherm data, type IV suggests the presence of numerous mesopores and macropores in RM with monolayer and multilayer adsorption.88 In contrast, the 3%K@RM hysteresis loop demonstrated a significant decrease in adsorption capacity. When the relative pressure was below 0.2, the adsorption and desorption curves were nearly superimposed, indicating the existence of micropores. Noticeably, BET surface area decreased gradually from 20 m2 g−1 to 11.2 m2 g−1 as the K loading increased to 5%. The adsorption of gas was probably facilitated in RM instead of K loaded RM (Table 2).
| Sample | Pore volume (cc g−1) | Surface area (m2 g−1) |
|---|---|---|
| RM | 0.029 | 20.36 |
| 1%K@RM | 0.026 | 14.19 |
| 3%K@RM | 0.023 | 15.26 |
| 5%K@RM | 0.019 | 11.2 |
3.2 Catalytic evaluation
The catalytic performance for CO2 hydrogenation process was performed in a fixed bed reactor with gas hourly space velocity (GHSV) of 4500 mL g−1 h−1. The catalytic process was performed at 375 °C and 30 bar using CO2:H2 mixture (1:3). As shown in Fig. 7a, the catalytic conversion of CO2 hydrogenation over pure RM promoted with different wt% of potassium was presented after 6 hours of reaction on stream. The conversion was found to have variation in catalytic activities. With pure RM acting as a control catalyst, 22% of CO2 conversion was accomplished. However, treating the pure Red Mud catalysts with HCl resulted in the reduction of the catalytic conversion to 16%, due to the role of acid in removing some of the elements such as aluminum from the catalytic structure and, consequentially, a chance for enhancing the presence of oxygen vacancies required for the reduction of CO2 to CO, but with less catalytic activity due to the limitation of metal active sites. By this treatment, it is proved that CO2 hydrogenation process requires large metal active sites as well as oxygen vacancies for the process of reverse-water gas shift reaction. Similarly, treating the RM catalyst with 3% potassium driven from potassium hydroxide source led to a reduction of the catalytic conversion to around 16% due to the role of potassium hydroxide in disturbing the crystallinity structure of silicon atoms within the catalyst bed. Although the catalytic conversion dropped in these two samples from 22 to 16%, the catalytic selectivity toward CO was enhanced by acid treatment and the catalytic selectivity for CO increased from 91% to 97%. On the other hand, selectivity dropped significantly from 91% to 64% by base treatment. The aforementioned data suggest that acid and base have a significant role in influencing both the metallic composition of the catalyst and the existence of oxygen vacancies, which are necessary for the conversion of CO2 to CO. Furthermore, acid and base treatment also had an impact on selectivity toward hydrocarbons. It was observed that the acid treatment led to a reduction in hydrocarbons from 9 to 3%, while base treatment led to an increase in the catalytic selectivity toward hydrocarbons from 9 to 36%, indicating that the base-treaded RM catalyst has enhanced the modified Fischer–Tropsch (FT) process. On the other hand, the selectivity of light olefins with the produced hydrocarbons was also affected by acid and base treatment. It was observed that the selectivity toward light olefins increased from 7 to 12% by acid treatment, while it decreased from 9% to 1% by base treatment. All the catalytic performance data has been summarized in Table 3 and comparative performance with conventional catalyst is presented in Table 4.
 | ||
| Fig. 7 CO2 conversion and olefins selectivity data of (a) pure RM and different wt% K-promoted RM (b) 3wt% different metal-promoted RM. | ||
| Sample | CO2 conversion | CO | Hydrocarbon | C2=4 | CH4 |
|---|---|---|---|---|---|
| a The converted CO2 was produced as CO hydrocarbons. The main products of those hydrocarbons were olefins and CH4. | |||||
| RM | 22 | 91 | 9 | 7 | 75 |
| HCl treated RM | 16 | 97 | 3 | 12 | 83 |
| 3%KOH@RM | 16 | 64 | 36 | 1 | 92 |
| 1%K@RM | 21 | 96 | 4 | 8 | 72 |
| 2%K@RM | 21 | 95 | 5 | 11 | 70 |
| 3%K@RM | 27 | 94 | 6 | 28 | 54 |
| 5%K@RM | 27 | 93 | 7 | 21 | 53 |
| 3%Na@RM | 26 | 92 | 8 | 3 | 82 |
| 3%Cs@RM | 27 | 93 | 7 | 5 | 77 |
| 3%Ba@RM | 26 | 91 | 9 | 3 | 80 |
| 3%Mg@RM | 25 | 95 | 5 | 6 | 77 |
| 3%Sr@RM | 25 | 90 | 10 | 3 | 84 |
| Promoter | Catalyst support | Synthesis method | Conditions | Performance | Ref. |
|---|---|---|---|---|---|
| a XCO2 = CO2 conversion efficiency, SCO = selectivity of CO, SCH4 = selectivity of CH4, and SC2–C4 = Selectivity of light olefins (C2–C4). | |||||
| 16.5% K and Ba | 1% Rh/Al2O3 | Flame spray pyrolysis | GHSV = 6000 h−1, H2/CO2 = 4/1 | XCO2 = 25%, SCH4 = 0% at 375 °C (16.5% K on 1% Rh/Al2O3) | 96 |
| 2% Pt | CeO2 | WHSV, 300 L g −1 h−1 | XCO2 = 21.7%, SCO = 100% at 290 °C | 97 | |
| Au@UIO-67 | MOF | Wet method | GHSV = 12000 mL h−1 gcat−1, CO2/H2 = 1:3, 2.0 Pa |
XCO2 = 30.2%, SCO = 28.6% at 408 °C | 98 |
| Ni | Ce–Zr–O | Co-precipitation | GHSV = 6000 mL h−1 gcat−1, CO2/H2 = 1:1, 1.0 MPa |
XCO2 = 34%, SCO = 19.7% at 550 °C | 99 |
| 2%K | 30%Fe3O4/ZrO2 | Dry mixing | GHSV = 15000 mL h−1 gcat−1, CO2/H2 = 1:4, 0.1 MPa |
XCO2 = 22%, SCO = 77%, SCH4 = 49%, SC2–C4 = 35%, at 375 °C | 100 |
| K | Red mud | Wet method | GHSV = 9600 mL h−1 gcat−1, CO2/H2 = 1:3, 30 bar |
XCO2 = 45%, SCO = 36% at 375 °C | 70 |
| RM | Dry mixing | GHSV = 4500 mL h−1 gcat−1, CO2/H2 = 1:3, 30 bar |
XCO2 = 22%, SCO = 91%, SCH4 = 75%, SC2–C4 = 7%, at 375 °C | This work | |
| 3%K (from KNO3) | XCO2 = 27%, SCO = 94%, SCH4 = 54%, SC2–C4 = 28%, at 375 °C | ||||
| 3% Na | XCO2 = 26%, SCO = 92%, SCH4 = 82%, SC2–C4 = 3%, at 375 °C | ||||
| 3% Cs | XCO2 = 27%, SCO = 93%, SCH4 = 77%, SC2–C4 = 5%, at 375 °C | ||||
| 3% Mg | XCO2 = 25%, SCO = 95%, SCH4 = 77%, SC2–C4 = 6%, at 375 °C | ||||
| 3% Ba | XCO2 = 26%, SCO = 91%, SCH4 = 80%, SC2–C4 = 3%, at 375 °C | ||||
| 3% Sr | XCO2 = 25%, SCO = 90%, SCH4 = 84%, SC2–C4 = 3%, at 375 °C | ||||
The higher olefins selectivity for HCl-treated RM and their subsequent DI-water washing may cause the removal of smaller metal cations from RM as shown in eqn (8) and enhanced the crystallinity of larger cations (as shown in HRTEM), which boosted the active sites of RM needed for the CO2 hydrogenation for olefins selectivity.
| Fe2O3 + 6 HCl → 2 FeCl3 + 3 H2O | (8) |
Heavy elements Sr and Ba favored methanation, but lighter elements Mg enhanced CO selectivity.89 At the same time, Mg enhanced the absorption of CO2 over the Mg-modified Iron based catalyst, resulting in increased methanation efficacy and highly active Fe carbides.90 RM contains around 29% Fe2O3, based on Table S1†'s XRF data. For RM, the CO2 conversion was 22%; for 3%Mg@RM, it was 25%. While the Na and K enhanced CO selectivity, they also increased the number of basic sites.91 CO2 adsorption on the surface was enhanced by larger size for monovalent cations like Cs (absorption of Cs+ > Na+), which directly affects CO2 conversion.92 That effect is clear from our observations presented in Table 3 as well.
The effect of varied concentrations of K (1, 2, 3, and 5 wt%) sourced from KNO3 was found to have a vital effect on the catalytic conversion of CO2 hydrogenation at high concentrations of K as shown in Fig. 7a. Though the addition of K can modify the electronic properties of RM support in a way that enhances the catalytic activity of transition metals (e.g., Fe, Al), the addition of 1% and 2% of K as a promoter (in dry mixing method) was found to have no role in improving the catalytic conversion. The interactions facilitate K+ production, which creates room for direct interactions of metal oxides present in RM. It increased the number of basic sites available on the RM surface for CO2 adsorption and activation. It produced CO from CO2 following the modified Fischer–Tropsch (FT) process39 and the reverse water-gas shift (RWGS) reaction (eqn (9)) by the endothermic reaction. The hydrogenation of CO can yield C2+ hydrocarbons, which are then subjected to the processes described in eqn (10)–(12):
| CO2 + H2 ⇌ CO + H2O; ΔH = +41.2 kJ mol−1 | (9) |
Besides the above-mentioned endothermic reaction can be turned into a spontaneous exothermic reaction by K impregnation as K → K+ + e−. These electrons will facilitate the production of CO as
| CO2 → CO* + O* | (10) |
| nCO + (2n + 1)H2 ⇌ CnH2n+1 + nH2O | (11) |
| nCO + 2nH2 ⇌ CnH2n + nH2O | (12) |
However, further increasing K concentration from 2% to 3 and 5% led to enhancing the catalytic conversion from 22% to 27%, indicating the role of potassium in enhancing the catalytic adsorption for CO2 molecules. Selectivity was found to be affected by the concentration of K. The higher loading concentration leads to the high dispersion of K+ ions, which could lead to better access for CO2 to interact with the K+ active sites. The catalysts with an appreciable amount of Fe, tend to form iron carbide after multiple reaction steps.
| αFe2O3 → Fe3O4 → FeO → Fe0 → Fe5C2 |
According to recent research, the phase transitions of Fe2O3 → Fe5C2 due to catalyst activation and Fe5C2 → Fe3O4 due to catalyst deactivation.93
Iron carbides (Fe5C2) on the catalyst surface improved CO activation and hydrocarbon chain development, leading to enhanced conversion and selectivity to olefins (Fig. 8). The carbides were more effective in CO hydrogenation, producing the required olefins.42 Hägg carbide (Fe5C2) improves CO2 conversion and olefin selectivity in the K–Fe15 catalyst. This phase is very active for CO activation and creation of C2–C4 olefins while reducing the generation of methane and longer-chain hydrocarbons.
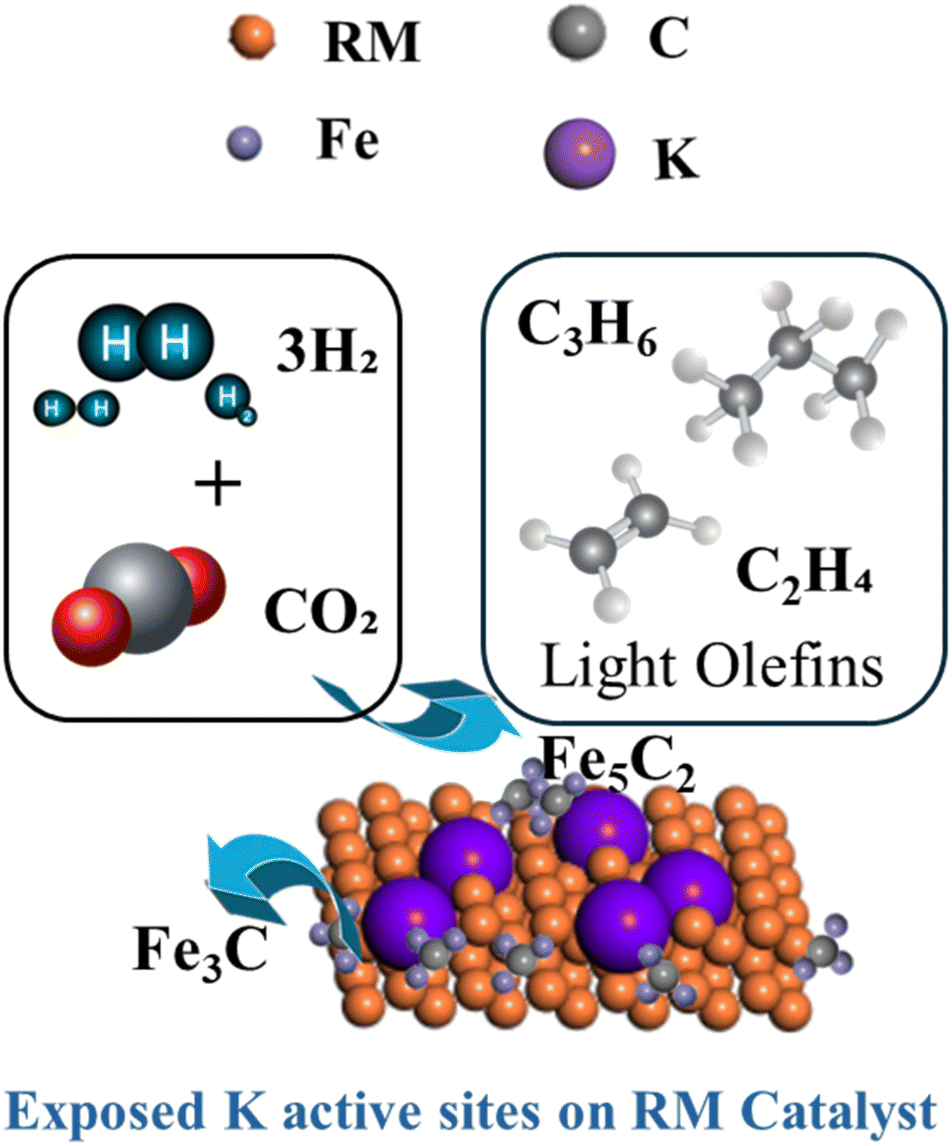 | ||
| Fig. 8 CO2 adsorption by the active sites on RM for higher conversion and olefins selectivity. | ||
It was observed that the CO selectivity has increased from 91 to 96% by increasing the potassium concentration by 1%. Further increase in K concentration was found to have a negative effect on CO selectivity, and the selectivity toward CO was reduced stepwise from 96% to 93% and hydrocarbon selectivity increased from 4 to 7% by increasing K concentration from 1 to 5%. Within hydrocarbons, the selectivity toward light olefins was enhanced from 8% to 28% by increasing K concentration from 1 to 3%. This effect is attributed to its promotion of CO2 adsorption and the suppression of further hydrogenation of produced olefins, which enhances selectivity.94 Beyond 3% of K, the selectivity toward light olefins starts to drop slightly from 27 to 21%, indicating the optimum concentration was obtained at 3% of K, which probably anticipated to attenuate the hydrogenation activity of Fe, consequently fostering the propensity for olefin preference formation.95 This, in turn, may facilitate the re-adsorption of olefins, thereby enabling extended chain growth.
A broader scope of the effect of alkali and alkaline earth metal promoters on CO2 hydrogenation is investigated under the optimized reaction condition. In this regard, a series of alkali or alkaline earth metals, such as Na, Cs, Ba, Mg, and Sr were employed in a fixed concentration (3%) and impregnated into the RM for CO2 hydrogenation. It was observed that the 3%Na loaded RM demonstrated comparable CO2 conversion (26%) with a very low olefins selectivity (3%) as shown in Fig. 7b. However, the Cs promoted RM, and retained its reactivity towards CO2 conversion as 3%K loaded RM. Notably, a trend in very low olefins selectivity (3–6%) was established with these alkali or alkaline earth metals with the RM.
The catalytic performance of red mud (RM) can be attributed to the synergistic roles of its key metal oxides, particularly Fe2O3 and TiO2. Fe2O3 plays a critical role in CO2 activation through its redox properties (Fe3+ ↔ Fe2+), enabling CO2 adsorption and dissociation, while TiO2 provides oxygen vacancies that act as additional active sites and enhances the dispersion of active phases. Together, these oxides create a unique catalytic environment that facilitates electron transfer and surface interactions essential for CO2 hydrogenation. The calcination process at 500 °C improves the crystallinity and distribution of these oxides, further boosting their activity. Additionally, the inclusion of potassium as a promoter modifies the electronic properties of Fe2O3 and TiO2, enhancing CO2 adsorption and hydrogenation efficiency.
Some significant factors affect the interacting mechanisms for CO2 hydrogenation. The catalyst preparation methods, the amount of loading of active metals, the source of support materials, and promoters including their precursor source play a vital role in this case. It is worth noting that the physical and chemical behavior of RM found in different geographical locations and other sources are different due to their different elemental composition. Besides optimal process conditions such as reaction temperature, pressure, and gas flow rate, reaction bed configuration, the relative ratio of CO2 and H2 mixture determines the olefin selectivity. The formation of iron carbides and its effect on catalytic inhibition and activation is also a major concern. The major challenge in the scientific community is to ensure higher CO2 conversion and greater selectivity for light olefin. Furthermore, understanding the feasibility of RM to be used as catalysts in processes such as CO2 hydrogenation is a smart approach to minimize the accumulation of this waste material produced due to the demands of mining and alumina production. This requires extensive studies to understand the relationship between the physiochemical properties of such kind of catalysts and reaction mechanisms. We understand from our studies that the presence of potassium over Red Mud can enhance the presence of light olefins. However, a limited amount of light olefins were obtained, which made room for us to further explore the possibilities of improving Red Mud in coming studies. Besides, to ensure higher CO2 conversion with appreciable hydrocarbon product selectivity, there is a huge space for research with other metals, especially transition metals such as Fe, Co, Cu, and so on. From the extended part of our study, it was observed that iron concentration has a great influence in tuning the catalytic conversion in CO2 hydrogenation and enhancing the catalytic selectivity for hydrocarbons. The investigation was observed for iron concentration ranging between 5 and 20 wt% over Red Mud (X%Fe@3%K@RM, X = 5, 10, or 20) as shown in ESI, Fig. S4.† The main finding was that these composites can lead to higher methane selectivity in the absence of light olefins, which is not the topic of interest here. This study is still in progress to find the desired product by tunable composition of metal active sites.
Our findings might be considered as a foundation of understanding the synergic effect of metals presence in natural materials.
4. Conclusion
In conclusion, RM-based catalysts, when paired with appropriately selected promoter materials, demonstrate a cost-effective solution for addressing the challenges posed by CO2. A straightforward catalyst preparation technique enhances environmental sustainability by minimizing the direct exposure of RM and contributes significantly to CO2 reduction efforts. RM calcined at 500 °C outperformed its HCl- and KOH-treated counterparts. Different effects on CO2 hydrogenation were found when several monovalent and bivalent metal promoters were used. Among them, 3% K-promoted RM emerged as the most effective, achieving 27% CO2 conversion and 28% olefin selectivity. Notably, the source of potassium played a crucial role, as 3% K in RM derived from NO3− and OH− sources yielded varying outcomes. This finding highlights the potential for further research to optimize catalytic performance by exploring diverse sources of the same metal promoter. Furthermore, our research indicates that one crucial factor that may be changed to attain desired olefin yields is the weight percentage of promoters. These insights cover the way for future advancements in designing efficient catalysts for CO2 hydrogenation.Data availability
The related data have been provided in the form of ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors gratefully acknowledge the support of Saudi Aramco for funding this work through grant number ORCP2390. The authors also thank the Saudi Arabian Mining Company (MAA’DEN) for providing the red mud.References
- L. J. R. Nunes, The Rising Threat of Atmospheric CO2: A Review on the Causes, Impacts, and Mitigation Strategies, Environments, 2023, 10(4), 66 CrossRef.
- T. Kuramochi, M. Roelfsema, A. Hsu, S. Lui, A. Weinfurter and S. Chan, et al., Beyond national climate action: the impact of region, city, and business commitments on global greenhouse gas emissions, Clim. Policy, 2020, 20(3), 275–291 CrossRef.
- A. Brdulak, G. Chaberek and J. Jagodziński, Development Forecasts for the Zero-Emission Bus Fleet in Servicing Public Transport in Chosen EU Member Countries, Energies, 2020, 13(16), 4239 CrossRef.
- J. M. Gilligan and M. P. Vandenbergh, A framework for assessing the impact of private climate governance, Energy Res. Soc. Sci., 2020, 60, 101400 CrossRef.
- Z. Xiaoping, S. Baran, W. Cel and Y. Cao, Sustainable Approach to Mitigation of CO2 Emission, Ecol. Chem. Eng. S, 2015, 21(4), 617–622 Search PubMed.
- W. N. Wang, J. Soulis, Y. J. Yang and P. Biswas, Comparison of CO2 Photoreduction Systems: A Review, Aerosol Air Qual. Res., 2014, 14(2), 533–549 CrossRef.
- Q. Zeng, T. Su, J. H. Bing, Y. Jiang, in Photocatalytic Reduction of Carbon Dioxide, 2015, pp. 61–98 Search PubMed.
- Y. Gao, K. Qian, B. Xu, Z. Li, J. Zheng and S. Zhao, et al., Recent advances in visible-light-driven conversion of CO2 by photocatalysts into fuels or value-added chemicals, Carbon Resour. Convers., 2020, 3, 46–59 CrossRef.
- J. Lin, S. Yan, C. Zhang, Q. Hu and Z. Cheng, Electroreduction of CO2 toward High Current Density, Processes, 2022, 10(5), 826 CrossRef.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang and X. Xue, et al., Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals, Adv. Sci., 2018, 5(1), 1700275 CrossRef.
- D. T. Whipple and P. J. A. Kenis, Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction, J. Phys. Chem. Lett., 2010, 1(24), 3451–3458 CrossRef.
- J. Lin, Z. Pan and X. Wang, Photochemical Reduction of CO2 by Graphitic Carbon Nitride Polymers, ACS Sustain. Chem. Eng., 2014, 2(3), 353–358 CrossRef.
- N. Ali, M. Bilal, M. S. Nazir, A. Khan, F. Ali and H. M. N. Iqbal, Thermochemical and electrochemical aspects of carbon dioxide methanation: A sustainable approach to generate fuel via waste to energy theme, Sci. Total Environ., 2020, 712, 136482 CrossRef PubMed.
- K. G. Burra, P. Chandna and A. K. Gupta, Thermochemical Solutions for CO2 Utilization to Fuels and Value-Added Products, Sustainable Development for Energy, Power, and Propulsion, 2021, pp. 59–89 Search PubMed.
- D. S. A. Simakov, in Thermocatalytic Conversion of CO2, 2017, pp. 1–25 Search PubMed.
- H. R. Jhong, S. Ma and P. J. Kenis, Electrochemical conversion of CO2 to useful chemicals: current status, remaining challenges, and future opportunities, Curr. Opin. Chem. Eng., 2013, 2(2), 191–199 CrossRef.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Recent progress in the electrochemical conversion and utilization of CO2, Catal. Sci. Technol., 2012, 2(1), 19–28 RSC.
- H. Yin, X. Mao, D. Tang, W. Xiao, L. Xing and H. Zhu, et al., Capture and electrochemical conversion of CO2 to value-added carbon and oxygen by molten salt electrolysis, Energy Environ. Sci., 2013, 6(5), 1538 RSC.
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki and C. Zeng, et al., Efficient Electrochemical CO2 Conversion Powered by Renewable Energy, ACS Appl. Mater. Interfaces, 2015, 7(28), 15626–15632 CrossRef.
- M. R. Gonçalves, A. Gomes, J. Condeço, R. Fernandes, T. Pardal and C. A. C. Sequeira, et al., Selective electrochemical conversion of CO2 to C2 hydrocarbons, Energy Convers. Manage., 2010, 51(1), 30–32 CrossRef.
- B. Lu, Z. Zhang, X. Li, C. Luo, Y. Xu and L. Zhang, High-efficiency CuCe(rod) catalysts for CO2 hydrogenation with high Cu content, Fuel, 2020, 276, 118135 CrossRef.
- A. Al-Fatesh, S. K. Singh, G. S. Kanade, H. Atia, A. H. Fakeeha and A. A. Ibrahim, et al., Rh promoted and ZrO2/Al2O3 supported Ni/Co based catalysts: High activity for CO2 reforming, steam–CO2 reforming and oxy–CO2 reforming of CH4, Int. J. Hydrogen Energy, 2018, 43(27), 12069–12080 CrossRef.
- C. S. He, L. Gong, J. Zhang, P. P. He and Y. Mu, Highly selective hydrogenation of CO2 into formic acid on a nano-Ni catalyst at ambient temperature: Process, mechanisms and catalyst stability, J. CO2 Util., 2017, 19, 157–164 CrossRef.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng and C. Wang, et al., Cooperative copper centres in a metal–organic framework for selective conversion of CO2 to ethanol, Nat. Catal., 2019, 2(8), 709–717 CrossRef.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, Highly Active and Selective Hydrogenation of CO2 to Ethanol by Ordered Pd–Cu Nanoparticles, J. Am. Chem. Soc., 2017, 139(20), 6827–6830 CrossRef PubMed.
- H. Zhang, H. Han, L. Xiao and W. Wu, Highly Selective Synthesis of Ethanol via CO2 Hydrogenation over CoMoCx Catalysts, ChemCatChem, 2021, 13(14), 3333–3339 CrossRef.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang and C. Yang, et al., CO2 Hydrogenation to Ethanol over Cu@Na-Beta, Chem, 2020, 6(10), 2673–2689 Search PubMed.
- D. Wang, Q. Bi, G. Yin, W. Zhao, F. Huang and X. Xie, et al., Direct synthesis of ethanol via CO2 hydrogenation using supported gold catalysts, Chem. Commun., 2016, 52(99), 14226–14229 RSC.
- Y. V. Kissin, Olefin Polymers, Introduction, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2015, pp. 1–11 Search PubMed.
- G. K. Min, K. Bjerglund, S. Kramer, T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Generation of Stoichiometric Ethylene and Isotopic Derivatives and Application in Transition-Metal-Catalyzed Vinylation and Enyne Metathesis, Chem.–Eur. J., 2013, 19(51), 17603–17607 CrossRef PubMed.
- G. A. Korneeva, YuG. Noskov, T. E. Kron, S. N. Rush, O. G. Karchevskaya and D. V. Marochkin, Hydroformylation of Low Molecular Olefins and Preparation of Oxosynthesis Products Based on C4–C5 Aldehydes in the Production of Ester Lubricating Oils. Part I. Catalysts and Industrial Processes, Chem. Technol. Fuels Oils, 2023, 636(2), 44–56 CrossRef.
- P. H. Ravenscroft and M. E. Turney, Detergents from synthetic olefins, J. Am. Oil Chem. Soc., 1955, 32(7), 418–420 CrossRef.
- L. Lloyd, in Olefin Polymerization Catalysts, 2011, pp. 311–350 Search PubMed.
- G. Leone, M. Mauri, F. Bertini, M. Canetti, D. Piovani and G. Ricci, Ni(II) α-Diimine-Catalyzed α-Olefins Polymerization: Thermoplastic Elastomers of Block Copolymers, Macromolecules, 2015, 48(5), 1304–1312 CrossRef.
- G. E. J. Reynolds, Vinyl acetate-ethylene copolymer dispersions, Br. Polym. J., 1969, 1(5), 233–238 CrossRef.
- G. K. Min, K. Bjerglund, S. Kramer, T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Generation of Stoichiometric Ethylene and Isotopic Derivatives and Application in Transition-Metal-Catalyzed Vinylation and Enyne Metathesis, Chem. - Eur. J., 2013, 19(51), 17603–17607 CrossRef CAS.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Propane to olefins tandem catalysis: a selective route towards light olefins production, Chem. Soc. Rev., 2021, 50(20), 11503–11529 RSC.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti and P. Forzatti, et al., CO2 hydrogenation to lower olefins on a high surface area K-promoted bulk Fe-catalyst, Appl. Catal., B, 2017, 200, 530–542 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu and X. Guo, et al., A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts, RSC Adv., 2018, 8(14), 7651–7669 RSC.
- T. Numpilai, T. Witoon, N. Chanlek, W. Limphirat, G. Bonura and M. Chareonpanich, et al., Structure–activity relationships of Fe-Co/K-Al2O3 catalysts calcined at different temperatures for CO2 hydrogenation to light olefins, Appl. Catal., A, 2017, 547, 219–229 CrossRef.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Light olefin synthesis from CO2 hydrogenation over K-promoted Fe–Co bimetallic catalysts, Catal. Today, 2015, 251, 34–40 CrossRef.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti and P. Forzatti, et al., CO2 hydrogenation to lower olefins on a high surface area K-promoted bulk Fe-catalyst, Appl. Catal., B, 2017, 200, 530–542 CrossRef.
- B. Pawelec, R. Guil-López, N. Mota, J. Fierro and R. Navarro Yerga, Catalysts for the Conversion of CO2 to Low Molecular Weight Olefins—A Review, Materials, 2021, 14(22), 6952 CrossRef.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang and S. Miao, et al., Highly Selective Conversion of Carbon Dioxide to Lower Olefins, ACS Catal., 2017, 7(12), 8544–8548 CrossRef.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity, Nat. Catal., 2019, 2(11), 955–970 CrossRef.
- J. Chen, S. Tian, J. Lu and Y. Xiong, Catalytic performance of MgO with different exposed crystal facets towards the ozonation of 4-chlorophenol, Appl. Catal., A, 2015, 506, 118–125 CrossRef.
- W. Liao, C. Tang, H. Zheng, J. Ding, K. Zhang and H. Wang, et al., Tuning activity and selectivity of CO2 hydrogenation via metal-oxide interfaces over ZnO-supported metal catalysts, J. Catal., 2022, 407, 126–140 CrossRef.
- X. Yuan, T. Pu, M. Gu, M. Zhu and J. Xu, Strong Metal–Support Interactions between Nickel and Iron Oxide during CO2 Hydrogenation, ACS Catal., 2021, 11(19), 11966–11972 CrossRef.
- F. C. F. Marcos, J. M. Assaf and E. M. Assaf, CuFe and CuCo supported on pillared clay as catalysts for CO2 hydrogenation into value-added products in one-step, Mol. Catal., 2018, 458, 297–306 CrossRef.
- W. K. Fan and M. Tahir, Structured clay minerals-based nanomaterials for sustainable photo/thermal carbon dioxide conversion to cleaner fuels: A critical review, Sci. Total Environ., 2022, 845, 157206 CrossRef PubMed.
- Q. Sun, B. W. J. Chen, N. Wang, Q. He, A. Chang and C. Yang, et al., Zeolite-Encaged Pd–Mn Nanocatalysts for CO2 Hydrogenation and Formic Acid Dehydrogenation, Angew. Chem., Int. Ed., 2020, 59(45), 20183–20191 CrossRef.
- I. Nezam, W. Zhou, G. S. Gusmão, M. J. Realff, Y. Wang and A. J. Medford, et al., Direct aromatization of CO2 via combined CO2 hydrogenation and zeolite-based acid catalysis, J. CO2 Util., 2021, 45, 101405 CrossRef.
- I. Graça, L. V. González, M. C. Bacariza, A. Fernandes, C. Henriques and J. M. Lopes, et al., CO2 hydrogenation into CH4 on NiHNaUSY zeolites, Appl. Catal., B, 2014, 147, 101–110 CrossRef.
- X. Yang, H. Duan, R. Wang, F. Zhao, F. Jin and W. Jiang, et al., Tailoring Zeolite L-Supported-Cu Catalysts for CO2 Hydrogenation: Insights into the Mechanism of CH3 OH and CO Formation, Inorg. Chem., 2023, 62(33), 13419–13427 CrossRef.
- W. Wang, C. Duong-Viet, L. Truong-Phuoc, J. M. Nhut, L. Vidal and C. Pham-Huu, Activated carbon supported nickel catalyst for selective CO2 hydrogenation to synthetic methane under contactless induction heating, Catal. Today, 2023, 418, 114073 CrossRef.
- E. Furimsky, CO2 Hydrogenation to Methanol and Methane over Carbon-Supported Catalysts, Ind. Eng. Chem. Res., 2020, 59(35), 15393–15423 CrossRef.
- F. Mateus, P. Teixeira, J. M. Lopes, C. Henriques and C. Bacariza, CO2 Methanation on Ni Catalysts Supported over Activated Carbons Derived from Cork Waste, Energy Fuels, 2023, 37(12), 8552–8562 CrossRef.
- Z. Xie, J. Hei, L. Cheng, J. Li, X. Yin and S. Meng, Influence of Cu/Al Ratio on the Performance of Carbon-Supported Cu/ZnO/Al2O3 Catalysts for CO2 Hydrogenation to Methanol, Catalysts, 2023, 13(5), 800 CrossRef.
- N. Koizumi, X. Jiang, J. Kugai and C. Song, Effects of mesoporous silica supports and alkaline promoters on activity of Pd catalysts in CO2 hydrogenation for methanol synthesis, Catal. Today, 2012, 194(1), 16–24 CrossRef.
- V. Srivastava, Mesoporous Silica Supported Ru Nanoparticles for Hydrogenation of CO2 Molecule, Lett. Org. Chem., 2017, 14(2), 74–79 CrossRef.
- S. Ni, J. Zhu, R. Roy, C. J. Li and R. B. Lennox, Catalytic hydrogenation of CO2 from air via porous silica-supported Au nanoparticles in aqueous solution, Green Chem., 2021, 23(10), 3740–3749 RSC.
- H. Cao, W. Wang, T. Cui, H. Wang, G. Zhu and X. Ren, Enhancing CO2 Hydrogenation to Methane by Ni-Based Catalyst with V Species Using 3D-mesoporous KIT-6 as Support, Energies, 2020, 13(9), 2235 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalón, A. Primo and H. García, Selective Gas-Phase Hydrogenation of CO2 to Methanol Catalysed by Metal-Organic Frameworks, Angew. Chem., Int. Ed., 2024, 63(3), e202311241 CrossRef CAS PubMed.
- S. A. Vali, J. Moral-Vico, X. Font and A. Sánchez, Cu/ZnO/CeO2 Supported on MOF-5 as a Novel Catalyst for the CO2 Hydrogenation to Methanol: A Mechanistic Study on the Effect of CeO2 and MOF-5 on Active Sites, Catal. Lett., 2024, 8, 1–17 Search PubMed.
- E. S. Gutterød, A. Lazzarini, T. Fjermestad, G. Kaur, M. Manzoli and S. Bordiga, et al., Hydrogenation of CO2 to Methanol by Pt Nanoparticles Encapsulated in UiO-67: Deciphering the Role of the Metal–Organic Framework, J. Am. Chem. Soc., 2020, 142(2), 999–1009 CrossRef.
- C. Zhou, Y. Nan, F. Zha, H. Tian, X. Tang and Y. Chang, Application of metal-organic frameworks in CO2 hydrogenation, J. Fuel Chem. Technol., 2021, 49(10), 1444–1457 CrossRef CAS.
- Q. Zhang, S. Wang, M. Dong and W. Fan, CO2 Hydrogenation on Metal-Organic Frameworks-Based Catalysts: A Mini Review, Front. Chem., 2022, 18, 10 Search PubMed.
- G. Yadav, N. Yadav, M. Sultana and M. Ahmaruzzaman, A comprehensive review on low-cost waste-derived catalysts for environmental remediation, Mater. Res. Bull., 2023, 164, 112261 CrossRef CAS.
- J. L. Cao, Z. L. Yan, Q. F. Deng, Z. Y. Yuan, Y. Wang and G. Sun, et al., Homogeneous precipitation method preparation of modified red mud supported Ni mesoporous catalysts for ammonia decomposition, Catal. Sci. Technol., 2014, 4(2), 361–368 RSC.
- A. Russkikh, G. Shterk, B. H. Al-Solami, B. A. Fadhel, A. Ramirez and J. Gascon, Turning Waste into Value: Potassium-Promoted Red Mud as an Effective Catalyst for the Hydrogenation of CO2, ChemSusChem, 2020, 13(11), 2981–2987 CrossRef PubMed.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu and M. Qiu, et al., Direct Production of Lower Olefins from CO2 Conversion via Bifunctional Catalysis, ACS Catal., 2018, 8(1), 571–578 CrossRef.
- X. Li, T. Zhang, G. Lv, K. Wang and S. Wang, Summary of Research Progress on Metallurgical Utilization Technology of Red Mud, Minerals, 2023, 13(6), 737 CrossRef.
- V. Mymrin, H. de Araújo Ponte, O. Ferreira Lopes and A. Vazquez Vaamonde, Environment-friendly method of high alkaline bauxite's Red Mud and Ferrous Slag utilization as an example of green chemistry, Green Chem., 2003, 5(3), 357–360 RSC.
- Y. Qi, The neutralization and recycling of red mud – a review, J. Phys.:Conf. Ser., 2021, 1759, 012004 CrossRef.
- Z. Karimi and A. Rahbar-Kelishami, Efficient utilization of red mud waste via stepwise leaching to obtain α-hematite and mesoporous γ-alumina, Sci. Rep., 2023, 13(1), 8527 CrossRef CAS PubMed.
- D. V. Ribeiro, J. A. Labrincha and M. R. Morelli, Potential use of natural red mud as pozzolan for Portland cement, Mater. Res., 2011, 14(1), 60–66 CrossRef CAS.
- Z. P. Hu, Z. M. Gao, X. Liu and Z. Y. Yuan, High-surface-area activated red mud for efficient removal of methylene blue from wastewater, Adsorpt. Sci. Technol., 2018, 36(1–2), 62–79 CrossRef CAS.
- S. J. Palmer and R. L. Frost, Characterisation of bauxite and seawater neutralised bauxite residue using XRD and vibrational spectroscopic techniques, J. Mater. Sci., 2009, 44(1), 55–63 CrossRef CAS.
- H. Qu, Z. Ling, X. Qi, Y. Xin, C. Liu and H. Cao, A Remote Raman System and Its Applications for Planetary Material Studies, Sensors, 2021, 21(21), 6973 CrossRef CAS.
- D. He, Y. Xiong, L. Wang, W. Sun, R. Liu and T. Yue, Arsenic (III) Removal from a High-Concentration Arsenic (III) Solution by Forming Ferric Arsenite on Red Mud Surface, Minerals, 2020, 10(7), 583 CrossRef CAS.
- S. Barroso-Solares, I. Mediavilla-Martinez, C. Sanz-Velasco, A. C. Prieto and J. Pinto, Pigments identification and analysis of the state of conservation of the decorative elements of the castle of Coca (XV-XVI AC, Segovia, Spain) by Raman Spectroscopy, J. Phys.:Conf. Ser., 2022, 2204(1), 012010 CrossRef CAS.
- S. Dai, Y. Wu, T. Sakai, Z. Du, H. Sakai and M. Abe, Preparation of Highly Crystalline TiO2 Nanostructures by Acid-assisted Hydrothermal Treatment of Hexagonal-structured Nanocrystalline Titania/Cetyltrimethyammonium Bromide Nanoskeleton, Nanoscale Res. Lett., 2010, 5(11), 1829–1835 CrossRef CAS PubMed.
- E. Di Iorio, H. G. Cho, Y. Liu, Z. Cheng, R. Angelico and C. Colombo, Arsenate retention mechanisms on hematite with different morphologies evaluated using AFM, TEM measurements and vibrational spectroscopy, Geochim. Cosmochim. Acta, 2018, 237, 155–170 CrossRef CAS.
- W. Gong, X. C. Li and B. Q. Zhu, Modeling Calculation and Synthesis of Alumina Whiskers Based on the Vapor Deposition Process, Materials, 2017, 10(10), 1192 CrossRef PubMed.
- P. Thomas, P. Sreekanth and K. E. Abraham, Nanosecond and ultrafast optical power limiting in luminescent Fe2O3 hexagonal nanomorphotype, J. Appl. Phys., 2015, 117(5), 053103 CrossRef.
- A. Pineau, N. Kanari and I. Gaballah, Kinetics of reduction of iron oxides by H2, Thermochim. Acta, 2006, 447(1), 89–100 CrossRef CAS.
- P. Li, Y. Li, J. Yu, P. Gao and Y. Han, Kinetics and microstructural changes during fluidized reduction of magnetite with hydrogen at low temperatures, Int. J. Hydrogen Energy, 2022, 47(73), 31140–31151 CrossRef CAS.
- B. Abebe, H. C. A. Murthy and E. Amare, Summary on Adsorption and Photocatalysis for Pollutant Remediation: Mini Review, J. Encapsulation Adsorpt. Sci., 2018, 08(04), 225–255 CrossRef.
- C. Liang, X. Hu, T. Wei, P. Jia, Z. Zhang and D. Dong, et al., Methanation of CO2 over Ni/Al2O3 modified with alkaline earth metals: Impacts of oxygen vacancies on catalytic activity, Int. J. Hydrogen Energy, 2019, 44(16), 8197–8213 CrossRef.
- Z. Baysal and S. Kureti, CO2 methanation on Mg-promoted Fe catalysts, Appl. Catal., B, 2020, 262, 118300 CrossRef.
- Z. Zhang, X. Zhang, L. Zhang, J. Gao, Y. Shao and D. Dong, et al., Impacts of alkali or alkaline earth metals addition on reaction intermediates formed in methanation of CO2 over cobalt catalysts, J. Energy Inst., 2020, 93(4), 1581–1596 CrossRef.
- M. C. Bacariza, R. Bértolo, I. Graça, J. M. Lopes and C. Henriques, The effect of the compensating cation on the catalytic performances of Ni/USY zeolites towards CO2 methanation, J. CO2 Util., 2017, 21, 280–291 CrossRef.
- Y. Zhang, C. Cao, C. Zhang, Z. Zhang, X. Liu and Z. Yang, et al., The study of structure-performance relationship of iron catalyst during a full life cycle for CO2 hydrogenation, J. Catal., 2019, 378, 51–62 CrossRef.
- A. Ramirez, L. Gevers, A. Bavykina, S. Ould-Chikh and J. Gascon, Metal Organic Framework-Derived Iron Catalysts for the Direct Hydrogenation of CO2 to Short Chain Olefins, ACS Catal., 2018, 8(10), 9174–9182 CrossRef.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich and H. U. Islam, et al., Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts, Nat. Commun., 2015, 6(1), 6451 CrossRef.
- R. Büchel, A. Baiker and S. E. Pratsinis, Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/Al2O3 catalysts for CO2 hydrogenation, Appl. Catal., A, 2014, 477, 93–101 CrossRef.
- A. Goguet, F. Meunier, J. P. Breen, R. Burch, M. I. Petch and A. Faur Ghenciu, Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (RWGS) reaction, J. Catal., 2004, 226(2), 382–392 CrossRef.
- H. Xu, Y. Li, X. Luo, Z. Xu and J. Ge, Monodispersed gold nanoparticles supported on a zirconium-based porous metal–organic framework and their high catalytic ability for the reverse water–gas shift reaction, Chem. Commun., 2017, 53(56), 7953–7956 RSC.
- F. M. Sun, C. F. Yan, Z. Da Wang, C. Q. Guo and S. L. Huang, Ni/Ce–Zr–O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGS), Int. J. Hydrogen Energy, 2015, 40(46), 15985–15993 CrossRef.
- H. S. Alghamdi, M. A. Sanhoob, A. M. Ajeebi, MdA. Aziz, M. Nasiruzzaman Shaikh and L. H. Voon, Ultrasmall Fe3O4 Nanoparticles on ZrO2 as Catalysts for CO2 Hydrogenation to Lower Olefins, Asian J. Org. Chem., 2024, e202400379 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08784d |
| This journal is © The Royal Society of Chemistry 2025 |