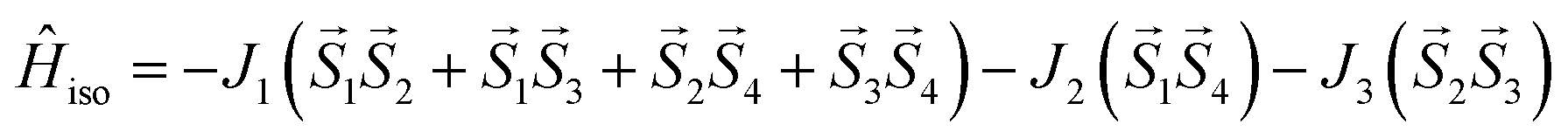DOI:
10.1039/D4RA08700C
(Paper)
RSC Adv., 2025,
15, 4250-4261
Ferromagnetically coupled tetranuclear Ni(II)-2-oxy-aceto- or benzo-phenonate complexes†‡
Received
11th December 2024
, Accepted 22nd January 2025
First published on 7th February 2025
Abstract
Reaction of 2-hydroxy-acetophenone (HL) or 2-hydroxy-benzophenone (HL′) with nickel(II) acetate provides the tetrakis-[(μ3-methanolato-κ3O:O:O)(methanol-κO)(2-oxyacetophenone-κ2O,O′)nickel(II)], [Ni(L)(μ3-CH3O)(CH3OH)]4 (1) or tetrakis-[(μ3-methanolato-κ3O:O:O)(aqua-κO)(2-oxybenzophenone-κ2O,O′)nickel(II)] monohydrate, [Ni(L′)(μ3-CH3O)(H2O)]4·H2O (2). Molecular structure determination demonstrates each nickel(II) ion is six-coordinate with a distorted octahedral geometry defined by three oxygen atoms from three methoxide fragments, a methanol (1) or water (2) molecule, and two oxygen atoms from the acetophenonate (L−) or benzophenonate (L′−) ligand, such that the four nickel atoms and four methoxide groups represent a cubane-type structural topology with each methoxide fragment bridging three of the metal centers. Stabilization of the cubane core occurs via intramolecular O–H⋯O hydrogen bonds. Solid-state magnetic measurements along with computational modeling confirm dominant ferromagnetic interactions for the compounds, attributed to their cubane topology and the Ni–O–Ni angles adopting values lower than 100°. Thermogravimetric analysis (TGA) suggests thermal decomposition of the complexes with successive release of the lattice water, coordinated solvents (MeOH or H2O), OCH3 groups and fragmented ligand species, supported by differential scanning calorimetry (DSC) studies. Cyclic voltammetry reveals a quasi-reversible two electrons charge transfer process in N,N-dimethylformamide.
Introduction
Polynuclear compounds containing paramagnetic metal centres show potential applications in various areas, including biomimetic systems to study multielectron transfer mechanisms and enzyme active sites,1,2 catalysis3,4 and in magnetic materials.5–12 Magnetic interaction among paramagnetic metal centers is noteworthy for conceptual purposes13 in particular for understanding the relationship between structural and magnetic features for the improvement of molecular magnetic materials including single-molecule magnets (SMMs).14–19 Nickel(II) complexes with high nuclearity have attracted substantial attention as models for metal-containing sites.20 Major efforts have been made to synthesize cubane-type Ni4O4 clusters, employing appropriate chelating ligands that can concurrently function as both terminal as well as bridging ligands for nickel(II) ions. Tetranuclear nickel(II) compounds containing diketones or diketone like salicylaldehydates have been well-established and have received considerable interest.21–27
Currently, there is rising curiosity regarding the architecture of active sites and catalytic functions of metalloenzymes and enzymatic transformation of urea in the domains of medical and agricultural chemistry that also include the nickel(II) ions.28–32 Salicylaldehyde and its derivatives possess significant coordination abilities with 3d transition metal ions, making them useful in various areas of pure and applied chemistry. The symmetrical Ni4O4 cores have been extensively studied to understand their crystal structures and magnetic properties. In Ni4O4 cubane type compounds, the exchange interaction has been found to be correlated with the Ni–O–Ni angle.33–37 Several theoretical models of super-exchange propose that the interaction between the spins of two d8 Ni(II) ions connected by an oxide ion and coordinated in an octahedral manner, will be positive (ferromagnetic) when the M–O–M angle is 90° and negative (antiferromagnetic) when it is 180°.34–37 The tetranuclear nickel cubane-like core (Ni4O4) compounds consist of four identical μ3-O bridges, which can arise from OCH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33,38–42 or OH40 moieties.
33,38–42 or OH40 moieties.
We report herein the syntheses, molecular structures and supramolecular analyses, thermal and electrochemical studies of the tetranuclear [Ni(L)(μ3-CH3O)(CH3OH)]4 (1) and [Ni(L′)(μ3-CH3O)(H2O)]4·H2O (2) (L− = 2-hydroxy-acetophenonate or L′− = 2-hydroxy-benzophenonate). Each nickel center is supported by a deprotonated ligand and three of the methoxide bridging units with either a methanol (1) or a water (2) molecule completing the coordination sphere, resulting in a distorted octahedral geometry around the metal ion. Each mononuclear unit is further connected to the other by Ni–Ni and Ni–O–Ni linkages, forming a tetranuclear cubane-type cluster. Although a similar structure of 2 was reported earlier,42b but it contains MeOH as coordinated solvent instead of H2O in 2. In addition, there is a lattice H2O molecule in the structure of 2, which connects two-symmetry related tetranuclear-Ni units via two intermolecular hydrogen bonds. The complexes are further tested for magnetic properties in the solid-state along with computational modeling.
Results and discussion
2-Hydroxy-acetophenone (HL) or 2-hydroxy-benzophenone (HL′) reacts with nickel(II) acetate to give the tetranuclear Ni(II)-complexes of tetrakis-[(μ3-methanolato-κ3O:O:O)(methanol-κO)(2-oxyacetophenone-κ2O,O′)nickel(II)], [Ni(L)(μ3-CH3O)(CH3OH)]4 (1) or tetrakis-[(μ3-methanolato-κ3O:O:O)(aqua-κO)(2-oxybenzophenone-κ2O,O′)nickel(II)] monohydrate, [Ni(L′)(μ3-CH3O)(H2O)]4·H2O (2) (Scheme 1). We trace the formation of methanolate (CH3O−) as a ligand in the tetranuclear clusters to the deprotonation by hydrogencarbonate. Although HCO3− is a weak base the acid–base equilibrium is irreversibly shifted to the side of deprotonated methanol by the escape of CO2 from equilibrium under reflux (CH3OH + HCO3− → CH3O− + H2O + CO2↑). Vibrational spectra feature main characteristic bands at 1616 (1) and 1604 (2) cm−1 for ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (Fig. S1‡). A weak broad band is found at 3449 cm−1 for ν(H2O) in 2. Electron impact (EI) mass spectra show the ion peak at m/z = 295 (1) and 452 (2), attributed to the monoligated species (Fig. S2‡). The very low molar conductance (Λm) values of 1.53 (1) and 1.37 (2) suggest non-electrolyte nature of the complexes in DMF at 25 °C.
O) (Fig. S1‡). A weak broad band is found at 3449 cm−1 for ν(H2O) in 2. Electron impact (EI) mass spectra show the ion peak at m/z = 295 (1) and 452 (2), attributed to the monoligated species (Fig. S2‡). The very low molar conductance (Λm) values of 1.53 (1) and 1.37 (2) suggest non-electrolyte nature of the complexes in DMF at 25 °C.
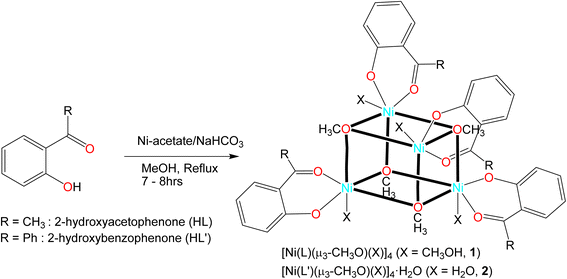 |
| | Scheme 1 Synthetic route to the formation of tetranuclear [Ni(L)(μ3-CH3O)(X)]4 and [Ni(L′)(μ3-CH3O)(X)]4·H2O (X = CH3OH, 1 or H2O, 2) complexes. | |
Electronic absorption spectra
Electronic absorption spectra (UV-vis) for 1 and 2 in chloroform (Fig. S3‡) feature several bands/shoulders below 450 nm, due to intra-ligand n → π*/π → π* electronic transitions (LL) for the coordinated 2-oxy-aceto- or benzo-phenonate ligands. The spectra further show a weak broad band in the visible region (ca. 550–950 nm), attributed to superposition of several metal-centered d–d electronic transitions (MM) for the nickel(II) d8-electrons (Fig. S3,‡ inset).43–47
X-ray crystal structure determination
X-ray diffraction confirmed the formation of the cubane-type cluster, with four Ni atoms occupying four vertices of the cube, and the remaining vertices being occupied by the oxygen atoms of triply-bridging methoxide groups (Fig. 1). Complex 1 crystallized in the monoclinic space group P21/c, and 2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In both complexes, each Ni atom is coordinated to one bidentate O^O-chelate from acetophenonate (L−) or benzophenonate (L′−) and three bridging μ3-methoxide groups, with a terminal non-bridging methanol (1) or water (2) molecule completing the six-coordinated distorted-octahedral geometry around the metal center, representing a cubane-type structural topology.48–51 Further, there is a lattice H2O molecule in the unit cell of the crystal structure of 2 (Fig. S5‡). On the contrary, a similar structure of compound 2 was reported earlier42b which contains MeOH as coordinated solvent instead of H2O in 2 with no lattice H2O. However, the Ni–Ni and Ni–O bond distances (Table 1) are in good agreement within the expected values found in related Ni4O4 cubane-type clusters.48–51
. In both complexes, each Ni atom is coordinated to one bidentate O^O-chelate from acetophenonate (L−) or benzophenonate (L′−) and three bridging μ3-methoxide groups, with a terminal non-bridging methanol (1) or water (2) molecule completing the six-coordinated distorted-octahedral geometry around the metal center, representing a cubane-type structural topology.48–51 Further, there is a lattice H2O molecule in the unit cell of the crystal structure of 2 (Fig. S5‡). On the contrary, a similar structure of compound 2 was reported earlier42b which contains MeOH as coordinated solvent instead of H2O in 2 with no lattice H2O. However, the Ni–Ni and Ni–O bond distances (Table 1) are in good agreement within the expected values found in related Ni4O4 cubane-type clusters.48–51
 |
| | Fig. 1 Molecular structures of 1 (a and b) and 2 (c and d) with thermal ellipsoids (in (a and c)) at 50% probability levels, and (b and d) emphasizing the cuboidal core by showing only the six-membered chelate ring part of L− and L′−, along with hydrogen bonds as dashed orange lines from CH3OH (1) or H2O (2) to the phenolate oxygen atom (H bond details are given in Tables S1 and S2 in ESI‡). The water molecule that was found to be a part of the lattice of 2 is not shown for clarity purposes (cf. Fig. S5‡). | |
Table 1 Selected bond lengths (Å) and angles (°) in 1 and 2
| Complex 1 |
Complex 2 |
| Bond lengths/Å |
| Ni1–O1 |
2.053(1) |
Ni1–O1 |
1.997(2) |
| Ni1–O3 |
2.037(1) |
Ni1–O2 |
2.022(2) |
| Ni1–O4 |
2.051(1) |
Ni1–O9 |
2.049(2) |
| Ni1–O11 |
1.985(1) |
Ni1–O10 |
2.055(2) |
| Ni1–O12 |
2.031(1) |
Ni1–O12 |
2.059(2) |
| Ni1–O13 |
2.123(1) |
Ni1–O13 |
2.130(2) |
| Ni2–O1 |
2.044(1) |
Ni2–O3 |
2.039(2) |
| Ni2–O2 |
2.060(1) |
Ni2–O4 |
2.017(2) |
| Ni2–O4 |
2.044(1) |
Ni2–O9 |
2.079(2) |
| Ni2–O8 |
1.994(1) |
Ni2–O11 |
2.042(2) |
| Ni2–O9 |
2.022(1) |
Ni2–O12 |
2.050(2) |
| Ni2–O10 |
2.131(2) |
Ni2–O17 |
2.146(2) |
| Ni3–O2 |
2.043(1) |
Ni3–O5 |
1.995(2) |
| Ni3–O3 |
2.063(1) |
Ni3–O6 |
2.016(2) |
| Ni3–O4 |
2.056(1) |
Ni3–O9 |
2.056(2) |
| Ni3–O14 |
1.981(1) |
Ni3–O10 |
2.046(2) |
| Ni3–O15 |
2.033(1) |
Ni3–O11 |
2.043(2) |
| Ni3–O16 |
2.125(2) |
Ni3–O14 |
2.162(2) |
| Ni4–O1 |
2.058(1) |
Ni4–O7 |
1.992(2) |
| Ni4–O2 |
2.055(1) |
Ni4–O8 |
2.034(2) |
| Ni4–O3 |
2.059(1) |
Ni4–O10 |
2.042(2) |
| Ni4–O5 |
2.010(1) |
Ni4–O11 |
2.071(2) |
| Ni4–O6 |
2.024(1) |
Ni4–O12 |
2.042(2) |
| Ni4–O7 |
2.114(1) |
Ni4–O15 |
2.140(2) |
|
| Intermetallic distances/Å |
| Ni1⋯Ni2 |
3.0503(5) |
Ni1⋯Ni4 |
3.0272(7) |
| Ni1⋯Ni3 |
3.0520(5) |
Ni2⋯Ni1 |
3.1353(7) |
| Ni1⋯Ni4 |
3.1294(4) |
Ni2⋯Ni3 |
3.0612(8) |
| Ni2⋯Ni3 |
3.1193(4) |
Ni2⋯Ni4 |
3.0500(7) |
| Ni2⋯Ni4 |
3.0627(5) |
Ni3⋯Ni1 |
3.0285(7) |
| Ni3⋯Ni4 |
3.0508(5) |
Ni3⋯Ni4 |
3.1092(9) |
|
| Bond angles/° |
| Ni1–O1–Ni2 |
96.24(5) |
Ni1–O10–Ni4 |
95.28(8) |
| Ni1–O1–Ni4 |
99.13(5) |
Ni1–O12–Ni4 |
95.13(8) |
| Ni2–O1–Ni4 |
96.58(5) |
Ni2–O9–Ni1 |
98.84(8) |
| Ni2–O2–Ni3 |
98.99(5) |
Ni2–O11–Ni3 |
97.10(8) |
| Ni2–O2–Ni4 |
96.21(5) |
Ni2–O11–Ni4 |
95.75(8) |
| Ni3–O2–Ni4 |
96.24(5) |
Ni2–O9–Ni3 |
95.54(8) |
| Ni1–O3–Ni3 |
96.21(5) |
Ni2–O12–Ni1 |
99.45(8) |
| Ni1–O3–Ni4 |
99.63(5) |
Ni2–O12–Ni4 |
96.37(8) |
| Ni3–O3–Ni4 |
95.48(5) |
Ni3–O9–Ni1 |
95.08(8) |
| Ni1–O4–Ni2 |
96.29(5) |
Ni3–O10–Ni1 |
95.22(8) |
| Ni1–O4–Ni3 |
95.97(5) |
Ni3–O10–Ni4 |
99.03(8) |
| Ni2–O4–Ni3 |
99.06(5) |
Ni3–O11–Ni4 |
98.20(8) |
X-ray analysis in 1 shows four strong charge-supported intramolecular hydrogen bonds between the alcoholic hydrogen (O–H) of coordinated methanol and the phenolate-oxygen atom in the cuboidal core (Fig. 1b and Table S1‡). Similarly, compound 2 contains four strong intramolecular hydrogen bonds between the hydrogen atom of coordinated H2O and the phenolate-oxygen atom (Fig. 1d and Table S2‡). In addition, two-symmetry related tetranuclear-Ni complexes are connected via two intermolecular hydrogen bonds from the hydrogen atom of coordinated H2O to the oxygen atom of lattice H2O molecule, and the phenolate-oxygen atom of the neighboring molecule to the same lattice H2O in 2 (Fig. S5 and Table S2‡).
The Hirshfeld surface analysis provides a quantitative assessment of the intermolecular interactions contributing to the crystal packing in the complexes. The 2D fingerprint plot of Hirshfeld surface using the program CrystalExplorer52,53 shows all contributions from closest contacts (Fig. 2, right: for a detailed breakdown see Fig. S6 and S7‡). The highest contribution to the crystal packing arises from H⋯H interactions (e.g., 77.9% for 1 and 65.5% for 2 of the total contacts), which highlights the prevalence of van der Waals forces between the hydrogen atoms in organizing the crystal structure. The second highest contribution includes the C⋯H contacts (19.0% for 1 and 27.4% for 2), an indicator of C–H⋯π and C–H⋯C interactions, which play a critical role in modulating the packing arrangements. The O⋯H interactions (2.9%) reflect a weak hydrogen bonding in 1, while 6.1% indicate an increased role of polar hydrogen bonding in 2 with the crystal water molecule. The comparative studies of Hirshfeld surface features distinct differences in the intermolecular interaction profiles of the two complexes. Complex 1 shows higher dependence on the nonpolar H⋯H interactions (van der Waals forces), in contrast, complex 2 proves a more diverse interactions scenario, with increased contributions from C⋯H and O⋯H contacts, which are indicative of enhanced polar interactions and hydrogen bonding. These differences are attributed to variations in molecular geometry, functional group/solvent distributions, or crystallographic environments between the two complexes. The Hirshfeld surface, mapped with the d-norm property, represents several red spots for the closest contacts and blue ones for the most distant contacts from interior and exterior to the surface, respectively (Fig. 2, left).
 |
| | Fig. 2 Hirshfeld surface mapped with the d-norm property (left side) and 2D fingerprint plot (right side) for 1 (a) and 2 (b), showing an overlay of all possible contributions from closest contacts (details breakdown of all relative contributions to Hirshfeld surface for closest contacts is shown in Fig. S6 and S7‡). The abscissa (di) and ordinate (de) are the distances from the surface to the nearest atoms interior and exterior to the surface, respectively. The large red spot on the Hirshfeld surface of 2 in (b) indicates the H-bond from the aqua ligand O13 to the lattice water O16 (cf. Fig. S5‡). | |
TGA and DSC analyses
Thermogravimetric analysis (Fig. 3) demonstrates that compound 1 undergoes an initial mass loss of ca. 3% at 35–100 °C, attributed to the elimination of one molecule of coordinated CH3OH (theor. 3.1%). Upon increasing the temperature, a mass loss of ca. 9% at 100–135 °C is detected due to release of three CH3OH groups (theor. 9.3%). An elimination of one salicylaldehydate and one CH3O is observed with a gradual mass loss of ca. 14% (theor. 14.7%) at 135–300 °C. However, the largest mass loss of ca. 44% is shown at 300–350 °C due to decomposition of three salicylaldehydates and three OCH3 groups (theor. 44.1%). The residual mass of ca. 27% at 1000 °C corresponds to a mixture of Ni and NiO (theor. Ni 22.8%, NiO 29.1%). Accordingly, compound 2 undertakes an initial mass loss of ca. 2% at 35–80 °C, attributed to the loss of the lattice H2O molecule (theor. 1.5%) and a subsequent mass loss of ca. 6% upon increasing temperature up to 130 °C due to the release of four molecules of coordinated H2O (theor. 5.8%). The mass loss between 130–500 °C of 70% correlates with the loss of the four phenyl-salicylaldehydate and four OCH3 groups (theor. 73.7%). The residual mass of ca. 17% at 1000 °C corresponds to Ni (theor. Ni 19.0%, NiO 24.1%).42b,c,54,55
 |
| | Fig. 3 Thermogravimetric curves (TGA) for 1 and 2 (heating rate 5 K min−1 under N2 atmosphere). | |
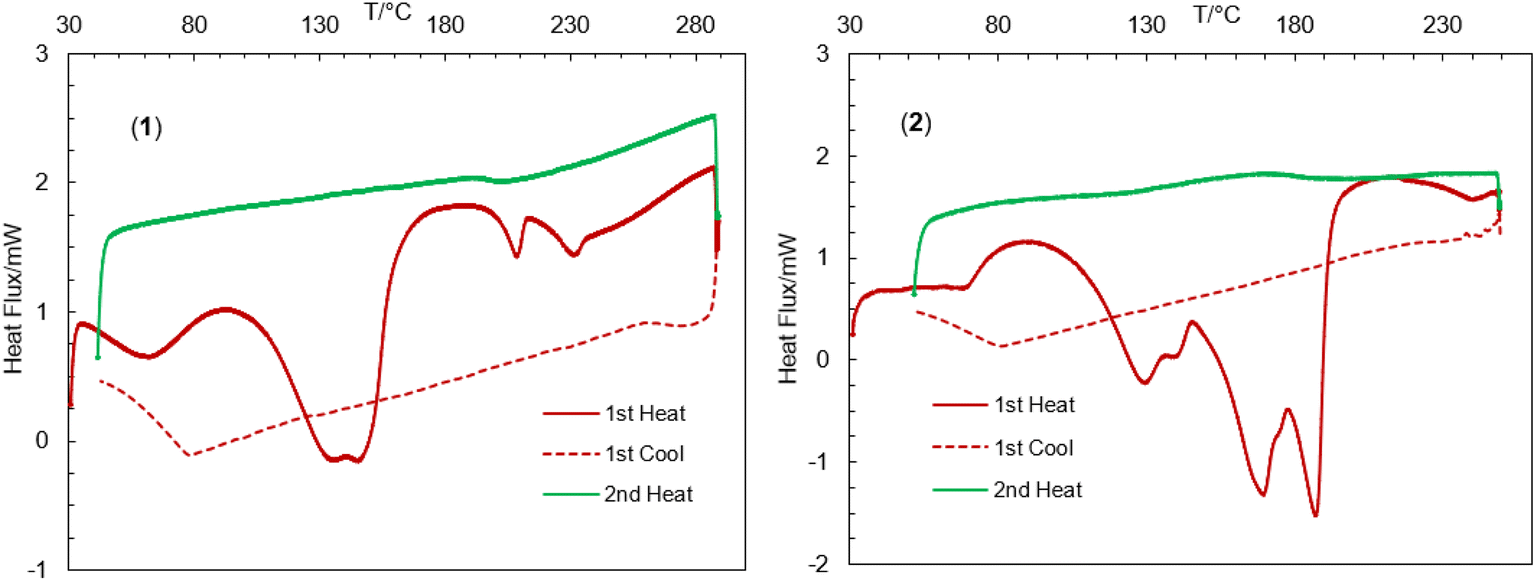
The thermal stability of the complexes was further investigated by differential scanning calorimetry (DSC) in the range of 30–300 °C (Fig. 4).44,45,47,56–59 The DSC heating curves feature several endothermic peaks at ca. 62, 145, 209 and 231 °C for 1 and ca. 67, 129, 169 and 187 °C for 2, correspond to the release of the lattice water, coordinated solvents (MeOH or H2O), OCH3 groups and fragmented ligand species resulting from thermal decomposition of the complexes, in parallel to TGA analyses. The DSC cooling curves display no peaks in the reverse direction, suggesting an irreversible phase transformation associated to the thermal decomposition.
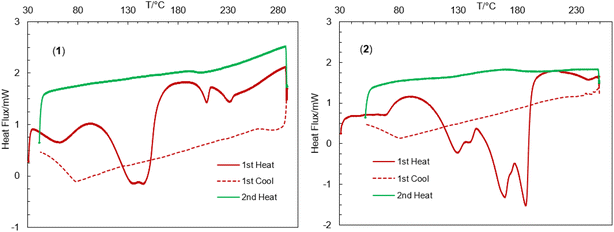 |
| | Fig. 4 Differential scanning calorimetry (DSC) heating and cooling curves for 1 and 2. | |
Electrochemistry
Cyclic voltammograms for 2 were recorded from −1.40 to 1.00 V vs. Ag/AgCl at varying scan rates in N,N-dimethylformamide (DMF) at 25 °C (Fig. 5). The reductive and oxidative scans showed a broad cathodic peak at ca. Ec = −0.89 V (Ic = +15.55 μA) and an anodic peak at ca. Ea = 0.73 V (Ia = −5.58 μA), respectively at a scan rate of 0.10 V s−1. The peak separation value (ΔEac) and E1/2 values are ca. 1.61 V and ca. −0.08 V, respectively. The larger peak separation value for Ni2+/Ni+ redox couple may arise from large distortions at the metal center following reduction to a Ni+ d9 species as it would become Jahn–Teller distorted. However, CV results suggest a one electron charge transfer process for the Ni2+/Ni+ couple during reductive wave and for the Ni+/Ni2+ couple during oxidative wave, diagnosing a quasi-reversible redox process as reported in the related Ni complexes.60–62 Analysis of the voltammograms at faster scan rates showed the cathodic and anodic peaks shifting to more negative and more positive potentials, respectively with an increasing peak intensity. The plots of the peak current vs. square root of the scan rate (i.e., Ic or Ia or Ia/Ic vs. ν1/2) display an increase of cathodic (Ic) as well as anodic (Ia) peak current, while a constant value of Ia/Ic ratio is maintained (Fig. 5, inset). The linear relationship of the plots suggests a diffusion-controlled electrochemical process in solution.
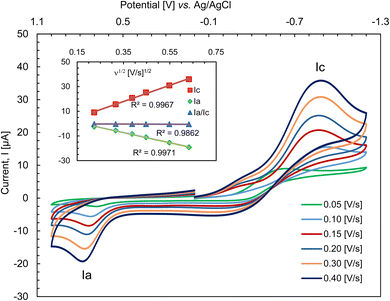 |
| | Fig. 5 Cyclic voltammograms for 2 (ca. 0.5 mM) at varying scan rates; electrolyte: TBAP (ca. 0.1 M) in DMF at 25 °C. | |
Decomplexation of compound 1 with NaCN and isolation of HL
Reaction of complex 1 with NaCN in methanol results in a decomplexation reaction followed by a color change from light green to orange-yellow. The product was isolated as solid and confirmed as 2-hydroxy-acetophenone (HL) by 1H NMR spectroscopy (Fig. S4,‡ details procedure and data are listed in the Experimental section).
Magnetic properties
The Ni4 compounds with cubane topology often exhibit interesting magnetic properties and can behave as magnetically ordered molecular magnets.63 Additionally, the Ni4 molecule can exhibit slow relaxation of magnetization without magnetic ordering, showing behavior typical of Single-Molecule Magnets (SMMs).64 This results from the cubane topology which implies the Ni–O–Ni angles within the cubane core to be close to 90 degrees. Previous works established several magneto-structural correlations for this class of complexes based on the Ni–O–Ni angle value with very similar outcomes: the Ni–O–Ni superexchange pathways possessing angles close to 90° results in ferromagnetic coupling, whereas pathways having angles larger than 99° result in antiferromagnetic coupling.65
This assumption can help to reduce the number of free parameters in the spin Hamiltonian used for fitting the static magnetic data.66 In this work, we decided to utilize the predictive power of ab initio calculations to estimate the strengths and nature of the exchange couplings between the Ni atom pairs by using Broken-Symmetry Density Functional Theory (BS-DFT) calculations. Thus, for both 1 and 2, the calculations on fragments involving only the complex molecule of the particular crystal structure. The BS-DFT calculations were performed for all six pairs of the Ni atoms within the cubane core, resulting in six calculations. In each case, two Ni atoms were substituted with diamagnetic d10 Zn(II) atoms to obtain the pair isotropic exchange interaction constant Jij (e.g. for J12 the fragment containing NiNiZnZn atoms) using the following spin Hamiltonian for two paramagnetic centers A and B: Ĥi = JSASB. The energies of high spin (HS, A↑B↑) and broken symmetry states (BS, A↑B↓) were evaluated using the approach of Ruiz et al. to calculate the J-values (for more details see the ESI‡).67 The scheme of magnetic couplings is shown in Fig. 6. For both compounds, all the calculated coupling constants are positive and thus ferromagnetic. They correlate well with the Ni–O–Ni angles, having the largest couplings calculated for the smallest Ni–O–Ni angles (Jij ≈ +12 cm−1 for ∠NiONi = 96–97°) and vice versa (Jij ≈ +3 cm−1 for ∠NiONi = 99–100°). The third exchange coupling constant should adopt Jij values of approximately +6 cm−1 for 1 and between +7 and +9 cm−1 for 2. Based on the BS-DFT results, and accounting for the general coupling scheme of cubane core (Fig. 6) we established the following isotropic exchange spin Hamiltonians for fitting of the magnetic data measured for 1 and 2:
| |
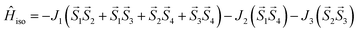 | (1) |
for
1 and,
| |
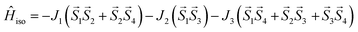 | (2) |
for
2, respectively.
 |
| | Fig. 6 General coupling scheme for cubane core. Red dots represent bridging oxygen atoms. | |
Then, the overall spin Hamiltonian including axial parameter of magnetic anisotropy D then reads:
| |
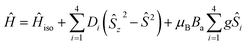 | (3) |
where the
a-direction of the magnetic field is defined as
Ba =
B(sin(
θ)cos(
φ), sin(
θ)sin(
φ), cos(
θ)). Such a form of general spin Hamiltonian was chosen to avoid overparametrization and possesses several simplifications such as same magnetic anisotropy for each cubane center with parallel axes of
D-tensor, no rhombicity and equal
g-values. We fitted both temperature dependence of magnetic moment (
T = 2–300 K) and isothermal molar magnetizations (
T = 2 and 5 K,
B = 0–7 T) simultaneously using the EasySpin package
68a and we opted to test both, positive and negative
D parameter in the fitting.
Both complexes exhibit effective magnetic moment (μeff/μB) values observed to be larger than the spin only value for four non-interacting Ni(II) atoms (S = 1, g = 2.0, μeff/μB = 5.66, μeff/μB > 7.5 observed for 1 and 2). For both compounds, the μeff/μB values increase with decreasing temperature, reaching their maximal values at approximately 15 K (Fig. S8 and S9‡). Upon further decreasing temperature, the μeff/μB values drop. The observed magnetic behaviour corresponds well to dominant ferromagnetic coupling among the Ni(II) atoms within the cubane core and is likely influenced by significant magnetic anisotropy resulting from zero-field splitting at the Ni centers and/or the presence of weaker antiferromagnetic exchange pathways alongside the dominant ferromagnetic ones. In 2, the high temperature dependence of magnetic moment differs from that observed for 1 significantly, and we observed a continuous increase in the μeff/μB value up to 8.1 at 300 K. This could correspond to a temperature independent paramagnetism and/or small amount of ferromagnetic impurity.
As a first attempt to fit the magnetic data we employed a simple model using only one “average” exchange interaction and thus the isotropic spin Hamiltonian was reduced to the following form
| |
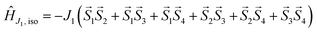 | (4) |
For
1, the simultaneous fitting of the field and temperature dependencies for both positive and negative
D parameters resulted in relatively reasonable fits (Fig. S7
‡), with small and ferromagnetic exchange coupling constants and relatively large magnetic anisotropy (+
D fit:
giso = 2.335,
D = +7.8 cm
−1,
J1 = +3.2 cm
−1; −
D fit:
giso = 2.324,
D = −7.6 cm
−1,
J1 = +3.5 cm
−1). The inclusion of the coupling scheme based on DFT calculations and thus three exchange coupling constants (
eqn (1)) led to a slight improvement of the fit, especially in the case of the fit for positive
D. On the other hand, a slightly worse fit with the negative
D parameter had all three coupling constants of positive value and thus ferromagnetic, which is in agreement with previously published magneto-structural correlations and results of BS-DFT calculations (+
D fit:
giso = 2.329,
D = +6.8 cm
−1,
J1 = +1.1 cm
−1,
J2 = −4.7 cm
−1,
J3 = +12.1 cm
−1; −
D fit:
giso = 2.324,
D = −7.5 cm
−1,
J1 = +2.9 cm
−1,
J2 = +3.8 cm
−1,
J3 = +5.6 cm
−1,
Fig. 7 and Table S3
‡).
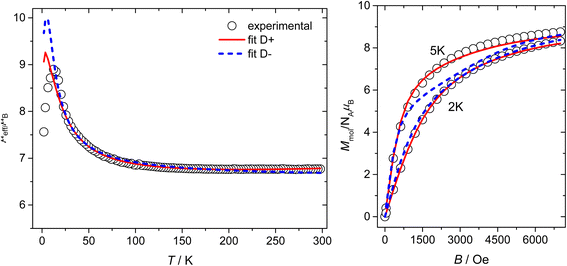 |
| | Fig. 7 Magnetic data for compound 1. Temperature dependence of the effective magnetic moment and the isothermal magnetizations measured at T = 2 and 5 K. The empty circles represent the experimental data points and the lines represent the best fits calculated by using eqn (1) and (3) with parameters listed in Table S3.‡ Full red line represents the best fits calculated for the positive D parameter. The dashed blue line represents the best fits calculated for the negative D parameter. | |
Fitting of magnetic data 2 did not result in satisfactory fits (Fig. S9‡). The results for the spin Hamiltonian with one isotropic exchange coupling constant (eqn (4)) resulted in relatively reasonable small values of the D parameters and large ferromagnetic J1 coupling (+D fit: giso = 2.054, D = +3.7 cm−1, J1 = +14.7 cm−1; −D fit: giso = 2.045, D = −2.7 cm−1, J1 = +14.5 cm−1). Even though the resulting fits reasonably reconstructed experimental data, we needed to use very large values of correction on temperature independent-paramagnetism, with values larger than 100 × 10−9 m3 mol−1 due to the presence of an increase in the μeff/μB values at higher temperatures. The maximal value should be up to 5 × 10−9 m3 mol−1 for 4 Ni atoms,68b indicating issues with the reliability of the experimental data. Therefore, we measured X-ray powder diffraction of the measured sample and found that it had lost its crystallinity and was amorphous, as no Bragg diffraction peaks were observed. We suspect that during the sample preparation the co-crystallized water molecule was lost leading to changes in the crystal structure that caused the compound to lose long-range periodicity. Probably, this is the reason why we cannot correlate its crystal structure with the measured magnetic properties. The fitting of magnetic data using eqn (2) and (3) did not lead to improvement and unreliably large exchange couplings (J3 > +40 cm−1) and again, very large correction on temperature-independent paramagnetism were observed (Table S4‡).
Experimental
Chemicals and instrumentation
IR spectra were recorded on a Nicolet iS10 (Thermo Scientific) spectrometer at ambient temperature. UV-vis spectra were obtained with a Shimadzu UV 1800 spectrophotometer in chloroform at 25 °C. 1H NMR spectra were recorded on a Bruker Avance DPX 400 spectrometer in DMSO-d6 and CDCl3 at 20 °C. Thermogravimetric analysis was carried out on a Netzsch TG 209 F3 Tarsus in the temperature range of 35 to 1000 °C with a heating rate of 5 K min−1 under nitrogen atmosphere. Differential Scanning Calorimetry (DSC) was performed on a Shimadzu DSC-60 at 30–300 °C (ca. 5 °C above the melting point) with a heating rate of 10 K min−1 under nitrogen gas. The molar conductance (Λm) was measured with a Mettler Toledo Fivego (Model F3) conductivity meter in N,N-dimethylformamide (DMF) at 25 °C. Electron Impact (EI) mass spectra were recorded with a Thermo-Finnigan TSQ 700 mass spectrometer. The spectra clearly show the isotopic distribution patterns for the 58/60Ni containing ions. Cyclic Voltammetry (CV) were ran with an Epsilon™ (BASi) instruments using the tetra-N-butylammonium-hexafluorophosphate (TBAP) as supporting electrolyte in DMF at 25 °C. A three electrodes system equipped with a platinum disc (working), platinum wire (auxiliary) and Ag/AgCl electrode (reference). Nitrogen gas was passed through the solution for ca. 10 minutes to avoid any air contamination.
Magnetic measurements
Herein reported magnetic investigation have been carried out on PPMS EverCool II (Quantum Design Inc., San Diego, CA, USA). The exact amount of sample was finely grinded and filled into the gelatine capsule, which has been used as the sample holder. The temperature dependency was recorded in the thermal range 2–300 K at B = 0.1 T using the 1 K min−1 sweeping rate, and field-dependency was measured at isothermal conditions in the range B = 0–7 T. Collected data were corrected for the diamagnetism of eicosane and gelatine capsule as well as for the molecular diamagnetic contribution.
Synthesis of the complexes (1 and 2)
Two equivalents of 2-hydroxy-acetophenone or 2-hydroxy-benzophenone (272.5 mg or 396.5 mg, 2.0 mmol) and NaHCO3 (168.12 mg, 2 mmol) were dissolved in 10 mL of methanol. This solution was stirred for 10 min at room temperature. One equivalent of Ni(II) acetate (249 mg, 1.0 mmol), dissolved in 5 mL methanol, was added to the solution which was then continued to stir under reflux. The color of the solution changed to green immediately and a greenish precipitate was formed within 2–3 hours. The reaction mixture was refluxed for about 7–8 hours under N2 gas. Green colored microcrystals were filtered off, washed 3 times with methanol (2 mL in each time) and dried in air for 2–3 days to obtain green microcrystals of 1 (for 2-hydroxy-acetophenone) or 2 (for 2-hydroxy-benzophenone). Single crystals suitable for X-ray measurements were grown via slow diffusion of methanol into a concentrated dichloromethane (DCM) solution of green microcrystals 1 or 2 after 2–3 days.
Compound 1. Tetrakis-[(μ3-methanolato-κ3O:O:O)(methanol-κO)(2-oxyacetophenone-κ2O,O′)nickel(II)], [Ni(L)(μ3-CH3O)(CH3OH)]4: yield: 0.375 g (72%). Anal. calcd for C40H56Ni4O16: C, 46.71; H, 5.49%. Found C, 46.25; H, 5.12%. IR (KBr, cm−1): ν = 3076, 3010, 2926w (C–H), 1616 vs. (CO) and 1531 vs. (CC). EI-MS: m/z = 295 (45) ([Ni(L)2–CH3OH–H]+), 251 (4) ([Ni(L)(CHO)2]+), 240 (100) ([Ni(L)(CHO)(OH) + H]+), 224 (45) ([Ni(L)(CHO) + H2]+), 210 (15) ([Ni(L)(OH)]+), 181 (20) ([Ni(C6H4)(O)(CHO) + H2]+), 136 (10) ([HL]+) and 121 (20) ([C6H5(COCH3)]+) {[Ni(L)2] = C16H14NiO4; (L)− = 2-oxy-acetophenonate; HL = C8H8O2}. Conductance (Λm) = 1.53 S m2 mol−1 in DMF at 25 °C.
Compound 2. Tetrakis-[(μ3-methanolato-κ3O:O:O)(aqua-κO)(2-oxybenzophenone-κ2O,O′)nickel(II)] monohydrate, [Ni(L′)(μ3-CH3O)(H2O)]4·H2O: yield: 0.503 g (78%). Anal. calcd for C56H56Ni4O16·H2O: C, 54.28; H, 4.69%. Found C, 53.86; H, 4.42%. IR (KBr, cm−1): ν = 3449wb (H2O), 3051, 3012, 2924w (C–H), 1604 vs. (CO) and 1573 vs. (CC). EI-MS: m/z = 452 (3) ([Ni(L′)2]+), 347 (8) ([Ni(L′)2–C6H5(CO)]+), 285 (10) ([Ni(L′)(CHO) + H]+), 257 (20) ([Ni(L′)2–L′ + 2H]+), 197 (100) ([HL′–H]+), 182 (15) ([HL′–OH]+), 121 (45) ([C7H5O2]+) and 105 (30) ([C7H5O]+) {[Ni(L′)2] = C26H18NiO4; (L′)− = 2-oxy-benzophenonate; HL′ = C13H10O2}. Conductance (Λm) = 1.37 S m2 mol−1 in DMF at 25 °C.
Decomplexation of compound 1 with NaCN and isolation of the HL
Complex 1 (40 mg) was dissolved in 5 mL methanol with an excess amount of NaCN. After 30 min of stirring, the solution's color changed from light green to orange-yellow. The solution was then filtered, the filtrate was collected and dried in vacuum using a rotary evaporator at 40 °C followed by in air. The crude products were again dissolved in 5 mL of dichloromethane (DCM), filtered and dried on a vacuum rotary evaporator at 40 °C followed by in air for 2–3 days to obtain the 2-hydroxy-acetophenone (HL), confirmed by 1H NMR spectrum (Fig. S3‡).
1H-NMR (400 MHz, DMSO-d6). δ/ppm = 2.44 (s, 3H, CH3), 5.88 (t, JHH = 7.2 Hz, 1H, H3), 6.25 (d, JHH = 8.4 Hz, 1H, H2), 6.86 (dt, JHH = 6.8, 4.8 Hz, 1H, H4) and 7.40 (dd, JHH = 6.0, 2.0 Hz, 1H, H5) (for hydrogen atom numbering see Fig. S3‡).
X-ray structure determination
X-ray crystal structure data were collected from multi-faceted crystals of suitable size and quality, selected from a representative sample of crystals of the same habit using an optical microscope. Crystals were mounted on MiTeGen loops and data collection for 1 was carried out at 298 K, for 2 in a cold stream of nitrogen (150 K). For 1 a Rigaku XtaLAB Synergy-S, Dualflex, HyPix diffractometer with a micro-focus X-ray tube with Cu-Kα radiation (λ = 1.54184 Å) was used; for 2 a Bruker D8 QUEST ECO. diffractometer was employed using the Bruker APEX4 software.69 For both 1 and 2 structure solution and refinement were performed using XS, XT and XL software, embedded within OLEX2.70 The non-hydrogen atom positions were refined with anisotropic temperature factors. Hydrogen atoms for aromatic CH, aliphatic CH and CH3 groups were positioned geometrically (C–H = 0.93 or 0.95 Å for aromatic CH, and 0.96 or 0.98 Å for CH3 in 1 or 2, respectively) and refined using a riding model (AFIX 43 for aromatic CH, AFIX 13 for aliphatic CH, AFIX 137 for CH3), with Uiso(H) = 1.2Ueq(CH) and Uiso(H) = 1.5Ueq(CH3). The protic hydrogen atoms for CH3OH in 1 were found and refined freely with Uiso(H) = 1.5Ueq(O). In 1, there is a positional disorder for the methoxy CH3 group on O10 which is not shown in Fig. 1a and b. For each structure, the absence of additional symmetry was confirmed using ADDSYM incorporated in the PLATON program.71 Crystal data and details on the structure refinement are given in Table 2. Graphics were drawn with the program DIAMOND.72 Computations on the supramolecular interactions were carried out with PLATON for Windows.73,74 The CCDC numbers are 2367079 and 2367080 for 1 and 2, respectively, and contain the ESI‡ crystallographic data reported in this paper.
Table 2 Crystal data and structure refinement for compounds 1 and 2
| R1 = [Σ(‖Fo| − |Fc‖)/Σ|Fo|]; wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. Goodness-of-fit S = [Σ[w(Fo2 − Fc2)2]/(n − p)]1/2. |
| Complexes |
1 |
2 |
| Code |
NiL104_2 |
ena015 |
| Empirical formula |
C40H56Ni4O16 |
C56H56Ni4O16·H2O |
| M (g mol−1) |
1027.68 |
1237.86 |
| Crystal size (mm) |
0.31 × 0.23 × 0.13 |
0.48 × 0.13 × 0.08 |
| Temperature (K) |
298.60(14) |
150.00 |
| θ range (°) |
2.31–76.11 |
2.35–27.50 |
| h; k; l range |
+24–23, +19–18, ±19 |
±15, ±17, ±23 |
| Crystal system |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P |
| a (Å) |
19.08705(9) |
11.6867(17) |
| b (Å) |
15.15100(7) |
13.294(3) |
| c (Å) |
15.82527(8) |
18.419(4) |
| α (°) |
90 |
76.144(9) |
| β (°) |
95.2175(5) |
74.388(10) |
| γ (°) |
90 |
82.908(7) |
| V (Å3) |
4557.51(4) |
2670.5(9) |
| Z |
4 |
2 |
| Dcalc. (g cm−3) |
1.498 |
1.539 |
| F(000) |
2144 |
1284 |
| μ (mm−1) |
2.417 |
1.461 |
| Max/min transmission |
0.744/0.521 |
0.7455/0.5951 |
| Refl. measured |
160640 |
80855 |
| Refl. unique |
9486 |
12233 |
| Rint |
0.0410 |
0.0257 |
| Parameters/restraints |
576/8 |
726/0 |
| Completeness |
1.000 |
1.000 |
| Largest diff. peak & hole (Δρ/e Å−3) |
0.381/-0.324 |
0.856/-0.624 |
| R1/wR2 [I > 2σ(I)]a |
0.0318/0.0967 |
0.0424/0.0815 |
| R1/wR2 (all reflect.)a |
0.0336/0.0980 |
0.0555/0.0911 |
| Goodness-of-fit on F2b |
1.069 |
1.192 |
| CCDC number |
2367079 |
2367080 |
Conclusion
Molecular structure for the titled tetranuclear [Ni(L)(μ3-CH3O)(CH3OH)]4 (1) and [Ni(L′)(μ3-CH3O)(H2O)]4·H2O (2) discovers each nickel(II) ion is six-coordinate with a distorted octahedral geometry comprising of three oxygen atoms from three methoxide fragments, a methanol (1) or water (2) molecule, and two oxygen atoms from the acetophenonate (L−) or benzophenonate (L′−), so that the four nickel atoms and four methoxide groups provide a cubane-type structure with each methoxide fragment bridging three of the metal centers. This cubane core structure is further stabilized via intramolecular O–H⋯O hydrogen bonds. The two-symmetry related tetranuclear-Ni species are connected via two intermolecular hydrogen bonds in 2. Studies of supramolecular packing are in parallel to Hirshfeld surface analyses. Solid-state magnetic measurements along with computational modelling authenticate dominant ferromagnetic interactions for the complexes, attributed to their cubane topology and the lower Ni–O–Ni angle values than 100°.
Data availability
The data presented in this study are available on request from the corresponding author.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors acknowledge the financial support from the Alexander von Humboldt Foundation (AvH), Bonn, Germany under the Research Group Linkage Program. We thank Professor Mominul Islam, Department of Chemistry, University of Dhaka, Bangladesh for CV measurements. Our sincere thanks to Mr Khairul Islam, SSO, WMSRC at Jahangirnagar University, Dhaka, Bangladesh. For computational resource, we acknowledge the computecanada.ca (https://ccdb.computecanada.ca) Ontario, Canada.
References
- W. H. Zhang, N. B. Sulaiman, P. S. Tio and T. A. Hor, CrystEngComm, 2011, 13(8), 2915–2922, 10.1039/C0CE00831A.
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- P. Sobota, Coord. Chem. Rev., 2004, 248, 1047–1060, DOI:10.1016/j.ccr.2004.06.002.
- J. Taquet, O. Siri, P. Braunstein and R. Welter, Inorg. Chem., 2006, 45, 4668–4676, DOI:10.1021/ic060019o.
- L. Zhu, K. L. Yao and Z. L. Liu, Appl. Phys. Lett., 2010, 96, 082115, DOI:10.1063/1.3319506.
- D. Gatteschi, R. Sessoli and A. Cornia, Chem. Commun., 2000, 9, 725–732, 10.1039/A908254I.
- M. Cavallini, J. Gómez-Segura, D. Ruiz-Molina, M. Massi, C. Albonetti, C. Rovira, J. Veciana and F. Biscarini, Angew. Chem., Int. Ed., 2005, 44, 888–892, DOI:10.1002/anie.200461554.
-
(a) D. Gatteschi, A. L. Barra, A. Caneschi, A. Cornia, R. Sessoli and L. Sorace, Coord. Chem. Rev., 2006, 250, 1514–1529, DOI:10.1016/j.ccr.2006.02.006;
(b) F. Dimakopoulou, C. G. Efthymiou, C. O'Malley, A. Kourtellaris, E. Moushi, A. Tasiopooulos, S. P. Perlepes, P. McArdle, E. Costa-Villén, J. Mayans and C. Papatriantafyllopoulou, Molecules, 2022, 27(15), 4701, DOI:10.3390/molecules27154701.
- N. E. Chakov, S. Lee, A. G. Harter, P. L. Kuhns, A. P. Reyes, S. O. Hill, N. S. Dalal, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2006, 128, 6975–6989, DOI:10.1021/ja060796n.
- J. Zhang, P. Teo, R. Pattacini, A. Kermagoret, R. Welter, G. Rogez, T. S. A. Hor and P. Braunstein, Angew. Chem., Int. Ed., 2010, 122, 4545–4548, DOI:10.1002/ange.201001412.
- L. Kayser, R. Pattacini, G. Rogez and P. Braunstein, Chem. Commun., 2010, 46, 6461–6463, 10.1039/C0CC01425G.
- S. Hameury, L. Kayser, R. Pattacini, G. Rogez, W. Wernsdorfer and P. Braunstein, Dalton Trans., 2013, 42, 5013–5024, 10.1039/C3DT32869D.
- D. Venegas-Yazigi, J. Cano, E. Ruiz and S. Alvarez, Phys. B, 2006, 384, 123–125, DOI:10.1016/j.physb.2006.05.169.
- A. Das, F. J. Klinke, S. Demeshko, S. Meyer, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 8141–8149, DOI:10.1021/ic300535d.
- S. Demeshko, G. Leibeling, S. Dechert and F. Meyer, Dalton Trans., 2006, 3458–3465, 10.1039/B517254C.
- M. Dey and M. Gogoi, Angew. Chem., Int. Ed., 2013, 52, 12780–12782, DOI:10.1002/anie.201304982.
- K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396–4415, 10.1039/C4CC00339J.
- W. F. Ruettinger, D. M. Ho and G. C. Dismukes, Inorg. Chem., 1999, 8, 1036–1037, DOI:10.1021/ic981145y.
- M. A. Halcrow, J. S. Sun, J. C. Huffman and G. Christou, Inorg. Chem., 1995, 34, 4167–4177, DOI:10.1021/ic00120a022.
- A. P. Ginsberg, J. A. Bertrand, R. I. Kaplan, C. E. Kirkwood, R. L. Martin and R. C. Sherwood, Inorg. Chem., 1971, 10, 240–246, DOI:10.1021/ic50096a006.
- W. L. Gladfelter, M. W. Lynch, W. P. Schaefer, D. N. Hendrickson and H. B. Gray, Inorg. Chem., 1981, 20, 2390–2397, DOI:10.1021/ic50222a007.
- C. C. Beedle, J. J. Henderson, P. C. Ho, T. Sayles, M. Nakano, J. R. O'Brie, K. J. Heroux, E. del Barco, M. B. Maple and D. N. Hendrickson, Inorg. Chem., 2010, 49, 5780–5782, DOI:10.1021/ic1003947.
- E. C. Yang, W. Wernsdorfer, L. N. Zakharov, Y. Karaki, A. Yamaguchi, R. M. Isidro, G. D. Lu, S. A. Wilson, A. L. Rheingold, H. Ishimoto and D. N. Hendrickson, Inorg. Chem., 2006, 45, 529–546, DOI:10.1021/ic050093r.
- M. Moragues-Canovas, M. Helliwell, L. Ricard, E. Riviere, W. Wernsdorfer, E. K. Brechin and T. Mallah, Eur. J. Inorg. Chem., 2004, 2004, 2219–2222, DOI:10.1002/ejic.200400129.
- M. SalahElFallah, E. Rentschler, A. Caneschi and D. Gatteschi, Inorg. Chim. Acta, 1996, 247, 231–235, DOI:10.1016/0020-1693(96)88184-9.
- G. S. Papaefstathiou, A. Escuer, F. A. Mautner, C. Raptopoulou, A. Terzis, S. P. Perlepes and R. Vicente, Eur. J. Inorg. Chem., 2005, 2005, 879–893, DOI:10.1002/ejic.200400670.
- R. Sen, K. Mondal, A. M. dos Santos, L. B. Escobar, P. Brandão, M. S. Reis and Z. Lin, J. Mol. Struct., 2023, 1274, 134412, DOI:10.1016/j.molstruc.2022.134412.
- R. M. Buchanan, M. S. Mashuta, K. J. Oberhausen, J. F. Richardson, Q. Li and D. N. Hendrickson, J. Am. Chem. Soc., 1989, 111, 4497–4498, DOI:10.1021/ja00194a054.
- M. A. Halcrow and G. Christou, Chem. Rev., 1994, 94, 2421–2481, DOI:10.1021/cr00032a008.
- J. Zhao, J. Ji, S. Wang, Y. Luo and Z. You, J. Coord. Chem., 2021, 74(21–24), 3127–3139, DOI:10.1080/00958972.2021.2024522.
- A. F. Kolodziej, Prog. Inorg. Chem., 1994, 41, 493, DOI:10.1002/9780470166420.ch7.
- P. A. Karplus, M. A. Pearson and R. P. Hausinger, Acc. Chem. Res., 1997, 30, 330–337, DOI:10.1021/ar960022j.
- S. H. Zhang, Y. D. Zhang, H. H. Zou, J. J. Guo, H. P. Li, Y. Song and H. Liang, Inorg. Chim. Acta, 2013, 396, 119–125, DOI:10.1016/j.ica.2012.10.032.
- M. Lalia-Kantouri, M. Gdaniec, T. Choli-Papadopoulou, A. Badounas, C. D. Papadopoulos, A. Czapik, G. D. Geromichalos, D. Sahpazidou and F. F. Tsitouroudi, J. Inorg. Biochem., 2012, 117, 25–34, DOI:10.1016/j.jinorgbio.2012.08.022.
-
(a) H. A. Rudbari, F. Lloret, M. Khorshidifard, G. Bruno and M. Julve, RSC Adv., 2016, 6, 7189–7194, 10.1039/C5RA25969J;
(b) M. Aryaeifar, H. A. Rudbari, E. Moreno-Pineda, J. V. Cuevas-Vicario, S. Paul, M. Schulze, W. Wernsdorfer, F. Lloret, N. Moini and O. Blacque, New J. Chem., 2024, 48(8), 3603–3613, 10.1039/D3NJ05585J.
-
(a) J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98, DOI:10.1016/0022-3697(59)90061-7;
(b) J. B. Goodenough, Magnetism and the Chemical Bond, Interscience, New York, 1963 Search PubMed;
(c) Z. You, Y. Luo, S. Herringer, Y. Li, S. Decurtins, K. W. Krämer and S. X. Liu, Crystals, 2020, 10(7), 592, DOI:10.3390/cryst10070592.
- J. E. Andrew and A. B. Blake, J. Chem. Soc. A, 1969, 1456–1461, 10.1039/J19690001456.
- J. E. Andrew and A. B. Blake, Chem. Commun., 1967, 1174–1176, 10.1039/C19670001174.
- L. Ballester, E. Coronado, A. Gutierrez, A. Monge, M. F. Perpinan, E. Pinilla and T. Rico, Inorg. Chem., 1992, 32, 2053–2056, DOI:10.1021/ic00037a014.
-
(a) B. Aurivillius, Acta Chem. Scand., 1977, 31a, 501–508, DOI:10.3891/acta.chem.scand.31a-0501;
(b) P. D. W. Boyd, R. L. Martin and G. Schwarzenbach, Aust. J. Chem., 1988, 41, 1449–1456, DOI:10.1071/CH9881449.
-
(a) A. G. Kruger and G. Winter, Aust. J. Chem., 1970, 23, 1–14, DOI:10.1071/CH9700001;
(b) J. A. Barnes and W. E. Hatfield, Inorg. Chem., 1971, 10, 2355–2357, DOI:10.1021/ic50104a063;
(c) A. J. Blake, E. K. Brechin, A. Codron, R. O. Gould, C. M. Grant, S. Parsons, J. M. Rawson and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1995, 1983–1985, 10.1039/C39950001983.
-
(a) J. M. Clemente-Juan, B. Chansou, B. Onnadieu and J. P. Tuchagues, Inorg. Chem., 2000, 39(24), 5515–5519, DOI:10.1021/ic0005442;
(b) A. Zianna, M. Šumar Ristović, A. Hatzidimitriou, C. D. Papadopoulos and M. Lalia-Kantouri, J. Therm. Anal. Calorim., 2014, 118, 1431–1440, DOI:10.1007/s10973-014-4035-5;
(c) J. C. Belmont-Sánchez, D. Choquesillo-Lazarte, R. Navarrete-Casas, A. Frontera, A. Castiñeiras, J. Niclós-Gutiérrez and A. Matilla-Hernández, Crystals, 2022, 13(1), 7, DOI:10.3390/cryst13010007.
- M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. J. Reiß and C. Janiak, Eur. J. Inorg. Chem., 2015, 2015(16), 2758–2768, DOI:10.1002/ejic.201500128.
- M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva and C. Janiak, Dalton Trans., 2016, 45, 667–680, 10.1039/C5DT03940A.
- O. Rotthaus, F. Thomas, O. Jarjayes, C. Philouze, E. Saint-Aman and J. L. Pierre, Chem.–Eur. J., 2006, 12(26), 6953–6962, DOI:10.1002/chem.200600258.
- A. Saadati, H. A. Rudbari, M. Aryaeifar, O. Blacque, I. Correia, M. K. Islam, D. Woschko, T. H. H. Sohi, C. Janiak and M. Enamullah, CrystEngComm, 2023, 25, 365–377, 10.1039/D2CE01311H.
- A. Mim, M. Enamullah, I. Haque, A. Mohabbat and C. Janiak, J. Mol. Struct., 2023, 1291, 135669, DOI:10.1016/j.molstruc.2023.135669.
- Y. Q. Yuan, F. L. Yuan, F. L. Li, Z. M. Hao, J. Guo, D. J. Young, W. H. Zhang and J. P. Lang, Dalton Trans., 2017, 46(22), 7154–7158, 10.1039/C7DT01579H.
-
(a) S. Petit, P. Neugebauer, G. Pilet, G. Chastanet, A.-L. Barra, A. B. Antunes, W. Wernsdorfer and D. Luneau, Inorg. Chem., 2012, 51(12), 6645–6654, DOI:10.1021/ic3001637;
(b) A. Sarkar, D. Basu, C. J. Gómez-García and H. P. Nayek, Eur. J. Inorg. Chem., 2023, 26(2), e202200565, DOI:10.1002/ejic.202200565.
- M. A. Nadeem, M. C. C. Ng, J. van Leusen, P. Kögerler and J. A. Stride, Chem.–Eur. J., 2020, 26(34), 7589–7594, DOI:10.1002/chem.202000867.
-
(a) Q. L. Zhang, Z. L. Wu, H. Xu, B. Zhai, Y. F. Wang, G. W. Feng and Y. L. Huang, Z. Anorg. Allg. Chem., 2016, 642, 414–418, DOI:10.1002/zaac.201500772;
(b) R. N. Patel, S. K. Patel, A. K. Patel, N. Patel and R. J. Butcher, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2022, 78(2), 98–102, DOI:10.1107/s2056989021012408.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, https://crystalexplorer.net Search PubMed.
-
(a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668, DOI:10.1107/S0108768104020300;
(b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392, 10.1039/B203191B;
(c) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816, 10.1039/B704980C.
- L. Wang, J. Wu, X. Su, J. Huang, P. Zhang, S. Zhao, B. Su and B. Xu, Transition Met. Chem., 2022, 47(7), 275–281, DOI:10.1007/s11243-022-00501-y.
- S. Shit, M. Nandy, G. Rosair, C. J. Gómez-García, J. J. B. Almenar and S. Mitra, Polyhedron, 2013, 61, 73–79, DOI:10.1016/j.poly.2013.05.029.
- M. Enamullah, T. Aziz, I. Haque, A. Mohabbat, A. Kacperkiewicz, D. E. Herbert and C. Janiak, J. Mol. Struct., 2024, 1312, 138509, DOI:10.1016/j.molstruc.2024.138509.
- M. Enamullah, A. Mim, I. Haque, B. K. Sidhu, A. Kacperkiewicz and D. E. Herbert, New J. Chem., 2023, 47, 21804–21814, 10.1039/D3NJ04915A.
- I. Haque, M. Enamullah, A. K. Resma, N. T. Jhumur, D. Woschko, A. Mohabbat, J. van Leusen, P. Kögerler and C. Janiak, Chem.–Asian J., 2024, 19(24), e202400915, DOI:10.1002/asia.202400915.
- I. Haque, M. S. Abdullah, M. K. Islam and M. Enamullah, Inorg. Chim. Acta, 2023, 550, 121430, DOI:10.1016/j.ica.2023.121430.
- J. Wang, X. Meng, W. Xie, X. Zhang, Y. Fan and M. Wang, J. Biol. Inorg. Chem., 2021, 26, 205–216, DOI:10.1007/s00775-020-01846-4.
- P. Bhunia, S. Maity, J. Mayans and A. Ghosh, New J. Chem., 2022, 46, 4363–4372, 10.1039/d1nj05638g.
- T. K. Ghosh, P. Mahapatra, S. Jana and A. Ghosh, CrystEngComm, 2019, 21, 4620–4631, 10.1039/c9ce00574a.
- R. Herchel, I. Nemec, M. Machata and Z. Trávníček, Dalton Trans., 2016, 45, 18622–18634, 10.1039/C6DT03520E.
- E.-C. Yang, W. Wernsdorfer, S. Hill, R. S. Edwards, M. Nakano, S. Maccagnano, L. N. Zakharov, A. L. Rheingold, G. Christou and D. N. Hendrickson, Polyhedron, 2003, 22, 1727–1733, DOI:10.1016/S0277-5387(03)00149-9.
- R. Herchel, I. Nemec, M. Machata and Z. Trávníček, New J. Chem., 2017, 41, 11258–11267, 10.1039/C7NJ02281F.
- A. N. Gusev, I. Nemec, R. Herchel, Y. I. Baluda, M. A. Kryukova, N. N. Efimov, M. A. Kiskin and W. Linert, Polyhedron, 2021, 196, 115017, DOI:10.1016/j.poly.2020.115017.
-
(a) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391–1400, DOI:10.1002/(sici)1096-987x(199910)20:13<1391::aid-jcc6>3.0.co;2-j;
(b) E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982–989, DOI:10.1002/jcc.10257.
-
(a) S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178(1), 42–55, DOI:10.1016/j.jmr.2005.08.013;
(b) O. Kahn, J. Chem. Educ., 1995, 72, A19, DOI:10.1021/ed072pA19.2.
- Bruker-AXS, APEX4 V2021.10-0, Madison, WI, USA, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- A. L. Spek, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2009, 65, 148–155, DOI:10.1107/S090744490804362X.
- K. Brandenburg, Diamond (Version 4.5), Crystal and Molecular Structure Visualization, Crystal Impact – K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2009–2022 Search PubMed.
- A. L. Spek, PLATON – A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2008 Search PubMed.
- L. J. Farrugia, Windows Implementation, Version 270519, University of Glasgow, Scotland, 2019 Search PubMed.
Footnotes |
| † Dedicated in memory of Professor ABP Lever, Department of Chemistry, York University, Toronto, Canada. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2367079 and 2367080. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra08700c |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mohammed Enamullah
a,
Mohammed Enamullah