
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of potential small molecule inhibitors against β-hCG: an in silico study with in vitro validation†
Shreya Sara Ittycheriaab,
Krishnankutty Chandrika Sivakumara,
Dipyaman Patra‡
a,
Bhavana Ramachandrana,
R. L. Neethaac,
Arathy V. Warrierac,
M. A. Aiswariyaad,
S. Kaviyaad,
Pankaj Sumane,
Nagarjun Narayanaswamya and
Priya Srinivas *ad
*ad
aCancer Research Program, BRIC-Rajiv Gandhi Centre for Biotechnology, Thycaud, Thiruvananthapuram 695014, Kerala, India. E-mail: priyasrinivas@rgcb.res.in; Tel: +91-471-2529495
bManipal Academy of Higher Education (MAHE), Manipal 576104, India
cResearch Centre, University of Kerala, Thiruvananthapuram, Kerala, India
dDBT-Regional Centre for Biotechnology (RCB), Faridabad 121001, Haryana, India
eNational Institute of Animal Biotechnology, Gachibowli, Hyderabad, Telangana 500032, India
First published on 10th June 2025
Abstract
Previous in vitro and in vivo studies have shown an association between BRCA1-defective cancers and the expression of β-hCG, where accelerated tumor progression has been observed in the presence of β-hCG due to its binding to and phosphorylation of TGFβR-II. Given the absence of reported small molecule inhibitors against β-hCG, the present study endeavours to identify the interacting residues of β-hCG with TGFβR-II. Through virtual screening, molecular docking and dynamic simulation studies, the investigation identified six potential small molecule inhibitors, namely, cefotetan, 2′,7′-dichlorofluorescein (DCF), 2′,7′-difluorofluorescein (DFF), F6658-4634, F0922-0590 and F0385-0029. Notably, all inhibitors exhibited interacting residues on β-hCG that coincided with the site to which it binds to TGFβR-II. Further in vitro MTT assays showed a reduction in proliferation in various cell lines, including the cell line developed in our laboratory, which was induced to have BRCA1 promoter hypermethylation using modified CRISPR technology. Binding studies such as Microscale Thermophoresis (MST) and Isothermal Titration Calorimetry (ITC) exhibited the binding of cefotetan to hCG, which could be indicative of the selective cytotoxic effect of cefotetan in β-hCG secreting breast cancer cell lines. The results indicate selective inhibition by these potential inhibitors in BRCA1-defective breast cancer cell lines, with cefotetan being the best among all with an IC50 of 32 μM. This is the first study to report the anticancer activity of cefotetan, an FDA-approved drug used to treat bacterial infections, and this drug could be repurposed as a targeted therapeutic against β-hCG expressing cancers. Findings of this study hold significant implications for developing potential therapeutic interventions, which target β-hCG in the context of BRCA1-defective breast cancers, aligning with the broader goal of advancing precision medicine in oncology.
1 Introduction
Breast cancer (BC) represents a significant global health concern, accounting for 23.8% of the global cancer burden and 15.4% of cancer-related deaths among women worldwide.1,2 Of note, the majority of BC cases occur sporadically, often associated with hypermethylation in the BRCA1 promoter and culminating in triple-negative breast cancers (TNBCs) in approximately 40% of cases.3 The presence of the beta subunit of human chorionic gonadotropin (hCG), a glycoprotein secreted by syncytiotrophoblasts within the placental endometrium, has been detected in some cancers.4 Extensive in vitro and in vivo investigations conducted in our laboratory have revealed the expression of β-hCG in BRCA1-defective cancers. The role of β-hCG in accelerating tumor progression and enhancing aggressiveness in BRCA1-defective cancer cells through the Smad signalling pathway indicates a direct relation between serum β-hCG levels and the disease progression in women with breast malignancies.10 β-hCG as a monomer (and not α-hCG) is reported to be expressed by various cancer cells and is considered stable; even free β-hCG is secreted through urine in pregnant women.Human chorionic gonadotropin (hCG) is a heterodimeric molecule consisting of α and β subunits, with the β-subunit spanning 145 amino acids, thus setting hCG apart from other glycoproteins. The α subunit of hCG is common to FSH, LH and TSH, but the β subunit is unique to hCG, which is secreted by certain cancers, thus setting hCG apart from other glycoproteins.11 In the realm of cancer therapeutics, the prospect of targeting the β-subunit of hCG holds potential by impeding its interaction with TGFβR-II, consequently mitigating tumor progression through the Smad signalling pathway in cancers expressing β-hCG. While the conventional approach of targeting the receptor, specifically TGFβR-II in BRCA1-defective cancers, may appear more intuitive, it is imperative to recognize the multifaceted role of TGFβ signalling. This encompasses pivotal biological pathways in both adult organisms and developing embryos, regulating cell growth, differentiation, migration, apoptosis, cellular homeostasis and other cellular functions.12 Disruption of the TGFβ signalling pathway, including inhibition of TGFβR-II, has the potential to perturb these essential cellular processes.
The atypical expression of β-hCG offers a viable therapeutic strategy in BRCA1-defective breast cancers. This approach not only streamlines the drug discovery process but also circumvents the long timelines and exorbitant costs associated with traditional drug development endeavours.
Drawing parallels with a study on the design of inhibitors against a secretory molecule akin to β-hCG, such as the design of IL-6 inhibitors utilizing in silico methodologies,13 the strategic importance of narrowing down potential drug candidates for subsequent in vitro validation cannot be ignored.
In light of the absence of identified chemical inhibitors against β-hCG to date, the present study leveraged in silico tools including ZDOCK,14 CastP,15 PrankWeb16 and the Schrödinger suite to facilitate the identification of prospective small molecules capable of inhibiting β-hCG. Validation of these findings was conducted in vitro utilizing the MTT assay, offering a novel avenue for the development of targeted therapies against BRCA1-defective cancers.
2 Results and discussion
2.1 Binding site residues of TGFβR-II on β-hCG
β-hCG interaction with TGFβR-II in promoting tumor progression via the Smad signalling pathway within BRCA1-defective cancers have been shown. The ZDOCK server was leveraged to discern the specific binding sites of TGFβR-II on β-hCG.Fig. 1a illustrates the selection process that was used to scrutinize the top 10 models generated by the ZDOCK server. To note are the residues highlighted in red, symbolizing the exact binding sites of TGFβR-II on β-hCG, illuminating the critical molecular interfaces essential for their interaction. The boxes within Fig. 1a serve as indicators of the prevalence of these residues across the top 10 models, offering insights into the consistency and significance of these binding sites. The numerical annotations at the bottom of Fig. 1a indicate the specific residue numbers along the β-hCG chain to help map the interacting residues. This analysis not only enriches our understanding of the molecular underpinnings of tumor progression in BRCA1-defective cancers but also lays a robust groundwork to explore therapeutic strategies that target the interaction between β-hCG and TGFβR-II.
 | ||
| Fig. 1 (a) Top 10 models of the β-hCG sequence, depicting the binding site residues of TGFβR-II (highlighted in red) as predicted by the ZDOCK server. The numerical annotations at the bottom of the figure corresponds to the residue numbers on the β-hCG chain. Rectangular boxes within the figure indicate the prevalence of these residues across the top 10 models. (b) Displays the frequency of occurrence of the binding site residues in the top 10 models of β-hCG with TGFβR-II, as anticipated by the ZDOCK server. (c) A comparative analysis of the predicted active site residues of β-hCG generated by different software tools, namely, PrankWeb, SiteMap (Schrödinger), and CastP. | ||
From Fig. 1b, it is evident that residues P-3, R-5, R-7, P-39, T-40, M-41, T-42, R-43, V-44, L-45, Q-46, L-52, P-53, V-55, R-60, Q-89 and C-90 exhibit a frequency of occurrence ≥50% in the binding site residues of TGFβR-II on β-hCG within the top 10 complexes. Notably, residues P-39, T-40, M-41 and R-43 demonstrate the highest frequency of occurrence at 90%. These identified residues are anticipated to play a direct role in the binding interaction between β-hCG and TGFβR-II. Given the significance of these residues in the β-hCG–TGFβR-II interaction, thereby promoting tumor progression, exploring small molecules that target these specific residues could offer a promising therapeutic approach.
2.2 Binding site prediction using PrankWeb, CastP and SiteMap tool
The software tools PrankWeb, CastP and SiteMap in Schrödinger Maestro version 13.5 were used to pinpoint potential active sites, as shown in Fig. 1c. Notably, certain residues such as L-16 (predicted by PrankWeb and CastP), V-56 (predicted by SiteMap and PrankWeb), Y-59 (predicted by PrankWeb and CastP), A-83 (predicted by PrankWeb and CastP), T-98 (predicted by PrankWeb and SiteMap) and P-107, L-108 (predicted by CastP and SiteMap) exhibit overlapping predictions. Conversely, while some residues do not coincide, they are positioned adjacent to the overlapping residues, such as A-14 and T-15 (predicted by Schrödinger's SiteMap) and L-16 (predicted by PrankWeb). From these results, it can be concluded that the prediction of active site residues by different software shows similar results. The results from the SiteMap software were used for further analysis.2.3 Virtual screening, molecular docking and MM-GBSA calculation
The suite of virtual screening and molecular docking tools within the Schrödinger software package, encompassing (HTVS) high throughput virtual screening, (SP) standard precision and (XP) extra precision, in conjunction with Desmond Prime for free energy calculations, facilitated the identification of the top five candidate molecules capable of potentially inhibiting β-hCG. Table 1 presents the top 5 molecules along with their docking scores and free energies (kcal mol−1). Additionally, Table 2 showcases the molecular formula, molecular weight (M.W.) and structures of these small molecules, while ESI Table 2† delineates the SMILES format of the small molecules. These molecules are expected to bind to β-hCG, impeding its interaction with TGFβRII. Several virtual libraries were used as mentioned in the Experimental section.| Sl no. | Small molecule | Docking score | MMGBSA (kcal mol−1) |
|---|---|---|---|
| 1 | Cefotetan | −5.17 | −45.44 |
| 2 | F0922-590 | −6.04 | −60.77 |
| 3 | F0385-0029 | −5.61 | −65.22 |
| 4 | F6658-4634 | −7.94 | −42.34 |
| 5 | 2,6,7-Trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one | −6.29 | −47.01 |
| Sl. no. | Name (molecular formula) and or (IUPAC name) | M.W. (a.u.) | Structure |
|---|---|---|---|
| 1 | Cefotetan (C17H15N7O8S4) (6R,7S)-7-[[4-(2-amino-1-carboxy-2-oxoethylidene)-1,3-dithietane-2-carbonyl]amino]-7-methoxy-3-[(1-methyltetrazol-5-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid | 573.607 | 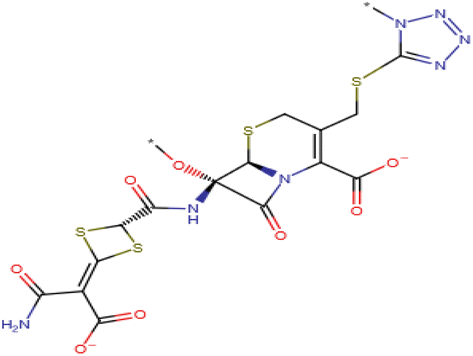 |
| 2 | F0922-0590 (C35H35N3O5) N-[2-(3,4-dimethoxyphenyl)ethyl]-4-({1-[(2,5-dimethylphenyl)methyl]-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl}methyl)benzamide | 577.686 |  |
| 3 | F0385-0029 (C28H22N4O8S4) 3-(4-{5-[(5E)-3-{3-[(3-carboxyphenyl)carbamoyl]propyl}-4-oxo-2-sulfanylidene-1,3-thiazolidin-5-ylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl}butanamido)benzoic acid | 670.766 |  |
| 4 | F6658-4634 (C16H20N8O2) 6-(4-{5,6-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl}piperazin-1-yl)-3-methyl-1,2,3,4-tetrahydropyrimidine-2,4-dione | 356.390 |  |
| 5 | 2,6,7-Trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one (C19H12O6) | 336.304 |  |
2.4 Bonding interactions of the identified inhibitors with β-hCG using molecular dynamics (MD) simulations
MD simulations were conducted over a duration of 100 ns, focusing on the interaction of five small molecules with β-hCG, including derivatives of compound 2,6,7-trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one, specifically 2′,7′-dichlorofluorescin and 2′,7′-difluorofluorescein, all of which exhibited stable binding with β-hCG.Among the small molecules identified, cefotetan demonstrated the most stable interaction with β-hCG. This compound established contacts with multiple residues of β-hCG, including CYS-93, GLN-54, ARG-95, LEU-52, GLN-46, CYS-100, GLY-36, CYS-57 and CYS-38 (Fig. 2a). From the top ZDOCK 10 models in Fig. 1a, even if one of the models contained a binding site residue of TGFβR-II on β-hCG, it would be considered, especially if it coincided with a residue maintaining hydrogen bonds (which are very important for drug design) with cefotetan. Therefore, residues GLN-54 (hydrogen bonding), LEU-52 (hydrogen bonding), GLN-46 (hydrogen bonding), CYS-100 (hydrogen bonding), GLY-36 (water bridges), CYS-57 (hydrogen bonding) and CYS-38 (water bridges) are mentioned (Fig. 1a and 2e). GLY-36 shows almost continuous contact throughout the set simulation with multiple points of contact in the contact timeline diagram of cefotetan (Fig. 2c), hence this residue is mentioned despite it forming only water bridges with cefotetan (Fig. 2e). At CYS-38, the RMSF shows a very stable value (within 1.6 Å) (Fig. 2d) and forms a point of contact indicated by a green vertical bar; therefore, this residue is being mentioned despite it forming only water bridges (Fig. 2e). ARG-95 was mentioned despite it not being a coinciding residue, as it contributes most significantly to the hydrogen bond fraction (Fig. 2e), and CYS-93 contributes to a significant fraction of contact in the interactions diagram (Fig. 2a). The formation of a stable complex between cefotetan and β-hCG was evidenced by the RMSD plot (Fig. 2b), where the difference between the protein (shown in blue colour) and ligand (shown in red colour) is shown; RMSD remained consistently within 3 Å, indicating a stable interaction. The contacts timeline (Fig. 2c) visually represents the duration and intensity of specific residue-ligand contacts, with a darker shade of orange in Fig. 2c, denoting multiple contacts with the ligand. Throughout the 100 ns simulation period, cefotetan maintained continuous contacts with residues GLY-36, GLN-54, CYS-57, CYS-93 and ARG-95, with intermittent breaks. The RMSF plot (Fig. 2d) highlights the protein residues interacting with the ligand, depicted by green vertical bars. Lastly, the types of bonds formed between the residues and the ligand are illustrated in Fig. 2e, emphasizing the prevalence of hydrogen bonds. Hydrogen bonds play a crucial role in drug design due to their strength and impact on drug specificity, metabolism and absorption. Notably, significant hydrogen bond interactions were observed at residues GLN-46, LEU-52, GLN-54, CYS-57, ARG-95 and CYS-100.
 | ||
| Fig. 2 Robust binding of cefotetan with β-hCG. (a) Structural configuration with the interacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds established with the interacting residues of β-hCG. | ||
Compound F0922-0590, exhibited notable interactions at residues ASN-13, THR-32, CYS-34 and ALA-83 (Fig. 3a). The binding of this compound to β-hCG displayed remarkable stability, as evidenced by the RMSD plot (Fig. 3b). Throughout the simulation period, consistent contacts were observed, particularly at residue ASN-13 and ALA-83, with intermittent breaks in between (Fig. 3c). The RMSF analysis (Fig. 3d) highlighted multiple points of contact, with a significant presence of hydrogen bonds at residues ASN-13, THR-32, CYS-34 and ALA-83 (Fig. 3e).
 | ||
| Fig. 3 Robust binding of the compound F0922-0590 with β-hCG. (a) Structural configuration with the interacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds established with the contacting residues of β-hCG. | ||
The interactions of compound F6658-4634, as illustrated in Fig. 4, demonstrate binding with the residues CYS-34, TYR-59, ALA-83 and ASP-99 of β-hCG (Fig. 4a). The binding between this compound and β-hCG exhibited stability, as indicated by the RMSD plot (Fig. 4b). Notably, a consistently stable contact was maintained throughout the simulation period at residue CYS-34 of β-hCG, while ALA-83 and ASN-99 residues displayed intermittent breaks but overall continuity (Fig. 4c). Analysis of the RMSF plot (Fig. 4d) revealed the establishment of several points of contact, with residue CYS-34 (Fig. 4e) exhibiting the most significant proportion of hydrogen bonding with the ligand.
 | ||
| Fig. 4 Robust binding of the compound F6658-4634 with β-hCG. (a) Structural configuration with the contacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds formed with the contacting residues of β-hCG. | ||
Compound F0385-0029 exhibited multiple points of contact on β-hCG at residues TYR-59, THR-98, ARG-94, ASN-58, GLY-101 and CYS-57, as illustrated in Fig. 5a. The RMSD plot in Fig. 5b indicated a stable interaction, albeit showing a slight deviation around 80 ns, potentially attributed to the bulky side chains of the ligand. The contacts timeline in Fig. 5c revealed continuous interactions at residues ASN-58 and TYR-59, with ARG-94 displaying recurrent contacts denoted by a darker shade of orange. The RMSF analysis (Fig. 5d) highlighted multiple points of contact. Notably, a significant proportion of hydrogen bonds were observed at residues CYS-57, ASN-58, ARG-94 and GLY-101, as depicted in Fig. 5e.
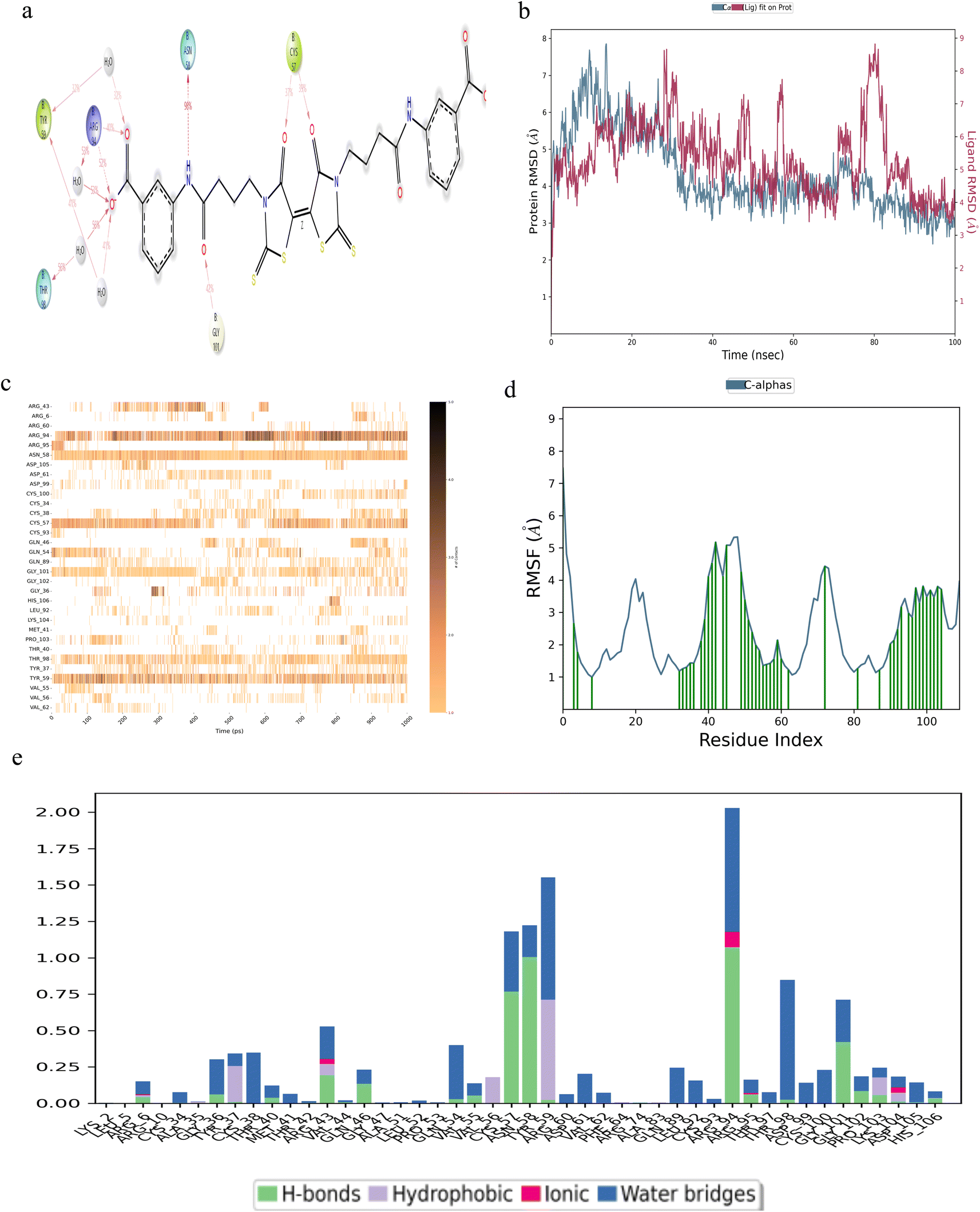 | ||
| Fig. 5 Robust binding of the compound F0385-0029 with β-hCG. (a) Structural configuration with the contacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds formed with the contacting residues of β-hCG. | ||
2,6,7-Trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one exhibited multiple points of contact with β-hCG at residues TYR-59, ASN-58, CYS-34 and ILE-33 (Fig. 6a). The RMSD plot illustrated a stable interaction of the compound with β-hCG, as shown in Fig. 6b. The contacts timeline in Fig. 6c demonstrated continuous contacts maintained at CYS-34. The RMSF analysis revealed several points of contact established, particularly around residue 100 of β-hCG (Fig. 6d). Notably, a significant proportion of hydrogen bonds are observed at residues CYS-34 and TYR-59, as depicted in Fig. 6e.
 | ||
| Fig. 6 Robust binding of the compound 2,6,7-trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one with β-hCG. (a) Structural configuration with the contacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds formed with the contacting residues of β-hCG. | ||
2′,7′-Dichlorofluorescin, a derivative of compound 2,6,7-trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one, exhibited contact at residue CYS-34 (Fig. 7a), a stable RMSD (Fig. 7b), continuous contacts maintained at CYS- 34 (Fig. 7c), several points of contact indicated by the RMSF plot (Fig. 7d) and a significant proportion of hydrogen bonds at CYS-34 (Fig. 7e).
 | ||
| Fig. 7 Stable binding of 2′,7′-dichlorofluorescin with β-hCG. (a) Structural configuration with the contacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds formed with the contacting residues of β-hCG. | ||
2′,7′-Difluorofluorescein, an additional derivative of compound 2,6,7-trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one, demonstrated points of contact at CYS-34 (Fig. 8a), a consistent RMSD profile (Fig. 8b), nearly uninterrupted contacts at CYS-34 (Fig. 8c), multiple points of contact highlighted by the RMSF plot (Fig. 8d), and a notable presence (>50%) of hydrogen bonds at residue CYS-34 (Fig. 8e).
 | ||
| Fig. 8 Stable binding of 2′,7′-difluorofluorescein with β-hCG. (a) Structural configuration with the contacting residues. (b) Protein–ligand RMSD plot. (c) Contact timeline. (d) RMSF visualization. (e) Delineation of the types of bonds formed with the contacting residues of β-hCG. | ||
From these in silico studies, it can be concluded that all six compounds could potentially bind and inhibit β-hCG, with cefotetan showing the most promise.
2.5 Coinciding residues
This study aims to identify inhibitors capable of binding β-hCG and impeding its interaction with TGFβR-II. It is essential to emphasize that each inhibitor targets specific sites on β-hCG, with at least one residue aligning with the TGFβR-II binding site on β-hCG. This alignment is based solely on hydrogen bonds due to their strength, with the exception of the small molecule F6658-4634, as detailed in Table 3. Notably, the small molecule referred to as F0385-0029, and subsequently cefotetan, demonstrated the highest number of overlapping residues during in silico simulations.| Sl no. | Inhibitor | Coinciding residues of TGFβR-II and inhibitor binding site on βhCG (only hydrogen bonds considered except for F6658-463) |
|---|---|---|
| 1 | Cefotetan | R43, L45, Q46, L52, Q54, C57, R94, C100, G101 |
| 2 | F0922-0590 | A83, K104, H106 |
| 3 | F6658-4634 | Y59 (hydrophobic), A83 (hydrophobic and water bridges), D99 (water bridges), D105 (water bridges) |
| 4 | F0385-0029 | R6, G36, T40, R43, Q46, Q54, V55, C57, R94, T98, G101, G102, P103, H106 |
| 5 | 2,6,7-Trihydroxy-9-(2-hydroxyphenyl)-3H-xanthen-3-one | Y59 |
| 6 | 2,7-Dichlorofluorescein | LEU-45, GLN-46, CYS-57, TYR-59, GLY-101 |
| 7 | 2,7-Difluorofluorescein | CYS-57, TYR-59, THR-98, ASN-99, CYS-100 |
2.6 β-hCG expression in cells
The media of various cell lines, such as MDA-MB-231, MDA-MB-231shBRCA1, MDA-MB-231 scrambled control, MDA-MB-231 BRCA1 sgRNA(1 + 2), MDA-MB-231 PCL-CTIG control and MDA-MB-468, were subjected to β-hCG ELISA (Abcam, ab178633), according to the manufacturer's instructions, alongside β-hCG standards. β-hCG was found to be secreted in the highest amount of 4.44 ng ml−1 of media in MDA-MB-231 BRCA1 sgRNA(1 + 2) cell line, followed closely by MDA-MB-468, which showed 4 ng ml−1 of β-hCG secretion in media and MDA-MB-231 shBRCA1 at a concentration of 1.88 ng ml−1. In the wild-type BRCA1 cell line, MDA-MB-231 and control for the knocked-down BRCA1 cell line MDA-MB-231 scrambled control, β-hCG was secreted at a concentration of 0.2 ng ml−1. The control for CRISPR modified cell line, MDA-MB-231 PCL-CTIG exhibited 0.4 ng ml−1 secretion of β-hCG in media (Fig. 9a). These results indicate that β-hCG expression is associated with the BRCA1 status in various breast cancer cell lines i.e., where the BRCA1 gene is defective, β-hCG expression was found to be present. | ||
| Fig. 9 (a) Secretion of β-hCG in the supernatant of various cell lines. (b) Cytotoxic impact of cefotetan on diverse cell lines. (c) Cytotoxic impact of 2′,7′-dichlorofluorescein diacetate (DCFDA) on different cell lines. (d) Cytotoxic effect of the β-hCG monoclonal antibody on different cell lines. | ||
2.7 Cell proliferation assay
The cell lines employed in this assay include MDA-MB-231, MDA-MB-shBRCA1, MDA-MB-231 scrambled control, MDA-MB-231 BRCA1 sgRNA(1 + 2), MDA-MB-231 PCL-CTIG control and MDA-MB-468.Cefotetan exhibited a notable decrease in viability in cell lines with defective BRCA1, as depicted in Fig. 9b. Specifically, MDA-MB-231 BRCA1 sgRNA 1 + 2, MDA-MB-231shBRCA1 and MDA-MB-468 displayed reduced viability with escalating concentrations of cefotetan, while MDA-MB-231, MDA-MB-231 PCL-CTIG and MDA-MB-231 scrambled control exhibited no significant viability alteration with increasing cefotetan concentrations.
2′,7′-Dichlorofluorescin diacetate was analyzed in the study, as 2′,7′-dichlorofluorescin alone lacks cell permeability. However, the diacetate salt can penetrate cells and undergo cleavage by intracellular esterases to generate 2′,7′-dichlorofluorescin. To maintain cell viability, the concentrations of DCFDA in the MTT assay were restricted to 10 μM, considering that higher DCFDA concentrations (>10 μM DCFDA) have been reported to compromise cell viability.24 Fig. 9c depicts a reduction in viability noted in BRCA1-defective cell lines, including MDA-MB-231 shBRCA1 and MDA-MB-231 BRCA1 sgRNA 1 + 2, as compared to their corresponding controls, namely MDA-MB-231 scrambled control and MDA-MB-231 PCL-CTIG control, respectively. These results were comparable to the control BRCA1-defective cell line, MDA-MB-468.
The MTT assay was also performed in various cell lines using the β-hCG monoclonal antibody (sc-271062, Santa Cruz). As can be seen in Fig. 9d, increasing concentrations of the antibody exhibited decreased viability in cell lines having the BRCA1-defective gene (namely MDA-MB-231 shBRCA1, MDA-MB-231 BRCA1 sgRNA 1 + 2 and MDA-MB-468), which also express β-hCG. However, their BRCA1 wild-type counterparts, namely the MDA-MB-231, MDA-MB-231 scrambled control and MDA-MB-231 PCL-CTIG control, did not show any alteration in viability.
Compounds F0922-0590, F6658-4634, F0385-0029, along with 2,7-difluoro-fluorescein, underwent cell viability testing exclusively in MDA-MB-231 BRCA1 sgRNA 1 + 2, its control, MDA-MB-231 PCL-CTIG and the parent cell line MDA-MB-231. The results revealed diminished viability for all four compounds, specifically in the MDA-MB-231 BRCA1 sgRNA 1 + 2 cell line, characterized by induced hypermethylation in the BRCA1 promoter leading to reduced BRCA1 expression, as depicted in Fig. 10a–d. Despite employing concentrations as high as 100 μM for each compound, viability reduction below 50% was not achieved.
 | ||
| Fig. 10 (a) Cytotoxic impact of the compound F0922-0590 on diverse cell lines. (b) Cytotoxic influence of the compound F6658-4634 on a range of cell lines. (c) Cytotoxic impact of the compound F0385-0029 on different cell lines. (d) Cytotoxic effect of 2,7-difluorofluorescein on different cell lines. | ||
All experiments were conducted using GraphPad Prism, where ‘ns’ denotes non-significant and **** indicates p < 0.0001. The β-hCG monoclonal antibody showed selective inhibition in BRCA1-defective cell lines.
2.8 Binding interaction
MST revealed that out of the six inhibitors obtained from in silico studies, the hCG molecule binds to cefotetan and F0922-0590 alone. Binding affinity determination revealed that hCG binds cefotetan with a KD = 4.6 ± 3 μM. The affinity of compound F0922-0590 was found to be KD = 0.388 ± 0.009 mM. ITC showed cefotetan to have a KD = 5.43 ± 2.99 μM (very close to that determined by MST). F0922-0590 did not show any binding with ITC (Fig. 11). These results confirm the fact that cefotetan binds β-hCG and causes a selective cytotoxic effect in β-hCG-expressing cells. | ||
| Fig. 11 (a) MST shows binding of six compounds to the hCG molecule. Only cefotetan and the compound F0922-0590 are seen to bind hCG. (b) Binding affinity of cefotetan to hCG with a KD = 4.6 ± 3 μM (c) and F0922-0590 with a KD = 0.388 ± 0.009 mM. (d) ITC shows binding of cefotetan to hCG with KD = 5.43 ± 32.99 μM. (e) No binding between F0922-0590 and hCG. | ||
2.9 Discussion
To identify the chemical inhibitors targeting β-hCG, which interacts with TGFβR-II and initiates tumor progression via the Smad signaling pathway in BRCA1-defective cancers, the interacting residues of β-hCG with TGFβR-II were identified from the top 10 models generated by ZDOCK. Subsequently, the active/druggable sites of β-hCG were delineated and compared utilizing various software tools. A comprehensive virtual screening of chemical molecules from multiple virtual libraries, totalling 127![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 846 molecules, was conducted against the identified active sites. Subsequently, docking simulations of these molecules were executed, followed by free energy calculations. The most promising complexes resulting from these analyses underwent a rigorous 100 ns simulation. This meticulous process culminated in the identification of the top six molecules.
846 molecules, was conducted against the identified active sites. Subsequently, docking simulations of these molecules were executed, followed by free energy calculations. The most promising complexes resulting from these analyses underwent a rigorous 100 ns simulation. This meticulous process culminated in the identification of the top six molecules.
These selected molecules then underwent cell viability assessments, such as MTT assays, across several BRCA1-defective and BRCA1 wild-type cell lines. Notably, among the BRCA1-defective cell lines evaluated, a novel cell line developed in our laboratory was included. This unique cell line was derived from the modification of the wild-type BRCA1 cell line MDA-MB-231, inducing hypermethylation at the BRCA1 promoter using modified CRISPR technology. Additionally, another BRCA1-defective cell line was established through stable knock-down of the BRCA1 gene.
Specifically, cefotetan was found to bind to residues R43, L45, Q46, L52, Q54, C57, R94, C100 and G101, which align with the predicted TGFβR-II binding site on β-hCG, as anticipated by ZDOCK. The outcomes of all MTT assays conducted consistently demonstrated that cefotetan exhibited the most notable reduction in cell viability in BRCA1-defective cell lines at a concentration as low as 30 μM. Table 4 represents the IC50 value of cefotetan and β-hCG monoclonal antibody as predicted by GraphPad Prism v10.2.2 using the Hill slope equation.
| Sl no. | Drug | IC50 |
|---|---|---|
| 1 | Cefotetan | 32.56 μM |
| 2 | β-hCG monoclonal antibody | 3.17 ng |
Although cefotetan showed a KD in the micromolar range, it is still significant, as we have used whole hCG instead of β-hCG. Most of the active site residues used for virtual screening, molecular docking and dynamic simulation fall in the region where β-hCG interlocks with α-hCG. There is a chance that antiproliferative activity of cefotetan may not be by binding to hCG but the fact that the reduction in cell viability was selectively seen particularly in the β-hCG-expressing cells when compared to cells that do not express β-hCG (Fig. 9a and b) and are comparable to the effects of the β-hCG monoclonal antibody (Fig. 9d). Therefore, there is a high chance that the selective cytotoxic effects of cefotetan could be due to its interaction with β-hCG. The MST/ITC techniques revealed that cefotetan would bind to hCG; however, to identify which specific residues on β-hCG would bind cefotetan, techniques like X-ray crystallography, NMR or SPR could be used.
Cefotetan, an FDA-approved bactericidal agent classified within the cephamycin subclass of second-generation cephalosporins,25 exerts its mechanism of action by impeding cell wall synthesis through disruption of peptidoglycan cross-linking mediated by penicillin-binding proteins.26 Additionally, cefotetan triggers the activation of bacterial autolysins, thereby facilitating cell lysis27 Notably, cefotetan monotherapy has demonstrated reduced rates of both inpatient and outpatient antibiotic utilization in comparison to non-cefotetan prophylaxis for Gustilo–Anderson type III open long bone fractures.28 The utilization of cefotetan as a standalone prophylactic measure may offer enhanced antibiotic stewardship for Gustilo–Anderson type III open long bone fractures when juxtaposed with traditional antibiotic prophylaxis regimens.29 Despite its established role as a bactericidal agent, there is a paucity of literature regarding the potential anti-cancer properties of cefotetan. This study is the first of its kind in exploring the cytotoxic effects of cefotetan in BRCA1-defective breast cancer cell lines compared to wild-type BRCA1 breast cancer cell lines. This study has been filed for US Provisional Patent. (Srinivas,Priya and Ittycheria,Shreya.2025. Repurposing FDA-approved Drug Cefotetan, as a Novel Anticancer Avenue Through Inhibition of β-hCG and its mechanism for the process of inhibition thereof. US Provisional Application Serial No. 63/799,963, filed May 5, 2025).
While PARP inhibitors have shown promise as a therapeutic avenue for BRCA1-defective cancers, a substantial proportion of patients (40-70%) have developed resistance over time.30 Given the existing FDA approval status of cefotetan as a bactericidal agent, the potential repurposing of this drug for the treatment of BRCA1-defective cancers, contingent upon efficacy in animal studies, emerges as a compelling prospect.
Numerous vaccines targeting β-hCG and α-hCG have been developed, including a conjugated toxoid,31 a recombinant chimeric antibody cPiPP, a curcumin-conjugated variant selectively toxic to β-hCG expressing cells,32 and Ovidrel, a recombinant α-hCG vaccine that has exhibited an impact on the RNA expression of BRCA1-defective breast cancer tissues.33 However, the production of antibodies is costly and may elicit adverse reactions in humans, particularly in pregnant women.34 In contrast, chemical inhibitors offer a distinct advantage by circumventing the occurrence of adverse reactions.
3 Experimental
3.1 In silico analysis
The computational analyses encompassing protein preparation, virtual screening, molecular docking, MM-GBSA calculation and molecular dynamic simulations were executed utilizing Maestro Schrödinger v13.5. These analyses were conducted on a Dell computer, equipped with a Windows 11 operating system, an Intel Core i7 processor and 16 GB RAM, ensuring optimal performance and reliability throughout the investigative process.846 small molecules sourced from diverse small molecule screening libraries, including ZINC (in which the “anticancer” filter was applied), NCI, Chembridge, ChemDB and LifeChemicals (in which the “anticancer compounds” filter was applied), were acquired in SDF format, details of which are given in ESI Table 1.† Ligand preparation was conducted utilizing the LigPrep module within Schrödinger Maestro version 13.5.21 This process involved the expansion of tautomeric and ionization states of the compounds, generation of ring conformations and production of stereoisomers to establish the ligand library (see ESI 2†).Prior to initiating the 100 ns simulations, a simulation box was constructed for the protein–ligand complexes of interest using the System Builder tool within Desmond. The system was solvated using the TIP3P water model, with an orthorhombic periodic boundary box shape set at a distance of 10 Å on both sides to ensure a consistent volume. To maintain system neutrality, appropriate ions such as Na+ and Cl− were introduced randomly into the solvated system at a salt concentration of 0.15 M. Subsequently, the system underwent a minimization and relaxation process following the default protocol integrated within the Desmond module, employing the OPLS4 force field parameters post the construction of the solvated system containing the protein–ligand complex.
Visualization aids, including plots and figures, were generated using the Simulation Interaction Diagram tool within Maestro. The assessment of the stability of the complex structure was conducted based on various parameters derived from the trajectory output, encompassing root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), protein–ligand contacts (P–L contact) and hydrogen bond interactions.
3.2 Experimental procedure
The dilution concentrations were entered into a dedicated MO affinity analysis software to determine KD.
For all the in vitro experiments, at least 3 biological replicates were done to calculate the standard deviation and the p values.
4 Conclusion
In this investigation, six potential small molecule inhibitors, out of 127846 small molecules, targeting β-hCG at sites coinciding with those of TGFβR-II's binding site on β-hCG, was identified through the employment of diverse in silico methodologies. Particularly, cefotetan exhibited the most promising outcomes in BRCA1-defective breast cancer cell lines compared to all other compounds. The FDA-approved antibacterial agent, cefotetan, though it showed a weak interaction with hCG in MST and ITC, is found to be promising as a cytotoxic agent against βhCG positive breast cancer cells compared to the β-hCG monoclonal antibody. The binding experiments show a possibility of cefotetan binding to β-hCG and could be potentially causing the inhibition in BRCA1-defective cell lines through binding β-hCG and preventing it from interacting with TGF β R-II. Binding experiments show that cefotetan may interact with β-hCG, preventing it from activating the TGFβ R-II signalling, which induces cytotoxicity in BRCA1-defective cancer cell lines. Future studies will include experiments such as affinity chromatography of the cell's contents coupled with mass spectra techniques to prove the interaction between cefotetan and β-hCG. Although the compounds DCF, DFF, F0922-0590, F6658-4634, and F0385-0029 demonstrated noteworthy efficacy in BRCA1-defective breast cancer cell lines, their individual impact did not reach a level of significance warranting consideration as standalone therapeutic modalities for BRCA1-defective breast cancers. Prior to advancing to clinical trials, it is imperative to conduct further investigations utilizing animal models to ascertain the safety and efficacy profiles of these compounds comprehensively.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Shreya Sara Ittycheria: investigation, methodology, software, validation, formal analysis, data curation, writing – original draft, and visualization; Krishnankutty Chandrika Sivakumar: software, and writing – review & editing; Nagarjun Narayanaswamy, Pankaj Suman: methodology, writing – review & editing; Dipyaman Patra: methodology; Bhavana Ramachandran, R. L. Neetha., Arathy V. Warrier M. A. Aiswarya and S. Kaviya: writing – review & editing; Priya Srinivas: conceptualization, investigation, resources, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
There is no interest to declare.Acknowledgements
The research obtained funding from CSIR [Sanction Order No. 37WS (0059)/2023-24/EMR-II/ASPIRE], ICMR [Sanction Order No. EMDR/SGll3l2023-2686] and RGCB Intramural. The authors wish to thank Dr Shijulal-Nelson Sathi and the Bioinformatics team at Rajiv Gandhi Centre for Biotechnology, Akkulam – 695011, for providing the necessary support for conducting the in silico study. The authors wish to express their gratitude to Didier Trono for the vector plasmid, pLVTHM (Addgene plasmid #12247), lentiviral plasmids psPax2 and pMD2.G (Addgene plasmid #12260 & Addgene plasmid #12259), vector, pLVTHM (Addgene plasmid #12247), lentiviral particles of TetO-dCas9-D3A (Addgene plasmid # 78254) and pCL-CTIG (Addgene plasmid # 149021), which are required for the development of BRCA1 promoter hyper-methylation induced cell line. The authors also wish to thank Ratheeshkumar Thankappan for helping with CRISPR cloning.References
- International Agency for Research on Cancer (IARC), Cancer statistics report worldwide, 2022, https://gco.iarc.fr/today/en/dataviz/pie?mode=cancer%26sexes=2, accessed 4 July, 2024 Search PubMed.
- International Agency for Research on Cancer (IARC), Cancer statistics report worldwide, 2022, https://gco.iarc.fr/today/en/dataviz/pie?mode=cancer%26sexes=2%26types=1, accessed 4 July, 2024 Search PubMed.
- X. Zhu, L. Shan, F. Wang, J. Wang, F. Wang, G. Shen, X. Liu, B. Wang, Y. Yuan, J. Ying and H. Yang, Hypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancer, Breast Cancer Res. Treat., 2015, 150, 4, DOI:10.1007/s10549-015-3338-y.
- R. K. Iles, Ectopic hCGβ expression by epithelial cancer: malignant behaviour, metastasis and inhibition of tumor cell apoptosis, Mol. Cell. Endocrinol., 2007, 260, 264–270, DOI:10.1016/j.mce.2006.02.019.
- S. K. Sengodan, R. Nadhan, R. S. Nair, S. K. Hemalatha, V. Somasundaram, R. R. Sushama, A. Rajan, N. R. Latha, G. R. Varghese, J. M. Kumar and A. Chil, BRCA1 regulation on β-hCG: a mechanism for tumorigenicity in BRCA1 defective breast cancer, Oncogenesis, 2017, 6(9), e376, DOI:10.1038/oncsis.2017.75.
- S. K. Sengodan, A. Rajan, S. K. Hemalatha, R. Nadhan, A. Jaleel and P. Srinivas, Proteomic profiling of β-hCG-induced spheres in BRCA1 defective triple negative breast cancer cells, J. Proteome Res., 2018, 17(1), 276–289, DOI:10.1021/acs.jproteome.7b00562.
- S. K. Sengodan, S. K. Hemalatha, R. Nadhan, T. Somanathan, A. P. Mathew, A. Chil, J. Kopczynski, R. S. Nair, J. M. Kumar and P. Srinivas, β-hCG-induced mutant BRCA1 ignites drug resistance in susceptible breast tissue, Carcinogenesis, 2019, 40(11), 1415–1426, DOI:10.1093/carcin/bgz070.
- R. Nadhan, J. V. Vaman, S. K. Sengodan, S. K. Hemalatha, N. Chellappan, S. Sadasivan, A. Pasuthottiyil Varkey, S. Yesodharan, K. Raji Sathyanpillai, A. K. Bhuvaneswari Venugopal and S. Prameelakumari Sreenivasan, BRCA1 promoter hypermethylation in human placenta: a hidden link with β-hCG expression, Carcinogenesis, 2020, 41(5), 611–624, DOI:10.1093/carcin/bgz117.
- G. R. Varghese, D. Patra, V. S. Jaikumar, A. Rajan, N. R. Latha and P. Srinivas, βhCG mediates immune suppression through upregulation of CD11b+ Gr1+ myeloid derived suppressor cells, CD206+ M2 macrophages, and CD4+ FOXP3+ regulatory T-cells in BRCA1 deficient breast cancers, Immunology, 2023, 170(2), 270–285, DOI:10.1111/imm.13673.
- A. Mohammed, T. Ahmed, R. R. Bhat, E. Mallik and A. Arulprakasam, Association of serum beta hCG levels in women with palpable malignant breast lesions, Sci. Rep., 2023, 13(1), 13208 CrossRef CAS PubMed.
- M. Ascoli, P. Narayan, J. L. Strauss and R. L. Barbieri, The gonadotropin hormones and their receptors, Yen and Jaffe's Reproductive Endocrinology, Saunders/Elsevier, Philadelphia, PA, 7th edn, 2013, pp. 27–44 Search PubMed.
- J. Massagué, TGFβ signalling in context, Nat. Rev. Mol. Cell Biol., 2012, 13(10), 616–630, DOI:10.1038/nrm3434.
- A. K. Swaroop, P. K. Namboori, M. Esakkimuthukumar, T. K. Praveen, P. Nagarjuna, S. K. Patnaik and J. Selvaraj, Leveraging decagonal in-silico strategies for uncovering IL-6 inhibitors with precision, Comput. Biol. Med., 2023, 163, 107231, DOI:10.1016/j.compbiomed.2023.107231.
- B. G. Pierce, K. Wiehe, H. Hwang, B. H. Kim, T. Vreven and Z. Weng, ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers, Bioinformatics, 2014, 30(12), 1771–1773, DOI:10.1093/bioinformatics/btu097.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, CASTp 3.0: computed atlas of surface topography of proteins, Nucleic Acids Res., 2018, 46(W1), W363–W367, DOI:10.1093/nar/gky473.
- L. Jendele, R. Krivak, P. Skoda, M. Novotny and D. Hoksza, PrankWeb: a web server for ligand binding site prediction and visualization, Nucleic Acids Res., 2019, 47(W1), W345–W349, DOI:10.1093/nar/gkz424.
- D. Patra, G. R. Varghese, V. S. Jaikumar, A. Rajan, N. Krishnan, K. Kuppuswamy, R. Thankappan and P. Srinivas, BRCA1 Hypermethylation In Sporadic Breast Cancers: Discovering A Novel Pathway To Tumorigenesis Via Coordinate NBR2 Deregulation And TNBC Transformation, BioRxiv, 2022, p. 2022-04 Search PubMed.
- SiteMap: Schrödinger Release 2024-2, Schrödinger, LLC, New York, NY, 2024 Search PubMed.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. Von Bargen and R. Abel, OPLS4: Improving force field accuracy on challenging regimes of chemical space, J. Chem. Theory Comput., 2021, 17(7), 4291–4300, DOI:10.1021/acs.jctc.1c00302.
- Glide: Schrödinger Release 2024-2, Schrödinger, LLC, New York, NY, 2024 Search PubMed.
- Schrödinger Release 2024-2: LigPrep, Schrödinger, LLC, New York, NY, 2024 Search PubMed.
- S. Sirin, R. Kumar, C. Martinez, M. J. Karmilowicz, P. Ghosh, Y. A. Abramov, V. Martin and W. Sherman, J. Chem. Inf. Model., 2014, 54, 2334–2346, DOI:10.1021/ci5002185.
- Desmond: Schrödinger Release 2024-2: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2024 Search PubMed.
- D. Figureueroa, M. Asaduzzaman and F. Young, Real time monitoring and quantification of reactive oxygen species in breast cancer cell line MCF-7 by 2′, 7′–dichlorofluorescin diacetate (DCFDA) assay, J. Pharmacol. Toxicol. Methods, 2018, 94, 26–33, DOI:10.1016/j.vascn.2018.03.007.
- A. Ward and D. M. Richards, Cefotetan: a review of its antibacterial activity, pharmacokinetic properties and therapeutic use, Drugs, 1985, 30, 382–426 CrossRef CAS PubMed.
- T. Bui and C. V. Preuss, Cephalosporins, in StatPearls [Internet], StatPearls Publishing, 2024 Search PubMed.
- P. Periti and T. Mazzei, Antibiotic-induced release of bacterial cell wall components in the pathogenesis of sepsis and septic shock: a review, J. Chemother., 1998, 10(6), 427–448, DOI:10.1179/joc.1998.10.6.427.
- G. G. Via and M. J. Prayson, Monotherapeutic cefotetan surgical prophylaxis: Current evidence and practice for orthopaedic surgeons, Journal of Orthopaedic Reports, 2023, 100215, DOI:10.1016/j.jorep.2023.100215.
- G. G. Via, D. A. Brueggeman, V. A. Murray, A. W. Froehle, S. D. Burdette and M. J. Prayson, Use of single agent Cefotetan for Gustilo-Anderson type III open fracture prophylaxis, Injury, 2023, 54(8), 110914, DOI:10.1016/j.injury.2023.110914.
- D. Kim and H. J. Nam, PARP inhibitors: clinical limitations and recent attempts to overcome them, Int. J. Mol. Sci., 2022, 23(15), 8412, DOI:10.3390/ijms23158412.
- G. P. Talwar, H. K. Vyas, S. Purswani and J. C. Gupta, Gonadotropin-releasing hormone/human chorionic gonadotropin β based recombinant antibodies and vaccines, J. Reprod. Immunol., 2009, 83(1–2), 158–163, DOI:10.1016/j.jri.2009.08.008.
- H. K. Vyas, R. Pal, R. Vishwakarma, N. K. Lohiya and G. P. Talwar, Selective killing of leukemia and lymphoma cells ectopically expressing HCGβ by a conjugate of curcumin with an antibody against HCGβ subunit, Oncology, 2009, 76(2), 101–111, DOI:10.1159/000188665.
- H. Depypere, Y. Su, N. Dang, B. Poppe, F. Stanczyk, J. Janssens and J. Russo, Prolonged recombinant pregnancy hormone use in BRCA1 and BRCA2 mutation carriers, Eur. J. Cancer Prev., 2021, 30(3), 195–203, DOI:10.1097/CEJ.0000000000000664.
- L. A. Cole, hCG, the wonder of today's science, Reprod. Biol. Endocrinol., 2012, 10, 1–18 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08663e |
| ‡ Current address: Department of Immunology, University of Pittsburgh, PA 15213, USA. |
| This journal is © The Royal Society of Chemistry 2025 |