
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitsunobu reaction: assembling C–N bonds in chiral traditional Chinese medicine
Xue Zhoua,
Liang Xua,
Zhanhui Maa,
Jin Cuia and
Bin Wang *ab
*ab
aCollege of Chinese Medicine, School of Pharmacy, Key Laboratory of Xin'an Medicine of the Ministry of Education, Anhui University of Chinese Medicine, Hefei, 230038, P. R. China. E-mail: bw5654@ahtcm.edu.cn; Fax: +86-551-65169371
bInstitute of Pharmaceutical Chemistry, Anhui Academy of Chinese Medicine, Hefei, 230038, P. R. China
First published on 17th February 2025
Abstract
The synthesis of chiral molecules has long been a central focus and challenge in medicinal chemistry research. The Mitsunobu reaction, developed by Japanese chemist Mitsunobu in 1967, is a widely utilized bimolecular nucleophilic substitution reaction that plays a vital role in synthesizing chiral natural products. In this reaction, alcohols react with nucleophilic reagents in the presence of a phosphine ligand to form an intermediate phosphonium salt. This intermediate enables the formation of various chemical bonds. The purpose of this review is to explore the applications of the Mitsunobu chemistry in constructing pivotal carbon–nitrogen bonds in traditional Chinese medicines (TCMs). Emphasis will be placed on the preparative synthetic applications of the Mitsunobu strategy as a key step in the total synthesis of naturally occurring biologically active products.
1. Introduction
Chirality refers to the property of objects or molecules in three-dimensional space characterized by non-superimposable mirror images.1 An object is considered chiral if it cannot perfectly align with its mirror image, like the relationship between left and right hands. In chemistry, chirality generally relates to the structural characteristics of molecules with one or more chiral centers.2 In chiral molecules, the two mirror-image forms are known as enantiomers. Although their physical and chemical properties are generally similar, their behavior can vary significantly when interacting with other chiral substances, including biomolecules. As a result, chirality is critically important in various fields, with its significance particularly pronounced in drug development, synthetic chemistry, and biochemistry.3 Biot identified the first scientific evidence of chirality in 1815 when he observed the optical rotation of camphor.4 Pasteur found, in 1848, that two tartaric acid molecules with identical properties exhibited opposite optical rotations; the specific rotation of L-tartaric acid is −12.6°, and that of D-tartaric acid is +12.6°, as depicted in Fig. 1.5 This groundbreaking discovery laid the foundation for stereochemistry, explicitly highlighting the concept of “chirality.”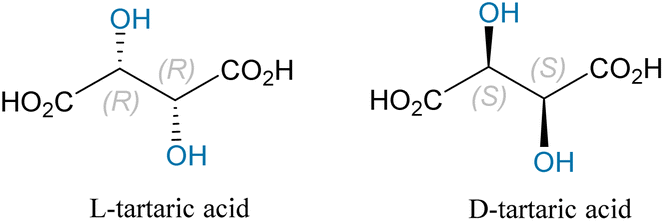 | ||
| Fig. 1 Schematic structures of L-tartaric acid and D-tartaric acid. | ||
In today's pharmaceutical landscape, more than half of the drugs on the market are chiral, with approximately 88% administered as racemic mixtures.6 It's important to note that the desired biological or pharmacological effects are often associated with a specific enantiomer. In contrast, the other enantiomer may have different effects, be inactive, or even present toxicity risks.7 For example, with well-known thalidomide, the R-enantiomer has sedative properties, whereas the S-enantiomer is associated with congenital disabilities.8 Therefore, separate pharmacological and toxicological evaluations are essential for each enantiomer, rather than relying solely on assessments of the racemic mixture. A thorough evaluation is needed to prevent exposure to inactive or harmful enantiomers. As a result, there has been a recent increase in the development of drugs marketed as individual enantiomers, highlighting the significance of using racemic molecules only when the enantiomers demonstrate complementary biological activities.
The Mitsunobu reaction is valuable for forming various chemical bonds, including C–O, C–N, C–S, C–C, and C–X bonds, using primary and secondary alcohols as substrates.9 While primary and secondary alcohols are well-suited for this reaction, tertiary alcohols are not suitable. Typically, this reaction involves the cooperative action of triphenylphosphine and diethyl azodicarboxylate, leading to the dehydration of the alcohol hydroxyl group with acidic compounds.10 When chiral alcohols are used as substrates, the resulting products usually exhibit configuration inversion, making this a valuable reaction for synthesizing chiral drugs. It is important to note that traditional Chinese medicine contains various chiral compounds, many demonstrating significant physiological activities. However, the extraction and purification of these chiral compounds can be challenging. Conventional methods often yield only small amounts of pure products, making them time-consuming and labor-intensive. Recently, there has been growing interest in utilizing appropriate chiral substrates to prepare natural chiral products efficiently through the Mitsunobu reaction. This approach not only proves effective but also enhances overall yield. In 2008, Yurovskaya et al. highlighted the Mitsunobu reaction's vital role in synthesizing nitrogen-containing heterocyclic compounds.11 Professor Pavan Kumar reviewed its applications from 1967 to 2009, establishing a historical context.12 Fletcher then examined its relevance in the 21st century,13 while Panday assessed the impact of improved Mitsunobu reagents.14 Additionally, Professor Mojzych emphasized its importance in natural product synthesis,15 and the Cai group explored advancements in catalytic Mitsunobu reactions.16 These contributions underscore the Mitsunobu reaction's versatility and significance in modern synthetic chemistry. These comprehensive reviews have deepened our understanding of the Mitsunobu reaction from various critical angles and propelled its advancement within the chemical community. In this context, as shown in Fig. 2, we assertively summarize the applications of the classic Mitsunobu reaction in synthesizing natural products, concentrating on constructing essential C–N bonds in compounds that typically contain chiral centers. The studies we cover span from 1978 to 2024.
 | ||
| Fig. 2 Utilizing the Mitsunobu strategy in the synthesis of active alkaloids from TCMs. | ||
2. Mechanism
The mechanism of the Mitsunobu reaction is controversial, as its complexity leads to debates regarding the intermediates and their roles.17 The specific reaction process is elucidated below using triphenylphosphine and diethyl azodicarboxylate, as shown in Fig. 3. | ||
| Fig. 3 The recognized mechanism of the Mitsunobu reaction. | ||
The entire process can be divided into four steps: in step 1, triphenylphosphine initiates the reaction by attacking diethyl azodicarboxylate (DEAD), forming an intermediate I called a phosphonium salt. These intermediate I capture a proton from the nucleophilic NucH, creating a cation II in step 2. Subsequently, in step 3, the hydroxyl functional group from the substrate alcohol then targets the phosphorus center of the cation II, forming an oxaphosphonium ion III.18 This step triggers the detachment of DEAD, producing hydrazine as a byproduct. Following this, an SN2 nucleophilic substitution reaction occurs, where the nucleophile Nuc− attacks the alkoxyl phosphonium intermediate III, inducing a configuration inversion, as depicted in step 4. Ultimately, the cleavage of the carbon–oxygen bond affords products with configuration inversion alongside triphenylphosphine oxide byproducts. This reaction is fundamentally redox in nature.
3. Reaction type
Herein, we summarize the applications of the classic Mitsunobu reaction in the synthesis of natural products. This reaction involves the formation of important C–N bonds using primary or secondary alcohols with azides (Scheme 1A), sulfonamides (Scheme 1B), amines (Scheme 1C), and others. | ||
| Scheme 1 Synthetic route to alkaloids by Mitsunobu strategy. | ||
3.1 Azide acts as nucleophiles
Lycopodium alkaloids (LAs) are derived from the medicinal plant Lycopodium serratum,19 recognized for their potent inhibitory effect on acetylcholinesterase. These compounds can significantly enhance cognitive function and have demonstrated improvements in learning and memory.20 Current research is significantly focused on applying LAs in treating Alzheimer's disease, which is expected to advance studies on synthesizing Lycopodium alkaloids (Fig. 4–6).21 | ||
| Fig. 4 Mitsunobu reaction using azide acts as nucleophiles. | ||
 | ||
| Fig. 5 Mitsunobu reaction using sulfonamide or sulfonyl hydrazine act as nucleophiles. | ||
 | ||
| Fig. 6 Mitsunobu reaction using various amines act as nucleophiles. | ||
A novel alkaloid named Lycoposerramine-X has been isolated from Lycopodium serratum. As depicted in Scheme 2, Takayama and co-workers conducted a reaction in 2009 using the chiral secondary alcohol 1 and nucleophile diphenylphosphoryl azide (DPPA) in tetrahydrofuran in the presence of Mitsunobu reagents (DEAD and PPh3), resulting in the formation of compound 2. Although the yield reached 80%, the low temperature of −20 °C adversely affected the formation of compound 2. Through 12 steps of reactions, the chiral piperidine derivative Lycoposerramine-X was successfully synthesized, with an overall yield of 25%.
 | ||
| Scheme 2 Takayama's synthesis of phlegmarine-type lycopodium alkaloids. | ||
One of the most significant challenges in treating malaria is the development of drug resistance to commonly used medications. As a result, researchers have gradually shifted their focus to natural products. Notably, certain indole alkaloids have shown high anti-malarial activity.22,23 Some of these active indole alkaloids are derived from two tryptophan units, with ditryptophenaline being a prominent example.
Ditryptophenaline has been isolated from several Aspergillus species. In 2001, Overman and colleagues realized the chemical synthesis of ditryptophenaline. They conducted a reaction at room temperature using C2-symmetric diol 3 and DPPA in toluene as the solvent in the presence of Mitsunobu reagents (DEAD and PPh3), as illustrated in Scheme 3. During this reaction, the hydroxyl groups in compound 3 were successfully replaced with azido groups, forming compound 4 with reversed configurations in 91% yields. Further transformations, consisting of 6 steps, can then lead to synthesizing the target compound, ditryptophenaline, with an overall yield of 20%.
 | ||
| Scheme 3 Overman's synthesis of diketopiperazine alkaloids. | ||
Batzelladine F is a marine natural product isolated from the red Jamaican sponge and plays a vital role in treating autoimmune diseases.24–26 As illustrated in Scheme 4, Overman and co-workers conducted a double substitution reaction in 2005 between chiral alcohol 5 and hydrazoic acid (HN3) using DEAD and PPh3 as auxiliary reagents. This reaction was followed by a palladium-catalyzed carbon–carbon bond reduction, which resulted in the formation of chiral diamine 6, with a yield of 80%. This diamine 6 undergoes 8 steps, resulting in an overall yield of 21% for the final product, batzelladine F.
 | ||
| Scheme 4 Overman's synthesis of batzelladine F. | ||
Cruentaren A is an antifungal benzolactone produced by the myxobacterium Byssovorax cruenta.27 It demonstrates high cytotoxicity against cancer cell.28,29 As shown in Scheme 5, Maier and co-workers 2007 conducted compound 7 in tetrahydrofuran with PPh3, DIAD, and the azide compound (PhO)2P(O)N3. This reaction effectively replaced the terminal hydroxyl group in the molecule with an azido group, yielding compound 8 with an impressive yield of 90%. The target compound, Cruentaren A, was ultimately synthesized with an overall yield of 34% through five subsequent steps that included reduction, acylation, and deprotection.
 | ||
| Scheme 5 Maier's synthesis of cruentaren A. | ||
McCarthy and his colleagues conducted a reaction, as shown in Scheme 6, using the chiral tertiary alcohol compound 9. They combined this with two equivalents of ADDP and PMe3 and two of the azide compound HN3 in tetrahydrofuran solvent for a duration exceeding 24 hours. This reaction replaced the hydroxyl group in compound 9 with an azido group, producing the chiral inverted compound 10. Namely, in compound 9, the hydroxyl functional group on the R-configured carbon atom effectively forms an active intermediate phosphonium salt with trimethyl phosphine. This intermediate then undergoes a well-defined SN2 nucleophilic substitution reaction with the azide anion, resulting in an inversion of configuration that definitively produces the S-configured compound 10. Subsequently, a simple reduction and hydrolysis were performed to synthesize the challenging chiral amino acid ethyl ester ditryptophenaline, with an overall yield of 56%.30 In the Mitsunobu reaction, it is essential to note that when the nucleophile HA has a pKa more significant than 11 or when the substrate is a tertiary alcohol, the product yield significantly diminishes. In many instances, the desired reaction may not occur at all. However, employing ADDP reagents decisively enhances the reaction's performance and can lead to successful outcomes.
 | ||
| Scheme 6 McCarthy's synthesis of disubstituted amino acids. | ||
Diospongins are a novel class of diaryl heptanoids isolated from the rhizomes of Dioscorea spongiosa.31 Due to their potent anti-osteoporotic activity demonstrated in in vitro cell experiments and their relatively simple structure, they have garnered significant interest from synthetic chemists.32
Scheme 7 shows Chandrasekhar and co-workers 2009 synthesized aza-(−)-diospongin A. They reacted chiral benzyl alcohol 11 with the hydrazoic acid in benzene at room temperature for 3 hours, using classical Mitsunobu reagents (PPh3 and DIAD). This reaction resulted in the substitution of the secondary alcohol hydroxyl group with an azido group, ultimately yielding compound 12 with a 40% overall yield, and successfully synthesized the 4-hydroxypiperidine derivative aza-(−)-diospongin A through 7 steps.
 | ||
| Scheme 7 Chandrasekhar's synthesis of aza-diospongin A. | ||
Quinolizidine alkaloids possess a range of biological activities, including anti-cancer, antibacterial, anti-inflammatory, antiviral, and anti-arrhythmic effects.33,34 These alkaloids can accumulate in various leguminous plants and have widespread applications in agriculture, pharmaceuticals, and the chemical industry.35 Research has been focusing on efficient stereoselective methods to construct these organic molecules.
As illustrated in Scheme 8, Santos and co-workers in 2010 employed acetone cyanohydrin 14 as the cyanation reagent in a classic Mitsunobu reaction, using DEAD and PPh3 as reagents. They conducted the reaction on the secondary chiral alcohol 13 in a mixed solvent of toluene and tetrahydrofuran, forming cyano compound 15 with a position at C-10 hydroxyl configuration inversion and an 81% yield. Gas chromatography analysis indicated that the reaction was completed without forming isomeric products. Similarly, using azidic acid as the nucleophilic reagent and in the presence of DEAD and PPh3, they obtained product 16 with a very high yield. Subsequent reduction and acylation, consisting of 2 steps, led to the synthesis of (−)-epiquinamide with an overall yield of 77% and the synthesis of (−)-lupinine with an overall yield of 85%, respectively.
 | ||
| Scheme 8 Santos's synthesis of quinolizidine alkaloids. | ||
Amaryllidaceae plants, with their unique secondary metabolites, the montanine-type alkaloids, stand out in disease treatment. These alkaloids, found exclusively in this plant family, have attracted widespread attention for their distinct anti-tumor activity. Research has revealed that Montanine Alkaloids, though present in low concentrations in plants, demonstrate significant inhibitory activity against cancer cells such as HeLa and A549.36,37
As depicted in Scheme 9, to obtain the target compound Ent brunsvigine, the authors successfully transformed the chiral enol 17 into the critical intermediate azide compound 18 with the collaborative action of DPPA, DIAD, and PPh3. After an additional 10 consecutive steps of reactions, Ent brunsvigine was obtained with an overall yield of 1.67%.
 | ||
| Scheme 9 Banwell's synthesis of ent brunsvigine. | ||
Mersicarpine is an aspidosperma alkaloid isolated from plants of the Kopsia genus. Its unique structural features have attracted significant attention in synthetic organic chemistry. Research has shown that it can induce apoptosis and exhibit specific inhibitory effects on protein synthesis. As a result, Mersicarpine is a novel translation inhibitor that induces apoptosis.38,39
As illustrated in Scheme 10, to obtain the target compound (−)-mersicarpine, in 2019, Wang and co-workers first performed a Mitsunobu reaction to convert the primary hydroxyl group in compound 19 into an azide-substituted compound 20, achieving this transformation with a high yield of 98%. Notably, the reaction yield was significantly influenced by temperature, as higher temperatures led to the formation of a substantial amount of intramolecular aziridination products. After completing four consecutive reaction steps, we obtained (−)-mersicarpine with a robust overall yield of 31%.
 | ||
| Scheme 10 Wang's synthesis of mersicarpine alkaloids. | ||
White Candida albicans is a common fungal infection source that often resists conventional drug treatments, leading to limited effectiveness or even treatment failure. However, animal experiments have indicated that batzelladine D could be a potential novel antifungal agent for Candida albicans infections.40,41
Evans group conducted a series of reactions on the 3,4-dihydropyridin-2-one derivative 21 to synthesize batzelladine D, as illustrated in Scheme 11. The process included a hydrosilylation reaction, an ester exchange reaction, and a classic Mitsunobu reaction. This sequence of reactions led to the formation of compound 22 by configuration inversion, achieving an overall yield of 95%. After completing seven consecutive reaction steps, batzelladine D was obtained with an overall yield of 24%.
 | ||
| Scheme 11 Evans' synthesis of tricyclic guanidine alkaloids-batzelladine D. | ||
3.2 Sulfonamide or sulfonyl hydrazine act as nucleophiles
The bark of the Angostura tree in South America is widely used in Venezuelan folk medicine for treating ailments such as indigestion, dysentery, chronic diarrhea, spinal motor nerve issues, and fever.42,43 Angustureine, an isoquinoline alkaloid extracted from the tree bark, is believed to be the main active component for treating the conditions above.44,45As depicted in Scheme 12, to synthesize the chiral isoquinoline angustureine, Nishida and co-workers conducted a reaction in 2004 using the secondary sulfonamide 23 and the chiral secondary alcohol 24 at room temperature with the involvement of DEAD and PPh3 for 2 hours. This resulted in the intermediate diene compound 25 yielding 78%. Compound 25 underwent decomposition reactions, methylation, and hydrolysis in four steps to produce the product (+)-(S)-angustureine with an overall yield of 69%.
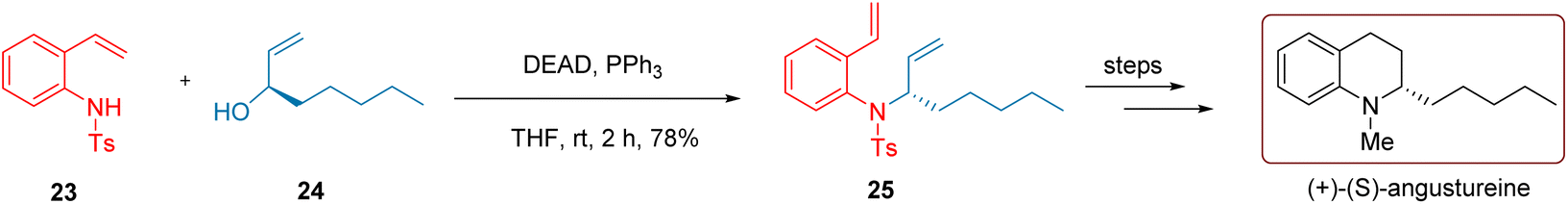 | ||
| Scheme 12 Nishida's synthesis of quinoline alkaloids. | ||
Deoxynupharidine is an alkaloid isolated from the rhizomes of Nuphar species and is a component of a larger class of terpene alkaloids. It is primarily used in treating rheumatoid arthritis and for alleviating back and leg pain and exhibits notable immunosuppressive activity.46,47
As shown in Scheme 13, obtaining deoxynupharidine, carried out by Harrity and co-workers in 2003, is a significant step in synthesizing this alkaloid. The reaction of the primary alcohol 26 in the presence of n-Bu3P and ADDP to form an active phosphonium intermediate, followed by the intramolecular nucleophilic attack of the sulfonamide group, led to the creation of the chiral cyclic sulfonamide compound 27 with an impressive yield of 80%. This research paves the way for further exploration and potential pharmacology and organic chemistry applications. After completing four consecutive reaction steps, deoxynupharidine was obtained with an overall yield of 35%.
 | ||
| Scheme 13 Harrity's synthesis of nuphar alkaloids. | ||
Over 300 alkaloids have been isolated from the Lycopodium genus, with the (+)-fawcettimine alkaloids being one of the most representative classes. They are known for their neuroprotective, anti-tumor, anti-inflammatory, antimicrobial, and antiviral properties. The unique spiral ring structure present in these alkaloids may be a key factor contributing to their distinct biological activities.48–51 For instance, Lycoposerramine-C is a novel alkaloid isolated from Lycopodium serratum, featuring a double bond at the C-6/C-7 positions of its molecule.
As illustrated in Scheme 14, Takayama et al. conducted a Mitsunobu reaction to develop synthesis methods for such alkaloids. They dissolved the primary alcohol 28 in tetrahydrofuran and allowed the reaction to proceed at room temperature, achieving a high yield of 91% for the macrocyclic compound 29. This transformation cleverly utilized the Mitsunobu reaction to facilitate intramolecular cyclization. Further deprotection steps ultimately led to the synthesis of Lycoposerramine-C and Phlegmariurine-A. The steps from compound 29 to Lycoposerramine-C involve five steps with an overall yield of 43.09%. In comparison, the steps from compound 29 to Phlegmariurine-A involve six steps with an overall yield of 40.93%.
 | ||
| Scheme 14 Takayama's synthesis of fawcettimine-type lycopodium alkaloids. | ||
Several years later, Taniguchi and co-workers (2014) utilized a cyclization reaction to assemble the intermediate compound 31, which is crucial for obtaining (+)-fawcettimine, as shown in Scheme 15. Substrate 30 was reacted in tetrahydrofuran using DMEAD (1.5 equiv.) and PPh3. This method successfully led to the synthesis of the nine-membered ring intermediate 31. Six steps are required to go from compound 31 to the final product, with an overall yield of 38%.
 | ||
| Scheme 15 Taniguchi's synthesis of lycopodium alkaloids. | ||
Serinolamide A and columbamide D are bioactive marine natural products. In recent years, the former was isolated from the cyanobacteria Lyngbya majuscula in Papua New Guinea.52,53
As illustrated in Scheme 16, Muthukrishnan and co-workers 2019 utilized a chiral glycerol derivative 32, protected at the C1 and C3 positions, and methyl naphthylsulfonamide 33 in tetrahydrofuran as the solvent. In the presence of PPh3 and DIAD, the reaction was conducted for 6 hours, transforming 32 to 34 with a high yield of 95%. The SN2 reaction ensured that the absolute configuration of the intermediate 34 was the S-configuration. Further transformations successfully synthesized Serinolamide A and Columbamide D. There are three steps from compound 34 to Serinolamide A, resulting in an overall yield of 71%. Additionally, there are three steps from compound 34 to Columbamide D, achieving an overall yield of 68%.
 | ||
| Scheme 16 Muthukrishnan's synthesis of serinolamide A and columbamide D. | ||
Epidithiodiketopiperazine natural products (ETPs) are significant fungal metabolites known for their unique structures and potent biological activities.54,55 These characteristics have drawn considerable interest from synthetic chemists and pharmacologists. Despite decades of research, only a limited number of total syntheses of ETPs have been accomplished, which has hindered their further profiling and applications. Scabrosins, a distinct subgroup of ETPs (a–e), were first isolated in 1978.56 After, their structures were revised based on 2D NMR and X-ray structural analysis.
As illustrated in Scheme 17, He and co-workers 2019 successfully constructed a polar inverted chiral C–N bond through a Mitsunobu reaction involving the ortho-hydroxy epoxide 35 in the presence of PPh3 and DIAD, reacting with sulfonamide 36. This reaction formed a critical intermediate 37 with a high yield of 91%. Through further synthetic transformations, the target compounds, scabrosins, were ultimately synthesized over six reaction steps, achieving an overall yield of approximately 31.18%.
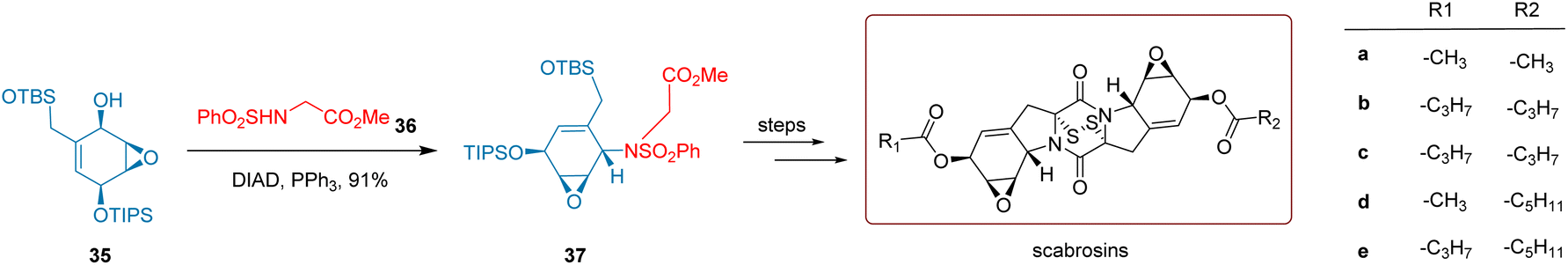 | ||
| Scheme 17 He's synthesis of scabrosins | ||
Lycopoclavamine-A is a structurally unique Lycopodium alkaloid with inhibitory effects on acetylcholinesterase, showing significant potential and promise for treating Alzheimer's disease (AD).57,58
To achieve the synthesis of Lycopoclavamine-A, Takayama and co-workers employed a two-step Mitsunobu reaction in 2019. As illustrated in Scheme 18, the authors first performed the initial Mitsunobu reaction, converting the primary alcohol 38 into compound 39 with a high yield of 92, using three equivalents of NsNH2. Subsequently, in the presence of DEAD and PPh3, compound 39 underwent an intramolecular Mitsunobu reaction, yielding the nine-membered ring product 40 with a yield of 74%. After six consecutive reaction steps, including removing the sulfonyl group, an intramolecular nucleophilic substitution was conducted, ultimately synthesizing the target compound Lycopoclavamine-A with an overall yield of 78%.
 | ||
| Scheme 18 Takayama's synthesis of lycopodium alkaloid. | ||
Morphine is classified as an essential medicine by the World Health Organization and is an effective opioid analgesic.59,60 Due to its synthetically challenging chiral molecular structure and significant clinical application value, the synthesis of morphine-type alkaloids has attracted considerable attention.
To synthesize (–)-morphine, Tu et al., 2019 utilized the differing reactivity of the alcohol hydroxyl groups in the substrates. They synthesized the critical intermediate 42 through a highly regioselective intermolecular Mitsunobu reaction between the fused ring compound 41 and methyl phenylacetamide, yielding the intermediate with a 50% yield. The results are illustrated in Scheme 19. It takes three steps from compound 42 to the final product, with an overall yield of 51%.
 | ||
| Scheme 19 Tu's synthesis of morphine-type alkaloids. | ||
Ar-macrocarpene is a naturally occurring irregular aromatic sesquiterpene with a 3,3,4′-trimethyl-1,1′-(bi-cyclohexyl) skeleton found in the leaves of Cupressus macrocarpa.61–63
In 2019, Bisai and co-workers reacted to synthesize (+)-ar-macrocarpene, using tetrahydrofuran as the solvent. They conducted the chiral enol 43 at low temperatures with PPh3, DIAD, and 2-nitrophenyl hydrazone 44, successfully constructing a C–N bond in compound 45. The results of this reaction are illustrated in Scheme 20. The transformation of compound 45 into the final product is achieved in three straightforward steps, yielding an impressive overall result of 64%.
 | ||
| Scheme 20 Bisai's synthesis of (+)-ar-macrocarpene. | ||
Daphniphyllum alkaloids are nitrogen-containing polycyclic natural products isolated from Daphniphyllum macropodum.64,65 Due to their complex chemical structures and specific anti-cancer and anti-HIV properties, these alkaloids have attracted widespread attention in the synthetic community.66
Sakakura and co-workers 2019 designed a reaction using Mitsunobu reagents (DIAD and PPh3) to synthesize the target daphniphyllum Alkaloid in toluene at room temperature. They replaced the chiral secondary alcohol in compound 46 with a naphthyl sulfonamide, achieving intramolecular cyclization and obtaining the pyrrole derivative 47. This synthetic route involves simple operations and applies to synthesizing other daphniphyllum Alkaloids, as illustrated in Scheme 21. Transforming compound 47 into the final product requires four steps, achieving an overall yield of 37%.
 | ||
| Scheme 21 Sakakura's synthesis of daphniphyllum alkaloids. | ||
Communesin A and B are two complex alkaloids isolated from algal-derived Penicillium fungus. They exhibit cytotoxicity against lymphocytes and leukemia cells.67,68 Movassaghi and co-workers 2019 first synthesized a pivotal intermediate to obtain these compounds. They utilized an amino-protected naphthyl sulfonamide reagent to successfully convert the hydroxyl group in compound 48 into an amino group in compound 49, laying a solid foundation for the further synthesis of the target compounds. The results of this transformation are illustrated in Scheme 22. Reaching Communesin A from compound 49 requires six steps and achieves an overall yield of 38%. In contrast, it takes seven steps to reach Communesin B from compound 49, resulting in a total yield of 35%.
 | ||
| Scheme 22 Movassaghi's synthesis of communesin alkaloids. | ||
Research has shown that Leuconodines D and E exhibit moderate cytotoxicity against KB cells and possess specific antimalarial activity against Plasmodium falciparum, generating significant interest among synthetic chemists.69–71
In 2019, Han group utilized 2-nitrophenylsulfonamide acetate 50 as a nucleophilic reagent in a mixed solvent of tetrahydrofuran and toluene. They performed a Mitsunobu reaction at room temperature for 10 hours with 1.5 equivalents of DEAD and PPh3 and 1.5 equivalents of primary alcohol 51, forming the indole derivative 52. Following this, a palladium–carbon-catalyzed hydrogenation reaction yielded Leuconodine E with an 88% yield. After oxidizing Leuconodine E, they successfully synthesized Leuconodine D with a yield of 66%. The results of these transformations are illustrated in Scheme 23. The synthesis of Leuconodine D from compound 52 requires 12 steps, achieving an overall yield of 9.2%. In comparison, synthesizing Leuconodine E from compound 52 involves only 10 steps, resulting in a significantly higher overall yield of 17%.
 | ||
| Scheme 23 Han's synthesis of leuconodines D and E. | ||
Calyciphylline B-type alkaloids are a subclass of calyciphylline alkaloids that belong to the more prominent family of Daphniphyllum alkaloids. Deoxycalyciphylline B is extracted from the stems of D. subverticillatum.72,73 These alkaloids exhibit various biological activities, including antioxidant and anticancer properties, and can promote nerve growth factors.74,75
Recently, Raghavan and co-workers attempted to synthesize a key intermediate using Mitsunobu reagents to obtain deoxycalyciphylline B. In the presence of DIAD and PPh3, they reacted in tetrahydrofuran as the solvent at room temperature for 24 hours, successfully converting from compound 53 to compound 54 with a yield of 65%. The results of this transformation are illustrated in Scheme 24.
 | ||
| Scheme 24 Raghavan's synthesis of deoxycalyciphylline B. | ||
Aspidosperma alkaloids are characterized by their polycyclic structures, and common compounds such as deoxoapodine and kopsifoline D belong to this class.76 Deoxoapodine is primarily isolated from Tabernae montana armeniaca, and its presence has also been discovered in Hazunta modesta. On the other hand, kopsifoline D was initially isolated from leaf extracts of the Malayan Kopsia species.77,78
Peng and co-workers conducted extensive experiments in 2019 to synthesize deoxoapodine and kopsifoline D. As illustrated in Scheme 25, in a tetrahydrofuran solvent, the primary alcohol 55 underwent a continuous reaction for 12 hours with Mitsunobu reagents (DIAD, PPh3) and sulfonylamide 56, ultimately converting to compound 57 with a high yield of 90%. The successful preparation of 57 laid a solid foundation for synthesizing deoxoapodine and kopsifoline D target compounds. The conversion from compound 57 to deoxoapodine requires precisely seven steps, yielding an overall return of 5.59%. In contrast, the process from compound 57 to kopsifoline D also involves seven steps, but produces a lower overall yield of 3.43%.
 | ||
| Scheme 25 Peng's synthesis of aspidosperma and kopsia alkaloids. | ||
Batzellines and isobatzellines are marine alkaloids that possess a pyrrole framework. Batzellines exhibit selective cytotoxicity towards specific pancreatic cancer cell lines. At the same time, isobatzellines show inhibitory effects on pancreatic cancer cells and have low toxicity towards normal cells, making them candidates for chemotherapy.79,80
Tokuyama and co-workers designed a new synthetic route to prepare these molecules in 2020 to synthesize the key intermediate 59. As shown in Scheme 26, they reacted the primary alcohol substrate 58 with NsNHBoc in the presence of DMEAD and PPh3 in a tetrahydrofuran solvent, successfully converting the hydroxyl functional group with a yield of 78%, completing the synthesis of intermediate 59. The pathway from compound 59 to isobatzelline B requires ten steps, just as it does to reach isobatzelline A. However, it only takes nine steps to convert to Batzellines A. The overall yield for these reactions ranges from 20% to 30%.
 | ||
| Scheme 26 Tokuyama's synthesis of marine alkaloids. | ||
Palhinine A is primarily isolated from the whole plant of Palhinhaea cernua L. (Lycopodiaceae) and belongs to the class of Lycopodium alkaloids. It exhibits acetylcholinesterase (AChE) inhibitory effects, which can improve cognitive impairments and alleviate symptoms of Parkinson's disease.81–83
To chemically synthesize Palhinine A, He and co-workers designed a two-step Mitsunobu reaction in 2020 to synthesize the critical intermediate 62. In this process, the selective protection of the two side chains of the primary alcohol substrate 60 underwent sequential intramolecular Mitsunobu reactions, silicon deprotection, and another intramolecular Mitsunobu reaction, successfully converting 60–62. The results of this transformation are illustrated in Scheme 27. To reach the final product from compound 62, two steps are needed, yielding 28%.
 | ||
| Scheme 27 He's synthesis of the skeleton of palhinine alkaloids. | ||
Over 400 Lycopodium alkaloids have been isolated and identified from the Lycopodiaceae family, with five novel Lycopodium alkaloids discovered in the whole plant of Huperzia serrata. Many of these alkaloids have been found to exhibit pharmacological activities such as acetylcholinesterase inhibition, neuroprotection, anti-tumor, and anti-inflammatory effects.84,85 These alkaloids' unique chemical structures and diverse pharmacological properties continue to captivate plant chemistry and synthesis experts.
Introducing a sulfonylamide group into the molecule and subsequently introducing a nitrogen atom through deprotection is a critical strategy in synthetic chemistry. As illustrated in Scheme 28, Qiu and co-workers successfully synthesized compound 64 using this approach in 2020. Specifically, under acidic conditions, the silicon-protecting group of compound 63 was cleaved to generate a primary alcohol. Subsequent intramolecular cyclization was achieved by treating Mitsunobu reagents (DEAD and PPh3). Notably, this step required high-temperature conditions at 80 °C and converted 63 to 64 with a moderate yield of 67%. Subsequent multi-step reactions ultimately led to the completion of the synthesis of (+)-fawcettimine. The process requires just one step to convert compound 64 into the final product, achieving a yield of 81%.
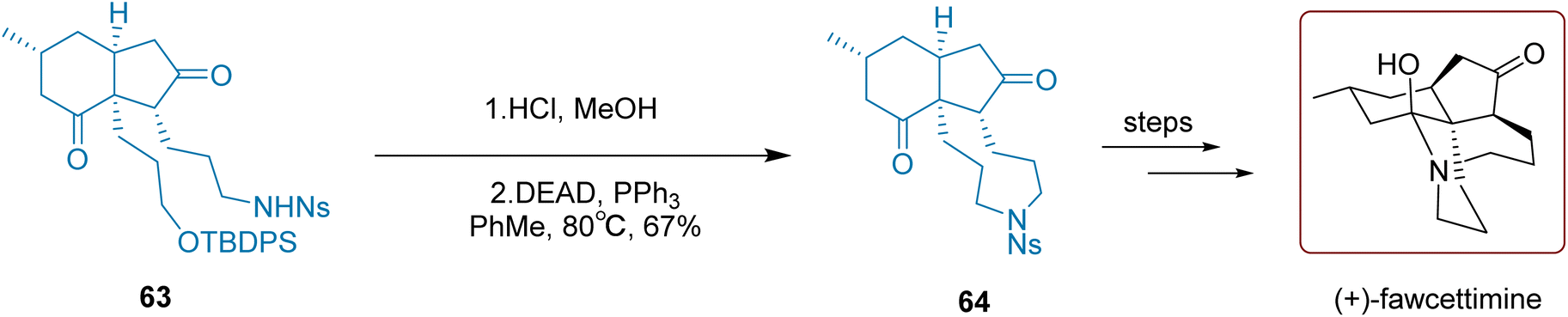 | ||
| Scheme 28 Qiu's synthesis of lycopodium alkaloids. | ||
Asperipin-2a is a bicyclic hexapeptide isolated from Aspergillus flavus,86,87 featuring eight chiral centers within its molecular structure. The chemical synthesis of such compounds poses significant challenges. As illustrated in Scheme 29, Hutton et al. utilized chiral secondary alcohol 65 and a tyrosine derivative 66 to perform a Mitsunobu reaction, forming the polar inverted compound 67. Subsequent deprotection and sulfonylation gave compound 68. Under the influence of Mitsunobu reagents (DIAD and PPh3), compound 68 underwent an intramolecular ortho-Mitsunobu reaction, forming aziridine 69 with a yield of 90%. This total synthesis of aspirin-2a involved two Mitsunobu reactions, highlighting the widespread applicability of the Mitsunobu reaction in the synthesis of natural products. The process from compound 69 to the final product consists of ten steps, achieving an overall yield of 6.47%.
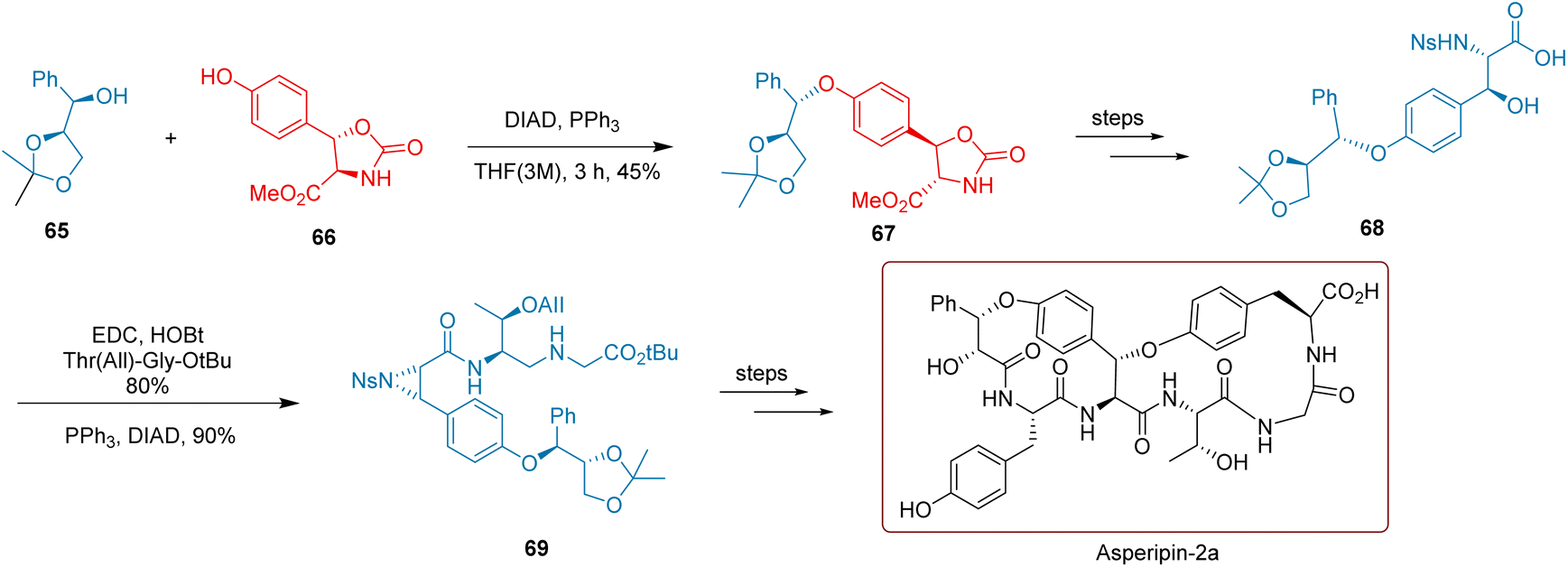 | ||
| Scheme 29 Hutton's synthesis of bicyclic hexapeptide. | ||
The discorhabdin alkaloid family is isolated from marine sponges and exhibits various bioactivities, including antibacterial,88,89 antiviral, and antitumor effects90 and potential therapeutic effects on neurodegenerative diseases.91 Due to their wide-ranging pharmacological activities and unique structures, the chemical synthesis of discorhabdin alkaloids has garnered significant interest in the synthetic community.92
As illustrated in Scheme 30, Tokuyama and co-workers synthesized compound 72 in 2021 by reacting a dihydroindole derivative with a chiral secondary alcohol 71 using Mitsunobu reagents (DIAD and PPh3). They removed the silyl-protecting groups, giving 73, and Ns-protecting groups, generating 74, to synthesize the intramolecular cyclic product discorhabdin V. There are seven steps required to transform compound 74 into the final product, resulting in an overall yield of 14%.
 | ||
| Scheme 30 Tokuyama's synthesis of discorhabdin alkaloids. | ||
Diterpenoid alkaloids are primarily isolated from the plant Aconitum septentrionale and are effective in treating pain and rheumatoid arthritis and exerting specific effects in antiarrhythmic therapy.93–95
In 2024, Liu and colleagues employed the classical sulfonylamide nucleophilic reagent in conjunction with alcohol substrates under Mitsunobu conditions to achieve the chemical synthesis of these alkaloids. As illustrated in Scheme 31, using toluene as the solvent, the naphthyl sulfonyl amide in compound 75 reacted with the intramolecular primary alcohol group in the presence of DIAD and PPh3, resulting in the construction of compound 76 with a yield of 80%. This compound is a crucial synthetic intermediate in the chemical synthesis of diterpenoid alkaloids. The conversion of compound 76 to the final product involves five essential steps, yielding an overall result of 22%.
 | ||
| Scheme 31 Liu's synthesis of A/E-ring fragment of C18-diterpenoid alkaloids. | ||
Manzamines are a class of bioactive compounds extracted from marine organisms. Sakai et al. first discovered them in 1986 in a marine sponge.96 These compounds exhibit various pharmacological effects, including anticancer and anti-inflammatory properties and significant effects on diseases such as hyperlipidemia and atherosclerosis.97
To synthesize manzamine C, as shown in Scheme 32, Cheng and colleagues 2024 utilized compound 77 in a Mitsunobu reaction involving DIAD and PPh3 to cyclize the molecule intramolecularly. Following purification by column chromatography, compound 78 was obtained with a yield of 53%. After removing the naphthyl sulfonyl group, intermediate 78 underwent nucleophilic substitution with an indole derivative to yield the desired manzamine C. There are two steps required to convert compound 78 into the final product, resulting in an overall yield of 70%.
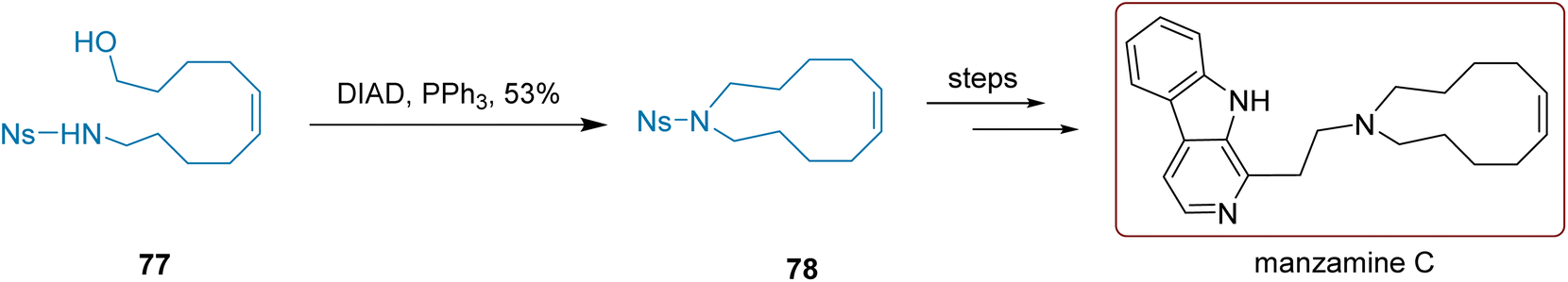 | ||
| Scheme 32 Cheng's synthesis of manzamine C. | ||
3.3 Fatty amines, aromatic amines, amides, nucleosides act as nucleophiles
Aspongopus chinensis (Pentatomidae) is primarily found in southern China. Aspongdopamine B is a compound isolated from the insect Aspongopus chinensis.98,99 As a TCM, it has been widely used to treat diseases such as cancer, severe stomach pain, and indigestion.Compound 81 is a critical intermediate in the design and synthesis route of aspongdopamine B. As illustrated in Scheme 33, Cheng and co-workers 2020 reacted the secondary alcohol substrate 79 with a purine derivative 80 in the presence of PPh3 and DIAD, using tetrahydrofuran as the reaction solvent, at room temperature for 30 minutes. This reaction successfully transformed compound 79 into 81 with a yield of 70%. It takes six steps to go from compound 81 to the final product, with an overall yield of 23.32%.
 | ||
| Scheme 33 Cheng's synthesis of aspongdopamines. | ||
In 2000, Kashman and co-workers successfully isolated and extracted polycitone A from marine ascidians, which exhibits potent antiviral effects.100,101 The main challenge in the chemical synthesis of polycitone A lies in connecting the para-hydroxyphenylethyl group to the pyrrole nitrogen atom within the molecule.
After, Steglich and co-workers ingeniously designed a one-step Mitsunobu reaction. They treated the pyrrole derivative 82 with 2-(4-acetoxyphenyl)ethanol 83, PPh3 (4 equiv.), and DEAD (4 equiv.), refluxed in tetrahydrofuran for 2 hours, purified by column chromatography, and finally obtained 84 with a yield of 64%. By removing the acetyl group using hydrazine hydrate, polycitone A was obtained with an 88% yield, as depicted in Scheme 34. The transformation from compound 84 to the final product occurs in a single step and delivers an impressive overall yield of 88%.
 | ||
| Scheme 34 Steglich's synthesis of marine pyrrole alkaloids. | ||
Lentiginosine possesses a trans-1,2-dihydroxyindolizidine skeleton and has been shown to induce apoptosis in tumor cells.102 Mechanistic studies suggest that this effect may be attributed to its ability to inhibit glycosidases, making it a promising candidate for new anticancer drugs.103
Bischoff and co-workers in 2007 employed a pyridine derivative 85 containing glycerol to synthesize such molecules. They first successfully constructed the C–N bond through an intramolecular Mitsunobu reaction, achieving the cyclized product 86 with a yield of 92%. Subsequently, they synthesized (−)-lentiginosine through reduction and polar inversion methods. The results are illustrated in Scheme 35. To reach the final product from compound 86, two steps are needed, resulting in an overall yield of 90.25%.
 | ||
| Scheme 35 Bischoff's synthesis of lentiginosine. | ||
The oriodin alkaloids are primarily isolated from marine sponges belonging to the orders Agelasida, Axinellida, and Halichondrida.104,105 They have demonstrated anti-inflammatory effects in a zebrafish inflammation model.106 Due to their complex structures and broad biological activities, these alkaloids continue to attract synthetic chemists' attention.107,108
As illustrated in Scheme 36, Lovely and co-workers conducted a Mitsunobu reaction in 2009 using tetrahydrofuran as the solvent. In this reaction, a dimethylamine 88 was successfully coupled with an imidazole substrate 87 containing primary alcohol facilitated by PPh3 and DIAD, forming compound 89 with an 80% yield. Further transformations of this compound led to the target molecule nagelamide D. It takes three definitive steps to convert compound 89 into the final product, achieving an overall yield of 22.70%.
 | ||
| Scheme 36 Lovely' s synthesis of nagelamide D. | ||
Tetrodotoxin (TTX) is the main toxic component responsible for pufferfish poisoning and is one of the most famous marine natural products.109 In toxic marine organisms, tetrodotoxin is believed to be produced by marine bacteria and accumulates through the food chain.110,111 Cep-212 and Cep-210 are crucial intermediates in the biosynthesis of tetrodotoxin.
To synthesize these two intermediates, Nishikawa and co-workers 2018 utilized Boc-protected urea derivatives 90 and 92 in separate intramolecular cyclization steps under the influence of Mitsunobu reagents (DIAD and PPh3) in tetrahydrofuran as the solvent. They obtained compounds 91 and 93 with 59% and 87% yields, respectively. Subsequent deprotection strategies led to the target molecules Cep-212 and Cep-210. The results are depicted in Scheme 37. The conversion of compound 91 to the product Cep-212 occurs in six definitive steps, resulting in an overall yield of 60.15%.
 | ||
| Scheme 37 Nishikawa's synthesis of cep-212 and cep-210. | ||
Caprazamycins are a class of nucleoside antibiotics produced by Streptomyces sp.112,113 Research has shown they can potentially treat tuberculosis.114 The construction of a seven-membered ring is thermodynamically less stable than that of a six-membered ring, making synthesizing such molecules require unique strategies.
To chemically synthesize caprazamycin A and related molecules, Takemoto and co-workers 2019 employed Mitsunobu reagents (DIAD (1.5 equiv.) and PPh3) to facilitate the intramolecular formation of a C–N bond in compound 94. This reaction successfully yielded compound 95 with an 84% yield. This step provided an essential intermediate for the further synthesis of the target molecule caprazamycin A. The results are illustrated in Scheme 38. The conversion of compound 95 to the final product demands eight precise steps, achieving an overall yield of 36.46%.
 | ||
| Scheme 38 Takemoto's synthesis of liponucleoside antibiotics. | ||
Morphine and its analogs, derived from opium poppy latex, are the oldest and most extensively studied alkaloids known to date.115,116 Due to their potent neurological and immunological activities, opioid analgesics such as morphine and fentanyl are widely used in the field of medicine. They are used as powerful pain relievers in the treatment of cancer pain in the third stage. Given the extensive applications of these molecules, a series of novel morphine-like derivatives have been synthesized.
As shown in Scheme 39, Dong and co-workers 2021 utilized a pre-synthesized polyhydroxy substrate 96 to undergo a classic Mitsunobu reaction at room temperature, converting to compound 97 with a yield of 52%. This reaction established a new carbon–nitrogen bond, facilitating intramolecular cyclization and providing important synthetic intermediates for the subsequent transformation and synthesis of thebainone A. The conversion of compound 97 to the final product involves six decisive steps, achieving an overall yield of 9.02%.
 | ||
| Scheme 39 Dong's synthesis of thebainone A. | ||
Multiple structurally unique alkaloids have been discovered in sponges, exhibiting promising anti-tumor activity.117 As demonstrated in Scheme 40, Nagasawa et al. employed a one-pot synthesis approach in 2001 to construct guanidine 99 from tetrahydropyran 98. Initially, the chiral secondary alcohol substrate 98 reacted with bis-Z-methylthiopseudourea under the influence of HgCl2 and triethylamine, generating an intermediate that underwent a reaction with DEAD, facilitating the creation of a new C–N bond through a Mitsunobu reaction. Finally, by selectively deprotecting the secondary alcohol hydroxyl group in 99 and deprotecting the guanidine to remove Cbz, the left-hand portion of batzelladine F was obtained with a yield of 67%.
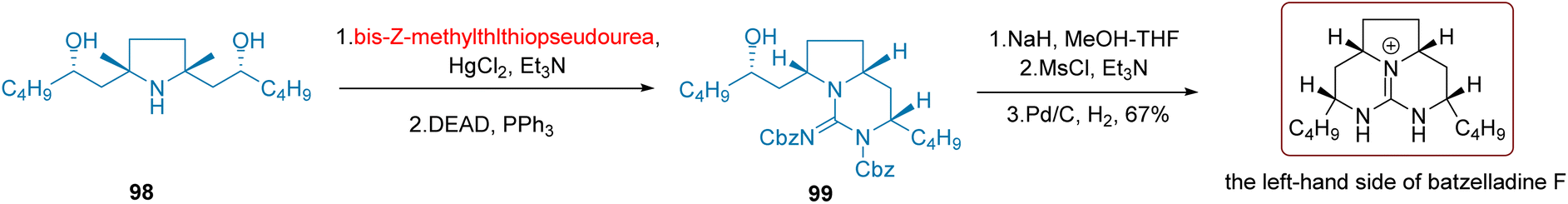 | ||
| Scheme 40 Nagasawa's synthesis of phlegmarine-type lycopodium alkaloids-the left-hand side of batzelladine F. | ||
3.4 Other nucleophiles
Batzelladine A, a type of polyguanidine marine natural product derived from the Caribbean sponge Batzella sp., displays inhibitory properties against the binding of human CD4 and HIV gp120.118,119To synthesize batzelladine A, researchers activated the pre-synthesized primary alcohol derivative 100 in toluene at room temperature using DIAD and PPh3, successfully facilitating an intramolecular cyclization reaction. The deprotected Boc group was then re-protected, yielding compound 101. This step provided an essential intermediate for synthesizing batzelladine A, as illustrated in Scheme 41. There are eight steps to transform compound 101 into the final product, yielding an overall return of 12.00%.
 | ||
| Scheme 41 Abdullah's synthesis of tricyclic guanidine alkaloids-batzelladine A. | ||
While numerous secondary metabolites with cytotoxic properties have been identified in marine invertebrates, compounds with antibacterial activity are less common. Merobatzelladines B was isolated by researchers from an extract of the sponge Monanchora sp., demonstrating antimicrobial effects against Vibrio anguillarum.120,121
As depicted in Scheme 42, Wolfe group synthesized (+)-merobatzelladine B in 2012, starting from 4-pentenal 102 and proceeding through 12 steps to yield compound 103. Subsequently, compound 103 underwent a palladium-catalyzed carbon–hydrogen reduction reaction, deprotection, and intramolecular cyclization using Mitsunobu reagents (DIAD and PPh3). After trifluoroacetic acid hydrolysis, (+)-merobatzelladine B was obtained. To reach the final product from compound 103, three steps are required, resulting in an overall yield of 41%.
 | ||
| Scheme 42 Wolfe's synthesis of merobatzelladine B. | ||
Kopsifolines are alkaloids extracted and isolated from the leaves of Malayan Kopsia species, containing several chiral centers within their molecular structure.122,123
As illustrated in Scheme 43, in 2021, Movassaghi and colleagues obtained (−)-kopsifoline E from the primary alcohol derivative 104 with a yield of 78% using tetrahydrofuran as the solvent and with the assistance of DIAD and PPh3. Furthermore, (−)-kopsifoline E can be further transformed into (−)-kopsifoline A. The Mitsunobu reaction, a classic synthetic method, is widely employed in synthesizing such alkaloids. It takes one step from kopsifoline E to kopsifoline A, with an overall yield of 73%.
 | ||
| Scheme 43 Movassaghi's synthesis of kopsifoline A. | ||
The Mitsunobu reaction is an essential tool for constructing C–N bonds and synthesizing C–O and C–S bonds. This capability provides critical strategic support that drives the advancement and success of natural product chemistry.
For example, the analogs of bactobolin A are crucial synthetic intermediates, which Sathyamoorthi et al. utilized to study and develop various analogs of bactobolin A. These analogs exhibit potential antimicrobial activity and serve as valuable tool compounds for in-depth investigations into the structure and function of bacterial ribosomes, as well as their role in antimicrobial mechanisms. With further structural optimization and bioactivity research, these analogs hold significant promise for the development of novel antibiotics.
As illustrated in Scheme 44, compound 1 undergoes a reaction with 3,5-dinitrobenzoic acid in the presence of PPh3 and DEAD during a Mitsunobu reaction. This process converts the C6 hydroxyl group of compound 105 into a 3,5-dinitrobenzoate, yielding compound 106 with a 65% yield. Subsequently, through ester hydrolysis and aza-Wacker cyclization, three additional steps were completed to synthesize more analogs of bactobolin A, resulting in a total yield of 14.09%. This demonstrates the diversity and complexity involved in synthesizing intricate organic molecules.124
 | ||
| Scheme 44 Sathyamoorthi's synthesis of analogs of bactobolin A. | ||
Xanthohumol is a powerful natural compound extracted from hops, renowned for its significant pharmacological activities, including potent antioxidant, anti-inflammatory, and anticancer effects.125,126 Given the compound's diverse biological activities, there is a compelling need for research into its synthesis to support in vivo efficacy and toxicity studies.
Erhardt et al. adeptly conducted a Mitsunobu reaction, successfully reacting the hydroxyl group of compound 107 with 3-methyl-2-buten-1-ol (108), achieving an impressive 80% yield of compound 109, as illustrated in Scheme 45. Following this, they efficiently carried out four subsequent steps, which included Claisen rearrangement, methylation, and deprotection of the MOM group, ultimately synthesizing the target compound, Xanthohumol, with a satisfactory total yield of 22.67%.
 | ||
| Scheme 45 Erhardt's synthesis of xanthohumol. | ||
Pyracurcasone exhibits powerful inhibitory effects on cancer cells by effectively targeting the BRAT1 protein. This inhibition significantly reduces cancer cell migration, proliferation, and the ability to repair DNA damage, thereby enhancing the sensitivity of cancer cells to DNA-damaging drugs.127,128 These compelling results establish pyracurcasone as a promising candidate with substantial potential for anticancer applications and a key player in developing new anticancer therapeutics.
Dai et al. successfully transformed compounds 110 and 111 into compound 112 using a Mitsunobu reaction that involved PPh3 and DEAD. They then synthesized Pyracurcasone through a meticulously executed series of seven steps, including Claisen rearrangement, addition, α-iodination, and α-methylation, resulting in an impressive total yield of 1.16%, as demonstrated in Scheme 46.
 | ||
| Scheme 46 Dai's synthesis of pyracurcasone. | ||
Phenylpropanoid monoglycerides display a range of pharmacological activities, such as antioxidant properties and anti-proliferative effects against tumor cells.129,130 As illustrated in Scheme 47, Ubukata et al. successfully synthesized compound 115 with a yield of 90% using a Mitsunobu reaction. They subsequently produced the final product, phenylpropanoid monoglycerides, with an impressive yield of 95% in a single step.
 | ||
| Scheme 47 Ubukata's synthesis of Phenylpropanoid Monoglycerides. | ||
Preussochromone A is a remarkable natural product isolated from fungi, distinguished by its unique tricyclic structure and notable cytotoxic activity. Its effectiveness against HeLa cells and another cell line clearly indicates substantial pharmacological potential, making further investigation into its applications in drug development essential.131,132
In a well-executed series of experiments, As illustrated in Scheme 48, Koert et al. converted compound 116 to compound 117, achieving an impressive yield of 71% through a Mitsunobu reaction employing DIAD and PPh3. They then synthesized Preussochromone A over 11 meticulously planned steps, including both addition and cyclization, ultimately securing an overall yield of 2.64%.
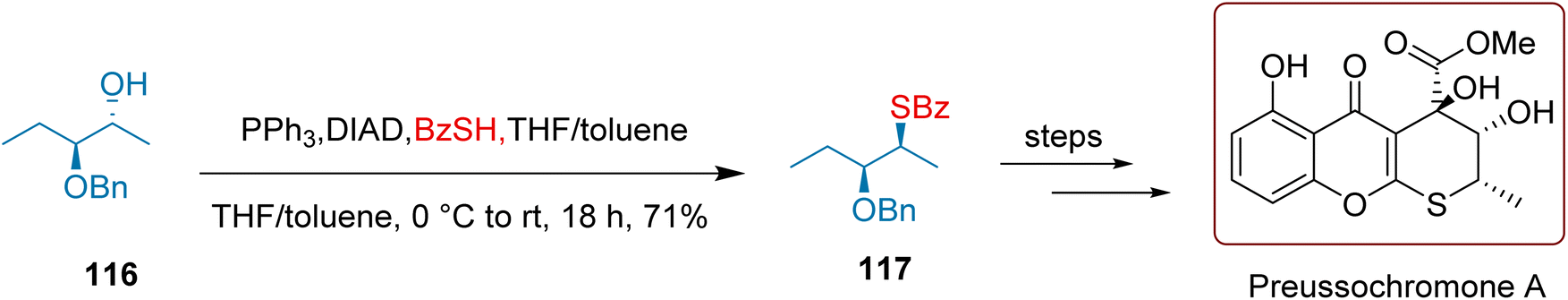 | ||
| Scheme 48 Koert's synthesis of Preussochromone A. | ||
In summary, nitrogen-containing compounds are essential in medicinal chemistry because they are critical in drug design and synthesis. Nitrogen atoms impart unique chemical properties and biological activities to these compounds, making them integral components of many pharmaceuticals. Consequently, various synthetic methodologies aim to incorporate nitrogen atoms into these molecular structures,133–136 with the Mitsunobu reaction being a noteworthy synthetic method that plays a crucial role in constructing C–N bond connections in nitrogen-containing compounds.
4. Conclusion
The Mitsunobu reaction is essential for introducing nitrogen-containing groups into chiral molecular frameworks, making it a powerful tool for designing specific chiral centers during the reaction process. This capability allows researchers to efficiently synthesize a variety of nitrogen-containing compounds with chiral centers, including amino alcohols, amino phenols, and amino acid derivatives. These compounds are critical in chiral drug design and discovery, firmly establishing the Mitsunobu reaction as a key method for constructing C–N bond connections, which supports groundbreaking advancements in medicinal chemistry—particularly in the total synthesis of natural products and the development of novel pharmaceuticals.Moreover, the Mitsunobu reaction presents several compelling advantages, including smooth inversion of configuration, short reaction times, mild conditions, and straightforward separation and purification techniques. Researchers have effectively utilized a diverse array of pronucleophiles, such as azides, sulfonamides, amines, and imides, to construct valuable chiral natural products, facilitating the formation of desired C–N bonds. However, it is crucial to acknowledge some limitations inherent in this method. For instance, the Mitsunobu reaction often struggles to achieve the desired yields in large-scale syntheses. Additionally, the reliance on stoichiometric amounts of oxidation and reducing reagents can be costly and cumbersome to manage during standard work-ups, which raises important concerns about the reaction's atom economy.
Significant progress has been made to overcome these challenges, with the development of a catalytic Mitsunobu reaction being hailed as the “Holy Grail” of Mitsunobu chemistry. Electroreduction is emerging as a promising pathway for realizing the catalytic cycle of organophosphorus compounds under greener and milder reaction conditions, effectively substituting chemical energy with electrical energy. These challenges point to both the obstacles that need to be addressed and the abundant opportunities for future advancements within the field. Ultimately, the Mitsunobu reaction's vital contributions to constructing C–N bonds, combined with its inherent advantages, unequivocally enhance its significance in medicinal chemistry and pharmaceutical innovation.
This review provides valuable insights into the Mitsunobu reaction and its applications. It offers an overview of the reaction's role in synthesizing nitrogen-containing heterocyclic compounds, referencing the work of Yurovskaya et al. (2008) and placing it in historical context with Professor Pavan Kumar's review from 1967 to 2009. Additionally, contemporary relevance is discussed, along with the improved Mitsunobu reagents identified by Panday and the importance of the reaction in natural product synthesis, as noted by Professor Mojzych. Advancements in catalytic Mitsunobu reactions presented by the Cai group are also highlighted. By synthesizing these contributions, the review demonstrates the versatility and significance of the Mitsunobu reaction in synthetic chemistry, particularly in constructing C–N bonds in chiral compounds. Covering studies from 1978 to 2024, this review is a valuable resource for researchers aiming to utilize the Mitsunobu reaction, offering insights into its history, current applications, and future directions.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.Author contributions
Xue Zhou: literature collection, formal analysis, writing-original draft; Liang Xu: literature collection, formal analysis; Zhanhui Ma: formal analysis; Jin Cui: formal analysis; Bin Wang: project administration, writing – reviewing, editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the Natural Science Foundation of Anhui province (2008085MH271), the Xin'an Medical Research Team for the Prevention and Treatment of Bi Syndrome(2024zyky05), the Key Program of the Anhui Provincial Department of Education (2023AH050831), the Open Fund of High-level Key Discipline of Chemistry of Chinese Medicine and Basic Theory of TCM of the State Administration of Traditional Chinese Medicine (ZYJCLLYB-06, HKDCCM2024015), and the Talent project of Anhui University of Chinese Medicine (2019rczd002).References
- J. Gal, Nat. Chem., 2017, 9, 604–605 CrossRef CAS PubMed.
- K. Qu, J. Li, Z. Lai and K. Chen, Univ. Chem., 2024, 39, 369–378 Search PubMed.
- A. Calcaterra and I. D. Acquarica, J. Pharm. Biomed. Anal., 2018, 147, 323–340 CrossRef CAS PubMed.
- R. Tamatam and D. Shin, Pharmaceuticals, 2023, 16, 339 CrossRef CAS PubMed.
- K. Qu, J. Li, Z. Lai and K. Chen, Univ. Chem., 2024, 39, 369–378 Search PubMed.
- R. Tamatam and D. Shin, Pharmaceuticals, 2023, 16, 339 CrossRef CAS PubMed.
- M. Jan, A. S. Sperling and B. L. Ebert, Nat. Rev. Clin. Oncol., 2021, 18, 401–417 CrossRef PubMed.
- N. Senkuttuvan, B. Komarasamy, R. Krishnamoorthy, S. Sarkar, S. Dhanasekarand and P. Anaikutti, RSC Adv., 2024, 14, 33429–33448 RSC.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC.
- B. K. Hordern, Chem. Soc. Rev., 2010, 39, 4466–4503 RSC.
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. Pavan Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef PubMed.
- S. Munawar, A. F. Zahoor, S. Ali, S. Javed, M. Irfan, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2022, 27, 6953 CrossRef CAS PubMed.
- K. Zhang, Y. Chen, L. Song and L. Cai, Asian J. Org. Chem., 2023, 12, e202200707 CrossRef CAS.
- S. K. Panday, Mini-Rev. Org. Chem., 2019, 16, 127–140 CrossRef CAS.
- N. E. Golantsov, A. V. Karchava and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2008, 44, 263–294 CrossRef CAS.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC.
- R. H. Beddoe, H. F. Sneddon and R. M. Denton, Org. Biomol. Chem., 2018, 16, 7774–7781 RSC.
- X. Wang, F. Yang, Z. Xue and X. Wang, Chin. J. Org. Chem., 2015, 35, 29–38 CrossRef CAS.
- T. Tanaka, N. Kogure, M. Kitajima and H. Takayama, J. Org. Chem., 2009, 74, 8675–8680 CrossRef CAS PubMed.
- W. Li, H. H. Zhu, X. Shen, J. L. Tan, Q. Tang, Z. P. Ling, H. Y. Zhao, Q. Lin, H. Sun, H. P. Zhang, Y. L. Li, G. C. Wang and Y. B. Zhang, Chem. Biodivers., 2023, 20, e202301024 CrossRef CAS PubMed.
- Z. J. Zhang, S. Jiang and Q. S. Zhao, Chem. Biodivers., 2024, 21, e202400954 CrossRef CAS PubMed.
- L. E. Overman and D. V. Paone, J. Am. Chem. Soc., 2001, 123, 9465–9467 CrossRef CAS PubMed.
- M. Frederich, M. Tits and L. Angenot, Trans. R. Soc. Trop. Med. Hyg., 2008, 102, 11–19 CrossRef CAS PubMed.
- F. Cohen and L. E. Overman, J. Am. Chem. Soc., 2006, 128, 2594–2603 CrossRef CAS PubMed.
- A. D. Patil, A. J. Freyer, P. B. Taylor, B. Carte, G. Zuber, R. K. Johnson and D. J. Faulkner, J. Org. Chem., 1997, 62, 1814–1819 CrossRef CAS.
- F. Cohen and L. E. Overman, J. Am. Chem. Soc., 2006, 128, 2604–2608 CrossRef CAS PubMed.
- V. V. Vintonyak and M. E. Maier, Angew. Chem., Int. Ed., 2007, 46, 5209–5211 CrossRef CAS PubMed.
- L. Yet, Chem. Rev., 2003, 103, 4283–4306 CrossRef CAS PubMed.
- M. R. Boyd, C. Farina, P. Belfiore, S. Gagliardi, J. W. Kim, Y. Hayakawa, J. A. Beutler, T. C. McKee, B. J. Bowman and E. J. Bowman, J. Pharmacol. Exp. Ther., 2001, 297, 114–120 CrossRef CAS PubMed.
- J. E. Green, D. M. Bender, S. Jackson, M. J. O. Donnell and J. R. McCarthy, Org. Lett., 2009, 11, 807–810 CrossRef CAS PubMed.
- S. Chandrasekhar, G. S. K. Babu and C. R. Reddy, Tetrahedron Asymmetry, 2009, 20, 2216–2219 CrossRef CAS.
- N. An, H. Zhang, L. Xu, S. Yang and Z. Zou, Food Chem., 2010, 119, 513–517 CrossRef CAS.
- L. S. Santos, Y. M. Gallardo, N. Shankaraiah and M. J. Simirgiotis, Synthesis, 2011, 1, 51–56 CrossRef.
- J. Zhang, Y. Q. Liu and J. Fang, Alkaloids - Chem. Biol., 2023, 89, 1–37 Search PubMed.
- D. Mancinotti, K. M. Frick and F. G. Flores, Nat. Prod. Rep., 2022, 39, 1423–1437 RSC.
- M. G. Banwell, N. Gao, B. D. Schwartz and L. V. White, Top. Curr. Chem., 2012, 309, 163–202 CrossRef CAS PubMed.
- D. Koutová, N. Maafi, R. Havelek, L. Opletal, G. Blunden, M. Řezáčová and L. Cahlíková, Molecules, 2020, 25, 2337 CrossRef PubMed.
- Y. Liu and H. Wang, Chem. Commun., 2019, 55, 3544–3547 RSC.
- T. Shiobara, Y. Nagumo, R. Nakajima, T. Fukuyama, S. Yokoshima and T. Usui, Biosci. Biotechnol. Biochem., 2021, 85, 92–96 CrossRef PubMed.
- P. A. Evans, J. Qin, J. E. Robinson and B. Bazin, Angew. Chem., Int. Ed., 2007, 46, 7417–7419 CrossRef CAS PubMed.
- L. T. S. Domingos, D. C. Moraes, M. F. C. Santos, J. A. R. Curvelo, B. B. Pacheco, E. A. Marquez, A. W. B. Martinez, R. G. S. Berlinck and A. F. Pereira, Mar. Drugs, 2024, 22, 502 CrossRef CAS PubMed.
- C. Theeraladanon, M. Arisawa, M. Nakagawa and A. Nishida, Tetrahedron Asymmetry, 2005, 16, 827–831 CrossRef CAS.
- I. Mester, Fitoterapia, 1973, 44, 123–152 CAS.
- J. Tummatorn, D. Muñoz and G. B. Dudley, Tetrahedron Lett., 2013, 54, 1312–1314 CrossRef CAS.
- I. J. Collet, F. B. Vical, A. Valentin, E. Stanislas, M. Mallié and I. Fourasté, Planta Med., 2002, 68, 68–69 CrossRef PubMed.
- W. J. Moran, K. M. Goodenough, P. Raubo and J. P. A. Harrity, Org. Lett., 2003, 5, 3427–3429 CrossRef CAS PubMed.
- H. Matsuda, H. Shimoda and M. Yoshikawa, Bioorg. Med. Chem., 2001, 9, 1031–1035 CrossRef CAS PubMed.
- A. Nakayama, N. Kogure, M. Kitajima and H. Takayama, Org. Lett., 2009, 11, 5554–5557 CrossRef CAS PubMed.
- Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679–729 CrossRef CAS PubMed.
- H. Zaimoku and T. Taniguchi, Chem.–Eur. J., 2014, 20, 9613–9619 CrossRef CAS PubMed.
- B. Wang, C. Guan and Q. Fu, Phytochem. Rev., 2022, 21, 1–79 CrossRef CAS.
- G. S. Ghotekar, M. Mujahid and M. Muthukrishnan, ACS Omega, 2019, 4, 1322–1328 CrossRef CAS PubMed.
- E. Mevers, T. Matainaho, M. Allara, V. D. Marzo and W. H. Gerwick, Lipids, 2014, 49, 1127–1132 CrossRef CAS PubMed.
- Y. Liu, Z. Wang, S. Banne, J. Guo and Y. He, J. Org. Chem., 2019, 84, 5838–5845 CrossRef CAS PubMed.
- E. Iwasa, Y. Hamashima and M. Sodeoka, Isr. J. Chem., 2011, 51, 420–433 CrossRef CAS.
- W. R. Begg, J. A. Elix and A. J. Jones, Tetrahedron Lett., 1978, 19, 1047–1050 CrossRef.
- H. Kaneko, S. Takahashi, N. Kogure, M. Kitajima and H. Takayama, J. Org. Chem., 2019, 84, 5645–5654 CrossRef CAS PubMed.
- E. S. Halldorsdottir, J. W. Jaroszewski and E. S. Olafsdottir, Phytochemistry, 2010, 71, 149–157 CrossRef CAS PubMed.
- Q. Zhang, F. M. Zhang, C. S. Zhang, S. Z. Liu, J. M. Tian, S. H. Wang, X. M. Zhang and Y. Q. Tu, Nat. Commun., 2019, 10, 2507 CrossRef PubMed.
- L. L. Christrup, Acta Anaesthesiol. Scand., 1997, 41, 116–122 CrossRef CAS PubMed.
- A. Khatua, S. Niyogi and V. Bisai, Org. Biomol. Chem., 2019, 17, 7140–7143 RSC.
- A. Khatua, A. Roy and V. Bisai, Tetrahedron, 2020, 76, 130918 CrossRef CAS.
- A. Khatua, K. Shaw and V. Bisai, Tetrahedron Lett., 2020, 61, 151736 CrossRef CAS.
- I. Hayakawa, R. Nagatani, M. Ikeda, D. Yoo, K. Saito, H. Kigoshi and A. Sakakura, Org. Lett., 2019, 21, 6337–6341 CrossRef CAS PubMed.
- Y. T. Di, H. P. He, C. S. Li, J. M. Tian, S. Z. Mu, S. L. Li, S. Gao and X. J. Hao, J. Nat. Prod., 2006, 69, 1745–1748 CrossRef CAS PubMed.
- L. D. Guo, Y. Zhang, J. Hu, C. Ning, H. Fu, Y. Chen and J. Xu, Nat. Commun., 2020, 11, 3538 CrossRef CAS PubMed.
- M. M. Pompeo, J. H. Cheah and M. Movassaghi, J. Am. Chem. Soc., 2019, 141, 14411–14420 CrossRef CAS PubMed.
- B. M. Trost and M. Osipov, Chem.–Eur. J., 2015, 21, 16318–16343 CrossRef CAS PubMed.
- J. Zhang and F. S. Han, J. Org. Chem., 2019, 84, 13890–13896 CrossRef CAS PubMed.
- C. Y. Gan, Y. Y. Low, N. F. Thomas and T. S. Kam, J. Nat. Prod., 2013, 76, 957–964 CrossRef CAS PubMed.
- A. E. Nugroho, Y. Hirasawa, W. C. Piow, T. Kaneda, A. H. A. Hadi, O. Shirota, W. Ekasari, A. Widyawaruyanti and H. Morita, J. Nat. Med., 2012, 66, 350–353 CrossRef CAS PubMed.
- B. S. Kumar and S. Raghavan, Synlett, 2019, 30, 2157–2160 CrossRef CAS.
- S. P. Yang and J. M. Yue, J. Org. Chem., 2003, 68, 7961–7966 CrossRef CAS PubMed.
- J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936–962 RSC.
- C. L. Hugelshofer, V. Palani and R. Sarpong, J. Org. Chem., 2019, 84, 14069–14091 CrossRef CAS PubMed.
- Y. G. Zhou, H. N. C. Wong and X. S. Peng, J. Org. Chem., 2019, 85, 967–976 CrossRef PubMed.
- B. M. Anne, C. D. Bhupesh and P. Pierre, Phytochemistry, 1980, 19, 1473–1475 CrossRef.
- T. S. Kam and Y. M. Choo, Helv. Chim. Acta, 2004, 87, 991–998 CrossRef CAS.
- Y. Yamashita, L. Poignant, J. Sakata and H. Tokuyama, Org. Lett., 2020, 22, 6239–6243 CrossRef CAS PubMed.
- E. A. Guzman, J. D. Johnson, M. K. Carrier, C. I. Meyer, T. P. Pitts, S. P. Gunasekera and A. E. Wright, Anti-Cancer Drugs, 2009, 20, 149–155 CrossRef CAS PubMed.
- C. Han, Y. Chen, Y. C. Ching, C. S. Lee and S. He, Org. Chem. Front., 2020, 7, 2243–2246 RSC.
- X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752–772 RSC.
- F. W. Zhao, Q. Y. Sun, F. M. Yang, G. W. Hu, J. F. Luo, G. H. Tang, Y. H. Wang and C. L. Long, Org. Lett., 2010, 12, 3922–3925 CrossRef CAS PubMed.
- X. Zeng, Z. Jia and F. G. Qiu, Tetrahedron Lett., 2020, 61, 152329 CrossRef CAS.
- Z. T. Deng, Y. C. Liu, Q. F. Zhu, S. Jiang, X. D. Wu and Q. S. Zhao, Chem. Biodivers., 2022, 19, e202200454 CrossRef CAS PubMed.
- S. Shabani, J. M. White and C. A. Hutton, Org. Lett., 2020, 22, 7730–7734 CrossRef CAS PubMed.
- N. Nagano, M. Umemura, M. Izumikawa, J. Kawano, T. Ishii, M. Kikuchi, K. Tomii, T. a. Kumagai, A. Yoshimi, M. Machida, K. Abe, K. Shin-ya and K. Asai, Fungal Genet. Biol., 2016, 86, 58–70 CrossRef CAS PubMed.
- T. Noro, J. Sakata and H. Tokuyama, Tetrahedron Lett., 2021, 81, 153333 CrossRef CAS.
- M. Na, Y. Ding, B. Wang, B. L. Tekwani, R. F. Schinazi, S. Franzblau, M. Kelly, R. Stone, X. C. Li, D. Ferreira and M. T. Hamann, Nat. Prod., 2010, 73, 383–387 CrossRef CAS PubMed.
- E. M. Harris, J. D. Strope, S. L. Beedie, P. A. Huang, A. K. L. Goey, K. M. Cook, C. J. Schofield, C. H. Chau, M. M. Cadelis, B. R. Copp, K. R. Gustafson and W. D. Figg, Mar. Drugs, 2018, 16, 241 CrossRef PubMed.
- T. Botic, A. Defant, P. Zanini, M. C. Zuzek, R. Frangez, D. Janussen, D. Kersken, Z. Knez, I. Mancini and K. Sepci, Eur. J. Med. Chem., 2017, 136, 294–304 CrossRef CAS PubMed.
- Y. Wada, H. Fujioka and Y. Kita, Mar. Drugs, 2010, 8, 1394–1416 CrossRef CAS PubMed.
- F. Lu, Y. Shao, S. Yan, D. Yang, H. Song, D. Zhang, X. Y. Liu and Y. Qin, J. Org. Chem., 2024, 89, 2807–2811 CrossRef CAS PubMed.
- A. Salehi, M. Ghanadian, B. Zolfaghari, A. R. Jassbi, M. Fattahian, P. Reisi, D. Csupor, I. A. Khan and Z. Ali, Pharmaceuticals, 2023, 16, 747 CrossRef CAS PubMed.
- F. P. Wang, Q. H. Chen and X. T. Liang, Alkaloids Chem. Biol., 2009, 67, 1–78 CAS.
- C. Zhang, S. Liu, Q. Xiong, L. Li and B. Cheng, J. Org. Chem., 2024, 89, 2064–2067 CrossRef CAS PubMed.
- A. M. S. Mayer, M. L. Hall, J. Lach, J. Clifford, K. Chandrasena, C. Canton, M. Kontoyianni, Y. Mun Choo, D. Karan and M. T. Hamann, Mar. Drugs, 2021, 19, 506 CrossRef CAS PubMed.
- W. Y. Ding, Y. M. Yan, X. H. Meng, La. A. Nafie, T. Xu, R. K. Dukor, H. B. Qin and Y. X. Cheng, Org. Lett., 2020, 22, 5726–5730 CrossRef CAS PubMed.
- Y. N. Shi, Z. C. Tu, X. L. Wang, Y. M. Yan, P. Fang, Z. L. Zuo, B. Hou, T. H. Yang and Y. X. Cheng, Bioorg. Med. Chem. Lett., 2014, 24, 5164–5169 CrossRef CAS PubMed.
- A. T. Kreipl, C. Reid and W. Steglich, Org. Lett., 2002, 4, 3287–3288 CrossRef CAS PubMed.
- R. Azzouz, C. Fruit, L. Bischoff and F. Marsais, J. Org. Chem., 2008, 73, 1154–1157 CrossRef CAS PubMed.
- F. M. Cordero, D. Giomi and A. Brandi, Curr. Top. Med. Chem., 2014, 14, 1294–1307 CrossRef CAS PubMed.
- A. Minutolo, S. Grelli, F. M. Merlo, F. M Cordero, A. Brandi, B. Macchi and A. Mastino, Cell Death Dis., 2012, 3, e358 CrossRef CAS PubMed.
- M. R. Bhandari, R. Sivappa and C. J. Lovely, Org. Lett., 2009, 11, 1535–1538 CrossRef CAS PubMed.
- H. Hoffmann and T. Lindel, Synthesis, 2003, 1753–1783 CAS.
- X. Liu, Q. Wang, Y. Zhang and H. Zhang, Mar. Drugs, 2024, 22, 477 CrossRef CAS PubMed.
- M. R. Bhandari, A. Herath, S. Rasapalli, M. Yousufuddin and C. J. Lovely, J. Org. Chem., 2020, 85, 12971–12987 CrossRef CAS PubMed.
- A. Araki, M. Tsuda, T. Kubota, Y. Mikami, J. Fromont and J. Kobayashi, Org. Lett., 2007, 9, 2369–2371 CrossRef CAS PubMed.
- M. Adachi, T. Miyasaka, Y. Kudo, K. Sugimoto, M. Y. Yamashita and T. Nishikawa, Org. Lett., 2018, 21, 780–784 CrossRef PubMed.
- M. Kono, T. Matsui, K. Furukawa, M. Y. Yamashita and K. Yamamori, Toxicon, 2008, 51, 1269–1273 CrossRef CAS PubMed.
- T. Yasumoto, D. Yasumura, M. Yotsu, T. Michishita, A. Endo and Y. Kotaki, Agric. Biol. Chem., 1986, 50, 793–795 CAS.
- H. Nakamura, C. Tsukano, T. Yoshida, M. Yasui, S. Yokouchi, Y. Kobayashi, M. Igarashi and Y. Takemoto, J. Am. Chem. Soc., 2019, 141, 8527–8540 CrossRef CAS PubMed.
- T. D. H. Bugg and R. V. Kerr, J. Antibiot., 2019, 72, 865–876 CrossRef CAS PubMed.
- S. Hirano, S. Ichikawa and A. Matsuda, Bioorg. Med. Chem., 2008, 16, 5123–5133 CrossRef CAS PubMed.
- Y. Wang, A. Hennig, T. Küttler, C. Hahn, A. Jäger and P. Metz, Org. Lett., 2020, 22, 3145–3148 CrossRef CAS PubMed.
- S. H. Hou, A. Y. Prichina and G. Dong, Angew. Chem., Int. Ed., 2021, 60, 13057–13064 CrossRef CAS PubMed.
- K. Nagasawa, H. Koshino and T. Nakata, Tetrahedron Lett., 2001, 42, 4155–4158 CrossRef CAS.
- N. Z. A. Rani, Y. K. Lee, S. Ahmad, R. Meesala and I. Abdullah, Mar. Drugs, 2022, 20, 579 CrossRef PubMed.
- S. G. Duron and D. Y. Gin, Org. Lett., 2001, 3, 1551–1554 CrossRef CAS PubMed.
- N. R. Babij and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 4128–4130 CrossRef CAS PubMed.
- S. Takishima, A. Ishiyama, M. Iwatsuki, K. Otoguro, H. Yamada, S. Omura, H. Kobayashi, R. W. M. Soest and S. Matsunaga, Org. Lett., 2009, 11, 2655–2658 CrossRef CAS PubMed.
- I. S. Myeong, N. H. Avci and M. Movassaghi, Org. Lett., 2021, 23, 9118–9122 CrossRef CAS PubMed.
- T. S. Kam and Y. M. Choo, Phytochemistry, 2004, 65, 2119–2122 CrossRef CAS PubMed.
- S. Nagamalla, D. K. Johnson and S. Sathyamoorthi, Med. Chem. Res., 2021, 30, 1348–1357 CrossRef CAS PubMed.
- R. S. Khupse and P. W. Erhardt, J. Nat. Prod., 2007, 70, 1507–1509 CrossRef CAS PubMed.
- J. F. Stevens and J. E. Page, Phytochemistry, 2004, 65, 1317–1330 CrossRef CAS PubMed.
- C. Cui, B. G. Dwyer, C. Liu, D. Abegg, Z. J. Cai, D. G. Hoch, X. Yin, N. Qiu, J. Q. Liu, A. Adibekian and M. Dai, J. Am. Chem. Soc., 2021, 143(11), 4379–4386 CrossRef CAS PubMed.
- J. Q. Liu, Y. F. Yang, X. Y. Li, E. Q. Liu, Z. R. Li, L. Zhou, Y. Li and M. H. Qiu, Phytochemistry, 2013, 96, 265–272 CrossRef CAS PubMed.
- D. I. Batovska, T. Kishimoto, V. S. Bankova, Z. G. Kamenarska and M. Ubukata, Molecules, 2005, 10, 552–558 CrossRef CAS PubMed.
- A. H. Banskota, T. Nagaoka, L. Y. Sumioka, Y. Tezuka, S. Awale, K. Midorikawa, K. Matsushige and S. Kadota, J. Ethnopharmacol., 2002, 80, 67–73 CrossRef CAS PubMed.
- J. Gao, P. Rao, K. Xu, S. Wang, Y. Wu, C. He and H. Ding, Total Synthesis of (−)-Preussochromone A, J. Org. Lett., 2020, 22, 6127–6631 CrossRef PubMed.
- F. Zhang, L. Li, S. Niu, Y. Si, L. Guo, X. Jiang and Y. Che, J. Nat. Prod., 2012, 75, 230–237 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, S. L. Homölle, B. Wang and L. Ackermann, Chem. Sci., 2024, 15, 1117–1122 RSC.
- L. Chen, Z. Wang, H. Liu, X. Li and B. Wang, Chem. Commun., 2022, 58, 9152–9155 RSC.
- B. Wang, Z. Yan, L. Liu, J. Wang, Z. Zha and Z. Wang, Green Chem., 2019, 21, 205–212 RSC.
- X. Li, Y. Fang, Y. Zhao, S. Luo, Y. Xue, T. Yong and B. Wang, Chem. Commun., 2024, 60, 13352–13355 RSC.
| This journal is © The Royal Society of Chemistry 2025 |