
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication and in vitro cytocompatibility evaluation of porous bone scaffold based on cuttlefish bone-derived nano-carbonated hydroxyapatite reinforced with polyethylene oxide/chitosan fibrous structure
Musyafa Riziq Habiburrohmana,
Muhammad Amir Jamilludin a,
Nilam Cahyatia,
Nendar Herdiantob and
Yusril Yusuf*a
a,
Nilam Cahyatia,
Nendar Herdiantob and
Yusril Yusuf*a
aDepartment of Physics, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia. E-mail: yusril@ugm.ac.id
bResearch Centre for Advanced Material, National Research and Innovation Agency (BRIN), South Tangerang 15314, Indonesia
First published on 17th February 2025
Abstract
A novel porous bone scaffold based on nano-carbonated hydroxyapatite reinforced with fibrous-like structured polyethylene oxide/chitosan network (nCHA/PEO/CS) was introduced and fabricated via freeze-drying. Prior to this, the nCHA was synthesized through a hydrothermal reaction based on cuttlefish bone (CFB, Sepia officinalis). The raw cuttlefish bone (raw-CFB) was first decomposed to obtain cuttlefish bone-derived calcium oxide (CaO-CFB) by calcination at 1000 °C, which was used for synthesizing nCHA. The chemical composition analysis showed that the nCHA formed AB-type CHA with a high carbonate content of 7.38 wt%, which is in the range of carbonate content in native bone (2–9 wt%). The Ca/P molar ratio of nCHA was 1.712, very close to the Ca/P of biological apatite of 1.71. Morphological analysis revealed that nCHA consists of nanosized particles, potentially offering a large surface area to volume to promote ion exchange and cell interaction. The excellent physicochemical and morphological properties of nCHA proposed suitability as a bone scaffold precursor combined with PEO and CS. The nCHA/PEO/CS scaffolds were freeze-dried with varying PEO/CS concentrations. Physicochemical analysis indicated that increasing the PEO/CS concentration decreased the crystallinity of the scaffold, causing it to be lower than the nCHA crystallinity, which may be beneficial for cell growth. Morphological analysis revealed that the scaffold structure comprised nCHA cross-linked within a fibrous-like structured PEO/CS network, which appropriately mimics the fibrous structure of extracellular matrix (ECM) in natural bone. However, the nCHA/PEO/CS-11 scaffold formed more appropriate pores with suitable porosity for cell development, blood vessel formation, and nutrient perfusion. The nCHA/PEO/CS-11 scaffold also demonstrated sufficient compressive strength and good swelling behavior, which may favor bone regeneration. The nCHA/PEO/CS-11 scaffold demonstrated high cytocompatibility and facilitated the adherence of MC3T3E1 cells on the scaffold surface. The nCHA/PEO/CS-11 scaffold also promoted cell osteogenic differentiation. Owing to its desirable and suitable characteristics, the nCHA/PEO/CS-11 scaffold is promising in bone tissue engineering.
1. Introduction
The world's median age is growing due to diminished fertility and increased life expectancy.1 The aging of the global population raises the incidence of osteoporosis and related fragility fractures, considerably influencing patient quality of life.2,3 Unfortunately, massive bone defects remain a challenge to be treated. Such autografts or xenografts have been widely considered the gold standard for treating bone defects.4 However, autografts caused several problems, such as inadequate availability, donor site morbidity, and prolonged surgery, leading to failure of bone replacement.5 Furthermore, xenografts can lead to complications, including displacement of the graft materials, foreign body reactions, chronic inflammations, soft-tissue fenestrations, and associated cysts.6 Thus, synthetic materials are required to overcome commercial bone graft drawbacks.Bone consists of an extracellular matrix (ECM) with a fibrous structure arising from the interaction between type-1 collagen and apatite mineral.7 Hence, synthetic materials should be reconstructed into a fibrous ECM-like structure with high porosity and surface area.8,9 Notably, since ECM in bone possesses multiscale pores, synthetic materials must form a three-dimensional (3D) scaffold that possesses macropores to provide a pathway for cell infiltration into the scaffold and micropores to create surface roughness for governing cell adhesion, proliferation, and differentiation, and enhancing protein adsorption, thereby promoting osteogenesis.10 Thus, the fibrous-like and porous structured scaffold, mimicking bone ECM, is desired for favorable regeneration of bone defects.
Various methods, including freeze-drying,11 porogen leaching,12 3D printing,13 and gas foaming,14 have been frequently used to fabricate a 3D scaffold. However, the freeze-drying method is preferable because it can produce a highly porous scaffold with interconnected pores formed by the sublimation of crystallized solvent that leaves a compact structure. The freeze-drying method involves a lyophilization process at a low temperature under vacuum conditions, which prevents contamination and carbon reaction.15 The freeze-dried scaffold also exhibits a unique pore structure, such as a fibrous-like, open-cell, and lamellar structure. These pore structures can mimic the native ECM structure that provides suitable curvatures for osteoblastic cell adhesion and migration, thereby enhancing osteogenesis and angiogenesis.16 Thus, the freeze-drying method is promising for developing a scaffold with suitable porous structures for bone regeneration.
In bone tissue engineering, synthetic polymers are commonly used as matrices or reinforcers when fabricating porous scaffolds.12,13,16,17 Polyethylene oxide (PEO) is one of the synthetic polymers clinically used in biomedical applications due to its biocompatibility and hydrophilicity.18 PEO has also been proven for its controllable biomechanical and biodegradable properties.19 PEO-based scaffolds can form a fibrous-like structure biomimetic to the native bone ECM.20 Since natural bone comprises collagenous ECM, using PEO alone as a scaffold base causes a restriction in bone repair. Hence, the scaffold must have a chemical structure similar to collagen in bone. However, shortcomings of collagen, including poor mechanical properties, high degradability, and lack of osteoconductivity, have been the limitations in clinical applications.21 To resolve the lack of type-1 collagen, chitosan (CS) is the best candidate due to its biocompatible, biodegradable, and osteoconductive properties with almost all tissue in the body.22,23 The combination of PEO and CS will produce a scaffold with controllable biodegradability, high mechanical strength, and suitable structure to promote bone tissue formation. However, bone comprises 60% apatite mineral, a natural calcium phosphate. Therefore, scaffold reconstruction using calcium phosphate-type materials, while reinforced with PEO/CS to enhance the scaffold integrity, is a necessity for favorable regeneration of bone tissue.24
Hydroxyapatite (HA; Ca10(PO4)6(OH)2) is a calcium phosphate that has been commonly employed along with polymeric composite due to its biocompatibility and osteoinductivity.25–27 The brittleness of HA can increase the biodegradability of polymer-based composites.28 Despite the osteogenic and osteoconductive properties of HA, its high stability has been a drawback due to its slow material resorption, which prevents synchronizing with bone ingrowth rate.29,30 Since biological bone apatite consists of 2–9 wt% carbonate content,31 carbonate content has been a prominent factor in the solubility of apatite under biological conditions.32,33 The higher content of carbonate ions in the apatite crystal causes a higher solubility, which leads to a higher resorption rate.34 Carbonated hydroxyapatite (CHA; Ca10−a(PO4)6−b(CO3)c(OH)2−d)) is another calcium phosphate that consists of high carbonate content in its apatite crystal, thereby demonstrating a high material resorption for a faster bone formation.35–37 Hence, CHA is preferably used due to its efficacy in bone regeneration.
Biogenic-derived calcium (Ca) sources have been quietly used in synthesizing CHA. Biogenic materials contain low concentrations of essential trace elements, which can enhance the properties of CHA. In this study, cuttlefish bone (CFB, Sepia officinalis) is used to synthesize CHA. CFB consists of aragonite, a polymorph of calcium carbonate (CaCO3) crystal, thus highly comprising Ca.38 CFB also lowly contains magnesium (Mg), sodium (Na), and strontium (Sr), which, when turned into CHA, have a significant effect on the bone healing process.38,39 Apart from its economic effectiveness, ecological friendliness, and wide availability, CHA based on CFB has been reported for its biocompatibility.40
Several methods for synthesizing CHA, including hydrothermal,41 co-precipitation,42 sol–gel,43 nano-emulsion,44 and mechanochemical methods,45 have been extensively studied. However, the hydrothermal method is a simple method that can produce nanosize-shaped CHA particles. This hydrothermal method involves a hydrothermally aging treatment at high temperatures, typically beyond the boiling point of water, within an autoclave.46 During the hydrothermal reaction, the nanocrystalline growth is highly influenced by the dissolution rate of materials, which can be controlled by adjusting the hydrothermal temperature.47,48 Therefore, in this study, the hydrothermal process is the preferred method for synthesizing CHA nanoparticles (nCHA) due to its ability to obtain an appropriate size and shape, resulting in a high surface area to volume ratio and aspect ratio of nCHA particles.49 nCHA has been reported for its superiority in osteoconduction and osteointegration processes in bone tissue repair.50,51
This study investigated the fabrication of a porous scaffold based on nCHA reinforced with a fibrous-like structured PEO/CS network for mimicking bone structure. The nCHA was synthesized using the hydrothermal method to mimic the apatite structure of bone, and CFB was used as a precursor. The nCHA/PEO/CS scaffolds were fabricated by varying the PEO/CS concentration. The effect elucidated by the PEO/CS incorporation was evaluated in terms of chemical-compositional, structural, mechanical properties, and swelling behavior. The in vitro cytocompatibility tests in terms of cell viability, adhesion, and osteogenic activity were conducted to determine the most desirable scaffold for bone regeneration.
2. Materials and methods
2.1. Materials
The CFB used as raw materials (raw-CFB) for the Ca source was purchased from the local fish market in Bandung, Indonesia. Diammonium hydrogen phosphate ((NH4)2HPO4) and ammonium bicarbonate (NH4HCO3) used as phosphate and carbonate sources, respectively, were purchased from Merck (USA). Ammonium hydroxide (NH4OH) 25% solution used for controlling pH was purchased from Merck (USA). PEO with a molecular weight of 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and CS with a medium molecular weight were purchased from Sigma-Aldrich (USA). Acetic acid 100% was purchased from Merck (Germany). Phosphate-buffered saline (PBS) was purchased from Sigma-Aldrich (USA).
000 and CS with a medium molecular weight were purchased from Sigma-Aldrich (USA). Acetic acid 100% was purchased from Merck (Germany). Phosphate-buffered saline (PBS) was purchased from Sigma-Aldrich (USA).
2.2. Methods
 | ||
| Fig. 1 Schematic methods for (a) the nCHA synthesis and (b) nCHA/PEO/CS scaffold fabrication. | ||
:CS was controlled at 7:3 (Table 1). The suspension was immediately stored in a deep freezer overnight at −40 °C before being lyophilized for 48 h at −55 °C to obtain 3D porous scaffolds (Fig. 1b). These scaffolds were characterized to assess their chemical-compositional, structural, mechanical properties, and swelling behavior.
2.3. Characterizations
 | (1) |
These compressive test procedures were repeated four times for each group (n = 4).
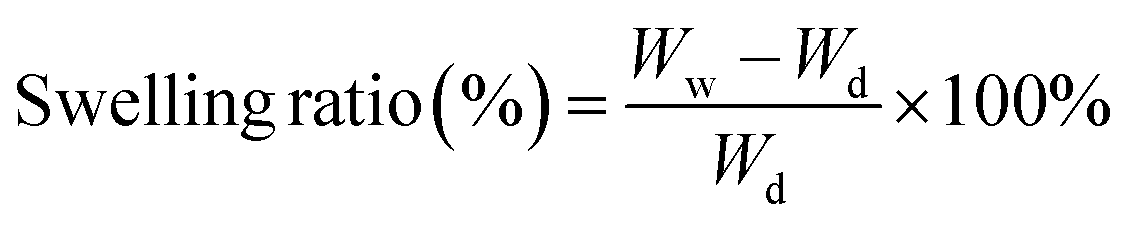 | (2) |
when the swelling ratio approached maximum, the swelling test procedures were repeated thrice for each group (n = 3).
The MTT assay was used to assess the cytocompatibility of the scaffold against MC3T3E1 cells.27 The cell viability was evaluated through the cell metabolic activity after 24 h of incubation. Briefly, the medium was gently aspirated from each well. Next, 100 μL of MTT solution (Biobasic, USA) with a concentration of 0.5 mg mL−1 was added to the well and incubated for 4 h. Then, 100 μL of dimethyl sulfoxide (DMSO, Merck, Germany) was added to each well. The absorbance at 570 nm was measured using a Tecan Spark® analyzer (Tecan, Switzerland). The cell viability was assessed by measuring the absorption value of the test cultures, expressed as a percentage of absorption for unstimulated control cultures.54 The cell viability was determined by the formula as follows in eqn (3):
 | (3) |
The half maximum inhibitory concentration (IC50) quantified the inhibitor amount required to suppress biological processes or components by 50%. Examining the IC50 determines the scaffold's safe dose to avoid cell growth inhibition. The IC50 value was analyzed using non-linear curve fitting.
3. Results and discussion
3.1. Properties of raw materials
The raw-CFB was first decomposed into CaO-CFB by calcination before being used as the Ca source in the nCHA synthesis. The heat energy received by CaCO3 polymorphic crystals in the raw CFB caused them to move faster and break their chemical bonds, causing the decomposition reaction to form CaO crystals, as follows in eqn (4). The endothermic transformation of CaCO3 to CaO phase commonly occurs at above 800 °C.55| CaCO3(s) → CaOs + CO2(g) | (4) |
The XRD patterns of the raw-CFB, CC-CFB, and CaO-CFB showed differences in the crystal structure (Fig. 2a). The XRD pattern of the raw-CFB corresponded to aragonite (PDF number 901-6527), which strongly exhibited at diffraction angles (2θ) of 26.2° and 45.8°. The CC-CFB diffraction pattern indicated peaks corresponding to calcite (PDF number 47-1743) that strongly exhibited at a 2θ of 29.4°. The unit cell volume, crystallite size, and crystallinity of the CC-CFB were higher than those of the raw-CFB (Table 2). Although the calcination at 600 °C caused a conversion of aragonite polymorphic crystal into calcite crystal, calcite has a highly stable phase, thereby unsuitable for using CC-CFB as an nCHA precursor. Contrarily, the CaO-CFB diffraction pattern showed diffraction peaks corresponding to CaO (PDF number 37-1497) with a main diffraction peak at 2θ of 37.4° and 53.8°. Although the unit cell volume of the CaO-CFB was lower than that of raw-CFB, the crystallite size and crystallinity of the CaO-CFB were higher than those of the raw-CFB (Table 2). These results suggest that at 1000 °C, the aragonite polymorphic crystal was decomposed into CaO crystal. CaO crystal is reportedly less stable than calcite crystal. Hence, CaO-CFB was more suitable as a precursor in synthesizing nCHA.
 | ||
| Fig. 2 (a) XRD patterns and (b) FTIR spectra of the raw-CFB, CC-CFB, and CaO-CFB. (c–e) Morphologies of the raw-CFB, CC-CFB, and CaO-CFB. Scale bars: 5 μm (c–e). | ||
| No. | Sample | Lattice parameter (Å) | Unit cell volume (cm3) (10−22) | Crystallite size (s ± Δs) (nm) | Microstrain (ε) (10−3) | Degree of crystallinity (%) | ||
|---|---|---|---|---|---|---|---|---|
| a = b | c | a/c | ||||||
| a ≠ b.b a = b = c. | ||||||||
| 1 | Raw-CFB | 5.752, 4.972a | 7.936 | — | 2.26 | 58.4 ± 3.41 | 1.55 ± 0.66 | 82.2 |
| 2 | CC-CFB | 4.989 | 17.08 | — | 3.68 | 82.5 ± 2.24 | 1.09 ± 0.72 | 91.8 |
| 3 | CaO-CFB | 4.813b | — | 1.11 | 70.9 ± 6.24 | 2.33 ± 0.61 | 84.6 | |
| 4 | nCHA | 9.433 | 6.892 | 0.730 | 5.31 | 56.7 ± 2.74 | 1.85 ± 0.14 | 79.9 |
The FTIR spectra of the raw-CFB, CC-CFB, and CaO-CFB showed transformations in functional groups (Fig. 2b). The raw-CFB and CC-CFB spectra displayed the main vibrational bonds of aragonite and calcite, respectively, which were the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds observed at 1784 and 1471–715 cm−1, respectively. In the CaO-CFB spectrum, the CaO bond was observed at 871 cm−1, which appeared as a consequence of a severe reduction in the C–O bond within the raw-CFB, thus suggesting that the aragonite in the raw-CFB was decomposed into CaO and released carbon dioxide (CO2) by calcination at 1000 °C. Although, in the CaO-CFB spectrum, the O–H stretching band appeared at 3637 cm−1 due to the hydration process of CaO,26 the diffraction data clarified that the CaO-CFB formed a CaO crystal structure.
O and C–O bonds observed at 1784 and 1471–715 cm−1, respectively. In the CaO-CFB spectrum, the CaO bond was observed at 871 cm−1, which appeared as a consequence of a severe reduction in the C–O bond within the raw-CFB, thus suggesting that the aragonite in the raw-CFB was decomposed into CaO and released carbon dioxide (CO2) by calcination at 1000 °C. Although, in the CaO-CFB spectrum, the O–H stretching band appeared at 3637 cm−1 due to the hydration process of CaO,26 the diffraction data clarified that the CaO-CFB formed a CaO crystal structure.
The morphologies of the raw-CFB, CC-CFB, and CaO-CFB showed changes in gross structure (Fig. 2c–e). The raw-CFB had a tiny irregular-shaped particle in its gross surface, creating a rough and brittle structure (Fig. 2c). The gross surface of the CC-CFB was more delicate than that of the raw-CFB with grown particles (Fig. 2d). These results suggest that the calcination at 600 °C caused the nucleation of aragonite polymorphic crystal that crystallized into calcite crystal to form a fine structure and agglomerated spherical-shaped particles. Although the CaO-CFB also exhibited delicate surfaces, it had a bulk-chunked structure (Fig. 2e), thus suggesting that the particle agglomeration in the time increment under 1000 °C occurred quickly before decomposing into the CaO phase. Hence, the structure of CaO-CFB indicated a stable phase, which was more appropriate as an nCHA precursor.
3.2. Properties of the nCHA based on CFB
The XRD pattern of the nCHA exhibited distinct peaks corresponding to apatite (PDF number 09-0432), with the main diffraction peaks observed at 2θ of 31.7°, 32.1°, 32.9°, and 34.0° attributed to the hexagonal lattice planes of (211), (112), (300), and (202), respectively (Fig. 3a). No secondary phases were detected in the nCHA diffraction pattern, thus suggesting that the nCHA had a high purity of apatite phase and avoided impurities. Both lattice parameters of a and c in the nCHA were larger than those of typically HA (a = 9.184 and c = 6.884 Å), respectively (Table 2). In general, A-type CHA forms due to CO32− substitution to the OH− site that induces contraction of the a-axis and expansion of the c-axis in the apatite lattice plane, while, in the B-type CHA, CO32− substitutes the PO43− site that does vice versa to the apatite lattice plane. The lattice parameters a and c of the nCHA showed that both the a- and c-axis were expanded, thus indicating the simultaneous substitution of CO32− to both the OH− and PO43− sites in the apatite lattice plane.56 These results suggest that the nCHA formed AB-type CHA. | ||
| Fig. 3 (a) XRD pattern, (b) FTIR spectrum, and (c) magnified FTIR spectrum of nCHA. (d) TEM, (e) FE-SEM images and (f) EDS spectrum of nCHA. Scale bars: 100 nm and 20 nm (d), and 100 nm (e). | ||
The FTIR spectrum of the nCHA showed the main characteristic bands of CHA (Fig. 3b and c). The peaks corresponding to the ν3 phosphate (P–O) asymmetric stretching, ν1 P–O stretching, ν4 P–O asymmetric bending, and ν2 P–O stretching bands were observed at 1094–1031, 961, 604–572, and 472 cm−1, respectively. The slight peaks observed at 3571 and 631 cm−1 corresponded to the O–H bending and stretching bands. The broad peaks at 3442 and 1639 cm−1 corresponded to the H2O absorption band. The carbonate (C–O) absorption bands were observed at 1546–876 cm−1. The peak at 1546 and 1457 cm−1 corresponded to the ν3 C–O bending band attributed to the CO32− substitution in the A-site (OH−) and B-site (PO43−), respectively. Previous studies pointed out that the ν2 C–O bending band at 876 cm−1 and the ν3 C–O stretching band at 1414 cm−1 were also attributed to A- and B-sites substitution, respectively. The intensity ratio between peaks at 1546 and 1414 cm−1 clearly showed the ratio of A-type to B-type CO3 content, which estimated that the peak intensity of B-type CO3 was higher than that of A-type CO3.30,36,56 These FTIR results deal with the XRD data, showing that the nCHA formed an AB-type CHA.
The morphologies of the nCHA clearly showed nanoparticles with rounded hexagonal shapes (Fig. 3d and e). The nanoparticle size of the nCHA was in the range of 32–76 nm (Fig. 3d and e), which is approximately close to the nanoparticle size of bone apatite.57 The nCHA particle size was ascribed to the nCHA crystallite size by diffraction analysis (Table 2). The nanosize of nCHA may provide a high surface area for ion exchange and interaction with cells and proteins. The elemental analysis revealed that the Ca/P molar ratio was 1.712, which is very close to the Ca/P molar ratio of biological apatite 1.71.27 The CO3 content amount within the nCHA was 7.38 wt%, which is considerably within the typical range of CO3 content in bone apatite, 2–9 wt%.57 Hence, the high CO3 content within the nCHA may promote a high material resorption.34
3.3. Properties of the nCHA/PEO/CS scaffolds
 | ||
| Fig. 4 (a) XRD patterns and (b) FTIR spectra of the nCHA/PEO/CS scaffolds. | ||
| No. | Sample | Degree of crystallinity (%) |
|---|---|---|
| 1 | nCHA | 79.9 |
| 2 | nCHA/PEO/CS-31 | 77.4 |
| 3 | nCHA/PEO/CS-21 | 72.8 |
| 4 | nCHA/PEO/CS-11 | 71.8 |
| 5 | nCHA/PEO/CS-15 | 63.2 |
The FTIR spectra of the nCHA/PEO/CS scaffolds showed differences in functional groups (Fig. 4b). In all scaffolds, the peaks at 2880, 1359–1344, 1277–1239, 1145, and 843 cm−1 corresponded to the C–H asymmetric stretching, CH2 wagging, CH2 twisting, C–O–C stretching, and CH2 asymmetric rocking vibrational bands, respectively, which were ascribed to the PEO functional groups.58 The slight peaks, corresponding to the CO stretching and NH2 stretching vibrational bands ascribed to the chitosan functional groups, appeared at 1644 and 1542 cm−1, respectively.59 The appearance of PEO and CS functional groups in the nCHA/PEO/CS spectra indicates that the PEO and CS molecules endured during lyophilization, and only the solvent underwent sublimation.15 The nCHA/PEO/CS scaffolds performed the carbonate, phosphate, and hydroxyl bands, thus suggesting that the scaffold contained nCHA. The ν3 P–O asymmetric stretching, ν1 P–O stretching, and ν4 P–O asymmetric bending bands were present at 1096–1020, 957, and 600–562 cm−1, respectively. The slight peak at 630 cm−1 was ascribed to the O–H bending band. The ν3 C–O bending and stretching bands remained observed at 1456 and 1412 cm−1, with the ν2 C–O bending band at 874 cm−1, thus suggesting that only B-type CO3 was detected. As the PEO/CS concentration increased, the peak intensity of PEO and CS functional groups increased, while the apatite functional groups decreased, thus dealing with the XRD data. Although the chemical composition of the nCHA/PEO/CS scaffolds was changed, the remaining CO3 content within the scaffold can mimic the chemical composition of bone.
The nCHA/PEO/CS scaffolds clearly showed their macro- and microstructure (Fig. 5). At the macroscale, the nCHA/PEO/CS scaffolds showed a compact and robust structure with heterogeneously and irregularly formed macropores (Fig. 5a–d). These macropores sizes involved in the nCHA/PEO/CS scaffolds were >50 μm and indicated increases in the size of the macropores as the PEO/CS concentration increased (Table 4). The nCHA/PEO/CS-11 and nCHA/PEO/CS-15 scaffolds exhibited macropores size >100 μm (Fig. 5c and d), the ideal macropore size for facilitating cell penetration and blood vessel formation.60 Although the nCHA/PEO/CS-11 and nCHA/PEO/CS-15 scaffolds had appropriate micropore sizes for bone regeneration, the nCHA/PEO/CS-15 lack in the nCHA content may turn to the low osteoconductivity. Hence, the nCHA/PEO/CS-11 is desirable in bone regeneration. At the microscale, the nCHA/PEO/CS scaffolds clearly showed the fibrous-like structured PEO/CS network that cross-linked the nCHA (Fig. 5e–h). The fibrous-like structure of the PEO/CS network within the scaffold mimics the fibrous structure of ECM in the bone that may provide a large surface area for cell recruitment and differentiation, thereby promoting endogenous tissue regeneration when filling it into the bone defect.7,17 In the nCHA/PEO/CS-15 scaffold, the nCHA particles were seemingly covered by crystallized polymeric chains that formed a dense structure, creating a flat surface curvature (Fig. 5h). On the contrary, in the nCHA/PEO/CS-31, nCHA/PEO/CS-21, and nCHA/PEO/CS-11 scaffolds, the nCHA particles were well-distributed and uncovered that created rough surface curvatures (Fig. 5e–g). The rough surface curvature may improve cell adhesion and migration on the scaffold surface, thereby promoting faster bone ingrowth.61 All nCHA/PEO/CS scaffolds involved micropores with size <10 μm that formed among the fibrous structure of PEO/CS that cross-linked nCHA particles (Fig. 5e–h), which indicated increases in the size of the micropores as the PEO/CS concentration increased (Table 4). The micropore size <10 μm can create a rough surface for facilitating cell adhesion, proliferation, and differentiation.62 In the nCHA/PEO/CS-31 and nCHA/PEO/CS-15 scaffolds, the micropores were irregular and diversely distributed (Fig. 5e and h). Meanwhile, the micropore distribution was heterogeneous in the nCHA/PEO/CS-21 scaffold (Fig. 5f). In contrast, the nCHA/PEO/CS-11 scaffold had regularly, homogeneously, and uniformly distributed micropores (Fig. 5g). The regular, homogeneous, and uniform micropores in the nCHA/PEO/CS-11 scaffold may encourage cells and cell nutrients to engage in the scaffold.63 These results suggest that the nCHA/PEO/CS-11 scaffold formed a fibrous-like and porous structure with suitable pores characteristics and surface curvatures, which may promote favorable bone regeneration.
 | ||
| Fig. 5 Morphologies of the nCHA/PEO/CS scaffolds in (a–d) macro- and (e–h) microscale. The characters “#,” “*,” orange and green arrowheads indicate nCHA, PEO/CS fibrous network, macropores, and micropores, respectively. Scale bars: 100 μm (a–d) and 10 μm (e–h). | ||
| No. | Sample | Macropore size (>50 μm) | Micropore size (<10 μm) | Macroporosity (%) | Microporosity (%) |
|---|---|---|---|---|---|
| 1 | nCHA/PEO/CS-31 | 56.9 ± 8.96 | 5.51 ± 1.20 | 51.3 | 59.4 |
| 2 | nCHA/PEO/CS-21 | 65.8 ± 13.0 | 2.49 ± 0.61 | 53.9 | 61.6 |
| 3 | nCHA/PEO/CS-11 | 115 ± 18.2 | 3.99 ± 0.86 | 57.6 | 64.8 |
| 4 | nCHA/PEO/CS-15 | 117 ± 24.2 | 5.60 ± 1.33 | 62.1 | 65.1 |
Coupled with the structural properties of the nCHA/PEO/CS scaffolds, the two-dimensional porosity in terms of macro- and microporosity of the nCHA/PEO/CS scaffolds showed increases with the increase in the PEO/CS concentration (Table 4). The ideal porosity of the scaffold suitable for cell penetration and nutrient perfusion was >60%.64 Hence, all nCHA/PEO/CS scaffolds had a microporosity close to 60%. However, only the nCHA/PEO/CS-11 and nCHA/PEO/CS-15 had a macroporosity close to 60%. Considering the most proper structural characteristics of the nCHA/PEO/CS-11 scaffold, the nCHA/PEO/CS-11 was preferable for bone regeneration.
 | ||
| Fig. 6 Compressive strengths of the nCHA/PEO/CS scaffolds. ***p < 0.001. | ||
 | ||
| Fig. 7 (a) Swelling profiles and (b) maximum swelling ratios of the nCHA/PEO/CS scaffolds. **p < 0.01 and ***p < 0.001. | ||
Related to the swelling behavior, the maximum swelling ratio of the nCHA/PEO/CS scaffolds increases with the increase in the PEO/CS concentration (Fig. 7b). The maximum swelling ratios of the nCHA/PEO/CS-31, nCHA/PEO/CS-21, nCHA/PEO/CS-11, and nCHA/PEO/CS-15 scaffolds were 93.4 ± 10.8, 114 ± 19.9, 120 ± 8.45, and 161 ± 20.3%, respectively. Hence, the maximum swelling ratios of the nCHA/PEO/CS scaffolds significantly increased due to the increase in the PEO/CS concentration (p < 0.01). These results were cohesive with the enhancement in the swelling ability of the nCHA/PEO/CS as the increase in the PEO/CS incorporation (Fig. 7a). Owing to the suitable swelling behavior and rapid swelling, the nCHA/PEO/CS-11 scaffold showed well-expected swelling properties that may encourage a rapid biodegradability of the scaffold for bone regeneration.66
The nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds exhibited increases in cell viability by the low scaffold concentrations after 24 h incubation (Fig. 8a). The cell viability values of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds were considerably high in all scaffold concentrations (Table 5). Pursuing ISO 10993-5, the cell viability values of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds perpetuated a cell viability value that exceeded the non-toxic density level of 80%,67 suggesting that the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds considerably had no cytotoxicity. The cell viability values of the nCHA/PEO/CS-11 scaffold were significantly higher than those of the nCHA/PEO/CS-21 (p < 0.001), suggesting that the higher content of chitosan within the nCHA/PEO/CS-11 scaffold enhanced the cytocompatibility since chitosan-based nanoparticles reportedly could increase the cell interaction by osteoconductive binding between positively charged surfaces of the amino group in the chitosan and negatively charged cell membrane.68 Hence, the nCHA/PEO/CS-11 scaffold induced better cell response for enhancing cell activity.
 | ||
| Fig. 8 (a) Viabilities of MC3T3E1 cells, (b) IC50 analysis by non-linear curve fitting, and (c) IC50 values of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds. ***p < 0.001. | ||
| No. | Scaffold concentration (μg mL−1) | Cell viability (%) (mean ± SD) | p-Value | |
|---|---|---|---|---|
| nCHA/PEO/CS-21 | nCHA/PEO/CS-11 | |||
| 1 | 7.81 | 89.5 ± 0.39 | 93.8 ± 0.28 | 0.001 |
| 2 | 15.6 | 88.8 ± 0.47 | 93.5 ± 0.17 | |
| 3 | 31.3 | 88.3 ± 0.49 | 93.1 ± 1.80 | |
| 4 | 62.5 | 87.9 ± 0.58 | 92.4 ± 1.27 | |
| 5 | 125 | 85.3 ± 0.41 | 90.8 ± 0.26 | |
| 6 | 250 | 84.4 ± 0.66 | 89.9 ± 0.58 | |
| 7 | 500 | 83.7 ± 0.29 | 89.8 ± 0.89 | |
| 8 | 1000 | 81.6 ± 1.33 | 85.9 ± 0.87 | |
For subsequence cytotoxicity analysis, the IC50 of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds were examined statistically using non-linear curve fitting. The fitted non-linear curve on the average cell viability of the nCHA/PEO/CS-11 scaffold precisely outpaced those of the nCHA/PEO/CS-21 scaffold in all diluted scaffold concentrations (Fig. 8b). The IC50 values of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds were estimated at 60.1 × 106 and 20.8 × 106 μg mL−1, respectively (Fig. 8c). Hence, the IC50 values of the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds were considerably high, suggesting that both scaffolds were highly safe to osteogenic cell for bone tissue repair. These results show that the highly osteoconductive nCHA in the scaffold could control the cytotoxic level in the high scaffold concentration.
The morphology of the well-connected MC3T3E1 cells formed a cell network in control and several diluted scaffold concentrations (Fig. 9a–h). Cell morphology in control formed several sub-confluent structures up to 80%, mostly clustered (Fig. 9a and e). As the scaffold concentration increased, most cells were still alive and highly viable with a fibroblastic structure, even though a few round-shaped cells appeared dead. The number of life cells in the nCHA/PEO/CS-11 scaffold (Fig. 9f–h) was seemingly higher than those in the nCHA/PEO/CS-21 scaffold (Fig. 9b–d), dealing with the cell viability values (Table 5). Hence, the nCHA/PEO/CS-11 was preferably suitable for use in bone regeneration. The higher content of biocompatible PEO/CS polymeric network within the nCHA/PEO/CS-11 scaffold triggered higher cell activity due to the presence of osteoconductive binding of amino group on the PEO/CS surface, thereby facilitating enhanced interaction with the cell membrane.69,70 Owing to its cytocompatibility, the nCHA/PEO/CS-11 scaffold is preferred in promoting bone regeneration.
 | ||
| Fig. 9 Morphology of MC3T3E1 cells after 24 incubation in conditioned medium with serial concentrations of (a–d) nCHA/PEO/CS-21 and (e–h) nCHA/PEO/CS-11 scaffolds. The white dashed circle, orange, purple, and blue arrowheads indicate cell cluster, life cells, dead cells, and scaffold particles, respectively. Scale bars: 100 μm (a–h). | ||
 | ||
| Fig. 10 Morphology of adherent MC3T3-E1 cells on the surfaces of (a1) nCHA/PEO/CS-21 and (b1) nCHA/PEO/CS-11 scaffolds. (a2 and b2) Cells highlighted in the panels a1 and b1. (c) Cell spreading area on the surfaces of nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds. Cytoskeleton of adherent cells on (d) nCHA/PEO/CS-21 and (e) nCHA/PEO/CS-11 scaffold surfaces. Scale bars: 20 μm (a1–b2,d,e). | ||
 | ||
| Fig. 11 ALP activity of MC3T3-E1 cells cultured with the nCHA/PEO/CS-21 and nCHA/PEO/CS-11 scaffolds. | ||
4. Conclusions
This research achieved the fabrication of a porous nCHA/PEO/CS bone scaffold comprising a fibrous structure for mimicking native bone structure. The synthesized nCHA based on CFB demonstrated excellent physicochemical and morphological properties that are very close to apatite in natural bone. When nCHA was combined with PEO and CS in various concentrations by freeze-drying technique, the nCHA/PEO/CS scaffolds were obtained. The nCHA/PEO/CS scaffold had low crystallinity, which may encourage cell growth. The structure of all nCHA/PEO/CS scaffolds revealed that the nCHA nanoparticles were cross-linked within a fibrous-like structured PEO/CS network, which mimics the fibrous structure of extracellular matrix (ECM) in native bone. However, only the nCHA/PEO/CS-11 scaffold formed appropriate macro- and micropores with suitable macro- and micro-porosity that may enhance cell development, blood vessel formation, and nutrient perfusion. The nCHA/PEO/CS-11 scaffold also had sufficient compressive strength and well-regulated swelling behavior that may favor bone regeneration. The nCHA/PEO/CS-11 demonstrated high cytocompatibility and facilitated the adherence of MC3T3E1 cells onto the scaffold surface. The nCHA/PEO/CS-11 also promoted cell osteogenic differentiation. All in all, the developed nCHA/PEO/CS-11 scaffold was considerably promising in bone tissue engineering.Data availability
All data underlying the results are available as part of this article.Author contributions
M. R. Habiburrohman: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review and editing. M. A. Jamilludin: conceptualization, data curation, writing – review and editing. N. Cahyati: conceptualization, data curation, writing – review and editing. N. Herdianto: funding acquisition, supervision. Y. Yusuf: validation, funding acquisition, supervision, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work in this article.Acknowledgements
The authors express their profound gratitude to the Research Scholarships for Research and Innovation Talents (BARISTA) 2023 program by the National Research and Innovation Agency (grant no. 108/II/HK/2023) and the Final Project Recognition (RTA) 2024 program by the Research Directorate of Universitas Gadjah Mada (grant no. 5286/UN1.P1/PT.01.03/2024) for funding this research. The authors acknowledge utilizing research instruments in the Material Physics and Electronics Laboratory, Department of Physics, Universitas Gadjah Mada, Indonesia. The authors are immensely thankful to the Integrated Laboratory for Research and Testing, Universitas Gadjah Mada, Indonesia, for facilitating technical support in conducting several characterizations and cell experiments.References
- M. Jakovljevic, N. Kumagai and S. Ogura, Public Health Front., 2023, 11, 1184950, DOI:10.3389/fpubh.2023.1184950.
- Y. Shen, X. Huang, J. Wu, X. Lin, X. Zhou, Z. Zhu, X. Pan, J. Xu, J. Qiao, T. Zhang, L. Ye, H. Jiang, Y. Ren and P.-F. Shan, Front. Endocrinol., 2022, 13, 882241, DOI:10.3389/fendo.2022.882241.
- P. J. Mitchell, D.-C. D. Chan, J.-K. Lee, I. Tabu and B. B. Alpuerto, Best Pract. Res., Clin. Rheumatol., 2022, 36, 101777, DOI:10.1002/jbmr.4821.
- P. Baldwin, D. J. Li, D. A. Auston, H. S. Mir, R. S. Yoon and K. J. Koval, J. Orthop. Trauma, 2019, 33, 203, DOI:10.1097/BOT.0000000000001420.
- Z. Sheikh, N. Hamdan, Y. Ikeda, M. Grynpas, B. Ganss and M. Glogauer, Biomater. Res., 2017, 21, 9, DOI:10.1186/s40824-017-0095-5.
- A. E. Rodriguez and H. Nowzari, J. Indian Soc. Periodontol., 2019, 23, 487–492, DOI:10.4103/jisp.jisp_656_18.
- M. Zhu, W. Li, X. Dong, X. Yuan, A. C. Midgley, H. Chang, Y. Wang, H. Wang, K. Wang, P. X. Ma, H. Wang and D. Kong, Nat. Commun., 2019, 10, 4620, DOI:10.1038/s41467-019-12545-3.
- F. Tao, Y. Cheng, X. Shi, H. Zheng, Y. Du, W. Xiang and H. Deng, Carbohydr. Polym., 2020, 230, 115658, DOI:10.1016/j.carbpol.2019.115658.
- B. Guo and P. X. Ma, Sci. China:Chem., 2014, 57, 490–500, DOI:10.1007/s11426-014-5086-y.
- M. A. Jamilludin, K. Hayashi, Y. Yusuf and K. Ishikawa, Ceram. Int., 2024, 50, 25988–25999, DOI:10.1016/j.ceramint.2024.04.341.
- C. Katrilaka, N. Karipidou, N. Petrou, C. Manglaris, G. Katrilakas, A. N. Tzavellas, M. Pitou, E. E. Tsiridis, T. Choli-Papadopoulou and A. Aggeli, Materials, 2023, 16, 4425, DOI:10.3390/ma16124425.
- I. K. H. Dinatha, M. A. Jamilludin, A. I. Supii, H. Wihadmadyatami, J. Partini and Y. Yusuf, J. Biomed. Mater. Res., Part B, 2024, 112, e35341, DOI:10.1002/jbm.b.35341.
- S. Tarafder and S. Bose, ACS Appl. Mater. Interfaces, 2014, 6, 9955–9965, DOI:10.1021/am501048n.
- S. M. Giannitelli, F. Basoli, P. Mozetic, P. Piva, F. N. Bartuli, F. Luciani, C. Arcuri, M. Trombetta, A. Rainer and S. Licoccia, Mater. Sci. Eng., C, 2015, 51, 329–335, DOI:10.1016/j.msec.2015.03.002.
- M. U. A. Khan, S. Haider, A. Haider, S. I. A. Razak, M. R. A. Kadir, S. A. Shah, A. Javed, I. Shakir and A. A. Al-Zahrani, Arabian J. Chem., 2021, 14, 102924, DOI:10.1016/j.arabjc.2020.102924.
- D. J. Patty, A. D. Nugraheni, I. D. Ana, A. Aminatun, Y. W. Sari, G. Gunawarman and Y. Yusuf, RSC Adv., 2023, 13, 34427–34438, 10.1039/D3RA07486B.
- A. H. Diputra, I. K. H. Dinatha, N. Cahyati, J. F. Fatriansyah, M. Taufik, H. Hartatiek and Y. Yusuf, Biomed. Mater., 2024, 19, 065021, DOI:10.1088/1748-605X/ad7e92.
- E. Waris, N. Ashammakhi, M. Lehtimäki, R.-M. Tulamo, P. Törmälä, M. Kellomäki and Y. T. Konttinen, Biomaterials, 2008, 29, 2509–2515, DOI:10.1016/j.biomaterials.2008.02.013.
- K. R. Remya, S. Chandran, S. Mani, A. John and P. Ramesh, J. Biomater. Sci. Polym. Ed., 2018, 29, 1444–1462, DOI:10.1080/09205063.2018.1465664.
- M. Oleksy, K. Dynarowicz and D. Aebisher, Molecules, 2023, 28, 6213, DOI:10.3390/molecules28176213.
- L. Fan, Y. Ren, S. Emmert, I. Vučković, S. Stojanovic, S. Najman, R. Schnettler, M. Barbeck, K. Schenke-Layland and X. Xiong, Int. J. Mol. Sci., 2023, 24, 3744, DOI:10.3390/ijms24043744.
- H. Wang, R. Sun, S. Huang, H. Wu and D. Zhang, Heliyon, 2024, 10, e25832, DOI:10.1016/j.heliyon.2024.e25832.
- S. L. Levengood and M. Zhang, J. Mater. Chem. B, 2014, 2, 3161–3184, 10.1039/C4TB00027G.
- M. M. Rahman, M. Shahruzzaman, M. S. Islam, M. N. Khan and P. Haque, J. Polym. Eng., 2019, 39, 134–142, DOI:10.1515/polyeng-2018-0103.
- M. A. Jamilludin, J. Partini, D. L. Kusindarta and Y. Yusuf, Results Mater., 2024, 22, 100580, DOI:10.1016/j.rinma.2024.100580.
- M. Sari, P. Hening, C. Chotimah, I. D. Ana and Y. Yusuf, Mater. Today Commun., 2021, 26, 102135, DOI:10.1016/j.mtcomm.2021.102135.
- I. K. Januariyasa and Y. Yusuf, J. Australas. Ceram. Soc., 2020, 8, 634–641, DOI:10.1080/21870764.2020.1770938.
- M. Alizadeh-Osgouei, Y. Li and C. Wen, Bioact. Mater., 2019, 4, 22–36, DOI:10.1016/j.bioactmat.2018.11.003.
- A. Ressler, A. Žužić, I. Ivanišević, N. Kamboj and H. Ivanković, Open Ceram., 2021, 6, 100122, DOI:10.1016/j.oceram.2021.100122.
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612, DOI:10.1002/jbm.10280.
- R. Florencio-Silva, G. R. D. A. S. Sasso, E. Sasso-Cerri, M. J. Simões and P. S. Cerri, Biomed. Res. Int., 2015, 2015, 421746, DOI:10.1155/2015/421746.
- Z. Z. Zyman, D. V. Rokhmistrov, V. I. Glushko and I. G. Ivanov, J. Mater. Sci.: Mater. Med., 2009, 20, 1389–1399, DOI:10.1007/s10856-009-3706-4.
- Q. Liu, J. P. Matinlinna, Z. Chen, C. Ning, G. Ni, H. Pan and B. W. Darvell, Ceram. Int., 2015, 41, 6149–6157, DOI:10.1016/j.ceramint.2014.11.062.
- K. Hayashi, T. Yanagisawa, M. Shimabukuro, R. Kishida and K. Ishikawa, Mater. Today Bio, 2022, 14, 100247, DOI:10.1016/j.mtbio.2022.100247.
- E. Landi, G. Celotti, G. Logroscino and A. Tampieri, J. Eur. Ceram. Soc., 2003, 23, 2931–2937, DOI:10.1016/S0955-2219(03)00304-2.
- E. Garskaite, K.-A. Gross, S.-W. Yang, T. C.-K. Yang, J.-C. Yang and A. Kareiva, CrystEngComm, 2014, 16, 3950–3959, 10.1039/C4CE00119B.
- J. P. Lafon, E. Champion and D. Bernache-Assollant, J. Eur. Ceram. Soc., 2008, 28, 139–147, DOI:10.1016/j.jeurceramsoc.2007.06.009.
- J. Venkatesan, P. D. Rekha, S. Anil, I. Bhatnagar, P. N. Sudha, C. Dechsakulwatana, S.-K. Kim and M. S. Shim, Biotechnol. Bioprocess Eng., 2018, 23, 383–393, DOI:10.1007/s12257-018-0169-9.
- D. Reinares-Fisac, S. Veintemillas-Verdaguer and L. Fernández-Díaz, CrystEngComm, 2016, 19, 110–116, 10.1039/C6CE01725H.
- S. Balu, M. V. Sundaradoss, S. Andra and J. Jeevanandam, Beilstein J. Nanotechnol., 2020, 11, 285–295, DOI:10.3762/bjnano.11.21.
- H. Ivankovic, G. G. Ferrer, E. Tkalcec, S. Orlic and M. Ivankovic, J. Mater. Sci.: Mater. Med., 2009, 20, 1039–1046, DOI:10.1007/s10856-008-3674-0.
- S. Kangkan, P. Arpornmaeklong and S. Ummartyotin, J. Met., Mater. Miner., 2020, 30, 136–141, DOI:10.55713/jmmm.v30i2.741.
- M. Baladi, M. Amiri, P. Mohammadi, K. S. Mahdi, Z. Golshani, R. Razavi and M. Salavati-Niasari, Arabian J. Chem., 2023, 16, 104646, DOI:10.1016/j.arabjc.2023.104646.
- A. Mushtaq, R. Zhao, D. Luo, E. Dempsey, X. Wang, M. Z. Iqbal and X. Kong, Mater. Des., 2021, 197, 109269, DOI:10.1016/j.matdes.2020.109269.
- A. C. Ferro and M. Guedes, Mater. Sci. Eng., C, 2019, 97, 124–140, DOI:10.1016/j.msec.2018.11.083.
- M. Sadat-Shojai, M.-T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591–7621, DOI:10.1016/j.actbio.2013.04.012.
- S. Kuśnieruk, J. Wojnarowicz, A. Chodara, T. Chudoba, S. Gierlotka and W. Lojkowski, Beilstein J. Nanotechnol., 2016, 7, 1586–1601, DOI:10.3762/bjnano.7.153.
- M. Manoj, R. Subbiah, D. Mangalaraj, N. Ponpandian, C. Viswanathan and K. Park, Nanobiomedicine, 2015, 2, 2, DOI:10.5772/60116.
- A. R. Noviyanti, N. Akbar, Y. Deawati, E. E. Ernawati, Y. T. Malik, R. P. Fauzia and R. Risdiana, Heliyon, 2020, 6, e03655, DOI:10.1016/j.heliyon.2020.e03655.
- P. Feng, R. Zhao, L. Yang, S. Chen, D. Wang, H. Pan and C. Shuai, Ceram. Int., 2022, 48, 33682–33692, DOI:10.1016/j.ceramint.2022.07.314.
- A. Aminatun, A. Supardi, Z. I. Nisa, D. Hikmawati and S. Siswanto, Int. J. Biomater., 2019, 2019, 1831208, DOI:10.1155/2019/1831208.
- L. Bauer, M. Antunović, A. Rogina, M. Ivanković and H. Ivanković, J. Mater. Sci., 2021, 56, 3947–3969, DOI:10.1007/s10853-020-05489-3.
- A. Jastram, T. Lindner, C. Luebbert, G. Sadowski and U. Kragl, Polymers, 2021, 13, 1834, DOI:10.3390/polym13111834.
- J. Zhu, Q. Kong, S. Zheng, Y. Wang, Z. Jiao, Y. Nie, T. Liu and K. Song, Int. J. Biol. Macromol., 2020, 158, 800–810, DOI:10.1016/j.ijbiomac.2020.04.267.
- K. S. P. Karunadasa, C. H. Manoratne, H. M. T. G. A. Pitawala and R. M. G. Rajapakse, J. Phys. Chem. Solids, 2019, 134, 21–28, DOI:10.1016/j.jpcs.2019.05.023.
- B. Li, X. Liao, L. Zheng, H. He, H. Wang, H. Fan and X. Zhang, Mater. Sci. Eng., C, 2012, 32, 929–936, DOI:10.1016/j.msec.2012.02.014.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116, DOI:10.1016/j.mser.2007.05.001.
- A. Dey, K. Das, S. Karan and S. K. De, Spectrochim. Acta, Part A, 2011, 83, 384–391, DOI:10.1016/j.saa.2011.08.050.
- H. U. Rehman, S. Cord-Landwehr, V. Shapaval, S. Dzurendova, A. Kohler, B. M. Moerschbacher and B. Zimmermann, Carbohydr. Polym., 2023, 302, 120428, DOI:10.1016/j.carbpol.2022.120428.
- R. Rial, M. González-Durruthy, Z. Liu and J. M. Ruso, Molecules, 2021, 26, 3190, DOI:10.3390/molecules26113190.
- J. Han, Z. Li, Y. Sun, F. Cheng, L. Zhu, Y. Zhang, Z. Zhang, J. Wu and J. Wang, Front. Bioeng. Biotechnol., 2022, 10, 888267, DOI:10.3389/fbioe.2022.888267.
- K. Zhang, Y. Fan, N. Dunne and X. Li, Regener. Biomater., 2018, 5, 115–124, DOI:10.1093/rb/rby001.
- G. Lutzweiler, A. N. Halili and N. E. Vrana, Pharmaceutics, 2020, 12, 602, DOI:10.3390/pharmaceutics12070602.
- H. Mohammadi, M. Sepantafar, N. Muhamad and A. B. Sulong, Adv. Eng. Mater., 2021, 23, 2100463, DOI:10.1002/adem.202100463.
- R. M. Felfel, M. J. Gideon-Adeniyi, K. M. Z. Hossain, G. A. F. Roberts and D. M. Grant, Carbohydr. Polym., 2019, 204, 59–67, DOI:10.1016/j.carbpol.2018.10.002.
- N. Abbasi, S. Hamlet, R. M. Love and N.-T. Nguyen, J. Sci.:Adv. Mater. Devices, 2020, 5, 1–9, DOI:10.1016/j.jsamd.2020.01.007.
- ISO 10993-5:2009, International Organization for Standardization, 3rd edn, 2009, online: https://www.iso.org/standard/36406.html.
- J. Frigaard, J. L. Jensen, H. K. Galtung and M. Hiorth, Front. Pharmacol., 2022, 13, 880377, DOI:10.3389/fphar.2022.880377.
- M. A. Jamilludin, I. K. H. Dinatha, A. I. Supii, J. Partini, D. L. Kusindarta and Y. Yusuf, RSC Adv., 2023, 13, 32444–32456, 10.1039/D3RA06165E.
- N. Cahyati, M. Sari and Y. Yusuf, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2024, 15, 035004, DOI:10.1088/2043-6262/ad6b7b.
- R. Shi, K. Hayashi and K. Ishikawa, Adv. Mater. Interfaces, 2020, 7, 2000636, DOI:10.1002/admi.202000636.
- Q. Yang, J. Guo, S. Zhang, F. Guan, Y. Yu, S. Feng, X. Song, D. Bao and X. Zhang, Int. J. Biol. Macromol., 2023, 236, 124004, DOI:10.1016/j.ijbiomac.2023.124004.
- I. K. Januariyasa, I. D. Ana and Y. Yusuf, Mater. Sci. Eng., C, 2020, 107, 110347, DOI:10.1016/j.msec.2019.110347.
| This journal is © The Royal Society of Chemistry 2025 |