
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Amino-4,6-diarylpyrimidines as potential chronic myeloid leukemia cell inhibitors targeting anti-ABL1 kinase: microwave-assisted synthesis, biological evaluation, molecular docking, and dynamics studies†
Thi-Anh-Truc Phanab,
Kim-Khanh-Huy Ngo a,
Thi-Cam-Thu Nguyena,
Thanh-Tan Maic,
Hai-Dang Nguyend,
Thu-Trang Duongd,
Le-Phu Tranab,
Thanh-Tuyen Duongab,
Thi-Kim-Chi Huynha,
Elena V. Korolevae,
Zhanna V. Ignatoviche,
Anastasiya L. Ermolinskayae,
Hoang-Phuc Nguyena,
Thi-Hong-An Nguyena,
Anh-Khoa Tona,
Tuong-Ha Dob and
Thi-Kim-Dung Hoang*a
a,
Thi-Cam-Thu Nguyena,
Thanh-Tan Maic,
Hai-Dang Nguyend,
Thu-Trang Duongd,
Le-Phu Tranab,
Thanh-Tuyen Duongab,
Thi-Kim-Chi Huynha,
Elena V. Korolevae,
Zhanna V. Ignatoviche,
Anastasiya L. Ermolinskayae,
Hoang-Phuc Nguyena,
Thi-Hong-An Nguyena,
Anh-Khoa Tona,
Tuong-Ha Dob and
Thi-Kim-Dung Hoang*a
aInstitute of Chemical Technology, Vietnam Academy of Science and Technology, No. 1A, TL29 St., Thanh Loc Ward, Dist. 12, Ho Chi Minh City 70000, Vietnam. E-mail: hoangthikimdung@gmail.com; htkdung@ict.vast.vn
bTon Duc Thang University, No. 19, Nguyen Huu Tho St., Tan Hung Ward, Dist. 7, Ho Chi Minh City 70000, Vietnam
cUniversity of Medicine and Pharmacy at Ho Chi Minh City, No. 41, Dinh Tien Hoang St., Ben Nghe Ward, Dist. 1, Ho Chi Minh City 70000, Vietnam
dUniversity of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, No. 18, Hoang Quoc Viet St., Nghia Do Ward, Cau Giay District, Ha Noi City 100000, Vietnam
eInstitute of Chemistry of New Materials, National Academy of Sciences of Belarus, 36, F. Skorina St., Minsk, 220141, Republic of Belarus
First published on 10th February 2025
Abstract
In this work, a simple and mild process was used to synthesize a series of 2-amino-4,6-diarylpyrimidine derivatives, 1a–1q, whose structures were verified by FTIR, 1D- and 2D-NMR, and HRMS techniques, to investigate and develop anticancer agents. Under microwave irradiation, a two-step process was carried out, consisting of aldol condensation of benzaldehydes and acetophenones to produce intermediate chalcones and ring closure condensation of chalcones and guanidine hydrochloride. Each generated compound's anticancer activity against the human chronic myelocytic leukemia K562 cancer cell line was investigated in vitro to determine the active compounds, which were subsequently evaluated for inhibiting the ABL1 tyrosine kinase. According to these findings, compound 1e demonstrated considerable inhibition against K562 cancer cells and ABL1 tyrosine kinase at IC50 values of 8.77 ± 0.55 μM and 3.35 ± 0.58 μM, respectively. The molecular docking on wild-type and mutant type ABL1 (PDB ID 2HYY and 5MO4) investigation indicated that 1e and 1g interacted with amino acids. It formed stable hydrogen bonds and π–π linkages with crucial residues in the active site of the enzyme. Moreover, the stability of these enzyme–ligand complexes was confirmed using molecular dynamics simulations. These findings suggested that compounds 1e and 1g can be considered promising cancer treatment agents.
1. Introduction
Cancer remains a formidable challenge for global health, standing as one of the major causes of death worldwide.1,2 The World Health Organization reports that roughly 14 million new cancer cases are diagnosed annually, resulting in around 9 million deaths.3 Among the diverse spectrum of cancer types, leukemia, which is defined by the malignant expansion of hematopoietic stem cells in the bone marrow, occupies a significant position due to its prevalence and clinical complexity.4,5 Notably, human chronic myeloid leukemia (CML) represents a subset of leukemia distinguished by the aberrant expansion of myeloid progenitor cells.6,7 Since the discovery of the presence of a unique and constant chromosomal abnormality more than four decades ago, significant achievements have been made in understanding the biological underpinnings of the disease.8,9 In leukemia research, the K562 cell line has emerged as a cornerstone for investigating the pathogenesis and therapeutic strategies pertinent to CML.10,11Tyrosine kinases (TKs) are the enzymes that initiate the transfer of phosphate groups from ATP to tyrosine, a vital process in cellular machinery. The K562 cell line, which expresses the BCR-ABL fusion gene, was discovered to be exceptionally resistant to apoptosis, regardless of the triggering stimulus, and scientists clarified the role of the BCR-ABL fusion protein and the Philadelphia chromosome in CML pathogenesis.12,13 Therefore, BCR-ABL inhibition is currently viewed as a significant molecular target in the treatment of CML. Imatinib and nilotinib (commercially known as Glivec and Tasigna, respectively) are kinase inhibitors that have emerged as a primary therapeutic agent for CML cancer treatment and represents a cornerstone of contemporary anticancer pharmacotherapy.14,15 Nonetheless, imatinib resistance linked to BCR/ABL1 mutation and the common adverse effects of marketed drug have emerged as a problem.16–18 Therefore, developing potential inhibitors with improved medication safety and efficacy is critical for treating CML patients. Recent reports have revealed that the structure of imatinib, nilotinib and their derivatives containing pyrimidine-2-amine showed inhibitory potential through the kinase pathway (Fig. 1).14,19,20 Additionally, different types of substituents linked to the C-4 and C-6 positions of pyrimidine-2-amine were found to have antiproliferative effects on diverse cell lines.21–24 This has led to our current research focusing on the synthesis of a pyrimidine-2-amine skeleton containing aryl substituents at the 4,6-position of the pyrimidine backbone, as well as the investigation of K562 cell line and ABL1 tyrosine kinase inhibitory activities.
 | ||
| Fig. 1 Structures of commercial anticancer drugs containing pyrimidin-2-amine moiety. | ||
With the importance and extensive range of biological activities associated with pyrimidine moieties, numerous methods for pyrimidine synthesis have been reported.25,26 Pyrimidine nucleus production is classified into three kinds according to the fundamental structure of the reactants combining (Fig. S1†), with type 1 being considered the most prevalent pyrimidine synthesis method.27 Chalcones are the most popular and highly stable precursors in the synthesis of numerous biologically active pyrimidines based on type 1 (ref. 28–30) because they could easily react with guanidine salts in basic conditions to produce 2-amino-4,6-diarylpyrimidines.31–34
2. Results and discussion
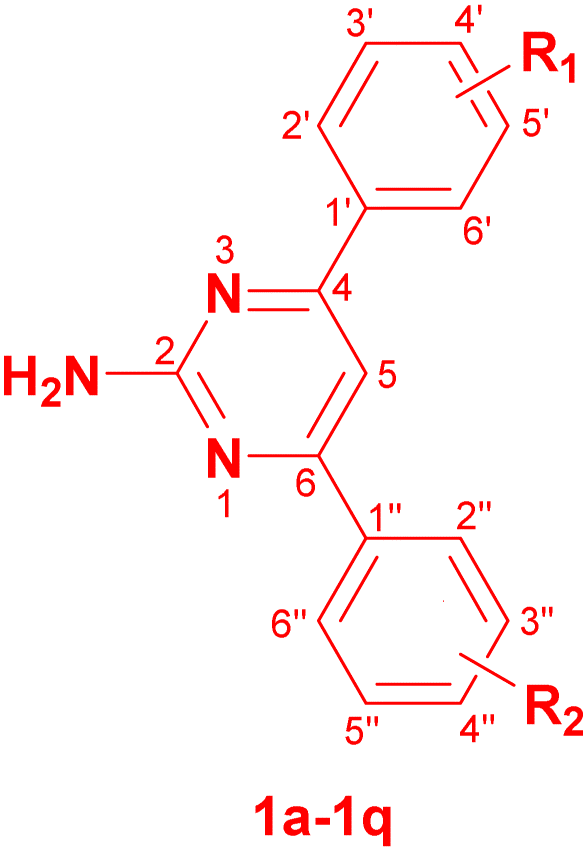
2.1. Chemistry
The target 2-amino-4,6-diarylpyrimidines 1a–1q were synthesized following the approach illustrated in Scheme 1. The one-pot method was attempted to cyclocondense the three-components among benzaldehydes, acetophenones, and guanidine hydrochloride (GHCl), but the results were not considered due to the excessive amount of impurities. Consequently, the synthesis procedure of the desired compounds was separated into two steps, which was carried out by utilizing the microwave (MW) technique. The intermediate chalcones were generated in the first stage using aldol condensation of substituted benzaldehyde and various acetophenone derivatives in ethylene glycol (EG) solvent with KOH catalysis via MW (80 °C, 100 W, 10–30 min). The second step of this work involved ring closure condensation of pure substituted chalcones, GHCl, and NaOH in pyridine under MW conditions (100 °C, 180 W, 5–10 min). The final compounds were produced with a yield ranging from 31% to 64% (Table 1) and the synthesis of pyrimidines that contained electron-withdrawing groups was listed in Table S1.† EG was utilized as a recyclable, non-volatile, and cost-effective solvent to efficiently dissolve reactants in the chalcone synthesis. This work-up procedure was convenient, straightforward, and prompt in preparing the final product and handling the post-reaction mixture. The structures of all synthesized derivatives were demonstrated carefully by FTIR, 1D- and 2D-NMR, and ESI-HRMS analyses. The novel compound 1h was obtained in the form of a pale brown powder. The FTIR of 1h indicated a sharp and medium peak at 3403 cm−1, which corresponded to the primary amine's N–H stretching vibration. Characteristic oscillations were also found at 3517, 1636, 1542, 1241, and 1037 cm−1 for the –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, C–N, and –OCH3 groups, respectively. Compound 1h's NMR spectra revealed nine aromatic protons at δH 6.87–8.09 ppm, corresponding to aromatic carbons at δC 100.93–164.12 ppm (Table 2). Additionally, the characteristic singlets at the chemical shift δH 3.85, 6.58, and 9.89 ppm were assigned to the protons of OCH3, NH2, and OH groups, respectively. This finding demonstrated the successful aldol condensation of 3-methoxybenzaldehyde with 4-acetophenone and cyclocondensation processes of the corresponding chalcone with GHCl. The HSQC spectrum revealed the direct interaction between methyl proton at 3.85 ppm (H-a) and methyl carbon at 55.22 ppm (C-a). Then, the correlation in the HMBC spectrum was determined between aromatic protons at 7.72 ppm (H-2′), 7.77 ppm (H-6′), and 7.57 ppm (H-5) and the same quaternary carbon at 164.12 ppm (C-4), between aromatic protons at 7.57 ppm (H-5) and 8.09 ppm (H-2′′/H-6), and the same carbon at 164.68 ppm (C-6). The ring closure sites at quaternary carbons C-4 and C-6 were discovered to generate compound 1h. The molecular formula of 1h was confirmed as C17H16N3O2 by ESI-HRMS analysis, showing a pseudo-molecular ion peak [M + H]+ m/z 294.1249 (calcd for C17H16N3O2, 294.1242). On the basis of the previously given spectrum data, 4-(2-amino-6-(3-methoxy-phenyl)-pyrimidin-4-yl)-phenol was identified as the structure of 1h.
N, CC, C–N, and –OCH3 groups, respectively. Compound 1h's NMR spectra revealed nine aromatic protons at δH 6.87–8.09 ppm, corresponding to aromatic carbons at δC 100.93–164.12 ppm (Table 2). Additionally, the characteristic singlets at the chemical shift δH 3.85, 6.58, and 9.89 ppm were assigned to the protons of OCH3, NH2, and OH groups, respectively. This finding demonstrated the successful aldol condensation of 3-methoxybenzaldehyde with 4-acetophenone and cyclocondensation processes of the corresponding chalcone with GHCl. The HSQC spectrum revealed the direct interaction between methyl proton at 3.85 ppm (H-a) and methyl carbon at 55.22 ppm (C-a). Then, the correlation in the HMBC spectrum was determined between aromatic protons at 7.72 ppm (H-2′), 7.77 ppm (H-6′), and 7.57 ppm (H-5) and the same quaternary carbon at 164.12 ppm (C-4), between aromatic protons at 7.57 ppm (H-5) and 8.09 ppm (H-2′′/H-6), and the same carbon at 164.68 ppm (C-6). The ring closure sites at quaternary carbons C-4 and C-6 were discovered to generate compound 1h. The molecular formula of 1h was confirmed as C17H16N3O2 by ESI-HRMS analysis, showing a pseudo-molecular ion peak [M + H]+ m/z 294.1249 (calcd for C17H16N3O2, 294.1242). On the basis of the previously given spectrum data, 4-(2-amino-6-(3-methoxy-phenyl)-pyrimidin-4-yl)-phenol was identified as the structure of 1h.
 | ||
| Scheme 1 Synthesis process of 2-amino-4,6-diarylpyrimidine derivatives 1a–1q. | ||
| Entry | Products | R1 | R2 | Yield (%) |
|---|---|---|---|---|
| 1 | 1a | 2′-OH | H | 41 |
| 2 | 1b | 2′-OH | 2′′-OH | 51 |
| 3 | 1c | 2′-OH | 3′′-OCH3 | 56 |
| 4 | 1d | 2′-OH | 4′′-OCH3 | 43 |
| 5 | 1e | 2′-OH | 4′′-Br | 33 |
| 6 | 1f | 2′-OH | 4′′-Cl | 31 |
| 7 | 1g | 3′-OCH3 | H | 36 |
| 8 | 1h | 3′-OCH3 | 4′′-OH | 48 |
| 9 | 1i | 3′-OCH3 | 4′′-OCH3 | 45 |
| 10 | 1j | 3′-OCH3 | 4′′-Br | 55 |
| 11 | 1k | 3′-OCH3 | 4′′-Cl | 48 |
| 12 | 1l | 4′-OCH3 | H | 47 |
| 13 | 1m | 4′-OCH3 | 4′′-OH | 42 |
| 14 | 1n | 4′-OCH3 | 4′′-OCH3 | 33 |
| 15 | 1o | 4′-OCH3 | 4′′-Br | 48 |
| 16 | 1p | 4′-OCH3 | 4′′-Cl | 64 |
| 17 | 1q | 4′-OH | H | 44 |
|
||||
|---|---|---|---|---|
| Position | 1H-NMR (ppm) | 13C-NMR (ppm) | Correlations | |
| HSQC | HMBC | |||
| 2 | — | 163.78 | — | — |
| 4 | — | 164.12 | — | — |
| 5 | 7.57 (1H, s) | 100.93 | C-5 | C-4, C-6, C-1′, C-1′′ |
| 6 | — | 164.68 | — | — |
| 1′ | — | 139.08 | — | — |
| 2′ | 7.72 (1H, s) | 112.00 | C-2′ | C-4, C-3′, C-4′, C-6′ |
| 3′ | — | 159.53 | — | — |
| 4′ | 7.07 (1H, dd, 2.4 Hz, 7.8 Hz) | 115.95 | C-4′ | C-2′, C-6′ |
| 5′ | 7.42 (1H, t, 7.8 Hz) | 129.56 | C-5′ | C-1′, C-3′ |
| 6′ | 7.77 (1H, d, 7.8 Hz) | 119.26 | C-6′ | C-4, C-2′, C-4′ |
| 1′′ | — | 127.99 | — | — |
| 2′′ | 8.09 (2H, d, 9.0 Hz) | 128.63 | C-2′′ | C-6, C-4′′, C-6′′ |
| 3′′ | 6.87 (2H, d, 9.0 Hz) | 115.25 | C-3′′ | C-1′′, C-2′′, C-4′′, C-5′′ |
| 4′′ | — | 159.74 | — | — |
| 5′′ | 6.87 (2H, d, 9.0 Hz) | 115.25 | C-5′′ | C-1′′, C-3′′, C-4′′, C-6′′ |
| 6′′ | 8.09 (2H, d, 9.0 Hz) | 128.63 | C-6′′ | C-6, C-2′′, C-4′′ |
| a | 3.85 (3H, s) | 55.22 | C-a | C-3′ |
| –NH2 | 6.58 (2H, s) | — | — | — |
| –OH | 9.89 (1H, s) | — | — | — |
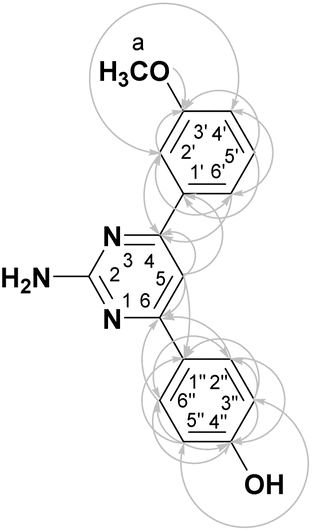
2.2. Cytotoxicity assay
|
||||
|---|---|---|---|---|
| Entry | Compound | R1 | R2 | IC50 ± SD (μM) |
| a ND: not determined. | ||||
| 1 | 1a | 2′-OH | H | 57.41 ± 1.76 |
| 2 | 1b | 2′-OH | 2′′-OH | ND |
| 3 | 1c | 2′-OH | 3′′-OCH3 | 82.04 ± 1.89 |
| 4 | 1d | 2′-OH | 4′′-OCH3 | ND |
| 5 | 1e | 2′-OH | 4′′-Br | 8.77 ± 0.55 |
| 6 | 1f | 2′-OH | 4-Cl | ND |
| 7 | 1g | 3′-OCH3 | H | 32.43 ± 1.59 |
| 8 | 1h | 3′-OCH3 | 4′′-OH | 48.42 ± 1.81 |
| 9 | 1i | 3′-OCH3 | 4′′-OCH3 | ND |
| 10 | 1j | 3′-OCH3 | 4-Br | 57.68 ± 1.73 |
| 11 | 1k | 3′-OCH3 | 4′′-Cl | ND |
| 12 | 1l | 4′-OCH3 | H | 61.94 ± 1.62 |
| 13 | 1m | 4′-OCH3 | 4′′-OH | 43.25 ± 1.42 |
| 14 | 1n | 4′-OCH3 | 4′′-OCH3 | ND |
| 15 | 1o | 4′-OCH3 | 4′′-Br | ND |
| 16 | 1p | 4′-OCH3 | 4′′-Cl | ND |
| 17 | 1q | 4′-OH | H | ND |
| 18 | Imatinib | 0.116 ± 0.002 | ||
When benzaldehydes and acetophenone (R2 = H) were combined, the compounds containing 2′-hydroxy (1a), 3′-methoxy (1g), and 4′-methoxy (1l) groups showed moderate anti-tumor activity (IC50 = 32.43–61.94 μM), whereas the 4-hydroxy group (1q) did not. It showed good activity in the four compounds mentioned above when a molecule was attached to only the 3′-methoxy group on the benzaldehyde ring of 4-position on the pyrimidine backbone. Therefore, the inhibitory activity against K562 cell line was further investigated by immobilizing R1 substituents (2′-hydroxy, 3′-methoxy, and 4′-methoxy) and modifying R2 substituted groups.
Methoxy, hydroxy, and halogen (chloro and bromo) groups were bonded to 4-position on the acetophenone skeleton and demonstrated their activity when compared with compounds 1a, 1g, and 1l. Unfortunately, the compounds with the methoxy group, such as 1d, 1i, and 1n, completely lost their anti-tumor activity. Compounds 1h and 1m exhibited a moderate antiproliferative effect in the presence of hydroxy groups, corresponding with IC50 values of 48.42 ± 1.81 and 43.25 ± 1.42 μM, respectively. The substitution of halogen groups such as chloro and bromo revealed a considerable variation in their bioactivity. Compounds with the chloro group (1f, 1k, and 1p) significantly decreased the inhibitory activity of compounds that contained the acetophenone ring only. Meanwhile, the presence of the bromo group (1e and 1i) resulted in moderate to good activity (IC50 = 8.77–57.68 μM), except for compound 1o. The antiproliferative effect of compound 1e was remarkably enhanced by the combination of 2′-hydroxy group on R1 and 4′′-bromo group on R2, as evidenced by its IC50 value of 8.77 ± 0.55 μM. Furthermore, adding 2′′-hydroxy (1b) and 3′′-methoxy (1c) groups to the acetophenone ring did not improve its inhibitory effect against the K562 cancer cell line.
In the summary, these findings revealed the correlation between the structure of 2-amino-4,6-diarylpyrimidine derivatives and bioactivity against the K562 cancer cell line. The substituted benzaldehydes (3′-OCH3, 4′-OCH3) at C-4 pyrimidine ring were combined with different acetophenones (3′′-OCH3, 4′′-OCH3, 4′′-Cl, 4′′-Br) at C-6 pyrimidine ring, which significantly reduced the anti-tumor activity. Significantly, the compounds were generated by combining the 4- and 6-position substituents (3-methoxybenzaldehyde and acetophenone, 2-hydroxybenzaldehyde and 4-bromoacetophenone) of the pyrimidine ring, thereby significantly boosting the antiproliferative activity against K562 cancer cell line. As a result, compounds 1e and 1g were chosen for further investigation into ABL1 tyrosine kinase inhibitory activity.
In this investigation, the concentration of ABL1 in the sample was measured by Human ABL1 Elisa Kit (FineTest, EH4452) according to the manufacturer's protocol. As seen in Fig. S102,† a significant decrease in ABL1 production was observed for all treated compounds compared with the control (p < 0.0001). As we can see in Table 4, compound 1e exhibited the strongest activity with 80% inhibition of ABL1 production at a concentration of 30 μM and IC50 value of 3.35 ± 0.58 μM. Compound 1g showed the weakest inhibition with an IC50 value of 35.16 ± 1.83 μM. The positive control imatinib revealed a significant decrease in ABL1 production in a dose-dependent manner. A similar trend was achieved in all tested compounds. On the basis of these findings, we can suggest that compound 1e has the potential for CML prevention and treatment. Several experiments, such as apoptosis, cell cycle arrest, and western blot, need to be conducted to determine the mechanism of action of this compound.
| Entry | Compound | IC50 ± SD (μM) |
|---|---|---|
| 1 | 1e | 3.35 ± 0.58 |
| 2 | 1g | 35.16 ± 1.83 |
| 3 | Imatinib | 0.069 ± 0.002 |
2.3. Computational analysis
 | ||
| Fig. 2 Results of redocking co-crystallized inhibitors onto ABL1 kinase. (A) Redocking of three imatinib conformations (in gray sticks) onto wild-type ABL1 showed a strong alignment with the co-crystallized ligand (in yellow sticks), with corresponding RMSD values. (B) Similarly, redocking of three nilotinib conformations (in gray sticks) onto mutant ABL1 (T334I-D382N) displayed a good match with the co-crystallized ligand (in blue sticks), along with their respective RMSD values. Zoomed-in views highlight the nearby residues and their interactions with the co-crystallized and re-docked ligands. | ||
The docking results of 17 2-amino-4,6-diarylpyrimidine derivatives on wild-type ABL1 are presented in Table 5. All investigated compounds exhibited docking affinities <−8.0 kcal mol−1, which were weaker than that of imatinib (−13.5 kcal mol−1). However, the docking score range of the synthesized compounds still suggested potential activity. The interaction diagrams of the best docking pose of each ligand are illustrated in Fig. S103.† Table 5 highlights the critical interactions of the ABL1–imatinib complex, which include hydrogen bonds with Glu286, Thr135, Met318, Ile360, and Asp381, as well as π–π interactions with Tyr253, Phe317, and Phe382. Similarly, most of our compounds exhibited π–π or hydrophobic interactions with Tyr253, Phe317, and Phe382, akin to imatinib. However, hydrogen bonding with Glu286 is crucial for the specific interactions that significantly contribute to the ligand–protein affinity and directly influence the inhibitory activity. Experimental studies have demonstrated that Glu286 is a conserved residue (in both wild-type and mutant variants) and forms an ion pair with Lys271 in the N-lobe of ABL1 to stabilize the phosphate group of ATP during phosphorylation. Consequently, many anti-ABL1 drugs, for both wild-type and mutant forms, form hydrogen bonds with Glu286. Notably, compound 1e, with an –OH substituent at 2′-position, was the only compound to form a hydrogen bond with Glu286 (with a distance of 1.74 Å), comparable to imatinib (2.32 Å) (Fig. S104†). Conversely, the orientation of the –OH or –OCH3 groups at 4′-position precluded hydrogen bonding with Glu286. In future studies, we aim to leverage these insights into structure–activity relationships (SARs) to design novel 2-amino-4,6-diarylpyrimidines capable of forming hydrogen bonds with Glu286 and other key residues such as Thr135, Met318, Ile360, and Asp381. According to the experimental and docking results, both compounds 1e and 1g that confirmed their ABL1 inhibitory activity in the above enzyme assay, were chosen for further analysis of binding stability to ABL1 through MD simulations and BFE calculations.
| Compound | IC50 (μM) (cell-based) | IC50 (μM) (enzyme) | ΔGvina (kcal mol−1) | Interaction with ABL1WT residues |
|---|---|---|---|---|
| a HBD: hydrogen bond donor, HBA: hydrogen bond acceptor, π–π: pi–pi stacking, Hyd: hydrophobic interaction. | ||||
| 1b | — | — | −9.0 | Thr315 (HBD), Glu316 (HBD, HBA), Met318 (HBD), Phe317 (π–π), Phe382 (π–π, Hyd), Leu248, Val256, Tyr253 (Hyd) |
| 1c | 82.04 ± 1.89 | — | −8.8 | Glu316 (HBD), Met318 (HBA), Phe317 (π–π), Phe382 (π–π, Hyd), Leu248, Tyr253, Val256 (Hyd) |
| 1a | 57.41 ± 1.76 | — | −8.8 | Thr315 (HBD), Met318 (HBA), Phe317 (π–π), Phe382 (π–π, Hyd), Leu248, Tyr253, Val256 (Hyd) |
| 1e | 8.77 ± 0.55 | 3.35 ± 0.58 | −8.6 | Glu286 (HBD), Ala269 (HBD, Hyd), Tyr253 (π–π), Phe382 (π–π, Hyd), Leu248, Met290, Val299 (Hyd) |
| 1f | — | — | −8.6 | Thr315 (HBD), Ala269 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Met290 (Hyd) |
| 1g | 32.43 ± 1.59 | 35.16 ± 1.83 | −8.6 | Phe317 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Val256, Gly321 (Hyd) |
| 1j | 57.68 ± 1.73 | — | −8.6 | Ala269 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Met290, Leu370 (Hyd) |
| 1o | — | — | −8.6 | Ala269 (HBD, Hyd), Tyr253 (π–π), Phe382 (π–π, Hyd), Leu248, Met290, Asp381 (Hyd) |
| 1d | — | — | −8.5 | Ala269 (HBD), Thr315 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Met290, Ala380, Asp381 (Hyd) |
| 1k | — | — | −8.5 | Ala269 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Met290, Leu370 (Hyd) |
| 1p | — | — | −8.5 | Ala269 (HBD, Hyd), Tyr253 (π–π), Phe382 (π–π, Hyd), Leu248, Ala269, Met290, Asp381 (Hyd) |
| 1i | — | — | −8.4 | Ala269 (HBD, Hyd), Tyr253 (π–π), Phe382 (π–π, Hyd), Leu248, Met290, Leu370, Ala380, Asp381 (Hyd) |
| 1n | — | — | −8.4 | Ala269 (HBD, Hyd), Tyr253 (π–π), Phe382 (π–π, Hyd), Leu248, Met290, Asp381 (Hyd) |
| 1h | 48.42 ± 1.81 | — | −8.3 | Ala269 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Leu370, Met290 (Hyd) |
| 1m | 43.25 ± 1.42 | — | −8.3 | Ala269 (HBD, Hyd), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Met290 (Hyd) |
| 1q | — | — | −8.2 | Thr315 (HBA), Phe317 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Gly321 (Hyd) |
| 1l | 61.94 ± 1.62 | — | −8.2 | Ala269 (HBD), Tyr253 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Ala269, Met290, Leu370, Ala380, Asp381 (Hyd) |
| Imatinib | 0.116 ± 0.002 | 0.069 ± 0.002 | −13.5 | Glu286 (HBD), Thr315 (HBD, Hyd), Met318 (HBA), Ile360 (HBD), Asp381 (HBA, Hyd), Tyr253 (π–π, Hyd), Phe317 (π–π, Hyd), Phe382 (π–π, Hyd), Leu248, Val256, Lys271, Val289, Met290, Leu370 (Hyd) |
 | ||
| Fig. 3 Analysis of MD simulation trajectories of ABL1 with imatinib (blue), 1e (orange), and 1g (green). (A) RMSD of the alpha carbon atoms of wild-type ABL1, (B) RMSF of ABL1 residues, with the red boxes indicating residues within the active site, (C) SASA, (D) radius of gyration of the enzyme, (E) RMSD of the heavy atoms of the ligands, (F) MM-GBSA binding free energy of the ligands to ABL1. | ||
The RMSF plot in Fig. 3B reflects the average fluctuations of the ABL1 residues. Here, the residues at the enzyme's active site are considered (Leu248–Met318 and Ile360–Phe382). Overall, most of the residues in the binding site of ABL1 in complexes with the three ligands exhibit similar fluctuations with amplitudes less than 2 Å, except for the Lys274–Glu279 loop (Fig. S105†). Interestingly, the presence of our synthetic compounds caused a decrease in the fluctuations of the residues in this loop compared with imatinib. However, the binding of 1g resulted in increased fluctuations of Arg307 within the Arg307–Phe311 loop.
Fig. 3E illustrates the ligands' fluctuations, as reflected by the RMSD values of the heavy atoms of the compounds. Both 1e and 1g exhibit motion amplitudes less than 1 Å, indicating high stability and the ability to maintain a consistent binding mode with ABL1 throughout the simulation (Fig. S106†). Imatinib shows a change in RMSD starting around 53 ns but eventually stabilizes toward the end of the trajectory. The stability of the ligands is further correlated with the binding free energy in Fig. 3F. Once imatinib reaches equilibrium, the ΔGMM-GBSA also becomes stable. Although 1e and 1g display higher BFE than imatinib does (−37.33 ± 2.77 and −35.72 ± 2.56 vs. −67.31 ± 5.25 kcal mol−1), their binding remains highly stable over the MD simulation time. The calculated BFE values were consistent with the experimental inhibitory activity of the compounds, as 1e exhibited a lower IC50 than 1g but a higher IC50 than imatinib.
The interaction fingerprints of 1e and 1g with ABL1 were analyzed using the MDAnalysis and ProLIF programs, resulting in the 2D diagrams shown in Fig. 4. Notably, the amino groups of the 2-amino-4,6-diarylpyrimidine derivatives play a crucial role in forming hydrogen bonds with residues in the enzyme's active site. Specifically, 1e forms hydrogen bonds with Ala269 and Ile313 with occupancies of 72% and 38%, respectively, while 1g maintains hydrogen bonding with Thr315 and Glu316 at occupancies of 85% and 96%. The phenyl groups of both compounds participate in π-stacking interactions with Tyr253, Phe317, and Phe382. Additionally, the ligands interact with several other hydrophobic residues within the binding site, such as Leu248, Val256, Lys271, Met290, Val299, Leu370, and Ala380. van der Waals interactions also significantly affect the ligand's binding affinity to the enzyme and are detailed in Table S5.†
 | ||
| Fig. 4 Occupancy of interactions between 1e and 1g with ABL1 along the 100 ns MD simulation trajectories. | ||
The results indicate that 1e and 1g are promising and stable inhibitors of wild-type ABL1. The IC50 and BFE values highlight the potential of these compounds and suggest that they could be further structurally developed to achieve stronger binding affinity and inhibitory activity.
3. Conclusions
In summary, 17 2-amino-4,6-diarylpyrimidine derivatives with various substituted groups at the 4- and 6-positions, including 1 novel compound 1h, were synthesized using a two-step approach, which provided several advantages such as time-saving and a simple and mild protocol. Aldol condensation (step 1) of benzaldehydes and acetophenones to produce intermediate chalcones and ring closure condensation (step 2) of chalcones and GHCl were carried out under microwave irradiation to obtain the desired 2-amino-4,6-diaryl-pyrimidines, with moderate yields and structure characterization by FTIR, 1D- and 2D-NMR, and HRMS analysis. The antiproliferative effect of the synthesized derivatives was then assessed in vitro against the CML K562 cell line using the MTT assay, and the active compounds were tested for ABL1 tyrosine kinase inhibition. Compound 1e had considerable cytotoxic activity against K562 cancer cells and ABL1 tyrosine kinase, with IC50 values of 8.77 ± 0.55 μM and 3.35 ± 0.58 μM, respectively. The molecular docking experiment and MD simulations demonstrated that compounds 1e and 1g are promising and stable inhibitors of wild-type ABL1 as well as potential inhibitors of the mutant ABL1 (Table 6).| Ligand | ΔGvina ABL-1 mutant (kcal mol−1) | ΔΔGdock (kcal mol−1) | BFEABL-1 mutant (ΔGbind MM-GBSA, kcal mol−1) | ΔΔGbind MM-GBSA (kcal mol−1) |
|---|---|---|---|---|
| PH2HBR | –8.6 | 0 | –36.49 | 0.84 |
| PH3MA | –9.5 | –0.9 | –36.08 | –0.36 |
| Imatinib | –12.4 | 1.1 | –58.15 | 9.16 |
4. Experimental
4.1. Materials and measurements
All the reaction reagents were purchased from Thermo Scientific Chemicals (Belgium). The solvent employed in the procedure required no further purification and was supplied from Chemsol (Vietnam). Merck silica gel 60 F254 plates were used for thin-layer chromatography (TLC). A CEM Discover 2.0 microwave synthesizer was employed in the reaction process. A Bruker Tensor 27 FTIR spectrometer (USA) was used to record Fourier-transform infrared (FTIR) data, with absorption bands measured in wave number (cm−1). A Bruker Advance Neo spectrometer was applied to record 1H (600 MHz) and 13C (125 MHz or 150 MHz) nuclear magnetic resonance (NMR) data, with dimethyl sulfoxide (DMSO) as the solvent. Chemical shifts (δ) were expressed in ppm, coupling constants (J) in hertz, and signal multiplicities were presented as s (singlet), d (doublet), t (triplet), dd (doublet of doublets), dt (doublet of triplets), and m (multiplet). Electrospray ionization (ESI)-high-resolution mass spectrometry (HRMS) data were recorded using ESI-quadrupole time-of-flight (QTOF)-mass spectrometry on a Sciex X500R QTOF system (USA).4.2. General procedure for the synthesis of 2-amino-4,6-diarylpyrimidine derivatives
Heterocyclic 2-amino-4,6-diarylpyrimidines were synthesized following two steps, as illustrated in Scheme 1. Initially, 17 intermediate chalcones were produced by the aldol condensation of benzaldehydes and acetophenones. The pure chalcones were then ring-closed with GHCl to generate desired derivatives.In the first step, a mixture of benzaldehydes (3 mmol), acetophenones (3 mmol), and potassium hydroxide (6 mmol) was added to a vessel along with 4 mL of EG. The reaction was conducted for 10 to 30 minutes under MW (100 W, 80 °C) and monitored using TLC to verify the process completion. The reaction mixture was then placed in cold water and neutralized to pH 7 by HCl, followed by overnight refrigeration. After that, the precipitate was removed by filtering, carefully washing with water, drying, and recrystallizing it from methanol to produce pure intermediates.
In the second step, a mixture of chalcone (1 mmol), GHCl (2 mmol), sodium hydroxide (2 M), and 2 mL of pyridine was introduced into a 25 mL vessel. The reaction was irradiated for 5 to 10 minutes under MW (180 W, 100 °C) and monitored by TLC. Following reaction completion, the mixture was added to ice-cooled water and evaporated in a hood fume. The crude product was then filtered, washed several times with water, vacuum-dried, and recrystallized from methanol, yielding the pure products 1a–1q.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v); FTIR (KBr, ν (cm−1)): 3499, 3446 (NH2), 3345 (OH), 1632 (CN), 1543 (CC), 1247 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.94 (m, 2H, H-3′, H-5′), 7.22 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.54 (m, 3H, H-3′′, H-4′′, H-5′′), 7.82 (s, 1H, H-5), 8.25 (m, 3H, H-6′, H-2′′, H-6′′), 13.99 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 99.89 (C-5), 117.51 (C-1′), 118.02 (C-3′), 118.62 (C-5′), 127.12 (C-2′′, C-6′′), 128.02 (C-4′′), 128.59 (C-3′′, C-5′′), 130.73 (C-6′), 132.60 (C-4′), 136.99 (C-1′′), 160.30 (C-2′), 161.29 (C-2), 165.19 (C-6), 165.33 (C-4); ESI-HRMS (MeOH): m/z = 264.1134, 264.1137 calcd [M + H]+ for C16H14N3O.:1 v/v); FTIR (KBr, ν (cm−1)): 3449, 3342 (NH2), 3219 (OH), 1646 (CN), 1548 (CC), 1234 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.96 (m, 4H, H-3′, H-5′, H-3′′, H-5′′), 7.41 (dt, 2H, J = 1.8 Hz, J = 8.4 Hz, H-4′, H-4′′), 7.63 (s, 2H, NH2), 7.91 (s, 1H, H-5), 8.29 (dd, 2H, J = 1.2 Hz, J = 8.4 Hz, H-6′, H-6′′), 13.75 (s, 2H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 98.26 (C-5), 117.39 (C-1′, C-1′′), 118.12 (C-3′, C-3′′), 118.71 (C-5′, C-5′′), 128.36 (C-6′, C-6′′), 132.95 (C-4′, C-4′′), 158.94 (C-2), 160.32 (C-2′, C-2′′), 165.62 (C-4, C-6); ESI-HRMS (MeOH): m/z = 280.1091, 280.1086 calcd [M + H]+ for C16H14N3O2.:3 v/v); FTIR (KBr, ν (cm−1)): 3458, 3341 (NH2), 3227 (OH), 1636 (CN), 1545 (CC), 1243 (C–N), 1043 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.87 (s, 3H, OCH3), 6.95 (m, 2H, H-3′, H-5′), 7.12 (dd, 1H, J = 2.4, J = 8.4 Hz, H-4′′), 7.21 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.46 (t, 1H, J = 8.4 Hz, H-5′′), 7.78 (s, 1H, H-2′′), 7.81 (s, 1H, H-5), 7.84 (d, 1H, J = 7.8 Hz, H-6′′), 8.26 (d, 1H, J = 8.4 Hz, H-6′), 13.98 (s, 1H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.26 (OCH3), 100.06 (C-5), 112.29 (C-2′′), 116.44 (C-4′′), 117.49 (C-1′), 117.99 (C-3′), 118.56 (C-5′), 119.53 (C-6′′), 128.08 (C-6′), 129.63 (C-5′′), 132.57 (C-4′), 138.50 (C-1′′), 159.56 (C-3′′), 160.29 (C-2′), 161.22 (C-2), 165.11 (C-6), 165.19 (C-4); ESI-HRMS (MeOH): m/z = 294.1247, 294.1243 calcd [M + H]+ for C17H16N3O2.:3 v/v); FTIR (KBr, ν (cm−1)): 3464, 3344 (NH2), 3219 (OH), 1579 (CN), 1538 (CC), 1250 (C–N), 1031 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.94 (m, 2H, H-3′, H-5′), 7.09 (m, 2H, H-3′′, H-5′′), 7.12 (s, 2H, NH2), 7.38 (dt, 1H, J = 1.8 Hz, J = 8.4 Hz, H-4′), 7.77 (s, 1H, H-5), 8.25 (m, 3H, H-6′, H-2′′, H-6′′), 14.08 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.31 (OCH3), 98.99 (C-5), 113.93 (C-3′′, C-5′′), 117.57 (C-1′), 117.98 (C-3′), 118.52 (C-5′), 127.90 (C-6′), 128.76 (C-2′′, C-6′′), 129.23 (C-1′′), 132.43 (C-4′), 160.30 (C-2′), 161.14 (C-6), 161.49 (C-2, C-4′′), 164.86 (C-4); ESI-HRMS (MeOH): m/z = 294.1248, 294.1243 calcd [M + H]+ for C17H16N3O2.:1 v/v); FTIR (KBr, ν (cm−1)): 3498, 3348 (NH2), 3215 (OH), 1638 (CN), 1545 (CC), 1253 (C–N), 579 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.95 (m, 2H, H-3′, H-5′), 7.25 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.75 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.85 (s, 1H, H-5), 8.24 (m, 3H, H-6′, H-2′′, H-6′′), 13.92 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 98.80 (C-5), 117.43 (C-1′), 118.02 (C-3′), 118.59 (C-5′), 124.42 (C-4′′), 128.07 (C-6′), 129.13 (C-2′′, C-6′′), 131.56 (C-3′′, C-5′′), 132.67 (C-4′), 136.14 (C-1′′), 160.29 (C-2′), 161.26 (C-2), 164.06 (C-6), 165.45 (C-4); ESI-HRMS (MeOH): m/z = 342.0239, 342.0242 calcd [M + H]+ for C16H13BrN3O.:3 v/v); FTIR (KBr, ν (cm−1)): 3503 (OH), 3345, 3215 (NH2), 1640 (CN), 1547 (CC), 1264 (C–N), 747 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.95 (m, 2H, H-3′, H-5′), 7.24 (s, 2H, NH2), 7.39 (t, 1H, J = 7.8 Hz, H-4′), 7.61 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.85 (s, 1H, H-5), 8.24 (d, 1H, J = 7.8 Hz, H-6′), 8.29 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′), 13.93 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 99.83 (C-5), 117.43 (C-1′), 118.01 (C-3′), 118.58 (C-5′), 128.07 (C-6′), 128.62 (C-3′′, C-5′′), 128.90 (C-2′′, C-6′′), 132.66 (C-4′), 135.52 (C-1′′), 135.78 (C-4′′), 160.29 (C-2′), 161.25 (C-2), 163.96 (C-6), 165.43 (C-4); ESI-HRMS (MeOH): m/z = 298.0753, 298.0747 calcd [M + H]+ for C16H13ClN3O.:3 v/v); FTIR (KBr, ν (cm−1)): 3450, 3429 (NH2), 1632 (CN), 1566 (CC), 1260 (C–N), 1038 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.73 (s, 2H, NH2), 7.09 (dd, 1H, J = 2.4 Hz, J = 7.8 Hz, H-4′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.52 (m, 3H, H-3′′, H-4′′, H-5′′); 7.69 (s, 1H, H-5), 7.76 (s, 1H, H-2′), 7.80 (d, 1H, J = 7.8 Hz, H-6′), 8.23 (m, 2H, H-2′′, H-6′′); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 102.02 (C-5), 112.13 (C-2′), 116.13 (C-4′), 119.36 (C-6′), 126.96 (C-2′′, C-6′′), 128.54 (C-3′′, C-5′′), 129.63 (C-5′), 130.38 (C-4′′), 137.30 (C-1′′), 138.86 (C-1′), 159.57 (C-3′), 163.92 (C-2), 164.67 (C-6), 164.88 (C-4); ESI-HRMS (MeOH): m/z = 278.1298, 278.1293 calcd [M + H]+ for C17H16N3O.:6 v/v); FTIR (KBr, ν (cm−1)): 3517 (OH), 3403 (NH), 1608 (CN), 1542 (CC), 1241 (C–N), 1037 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.58 (2H, s, NH2), 6.87 (d, 2H, J = 9.0 Hz, H-3′′, H-5′′), 7.07 (dd, 1H, J = 2.4 Hz, J = 7.8 Hz, H-4′), 7.42 (t, 1H, J = 7.8 Hz, H-5′), 7.57 (s, 1H, H-5), 7.72 (s, 1H, H-2′), 7.77 (d, 1H, J = 7.8 Hz, H-6′), 8.09 (d, 2H, J = 9.0 Hz, H-2′′, H-6′′), 9.89 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.22 (OCH3), 100.93 (C-5), 112.00 (C-2′), 115.25 (C-3′′, C-5′′), 115.95 (C-4′), 119.26 (C-6′), 127.99 (C-1′′), 128.63 (C-2′′, C-6′′), 129.56 (C-5′), 139.08 (C-1′), 159.53 (C-3′), 159.74 (C-4′′), 163.78 (C-2), 164.12 (C-4), 164.68 (C-6); ESI-HRMS (MeOH): m/z = 294.1249, 294.1242 calcd [M + H]+ for C17H16N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3428, 3310 (NH2), 1641 (CN), 1566 (CC), 1245 (C–N), 1032 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.64 (s, 2H, NH2), 7.09 (m, 3H, H-4′, H-3′′, H-5′′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.64 (s, 1H, H-5), 7.75 (s, 1H, H-2), 7,79 (d, 1H, J = 7.8 Hz, H-6′), 8,21 (d, 2H, J = 7.2 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.21 (OCH3), 55.25 (OCH3), 101.21 (C-5), 112.09 (C-2′), 113.86 (C-3′′, C-5′′), 115.95 (C-4′), 119.28 (C-6′), 128.52 (C-2′′, C-6′′), 129.55 (C-5′), 129.57 (C-1′′), 138.99 (C-1′), 159.52 (C-3′), 161.19 (C-4′′), 163.80 (C-2), 164.31 (C-4), 164.39 (C-6); ESI-HRMS (MeOH): m/z = 308.1400, 308.1399 calcd [M + H]+ for C18H18N3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3459, 3318 (–NH2), 1638 (CN), 1542 (CC), 1260 (C–N), 1034 (OCH3), 593 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.86 (s, 3H, OCH3), 6.77 (s, 2H, NH2), 7.10 (dd, J = 2.4 Hz, J = 6.0 Hz, 1H, H-4′), 7.45 (t, 1H, J = 7.8 Hz, H-5′), 7.73 (m, 3H, H-5, H-3′′, H-5′′), 7.76 (s, 1H, H-2), 7.81 (d, 1H, J = 7.8 Hz, H-6′), 8.20 (d, 2H, J = 7.2 Hz, H-2′′, H-6′′); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.32 (OCH3), 101.94 (C-5), 112.22 (C-2′), 116.29 (C-4′), 119.45 (C-6′), 124.11 (C-4′′), 129.08 (C-2′′, C-6′′), 129.73 (C-5′), 131.61 (C-3′′, C-5′′), 136.51 (C-1′′), 138.75 (C-1′), 159.63 (C-3′), 163.73 (C-6), 163.95 (C-2), 164.99 (C-4); ESI-HRMS (MeOH): m/z = 356.0397, 356.0398 calcd [M + H]+ for C17H15BrN3O.:5 v/v); FTIR (KBr, ν (cm−1)): 3319, 3204 (NH2), 1639 (CN), 1544 (CC), 1261 (C–N), 1035 (OCH3), 784 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.76 (s, 2H, NH2), 7.09 (dd, J = 2.4 Hz, J = 7.8 Hz, 1H, H-4′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.59 (d, 2H, J = 6.6 Hz, H-3′′, H-5′′), 7.72 (s, 1H, H-5), 7.76 (s, 1H, H-2′), 7.80 (d, 1H, J = 7.8 Hz, H-6′), 8.26 (d, 2H, J = 6.6 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.26 (OCH3), 101.91 (C-5), 112.19 (C-2′), 116.21 (C-4′), 119.40 (C-6′), 128.60 (C-3′′, C-5′′), 128.77 (C-2′′, C-6′′), 129.65 (C-5′), 135.19 (C-4′′), 136.11 (C-1′′), 138.73 (C-1′), 159.58 (C-3′), 163.57 (C-6), 163.90 (C-2), 164.90 (C-4); ESI-HRMS (MeOH): m/z = 312.0902, 312.0904 calcd [M + H]+ for C17H15ClN3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3364, 3326 (NH2), 1644 (CN), 1587 (CC), 1258 (C–N), 1031 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 6.64 (s, 2H, NH2), 7.08 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.54 (m, 3H, H-3′′, H-4′′, H-5′′), 7.65 (s, 1H, H-5), 8.22 (m, 4H, H-2′, H-6′, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.30 (OCH3), 101.07 (C-5), 113.93 (C-3′, C-5′), 126.90 (C-2′, C-6′), 128.53 (C-2′′, C-6′′), 128.54 (C-3′′, C-5′′), 129.62 (C-1′), 130.28 (C-4′′), 137.48 (C-1′′), 161.23 (C-4′), 163.90 (C-2), 164.42 (C-6), 164.56 (C-4); ESI-HRMS (MeOH): m/z = 278.1298, 278.1293 calcd [M + H]+ for C17H16N3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3504 (OH), 3387 (NH), 1568 (CN), 1537 (CC), 1233 (C–N), 1176 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.83 (s, 3H, OCH3), 6.49 (s, 2H, NH2), 6.88 (d, 2H, J = 6.6 Hz, H-3′′, H-5′′), 7.05 (d, 2H, J = 6.6 Hz, H-3′, H-5′), 7.53 (s, 1H, H-5), 8.08 (d, 2H, J = 6.6 Hz, H-2′′, H-6′′), 8.17 (d, 2H, J = 6.6 Hz, H-2′, H-6′), 9.90 (s, 1H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 99.97 (C-5), 113.84 (C-3′, C-5′), 115.24 (C-3′′, C-5′′), 128.15 (C-1′′), 128.38 (C-2′, C-6′), 128.51 (C-2′′, C-6′′), 129.82 (C-1′), 159.64 (C-4′′), 161.05 (C-4′), 163.74 (C-4), 163.89 (C-6), 164.38 (C-2); ESI-HRMS (MeOH): m/z = 294.1244, 294.1243 calcd [M + H]+ for C17H16N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3331, 3202 (NH2), 1648 (CN), 1584 (CC), 1236 (C–N), 1028 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 6H, OCH3), 6.55 (s, 2H, NH2), 7.07 (d, 4H, J = 9.0 Hz, H-3′, H-5′, H-3′′, H-5′′), 7.59 (s, 1H, H-5), 8.20 (d, 4H, J = 9.0 Hz, H-2′, H-6′, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 100.24 (C-5), 113.84 (C-3′, C-5′, C-3′′, C-5′′), 128.43 (C-2′, C-6′, C-2′′, C-6′′), 129.74 (C-1′, C-1′′), 161.09 (C-4′, C-4′′), 163.77 (C-2), 164.07 (C-4, C-6); ESI-HRMS (MeOH): m/z = 308.1400, 308.1399 calcd [M + H]+ for C18H18N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3367, 3212 (NH2), 1645 (CN), 1588 (CC), 1255 (C–N), 1033 (OCH3), 592 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.89 (s, 3H, OCH3), 6.73 (s, 2H, NH2), 7.12 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.73 (s, 1H, H-5), 7.77 (d, 2H, J = 9.0 Hz, H-3′′, H-5′′), 8.23 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′), 8.26 (d, 2H, J = 9.0 Hz, H-2′, H-6′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.29 (OCH3), 100.09 (C-5), 113.91 (C-3′, C-5′), 123.89 (C-4′′), 128.56 (C-2′, C-6′), 128.93 (C-2′′, C-6′′), 129.47 (C-1′), 131.51 (C-3′′, C-5′′), 136.64 (C-1′′), 161.29 (C-4′), 163.29 (C-6), 163.86 (C-2), 164.65 (C-4); ESI-HRMS (MeOH): m/z = 356.0397, 356.0398 calcd [M + H]+ for C17H15BrN3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3331, 3210 (NH2), 1644 (CN), 1562 (CC), 1238 (C–N), 1032 (OCH3), 815 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 6.68 (s, 2H, NH2), 7.07 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.58 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.67 (s, 1H, H-5), 8.21 (d, 2H, J = 9.0 Hz, H-2′, H-6′), 8.25 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.27 (OCH3), 100.93 (C-5), 113.89 (C-3′, C-5′), 128.55 (C-2′′, C-3′′, C-5′′, C-6′′), 128.67 (C-2′, C-6′), 129.47 (C-1′), 135.02 (C-4′′), 136.26 (C-1′′), 161.27 (C-4′), 163.19 (C-6), 163.84 (C-2), 164.61 (C-4); ESI-HRMS (MeOH): m/z = 312.0908, 312.0904 calcd [M + H]+ for C17H15ClN3O.:1 v/v); FTIR (KBr, ν (cm−1)): 3506, 3399 (NH2), 3057 (OH), 1602 (CN), 1539 (CC), 1231 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.59 (s, 2H, NH2), 6.89 (d, 2H, J = 8.4 Hz, H-3′, H-5′), 7.51 (m, 3H, H-3′′, H-4′′, H-5′′); 7.59 (s, 1H, H-5), 8.10 (d, 2H, J = 9.0 Hz, H-2′, H-6′), 8.19 (m, 2H, H-2′′, H-6′′), 9.91 (s, 1H, OH), 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 100.78 (C-5), 115.28 (C-3′, C-5′), 126.82 (C-2′′, C-6′′), 128.03 (C-1′), 128.49 (C-3′′, C-5′′), 128.58 (C-2′, C-6′), 130.17 (C-4′′), 137.54 (C-1′′), 159.72 (C-4′), 163.84 (C-2), 164.34 (C-6), 164.69 (C-4); ESI-HRMS (MeOH): m/z = 264.1141, 264.1137 calcd [M + H]+ for C16H14N3O.
:3 v/v); FTIR (KBr, ν (cm−1)): 3499, 3446 (NH2), 3345 (OH), 1632 (CN), 1543 (CC), 1247 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.94 (m, 2H, H-3′, H-5′), 7.22 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.54 (m, 3H, H-3′′, H-4′′, H-5′′), 7.82 (s, 1H, H-5), 8.25 (m, 3H, H-6′, H-2′′, H-6′′), 13.99 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 99.89 (C-5), 117.51 (C-1′), 118.02 (C-3′), 118.62 (C-5′), 127.12 (C-2′′, C-6′′), 128.02 (C-4′′), 128.59 (C-3′′, C-5′′), 130.73 (C-6′), 132.60 (C-4′), 136.99 (C-1′′), 160.30 (C-2′), 161.29 (C-2), 165.19 (C-6), 165.33 (C-4); ESI-HRMS (MeOH): m/z = 264.1134, 264.1137 calcd [M + H]+ for C16H14N3O.:1 v/v); FTIR (KBr, ν (cm−1)): 3449, 3342 (NH2), 3219 (OH), 1646 (CN), 1548 (CC), 1234 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.96 (m, 4H, H-3′, H-5′, H-3′′, H-5′′), 7.41 (dt, 2H, J = 1.8 Hz, J = 8.4 Hz, H-4′, H-4′′), 7.63 (s, 2H, NH2), 7.91 (s, 1H, H-5), 8.29 (dd, 2H, J = 1.2 Hz, J = 8.4 Hz, H-6′, H-6′′), 13.75 (s, 2H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 98.26 (C-5), 117.39 (C-1′, C-1′′), 118.12 (C-3′, C-3′′), 118.71 (C-5′, C-5′′), 128.36 (C-6′, C-6′′), 132.95 (C-4′, C-4′′), 158.94 (C-2), 160.32 (C-2′, C-2′′), 165.62 (C-4, C-6); ESI-HRMS (MeOH): m/z = 280.1091, 280.1086 calcd [M + H]+ for C16H14N3O2.:3 v/v); FTIR (KBr, ν (cm−1)): 3458, 3341 (NH2), 3227 (OH), 1636 (CN), 1545 (CC), 1243 (C–N), 1043 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.87 (s, 3H, OCH3), 6.95 (m, 2H, H-3′, H-5′), 7.12 (dd, 1H, J = 2.4, J = 8.4 Hz, H-4′′), 7.21 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.46 (t, 1H, J = 8.4 Hz, H-5′′), 7.78 (s, 1H, H-2′′), 7.81 (s, 1H, H-5), 7.84 (d, 1H, J = 7.8 Hz, H-6′′), 8.26 (d, 1H, J = 8.4 Hz, H-6′), 13.98 (s, 1H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.26 (OCH3), 100.06 (C-5), 112.29 (C-2′′), 116.44 (C-4′′), 117.49 (C-1′), 117.99 (C-3′), 118.56 (C-5′), 119.53 (C-6′′), 128.08 (C-6′), 129.63 (C-5′′), 132.57 (C-4′), 138.50 (C-1′′), 159.56 (C-3′′), 160.29 (C-2′), 161.22 (C-2), 165.11 (C-6), 165.19 (C-4); ESI-HRMS (MeOH): m/z = 294.1247, 294.1243 calcd [M + H]+ for C17H16N3O2.:3 v/v); FTIR (KBr, ν (cm−1)): 3464, 3344 (NH2), 3219 (OH), 1579 (CN), 1538 (CC), 1250 (C–N), 1031 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.94 (m, 2H, H-3′, H-5′), 7.09 (m, 2H, H-3′′, H-5′′), 7.12 (s, 2H, NH2), 7.38 (dt, 1H, J = 1.8 Hz, J = 8.4 Hz, H-4′), 7.77 (s, 1H, H-5), 8.25 (m, 3H, H-6′, H-2′′, H-6′′), 14.08 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.31 (OCH3), 98.99 (C-5), 113.93 (C-3′′, C-5′′), 117.57 (C-1′), 117.98 (C-3′), 118.52 (C-5′), 127.90 (C-6′), 128.76 (C-2′′, C-6′′), 129.23 (C-1′′), 132.43 (C-4′), 160.30 (C-2′), 161.14 (C-6), 161.49 (C-2, C-4′′), 164.86 (C-4); ESI-HRMS (MeOH): m/z = 294.1248, 294.1243 calcd [M + H]+ for C17H16N3O2.:1 v/v); FTIR (KBr, ν (cm−1)): 3498, 3348 (NH2), 3215 (OH), 1638 (CN), 1545 (CC), 1253 (C–N), 579 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.95 (m, 2H, H-3′, H-5′), 7.25 (s, 2H, NH2), 7.39 (dt, 1H, J = 1.2 Hz, J = 8.4 Hz, H-4′), 7.75 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.85 (s, 1H, H-5), 8.24 (m, 3H, H-6′, H-2′′, H-6′′), 13.92 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 98.80 (C-5), 117.43 (C-1′), 118.02 (C-3′), 118.59 (C-5′), 124.42 (C-4′′), 128.07 (C-6′), 129.13 (C-2′′, C-6′′), 131.56 (C-3′′, C-5′′), 132.67 (C-4′), 136.14 (C-1′′), 160.29 (C-2′), 161.26 (C-2), 164.06 (C-6), 165.45 (C-4); ESI-HRMS (MeOH): m/z = 342.0239, 342.0242 calcd [M + H]+ for C16H13BrN3O.:3 v/v); FTIR (KBr, ν (cm−1)): 3503 (OH), 3345, 3215 (NH2), 1640 (CN), 1547 (CC), 1264 (C–N), 747 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.95 (m, 2H, H-3′, H-5′), 7.24 (s, 2H, NH2), 7.39 (t, 1H, J = 7.8 Hz, H-4′), 7.61 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.85 (s, 1H, H-5), 8.24 (d, 1H, J = 7.8 Hz, H-6′), 8.29 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′), 13.93 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 99.83 (C-5), 117.43 (C-1′), 118.01 (C-3′), 118.58 (C-5′), 128.07 (C-6′), 128.62 (C-3′′, C-5′′), 128.90 (C-2′′, C-6′′), 132.66 (C-4′), 135.52 (C-1′′), 135.78 (C-4′′), 160.29 (C-2′), 161.25 (C-2), 163.96 (C-6), 165.43 (C-4); ESI-HRMS (MeOH): m/z = 298.0753, 298.0747 calcd [M + H]+ for C16H13ClN3O.:3 v/v); FTIR (KBr, ν (cm−1)): 3450, 3429 (NH2), 1632 (CN), 1566 (CC), 1260 (C–N), 1038 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.73 (s, 2H, NH2), 7.09 (dd, 1H, J = 2.4 Hz, J = 7.8 Hz, H-4′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.52 (m, 3H, H-3′′, H-4′′, H-5′′); 7.69 (s, 1H, H-5), 7.76 (s, 1H, H-2′), 7.80 (d, 1H, J = 7.8 Hz, H-6′), 8.23 (m, 2H, H-2′′, H-6′′); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 102.02 (C-5), 112.13 (C-2′), 116.13 (C-4′), 119.36 (C-6′), 126.96 (C-2′′, C-6′′), 128.54 (C-3′′, C-5′′), 129.63 (C-5′), 130.38 (C-4′′), 137.30 (C-1′′), 138.86 (C-1′), 159.57 (C-3′), 163.92 (C-2), 164.67 (C-6), 164.88 (C-4); ESI-HRMS (MeOH): m/z = 278.1298, 278.1293 calcd [M + H]+ for C17H16N3O.:6 v/v); FTIR (KBr, ν (cm−1)): 3517 (OH), 3403 (NH), 1608 (CN), 1542 (CC), 1241 (C–N), 1037 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.58 (2H, s, NH2), 6.87 (d, 2H, J = 9.0 Hz, H-3′′, H-5′′), 7.07 (dd, 1H, J = 2.4 Hz, J = 7.8 Hz, H-4′), 7.42 (t, 1H, J = 7.8 Hz, H-5′), 7.57 (s, 1H, H-5), 7.72 (s, 1H, H-2′), 7.77 (d, 1H, J = 7.8 Hz, H-6′), 8.09 (d, 2H, J = 9.0 Hz, H-2′′, H-6′′), 9.89 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.22 (OCH3), 100.93 (C-5), 112.00 (C-2′), 115.25 (C-3′′, C-5′′), 115.95 (C-4′), 119.26 (C-6′), 127.99 (C-1′′), 128.63 (C-2′′, C-6′′), 129.56 (C-5′), 139.08 (C-1′), 159.53 (C-3′), 159.74 (C-4′′), 163.78 (C-2), 164.12 (C-4), 164.68 (C-6); ESI-HRMS (MeOH): m/z = 294.1249, 294.1242 calcd [M + H]+ for C17H16N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3428, 3310 (NH2), 1641 (CN), 1566 (CC), 1245 (C–N), 1032 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.64 (s, 2H, NH2), 7.09 (m, 3H, H-4′, H-3′′, H-5′′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.64 (s, 1H, H-5), 7.75 (s, 1H, H-2), 7,79 (d, 1H, J = 7.8 Hz, H-6′), 8,21 (d, 2H, J = 7.2 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.21 (OCH3), 55.25 (OCH3), 101.21 (C-5), 112.09 (C-2′), 113.86 (C-3′′, C-5′′), 115.95 (C-4′), 119.28 (C-6′), 128.52 (C-2′′, C-6′′), 129.55 (C-5′), 129.57 (C-1′′), 138.99 (C-1′), 159.52 (C-3′), 161.19 (C-4′′), 163.80 (C-2), 164.31 (C-4), 164.39 (C-6); ESI-HRMS (MeOH): m/z = 308.1400, 308.1399 calcd [M + H]+ for C18H18N3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3459, 3318 (–NH2), 1638 (CN), 1542 (CC), 1260 (C–N), 1034 (OCH3), 593 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.86 (s, 3H, OCH3), 6.77 (s, 2H, NH2), 7.10 (dd, J = 2.4 Hz, J = 6.0 Hz, 1H, H-4′), 7.45 (t, 1H, J = 7.8 Hz, H-5′), 7.73 (m, 3H, H-5, H-3′′, H-5′′), 7.76 (s, 1H, H-2), 7.81 (d, 1H, J = 7.8 Hz, H-6′), 8.20 (d, 2H, J = 7.2 Hz, H-2′′, H-6′′); 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 55.32 (OCH3), 101.94 (C-5), 112.22 (C-2′), 116.29 (C-4′), 119.45 (C-6′), 124.11 (C-4′′), 129.08 (C-2′′, C-6′′), 129.73 (C-5′), 131.61 (C-3′′, C-5′′), 136.51 (C-1′′), 138.75 (C-1′), 159.63 (C-3′), 163.73 (C-6), 163.95 (C-2), 164.99 (C-4); ESI-HRMS (MeOH): m/z = 356.0397, 356.0398 calcd [M + H]+ for C17H15BrN3O.:5 v/v); FTIR (KBr, ν (cm−1)): 3319, 3204 (NH2), 1639 (CN), 1544 (CC), 1261 (C–N), 1035 (OCH3), 784 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.85 (s, 3H, OCH3), 6.76 (s, 2H, NH2), 7.09 (dd, J = 2.4 Hz, J = 7.8 Hz, 1H, H-4′), 7.44 (t, 1H, J = 7.8 Hz, H-5′), 7.59 (d, 2H, J = 6.6 Hz, H-3′′, H-5′′), 7.72 (s, 1H, H-5), 7.76 (s, 1H, H-2′), 7.80 (d, 1H, J = 7.8 Hz, H-6′), 8.26 (d, 2H, J = 6.6 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.26 (OCH3), 101.91 (C-5), 112.19 (C-2′), 116.21 (C-4′), 119.40 (C-6′), 128.60 (C-3′′, C-5′′), 128.77 (C-2′′, C-6′′), 129.65 (C-5′), 135.19 (C-4′′), 136.11 (C-1′′), 138.73 (C-1′), 159.58 (C-3′), 163.57 (C-6), 163.90 (C-2), 164.90 (C-4); ESI-HRMS (MeOH): m/z = 312.0902, 312.0904 calcd [M + H]+ for C17H15ClN3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3364, 3326 (NH2), 1644 (CN), 1587 (CC), 1258 (C–N), 1031 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 6.64 (s, 2H, NH2), 7.08 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.54 (m, 3H, H-3′′, H-4′′, H-5′′), 7.65 (s, 1H, H-5), 8.22 (m, 4H, H-2′, H-6′, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.30 (OCH3), 101.07 (C-5), 113.93 (C-3′, C-5′), 126.90 (C-2′, C-6′), 128.53 (C-2′′, C-6′′), 128.54 (C-3′′, C-5′′), 129.62 (C-1′), 130.28 (C-4′′), 137.48 (C-1′′), 161.23 (C-4′), 163.90 (C-2), 164.42 (C-6), 164.56 (C-4); ESI-HRMS (MeOH): m/z = 278.1298, 278.1293 calcd [M + H]+ for C17H16N3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3504 (OH), 3387 (NH), 1568 (CN), 1537 (CC), 1233 (C–N), 1176 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.83 (s, 3H, OCH3), 6.49 (s, 2H, NH2), 6.88 (d, 2H, J = 6.6 Hz, H-3′′, H-5′′), 7.05 (d, 2H, J = 6.6 Hz, H-3′, H-5′), 7.53 (s, 1H, H-5), 8.08 (d, 2H, J = 6.6 Hz, H-2′′, H-6′′), 8.17 (d, 2H, J = 6.6 Hz, H-2′, H-6′), 9.90 (s, 1H, OH); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 99.97 (C-5), 113.84 (C-3′, C-5′), 115.24 (C-3′′, C-5′′), 128.15 (C-1′′), 128.38 (C-2′, C-6′), 128.51 (C-2′′, C-6′′), 129.82 (C-1′), 159.64 (C-4′′), 161.05 (C-4′), 163.74 (C-4), 163.89 (C-6), 164.38 (C-2); ESI-HRMS (MeOH): m/z = 294.1244, 294.1243 calcd [M + H]+ for C17H16N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3331, 3202 (NH2), 1648 (CN), 1584 (CC), 1236 (C–N), 1028 (OCH3); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 6H, OCH3), 6.55 (s, 2H, NH2), 7.07 (d, 4H, J = 9.0 Hz, H-3′, H-5′, H-3′′, H-5′′), 7.59 (s, 1H, H-5), 8.20 (d, 4H, J = 9.0 Hz, H-2′, H-6′, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.25 (OCH3), 100.24 (C-5), 113.84 (C-3′, C-5′, C-3′′, C-5′′), 128.43 (C-2′, C-6′, C-2′′, C-6′′), 129.74 (C-1′, C-1′′), 161.09 (C-4′, C-4′′), 163.77 (C-2), 164.07 (C-4, C-6); ESI-HRMS (MeOH): m/z = 308.1400, 308.1399 calcd [M + H]+ for C18H18N3O2.:2 v/v); FTIR (KBr, ν (cm−1)): 3367, 3212 (NH2), 1645 (CN), 1588 (CC), 1255 (C–N), 1033 (OCH3), 592 (C–Br); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.89 (s, 3H, OCH3), 6.73 (s, 2H, NH2), 7.12 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.73 (s, 1H, H-5), 7.77 (d, 2H, J = 9.0 Hz, H-3′′, H-5′′), 8.23 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′), 8.26 (d, 2H, J = 9.0 Hz, H-2′, H-6′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.29 (OCH3), 100.09 (C-5), 113.91 (C-3′, C-5′), 123.89 (C-4′′), 128.56 (C-2′, C-6′), 128.93 (C-2′′, C-6′′), 129.47 (C-1′), 131.51 (C-3′′, C-5′′), 136.64 (C-1′′), 161.29 (C-4′), 163.29 (C-6), 163.86 (C-2), 164.65 (C-4); ESI-HRMS (MeOH): m/z = 356.0397, 356.0398 calcd [M + H]+ for C17H15BrN3O.:2 v/v); FTIR (KBr, ν (cm−1)): 3331, 3210 (NH2), 1644 (CN), 1562 (CC), 1238 (C–N), 1032 (OCH3), 815 (C–Cl); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 3.84 (s, 3H, OCH3), 6.68 (s, 2H, NH2), 7.07 (d, 2H, J = 9.0 Hz, H-3′, H-5′), 7.58 (d, 2H, J = 8.4 Hz, H-3′′, H-5′′), 7.67 (s, 1H, H-5), 8.21 (d, 2H, J = 9.0 Hz, H-2′, H-6′), 8.25 (d, 2H, J = 8.4 Hz, H-2′′, H-6′′); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 55.27 (OCH3), 100.93 (C-5), 113.89 (C-3′, C-5′), 128.55 (C-2′′, C-3′′, C-5′′, C-6′′), 128.67 (C-2′, C-6′), 129.47 (C-1′), 135.02 (C-4′′), 136.26 (C-1′′), 161.27 (C-4′), 163.19 (C-6), 163.84 (C-2), 164.61 (C-4); ESI-HRMS (MeOH): m/z = 312.0908, 312.0904 calcd [M + H]+ for C17H15ClN3O.:1 v/v); FTIR (KBr, ν (cm−1)): 3506, 3399 (NH2), 3057 (OH), 1602 (CN), 1539 (CC), 1231 (C–N); 1H-NMR (600 MHz, DMSO-d6, δ (ppm)): 6.59 (s, 2H, NH2), 6.89 (d, 2H, J = 8.4 Hz, H-3′, H-5′), 7.51 (m, 3H, H-3′′, H-4′′, H-5′′); 7.59 (s, 1H, H-5), 8.10 (d, 2H, J = 9.0 Hz, H-2′, H-6′), 8.19 (m, 2H, H-2′′, H-6′′), 9.91 (s, 1H, OH), 13C-NMR (125 MHz, DMSO-d6, δ (ppm)): 100.78 (C-5), 115.28 (C-3′, C-5′), 126.82 (C-2′′, C-6′′), 128.03 (C-1′), 128.49 (C-3′′, C-5′′), 128.58 (C-2′, C-6′), 130.17 (C-4′′), 137.54 (C-1′′), 159.72 (C-4′), 163.84 (C-2), 164.34 (C-6), 164.69 (C-4); ESI-HRMS (MeOH): m/z = 264.1141, 264.1137 calcd [M + H]+ for C16H14N3O.4.3. Cytotoxicity assay
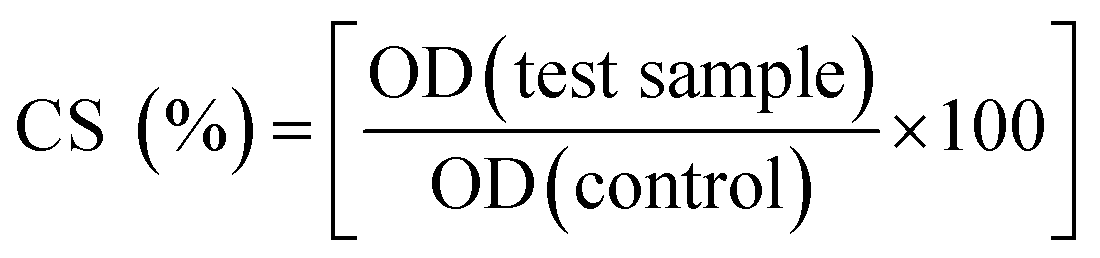
For the compounds that have CS values less than 50% in the first screening, the IC50 values (μM) were determined using nonlinear regression analysis in GraphPad Prism 5.0.
4.4. Computational analysis
Although imatinib remains the first-line treatment for CML, drug-resistant mutations, most notably ABL1 T334I, have significantly reduced its long-term efficacy.39 Therefore, the investigated compounds are evaluated for their potential inhibition against drug-resistant CML. We also searched for PDB structures of ABL1 kinase mutants for simulation purposes. However, no crystal structure of the mutant enzyme–imatinib complex exists at the ATP-binding site. As a result, the co-crystallized structure of the ABL1 T334I-D382N mutant with nilotinib (PDB ID 5MO4)40 was employed for docking studies. In this study, nilotinib was used exclusively to validate the docking model for mutant ABL1. The binding affinity of 2-amino-4,6-diarylpyrimidine derivatives at the active site of the ABL1 T334I mutant was then evaluated using imatinib as the reference.
| ΔGbind = ΔGcomplex − (ΔGenzyme + ΔGinhibitor) | (1) |
The occupancy of interactions between ligands and enzyme residues during MD simulations was analyzed using interaction fingerprints with the ProLIF and MDAnalysis tools.48,49
Data availability
The data supporting this article have been included as part of the ESI,† including the FTIR, HRMS, 1D-, 2D-NMR spectra of synthesized derivatives, the cell survival results of K562 cytotoxic activity, and the computational data of compounds and Imatinib on ABL1 tyrosine kinase.Author contributions
Thi-Anh-Truc Phan and Kim-Khanh-Huy Ngo: conceptualization, data curation, formal analysis, methodology, validation, visualization, writing – original draft, and writing – review & editing, Thi-Cam-Thu Nguyen: writing – original draft, and writing – review & editing, Thanh-Tan Mai: data curation, formal analysis, methodology, software, validation, visualization, and writing – review & editing, Hai-Dang Nguyen and Thu-Trang Duong: data curation, formal analysis, methodology, validation, visualization, and writing – review & editing, Le-Phu Tran, Thanh-Tuyen Duong, Thi-Kim-Chi Huynh, Elena V. Koroleva, Zhanna V. Ignatovich, Anastasiya L. Ermolinskaya, Hoang-Phuc Nguyen, Thi-Hong-An Nguyen, Anh-Khoa Ton, Tuong-Ha Do: formal analysis, and writing – review & editing, Thi-Kim-Dung Hoang: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the Vietnam Academy of Science and Technology under grant number QTBY01.04/23-24.References
- W. Peng, G. A. de Tuya, A. A. Eduardo, J. A. Vishny and Q. Huang, Publ. Understand. Sci., 2022, 31, 53–69 CrossRef PubMed.
- J. D. Mizrahi, R. Surana, J. W. Valle and R. T. Shroff, Lancet, 2020, 395, 2008–2020 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- A. S. Davis, A. J. Viera and M. D. Mead, Am. Fam. Physician, 2014, 89, 731–738 Search PubMed.
- D. G. Gilliland, C. T. Jordan and C. A. Felix, Hematol. Am. Soc. Hematol. Educ. Program, 2004, 2004, 80–97 CrossRef PubMed.
- J. V. Melo, T. P. Hughes and J. F. Apperley, Hematol. Am. Soc. Hematol. Educ. Program, 2003, 2003, 132–152 CrossRef PubMed.
- M. W. Deininger, J. M. Goldman and J. V. Melo, Blood, 2000, 96, 3343–3356 CrossRef CAS PubMed.
- A. Quintás-Cardama and J. E. Cortes, Mayo Clin. Proc., 2006, 81, 973–988 CrossRef PubMed.
- M. Baccarani and R. P. Gale, Leukemia, 2021, 35, 2199–2204 CrossRef CAS PubMed.
- E. Klein, F. Vánky, H. Ben-Bassat, H. Neumann, P. Ralph, J. Zeuthen and A. Polliack, Int. J. Cancer, 1976, 18, 421–431 CrossRef CAS PubMed.
- F. Luchetti, A. Gregorini, S. Papa, S. Burattini, B. Canonico, M. Valentini and E. Falcieri, Haematologica, 1998, 83, 974–980 CAS.
- B. Druker, N. Engl. J. Med., 2001, 345, 232 Search PubMed.
- M. Okada, S. Adachi, T. Imai, K.-i. Watanabe, S.-y. Toyokuni, M. Ueno, A. S. Zervos, G. Kroemer and T. Nakahata, Blood, 2004, 103, 2299–2307 CrossRef CAS PubMed.
- E. Koroleva, Z. I. Ignatovich, Y. V. Sinyutich and K. Gusak, Russ. J. Org. Chem., 2016, 52, 139–177 CrossRef CAS.
- S. S. Ganguly and R. Plattner, Genes Cancer, 2012, 3, 414–425 CrossRef CAS PubMed.
- S. A. Rosenzweig, Adv. Cancer Res., 2018, 138, 71–98 CrossRef CAS PubMed.
- J. V. Melo and C. Chuah, Cancer Lett., 2007, 249, 121–132 CrossRef CAS PubMed.
- V. Nardi, M. Azam and G. Q. Daley, Curr. Opin. Hematol., 2004, 11, 35–43 CrossRef CAS PubMed.
- A. Rahim, R. Syed, Y. Poornachandra, M. S. Malik, C. V. R. Reddy, M. Alvala, K. Boppana, B. Sridhar, R. Amanchy and A. Kamal, Med. Chem. Res., 2019, 28, 633–645 CrossRef CAS.
- J. Dietrich, C. Hulme and L. H. Hurley, Bioorg. Med. Chem. Lett., 2010, 18, 5738–5748 CrossRef CAS PubMed.
- J. Lee, K.-H. Kim and S. Jeong, Bioorg. Med. Chem. Lett., 2011, 21, 4203–4205 CrossRef CAS PubMed.
- K. P. Cheremnykh, V. A. Savelyev, M. A. Pokrovskii, D. S. Baev, T. G. Tolstikova, A. G. Pokrovskii and E. E. Shults, Med. Chem. Res., 2019, 28, 545–558 CrossRef CAS.
- R. Ramajayam, R. Giridhar, M. R. Yadav, H. Djaballah, D. Shum and C. Radu, J. Enzyme Inhib. Med. Chem., 2007, 22, 716–721 CrossRef CAS PubMed.
- F. Giraud, G. Alves, E. Debiton, L. Nauton, V. Théry, E. Durieu, Y. Ferandin, O. Lozach, L. Meijer and F. Anizon, J. Med. Chem., 2011, 54, 4474–4489 CrossRef CAS PubMed.
- F. Jubeen, S. Z. Iqbal, N. Shafiq, M. Khan, S. Parveen, M. Iqbal and A. Nazir, Synth. Commun., 2018, 48, 601–625 CrossRef CAS.
- S. Wang, X.-H. Yuan, S.-Q. Wang, W. Zhao, X.-B. Chen and B. Yu, Eur. J. Med. Chem., 2021, 214, 113218 CrossRef CAS PubMed.
- I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1–50 CrossRef CAS PubMed.
- S. A. Rostom, M. H. Badr, H. A. A. E. Razik, H. M. Ashour and A. E. A. Wahab, Arch. Pharm., 2011, 344, 572–587 CrossRef CAS PubMed.
- S. Farooq and Z. Ngaini, J. Heterocycl. Chem., 2021, 58, 1209–1224 CrossRef CAS.
- N. Kahriman, K. Peker, V. Serdaroğlu, A. Aydın, A. Usta, S. Fandaklı and N. Yaylı, Bioinorg. Chem., 2020, 99, 103805 CrossRef CAS PubMed.
- A. E. Aiwonegbe, J. U. Iyasele and C. O. Usifoh, ChemSearch J., 2024, 15, 112–124 Search PubMed.
- S. Prasad, V. Radhakrishna and T. K. Ravi, Arabian J. Chem., 2019, 12, 3943–3947 CrossRef CAS.
- M. H. Ahmed, M. A. El-Hashash, M. I. Marzouk and A. M. El-Naggar, J. Heterocycl. Chem., 2020, 57, 3412–3427 CrossRef CAS.
- A. R. Dwivedi, V. Kumar, H. Kaur, N. Kumar, R. P. Yadav, R. Poduri, S. Baranwal and V. Kumar, Bioorg. Med. Chem. Lett., 2020, 30, 127468 CrossRef PubMed.
- X. An, A. K. Tiwari, Y. Sun, P.-R. Ding, C. R. Ashby Jr and Z.-S. Chen, Leuk. Res., 2010, 34, 1255–1268 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- S. K. Burley, C. Bhikadiya, C. Bi, S. Bittrich, H. Chao, L. Chen, P. A. Craig, G. V. Crichlow, K. Dalenberg, J. M. Duarte, S. Dutta, M. Fayazi, Z. Feng, J. W. Flatt, S. Ganesan, S. Ghosh, D. S. Goodsell, R. K. Green, V. Guranovic, J. Henry, B. P. Hudson, I. Khokhriakov, C. L. Lawson, Y. Liang, R. Lowe, E. Peisach, I. Persikova, D. W. Piehl, Y. Rose, A. Sali, J. Segura, M. Sekharan, C. Shao, B. Vallat, M. Voigt, B. Webb, J. D. Westbrook, S. Whetstone, J. Y. Young, A. Zalevsky and C. Zardecki, Nucleic Acids Res., 2023, 51, D488–D508 CrossRef CAS PubMed.
- S. W. Cowan-Jacob, G. Fendrich, A. Floersheimer, P. Furet, J. Liebetanz, G. Rummel, P. Rheinberger, M. Centeleghe, D. Fabbro and P. W. Manley, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2007, 63, 80–93 CrossRef CAS PubMed.
- K. A. Rygiel and J. M. Elkins, Curr. Opin. Struct. Biol., 2023, 82, 102665 CrossRef CAS PubMed.
- A. A. Wylie, J. Schoepfer, W. Jahnke, S. W. Cowan-Jacob, A. Loo, P. Furet, A. L. Marzinzik, X. Pelle, J. Donovan, W. Zhu, S. Buonamici, A. Q. Hassan, F. Lombardo, V. Iyer, M. Palmer, G. Berellini, S. Dodd, S. Thohan, H. Bitter, S. Branford, D. M. Ross, T. P. Hughes, L. Petruzzelli, K. G. Vanasse, M. Warmuth, F. Hofmann, N. J. Keen and W. R. Sellers, Nature, 2017, 543, 733–737 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- K. Stierand and M. Rarey, ACS Med. Chem. Lett., 2010, 1, 540–545 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, N. Forouzesh, G. Giambaşu, M. K. T. Giese, H. G. Gilson, A. W. Goetz, J. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, J. Wang, H. Wei, X. Wu, Y. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao, Q. Zhu and P. A. Kollman, https://ambermd.org/, Accessed September 2024.
- T. T. Mai, T.-P. Lam, L.-H. D. Pham, K.-H. Nguyen, Q.-T. Nguyen, M.-T. Le and K.-M. Thai, J. Phys. Chem. B, 2024, 128, 8362–8375 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- C. Bouysset and S. Fiorucci, J. Cheminf., 2021, 13, 72 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08330j |
| This journal is © The Royal Society of Chemistry 2025 |