
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible color changing response of pyrenylated charge-transfer probes towards Hg2+: linker-driven modulation of sensitivity and selectivity†
Nilanjan Dey *
*
Department of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad, Telangana-500078, India. E-mail: nilanjan@hyderabad.bits-pilani.ac.in
First published on 28th March 2025
Abstract
Two easily synthesizable prenylated charge transfer probes with a terminal pyridine unit were designed. The compounds differ based on the nature of the spacer: rigid olefinic (1) and flexible hydrazo (2). Both compounds show exclusive interaction with Hg2+ ions, leading to changes in visible color. Experimental analyses showed that compound 1 outperformed compound 2 in terms of sensitivity (A/A0 ∼ 13-fold vs. ∼2-fold) and detection limit (1.2 ppm vs. 5.8 ppm), attributed to its rigid linker and enhanced binding affinity. Moreover, ratiometric responses (r2 > 0.99) and excellent selectivity for Hg2+ were demonstrated. This study highlights the development of robust, reusable, and cost-effective tools for mercury detection, thus addressing critical environmental and health concerns associated with mercury contamination. Environmental robustness of compound 1 was validated across solvents, counter-ions, and water mixtures, with decreasing efficacy in high-water-content systems owing to solvation effects and probe aggregation. Mechanistic studies confirmed pyridine nitrogen as the primary coordination site with reversible binding, enabling probe reuse. Practical applications were explored using paper strips modified with the probes, which facilitated rapid, cost-effective, and quantitative detection of Hg2+ via image analysis.
Introduction
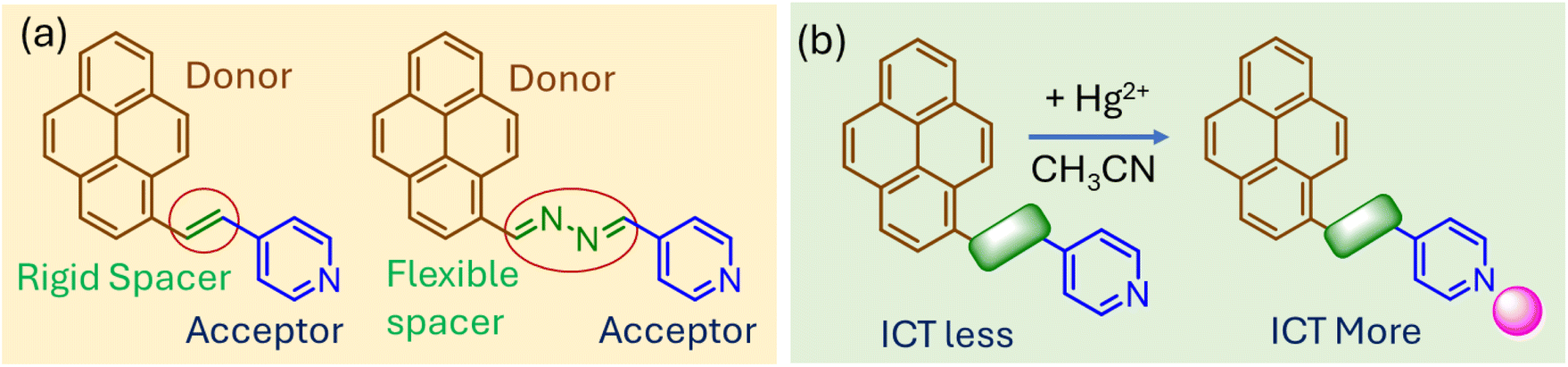
Charge transfer (CT)-based probes are invaluable for the optical sensing of heavy metal pollutants because of their high sensitivity, selectivity, and rapid response, enabling detection at concentrations as low as nanomolar levels, often below regulatory limits for pollutants such as Hg2+ (2 ppb set by the EPA) or Pb2+ (15 ppb set by the EPA).1 Tailored with specific functional groups, these probes offer exceptional selectivity for metals such as Hg2+, Pb2+, or Ag+ and produce rapid, often visible optical changes with detection times typically under minutes, facilitating on-site detection without complex equipment. Their versatility allows them to function across diverse conditions, such as pH ranges of 4–10 or in high ionic strength environments, providing mechanistic insights into metal–probe interactions and advancing sensor design.2 Moreover, CT-based probes can be integrated into portable formats such as paper strips or hydrogels, achieving detection limits as low as 10−9 M.3 Recent innovations incorporate smartphone-enabled imaging and machine learning for enhanced data interpretation.4The spacer group in charge transfer (CT)-based probes plays a pivotal role in modulating their electronic and optical properties, directly influencing their colorimetric sensing capabilities. The nature of the spacer dictates how efficiently the donor and acceptor groups interact. Conjugated linkers often enhance electronic communication, leading to red-shifted absorption spectra and a more sensitive response to analytes. However, non-conjugated linkers reduce electronic coupling, weakening CT efficiency but often increasing selectivity towards target analytes by minimizing background interference.5 Similarly, flexibility of the spacer can affect sensing performance. Flexible spacers allow the probe to adopt various conformations, potentially improving analyte binding. However, this flexibility may result in less stable and reproducible responses. Rigid spacers, on the contrary, maintain a fixed spatial arrangement of donor and acceptor groups, ensuring consistent CT behavior and reliable colorimetric response.6 Moreover, the length of the spacer influences the strength of electronic interactions. Short spacers enhance CT efficiency by facilitating strong electronic overlap between donor and acceptor groups, resulting in intense optical changes upon analyte binding. In contrast, long spacers reduce electronic overlap, weakening CT and shifting absorption or emission to shorter wavelengths.7 Moreover, longer spacers can improve selectivity by spatially separating reactive sites, reducing nonspecific interactions.
Despite such important roles of spacers in the optical properties of CT probes and sensing efficacy, not many systematic investigations have been carried out focusing on this aspect. Therefore, herein, we designed pyrene-based, easily synthesizable charge-transfer probes with pyridine as the metal ion binding site (Fig. 1a). Two compounds with different spacers (olefinic vs. hydrazo) were considered with the same donor (pyrene) and acceptor (pyridine) moieties. Such variations in the spacer unit showed distinct influence on the optical properties of the compounds and efficacy towards Hg2+ sensing. Binding with Hg2+ ions would facilitate charge transfer from the donor to acceptor moiety, leading to changes in visible color (Fig. 1b). In addition to solution-based analysis, the paper attempted rapid, on-location detection of Hg2+ using chemically modified paper strips. The quantitative analysis in this case was achieved using ImageJ (free version).
 | ||
| Fig. 1 (a) Chemical structures of probe molecules involved in the present study. (b) Shows plausible binding interaction with Hg2+ ions. | ||
Results and discussion
Design and synthesis
In this work, we designed two easily synthesizable prenylated probes with pyridine as the terminal functional group.8 The two compounds differed based on the nature of the linker unit connecting the donor and acceptor sites. In case of compound 1, the electron-rich pyrene unit is linked with electron-deficient pyridine residue via a trans olefinic bond, while the same units are connected via a hydrazo spacer in compound 2. Both compounds showed a color in the visible region, attributed to intramolecular charge transfer from pyrene to pyridine unit. Therefore, any change in the electronic nature of the pyridine unit would be expected to influence the optical response of the probe molecules. The olefinic linker, with its conjugated and planar structure, is expected to promote efficient π-electron delocalization between the donor and acceptor units, resulting in enhanced charge transfer (CT) and higher sensitivity to changes in the electronic environment caused by metal ion binding. However, the hydrazo linker, containing lone pair-bearing nitrogen atoms, disrupts π-conjugation but provides an additional coordination site for metal ions. The rigid structure of the olefinic linker supports higher fluorescence quantum yields, while the flexible hydrazo linker allows for conformational adaptability, enhancing metal ion chelation.The energy optimization of compounds 1 and 2 was carried out using B3LYP level of theory with the 6-31G* basis set.9 Both compounds demonstrated a nearly planar structure with dihedral angles of <0.2° between the spacer and donor (or acceptor) moieties (Fig. 2a). The HOMO–LUMO energy gap was calculated for 1 and 2 and was found to be in the similar range (ΔE ∼ 0.12 eV) (Fig. 2b). Further, we calculated Mulliken charge distribution in the pyridine ring. For both compounds, the pyridyl nitrogen ends appeared to have high negative charge density, suitable for metal ion coordination (Fig. 2c and d).
 | ||
| Fig. 2 (a) Energy-optimized structures of 1 and 2 using B3LYP/6-31G* level of theory. (b) Schematic showing relative HOMO–LUMO energy levels for compounds 1 and 2. Mulliken charge distribution on the pyridine ring for compounds (c) 1 and (d) 2. | ||
Spectroscopic response in the solution phase
The UV-visible spectra of compounds 1 and 2 were recorded in acetonitrile. The UV-visible spectrum of compound 1 showed two distinct absorption bands at 287 (ε = 2.58 × 105 M−1 cm−1) and 373 (ε = 4.05 × 105 M−1 cm−1) nm owing to π–π* transition and charge transfer interaction, respectively. For compound 2, both these bands appeared to be slightly red-shifted and were observed at 291 (ε = 2.90 × 105 M−1 cm−1) and 380 (ε = 3.39 × 105 M−1 cm−1) nm, respectively. Moreover, an additional absorption band was found at a lower energy region, centered at 408 nm (ε = 2.64 × 105 M−1 cm−1) (Fig. 3a). The lone pairs on the nitrogen atoms of the hydrazo unit could facilitate charge-transfer interaction, leading to the formation of a red-shifted absorption band.10 Moreover, the hydrazo compound can form hydrogen bonding with polar protic solvents and form self-assembled aggregated structures. However, the fluorescence spectrum of 1 showed broad emission band in the 457 nm region. Although emission maximum was observed at a similar position for compound 2, the intensity was found to be significantly less (∼4.6-fold) (Fig. 3b). The higher flexibility of the hydrazo linker probably enhances the possibilities of non-radiative decay, which resulted in low fluorescence intensity.11 Furthermore, to compare the metal ion binding efficiency of 1 and 2, we recorded 1H-NMR spectra of both compounds in DMSO-d6 (Fig. 3c). We could not record the spectrum in CD3CN owing to limited solubility at room temperature. The aromatic protons of compound 2 adjacent to pyridine nitrogen ends (a and b) appeared in the deshielded region compared to compound 1. This suggested that the pyridine ring in the case of compound 2 is relatively electron deficient. Moreover, the imine protons in 2 expectedly showed a considerable downfield shift compared to olefinic protons of 1. Consequently, there is a possibility that probe 2 would not bind with metal ions as effectively as compound 1. | ||
| Fig. 3 (a) UV-visible spectra of 1 and 2 (10 μM) in CH3CN. (b) Fluorescence spectra of 1 and 2 (10 μM, λex = 375 nm) in CH3CN. (c) Partial 1H-NMR spectra of 1 and 2 (5 mM) in DMSO-d6 (inset shows structures of compounds 1 and 2 with assigned protons). | ||
Chromogenic response towards Hg2+ ions
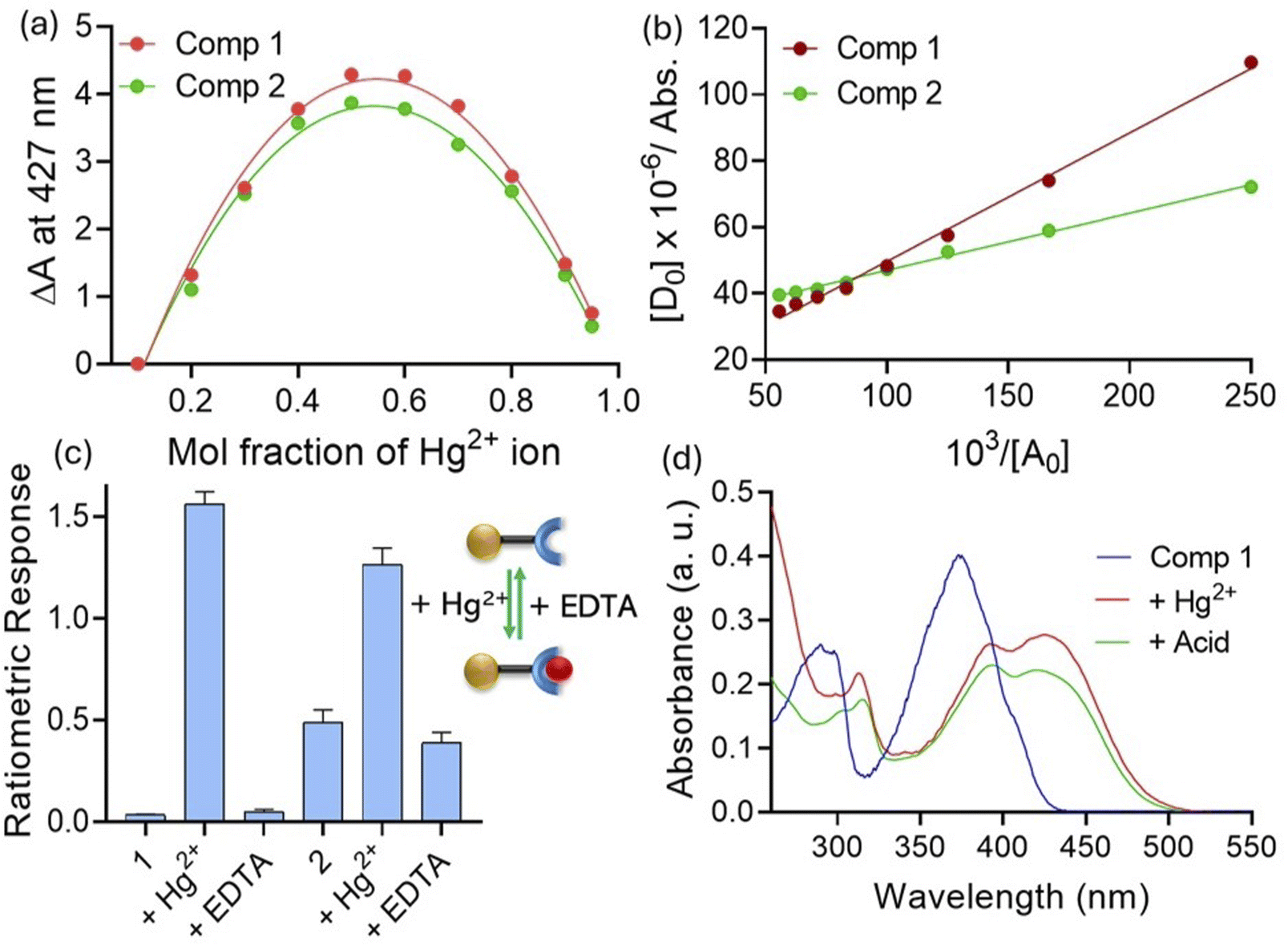
We investigated the metal ion binding properties of compounds in acetonitrile. When compound 1 was exposed to Hg2+ ions, the color of the solution immediately changed to yellow. Expectedly, the UV-visible spectrum with Hg2+ showed formation of a new band at the 427 nm region. Titration studies showed that the absorbance at 427 nm enhanced with an increasing concentration of Hg2+ ions at the expense of the 375 nm band (Fig. 4a). The isosbestic points were found at 328 and 392 nm, indicating a one-to-one equilibrium between 1 and Hg2+ ions. When compound 2 was subjected to Hg2+ ions, we witnessed a new charge transfer band at 423 nm. Here, titration studies showed that absorbance at the 423 nm band enhanced concomitantly with quenching at 378 and 404 nm bands (Fig. 4b). In this case, isosbestic points were observed at 343 and 410 nm. Interestingly, comparison studies indicated that the charge transfer band for compound 1 was found to be more prominent (A/A0 ∼ 13 fold at 427 nm) than that witnessed with compound 2 (A/A0 ∼ 2 fold at 423 nm). Furthermore, we plotted the ratiometric responses of compounds 1 (Abs428 nm/Abs373 nm) and 2 (Abs423 nm/Abs380 nm) against the concentrations of Hg2+ added (Fig. 4c). In both cases, we observed linear regression (r2 > 0.99), which indicated the ratiometric nature of the probe molecules. This is particularly important for real-life sample analysis as ratiometric probes by considering responses at different wavelengths can minimize background interferences and improve the signal-to-noise ratio.12 Moreover, we noted that changes in the response were prominent with compound 1 compared with compound 2. Since selectivity is one of the most important criteria for quantitative analysis, we analysed interactions of compounds 1 and 2 with other metal ions under similar conditions. For both compounds, no other metal ions could show changes in the absorption signal as prominently as that observed with Hg2+ ions (Fig. 4d). This indicated that both sensors 1 and 2, irrespective to the differences in the sensitivity, showed good selectivity towards Hg2+ ions in the solution phase. Furthermore, UV-visible titration studies indicated that the limits of detection for Hg2+ ions were 0.012 and 0.02 ppm with probes 1 and 2, respectively. | ||
| Fig. 4 UV-visible titration of (a) 1 and (b) 2 (10 μM) with Hg2+ (0–20 μM) in a CH3CN medium. (c) Ratiometric responses of 1 and 2 (10 μM) towards Hg2+ ions (0–20 μM) in a CH3CN medium. (d) Ratiometric responses of 1 and 2 (10 μM) in the presence of various metal ions (25 μM) in a CH3CN medium [each experiment was independently performed thrice for reproducibility]. | ||
Time-dependent studies indicate that the perceptible color change of 1 with Hg2+ ions could be seen within 30 s of Hg2+ ion addition. However, the color response was found to be quite stable and no further change in absorbance was noted even after 1 h of Hg2+ addition (Fig. S1†). This indicated that the present method is robust and can produce reliable spectroscopic signals for Hg2+ ions. Considering that traditional solution-based sensing studies need sophisticated instrumental facilities and trained technicians, we developed an alternative strategy for Hg2+ detection using chemically modified precoated paper strips.13 The paper strips in the native state showed no color under day-light. However, when the paper strip was exposed to Hg2+ ions, the color of the strip changed to yellow (Fig. 5a). This observation indicated that detection of Hg2+ was indeed possible on a solid surface. Furthermore, the changes in the color of the paper strips were quantitatively assessed by ImageJ.14 It was observed that the paper strips spiked with Hg2+-contaminated solution can be reused upon washing with diluted EDTA solution. Since this method does not involve multi-step sample preparation, involvement of skilled technicians or analysis of complex signal output, people with limited knowledge of science or technology will be able to use it without much difficulty.
 | ||
| Fig. 5 (a) Reusable paper strips (coated with compound 1) for detection of Hg2+ ions. (b) Changes in absorbance of 1 (10 μM) upon addition of Hg2+ (0–20 μM) in different organic media. (c) Changes in absorbance at the 427 nm band with Hg2+ ions in different CH3CN–H2O mixtures. (d) UV-visible spectra of 1 (10 μM) with different Hg2+ salts (25 μM) in a CH3CN medium [each experiment was independently performed thrive for reproducibility]. | ||
Effect of the microenvironment on Hg2+-ion sensing
Considering the selective interaction, we further investigated effects of different environmental and other factors on the efficacy of Hg2+ detection by compound 1. First, we checked the interaction of compound 1 with different Hg2+ salts (with –NO3 and –ClO4 counter anions) in a CH3CN medium (Fig. 5d). Such variation in counter ions showed no detectable impact on the response of 1 towards Hg2+ ions. This is indeed an important observation, as in many cases, interference from counter anions can hinder the accuracy of the sensor molecules. Since Hg2+ contamination can be encountered in various types of industrial waste materials, we examined the responses of compound 1 towards Hg2+ ions in different organic solvents, such as CH3CN, THF, and DMF (Fig. 5b). The degree of response, as denoted by changes in absorbance at the 427 nm band, were found to be maximum in THF, followed by CH3CN and DMF. In less polar solvents such as THF, the interaction between Hg2+ ions and the probe will be stronger because Hg2+ ions are less solvated, making them more available to bind with the probe. However, DMF and CH3CN could form Hg2+-solvent complexes owing to the lone pairs on oxygen (DMF) or nitrogen (CH3CN) atoms, which significantly reduce the interaction with Hg2+ ions.15Since we studied interactions in a pure CH3CN medium, we were interested to investigate the effect of water on Hg2+ sensing. It was observed that with increasing water content in the mixture, the response towards Hg2+ ions gradually decreased (Fig. 5c). The changes in absorbance (A/A0) at the 427 nm band was found to be ∼10-fold in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN–H2O mixture, while it was ∼1.5-fold in pure water. This might be due to the extensive solvation of Hg2+ ions in water and aggregation of probe molecules through hydrogen bonding interaction.16
:1 CH3CN–H2O mixture, while it was ∼1.5-fold in pure water. This might be due to the extensive solvation of Hg2+ ions in water and aggregation of probe molecules through hydrogen bonding interaction.16
Mechanistic investigation with Hg2+ ions
The mode of binding with Hg2+ was unveiled using a series of spectroscopic investigations. The superior response of compound 1 towards Hg2+ compared to 2 ruled out the possible involvement of the hydrazo linker in the coordination with Hg2+ ions. Thus, it could be anticipated that Hg2+ in both cases is coordinated through the pyridyl nitrogen end. Irrespective of spectral response, both compounds showed 1:1 binding stoichiometry with Hg2+ ions in a CH3CN medium (Fig. 6a). The Benesi–Hildebrand equation for the 1:1 model was utilized for calculation of binding constants (Fig. 6b). Compound 1 showed larger binding affinity (logK = 4.84 ± 0.03) towards Hg2+ ions than compound 2 (logK = 3.95 ± 0.02), which justified the larger optical response observed with the former probe. The reversible nature of Hg2+ coordination was verified using EDTA as the chelator agent.17 First, Hg2+ (2 equiv.) was added to the solution of 1 (or 2) and to that the same equivalent of EDTA was added, which completely revived the original spectrum of 1 (or 2) (Fig. 6c). This suggested a reversible coordination between the probe molecules and Hg2+ ions. Moreover, this result indicated that a single solution of the probe (either 1 or 2) can be used multiple times for the detection of Hg2+ ions. Since protonation of the pyridine moiety can facilitate charge transfer interaction, we compared the spectral response in the presence of both Hg2+ and H+ (HCl) (Fig. 6d).18 Both spectra resembled each other, which indicated that Hg2+ ions coordinated through the pyridine unit of the compound.
 | ||
| Fig. 6 (a) Job's plot of compounds 1 and 2 in the presence of Hg2+ ions. (b) Determination of binding constants of compounds 1 and 2 with Hg2+ ions. (c) Recovery experiment with EDTA showing the reversible interaction of 1 and 2. (d) UV-visible spectra of 1 (10 μM) with different Hg2+ salts (25 μM) in a CH3CN medium [each experiment was independently performed thrice for reproducibility]. | ||
Conclusion
This study highlights the design, synthesis, and evaluation of two prenylated probes for selective detection of Hg2+, emphasizing the role of structural and electronic factors in determining sensing efficiency. The two probes, differing in their linker units, olefinic (compound 1) and hydrazo (compound 2), exhibited charge transfer interactions leading to visible color changes upon interaction with Hg2+ ions. Computational studies revealed planar structures and similar HOMO–LUMO energy gaps (∼0.12 eV), while experimental UV-visible and fluorescence spectroscopy identified distinct optical responses. Compound 1 demonstrated superior sensitivity (A/A0 ∼ 13-fold) and a lower detection limit (1.2 ppm) compared to compound 2 (A/A0 ∼ 2-fold, 5.8 ppm), possibly owing to its less flexible linker and higher binding affinity for Hg2+ ions. Ratiometric responses (r2 > 0.99) and excellent selectivity towards Hg2+ over other metal ions further established the efficacy of these probes for quantitative analysis. The environmental robustness of the probes was evaluated under varying conditions, including solvent polarity, water content, and counter-ion effects. Sensing efficiency decreased in high-water-content systems because of solvation effects and probe aggregation but remained consistent across different Hg2+ salts. Mechanistic investigations confirmed pyridine nitrogen as the primary coordination site, with 1:1 stoichiometry and reversible binding validated by EDTA recovery. Practical applicability was demonstrated using chemically modified paper strips, enabling cost-effective, user-friendly Hg2+ detection with quantitative image analysis. These findings underline the potential of structurally tuneable charge-transfer-based probes for developing reusable, sensitive, and selective optical sensors for heavy metal pollutants in diverse environments.
Data availability
Data are available from the author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. D. thanks DST for the SYST grant (SP/YO/2021/1632) and also thanks the Ministry of Education (MOE) for STARS grant (STARS-2/2023-0300).References
- A. Taha, N. Farooq, N. Singh and A. A. Hashmi, J. Mol. Liq., 2024, 401, 124678 CrossRef CAS.
- X. Wang, C. Shen, C. Zhou, Y. Bu and X. Yan, Chem. Eng. J., 2021, 417, 129125 CrossRef CAS.
- (a) S. Shakya and I. M. Khan, J. Hazard. Mater., 2021, 403, 123537 CrossRef CAS PubMed; (b) R. S. Fernandes and N. Dey, J. Mol. Liq., 2022, 367, 120369 CrossRef CAS.
- (a) D. Shen, W.-C. Chen, M.-F. Lo and C.-S. Lee, Mater. Today Energy, 2021, 20, 100644 CrossRef CAS; (b) R. S. Fernandes and N. Dey, Ind. Eng. Chem. Res., 2023, 62, 21536–21545 CAS.
- J. Jia, X. Wu, X. Zhang, Y. Wang, J. Yang, Y. Fang and Y. Song, Phys. Chem. Chem. Phys., 2022, 24, 955–965 RSC.
- M. Baharfar, A. C. Hillier and G. Mao, Adv. Mater., 2024, 36, 2406083 CAS.
- S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- (a) Y. Singh, A. Gulyani and S. Bhattacharya, FEBS Lett., 2003, 541, 132–136 CAS; (b) N. Dey and S. Bhattacharya, Dalton Trans., 2018, 47, 2352–2359 CAS.
- H. Oberhofer, K. Reuter and J. Blumberger, Chem. Rev., 2017, 117, 10319–10357 CAS.
- S. Kumar, L. G. Franca, K. Stavrou, E. Crovini, D. B. Cordes, A. M. Z. Slawin, A. P. Monkman and E. Zysman-Colman, J. Phys. Chem. Lett., 2021, 12, 2820–2830 CAS.
- (a) F. de Moliner, F. Nadal-Bufi and M. Vendrell, Curr. Opin. Chem. Biol., 2024, 80, 102458 Search PubMed; (b) N. Dey, Dalton Trans., 2021, 50, 12563–12569 CAS.
- X. Huang, J. Song, B. C. Yung, X. Huang, Y. Xiong and X. Chen, Chem. Soc. Rev., 2018, 47, 2873–2920 CAS.
- A. Ko and C. Liao, Anal. Methods, 2023, 15, 4377–4404 Search PubMed.
- (a) S. Patel, R. Jamunkar, D. Sinha, M. Patle, T. K. Patle, T. Kant, K. Dewangan and K. Shrivas, Trends Environ. Anal. Chem., 2021, 31, e00136 Search PubMed; (b) B. Maiti, N. Dey and S. Bhattacharya, ACS Appl. Bio Mater., 2019, 2, 2365–2373 CAS.
- (a) H. Li, J. Li, Z. Pan, T. Zheng, Y. Song, J. Zhang and Z. Xiao, Spectrochim. Acta, Part A, 2023, 291, 122379 CAS; (b) H. V. Barkale and N. Dey, Asian J. Org. Chem., 2024, 13, e202300657 CAS.
- M. Bogunia and M. Makowski, J. Phys. Chem. B, 2020, 124, 10326–10336 CrossRef CAS PubMed.
- (a) P. Zhou, Q. Meng, G. He, H. Wu, C. Duan and X. Quan, J. Environ. Monit., 2009, 11, 648–653 RSC; (b) N. Dey, N. Kumari, D. Biswakarma, S. Jha and S. Bhattacharya, Inorg. Chim. Acta, 2019, 487, 50–57 CrossRef CAS.
- S. Saha, A. De, A. Ghosh, A. Ghosh, K. Bera, K. S. Das, S. Akhtar, N. C. Maiti, A. K. Das, B. B. Das and R. Mondal, RSC Adv., 2021, 11, 10094–10109 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08302d |
| This journal is © The Royal Society of Chemistry 2025 |