
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective arrangement of three types of calcium–terbium tetranuclear cores by a thiacalixarene ligand using thermodynamic and kinetic strategies†
Ryunosuke Karashimada *,
Hironori Matsuoka and
Nobuhiko Iki*
*,
Hironori Matsuoka and
Nobuhiko Iki*
Graduate School of Environmental Studies, Tohoku University, 6-6-07, Aramaki-Aoba, Aoba-ku, Sendai 980-8579, Japan. E-mail: karashimada@tohoku.ac.jp; iki@tohoku.ac.jp
First published on 5th February 2025
Abstract
In this study, we report thermodynamic and kinetic strategies for arranging three types of Ca–Tb heterotetranuclear cores in a square configuration sandwiched by thiacalix[4]arene-p-tetrasulfonate (TCAS) ligands in aqueous solutions. In the thermodynamic strategy, the components were mixed under optimum pH conditions to afford a complex with a desired ratio of Ca:Tb:TCAS. In the kinetic strategy, the Ca1Tb3TCAS2 complex was formed via mixing kinetically stable Tb3TCAS2 with Ca2+. Interestingly, the resulting complexes (CaxTb4−xTCAS2, x = 1–3) exhibited Tb-centered luminescence upon excitation of the TCAS center with a high quvdantum yield (ϕ = 0.11–0.14) and a long luminescence lifetime (approximately 1.2 ms). The thermodynamic strategy can be applied to Sr2+ instead of Ca2+, but it is not suitable for first transition metal ions. However, the kinetic strategy is versatile and can be applied to first transition metal ions to afford M1Tb3TCAS2 (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+).
Introduction
The functionality of metal complexes arises from the ligand and the metal center. Introducing different metal centers to a complex to form a heteronuclear complex can result in novel functions imparted by metal–metal interactions and are of significant interest to chemists. Many heteronuclear complexes have been used as luminescent materials, magnetic materials (single molecule magnets), and catalysts.1Lanthanide (Ln) complexes exhibit potential as bio-imaging materials, such as luminescence probes and contrast agents for magnetic resonance imaging (MRI).2 To enhance the performance of Ln-based probes, the design of a heteronuclear complex featuring different metal centers (d–f or f–f metal centers) can be useful.1a,3 The d–f and f–f heteronuclear complexes can provide multifunctionality and novel functions through interactions between the metal centers, such as dual luminescence, up-conversion, and multimodalities (e.g., luminescence imaging and MRI).4–7
Thermodynamic and kinetic strategies (Fig. 1) are known approaches for the selective synthesis of a single heteronuclear complex.3a In the thermodynamic strategy, two coordination sites of a ligand are designed to provide selectivity towards two metal ions, enabling the binding of distinct metal centers. This thermodynamic strategy is straightforward to implement because of the simple mixing of the ligand and metal ions. Furthermore, the metal centers are in close proximity for their interactions. However, the chemical properties of the metal ions should be significantly different; consequently, the combination of metal ions is limited to d–d or d–f. In the f–f combination, obtaining site-selective coordination is impossible. Moreover, the resulting complex should be kinetically stable, otherwise the Ln and Ln′ ions in the formed complex shuffle between different coordination sites.
 | ||
| Fig. 1 Schematics of the (a) thermodynamic and (b) kinetic strategies for the selective synthesis of a heteronuclear complex. | ||
Two methods are used to implement the kinetic strategy. The first method is the step-wise method, which involves the initial synthesis of a mononuclear complex. The mononuclear complex subsequently reacts with a different metal ion to form a heteronuclear complex. The other method is the covalent joining of two mononuclear complexes. These methods require kinetic stability of the mononuclear intermediates such that they do not release the metal center. Therefore, an endo-type ligand, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) is usually used as a coordination moiety (The definition of an endo-type ligand is one that can encapsulate a metal ion inside the ligand.10b). However, an endo-type ligand and linker moiety induce the opening of a large distance between the metal centers, which causes weakening of the f–f interaction due to wrapping of the metal centers by the endo-type ligand and separation by the linker moiety.
A coordination bond is dynamic, and its timescale significantly depends on the kinetic property of the metal center. Therefore, the kinetic stability of a heteronuclear complex is crucial in determining the formation of heteronuclear complexes. If a heteronuclear complex is kinetically unstable, the metal centers shuffle between the coordination sites, resulting in a decrease in the intended heteronuclear complex and the formation of unwanted homo- and heteronuclear complexes. Furthermore, the synthesis of a single heteronuclear complex is difficult due to the low selectivity of the coordination moiety of the ligand. This low selectivity of the coordination moiety can also result in the concomitant formation of unwanted homo- and heteronuclear complexes. Therefore, the development of selective synthesis methods and kinetically stable single heteronuclear complexes in aqueous solutions remains a significant challenge in research.
In a previous study, we reported that thiacalix[4]arene-p-tetrasulfonate (TCAS, Fig. 2) can be a ligand for complexes containing multiple Ln and M–Ln cores.8 For example, Ln ions self-assemble to form a trinuclear Ln–TCAS complex (Ln3TCAS2, Fig. 2).9 The TCAS ligand functions as an exo-type ligand, coordinating to the tri-Ln core through O,S,O donor sets located on the surface of TCAS. (The definition of an exo-type ligand is one that binds to a metal center at the surface of the ligand.10b). The Ln3TCAS2 complex exhibits Ln-centered luminescence (Ln = Tb, Yb, and Nd) through an energy-transfer mechanism with good luminescence properties, and possesses high proton relaxivity, and thus functions as a suitable contrast agent for MRI with kinetic stability.9,10 Furthermore, d–f and f–f heteronuclear complexes, such as Ag–Ln–TCAS, Cd–Ln–TCAS, and Ln–Ln′–TCAS systems, have been successfully synthesized to attain distinctive functions that have not been achieved by mononuclear Ln1TCAS1.7,11
 | ||
| Fig. 2 Two-dimensional representations of TCAS, Ln1TCAS1, Ln3TCAS2, and heterotetranuclear Ca–Tb–TCAS complexes. | ||
Simply mixing the components at pH 10 according to the thermodynamic strategy afforded Ag4Tb1TCAS2, which showed an exceptionally long-lived Tb-centered luminescent lifetime (4.6 ms) in an aqueous solution by eliminating coordinated water molecules through the O8-cubic coordination geometry provided by the two TCAS ligands.11a,b In another example, the Tb–Yb–TCAS system provided a mixture of homo- and heterotrinuclear complexes (TbxYb3−xTCAS2, x = 0–3), in which heterotrinuclear complexes (TbxYb3−xTCAS2, x = 1, 2) exhibited a Tb→Yb energy transfer, that is, f–f communication.7 In contrast, following the kinetic strategy, the Tb1Yb2TCAS2 complex can be selectively synthesized by isolating an intermediate complex (Ln1TCAS1, Fig. 2).7 Thus, TCAS is a suitable ligand for the formation of d–f and f–f heteronuclear complexes.
In a recent experiment aimed at preparing TbxYb3−xTCAS2 (x = 1, 2), we accidently found new heteronuclear complexes containing TCAS, Ca2+, and Tb3+ (Fig. 2) in the reaction mixture. The Ca–Tb–TCAS system modulates the stoichiometry of the Ca–Tb core by manipulating the Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Tb ratio and pH condition, which results in energy-transfer luminescence. Calcium ions play significant roles in cell membrane responses, serving as a biomaterial component, with applications for cancer therapy.12
:Tb ratio and pH condition, which results in energy-transfer luminescence. Calcium ions play significant roles in cell membrane responses, serving as a biomaterial component, with applications for cancer therapy.12
In previous research on heteronuclear complexes,4–6 a precise design and complicated synthesis of the ligands were required for the selective formation of heteronuclear complexes. For example, Piguet et al. reported some d-f heteronuclear complexes with a precisely designed ligand providing different coordination sites of 6 and 9 coordination numbers to d and f ions, respectively.4 In contrast, our system only involves mixing the components (Ca, Tb, and TCAS) while controlling the pH condition for the formation of Ca–Tb–TCAS complexes without modification of the TCAS ligand.
In this study, we demonstrate the selective synthesis of three types of heterotetranuclear Ca–Tb–TCAS complexes using thermodynamic and kinetic strategies. Moreover, the study reveals the luminescent properties of the resulting complexes, and extending the strategies to metal ions other than Ca2+ is also successfully demonstrated.
Results and discussion
Self-assembly of heterotetranuclear Ca–Tb–TCAS complexes using the thermodynamic strategy
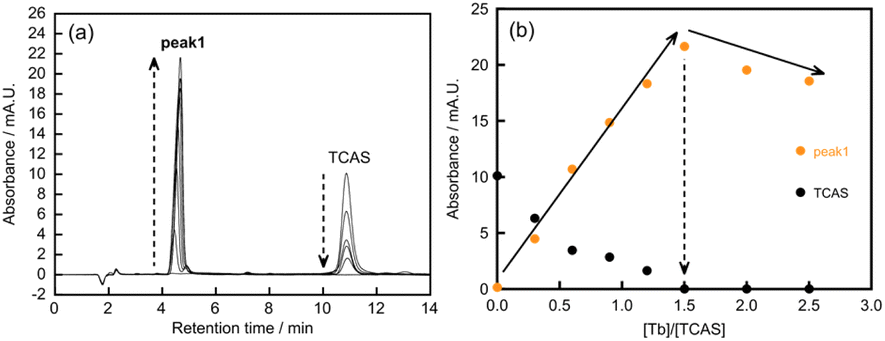
In our previous study, we found an unknown trace in a reaction mixture used for selective synthesis of heterotrinuclear complexes (TbxYb3−xTCAS2, x = 1, 2).7b It was confirmed that the unknown trace was a heteronuclear complex containing Tb Tb3+, TCAS (H4tcas4−), and Ca2+. Then, to synthesize Ca–Tb–TCAS complexes, the ratios of Ca:Tb:TCAS and pH conditions were varied. As a result, three types of heterotetranuclear Ca–Tb–TCAS (CaxTb4−xTCAS2, x = 1−3) were selectively formed under the appropriate conditions, as confirmed by high-performance liquid chromatography (HPLC) peaks (peak 1, peak 2, and peak 3) with different retention times (Fig. 3). The HPLC conditions were 45 wt% CH3CN in H2O containing 10 mmol per kg HEPES (apparent pH 7.4) and 30 mmol per kg TBABr (HEPES = 2-[4-(2-hydroxyethyl)-1-pyperadinyl]ethanesulfonic acid, TBABr = tetrabutylammonium bromide). | ||
| Fig. 3 Chromatograms of Ca–Tb–TCAS systems at different pH conditions. pH 6.0 condition: [Ca2+] = 10 μM, [Tb3+] = 30 μM, [TCAS] = 20 μM, [MES] = 20 mM, pH 6.0, t = 24 h, 60 °C; pH 10 condition: [Ca2+] = [Tb3+] = [TCAS] = 20 μM, [CAPS] = 20 mM, pH 10, t = 24 h, 60 °C; pH 11 condition: [Ca2+] = 30 μM, [Tb3+] = 10 μM, [TCAS] = 20 μM, pH 11 (adjusted with NH3), t = 1 h, 60 °C. | ||
Because previously reported Ln3TCAS2 complexes showed high kinetic stability under strongly acidic conditions (Ln = Yb: t1/2 = 1.53 h at pH 1.16 and Ln = Gd: t1/2 = 2.4 h at pH 2),10a,b it was expected that the heterotetranuclear Ca–Tb–TCAS complexes would exhibit high kinetic stability, implying that the dissociation reaction during the HPLC measurement is negligibly slow. The stoichiometry of the heterotetranuclear Ca–Tb–TCAS complexes was determined by employing the molar ratio method and electro-spray ionization-mass spectrometry (ESI-MS) analysis. Hereinafter, we describe the self-assembly of the ternary complex at the optimum pH (6.0, 10, and 11). In addition, conditions with different pH values resulted in a mixture of the complexes and/or TCAS ligand (as an example, at pH 7.4, as shown in Fig. S1†).
At pH 6.0, the molar ratio of Tb3+/TCAS was fixed at 1.5, and the concentration of Ca2+ was varied to investigate the height of peak 1 (Fig. 4). In zero- or low-concentration regions of Ca2+, the peak of Tb3TCAS2 was observed. As the concentration of Ca2+ increased, peak 1 appeared, and its height increased, reaching a maximum at the ratio of Ca2+/TCAS = 0.5. In contrast, when the ratio of Ca2+/TCAS was fixed at 0.5 and the concentration of Tb3+ was varied, the peak of TCAS was observed at zero or low concentrations of Tb3+ (Fig. 5). As the concentration of Tb3+ increased, the height of peak 1 reached a maximum at Tb3+/TCAS = 1.5.
 | ||
| Fig. 4 (a) Chromatograms of the molar ratio method and (b) molar ratio curves between [Ca2+] and [TCAS] for the Ca–Tb–TCAS system at pH 6.0. [Ca2+] = 0–16 μM, [Tb3+] = 30 μM, [TCAS] = 20 μM, [MES] = 20 mM, pH 6.0, λ = 316 nm, t = 24 h, 60 °C. | ||
 | ||
| Fig. 5 (a) Chromatograms of the molar ratio method and (b) molar ratio curves between [Tb3+] and [TCAS] for the Ca–Tb–TCAS system at pH 6.0. [Ca2+] = 20 μM, [Tb3+] = 0–0.10 mM, [TCAS] = 40 μM, [MES] = 20 mM, pH 6.0, λ = 316 nm, t = 24 h, 60 °C. | ||
These results suggest that the stoichiometry of the Ca–Tb–TCAS complex for peak 1 was Ca:Tb:TCAS = 0.5:1.5:1.0, implying the formation of the Ca1Tb3TCAS2 complex. To confirm the formation of Ca1Tb3TCAS2, ESI mass spectrometry was also performed at pH 6.0, adjusted with NH3 solution, and CH3CN was added to the sample solution to facilitate the ionization process (Fig. 6 and S2†). The mass spectra exhibited intense peaks at m/z 454.461738, which could be assigned to [Ca2+ + 3Tb3+ + 2tcas8− + 2Na+ + 2OH− + NH3 + CH3CN]5− (m/z 454.4581). All assignment is included in Fig. S2,† and other products with different metal ratios of Ca–Tb–TCAS complexes were not detected.
 | ||
| Fig. 6 ESI mass spectra of the Ca–Tb–TCAS system at pH 6.0. (a) Observed isotopic distribution at m/z 454.461738 under the following conditions: [Ca2+] = 10 μM, [Tb3+] = 30 μM, [TCAS] = 20 μM, pH 6.0 (adjusted with NH3), t = 24 h, 60 °C. (b) Calculated isotopic distribution for [Ca2+ + 3Tb3+ + 2tcas8− + 2Na+ + 2OH− + NH3 + CH3CN]5− at m/z 454.45818−. | ||
In previous studies, the results of ESI mass spectrometry showed many adducts in the metal complexes using TCAS ligand.7,9–11 In our case, the adducts of CH3CN and NH3 presenting as guest molecules in the TCAS ligand of the Ca–Tb–TCAS complexes is suggested,13 and there is also the possibility for a coordinated NH3 molecule with the latter because the Tb center of the Ca–Tb–TCAS complexes contains two coordinated water molecules (the details of this result will be discussed later, and see Table 1). Moreover, for other compounds with O2− and H+, we assigned these compounds as consisting of H2O or OH−. Thus, the Ca1Tb3TCAS2 complex can be selectively formed at pH 6.0.
At pH 10, the molar ratio method at a fixed ratio of Tb3+/TCAS = 1.0 with varying concentrations of Ca2+ showed that the height of peak 2 reached a maximum at Ca2+/TCAS = 1.0 (Fig. S3†). However, when the ratio of Ca2+/TCAS was fixed at 1.0, and the concentration of Tb3+ was varied, the height of peak 2 reached a maximum at Tb3+/TCAS = 1.0 (Fig. S4†). These observations indicate that the stoichiometry of the complex is CaiTbiTCASi (i: natural number). Additionally, the ESI mass spectra of the sample solution at pH 10, which was adjusted with NH3 solution and contained CH3CN, showed intense peaks at m/z 708.782527, corresponding to the components of [2Ca2+ + 2Tb3+ + 2tcas8− + Na+ + 2H+ + 3H2O + 2NH3]3− (m/z 708.7946) and other assignments, as shown in Fig. S5.† These results indicate that the stoichiometry of the Ca–Tb–TCAS system at pH 10 was Ca2Tb2TCAS2.
At pH 11, the molar ratio plot of peak 3 showed a maximum at Ca2+/TCAS = 1.5 when the Tb3+/TCAS ratio was fixed at 0.5 (Fig. S6†). Furthermore, a maximum of peak 3 appeared at Tb3+/TCAS = 0.5 when the Ca2+/TCAS ratio was fixed at 1.5 (Fig. S7†). These results suggest that the Ca3Tb1TCAS2 complex is primarily formed at pH 11; however, peaks of unwanted impurities, such as Ca2Tb2TCAS2 and TCAS, were also observed. Therefore, a more suitable condition for the selective synthesis of the Ca3Tb1TCAS2 complex was necessary.
Considering the success in enhancing the selectivity by adding TBABr during the synthesis of the Ln–Ln′–TCAS heterotrinuclear complex,7b we added tetrabutylammonium (TBA+) bromide (TBABr) salt to the mixture of Ca2+, Tb3+, and TCAS to enhance the selectivity. Upon the addition of ≥0.2 M TBABr, the proportion of Ca3Tb1TCAS2 increased, and that of Ca2Tb2TCAS2 significantly decreased (Fig. S8†). The molar ratio method under the conditions of 0.2 M TBABr and pH 11 resulted in maxima at Ca2+/TCAS = 1.5 (Fig. S9†) and at Tb3+/TCAS = 0.5 (Fig. S10†). The effect of TBABr salt on the reaction is currently unclear and is still under investigation. We briefly described the effect of the TBABr salt as a hypothesis based on our previous findings.7b The presence of TBABr or TBA+ affects the rate constant of the complexation reaction during the formation of a sandwich structure, leading to a decrease in the rate of the reaction step:
| 2Ln1TCAS1 → Ln2TCAS2 | (1) |
:2 complexes, as shown below:| 2Tb1TCAS1 → Tb2TCAS2 | (1′) |
| 2Ca1TCAS1 → Ca2TCAS2 | (2) |
| Tb1TCAS1 + Ca1TCAS1 → Ca1Tb1TCAS2 | (3) |
We assume that the presence of TBABr or TBA+ selectively decreases the reaction rate of eqn (1′). The ESI-MS analysis of the sample solution of the Ca–Tb–TCAS system, which was adjusted to pH 11 with NH3 solution and contained CH3CN, exhibited intense peaks at m/z 382.670041 and 382.8715772. This isotopic distribution corresponds to the components of [3Ca2+ + Tb3+ + 2tcas8− + H+ + H2O]5− (m/z 382.6698) and [3Ca2+ + Tb3+ + 2tcas8− + 2H+ + H2O]5− (m/z 382.8714), confirming the formation of the Ca3Tb1TCAS2 complex at pH 11. Additionally, other signals and their assignments are shown in Fig. S11.†
We attempted to reveal the structure of these Ca–Tb–TCAS complexes. Despite some crystallization experiments, the single crystal suitable for X-ray diffraction analysis has not yet been obtained and is in progress. We preliminarily investigated isolation of a solid sample of the Ca–Tb–TCAS complexes as a salt with hexadecyltrimethylammonium ion (CTMA+). We successfully obtained precipitates produced by the mixing of the Ca–Tb–TCAS complexes and CTMA+.
In the mixture of the Ca1Tb3TCAS2 complex and the CTMA+ system, the CHN elemental analysis indicated that the composition of the salt was CTMA6Ca1Tb3TCAS2(H2O)10Cl1. This result was consistent with the salt for the Tb3TCAS2 complex with quaternary ammonium ions in the previous study, for example, CTMA7Tb3TCAS2(H2O)10.16b The other two complexes, Ca2Tb2TCAS2 and Ca3Tb1TCAS2, also showed similar compositions: CTMA10Ca2Tb2TCAS2(H2O)18(NO3)3 and CTMA10Ca3Tb1TCAS2(H2O)20Cl3. These CTMA–Ca–Tb–TCAS salts contained H2O molecules as coordinated water (see Table 1) and crystal solvent, and the coprecipitation of CTMACl or CTMANO3 salts was suggested due to the experimental conditions (addition of a small excess amount of CTMACl and cooling before filtration).
By simply mixing the components (Ca2+, Tb3+, and TCAS) at an appropriate ratio and appropriate pH (6.0, 10, and 11), selective formation of the Ca1Tb3TCAS2, Ca2Tb2TCAS2, and Ca3Tb1TCAS2 complexes can be achieved using the thermodynamic strategy. The number of Tb3+ in the Ca–Tb–TCAS complexes decreased at high pH, which may be attributed to the fact that Tb3+ is more susceptible to hydrolysis compared to Ca2+. This phenomenon is influenced by the valence of the metal ions.
The results of the present work show that the ratio of Tb3+ and Ca2+ (Ca1Tb3, Ca2Tb2, and Ca3Tb1) could be completely controlled by pH conditions despite their properties of chemical similarity. These systems enabled selective synthesis of heteronuclear complexes without the need for precise design or modification of the ligand. In addition, this observation is remarkably unique when compared to previous studies in which TCAS or TCA was used as a ligand containing a M–Ln core, and the chemical properties of M (Ag+, Cd2+, Mn2+, Zn2+) and Ln (Tb3+, Yb3+, Nd3+, Gd3+, Dy3+, Ho3+) are significantly different.11b–d,14
We assume that the Ca–Tb–TCAS complexes possess a double-cone structure in which the tetrametal core is sandwiched by two conical TCAS anions, as exemplified by Ln3TCAS2 and Ln4TCA2.9,15 First, Ca1Tb3TCAS2 can also be formed by mixing Ca2+ and Tb3TCAS2 complex in the kinetic strategy (vide infra), suggesting that Ca1Tb3TCAS2 adopts the double-cone structure. Second, the luminescence properties as well as the number of coordinated water molecules of the Ca2Tb2TCAS2 and Ca3Tb1TCAS2 complexes are very similar to those of Ln3TCAS2 and Ca1Tb3TCAS2, indicating the structural similarity among these complexes. Third, the chromatographic behavior of those complexes is very similar. The number of coordination bonds connecting the metal core and TCAS in the Ca–Tb–TCAS complexes is larger than that in the Ln3TCAS2 complex. Consequently, the kinetic stability of the heterotetranuclear Ca–Tb–TCAS complexes is expected to be greater than that of the Tb3TCAS2 complex.
Time dependence of the self-assembly of the heterotetranuclear Ca–Tb–TCAS complexes
The self-assembly of the heterotetranuclear Ca–Tb–TCAS complexes was measured as a function of time under specific ratios of the components (Ca2+, Tb3+, and TCAS) at optimum pH (Fig. 7). At pH 6.0, peak 1 (Ca1Tb3TCAS2) immediately appeared upon mixing the components, and reached a plateau after 120 min (Fig. 7(a)). The byproducts (Tb1TCAS1, Tb3TCAS2, and Ca2Tb2TCAS2) decreased and eventually disappeared at 120 min, indicating the formation of a single Ca1Tb3TCAS2 complex. There was an appearance by peak 2 (Ca2Tb2TCAS2), which reached a plateau at 240 min, while the peaks of the byproducts Ca1Tb3TCAS2 and Ca3Tb1TCAS2 disappeared at pH 10 (Fig. 7(b)), indicating the formation of a single Ca2Tb2TCAS2 complex. In the absence of TBABr at pH 11, not only peak 3 (Ca3Tb1TCAS2) but also significant amounts of unwanted peak 2 Ca2Tb2TCAS2 (approximately 25%) and TCAS appeared and persisted (Fig. S12†). In the presence of TBABr at pH 11, peak 2 and TCAS were substantially suppressed, and peak 3 was predominant for 150 min (Fig. 7(c)). | ||
| Fig. 7 Time dependence of the height of the HPLC peaks of the Ca–Tb–TCAS complexes during self-assembly. (a) [Ca2+] = 0.10 mM, [Tb3+] = 0.30 mM, [TCAS] = 0.20 mM, [MES] = 40 mM, pH 6.0, 60 °C. (b) [Ca2+] = 2.0 mM, [Tb3+] = 2.0 mM, [TCAS] = 2.0 mM, pH 10 (adjusted with NH3), 60 °C. (c) [Ca2+] = 0.15 mM, [Tb3+] = 50 μM, [TCAS] = 0.10 mM, [TBABr] = 0.20 M, pH 11 (adjusted with NH3), 60 °C. | ||
The time scale of the formation of each Ca–Tb–TCAS complex should be governed by a rate-determining step (RDS), which is different among the ternary complexes. For example, in Fig. 7(a), the increasing rate of peak 1 and decreasing rate of Tb3TCAS2 were the same, implying that the RDS is:
| Ca2+ + Tb3TCAS2 → Ca1Tb3TCAS2 | (4) |
Synthesis of a heterotetranuclear Ca–Tb–TCAS complex using the kinetic strategy
Considering eqn (4), we prepared Ca1Tb3TCAS2 by introducing Ca2+ into Tb3TCAS2. One equivalent of Ca2+ was added to a solution containing Tb3TCAS2 obtained by a previously reported method,15 and the self-assembly process of the complexation was monitored. The peak of Tb3TCAS2 was dominant immediately after the addition of Ca2+ (Fig. 8). Subsequently, the peak of Tb3TCAS2 decreased; however, peak 1 (Ca1Tb3TCAS2) emerged and increased. Furthermore, peak 1 reached a plateau at 240 min, and the peak of Tb3TCAS2 disappeared. Thus, mere mixing of the Tb3TCAS2 complex and Ca2+ afforded Ca1Tb3TCAS2, which formed due to the kinetic stability of Tb3TCAS2; thus, this approach can be classified as a kinetic strategy. The times for reaching the plateau were not the same (120 min in Fig. 7(a) and 240 min in Fig. 8), which can be attributed to the difference in the pH conditions.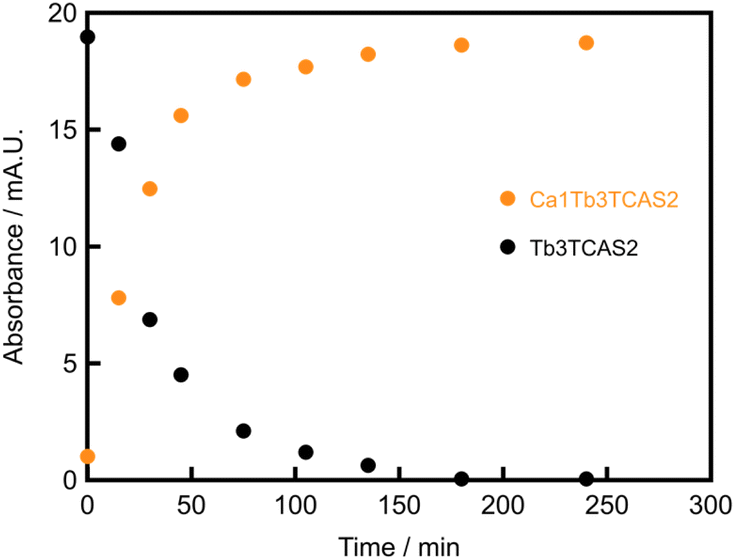 | ||
| Fig. 8 Time dependence of the height of the HPLC peaks of the Ca1Tb3TCAS2 complex in the mixture of the Tb3TCAS2 complex and Ca2+. [Ca2+] = 0.10 mM, [Tb3TCAS2] = 0.10 mM, pH 10 (adjusted with NH3), 60 °C. | ||
Luminescence properties of the heterotetranuclear Ca–Tb–TCAS complexes
The excitation spectra of the three heterotetranuclear Ca–Tb–TCAS complexes (CaxTb4−xTCAS2, x = 1–3) exhibited a broad peak, attributed to the π–π* transition of ligand-centered absorption (Fig. 9(a)). The wavelengths of the excitation peaks were disparate, with values of 314 nm for Ca1Tb3TCAS2, 320 nm for Ca2Tb2TCAS2, and 326 nm for Ca3Tb1TCAS2. The wavelength of the peaks depended on the charge of the metal core; that is, the smaller the number of the charges of the tetrametal core (i.e., +11 for Ca1Tb3, +10 for Ca2Tb2, and +9 for Ca3Tb1), the longer the excitation wavelength. The emission spectra of the complexes CaxTb4−xTCAS2 (x = 1–3) exhibited sharp emission bands, corresponding to Tb-centered luminescence (5D4 → 7FJ, J = 6–3). The ligand-centered absorption and the Tb-centered luminescence suggest that all heterotetranuclear Ca–Tb–TCAS complexes (CaxTb4−xTCAS2, x = 1–3) exhibited luminescence through energy transfer from TCAS to Tb3+. | ||
| Fig. 9 (a) Excitation and emission spectra and (b and c) the fine-structure of Tb-centered luminescence for the Ca–Tb–TCAS systems. The Ca1Tb3TCAS2 system: [Ca1Tb3TCAS2] = 1.0 μM, [CAPS] = 10 mM, pH 10, λex = 314 nm, λem = 550 nm, t = 24 h, 60 °C; the Ca2Tb2TCAS2 system: [Ca2Tb2TCAS2] = 1.0 μM, [CAPS] = 10 mM, pH 10, λex = 320 nm, λem = 550 nm, t = 24 h, 60 °C; and the Ca3Tb1TCAS2 system: [Ca3Tb1TCAS2] = 1.0 μM, [TBABr] = 20 mM, pH 10.5 (adjusted with NH3), λex = 326 nm, λem = 550 nm, t = 1 h, 60 °C. (a) The width of the excitation and emission slits was 5 nm. (b and c) The width of the excitation slit was 20 nm, and the width of the emission slit was 1 nm. Normalized at 550 nm. | ||
In the fine structure of Tb-centered luminescence (5D4 → 7FJ, J = 5, 4), the shapes of the bands for the Ca1Tb3TCAS2, Ca2Tb2TCAS2, and Ca3Tb1TCAS2 complexes were not identical (Fig. 9(b and c)). Notably, the luminescence spectra of the Ca3Tb1TCAS2 complex exhibited significantly different shapes compared to those of the Ca1Tb3TCAS2 and Ca2Tb2TCAS2 complexes. In general, the fine structure of the Tb-centered luminescence depends on the coordination environment of the Tb3+ center. Assuming a sandwich-type structure for all Ca–Tb–TCAS complexes, the difference in the coordination environment of the Tb3+ center can be understood by the electron density of μ2-O, a phenol oxygen atom of TCAS (OTCAS), bridging two metal centers according to Ca-OTCAS-Ca, Ca-OTCAS-Tb, and Tb-OTCAS-Tb (Fig. 10).
 | ||
| Fig. 10 Arrangements of the Ca–Tb core and three types of phenol oxygen atom of TCAS in the heterotetranuclear complexes. | ||
The electron density of μ2-O decreases in the following order: Ca-OTCAS-Ca > Ca-OTCAS-Tb > Tb-OTCAS-Tb. Considering this order, the Tb3+ center in the Ca3Tb1TCAS2 is coordinated by two μ2-O (Ca-OTCAS-Tb) with an intermediate electron density. For Ca1Tb3TCAS2, two of the Tb3+ centers are coordinated by μ2-O of Ca-OTCAS-Tb, but one is coordinated by the most electron-deficient μ2-O (Tb-OTCAS-Tb). Therefore, it is likely that the difference between the fine structures of the Tb-centered luminescence of the Ca1Tb3TCAS2 and Ca3Tb1TCAS2 complexes originated from the presence or the absence of the Tb-OTCAS-Tb type of μ2-O.
For Ca2Tb2TCAS2, there are two possible configurations: a diagonal arrangement (Ca–Tb–Ca–Tb disposition) or an arrangement adjacent to an identical metal center (Ca–Ca–Tb–Tb disposition) (Fig. 10). These configurations represent distinct types of μ2-O: the Ca–Tb–Ca–Tb disposition has only one type of μ2-O, that is, Ca–OTCAS-Tb; however, the Ca–Ca–Tb–Tb disposition has two types of μ2-O, that is, Ca–OTCAS–Tb and Tb–OTCAS–Tb. Because the shape of the luminescence bands of Ca2Tb2TCAS2 is similar to that of Ca1Tb3TCAS2 with Tb–OTCAS–Tb, the arrangement of the tetrametal core in Ca2Tb2TCAS2 is possibly Ca–Ca–Tb–Tb.
This hypothesis is reasonable by considering the coordination bond length of M–OTCAS or M–S, which should be different among M = Ca2+ and Tb3+. The Ca–Ca–Tb–Tb disposition is more tolerant than the Ca–Tb–Ca–Tb disposition toward sandwiching the metal core while maintaining the optimal M–OTCAS and M–S length. Consequently, two conical TCAS ligands were not in parallel alignment, but rather, were aligned in a slightly tilted manner. In fact, examples of such M–M–Ln–Ln arrangements in M–Ln–thiacalixarene systems have been previously reported;11b–d,14a however, no arrangements of M–Ln–M–Ln have been reported.
Table 1 summarizes the photophysical properties of the heterotetranuclear Ca–Tb–TCAS complexes (CaxTb4−xTCAS2, x = 1–3) and the previously reported Tb3TCAS2 complex. The Tb-centered luminescence lifetime for the three heterotetranuclear complexes was long, with equal values of approximately 1.2 ms. The number of coordinated water molecules was estimated to be two for each heterotetranuclear complex by Horrocks' equation17 and the luminescence lifetimes in H2O and D2O. The quantum yield of the three heterotetranuclear complexes was 0.11–0.14. The luminescence properties of the heterotetranuclear complexes were as high as those of the previously reported homotrinuclear Tb3TCAS2 complex,16 indicating that they possess a similar sandwich-type structure and octa-coordination environment of the Tb3+ center by two O, S, O donor sets from two TCAS ligands and two oxygen atoms from water.9
| ϕ/— | τH/ms | τD/ms | q | |
|---|---|---|---|---|
| a [complex] = 0.25 μM, [CAPS] = 10 mM, pH 10, λex = 314 nm for Ca1Tb3TCAS2, 320 nm for Ca2Tb2TCAS2, 326 nm for Ca3Tb1TCAS2, t = 24 h for Ca1Tb3TCAS2 and Ca2Tb2TCAS2, 1 h for Ca3Tb1TCAS2.b For the decay curves, see Fig. S13–15 in ESI. Subscripts H and D for τ denote H2O and D2O solvents, respectively.c Estimated by Horrocks' equation. q = 4.19(τH−1 – τD−1).d See ref. 16. | ||||
| Ca3Tb1TCAS2 | 0.11 | 1.19 | 3.50 | 2.3 |
| Ca2Tb2TCAS2 | 0.13 | 1.19 | 2.81 | 2.1 |
| Ca1Tb3TCAS2 | 0.14 | 1.22 | 2.90 | 2.0 |
| Tb3TCAS2d | 0.14 | 1.12 | 3.22 | 2.4 |
Thermodynamic and kinetic strategies for other metal ions to form heterotetranuclear M–Tb–TCAS complexes
Inspired by the success of the selective preparation of three types of Ca–Tb–TCAS heterotetranuclear complexes using the thermodynamic strategy, and the preparation of Ca1Tb3TCAS2 using the kinetic strategy, we extended the strategy to other alkaline earth and transition metal ions (M) (Fig. S16†). For example, the chromatograms of the reaction mixture in the thermodynamic strategy (M:Tb:TCAS = 1:3:2, pH 6.0 and M:Tb:TCAS = 2:2:2, pH 10) showed a single peak in the presence of TCAS and Tb3+ with M = Sr2+ at the retention time (tR = 4.53 min for Sr:Tb:TCAS = 1:3:2 at pH 6.0, 6.56 min for M:Tb:TCAS = 2:2:2 at pH 10), which was similar to that for one of the Ca–Tb–TCAS complexes (tR = 4.67 min for Ca1Tb3TCAS2, 6.86 min for Ca2Tb2TCAS2). In contrast, the chromatograms of the transition metal systems (M = Ti3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) showed several peaks corresponding to various M–Tb–TCAS and Tb–TCAS complexes. This may be caused by many factors, including the affinity for the TCAS ligand, complexation kinetics, and the tendency towards hydrolysis of transition metal ions other than Ca2+. Hence, the optimum conditions for the formation of heterotetranuclear M–Tb–TCAS complexes should be different from those for the formation of Ca–Tb–TCAS.
In contrast, the kinetic strategy using the reaction between Tb3TCAS2 and metal ions (M) resulted in a single peak in all systems, and M1Tb3TCAS2 complexes (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) were successfully formed. The peak of the Sr–Tb3TCAS2 system appeared at tR = 4.35 min, which is similar to that of the Ca1Tb3TCAS2 complex (tR = 4.67 min). For the transition metal-Tb3TCAS2 systems, a single peak appeared at tR = 5.22–5.35 min, which is different than that of the Ca1Tb3TCAS2 complex. This may have been caused by the difference in the charge of the M and/or in the deprotonation of the coordinating water to M. Thus, the kinetic strategy can be applied not only to Ca2+, but also to various metal ions.
We further investigated the ESI mass spectrometry for the mixture of M and the Tb3TCAS2 solution to confirm the formation of the M1Tb3TCAS2 complex (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) (Fig. S17–23†). For example, the mixture of Sr2+ and Tb3TCAS2 solution showed isotopic distribution at m/z 443.24429, which could be assigned to [Sr2+ + 3Tb3+ + 2tcas8− + 2H2O]5− (m/z 443.2449) (Fig. S17†). This result indicates the formation of the Sr1Tb3TCAS2 complex. The others also showed mass spectra corresponding to [M2+ + 3Tb3+ + 2tcas8− + 2H2O]5− (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Zn2+) and [Cu2+ + 3Tb3+ + 2tcas8− + H2O]5−. These ESI-MS measurement results are consistent with the above HPLC measurements, implying that the formation of the M1Tb3TCAS2 complex could be possible by the mixture of M and Tb3TCAS2 solution at pH 6.0. Moreover, the mixture of M and Tb3TCAS2 emitted Tb-centered luminescence due to energy transfer from TCAS ligand, but the intensity at 550 nm was different depending on the type of metal ion (M) (Fig. S24†).
In the case of M = Sr2+ or Zn2+, the intensity at 550 nm was the same as that of the Tb3TCAS2 complex. In contrast, in the case of M = Mn2+, Fe2+, Co2+, Ni2+, or Cu2+, the Tb-centered luminescence was diminished, indicating paramagnetic quenching. According to the above findings, the mixture of M and Tb3TCAS2 produces M1Tb3TCAS2 complexes (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+).
Conclusions
Herein, we presented a facile method to selectively prepare three luminescent heterotetranuclear complexes (CaxTb4−xTCAS2, x = 1–3), in which heterometal cores are arranged in a square form. In the thermodynamic strategy, three complexes (CaxTb4−xTCAS2, x = 1–3) were formed by simply mixing Ca, Tb, and TCAS at an appropriate proportion with an optimized pH. The selectivity of the formation of heterotetranuclear complexes seemingly depends upon the fact that the tendency of Tb3+ to hydrolyze is greater than that of Ca2+ (Fig. 11). This result suggests that the difference in the susceptibility to hydrolysis between heterometal ions is a useful factor to control the heterometal core.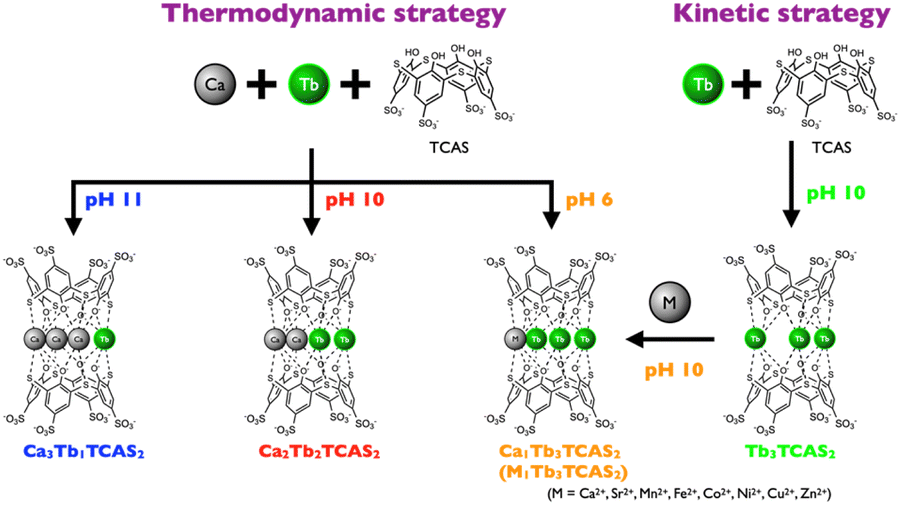 | ||
| Fig. 11 Schematic of the formation of the heterotetranuclear Ca–Tb–TCAS complexes. | ||
Moreover, this method can be applied not only to the TCAS ligand but also to many other ligands. In the kinetic strategy, Ca1Tb3TCAS2 was afforded by simple addition of Ca2+ to Tb3TCAS2 in solution (Fig. 11). All heterotetranuclear complexes (CaxTb4−xTCAS2, x = 1–3) exhibited Tb-centered luminescence by an energy-transfer mechanism with nearly the same lifetime and quantum yield. However, the heterotetranuclear complex is kinetically more stable than the Ln3TCAS2 complex because of the multi-coordination bond by the tetranuclear core. Finally, we extended the strategies to the preparation of M–Tb–TCAS complexes (M = Sr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+). Because thiacalixarene ligands can form a metal complex with all Ln3+ species9 and various transition metal ions,11d the strategy may be applicable to other metal–lanthanide–TCAS complexes. Thus, the strategies mentioned in this study are promising, and will pave the way for the application of heterometal complexes in multimodal probes and multifunctional materials.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ryunosuke Karashimada: conceptualization, data curation, funding acquisition, investigation, project administration, resources, supervision, visualization, writing–original draft, writing–review and editing; Hironori Matsuoka: data curation, investigation, validation, visualization; Nobuhiko Iki: funding acquisition, project administration, resources, supervision, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ESI-MS measurements were supported by Mr Hiroyuki Momma and Shunsuke Kayamori. This work was partly supported by JSPS KAKENHI (Grant Numbers 18K14248, 20K15308, and 23K04801).References
- (a) F. Artizzu, F. Quochi, A. Serpe, E. Sessini and P. Deplano, Inorg. Chem. Front., 2015, 2, 213–222 RSC; (b) J.-R. Jiménez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 513750–513769 CrossRef; (c) X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374–6458 CrossRef CAS PubMed; (d) Z. Zhu and J. Tang, Chem. Soc. Rev., 2022, 51, 9469–9481 RSC; (e) Y. Peng, H. Kaemmerer and A. K. Powell, Chem. Eur J., 2021, 27, 15043–15065 CAS; (f) Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC; (g) Z.-H. Pan, Z.-Z. Weng, X.-J. Kong, L.-S. Long and L.-S. Zheng, Coord. Chem. Rev., 2022, 457, 214419–214440 CrossRef CAS.
- (a) J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed; (b) J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef; (c) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC; (d) M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonnière, Chem. Commun., 2016, 52, 5080–5095 RSC.
- (a) N. Iki, Supramol. Chem., 2011, 23, 160–168 CrossRef CAS; (b) A. M. Nonat and L. J. Charbonnière, Coord. Chem. Rev., 2020, 409, 213192–213208 CrossRef CAS.
- (a) L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J.-C. G. Bünzli, P.-Y. Morgantini, K. R. Kittilstved, A. Hauser, S. V. Eliseeva, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2012, 134, 12675–12684 CrossRef CAS PubMed; (b) D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, Dalton Trans., 2015, 44, 2529–2540 RSC; (c) D. Zare, Y. Suffren, H. Nozary, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2017, 56, 14612–14617 CrossRef CAS PubMed.
- (a) F. Artizzu, F. Quochi, L. Marchiò, E. Sessini, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, J. Phys. Chem. Lett., 2013, 4, 3062–3066 CrossRef CAS; (b) F. Artizzu, F. Quochi, L. Marchiò, R. F. Correia, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, Chem. Eur J., 2014, 21, 3882–3885 CrossRef PubMed; (c) F. Artizzu, F. Quochi, L. Marchiò, C. Figus, D. Loche, M. Atzori, V. Sarritzu, A. M. Kaczmarek, R. V. Deun, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, Chem. Mater., 2015, 27, 4082–4092 CrossRef CAS; (d) F. Artizzu, A. Serpe, L. Marchiò, M. Saba, A. Mura, M. L. Mercuri, G. Bongiovanni, P. Deplano and F. Quochi, J. Mater. Chem. C, 2015, 3, 11524–11530 RSC.
- (a) N. Souri, P. Tian, C. Platas-Iglesias, K.-L. Wong, A. Nonat and L. J. Charbonnière, J. Am. Chem. Soc., 2017, 139, 1456–1459 CrossRef CAS PubMed; (b) J. Salaam, L. Tabti, S. Bahamyirou, A. Lecointre, O. H. Alba, O. Jeannin, F. Camerel, S. Cianférani, E. Bentouhami, A. M. Nonat and L. J. Charbonnière, Inorg. Chem., 2018, 57, 6095–6106 CrossRef CAS PubMed; (c) A. Nonat, S. Bahamyirou, A. Lecointre, F. Przybilla, Y. Mély, C. Platas-Iglesias, F. Camerel, O. Jeannin and L. J. Charbonnière, J. Am. Chem. Soc., 2019, 141, 1568–1576 CrossRef CAS PubMed; (d) R. C. Knighton, L. K. Soro, T. Troadec, V. Mazan, A. M. Nonat, M. Elhabiri, N. Saffon-Merceron, S. Djenad, R. Tripier and L. J. Charbonnière, Inorg. Chem., 2020, 59, 10311–10327 CrossRef CAS PubMed; (e) R. C. Knighton, L. K. Soro, A. Lecointre, G. Pilet, A. Fateeva, L. Pontille, L. Francés-Soriano, N. Hildebrandt and L. J. Charbonnière, Chem. Commun., 2021, 57, 53–56 RSC; (f) L. K. Soro, C. Charpentier, F. Przybilla, Y. Mély, A. M. Nonat and L. J. Charbonnière, Chem. Eur J., 2021, 3, 1037–1046 CAS; (g) R. C. Knighton, L. K. Soro, L. Francés-Soriano, A. Rodríguez-Rodríguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hilderbrandt and L. J. Charbonnière, Angew. Chem., Int. Ed., 2022, 61, e202113114 CrossRef CAS PubMed.
- (a) R. Karashimada and N. Iki, Chem. Commun., 2016, 52, 3139–3142 RSC; (b) R. Karashimada, K. Musha and N. Iki, Bunseki Kagaku, 2022, 71, 145–151 CrossRef CAS.
- N. Morohashi and N. Iki, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 2022, vol. 326, pp. 1–280 Search PubMed.
- N. Iki, T. Tanaka, S. Hiro-oka and K. Shinoda, Eur. J. Inorg. Chem., 2016, 5020–5027 CrossRef CAS.
- (a) N. Iki, E. Boros, M. Nakamura, R. Baba and P. Caravan, Inorg. Chem., 2016, 55, 4000–4005 CrossRef CAS PubMed; (b) N. Iki, S. Hiro-oka, M. Nakamura, T. Tanaka and H. Hoshino, Eur. J. Inorg. Chem., 2012, 3541–3545 CrossRef CAS; (c) N. Iki, S. Hiro-oka, T. Tanaka, C. Kabuto and H. Hoshino, Inorg. Chem., 2012, 51, 1648–1656 CrossRef CAS PubMed.
- (a) T. Tanaka, N. Iki, T. Kajiwara, M. Yamashita and H. Hoshino, J. Inclusion Phenom. Macrocyclic Chem., 2009, 64, 379–383 CrossRef CAS; (b) N. Iki, M. Ohta, T. Horiuchi and H. Hoshino, Chem. Asian J., 2008, 3, 849–853 CrossRef CAS PubMed; (c) N. Iki, M. Ohta, T. Tanaka, T. Horiuchi and H. Hoshino, New J. Chem., 2009, 33, 23–25 RSC; (d) N. Iki, T. Tanaka and H. Hoshino, Inorg. Chim. Acta, 2013, 397, 42–47 CrossRef CAS.
- Y. Kang, L. Xu, J. Dong, Y. Huang, X. Yuan, R. Li, L. Chen, Z. Wang and X. Ji, Coord. Chem. Rev., 2023, 481, 215050–215068 CrossRef CAS.
- N. Iki, T. Fujimoto and S. Miyano, Chem. Lett., 1998, 27, 625–626 CrossRef.
- (a) Y. Bi, Y. Li, W. Liao, H. Zhang and D. Li, Inorg. Chem., 2008, 47, 9733–9735 CrossRef CAS PubMed; (b) K. Su, F. Jiang, J. Qian, M. Wu, K. Xiong, Y. Gai and M. Hong, Inorg. Chem., 2013, 52, 3780–3786 CrossRef CAS PubMed.
- A. Bilyk, J. W. Dunlop, R. O. Fuller, A. K. Hall, J. M. Harrowfield, M. W. Hosseini, G. A. Koutsantonis, I. W. Murray, B. W. Skelton, A. N. Sobolev, R. L. Stamps and A. H. White, Eur. J. Inorg. Chem., 2010, 2127–2152 CrossRef CAS.
- (a) N. Shiraishi, R. Karashimada and N. Iki, Bull. Chem. Soc. Jpn., 2019, 92, 1847–1852 CrossRef CAS; (b) N. Shiraishi, D. Iikura, R. Karashimada and N. Iki, J. Lumin., 2024, 269, 120521–120529 CrossRef CAS.
- W. D. H. Jr and D. R. Sundnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08259a |
| This journal is © The Royal Society of Chemistry 2025 |