
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale drug formulations for the treatment of Alzheimer's disease progression
Liqin Liuad,
Haini He*ad,
Bin Du *b and
Yang He*cd
*b and
Yang He*cd
aDepartment of Pediatrics of Neurology Nursing, West China School of Nursing, West China Second University Hospital, Sichuan University, Chengdu 610000, China. E-mail: 2398547353@qq.com
bState Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610000, China. E-mail: bin.du@scu.edu.cn
cDepartment of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu 610000, China. E-mail: heyang1235@126.com
dKey Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu 610000, China
First published on 7th February 2025
Abstract
Alzheimer's disease (AD) is a devastating neurodegenerative disorder with no effective disease-modifying treatments. The blood–brain barrier hinders drug delivery to the brain, limiting therapeutic efficacy. Nanoparticle-based systems have emerged as promising tools to overcome these challenges. This review highlights recent advances in nanoparticle technologies for AD treatment, including liposomes, polymeric, inorganic, and biomimetic nanoparticles. These nanoparticles improve drug delivery across the blood–brain barrier, improve stability and bioavailability, and enable targeted delivery to affected brain regions. Functionalization strategies further enhance their therapeutic potential. Multifunctional nanoparticles combining therapeutic and diagnostic properties offer theranostic approaches. While progress has been made, challenges related to safety, targeting precision, and clinical translation remain. Future perspectives emphasize the need for collaborative efforts to optimize nanoparticle design, conduct rigorous studies, and accelerate the development of effective nanotherapeutics. With continued innovation, nanoparticle-based delivery systems hold great promise for revolutionizing AD treatment.
1. Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative condition that progressively inhibits cognitive functions, memory, and behavior.1 It is the most prevalent form of dementia, contributing to 60–80% of all dementia cases globally. As populations age, AD has become a significant public health concern, creating a substantial healthcare burden worldwide. In 2019, official death certificates in the USA recorded 121![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 499 deaths due to AD, ranking it as the sixth leading cause of mortality in the country. The financial implications are staggering, with the total cost of care for AD patients estimated at $305 billion in 2020. This cost is projected to exceed $1 trillion as the number of affected individuals is expected to reach 12.7 million by 2050.2
499 deaths due to AD, ranking it as the sixth leading cause of mortality in the country. The financial implications are staggering, with the total cost of care for AD patients estimated at $305 billion in 2020. This cost is projected to exceed $1 trillion as the number of affected individuals is expected to reach 12.7 million by 2050.2
The pathogenesis of AD is complex and multifactorial.3,4 A hallmark of AD is the accumulation of extracellular amyloid-beta (Aβ) plaques and the presence of intracellular neurofibrillary tangles (NFTs) made of hyperphosphorylated tau protein. These aggregates compromise neuronal function by disrupting microtubule stability and impairing synaptic communication. Additionally, oxidative stress and mitochondrial dysfunction contribute to neuronal damage. Aβ can induce oxidative damage, leading to abnormal mitochondrial development and heightened production of reactive oxygen species, which further exacerbate neuronal degeneration.
AD is a progressive neurodegenerative disorder characterized by hallmark pathological features. Despite significant progress in understanding genetic risk factors and clinical manifestations, the primary cause of AD remains elusive. This uncertainty underscores the need for therapeutic interventions that can address both symptomatic relief and potential disease-modifying mechanisms. However, existing treatments focus primarily on symptomatic relief rather than addressing the underlying causes of the disease. The U.S. Food and Drug Administration (FDA) has approved four medications: donepezil, rivastigmine, galantamine, and memantine. Donepezil, rivastigmine, and galantamine are cholinesterase inhibitors that aim to enhance cholinergic function, while memantine is an N-methyl-D-aspartate (NMDA) receptor antagonist that modulates glutamatergic activity. These drugs may provide modest cognitive benefits but do not halt or reverse disease progression. In recent developments, aducanumab, a monoclonal antibody targeting Aβ plaques, was approved by the FDA in June 2021 as the first disease-modifying therapy for AD.5–7 Lecanemab and Donanemab, administered as intravenous antibody treatments, are newly developed therapies that have been clinically proven to slow the rate of cognitive decline.8 However, its clinical efficacy and impact on long-term outcomes remain subjects of ongoing debate and investigation. The challenges in treating AD are compounded by the presence of the blood–brain barrier (BBB), which restricts the delivery of therapeutic agents to the central nervous system.9–12 Over 98% of small-molecule drugs and nearly all large biological molecules are unable to cross the BBB, limiting the effectiveness of potential treatments.13
Given these obstacles, there is a pressing need for innovative therapeutic strategies that can effectively target the pathological mechanisms of AD while overcoming delivery challenges.9,11,12 Nanoparticle (NP)-based drug delivery systems have emerged as a promising approach to address these issues. Nanoparticles can aid in transporting therapeutic agents across the BBB, enhance drug stability, and enable targeted delivery to affected neuronal tissues.12,14–16 By leveraging nanotechnology, it may be possible to develop more effective treatments that not only alleviate symptoms but also modify the course of the disease.
In summary, NP-based systems present a versatile and promising approach to overcoming the challenges posed by the BBB in AD treatment. By enhancing drug stability, targeting ability, and penetration into the brain, nanoparticles hold the potential to significantly improve therapeutic outcomes for patients with Alzheimer's disease.17 In summarizing the recent advancements in NP-based systems, this review highlights how these innovative strategies contribute to a better understanding of AD pathogenesis. By exploring the interactions between nanoparticles and the brain microenvironment, we gain insights into disease mechanisms and identify new avenues for intervention. The integration of nanotechnology into AD research represents a significant step toward more effective therapies and diagnostic techniques, potentially transforming the landscape of neurodegenerative disease management.18
2. AD pathology
The fundamental neuropathological features of AD include the buildup of extracellular Aβ plaques and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein.4,19,20 These pathological features initiate a cascade of neurodegenerative processes that ultimately lead to synaptic dysfunction, neuronal loss, and brain atrophy.2.1. Amyloid hypothesis
The abnormal processing of amyloid precursor protein (APP) is a critical factor in the development of AD.21–23 Normally, APP is cleaved by α-secretase, resulting in non-toxic peptides. However, in AD, there is a shift towards amyloidogenic processing, where APP is sequentially cleaved by β-secretase and γ-secretase. This alternative pathway produces Aβ peptides, specifically Aβ40 and Aβ42. Aβ42, due to its hydrophobic nature, is particularly prone to aggregation. These Aβ peptides accumulate extracellularly, forming soluble oligomers and eventually insoluble amyloid plaques.24–26 The plaques disrupt neuronal signaling by interfering with synaptic function and plasticity. Aβ oligomers can bind to neuronal receptors or integrate into cell membranes, leading to impaired signal transduction pathways and altered neuronal activity.27 This disruption contributes to cognitive deficits observed in AD patients. The accumulation of Aβ peptides also triggers the hyperphosphorylation of the microtubule-associated protein tau. Under normal conditions, tau stabilizes microtubules essential for axonal transport. Hyperphosphorylated tau detaches from microtubules, causing their disassembly. Detached tau proteins aggregate to form NFTs within neurons.28–30 The formation of NFTs further disrupts intracellular transport and leads to neuronal dysfunction and death.Moreover, Aβ-induced tau pathology is thought to have a prion-like spreading mechanism, propagating tau abnormalities across connected neuronal networks.31,32 This synergistic interaction between extracellular Aβ plaques and intracellular tau tangles accelerates neurodegeneration. The combined effects of Aβ accumulation, synaptic disruption, and tau pathology constitute key elements in the pathophysiological cascade of AD.33
2.2. Tau hypothesis
While Aβ plaques have been extensively studied, mounting evidence highlights the critical role of tau protein hyperphosphorylation in AD progression. Tau is a microtubule-associated protein that stabilizes microtubule structures in neuronal axons, facilitating proper intracellular transport.34 In AD, aberrant hyperphosphorylation of tau reduces its affinity for microtubules, causing it to detach and destabilize the microtubule network. Detached tau proteins undergo conformational changes and aggregate into insoluble NFTs within the neuron's cytoplasm. These NFTs obstruct cytoplasmic functions by disrupting the cytoskeletal framework and hindering essential intracellular transport mechanisms. The culmination of these disruptions leads to neuronal dysfunction and ultimately cell death.The formation of NFTs not only impairs individual neurons but also contributes to a broader network failure within the brain. The loss of functional neurons and synaptic connections correlates with the severity of cognitive symptoms observed in AD patients. Moreover, hyperphosphorylated tau exhibits a “prion-like” behavior, spreading to anatomically connected regions and promoting tau pathology in neighboring neurons.31 This propagation exacerbates neuronal damage and accelerates disease progression. Importantly, toxic tau species have been shown to enhance Aβ toxicity. The interplay between hyperphosphorylated tau and Aβ creates a detrimental feedback loop, where each pathology amplifies the other's neurotoxic effects. This synergistic relationship underscores the necessity of targeting both tau and Aβ in therapeutic strategies.33
2.3. Other contributing factors
While Aβ aggregation and tau protein hyperphosphorylation are central to AD pathology, several other factors contribute significantly to the disease progression, including oxidative stress, cellular and vascular dysfunction, cholesterol imbalance, inflammation, and metal ion dysregulation.35 Oxidative stress plays a pivotal role in neuronal damage associated with AD.36,37 The brain consumes a substantial amount of oxygen and has high lipid content, making it particularly vulnerable to reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), hydroxyl radicals (OH˙), and superoxide radicals (O2˙−). These ROS are byproducts of mitochondrial electron transport and can lead to protein oxidation, lipid peroxidation, and DNA damage. Accumulation of Aβ can further induce oxidative stress, creating a detrimental cycle that exacerbates neuronal injury and promotes AD progression.Cellular and vascular dysfunction are also critical contributors to AD.38,39 Cerebral hypoperfusion resulting from microvascular pathology reduces the delivery of oxygen and nutrients to the brain and impairs the clearance of metabolic waste products. This hypoperfusion can disrupt endothelial nitric oxide (NO) production, affecting vascular tone and leading to capillary degeneration. Vascular impairment also activates astrocytes and microglia, triggering chronic inflammation that damages neuronal networks.
Cholesterol imbalance is intimately linked with AD pathology.22,40,41 The brain contains approximately 25% of the body's total cholesterol, synthesized locally within the central nervous system.42 Dysregulation of cholesterol homeostasis can promote neurite degeneration, tau hyperphosphorylation, and enhance the amyloidogenic processing of APP.22 Elevated brain cholesterol levels have been associated with increased Aβ formation.22,41 The apolipoprotein E (APOE) ε4 allele, a major genetic risk factor for AD, affects cholesterol transport and metabolism, influencing Aβ production and clearance.43–45
Inflammation is a significant factor in AD development.46 Aβ aggregates can activate microglia and astrocytes, prompting them to express major histocompatibility complex II (MHC II) and secrete pro-inflammatory cytokines, prostaglandins, and other inflammatory mediators. This neuroinflammatory response contributes to neuronal dysfunction and synaptic loss. Chronic inflammation may also result from impaired clearance mechanisms due to vascular dysfunction, further promoting neurodegeneration.
Metal ion imbalance, particularly involving copper (Cu), iron (Fe), and zinc (Zn), has been implicated in AD.47,48 These metals can interact with Aβ peptides, promoting their aggregation and enhancing oxidative stress through the generation of ROS. Excess Cu and Fe can catalyze Fenton reactions, leading to cellular damage. Zinc, while essential for normal brain function, can induce Aβ deposition when dysregulated. Metal ions also influence intracellular signaling pathways, affecting kinase and phosphatase activities that modulate tau phosphorylation. Therapeutic strategies targeting metal chelation are being explored to restore metal homeostasis and mitigate AD pathologies.
Understanding these additional factors is crucial for developing comprehensive approaches to AD treatment. Targeting oxidative stress, improving vascular function, regulating cholesterol levels, modulating inflammation, and correcting metal ion imbalances hold promise for slowing or preventing the progression of AD.
3. BBB
The BBB is a highly selective barrier formed by endothelial cells connected by tight junctions, limiting the passive diffusion of substances from the bloodstream into the brain.49,50 Only small, lipid-soluble molecules with a molecular weight under 400 Da or containing fewer than eight hydrogen bonds can passively cross the BBB.51 This restriction poses a hurdle for the delivery of larger or hydrophilic contrast agents. BBB maintains an optimal environment for neuronal function by controlling ionic balance and supplying essential nutrients.50,52,53 Ion channels and transporters within the BBB endothelial cells are crucial for regulating the brain microenvironment.52,53 Key ions like Na+, K+, Ca2+, and Cl− are tightly controlled to preserve the electrochemical gradients necessary for neural signaling. The BBB is equipped with specialized transport mechanisms to maintain brain homeostasis, including active efflux transporters, carrier-mediated transporters (CMT), receptor-mediated transporters (RMT), absorptive-mediated transporters (AMT), and ion transporters52–55 (Fig. 1). Solute carrier (SLC) transporters and ATP-binding cassette (ABC) transporters manage the influx and efflux of metabolites, nutrients, and xenobiotics.56 For instance, glucose transporter 1 (GLUT1) enables glucose entry into the brain, while P-glycoprotein (Pgp) and other ABC transporters actively expel potentially harmful compounds.55,57–59 These transporters play a critical role in preventing the accumulation of xenobiotics and certain drugs within the central nervous system, presenting a barrier to contrast agent delivery.55 Carrier-mediated transporters are highly selective proteins that facilitate the movement of essential nutrients and metabolites into the brain. | ||
| Fig. 1 Mechanisms of substance transport across endothelial cells. (A) Passive diffusion of a limited number of small molecules (blue) across endothelial cells. (B) Paracellular transport of certain water-soluble agents (pink) between endothelial cells through tight junction proteins. (C) Active efflux transporters (yellow), predominantly ATP-binding cassette (ABC) transporters such as P-glycoprotein (Pgp), multidrug resistance proteins (MRPs), and breast cancer resistance protein (BCRP) (purple), eliminate drugs and substances from the brain. (D) Carrier-mediated transport, which can occur bidirectionally and may involve clathrin-dependent endocytosis, includes major transporters like glucose transporter 1 (GLUT1), L-type amino acid transporters 1 and 2 (LAT1/2), cationic amino acid transporters 1 and 3 (CAT1/3), monocarboxylic acid transporters 1 and 8 (MCT1/8), organic anion-transporting polypeptide 1C1 (OATP1C1), fatty acid transport proteins 1 and 4 (FATP1/4), sodium-independent concentrative nucleoside transporter 2 (CNT2), organic anion transporter 3 (OAT3), organic anion-transporting polypeptides 1A4 and 2B1 (OATP1A4 and OATP2B1), and organic cation transporter 2 (OCTN2). (E) Receptor-mediated transport relies on interactions between ligands (green) and receptors to translocate larger molecules through endothelial cells; key receptors include transferrin receptor (TfR), insulin receptor (IR), leptin receptor (LEP-R), lipoprotein receptors 1 and 2 (LRP1/2), and receptor for advanced glycation end products (RAGE). (F) Adsorptive-mediated transport is caveolin-mediated endocytosis dependent on interactions between ligands (orange) and the endothelial glycocalyx. (G) Ion transporters (turquoise), including sodium pumps, calcium transporters, and potassium channels, regulate ion exchange across the barrier.54 | ||
RMT involves the binding of a ligand to a specific receptor on the endothelial cell surface, triggering endocytosis and transcytosis into the brain. Receptors such as the transferrin receptor (TfR) and insulin receptor (IR) have been exploited using the “Trojan horse” strategy to ferry therapeutic agents across the BBB.60 Conjugating contrast agents to ligands that target these receptors can enhance their brain uptake.61 AMT is another mechanism that can be harnessed, though it is less prominent in the BBB. This pathway relies on electrostatic interactions between positively charged molecules and the negatively charged glycocalyx of endothelial cells, leading to caveolae-mediated endocytosis. Modifying contrast agents to increase their charge interactions may improve their transcytosis across the BBB.
NPs can traverse the BBB via transcellular pathways, specifically exploiting RMT, AMT, and CMT.62 AMT relies on electrostatic interactions between the NPs and the endothelial cell membrane of the BBB. The luminal surface of endothelial cells carries a negative charge due to the presence of glycoproteins and proteoglycans. By functionalizing NPs with positively charged ligands-such as cell-penetrating peptides (CPPs), lectins, or cationic polymers-they can interact electrostatically with the negatively charged membrane. This interaction induces endocytosis and facilitates the transcellular transport of NPs into the brain. AMT is advantageous for its ability to enhance the uptake of a broad range of molecules without the need for specific receptor-ligand recognition. CMT involves NPs that are functionalized with molecules recognized by specific transporters overexpressed on BBB endothelial cells. Endogenous substances like glucose and amino acids cross the BBB via transporters such as glucose transporters (GLUT1 and GLUT3) and large neutral amino acid transporters (LAT1). By attaching glucose, mannose, or amino acids to the surface of NPs, they can mimic these natural substrates and be transported into the brain via facilitated diffusion or active transport mechanisms. For example, glucose-coated NPs can engage GLUT1 transporters to cross the BBB, enabling the delivery of therapeutic compounds to target sites affected by AD.63,64
Moreover, local delivery routes enable AD therapeutics to reach the CNS without traversing the BBB. Methods such as intracerebral, intracerebroventricular (ICV), intrathecal, and intranasal administrations provide direct or indirect access to the brain, potentially enhancing drug efficacy and specificity.65 Intracerebral administration involves the direct injection of therapeutics into brain tissue, allowing immediate effect at the target site. However, drug diffusion via this route is limited to areas adjacent to the injection point. ICV and intrathecal administrations involve direct injection into the cerebral ventricles or lumbar subarachnoid space, respectively. The ICV route is utilized to introduce compounds such as colchicine, streptozotocin, and amyloid-beta peptides directly into the lateral ventricles, often to simulate AD-like pathology in animal models. Intrathecal delivery, on the other hand, targets the subarachnoid space where cerebrospinal fluid circulates, facilitating widespread distribution throughout the CNS. Despite their efficacy, these invasive procedures carry significant risks of infection and neurotoxicity, limiting their clinical application. One non-invasive physical method is focused ultrasound (FUS) sonication. When combined with microbubble contrast agents, FUS can temporarily open the BBB without causing permanent damage to the brain tissue.66 The ultrasound waves cause the microbubbles to oscillate, inducing mechanical stress on the endothelial cells of the BBB. This stress leads to a transient opening of tight junctions between cells, allowing nanoparticles to pass through and reach the brain parenchyma.
These techniques provide valuable strategies for improving the delivery of NP-based therapeutics to the brain. By temporarily and safely opening the BBB, it becomes possible to target the pathological processes of AD more effectively. Combining these BBB modulation methods with advanced nanoparticle design holds promise for enhancing treatment efficacy and patient outcomes.
4. Current AD treatments
Current pharmacological treatments focus on symptom management rather than curing the disease. Cholinesterase inhibitors, such as donepezil, rivastigmine, and galantamine, are commonly prescribed to boost cholinergic function in the brain. Clinical trials have shown that these medications offer moderate improvements in cognitive performance, with effect sizes ranging from 0.27 to 0.49. Rivastigmine, for example, has demonstrated cognitive improvements on the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-cog), showing a mean treatment difference of −1.42 compared to placebo.67 Memantine, an NMDA receptor antagonist, is another approved medication for AD. It works by regulating glutamatergic neurotransmission to prevent excitotoxicity. Meta-analyses of randomized controlled trials have indicated that memantine leads to significant reductions in behavioral symptoms and cognitive decline, with effect sizes of 0.20 for cognitive function and 0.18 for overall function.Advancements in therapeutics are targeting the underlying pathology of AD.68 Monoclonal antibodies like aducanumab (Aduhelm) have been developed to reduce Aβ plaque accumulation in the brain.6 Phase 3 clinical trials have shown that aducanumab significantly decreases amyloid plaque levels in a dose-dependent fashion, as confirmed by amyloid positron emission tomography (PET) imaging.6 Patients receiving high doses also exhibited cognitive benefits on the Clinical Dementia Rating-Sum of Boxes (CDR-SB) scale compared to placebo. Lecanemab (Leqembi) is another monoclonal antibody under investigation. Phase 2 studies reported a statistically significant reduction in Aβ plaque levels, with PET imaging confirming reduced amyloid burden. While Lecanemab does not directly treat symptoms, it has been observed to delay cognitive and functional deterioration in early-stage AD patients. Cognitive assessments using the CDR-SB and ADAS-cog tests indicated that the treatment group performed significantly better than the placebo group.69
Targeting tau protein aggregation is an emerging therapeutic strategy.70,71 Tau inhibitors aim to prevent the formation of neurofibrillary tangles, which contribute to neurodegeneration in AD. Preliminary clinical trials have shown promise, with reductions in tau levels and improvements in cognitive outcomes.72 Repositioned drugs offer additional therapeutic avenues. Pioglitazone, an anti-diabetic medication and peroxisome proliferator-activated receptor (PPAR) agonist, has demonstrated potential in preclinical AD models. It appears to reduce neuroinflammatory responses and support synaptic plasticity, leading to enhanced cognitive function. While existing commercial drugs provide symptomatic relief, these emerging therapeutics, including monoclonal antibodies and repositioned drugs, hold potential for modifying disease progression. Innovative delivery systems like nanoparticles may overcome BBB obstacles, offering new hope for effective AD treatments.12
4.1. Limitations of current treatments
Current therapeutic options for AD are limited and mainly provide symptomatic relief.73 Acetylcholinesterase inhibitors (AChEIs) like donepezil, rivastigmine, and galantamine aim to enhance cholinergic neurotransmission but offer only modest benefits. Common side effects, such as gastrointestinal disturbances, often limit their tolerability.74 Memantine, an NMDA receptor antagonist, is used to mitigate glutamate-induced excitotoxicity in moderate to severe AD cases.75 However, its efficacy in improving cognitive function is limited, and it does not prevent disease progression.In recent years, considerable attention has been directed toward anti-amyloid monoclonal antibodies designed to target the amyloid-beta (Aβ) plaques characteristic of AD pathology. Aducanumab was the first such antibody granted accelerated approval by the FDA in 2021.6,76 Despite its ability to reduce amyloid plaques, aducanumab's clinical efficacy has been highly controversial. Clinical trials yielded inconsistent results, with one showing minimal cognitive benefits and another failing to demonstrate effectiveness. Concerns over serious adverse effects, including amyloid-related imaging abnormalities (ARIA) such as cerebral edema and microhemorrhages, have further clouded its therapeutic value. The high cost of treatment and the need for regular monitoring with MRI add to the challenges of its clinical use. Lecanemab, another anti-Aβ antibody, received accelerated FDA approval in 2023 after showing a modest slowing of cognitive decline in patients with mild cognitive impairment or early-stage AD.77,78 While it reduced amyloid plaques and showed a 27% slowing of disease progression as measured by clinical scales, serious side effects and the incidence of ARIA remained significant concerns. The modest efficacy, combined with high treatment costs and potential risks, underscores the ongoing controversy surrounding anti-Aβ therapies.
Despite extensive research efforts targeting these hallmark features, treatments have shown limited efficacy in clinical trials. One of the significant challenges impeding the success of these therapies is the difficulty in delivering drugs effectively to the brain due to the highly selective nature of the BBB as discussed earlier.9–12 Multiple strategies have been explored to overcome the challenges posed by the BBB.79 These approaches can be categorized into methods that involve “crossing”, “avoiding”, or “disrupting” the barrier. Crossing the BBB entails utilizing existing physiological pathways such as transcellular lipophilic transport or receptor-mediated transcytosis to facilitate drug entry into the brain.80 Avoiding the BBB involves alternative delivery routes that bypass the barrier altogether, including intracerebroventricular, intrathecal, or direct intracerebral administration.81 Disrupting the BBB seeks to transiently increase its permeability through techniques like focused ultrasound sonication, radiation, or the use of hyperosmotic agents and surfactants. Despite these efforts, safely and effectively delivering therapeutic agents across the BBB remains a formidable challenge in the treatment of AD.9–12 The limited success of conventional pharmacological interventions underscores the necessity for innovative drug delivery systems. Advances in nanotechnology offer promising solutions, but the complexity of the BBB and the need for precise targeting continue to necessitate further research. Overcoming the BBB's protective functions without compromising its essential role in CNS homeostasis is crucial for the development of effective AD treatments.9–12
These limitations highlight the urgent need for novel therapeutic strategies that can effectively address the complex pathology of AD with improved safety and efficacy. Nanoparticle-based therapies have emerged as a promising avenue, offering the potential to cross the blood–brain barrier, deliver drugs directly to neuronal targets, and modulate pathological processes at the molecular level. By exploiting the unique properties of nanoparticles, such as their small size and surface modifiability, it may be possible to enhance drug delivery, reduce side effects, and achieve better clinical outcomes for patients with Alzheimer's disease.
4.2. Potential of nanoparticles
Nanoscale delivery systems offer unique advantages due to their distinct physicochemical properties and the ability to modify their surfaces. NPs can encapsulate therapeutic molecules, protecting them from enzymatic degradation and prolonging their circulation time. This encapsulation also allows for controlled drug release and targeted delivery to specific brain regions. By adjusting the size, surface charge, and functional groups of NPs, they can be engineered to interact with specific receptors or transporters on BBB endothelial cells. Surface modifications of NPs enable them to exploit endogenous transport mechanisms across the BBB, such as receptor-mediated transcytosis and adsorptive-mediated transcytosis. For instance, attaching ligands or antibodies to the NP surface can facilitate binding to receptors like transferrin or low-density lipoprotein receptors, enhancing their uptake into the brain. Additionally, tailoring the chemical composition of NPs improves their solubility, biocompatibility, and stability, ensuring safe passage across the BBB while maintaining the integrity of the therapeutic payload.Various types of NPs have been investigated for AD treatment, including lipid-based nanoparticles like nanoliposomes and extracellular vesicles (EVs), polymeric nanoparticles such as poly(lactic-co-glycolic acid) (PLGA) nanoparticles, and inorganic nanoparticles like gold nanoparticles (AuNPs) and quantum dots (QD). Nanoliposomes and EVs are particularly effective due to their biocompatibility and ability to fuse with cellular membranes, facilitating drug delivery to target tissues.82,83 Poly(amidoamine) (PAMAM) dendrimers, with their highly branched structure and customizable surface groups, have shown potential as targeting vectors capable of crossing the BBB due to their optimal size of less than 10 nanometers.84 The use of NPs also opens the possibility of combining therapeutic and diagnostic functions, known as theranostics.85 This approach is valuable in AD, where early detection and treatment are crucial. NPs can be loaded with diagnostic agents alongside therapeutics, enabling simultaneous imaging and treatment of AD pathology.
5. Nano delivery system for the treatment of AD
AD is a progressive neurodegenerative disorder characterized by cognitive decline and memory loss.86,87 The accumulation of Aβ plaques and tau protein tangles in the brain disrupts neuronal communication and function. A significant challenge in treating AD is the delivery of therapeutic agents across the BBB, a selective barrier that protects the brain but also limits drug entry.9–12 NPs have emerged as a promising tool to overcome this hurdle, offering targeted delivery and controlled release of therapeutics. Recent developments in nanoparticle technology aim to enhance BBB penetration, improve target specificity, and reduce side effects. Various nanoparticle systems have been designed, each with unique properties suited for different therapeutic strategies.5.1. Liposomes
Liposomes are spherical, double-layered vesicles composed of phospholipid bilayers that have emerged as versatile drug delivery systems for Alzheimer's disease treatment.88–91 Their unique structure allows them to encapsulate both hydrophilic and lipophilic therapeutic agents: hydrophilic drugs are enclosed within the aqueous inner compartment, while lipophilic drugs are incorporated into the lipid bilayer.92 This dual capability enables the simultaneous delivery of multiple drugs with differing solubility profiles, enhancing therapeutic efficacy.He et al. (2024) developed a liposomal nanodrug (felodipine@LND) encapsulating felodipine, a calcium channel antagonist, to restore intracellular calcium homeostasis in AD neurons.93 Utilizing low-intensity pulsed ultrasound (LIPUS) to enhance BBB permeability, felodipine@LND was effectively delivered to the brains of 5 × FAD transgenic mice. The treatment modulated the endoplasmic reticulum unfolded protein response toward antioxidant signaling via activation of the PERK-Nrf2 pathway, inhibited NLRP3 inflammasome activation, reduced Aβ aggregation, and promoted mitophagy, collectively attenuating neuronal apoptosis. Behavioral assessments revealed significant improvements in anxiety-like behavior and cognitive function, with treated mice showing enhanced performance in the open field test, object recognition test, and Morris water maze. Histological analyses confirmed a reduction in Aβ plaques in both the cortex and hippocampus. Importantly, no toxicity was observed in major organs, underscoring the therapeutic potential of felodipine@LND as a novel approach to AD treatment.
In another approach, the use of transferrin-modified liposomes was explored to deliver pantothenate, aiming to modulate CRM1-mediated PKM2 nuclear translocation, a key mechanism in AD pathology.94 The pantothenate-loaded, transferrin-modified liposomal nanoparticles (Pan@TRF@Liposome NPs) demonstrated efficient BBB penetration and biocompatibility, with no observed toxicity in vivo. These nanoparticles inhibited PKM2 nuclear translocation, reduced neuroinflammation, and decreased neuronal apoptosis in cellular models. When combined with exercise, which induces beneficial metabolic alterations, the treatment improved neurofunctional outcomes and cognitive performance in AD animal models, suggesting a synergistic therapeutic effect.
Gu et al. (2024) addressed the challenge of drug solubility and brain penetration by developing polyethylene glycol-modified liposomal nanoparticles (PEG–ATX@NPs) encapsulating astaxanthin (ATX), a potent antioxidant capable of scavenging endogenous formaldehyde (FA).95 The PEGylation of liposomes improved ATX solubility and stability, facilitating its delivery to the brain. In vitro, PEG–ATX@NPs reduced Aβ neurotoxicity by degrading FA and inhibiting FA-induced Aβ assembly. In APPswe/PS1dE9 transgenic mice, the nanoparticles decreased brain FA levels, attenuated oxidative stress, reduced Aβ oligomerization and plaque formation, and improved spatial learning and memory. The study demonstrates the potential of ATX-loaded PEGylated liposomes as a disease-modifying therapy for AD.
Collectively, these studies underscore the versatility and efficacy of liposomal nanoparticles in targeting various pathological aspects of AD, including calcium dysregulation, Aβ aggregation, metabolic imbalances, and oxidative stress. The ability of liposomes to encapsulate diverse therapeutic agents, enhance BBB penetration, and provide targeted delivery makes them a promising platform for the development of effective AD treatments.88–90
However, several limitations impede their clinical translation.96 A primary challenge is the stability of liposomes under physiological conditions. Factors such as osmolarity, salinity, pH, and temperature can adversely affect liposome integrity, leading to aggregation, coalescence, or leakage of the encapsulated drug. Instability may result in premature release of the therapeutic agent, reducing efficacy and potentially causing off-target effects. Scaling up the manufacture of liposomes presents another significant hurdle. Many production methods, like extrusion and hydration techniques, are well-established at the laboratory scale but are difficult to translate into large-scale processes. These methods often yield liposomes with inconsistent size and lamellarity due to poorly controlled mechanical and chemical conditions during formation. Microfluidic approaches offer improved control over liposome characteristics but typically operate with very low solution volumes, making them unsuitable for mass production. Additionally, some methods are cumbersome to set up and require precise control over multiple parameters, complicating the scaling process. Reproducibility between batches is also a concern in liposome production. Variability in liposome preparations can lead to significant differences in pharmacokinetics and biodistribution, which are critical for the effective treatment of Alzheimer's disease. Ensuring consistent encapsulation efficiency and release profiles is challenging, especially when dealing with complex therapeutic agents like nucleic acids or proteins.
5.2. Polymeric NPs (PNPs)
Polymeric nanoparticles (PNPs) have gained significant attention as drug delivery systems for treating neurological disorders such as Alzheimer's disease. These solid particles are formulated from various polymeric materials, allowing for controlled drug release and enhanced stability of therapeutic agents.Nanoparticle-based drug delivery systems targeting mitochondrial dysfunction was also reported (Fig. 2).98 Given the early occurrence of mitochondrial dysfunction in neurons during AD and the need for multi-pathway regulation beyond antioxidative monotherapy, a multifunctional hybrid peptide, HNSS was developed, combining the antioxidant peptide SS31 with the neuroprotective peptide S14G-Humanin (HNG). To effectively deliver HNSS to the brain and target cholinergic neurons, nanoparticles made of citraconylation-modified poly(ethylene glycol)–poly(trimethylene carbonate) polymer (PEG–PTMC(Cit)) were engineered, exhibiting high HNSS loading capacity through electrostatic interactions. These nanoparticles were further modified with the FGL peptide, an FGFR1 ligand, to exploit FGFR1 overexpression at the blood–brain barrier and in cholinergic neurons, resulting in a 4.8-fold increase in brain accumulation and preferential distribution to cholinergic neurons in diseased regions. The acid-sensitive nature of PEG–PTMC(Cit) enabled lysosomal escape and intracellular release of HNSS via charge switching, enabling mitochondrial enrichment of HNSS through the SS31 moiety. In 3 × Tg-AD mice, treatment with FGL-NP(Cit)/HNSS effectively restored mitochondrial function by activating of PGC-1α and STAT3 pathways, reduced amyloid-β deposition and tau hyperphosphorylation, and improved memory deficits and cholinergic neuronal damage. Notably, FGL-NP(Cit)/HNSS increased the ratio of p-STAT3/STAT3 to 118% of wild-type levels and raised antioxidative enzyme activity by 76.1% compared to saline-treated AD mice. The design of FGL-NP(Cit)/HNSS integrates targeted delivery, responsive drug release, and mitochondrial targeting mechanisms, resulting in significant therapeutic outcomes in AD models with good biocompatibility and minimal in vivo toxicity.
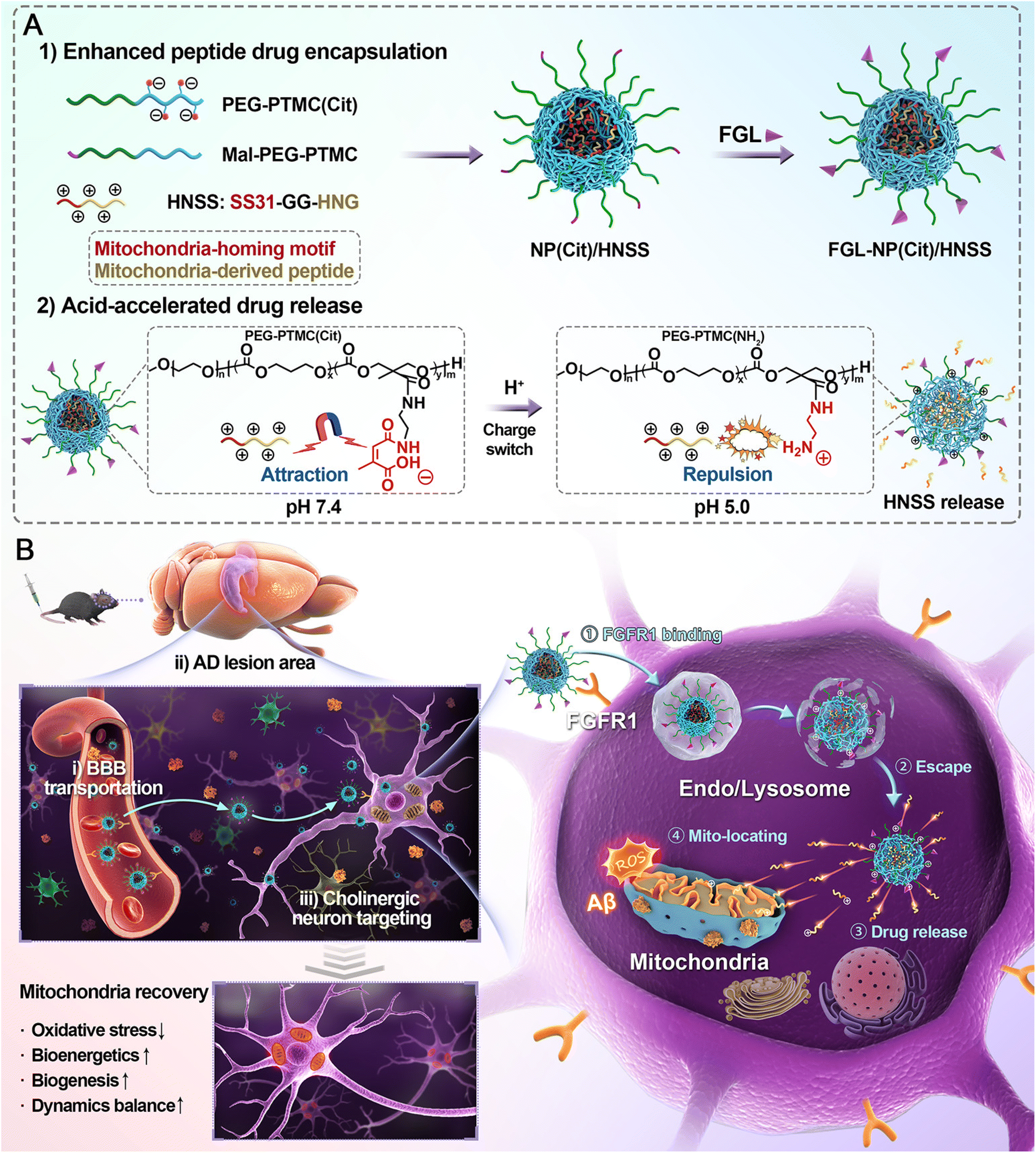 | ||
| Fig. 2 (A) Schematic illustration of the construction of FGL-NP(Cit)/HNSS nanoparticles and their acid-responsive features, including charge reversal and drug release. (B) FGL-NP(Cit)/HNSS penetrates the blood–brain barrier and specifically targets cholinergic neurons in Alzheimer's disease lesion areas with high FGFR1 expression. After neuronal endocytosis and lysosomal transfer, FGL-NP(Cit)/HNSS undergoes charge reversal in the acidic microenvironment, facilitating rapid lysosomal escape and complete intracellular release of HNSS. HNSS efficiently targets mitochondria via the SS31 peptide and modulates mitochondrial function through multiple pathological mechanisms, thereby promoting cholinergic neuron survival and exerting protective effects on cognitive function.98 | ||
Several nanoparticle systems have been developed to co-deliver anti-inflammatory agents, siRNA, peptides, or antioxidants directly to affected brain regions.12 For instance, zwitterionic poly(carboxybetaine)-based nanoparticles and citraconylation-modified PEG–PTMC nanoparticles have normalized dysfunctional microglia,97,98 reduced proinflammatory cytokines, and improved mitochondrial function. Anthocyanin-loaded PLGA–PEG nanoparticles and PEGylated PLGA nanoparticles encapsulating epigallocatechin-3-gallate and ascorbic acid enhanced the stability and bioavailability of antioxidant compounds, resulting in reduced oxidative stress and neuroinflammation.99,100
Advancements in nanoparticle design have focused on targeting specific cellular mechanisms and enhancing BBB penetration.12 Oxytocin-loaded angiopep-2-modified chitosan nanogels inhibited microglia-mediated neuroinflammation,101 while melanin-like polydopamine nanoparticles modified with the KLVFF peptide chelated metal ions and scavenged reactive oxygen species, mitigating Aβ aggregation.102 Sugar-based amphiphilic nanoparticles targeting microglial scavenger receptors and reactive oxygen species-responsive dendrimer–peptide conjugates have also shown efficacy in modulating neuroinflammation and reducing Aβ burden.103,104
Further, dual-ligand fusion peptide-modified nanoparticles and multifunctional nanoprodrugs conjugating curcumin to hybrid peptides improved BBB penetration and targeted delivery to neurons and pericytes, respectively, resulting in enhanced cognitive functions and reduced pathological markers.105,106 Self-destructive nanosweepers composed of multifunctional peptide-polymers and nanoparticles encapsulating α-mangostin demonstrated the ability to capture and degrade Aβ, promote its uptake and degradation, and reverse behavioral deficits.107,108 Additionally, amorphous PDLLA-dextran bottlebrush copolymers effectively delivered hydrophilic antioxidants, ameliorating AD symptoms in mice.109
Collectively, these studies underscore the therapeutic potential of polymeric nanoparticles in targeting neuroinflammation and other pathological mechanisms in Alzheimer's disease.17 The versatility and multifunctionality of these nanoparticles offer promising avenues for future AD therapies, though further research is needed to evaluate their long-term safety, efficacy, and clinical applicability.
| Nanoparticle design | Function of nanoparticle | Reference |
|---|---|---|
| Rabies virus glycoprotein peptide-modified mesenchymal stem cell-derived exosomes as shell and ROS-responsive polymer loaded with siRNAs as core | Targeted delivery and controlled release of siRNAs to ameliorate neurological injury | 110 |
| Glutathione (GSH)-responsive silica nanocapsules (SNCs) conjugated with glucose and rabies virus glycoprotein peptide | Brain-targeted delivery of biologics via systemic administration, bypassing the blood–brain barrier | 111 |
| Carboxylated graphene oxide nanosheets functionalized with PEG and PEI | Delivery of GSK3β siRNA | 112 |
| Integrated ceria nanozymes into MOFs loaded with siSOX9 and RA | Promotes neuron differentiation and eliminates ROS | 113 |
| CRISPR-Cas9 nanocomplexes | In vivo gene editing | 114 |
| Traceable nano-biohybrid complexes loaded with CRISPR/Cas9 plasmids | Efficient delivery of CRISPR-chem drugs into brain lesions and accurate imaging | 115 |
| Electrostatically driven r8-C12 RNA nanocomplexes enveloped with PEG–PGA or hyaluronic acid | Enhance nose-to-brain delivery and protect RNA | 116 |
| Tetrahedral DNA framework-based nanoparticles modified with TPP, cholesterol, and antisense oligonucleotide | Cross blood–brain barrier and target mitochondria for AD diagnosis and gene silencing | 117 |
| DNA nanoflowers modified with RVG29 peptide and loaded with miR-124 and Rutin | Delivery of miR-124 and Rutin across the blood–brain barrier and targeting neurons | 118 |
| PBAE–PLGA–Ag2S S–RA–siSOX9 (PPAR–siSOX9) nanoformulation | High gene/drug deliverability to overcome AD microenvironment-associated adverse effects and promote neuronal differentiation of NEP-expressing NSCs | 119 |
| PEGylated dendrigraft poly-L-lysines with brain-targeted ligand modification | Co-delivery of therapeutic gene and peptide to the brain | 120 |
| Cyclodextrin-appended cationic dendrimer (CDE) | Delivery of shRNA to suppress amyloid protein production, inhibit amyloid formation, and disrupt existing amyloid fibrils | 121 |
| Positively charged polyprodrug amphiphiles loaded with SPIONs and let-7b antisense oligonucleotide | Traceable co-delivery of therapeutic agents with controlled release and MRI tracking | 122 |
| PEG–PDMAEMA modified with CGN and Tet1 peptides | Delivery of BACE1-targeting siRNA to neurons | 123 |
| Disulfide-linked poly(β-L-malic acid-trileucine)-copolymer conjugated with D3-peptide | Neuron-selective delivery of miRNA and antisense RNA across the BBB | 124 |
| Poly(D,L-lactic-co-glycolic acid) (PLGA) nanoparticles | Deliver siRNA to microglia and control microglial reactivity | 125 |
| PEGylated dendrigraft poly-L-lysines (DGLs) modified with Aleuria aurantia lectin (AAL) and β-amyploid (Aβ)-binding peptides (KLVFF) | Co-delivery of BACE1 siRNA and rapamycin into the brain | 126 |
| Lipid nanoparticle (MG-LNP) | Efficient RNA delivery to activated microglia | 127 |
One approach involves the use of dendrigraft poly-L-lysines (DGLs) modified with targeting ligands and functional peptides. For instance, PEGylated DGLs conjugated with the brain-targeting ligand RVG29 and a therapeutic D-peptide (D-TLKIVW) were developed to co-deliver a non-coding RNA plasmid targeting BACE1-AS and the peptide to the brain via systemic administration.120 This multifunctional nanocarrier successfully down-regulated BACE1 mRNA levels, reduced amyloid plaque deposition, decreased phosphorylated tau levels, and improved cognitive performance in transgenic AD mice, demonstrating the potential of combining gene and peptide therapy in a single platform.
Another strategy focuses on dual-targeting nanoparticles to enhance specificity and efficiency of delivery. Nanocarriers composed of PEGylated poly(2-(N,N-dimethylamino) ethyl methacrylate) (PEG–PDMAEMA) were modified with both the CGN peptides for BBB penetration and Tet1 peptides for neuron-specific targeting.123 These nanocomplexes effectively delivered BACE1 siRNA to central neurons via systemic administration, resulting in significant reduction of BACE1 mRNA expression, decreased amyloid plaque burden, and restored cognitive performance in APP/PS1 transgenic mice. The dual-targeting design leveraging both BBB penetration and neuron-specific ligands exemplifies an advanced strategy for targeted gene therapy in AD.
Cyclodextrin-appended cationic dendrimers (CDE) complexed with short hairpin RNA (shRNA) have been utilized to simultaneously target multiple pathological steps of amyloidosis, including precursor protein production, amyloid formation, and deposition.121 The CDE/shRNA complex demonstrated significant suppression of amyloidogenic protein production via RNA interference, inhibition of amyloid formation, and disruption of existing amyloid fibrils both in vitro and in vivo. This multifunctional approach was effective in reducing amyloid deposition and improving cognitive function in animal models, highlighting the potential of targeting multiple pathways in AD therapy.
Intranasal delivery of multifunctional nanocarriers presents an alternative route to bypass the BBB.81,128 A nanocarrier system comprising rapamycin and BACE1 siRNA encapsulated in PEGylated DGLs modified with Aleuria aurantia lectin (AAL) and the β-amyloid-binding peptide KLVFF was developed for intranasal administration.126 This system enhanced nasal-to-brain transport, targeted Aβ aggregates, inhibited Aβ aggregation, downregulated BACE1 mRNA, and induced autophagy in the hippocampus. Treated transgenic AD mice showed improved cognitive performance, reduced Aβ deposition, and decreased tau protein levels, demonstrating the efficacy of combining autophagy induction with gene therapy.
Biodegradable amphiphilic nanopolymers based on poly(β-L-malic acid-trileucine) (PMLA/LLL) conjugated with D-peptides targeting the LRP-1 transcytosis pathway have been developed to facilitate efficient BBB crossing and neuron-specific delivery of microRNA and antisense RNA.124 These nanodrugs achieved significant neuronal uptake and accumulation within extracellular amyloid plaques in AD mice, leading to modulation of AD-related gene expression without adverse effects, highlighting the potential of D-peptide-conjugated nanopolymers in neuron-selective gene therapy.
Targeting microglial senescence is another therapeutic avenue explored using polymeric nanoparticles. Poly(D,L-lactic-co-glycolic acid) (PLGA) nanoparticles were designed to deliver siRNA targeting cyclin-dependent kinase inhibitor 2A (CDKN2A) to microglia.125 Downregulation of CDKN2A rejuvenated microglia, enhanced their phagocytic capacity for Aβ, reduced amyloid plaque formation, and reversed cognitive deficits in 5 × FAD mice. This approach underscores the potential of modulating microglial function via nanoparticle-mediated gene therapy in AD.
Peptides have been designed to deliver CRISPR-Cas9 nanocomplexes for efficient in vivo gene editing of post-mitotic neurons in adult mice, targeting the Bace1 gene, which is critical for amyloid beta (Aβ) peptide production implicated in Alzheimer's pathology (Fig. 3).114 The nanocomplexes were formulated by assembling Cas9-sgRNA ribonucleoproteins with an amphiphilic R7L10 peptide, creating stable spherical nanoparticles approximately 125 nm in diameter, as characterized by electron microscopy, dynamic light scattering, and atomic force microscopy (Fig. 3A). In vitro, these nanocomplexes achieved indel frequencies up to 45% in primary neurons for Bace1 and tyrosine hydroxylase (Th) genes, with minimal cytotoxicity observed at concentrations up to 10 μM. For in vivo studies, nanocomplexes were injected into the cerebral cortex and hippocampus of 6 months-old 5 × FAD transgenic Alzheimer's disease mice and wild-type mice. Treatment resulted in a significant reduction of Bace1 expression by approximately 70% in the CA3 hippocampal region and a 34% decrease in Aβ42 levels, along with a reduction in Aβ plaque accumulation by over 50% (Fig. 3B and C). Behavioral assessments demonstrated that treated mice exhibited enhanced cognitive function, including a significant increase in freezing behavior during fear conditioning tests (from 20% to 60%) and improved performance in the Morris water maze, with escape latencies decreasing from 40 s to 20 s over training days. The nanocomplex design ensured minimal off-target effects, as whole-genome sequencing and Digenome-seq analysis revealed no significant increase in mutation rates or genomic rearrangements compared to controls, and no significant inflammation or apoptosis was detected. However, limitations include challenges in achieving widespread nanoparticle delivery throughout the brain to address diffuse neural pathology and ensuring the long-term safety and specificity of gene editing to prevent rare but potentially harmful genomic alterations.
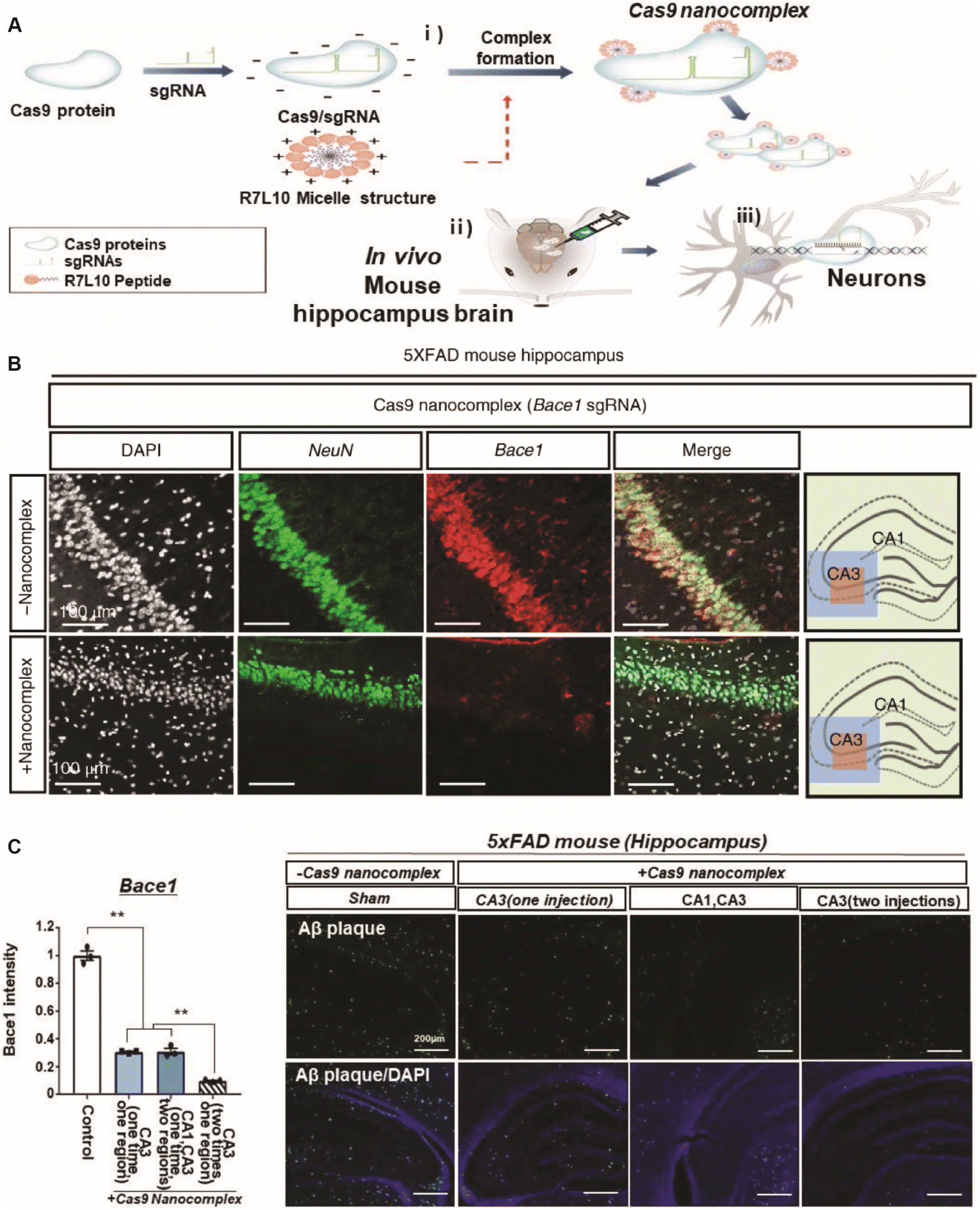 | ||
| Fig. 3 (A) Schematic representation of the CRISPR-Cas9 nanocomplex delivery system: (i) formation of CRISPR-Cas9 nanocomplexes; (ii) delivery of Cas9 nanocomplexes into the in vivo brain; and (iii) gene editing in post-mitotic neurons using Cas9 nanocomplexes. (B) Immunohistochemical staining for NeuN (green), Bace1 (red), and DAPI (white) in the hippocampus of 6 months-old 5 × FAD mice treated with Cas9 nanocomplexes containing Bace1 sgRNA. (C) Quantification of Bace1 immunofluorescence intensity and the number of Bace1-positive cells. Data are expressed as mean ± SEM, n = 3. p < 0.01, ANOVA with Tukey's post hoc test.114 | ||
In summary, polymeric nanoparticles offer a promising platform for gene therapy in AD by enabling targeted delivery of therapeutic nucleic acids and peptides across the BBB. Various strategies, including the use of targeting ligands, dual-targeting designs, multifunctional nanocarriers, and alternative administration routes like intranasal delivery, have been employed to enhance delivery efficiency, specificity, and therapeutic efficacy. These advances demonstrate significant potential for polymeric nanoparticle-based gene therapy in AD treatment, although further studies are needed to address challenges related to long-term safety, immunogenicity, and translation to clinical applications.
Safety is a paramount concern in developing gene therapies for AD, particularly concerning delivery methods and potential off-target effects.129 One major safety consideration is the potential for off-target genetic modifications, which could lead to unintended gene disruption or activation. Strategies to enhance specificity include optimizing nanoparticle formulations for targeted delivery and using precision gene-editing tools with high fidelity. Nanoparticles can be engineered to deliver therapeutic genes or gene-editing components directly to affected neurons, reducing systemic exposure and the risk of off-target effects. Insertional mutagenesis is a risk associated with integrating viral vectors traditionally used in gene therapy. Nanoparticles offer a non-viral delivery alternative that reduces this risk, as they can deliver non-integrating genetic material such as mRNA. This transient expression reduces the likelihood of long-term genomic alterations but may necessitate repeated administrations, which brings its own safety considerations. Moreover, immune responses to both the nanoparticle carriers and the delivered genetic material pose another safety concern. The immune system may recognize nanoparticles or therapeutic agents as foreign, leading to inflammation or other adverse effects. To mitigate this risk, nanoparticles can be designed using biocompatible materials such as lipids or polymers that are less likely to elicit an immune response.
One primary approach focuses on nanoparticles designed to inhibit Aβ aggregation and promote disassembly of existing fibrils. Native poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles have demonstrated the ability to suppress spontaneous aggregation of Aβ1–42 and disassemble preformed aggregates without the need for additional drug conjugation.130,131 These nanoparticles interact with the hydrophobic domains of Aβ1–42, preventing its conformational shift toward β-sheet structures, thereby reducing neurotoxicity in neuronal cultures and animal models. The researchers demonstrated that native PLGA nanoparticles, at concentrations of 25–50 μM, could suppress spontaneous aggregation of 10 μM Aβ1–42 and induce the disassembly of preformed Aβ aggregates (Fig. 4).130 Spectroscopic studies, molecular dynamics simulations, and biochemical analyses revealed that PLGA interacts with the hydrophobic domain of Aβ1–42, particularly residues Lys16 to Ala21, preventing its conformational shift to a β-sheet structure and thereby inhibiting the formation and promoting disassembly of aggregates. PLGA-treated Aβ samples enhanced neuronal viability in mouse cortical neurons by reducing tau protein phosphorylation and its related signaling pathways, including decreased activation of ERK1/2 and GSK-3β pathways. In the 5 × FAD mouse model of AD, intracerebroventricular administration of PLGA at a concentration achieving 25 μM in cerebrospinal fluid over 28 days attenuated memory deficits, as measured by novel-object recognition tests, and reduced cortical Aβ levels and plaque load without observable toxicity. Furthermore, PLGA protected induced pluripotent stem cell (iPSC)-derived neurons from AD patients against Aβ-induced toxicity by decreasing tau phosphorylation and improving cell viability. The design of native PLGA nanoparticles allows them to target different facets of the Aβ axis without the need for drug conjugation, offering a unique therapeutic mechanism with demonstrated safety and efficacy in both cell and animal models. This study highlights the novel significance of native PLGA nanoparticles as a potential disease-modifying treatment for AD pathology.132 However, the impact of PLGA on neurofibrillary tangles remains to be elucidated. Further research is necessary to confirm these findings and to determine its impact on other aspects of cognitive function and pathology, as well as its efficacy in the complex human brain environment.
 | ||
| Fig. 4 PLGA nanoparticles inhibit Aβ aggregation through interactions with hydrophobic domains, enhancing neuronal viability in mouse neurons. PLGA-mediated inhibition of Aβ aggregation improves cognitive function and reduces pathology in the 5 × FAD Alzheimer's disease mouse model. Additionally, PLGA protects iPSC-derived neurons from Alzheimer's disease patients against Aβ-induced toxicity.130 | ||
Functionalization of polymeric nanoparticles with specific ligands enhances their targeting capabilities and inhibitory effects. Copolymeric nanoparticles composed of N-isopropylacrylamide and N-tert-butylacrylamide have been shown to retard Aβ fibrillation by prolonging the nucleation lag phase through binding to monomeric and oligomeric Aβ species.133 Similarly, PEGylated poly(alkyl cyanoacrylate) nanoparticles functionalized with curcumin derivatives or anti-Aβ1–42 antibodies exhibit high affinity for Aβ peptides, effectively inhibiting aggregation and reducing cytotoxicity in neuronal cells.134,135 Peptide-functionalized nanoparticles, such as those conjugated with the modified peptide Ac-LVFFARK-NH2 (LK7) onto PLGA nanoparticles, inhibit Aβ42 aggregation while mitigating the cytotoxicity associated with peptide self-assembly.136 Additionally, iminodiacetic acid-conjugated nanoparticles (IDA-NP) function as bifunctional modulators by chelating metal ions like Zn2+, which facilitate Aβ aggregation, and directly inhibiting Aβ42 fibrillation, thereby protecting neuronal cells from cytotoxicity.137
Targeting tau protein aggregation represents another critical strategy. A tau-targeted multifunctional nanoinhibitor was developed using self-assembled polymeric micelles decorated with a tau-binding peptide, effectively inhibiting tau aggregation, blocking the seeding activity of extracellular tau aggregates, and promoting their proteolytic degradation.138 This approach addresses the neurotoxicity and propagation of tau aggregates, offering a potential therapeutic avenue for tau pathology in AD.
Multifunctional nanoparticles (dcHGT NPs) were developed by co-encapsulating clioquinol, a metal-ion chelator, and donepezil, an acetylcholinesterase inhibitor, within human serum albumin nanoparticles, which were further modified with transcriptional activator protein (TAT) and monosialotetrahexosylganglioside (GM1).139 The dcHGT NPs had an average diameter of approximately 15 nm and demonstrated drug-loading efficiencies of 41% for clioquinol and 35% for donepezil, with sustained drug release over 10 days (27% clioquinol and 15% donepezil released). In vitro, dcHGT NPs significantly inhibited and disaggregated Aβ fibrils induced by Cu2+ ions, and reduced Aβ-mediated inflammation in microglial cells by decreasing TNF-α levels from 52.0% to 14.1% and IFN-γ levels from 10.2% to 3.82%. The nanoparticles also protected primary neurons from Aβ oligomer-induced neurotoxicity, increasing neuron survival by 227% compared to Aβ-treated controls and preserving neurite length and root number. In vivo, intranasal administration of dcHGT NPs in APP/PS1 transgenic mice resulted in efficient brain accumulation and retention for up to 96 hours, with brain fluorescence intensity 1.9 times higher than controls at 5 minutes and maintained at 96 hours. Treated mice exhibited significant improvements in spatial learning and memory, with a 68.9% increase in target quadrant exploration time in the Morris water maze test, and EEG analyses showed amelioration of acetylcholine imbalance, evidenced by increased high-frequency α and β wave activity and decreased low-frequency θ waves. Histological analyses revealed reduced Aβ deposition, amelioration of neuronal morphological changes, a 2.2-fold increase in synapse number compared to controls, and improved neuronal viability and activity. The dcHGT NPs leverage the synergistic effects of metal-ion chelation and acetylcholinesterase inhibition, combined with enhanced brain targeting via GM1 and TAT modifications, offering a novel and highly efficient combination therapy for AD with demonstrated safety and therapeutic benefits in cellular and animal models. Similarly, dual-functional nanoparticles modified with BBB-penetrating and Aβ-targeting peptides were developed to enhance the delivery of therapeutic agents like the β-sheet breaker peptide H102 across the BBB and specifically target Aβ plaques, resulting in improved spatial learning and memory in AD model mice.140
Multifunctional nanoparticles were designed by co-assembling guanidinium-modified calixarene (GCA) with ascorbyl palmitate (AP) and loading dipotassium phytate (IP6) within the calixarene cavity, utilizing supramolecular strategies based on molecular recognition and self-assembly (Fig. 5).141 These nanoparticles simultaneously inhibited β-amyloid (Aβ) production and aggregation, disintegrated Aβ fibrils, accelerated Aβ metabolic clearance, and regulated oxidative stress. In vitro experiments demonstrated that the nanoparticles effectively inhibited Aβ fibrillation, reducing thioflavin T fluorescence to 0.7% of the control after 96 hours, and promoted disintegration of preformed Aβ fibrils, decreasing fluorescence to 2.1%. In BV-2 microglial cells, the nanoparticles enhanced phagocytic uptake of Aβ42 by up to 2.5-fold compared to control. In vivo studies using 5 × FAD mice showed significant amelioration of cognitive impairment, evidenced by a 66% increase in nesting score and a 69% increase in discrimination index in the novel object recognition test compared to untreated mice. Additionally, the area fraction of thioflavin S-stained Aβ plaques in the hippocampus was reduced by 87%, and levels of oxidative stress markers and neuroinflammation were substantially decreased. The design leveraged the dynamic reversibility of supramolecular self-assembly, allowing flexible component substitution and ratio adjustment, resulting in a versatile platform for AD combinational therapy with favorable safety profiles. The novelty of this work lies in its adaptable supramolecular approach to effectively integrate multiple therapeutic functions into a single nanoparticle system, potentially expediting advancements in AD treatment. However, a major limitation is the need for further studies to evaluate long-term efficacy and safety in clinical settings.
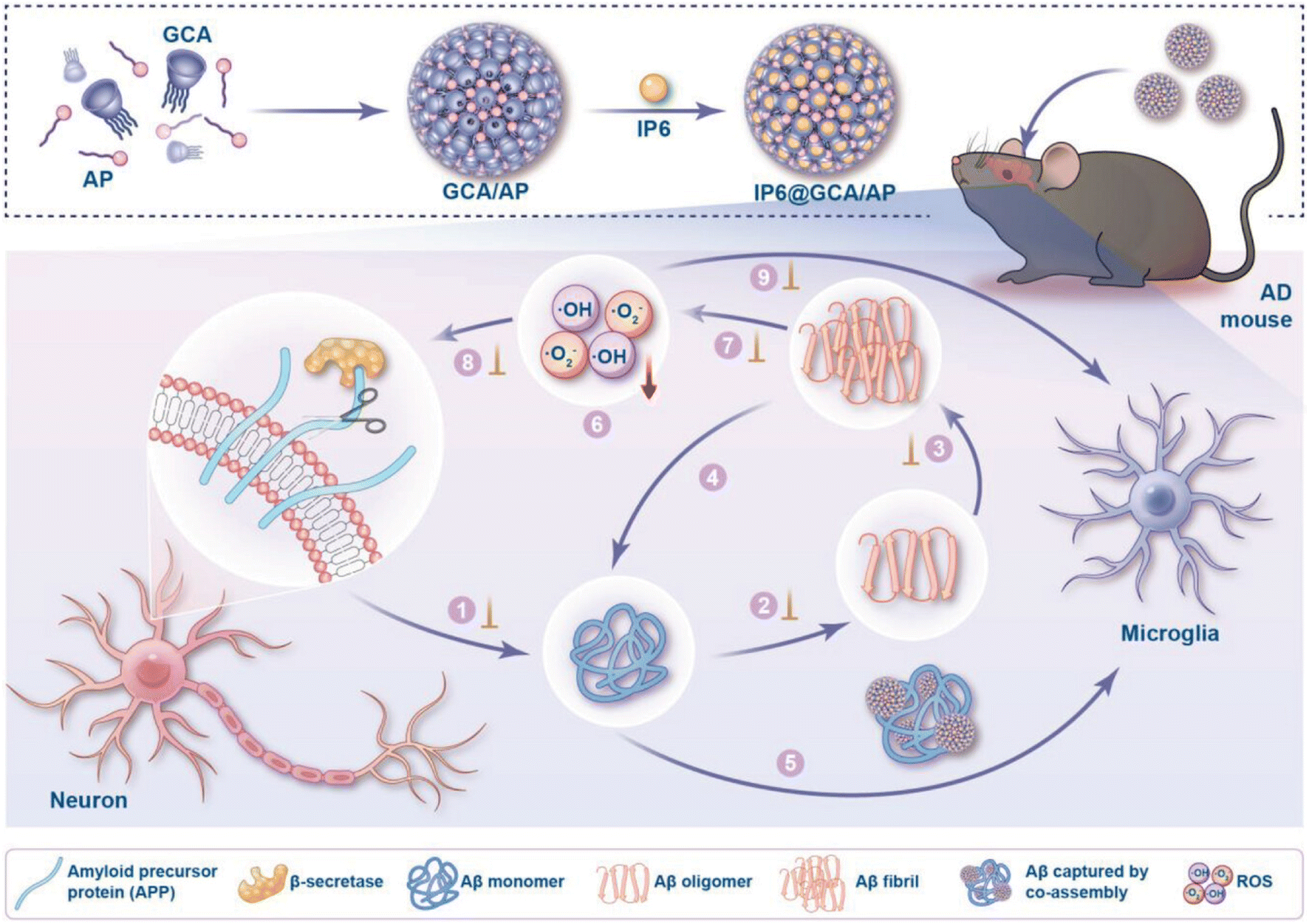 | ||
| Fig. 5 Schematic illustration of the construction of IP6@GCA/AP co-assembly and its comprehensive intervention in both amyloid-β (Aβ) fibrillation and oxidative stress pathological processes. The co-assembly was fabricated by combining the two amphiphilic components, glycyrrhetinic acid (GCA) and ascorbyl palmitate (AP), with inositol hexaphosphate (IP6) loaded into the cavity of GCA. Key events in the targeted pathways are addressed, including: (1) inhibiting Aβ generation by reducing β-secretase activity; inhibiting Aβ aggregation into (2) oligomers and (3) fibrils; (4) disintegrating pre-existing Aβ fibrils; (5) accelerating Aβ clearance via microglial phagocytosis; and (6) scavenging reactive oxygen species (ROS) to alleviate oxidative stress. Collectively, these interventions aim to prevent (7) Aβ-induced ROS production, (8) ROS-induced enhancement of β-secretase activity, and (9) ROS-induced impairment of microglial phagocytosis, ultimately disrupting the vicious cycle between Aβ pathology and oxidative stress.141 | ||
These studies collectively highlight the versatility and potential of polymeric nanoparticles in developing inhibitor therapies for AD. By targeting specific pathological features such as Aβ and tau aggregation, and enhancing drug delivery across the BBB, these nanoparticle-based approaches offer promising strategies for treating AD. Further research is necessary to translate these findings into clinical applications, addressing challenges like long-term safety, immunogenicity, and the scalability of nanoparticle synthesis.
Researchers also developed traceable nanoparticles composed of poly(2-hydroxyethyl methacrylate)–retinoic acid–poly(carboxybetaine)–cell-penetrating peptide (PHEMA–RA–PCB–CPP) polymers to control the differentiation of neural stem cells (NSCs) into neurons.142 These nanoparticles encapsulated superparamagnetic iron oxide nanoparticles (SPIONs) for magnetic resonance imaging (MRI) tracking and complexed small interfering RNA (siSOX9) to downregulate the SOX9 protein, which suppresses neuronal gene expression. The charge-reversible PCB allowed for the temporal release of siSOX9 and RA, with siSOX9 released first in the acidic environment of endosomes/lysosomes and RA released later in the cytoplasm. In vitro experiments demonstrated efficient cellular uptake by NSCs, with a mean fluorescence intensity 1.6 times higher than nanoparticles without CPP modification, and a 52.3% knockdown of SOX9 mRNA expression. Neuronal differentiation was significantly enhanced, with microtubule-associated protein 2 (MAP-2) expression reaching 76.8% compared to 11.0% in controls. In vivo, transplanted NSCs treated with nanoparticles resulted in improved cognitive function in AD mice, evidenced by shorter escape latencies and increased time spent in the target quadrant during Morris water maze tests. The nanoparticles exhibited an r2 relaxivity value of 171.05 mM−1 s−1, enabling real-time MRI tracking of NSC migration for up to five weeks. This work presents a novel approach that combines temporally controlled delivery of siRNA and RA with MRI traceability to enhance NSC therapy for AD. However, limitations include the complexity of the nanoparticle system and the need for further studies to assess long-term safety and efficacy before clinical application.
Researchers developed targeted multimodal polypeptide-based nanoconjugates composed of polyglutamic acid (PGA) carriers bearing neuroprotective propargylamine moieties and conjugated with either bisdemethoxycurcumin (BDMC) or genistein.143 The nanoconjugates were further modified with Angiopep-2 (ANG), a targeting ligand for the low-density lipoprotein receptor-related protein 1 (LRP1), to enhance BBB transcytosis. In vitro studies demonstrated that these nanoconjugates provided neuroprotection and increased dendritic density of pyramidal neurons in organotypic hippocampal cultures, with significant reductions in cell death (nearly threefold decrease compared to untreated controls at 0.05 μM drug-equivalents). In vivo, the ANG-modified nanoconjugates effectively crossed the BBB, accumulated in neurogenic brain regions such as the olfactory bulb, and were internalized by neurons, astrocytes, and microglia in APP/PS1 transgenic AD model mice. Treatment with the nanoconjugates significantly reduced brain levels of neurotoxic β-amyloid aggregates (Aβ1–40 levels decreased by approximately 25%) and rescued impairments in olfactory memory and object recognition, restoring performance to levels similar to wild-type mice. Safety assessments showed no significant toxicity, with normal plasma levels of lactate dehydrogenase, creatinine, and liver enzymes in treated animals.143
5.3. Inorganic NP
Inorganic nanoparticles present a unique category where their role can vary from passive carries of active therapeutic agents to active participants in therapeutic processes. These particles have emerged as promising therapeutic agents for AD due to their unique physicochemical properties that enable them to interfere with Aβ aggregation, cross the BBB, and reduce neurotoxicity. Various studies have explored different types of inorganic nanoparticles, such as quantum dots, gold nanoparticles (AuNPs), selenium nanoparticles (SeNPs), polyoxometalate-peptide hybrids, and others, demonstrating their potential in modulating Aβ aggregation pathways and mitigating AD-related pathology (Table 2). Additionally, these nanoparticles can act as nanocarriers, further enhancing their therapeutic versatility. This dual functionality exemplifies the potential for inorganic nanoparticles to serve as both nanocarriers and nanotherapeutics.| Nanoparticle design | Function of nanoparticle | Reference |
|---|---|---|
| RBC membrane encapsulating carbon quantum dots and polydopamine | Evade immune clearance, mitigate oxidative stress, and chelate metal ions | 144 |
| Erythrocyte membrane-modified core–shell upconversion nanoparticle loaded with curcumin | Biomimetic nanobait to improve photodynamic therapy efficiency | 145 |
| Red blood cell membrane-templated cerium oxide nanocrystals encapsulated with carbon quantum dots (CQD–Ce–RBC) | Biocompatible nanocomposite with antioxidant properties, copper ion chelating, Aβ aggregation prevention, and photothermal effects to break down Aβ fibers and enhance blood–brain barrier permeability | 146 |
| Composite nanometer system of red blood cell membranes-encapsulated Prussian blue nanoparticles (PB/RBC) | Chelate Cu2+, reduce ROS, photothermally open BBB, depolymerize Aβ deposits | 147 |
| Macrophage membrane (RAW-M) encapsulated nitrogen-doped carbon quantum dots | Capture excess Cu2+, inhibit Aβ aggregation, depolymerize Aβ fibrils with photothermal properties, enhance BBB permeability | 148 |
| MoS2 QDs/MM | Elimination of ROS and anti-Aβ1–42 deposition | 149 |
| Design includes upconversion nanoparticles and a chelating agent to create a nanoprobe capable of detecting and capturing Cu(2+) ions | Detects and captures Cu(2+) ions and enables upconversion luminescence imaging | 150 |
| Mesoporous silica nanomaterials loaded with curcumin and IR780, grafted with cerium oxide nanoparticles and peptide K | Inhibiting β-amyloid aggregation and scavenging reactive oxygen species (ROS) | 151 |
| RVG29-modified biodegradable mesoporous silica nanoparticles loaded with ultra-small cerium oxide nanocrystals and conjugated with antibody 1F12 | Inhibits Aβ42 misfolding and aggregation, accelerates Aβ42 clearance, and scavenges reactive oxygen species | 152 |
| N-acetyl-L-cysteine capped quantum dots | Inhibit amyloid fibrillation by quenching nucleation and elongation | 153 |
| MSiO2@SiCDs nanocomposites | Efficient Cu2+ chelator and inhibitor of Aβ aggregation | 154 |
| Porous silicon nanoparticles functionalized with biotin-polyethylene glycol and loaded with ANA | Targeted delivery to AD brain and amyloid-beta plaque disaggregation | 155 |
| Penetratin peptide loaded PEG-stabilized gold nanostars modified with ruthenium complex (Ru@Pen@PEG–AuNS) | Inhibit and dissociate amyloid-beta fibrils under near-infrared irradiation | 156 |
| 3.3 nm L- and D-glutathione stabilized gold nanoparticles (L3.3 and D3.3) | Inhibit aggregation of Aβ42 and cross the blood–brain barrier without noticeable toxicity | 157 |
| KLVFF@Au–CeO2 (K-CAC) nanocomposites consisting of gold nanorods coated with CeO2 nanoparticles and modified with Aβ-targeted inhibitory peptides | Photocatalysis and photothermal therapy for enhancing redox performance and BBB permeability | 158 |
| RPOMs@MSNs@copolymer | Photothermal disaggregation of Aβ fibrils and ROS scavenging | 159 |
| Custom-made peptide dendrimers conjugated to star-shaped and spherical gold nanostructures (H3/H6–AuNS/AuNP) | Delivery of neuroprotectants and protection of neurons | 160 |
| Gold nanoparticles with negative surface potential | Inhibit and redirect amyloid-β fibrillization | 161 |
| Carboxylated graphene oxide nanosheets functionalized with PEG and PEI | Delivery of GSK3β siRNA | 112 |
| Functionalized-Gd@C82 nanoparticles with hydrogen-binding sites and charged groups | Redirect Aβ peptide self-assembly and disaggregate amyloid fibrils | 162 |
| Chiral Au nanoparticles | Restored cognitive abilities and ameliorated amyloid-β and hyperphosphorylated tau pathologies | 163 |
| RB-loaded upconverting nanocomposites with rattle-structured organosilica shell on NaYF4:Yb, Er nanocrystals | NIR-responsive inhibitor of Aβ aggregation and suppressor of Aβ-induced cytotoxicity | 164 |
| Borneol (Bor)-modified octahedral palladium (Pd@PEG@Bor) nanozyme platform | Eliminate intracellular reactive oxygen species (ROS) and elevate epithelial cell penetrability | 165 |
| Beta casein-coated iron oxide nanoparticles synthesized via a BPA-P(OEGA-b-DBM) block copolymer linker | Inhibition of amyloid aggregation | 166 |
| EGCG-stabilized selenium nanoparticles coated with Tet-1 peptide (Tet-1-EGCG@Se) | Inhibits Aβ fibrillation and disaggregates Aβ fibrils | 167 |
| Hybrid peptide VVIACLPFFD conjugated to gold nanoparticles | Inhibition of amyloid-β aggregation and reduction of cytotoxicity | 168 |
| Graphene oxide (GO) based nanomaterials | Reduces amyloid-β levels and improves cognitive function | 169 |
| Small-sized Pd hydride (PdH) nanoparticles | High payload and sustained release of hydrogen | 170 |
| Res-selenium-peptide nanocomposite (TGN-Res@SeNPs) | Eliminates Aβ aggregate-induced neurotoxicity and mitigates gut microbiota imbalance | 171 |
| MoO3−x nanodots synthesized by pulsed laser ablation in MoS2 nanosheets | Dual enzyme mimic activities (catalase and SOD) and modulation of Aβ fibrillation | 172 |
| Gold nanorods loaded with scFv 12B4 and APH ST0779 (GNRs-APH-scFv, GAS) | Rapid detection of Aβ aggregates and NIR photothermal disassembly | 173 |
| Isomeric gold nanoclusters modified with p-MBA, m-MBA, and o-MBA | Inhibition of Aβ40 misfolding, aggregation, and fibrillation | 174 |
| Mitochondria-targeted nanozymes known as (3-carboxypropyl)triphenyl-phosphonium bromide-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]-functionalized molybdenum disulfide quantum dots (TPP–MoS2 QDs) | Mitigate Aβ aggregate-mediated neurotoxicity and eliminate Aβ aggregates by switching microglia from M1 to M2 phenotype | 175 |
| Mesoporous nano-selenium (MSe) release delivery system (MSe-Res/Fc-β-CD/Bor) based on borneol target and β-cyclodextrin nanovalves | Controlled release and targeted delivery across the blood–brain barrier | 176 |
| SA-modified selenium nanoparticles conjugated with B6 peptide (B6-SA-SeNPs) | High permeability across the blood–brain barrier and inhibition/disaggregation of amyloid-β aggregation | 177 |
| Magnetic nanoparticles (MNPs) selectively attached to amyloid-β aggregates for efficient hysteretic power dissipation | Magnetothermal disruption to break up amyloid-β aggregates | 178 |
| Magnetic nanoparticles conjugated with ΛA | Aβ clearance | 179 |
| Gold nanoparticles conjugated to CLPFFD and THRPPMWSPVWP | Destroy toxic β-amyloid aggregates and enhance permeability across the blood–brain barrier | 180 |
| Selenium quantum dots (SeQDs) with ultrasmall size | Diagnose and track AD via fluorescence, scavenge free radicals, inhibit Aβ aggregation, reduce tau phosphorylation, protect nerve cells | 181 |
| Brain-penetrating manganese dioxide nanoparticles | Reduce hypoxia, neuroinflammation, oxidative stress, and amyloid β plaques | 182 |
| H2O2 responsive controlled-release mesoporous silica nanoparticles (MSNs) | Controlled release of AD therapeutic metal chelator in response to H2O2 levels | 183 |
| Polyoxometalate–peptide (POM@P) hybrid particles | Bifunctional Aβ inhibitors and fluorescent probes | 184 |
| Tg–CS/DMY@SeNPs | Inhibit Aβ aggregation and reduce inflammatory cytokines | 185 |
| Magnetoelectric BiFeO3-coated CoFe2O4 (BCFO) nanoparticles | Dissociation of β-amyloid (Aβ) aggregates | 186 |
| Quercetin modified polysorbate 80-coated AuPd core–shell structure | Activate autophagy and promote amyloid-β clearance | 187 |
| Gold nanoparticles surface-functionalized with mimosine | Suppress Aβ aggregation and disassemble Aβ fibers | 188 |
| Ultrasmall MoS2 quantum dots | Potent inhibitor of Aβ amyloid aggregation and recovery of membrane fluidity | 189 |
| Gold nanoparticles (AuNP) selectively attached to aggregates | Deliver local heat to remove and dissolve amyloid-beta aggregates | 190 |
| Dual-targeted magnetic mesoporous silica nanoparticle (HA-MMSN-1F12) with surface-coupled Aβ42-targeting antibody 1F12 and CD44-targeting hyaluronic acid (HA) | Crosses BBB to degrade brain Aβ plaques, avoids hepatic uptake, and facilitates excretion of Aβ through intestinal metabolism | 191 |
| Casein coated-gold nanoparticles (βCas AuNPs) | Translocate across blood brain barrier and sequester amyloid beta in a chaperone-like manner | 192 |
| Gold and platinum nanoparticles coated with multiple ligands | Increase binding affinity of Aβ-specific small molecules to inhibit Aβ peptide aggregation | 193 |
| Protoporphyrin IX (PX)-modified oxidized mesoporous carbon nanospheres (PX@OMCN@PEG(OP)@RVGs) | Inhibits tau phosphorylation and amyloid beta aggregation, enhances blood–brain barrier permeability | 194 |
| Mesoporous silica nanospheres immobilized on Bifidobacterium (MSNs-Bi) | Intranasal delivery to transport nanoparticles through brain to peripheral intestine, inhibit intestinal inflammation, reduce brain Aβ burden, improve olfactory sensitivity | 195 |
| UCNP@C60-pep (upconversion nanoparticle and Aβ-target peptide KLVFF) | Near-infrared-switchable ROS producer and scavenger, Aβ-targeting, and imaging capabilities | 196 |
| Gold nanoparticle–capped mesoporous silica (MSN–AuNPs)-based H2O2-responsive controlled release system | Targeted delivery of metal chelator CQ and inhibition of Aβ aggregation | 197 |
| Cysteine–Aβ peptide–conjugated gold nanoparticles (Cys–Aβ@AuNP) | Detection of subfemtomolar Aβ peptides and early-stage Aβ oligomerization | 198 |
| Chiral amide-gel-directed synthesis of molecularly chiral mesoporous silica nanospheres | Inhibits β-amyloid aggregation and reduces cytotoxicity | 199 |
| Polyvinylpyrrolidone-functionalized MoS2 nanoparticles fabricated by pulsed laser ablation | Inhibits Aβ aggregation, destabilizes Aβ fibrils, alleviates oxidative stress and cell toxicity, blocks Ca2+ channel formation | 200 |
| Superparamagnetic iron oxide nanoparticle conjugated with Aβ oligomer-specific scFv antibody W20 and class A scavenger receptor activator XD4 (W20/XD4-SPIONs) | Inhibiting Aβ aggregation, attenuating AβO-induced cytotoxicity, and increasing microglial phagocytosis of Aβ | 201 |
| Ceria/Polyoxometalates hybrid (CeONP@POMs) | Degrades amyloid-β aggregates and reduces reactive oxygen species (ROS) | 202 |
| Cyclic dipeptide-based copolymer interacting with gold nanoparticles and polyoxometalate | Inhibits β-amyloid aggregation, dissolves preformed aggregates, scavenges reactive oxygen species | 203 |
| Curcumin and SPIO nanoparticles encapsulated by DSPE–PEG and modified with CRT and QSH peptides | Early diagnosis via MRI and therapeutic intervention by reducing β-amyloid plaque burden | 204 |
| Chiral penicillamine-capped selenium nanoparticles | Chiral amyloid-β (Aβ) inhibitors | 205 |
| Chiral L/D-FexCuySe nanoparticles | Interfere with Aβ42 self-assembly and disrupt fibrils under 808 nm near-infrared illumination | 206 |
| HSA-embedded ultrasmall copper nanoclusters (CuNCs@HSA) | Elimination of ROS, inhibition of Aβ aggregation, and mitigation of neuroinflammation | 207 |
| Porphyrinic metal–organic framework (MOF) PCN-224 nanoparticles synthesized by coordinating TCPP ligands with zirconium | Suppress aggregation of amyloid-β peptide and reduce cytotoxicity under near-infrared light | 208 |
| Metal–organic framework-derived carbon (MOFC) with defect-rich and entangled graphitic layers | Photoacoustic dissociation of beta-amyloid aggregates | 209 |
| Graphene quantum dots (GQDs) conjugated with neuroprotective peptide glycine–proline–glutamate (GQDG) | Inhibit aggregation of Aβ1–42 fibrils | 210 |
| Inhibitor-conjugated NIR laser-propelled Janus nanomotor (JNM-I) | Modulation of amyloid-β aggregation | 211 |
| Congo red-derived carbon dots synthesized from Congo red and citric acid with variants CRCD1-3 | Dual inhibitors of tau and amyloid-beta aggregation and act as nanocarriers with BBB permeability | 212 |
| Nanovehicles (nanoparticles-IgG4.1) loaded with imaging agents and therapeutic agents | Target cerebrovascular amyloid deposits for diagnostic imaging and drug delivery | 213 |
| Chiral nanoparticles (L-type and D-type) | Accelerate differentiation of neural stem cells into neurons and clear amyloid and p-tau proteins under NIR light | 214 |
| C3N nanodots | Aβ peptide aggregation inhibitor | 215 |
| Stepwise metal-phenolic coordination of rhein and polydopamine to create K8@Fe–Rh/Pda nanoparticles | Inhibit Aβ aggregation, repair neuronal damage, promote mitochondrial biogenesis, and inhibit neuronal apoptosis | 216 |
| Biocompatible metal-phenolic network (MPN) with EGCG and Zn(II) on gold nanoparticles | Inhibits amyloid beta aggregation and toxicity, crosses blood–brain barrier | 217 |
| Iminodiacetic acid-conjugated nanoparticles | Modulate Aβ42 aggregation and reduce the cytotoxicity accelerated by Zn2+ | 218 |
| EMT-type zeolite nanoparticles with particle size of 10–20 nm and external surface area of 200 m2 g−1 | Inhibit Aβ-fibrinogen interactions and prevent abnormal clot formation | 219 |
| NiM@P hybrid particles | Bifunctional Aβ inhibitors | 220 |
| PBAE–PLGA–Ag2S S–RA–siSOX9 (PPAR–siSOX9) nanoformulation | High gene/drug deliverability to overcome AD microenvironment-associated adverse effects and promote neuronal differentiation of NEP-expressing NSCs | 119 |
| Prussian blue/polyamidoamine (PAMAM) dendrimer/Angiopep-2 (PPA) nanoparticles | Superior BBB permeability, ROS scavenging, restoration of mitochondrial function, regulation of microglia mitophagy | 84 |
Ultra-small C3N nanodots were developed as inhibitors of Aβ42 peptide aggregation.215 These nanodots, with an average lateral size of 4.5 ± 0.4 nm, demonstrated the ability to inhibit Aβ42 aggregation and disaggregate mature fibrils in vitro, as confirmed by Thioflavin T fluorescence assay, dot blotting, atomic force microscopy, transmission electron microscopy, and circular dichroism spectroscopy. In primary mouse neurons, C3N nanodots alleviated aggregation-induced cytotoxicity, increasing cell viability from approximately 29.89% (Aβ42 alone) to 65.52% (Aβ42 with 500 μg mL−1 C3N nanodots). Importantly, in vivo studies showed that intraperitoneal administration of C3N nanodots at 1 mg per kg per day for six months to APP/PS1 double transgenic male AD mice significantly reduced cerebral Aβ plaque levels by about 60%, decreased total Aβ42/Aβ40 levels by 36%/50%, respectively, and restored synaptic protein expression. Behavioral assessments revealed that treated mice exhibited improved cognitive function, with escape latency in the Morris water maze test reduced from approximately 42.4 ± 6.9 s (control mice) to 19.2 ± 2.3 s (treated mice) on day five. The therapeutic efficacy of C3N nanodots is attributed to their ability to interact with Aβ peptides via van der Waals and electrostatic interactions, hydrophobic interactions, hydrogen bonding, and π–π stacking, as revealed by molecular dynamics simulations, thereby preventing β-sheet formation and peptide aggregation. Safety evaluations indicated that C3N nanodots exhibited minimal toxicity, with no significant pathological damage observed in vital organs and normal inflammation and liver and kidney function indices after six months of treatment.
Functionalization of nanoparticles with targeting peptides has been employed to enhance BBB permeability and therapeutic efficacy. For instance, AuNPs conjugated with a transferrin receptor-interacting peptide (THR) and an Aβ-binding peptide (CLPFFD) demonstrated enhanced BBB crossing and effectively inhibited Aβ aggregation without significant cytotoxicity.180 Polyoxometalate–peptide (POM@P) hybrid nanoparticles, combining a Wells–Dawson-type phosphotungstate with an Aβ-targeted peptide, showed enhanced inhibition of Aβ1–40 aggregation and reduced cytotoxicity in neuronal cells, leveraging both targeted binding and electrostatic interactions.184 Selenium nanoparticles have also been explored for their neuroprotective properties.222 EGCG-stabilized SeNPs coated with Tet-1 peptide (Tet-1-EGCG@Se) effectively inhibited Aβ fibrillation, disaggregated preformed fibrils, and reduced reactive oxygen species levels, thereby mitigating Aβ-induced cytotoxicity.167 Sialic acid-modified SeNPs conjugated with B6 peptide (B6-SA-SeNPs) enhanced BBB permeability, inhibited Aβ aggregation, and protected neuronal cells from Aβ-induced apoptosis, demonstrating the potential of SeNPs in AD therapy.177
Multivalent ligand presentation on nanoparticles has been utilized to enhance binding affinity to Aβ peptides. Gold and platinum nanoparticles coated with Aβ-specific ligands exhibited significantly increased binding affinity and more effectively inhibited Aβ aggregation compared to free ligands, offering a strategy to overcome the lower affinity of small molecules.193 Additionally, peptide inhibitors conjugated onto AuNPs showed greatly enhanced inhibition of Aβ42 aggregation and cytotoxicity, emphasizing the synergistic effects of nanoparticle conjugation.168 A recent study reported how the core/ligands interfacial anchor structures of AuNPs influence their ability to regulate the spatial conformation of amyloid peptides, providing insights for the precise design of artificial nano-chaperones.223 By synthesizing three types of AuNPs-Au@ABA NPs (Au–NH bond) using 4-aminobenzoic acid, Au@MBA NPs (Au–S bond) using 4-mercaptobenzoic acid, and Au@EBA NPs (Au–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond) using 4-ethynylbenzoic acid-with similar core sizes (∼3 nm) and identical benzoic acid-exposed surfaces, the study investigated their interactions with Aβ40 and amylin peptides. Thioflavin-T assays demonstrated that all three AuNPs inhibited Aβ40 fibrillization in a dose-dependent manner, with Au@EBA NPs showing the most substantial reduction in fluorescence intensity at high concentrations, indicating stronger inhibition. Circular dichroism spectroscopy revealed that Au@EBA NPs and Au@ABA NPs prevented the conformational transition of Aβ40 from random coil to β-sheet structure, while Au@MBA NPs only partially inhibited this transformation. Atomic force microscopy and transmission electron microscopy confirmed the absence of fibril formation in the presence of Au@EBA NPs and Au@ABA NPs. In contrast, for amylin, Au@ABA NPs inhibited fibrillization, whereas Au@MBA NPs and Au@EBA NPs promoted misfolding and fibrillization, as evidenced by spectroscopic analyses and imaging techniques. Molecular dynamics simulations and isothermal titration calorimetry indicated that different core/ligand anchors led to variations in electrostatic potential distributions and binding energies, affecting the nanoparticles' binding sites and strengths on the peptides. Unlike previous research that focused on ligand effects, this work emphasizes the critical role of core/ligands interfacial anchors in modulating nanoparticle–peptide interactions, advancing the understanding of artificial nano-chaperone design. Several other studies have also reported similar approaches using AuNPs to inhibit Aβ formation.224,225
C bond) using 4-ethynylbenzoic acid-with similar core sizes (∼3 nm) and identical benzoic acid-exposed surfaces, the study investigated their interactions with Aβ40 and amylin peptides. Thioflavin-T assays demonstrated that all three AuNPs inhibited Aβ40 fibrillization in a dose-dependent manner, with Au@EBA NPs showing the most substantial reduction in fluorescence intensity at high concentrations, indicating stronger inhibition. Circular dichroism spectroscopy revealed that Au@EBA NPs and Au@ABA NPs prevented the conformational transition of Aβ40 from random coil to β-sheet structure, while Au@MBA NPs only partially inhibited this transformation. Atomic force microscopy and transmission electron microscopy confirmed the absence of fibril formation in the presence of Au@EBA NPs and Au@ABA NPs. In contrast, for amylin, Au@ABA NPs inhibited fibrillization, whereas Au@MBA NPs and Au@EBA NPs promoted misfolding and fibrillization, as evidenced by spectroscopic analyses and imaging techniques. Molecular dynamics simulations and isothermal titration calorimetry indicated that different core/ligand anchors led to variations in electrostatic potential distributions and binding energies, affecting the nanoparticles' binding sites and strengths on the peptides. Unlike previous research that focused on ligand effects, this work emphasizes the critical role of core/ligands interfacial anchors in modulating nanoparticle–peptide interactions, advancing the understanding of artificial nano-chaperone design. Several other studies have also reported similar approaches using AuNPs to inhibit Aβ formation.224,225
Chiral nanoparticles have demonstrated enantioselective inhibition of Aβ aggregation.157 Chiral penicillamine-capped SeNPs, particularly the D-enantiomer (D-Pen@Se NPs), effectively inhibited Zn2+-induced Aβ40 fibrillation and ameliorated cognitive impairments in AD mouse models, highlighting the significance of nanoscale chirality in therapeutic applications.205 Similarly, L- and D-glutathione-stabilized gold nanoparticles inhibited Aβ42 aggregation and crossed the BBB, with the D-enantiomer showing higher brain biodistribution and more pronounced cognitive rescue in AD mice.157
Other inorganic nanoparticles, such as graphene oxide (GO) and molybdenum disulfide quantum dots, have been shown to reduce Aβ production, enhance its degradation, and restore membrane fluidity disrupted by Aβ oligomers, offering multifaceted therapeutic mechanisms.189 Chiral metallohelices and chiral mesoporous silica nanospheres have also been utilized to enantioselectively inhibit Aβ aggregation and mitigate cytotoxicity, providing insights into the role of molecular chirality in nanoparticle design.199 A mesoporous nano-selenium (MSe) delivery system, incorporating resveratrol and responsive to redox stimuli, achieved targeted delivery and significant therapeutic benefits in AD mouse models.176
Furthermore, nanoparticles have been employed for both therapeutic and diagnostic purposes. Multifunctional nanoparticles combining curcumin and superparamagnetic iron oxide, modified with peptides for BBB penetration and Aβ targeting, enabled noninvasive MRI detection of Aβ plaques and improved cognitive deficits in AD mice.135,204,226 Nanoparticles conjugated with D-enantiomeric peptides demonstrated the ability to fragment tau fibrils, reduce tau pathology, and improve cognitive function in AD mouse models.227
| Nanoparticle design | Function of nanoparticle | Reference |
|---|---|---|
| Macrophage membrane-coated solid lipid nanoparticles with RVG29 and TPP | Targeted delivery of antioxidants to neuronal mitochondria across the blood–brain barrier | 228 |
| RBC membrane encapsulating carbon quantum dots and polydopamine | Evade immune clearance, mitigate oxidative stress, and chelate metal ions | 144 |
| RBC membrane-camouflaged human serum albumin nanoparticles bearing T807 and TPP | Targeted delivery of antioxidants to neuronal mitochondria | 229 |
| Red blood cell membrane-templated cerium oxide nanocrystals encapsulated with carbon quantum dots (CQD–Ce–RBC) | Biocompatible nanocomposite with antioxidant properties, copper ion chelating, Aβ aggregation prevention, and photothermal effects to break down Aβ fibers and enhance blood–brain barrier permeability | 146 |
| Composite nanometer system of red blood cell membranes-encapsulated Prussian blue nanoparticles (PB/RBC) | Chelate Cu2+, reduce ROS, photothermally open BBB, depolymerize Aβ deposits | 147 |
| MoS2 QDs/MM | Elimination of ROS and anti-Aβ1–42 deposition | 149 |
| Cerium oxide nanoparticles that switch between Ce(3+) and Ce(4+) states | Scavenge reactive oxygen and nitrogen species, internalize in neurons and accumulate at mitochondrial and plasma membranes | 230 |
| Mesoporous silica nanomaterials loaded with curcumin and IR780, grafted with cerium oxide nanoparticles and peptide K | Inhibiting β-amyloid aggregation and scavenging reactive oxygen species (ROS) | 151 |
| RVG29-modified biodegradable mesoporous silica nanoparticles loaded with ultra-small cerium oxide nanocrystals and conjugated with antibody 1F12 | Inhibits Aβ42 misfolding and aggregation, accelerates Aβ42 clearance, and scavenges reactive oxygen species | 152 |
| MSiO2@SiCDs nanocomposites | Efficient Cu2+ chelator and inhibitor of Aβ aggregation | 154 |
| KLVFF@Au–CeO2 (K-CAC) nanocomposites consisting of gold nanorods coated with CeO2 nanoparticles and modified with Aβ-targeted inhibitory peptides | Photocatalysis and photothermal therapy for enhancing redox performance and BBB permeability | 158 |
| RPOMs@MSNs@copolymer | Photothermal disaggregation of Aβ fibrils and ROS scavenging | 159 |
| Custom-made peptide dendrimers conjugated to star-shaped and spherical gold nanostructures (H3/H6–AuNS/AuNP) | Delivery of neuroprotectants and protection of neurons | 160 |
| Iron oxide nanoparticles | Mimic catalase and decompose reactive oxygen species (ROS) | 231 |
| Borneol (Bor)-modified octahedral palladium (Pd@PEG@Bor) nanozyme platform | Eliminate intracellular reactive oxygen species (ROS) and elevate epithelial cell penetrability | 165 |
| Beta casein-coated iron oxide nanoparticles synthesized via a BPA-P(OEGA-b-DBM) block copolymer linker | Inhibition of amyloid aggregation | 166 |
| EGCG-stabilized selenium nanoparticles coated with Tet-1 peptide (Tet-1-EGCG@Se) | Inhibits Aβ fibrillation and disaggregates Aβ fibrils | 167 |
| Small-sized Pd hydride (PdH) nanoparticles | High payload and sustained release of hydrogen | 170 |
| Res-selenium-peptide nanocomposite (TGN-Res@SeNPs) | Eliminates Aβ aggregate-induced neurotoxicity and mitigates gut microbiota imbalance | 171 |
| MoO3−x nanodots synthesized by pulsed laser ablation in MoS2 nanosheets | Dual enzyme mimic activities (catalase and SOD) and modulation of Aβ fibrillation | 172 |
| UCNP@SiO2@Cur/CQ | Controlled sequential drug release, Cu2+ chelation, and ROS removal | 232 |
| Mitochondria-targeted nanozymes known as (3-carboxypropyl)triphenyl-phosphonium bromide-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]-functionalized molybdenum disulfide quantum dots (TPP–MoS2 QDs) | Mitigate Aβ aggregate-mediated neurotoxicity and eliminate Aβ aggregates by switching microglia from M1 to M2 phenotype | 175 |
| Mesoporous nano-selenium (MSe) release delivery system (MSe-Res/Fc-β-CD/Bor) based on borneol target and β-cyclodextrin nanovalves | Controlled release and targeted delivery across the blood–brain barrier | 176 |
| SA-modified selenium nanoparticles conjugated with B6 peptide (B6-SA-SeNPs) | High permeability across the blood–brain barrier and inhibition/disaggregation of amyloid-β aggregation | 177 |
| Selenium quantum dots (SeQDs) with ultrasmall size | Diagnose and track AD via fluorescence, scavenge free radicals, inhibit Aβ aggregation, reduce tau phosphorylation, protect nerve cells | 181 |
| Brain-penetrating manganese dioxide nanoparticles | Reduce hypoxia, neuroinflammation, oxidative stress, and amyloid β plaques | 182 |
| Triphenylphosphonium-conjugated ceria nanoparticles | Recyclable ROS scavengers | 233 |
| Gold nanoparticles surface-functionalized with mimosine | Suppress Aβ aggregation and disassemble Aβ fibers | 188 |
| Magnetite core, ceria shell | Remove Aβ peptides and scavenge reactive oxygen species | 234 |
| Congo red/Rutin-MNPs | Diagnosis and treatment of Alzheimer's disease | 235 |
| UCNP@C60-pep (upconversion nanoparticle and Aβ-target peptide KLVFF) | Near-infrared-switchable ROS producer and scavenger, Aβ-targeting, and imaging capabilities | 196 |
| Gold nanoparticle–capped mesoporous silica (MSN–AuNPs)-based H2O2-responsive controlled release system | Targeted delivery of metal chelator CQ and inhibition of Aβ aggregation | 197 |
| Polyvinylpyrrolidone-functionalized MoS2 nanoparticles fabricated by pulsed laser ablation | Inhibits Aβ aggregation, destabilizes Aβ fibrils, alleviates oxidative stress and cell toxicity, blocks Ca2+ channel formation | 200 |
| Ceria/polyoxometalates hybrid (CeONP@POMs) | Degrades amyloid-β aggregates and reduces reactive oxygen species (ROS) | 202 |
| Cyclic dipeptide-based copolymer interacting with gold nanoparticles and polyoxometalate | Inhibits β-amyloid aggregation, dissolves preformed aggregates, scavenges reactive oxygen species | 203 |
| Curcumin and SPIO nanoparticles encapsulated by DSPE–PEG and modified with CRT and QSH peptides | Early diagnosis via MRI and therapeutic intervention by reducing β-amyloid plaque burden | 204 |
| HSA-embedded ultrasmall copper nanoclusters (CuNCs@HSA) | Elimination of ROS, inhibition of Aβ aggregation, and mitigation of neuroinflammation | 207 |
| Integrated ceria nanozymes into MOFs loaded with siSOX9 and RA | Promotes neuron differentiation and eliminates ROS | 113 |
| Hollow manganese Prussian white nanocapsules (HMPWCs) self-assembled with manganese Prussian white analogue and bovine serum albumin via novel biomimetic mineralization | Relieving oxidative stress, inhibiting tau neuropathology, and counteracting neuroinflammation | 236 |
| Co-assembled guanidinium-modified calixarene with ascorbyl palmitate and dipotassium phytate | Simultaneous inhibition and disintegration of β-amyloid fibrils and regulation of oxidative stress | 141 |
| Stepwise metal-phenolic coordination of rhein and polydopamine to create K8@Fe–Rh/Pda nanoparticles | Inhibit Aβ aggregation, repair neuronal damage, promote mitochondrial biogenesis, and inhibit neuronal apoptosis | 216 |
| C70-derived graphene acid quantum dots (GAQDs) | Inhibition of amyloid fibril formation, scavenging reactive oxygen species | 237 |
| Human serum albumin encapsulated quercetin (HSA@QC) nanoparticles | Natural phyto-antioxidant albumin nanoagent for treating advanced Alzheimer's disease | 238 |
| Polydopamine nanoparticles decorated with KLVFF peptide | Metal ion chelating, ROS scavenging, and enhanced blood–brain barrier crossing | 102 |
| Poly(lactide-co-glycolide) (PLGA) and polyethylene glycol (PEG)-2000 based biodegradable nanoparticles | Encapsulation of anthocyanins to enhance bioavailability and stability | 99 |
| TPL comprising BBB-penetrating peptide TGN and neuron-targeting peptide Tet1 via four-glycine linker | Targeted delivery of neuroprotective peptide NAP across BBB to neurons | 105 |
| Combination of chitosan and graphene quantum dots into ultrasmall nanoparticles via microfluidic-based synthesis | Theranostic agents for brain targeting, transcellular transfer, and bioimaging | 239 |
| Mitochondria-targeted polymeric nanoparticle system based on PLGA-b-PEG–TPP blended with PLGA-b-PEG–OH or PLGA–COOH | Efficient delivery of mitochondria-acting therapeutics to the mitochondrial matrix | 240 |
| Citraconylation-modified poly(ethylene glycol)–poly(trimethylene carbonate) polymer (PEG–PTMC(Cit)) with FGL peptide modification | Targeted delivery of HNSS peptide to mitochondria in cholinergic neurons | 98 |
| Chitosan/TPP nanoparticles loaded with Resveratrol and modified with TG peptide | Delivery of Resveratrol to the brain | 241 |
| Prussian blue/polyamidoamine (PAMAM) dendrimer/Angiopep-2 (PPA) nanoparticles | Superior BBB permeability, ROS scavenging, restoration of mitochondrial function, regulation of microglia mitophagy | 84 |
| Hybrid peptide VLC (VHS + COG1410) conjugated to curcumin via phenylboronic ester bond | Targeted delivery to pericyte lesions and release upon ROS stimulation | 106 |
| ROS-responsive dendrimer–peptide conjugate (APBP) | Scavenges ROS, promotes Aβ phagocytosis, and normalizes glial cell phenotype | 104 |
| Amorphous PDLLA-dextran bottlebrush with controlled graft density and side chain length forming micelles, vesicles, and compound vesicles | Codelivery of hydrophilic antioxidants (citric acid, vitamin C, gallic acid) | 109 |
Iron oxide nanoparticles have also been explored for their antioxidative properties and multifunctionality in AD treatment. Zhang et al. synthesized iron oxide (Fe3O4) nanoparticles averaging 20 nm in diameter that demonstrated catalase-like activity, decomposing hydrogen peroxide (H2O2) into water and oxygen under neutral pH conditions.231 These nanoparticles reduced intracellular H2O2 levels, protected against oxidative stress-induced apoptosis in neuronal cells, and ameliorated neurodegeneration in Drosophila models. In a complementary approach, Hu et al. developed ultrasmall superparamagnetic iron oxide nanoparticles (USPIONs) conjugated with Congo red and Rutin (Congo red/Rutin-MNPs) for targeted imaging and antioxidant therapy (Fig. 6A).235 These nanoparticles were designed to release Rutin, a natural antioxidant, in response to elevated H2O2 levels characteristic of AD pathology. In vitro studies demonstrated that the nanoparticles inhibited Aβ-induced cytotoxicity and reduced ROS levels in neuronal cells. In vivo, treatment with Congo red/Rutin-MNPs improved cognitive function and reduced Aβ deposition in transgenic AD mice. These studies highlight the dual functionality of iron oxide nanoparticles in both ROS scavenging and serving as imaging agents.
 | ||
| Fig. 6 (A) Schematic illustration of the preparation of Congo red and rutin-loaded magnetic nanoparticles (Congo red/Rutin-MNPs). DSPE–PEG–Congo red and DSPE–PEG–phenylboronic acid were utilized to enhance the biocompatibility of magnetic nanoparticles through micelle formation. Rutin was grafted onto the nanoparticle surface via the formation of boronate ester bonds between vicinal diols and phenylboronic acid. (B) Illustration of the synthesis strategies and anti-Alzheimer's disease (AD) application of palladium hydride (PdH) nanoparticles. (C) Depiction of the brain microenvironment in AD: untreated or vehicle-treated brains exhibit denser and larger Aβ plaques, higher oxidative stress, increased neuroinflammation, and abnormal vasculature, whereas brains treated with Ab-TP-MDNPs show fewer Aβ plaques, normalized vascular structure, and reduced oxidative stress and neuroinflammation.170,182,235 | ||
Palladium-based nanoparticles have emerged as effective nanozymes for ROS scavenging due to their intrinsic enzyme-like activities. Zhang et al. developed small-sized palladium hydride (PdH) nanoparticles capable of sustained release of bio-reductive hydrogen in the AD brain (Fig. 6B).170 The PdH nanoparticles selectively scavenged highly cytotoxic ROS, such as hydroxyl radicals, restored mitochondrial function, inhibited Aβ generation and aggregation, and improved cognitive function in a transgenic AD mouse model. Similarly, octahedral palladium nanoparticles (Pd NPs) functionalized with polyethylene glycol and borneol (Pd@PEG@Bor) were designed to enhance BBBpermeability and leverage the antioxidative properties of Pd NPs.165 The borneol modification facilitated BBB penetration, allowing the nanoparticles to accumulate in the brain. In vitro, Pd@PEG@Bor nanoparticles reduced intracellular ROS levels, protected mitochondrial integrity, and decreased neuroinflammation. In vivo, they reduced Aβ plaque deposition and improved cognitive function in AD mice. These palladium-based nanozymes offer a promising strategy for ROS scavenging and neuroprotection in AD.
Manganese dioxide (MnO2) nanoparticles have been investigated for their ability to modulate the oxidative microenvironment in the AD brain. Park et al. designed bioactive MnO2-polymer-lipid hybrid nanoparticles functionalized with anti-Aβ antibodies (Ab-TP-MDNPs) (Fig. 6C).182 These nanoparticles were engineered to cross the BBB, target Aβ plaques, and react with ROS to generate oxygen, thereby alleviating hypoxia and oxidative stress. In a transgenic AD mouse model, treatment with Ab-TP-MDNPs reduced hypoxia markers, decreased neuroinflammation, improved cerebral blood flow, reduced Aβ plaque burden, and enhanced cognitive function. By simultaneously addressing multiple pathological factors, MnO2 nanoparticles demonstrate significant potential in remodeling the brain microenvironment in AD.
Collectively, these studies illustrate the versatility and efficacy of inorganic nanoparticles in scavenging ROS and mitigating oxidative stress in Alzheimer's disease. By exploiting properties such as enzyme-mimetic activity, targeted mitochondrial localization, and responsive release mechanisms, nanoparticles like cerium oxide, iron oxide, palladium, and manganese dioxide offer innovative therapeutic approaches. Their ability to cross the BBB and target specific pathological sites enhances their clinical potential. Future research should focus on optimizing these nanoparticle systems for safety, biocompatibility, and efficacy in long-term studies to facilitate their translation into clinical applications for AD treatment.
A cyclic dipeptide-based copolymer (CDP-CP) was synthesized to self-assemble into anisotropic architectures and interact with gold nanoparticles (GNPs) and polyoxometalate (POM), forming nanocomposites CP-GNP and CP-POM, respectively (Fig. 7A).203 These nanocomposites effectively inhibited Aβ42 fibril formation by 55–75% at concentrations as low as 100 nM and dissolved preformed aggregates by up to 50%. They also exhibited significant ROS scavenging activity, reducing intracellular ROS levels by up to 45%, and protected neuronal cells from Aβ-induced toxicity. The hierarchical organization of CDP-CP into organic–inorganic hybrids enhanced their therapeutic efficacy while maintaining minimal cytotoxicity, highlighting their potential in combating multifaceted amyloid toxicity in AD.
 | ||
| Fig. 7 (A) Representation of the CDP-CP copolymer and its molecular assembly into multifunctional material architectures with biomedical properties. The copolymer exhibits solvent polarity and concentration-dependent structural tailorability, enabling the development of CDP-CP-based gold nanoparticle (GNP) and polyoxometalate (POM) nanocomposites (CP-GNP and CP-POM) to protect neuronal cells from amyloid toxicity. (B) Illustration of RVG29-bioactive mesoporous silica nanoparticles loaded with Ce–1F12 (RVG29-bMSNs@Ce–1F12) for combinational therapy of Alzheimer's disease.152,203 | ||
Ceria/polyoxometalate hybrid nanoparticles (CeONP@POMD) were designed as artificial nanozymes exhibiting both proteolytic and superoxide dismutase (SOD) activities.202 These nanoparticles, approximately 5 nm in size, degraded Aβ monomers and fibrils, inhibited Aβ-induced cytotoxicity, and reduced intracellular ROS levels by 68%. They demonstrated the ability to cross the BBB and inhibited Aβ-induced activation of microglial cells. In vivo studies confirmed their good biocompatibility and potential as multifunctional therapeutic agents against Aβ neurotoxicity, offering a novel approach to overcome the limitations of natural proteases in AD treatment. Furthermore, a multifunctional nanocomposite, RVG29-bMSNs@Ce–1F12, was developed for ROS scavenging and Aβ42 targeting. This composite consists of biodegradable mesoporous silica nanoparticles (bMSNs) loaded with ultra-small cerium oxide nanocrystals (CeNPs) (Fig. 7B).152 Functionalized with the anti-Aβ42 antibody 1F12 and brain-targeting rabies virus glycoprotein 29 (RVG29), this nanocomposite effectively inhibited Aβ42 aggregation, promoted depolymerization of Aβ42 fibrils, and reduced ROS levels in neuronal cells. In vivo, it decreased soluble Aβ42 levels in plasma and brain tissues, reduced hyperphosphorylated tau burden, mitigated microglial activation, and enhanced cognitive function in APP/PS1 transgenic mice. The design offers a synergistic therapeutic strategy by simultaneously targeting Aβ aggregates and ROS, demonstrating significant potential for AD treatment.
Polyvinylpyrrolidone-functionalized molybdenum disulfide (MoS2) nanoparticles were fabricated via a pulsed laser ablation method and evaluated for their effects on Aβ peptides.200 The MoS2 nanoparticles inhibited Aβ aggregation, decreasing Thioflavin T fluorescence intensity by up to 60%, and destabilized preformed Aβ fibrils. They alleviated Aβ-induced oxidative stress and cytotoxicity in neuronal cells, improving cell viability by up to 20%. Additionally, MoS2 nanoparticles blocked the formation of calcium channels induced by Aβ fibrils in neuronal cell membranes, significantly reducing intracellular calcium accumulation. This multifunctional activity positions MoS2 nanoparticles as promising therapeutic agents against amyloid-related neurodegenerative diseases.
MoO3−x nanodots with dual enzyme-mimetic activities were synthesized as multifunctional modulators of Aβ fibrillation and neurotoxicity.172 These nanodots exhibited excellent catalase-like and SOD-like activities due to efficient charge transitions between Mo5+/Mo6+ on their surfaces. They effectively inhibited Aβ42 aggregation, altered the peptide's secondary structure from β-sheet to α-helix and random coil, and destabilized preformed Aβ fibrils. The nanodots also protected neuronal cells from Aβ-induced apoptosis and alleviated oxidative stress by decomposing H2O2 and scavenging superoxide radicals. Their biocompatibility and stability make MoO3−x nanodots a promising therapeutic candidate for AD treatment.
Gold nanoparticles (AuNPs) surface-functionalized with mimosine (Mimo-AuNPs), a plant-based amino acid capable of crossing the BBB, were developed to inhibit Aβ fibrillization and reduce neurotoxicity in AD.188 Mimo-AuNPs exhibited enhanced permeability across an in vitro BBB model and dose-dependently inhibited spontaneous and seed-induced aggregation of Aβ1–42, achieving over 90% inhibition at a 1:30 molar ratio. They also promoted disassembly of mature Aβ1–42 fibrils and reduced aggregation of other Aβ isoforms, including familial mutants. In neuronal cells, Mimo-AuNPs mitigated Aβ-induced cytotoxicity, reduced tau phosphorylation, and decreased oxidative stress without eliciting cytotoxic effects, introducing a novel multifunctional nanotherapeutic approach for AD therapy.
Human serum albumin (HSA)-embedded ultrasmall copper nanoclusters (CuNCs@HSA) were also synthesized as multifunctional nanomaterials with remarkable multienzyme-like activities.207 These nanoclusters exhibited superoxide dismutase, catalase, and glutathione peroxidase activities, alongside hydroxyl radical scavenging ability. CuNCs@HSA inhibited Aβ fibrillization with an inhibitory potency 2.5-fold higher than native HSA. They significantly increased the viability of Aβ-treated neuronal cells and mitigated oxidative stress. In vivo studies using transgenic Caenorhabditis elegans demonstrated that CuNCs@HSA effectively suppressed Aβ plaque formation, reduced ROS levels, and extended lifespan. This multifunctional nanomaterial simultaneously inhibits Aβ aggregation, scavenges ROS, and mitigates neuroinflammation, offering a promising therapeutic strategy for AD.
Collectively, these studies highlight the potential of inorganic nanoparticles as multifunctional therapeutic agents for AD, combining inhibitory effects on Aβ aggregation with ROS scavenging capabilities. The integration of nanozyme activities and targeted delivery strategies in these nanoparticles offers promising avenues for the development of effective treatments for AD, addressing multiple pathological aspects of the disease.
Chiral L/D-FexCuySe nanoparticles (NPs), functionalized with D- or L-penicillamine (Pen), were synthesized to interfere with the self-assembly of Aβ42 monomers and promote the disaggregation of dense Aβ42 fibrils into looser monomers under 808 nm NIR illumination (Fig. 8).206 The D-FexCuySe NPs exhibited a binding constant (K) of 2.67 × 105 M−1 to Aβ42 fibrils, which is two times higher than that of L-FexCuySe NPs (1.13 × 105 M−1), indicating a higher affinity due to stereoselective interactions. Under NIR-light irradiation, these chiral FexCuySe NPs generated significant amounts of ROS, including singlet oxygen (1O2) and hydroxyl radicals (˙OH), facilitating rapid disaggregation of Aβ42 fibrils within 10 minutes without photothermal effects. In MN9D neuronal cells, D-NPs attenuated the adhesion of Aβ42 to cell membranes and prevented neuron loss after NIR treatment, resulting in normal neurite growth of 70 ± 10 μm and higher expression levels of neuronal markers TuJ1 and Map2 compared to other treatment groups. In vivo, D-FexCuySe NPs effectively protected against neuronal damage caused by Aβ42 deposition and alleviated symptoms in an AD mouse model, reducing the concentration of Aβ42 in cerebrospinal fluid from 21.06 ng mL−1 to 6.87 ng mL−1-comparable to levels in wild-type mice (6.52 ng mL−1) and facilitated the recovery of cognitive function. The unique design of chiral recognition combined with NIR-triggered ROS generation by the D-FexCuySe NPs offers a safe and efficient therapeutic strategy for AD without observable toxicity in vitro or in vivo. This study presents a novel approach that rapidly disassembles Aβ42 fibrils through chiral recognition and NIR-induced ROS production, achieving therapeutic effects in AD models with unprecedented efficiency. While the results are promising, further investigation is required to assess the long-term effects and potential clinical applicability of these chiral nanoparticles.
 | ||
| Fig. 8 Synthesis of penicillamine-modified FexCuySe nanoparticles (D-Pen FexCuySe) and schematic illustration of their inhibitory and disassembly effects on Aβ42 aggregation, along with the mitigation of potential neurotoxicity in an Alzheimer's disease mouse model.206 | ||
Another approach involved the synthesis of penetratin peptide-loaded polyethylene glycol-stabilized gold nanostars (AuNS) modified with a ruthenium(II) complex (Ru@Pen@PEG–AuNS).156 These multifunctional nanoparticles were designed to inhibit Aβ fibril formation, disaggregate preformed fibrils under NIR irradiation, and improve blood–brain barrier permeability. Upon NIR irradiation, Ru@Pen@PEG–AuNS effectively disaggregated Aβ fibrils and reduced Aβ-induced cytotoxicity in neuronal cells, highlighting their potential as therapeutic agents for AD.
Rose bengal-loaded upconverting nanocomposites were developed as NIR-responsive inhibitors of Aβ aggregation.164 These nanocomposites utilized rattle-structured organosilica shells coated on NaYF4:Yb, Er upconversion nanoparticles (UCNPs) to achieve high rose bengal loading efficiency. Upon 980 nm NIR irradiation, the UCNPs activated rose bengal to generate singlet oxygen, effectively inhibiting Aβ self-assembly and reducing Aβ-induced cytotoxicity in neuronal cells. Moreover, a near-infrared responsive drug delivery system, UCNP@SiO2@Cur/CQ, was designed to enhance the efficacy of polyphenol compounds in AD therapy.232 This system utilized UCNPs coated with silica, onto which curcumin and clioquinol were attached. Sequential release of clioquinol and curcumin was triggered by NIR irradiation, effectively overcoming metal ion interference and enhancing the inhibition of Aβ aggregation and reduction of ROS. In vitro experiments showed that treatment reduced Aβ-Cu2+ aggregate size and decreased intracellular ROS levels. In vivo studies confirmed that nanoparticles crossed the blood–brain barrier and accumulated in the brain. In addition, a NIR-switchable nanoplatform, UCNP@C60-pep, was designed by conjugating upconversion nanoparticles with fullerene C60 and an Aβ-targeting peptide.196 Under NIR light, the UCNPs activated C60 to produce ROS, which oxygenated Aβ, inhibited its aggregation, and reduced cytotoxicity. In the absence of light, UCNP@C60-pep scavenged overproduced ROS, protecting against oxidative stress. In vivo studies in a _C. elegans_ model demonstrated decreased Aβ deposits and improved neurological function. This dual-functionality offers a novel approach by integrating C60's properties in a single system for synergistic AD therapy.
A multifunctional theranostic complex (GAS) was developed by conjugating gold nanorods with thermophilic acylpeptide hydrolase and a single-chain variable fragment antibody to target and degrade Aβ aggregates.173 The GAS complex leveraged the NIR absorption properties of gold nanorods to enable photothermal disassembly of Aβ fibrils, while the enzyme degraded Aβ monomers, and the antibody fragment specifically bound to Aβ oligomers and fibrils to inhibit aggregation. In vitro and in vivo studies demonstrated significant inhibition of Aβ fibril formation and protective effects against Aβ-induced toxicity. A redox-activated NIR-responsive polyoxometalate-based nanoplatform (rPOMDs@MSNs@copolymer) was also developed with high photothermal effect and antioxidant activity.159 Upon NIR laser irradiation, the nanoplatform generated localized hyperthermia, effectively disaggregating Aβ fibrils. The released polyoxometalates exhibited antioxidant activity, scavenging Aβ-induced ROS and inhibiting Aβ aggregation, demonstrating both safety and therapeutic efficacy in vitro. This study is significant as it is the first to employ reduced polyoxometalates for NIR photothermal treatment of AD.
A multifunctional nanoparticle CICe@M–K was developed, incorporating curcumin (Cur), IR780, cerium oxide nanoparticles (CeO2 NPs), and a short peptide K (CKLVFFAED), to simultaneously inhibit β-amyloid (Aβ) aggregation and scavenge ROS (Fig. 9).151 Mesoporous silica nanoparticles (MSNs) served as carriers for Cur and IR780, while CeO2 NPs and peptide K were grafted onto the surface. In vitro assays demonstrated that CICe@M–K effectively inhibited Aβ aggregation, with Thioflavin T fluorescence intensity reduced by approximately 50%, and prevented the formation of Aβ fibrils as observed via electron microscopy. The nanoparticles also exhibited significant antioxidant activity, reducing ROS levels in PC12 cells by over 60% compared to untreated controls, and increased cell viability to nearly 90% by mitigating oxidative stress-induced apoptosis, with the cell apoptosis rate decreasing to 6.54% after treatment. In vivo imaging showed that CICe@M–K crossed the BBB efficiently and accumulated in the brain, liver, and kidneys. In APP/PS1 transgenic AD mouse models, treatment with CICe@M–K improved cognitive abilities, as demonstrated by a reduction in escape latency time from over 50 seconds to approximately 30 seconds in the Morris water maze test, and decreased Aβ deposits and oxidative stress markers in brain tissues. The CICe@M–K nanoparticles, designed to cross the BBB via peptide K modification and release Cur under near-infrared irradiation, function by inhibiting Aβ aggregation and scavenging ROS, leading to improved cognitive function in AD mouse models, as evidenced by significant reductions in Aβ deposition and oxidative stress, while demonstrating good biocompatibility and safety with negligible toxicity in major organs. The significance of this work lies in the development of a multifunctional nanoparticle that addresses multiple pathological factors in AD simultaneously, offering a potential new avenue for treatment. However, further studies are needed to evaluate the long-term safety and efficacy of CICe@M–K in clinical settings.
 | ||
| Fig. 9 Schematic illustration of CICe@M–K nanoparticles in Alzheimer's disease treatment, highlighting their dual role in inhibiting amyloid-β (Aβ) aggregation and scavenging reactive oxygen species (ROS) after crossing the blood–brain barrier.151 | ||
Lastly, KLVFF@Au–CeO2 nanocomposites were developed by coating gold nanorods with ceria nanoparticles and modifying them with an Aβ-targeted inhibitory peptide.158 The spatial separation of ceria nanoparticles improved photocatalytic activity and photothermal conversion efficiency under NIR irradiation. These nanocomposites significantly reduced ROS levels, inhibited Aβ aggregation, and improved cognitive function in an AD mouse model. The design integrates enhanced catalytic activity of CeO2 under NIR irradiation, photothermal therapy, and targeted inhibition of Aβ aggregation, demonstrating safety and therapeutic efficacy. These studies collectively demonstrate the potential of inorganic nanoparticle-based phototherapy approaches in treating Alzheimer's disease. By utilizing the unique properties of inorganic nanoparticles activated by near-infrared light, these strategies offer targeted inhibition or disassembly of Aβ aggregates, reduction of oxidative stress, and improved neuronal survival. Further research and clinical trials are necessary to evaluate the long-term safety and therapeutic efficacy of these promising nanotechnologies.
The researchers developed a library of glutathione (GSH)-responsive silica nanocapsules (SNCs) engineered for brain-targeted delivery of CRISPR genome editors via systemic administration (Fig. 10).111 These SNCs were designed with a GSH-responsive silica network for intracellular release and were surface-functionalized with glucose and rabies virus glycoprotein peptide under glycemic control to facilitate BBB crossing and neuronal targeting. In vivo studies demonstrated that systemically administered SNCs efficiently bypassed the intact BBB, enabling brain-wide delivery of various biologics. Notably, the SNCs achieved up to 28% neuronal editing through systemic delivery of Cre mRNA in Ai14 reporter mice. In wild-type mice, they accomplished up to 6.1% editing of the App gene, resulting in a 19.1% reduction in intact APP expression, and up to 3.9% editing of the tyrosine hydroxylase (Th) gene, leading to a 30.3% decrease in TH expression levels. The safety profile of the SNCs was favorable, with no significant toxicity or inflammatory responses observed in treated animals, as evidenced by histological analysis and cytokine expression levels. The design of these GSH-responsive SNCs, capable of systemic administration for efficient BBB crossing and targeted gene editing, represents a significant advancement in nanoparticle-mediated gene therapy for central nervous system disorders, providing a novel strategy for diseases such as Alzheimer's. However, a major limitation identified was the off-target delivery to peripheral organs and non-neuronal brain cells, indicating a need for further optimization to enhance specificity for neuronal cells and mitigate potential side effects.
 | ||
| Fig. 10 Design of the silica nanoconjugate (SNC) formulation and brain-targeting strategy. (A and B) Schematic illustrations of SNCs formed by silica precursors with (A) four active arms (SNC1) or (B) three active arms (SNC2–8), where R represents a nonhydrolyzable inactive arm. (C) Illustration of the systemic delivery of SNCs into the brain using a dual-targeting ligand strategy.111 | ||
Multifunctional SPIONs were engineered by conjugating an amyloid-beta oligomer (AβO)-specific single-chain variable fragment antibody W20 and a class A scavenger receptor activator peptide XD4 (W20/XD4-SPIONs), aiming to target and clear neurotoxic AβOs.201 The conjugation efficiencies for W20 and XD4 were 50% and 21.3%, respectively, resulting in nanoparticles with a mean diameter of 10.1 ± 1.5 nm. In vitro, W20/XD4-SPIONs inhibited Aβ42 aggregation and attenuated AβO-induced cytotoxicity, increasing SH-SY5Y cell viability by 52.9% compared to AβO-treated controls; they also enhanced microglial phagocytosis of Aβ, reducing inflammatory cytokine production. In APP/PS1 transgenic AD mice, intraperitoneal administration of W20/XD4-SPIONs over 28 days significantly rescued cognitive deficits in Y-maze and Morris water maze tests and alleviated neuropathology. Specifically, W20/XD4-SPIONs reduced GFAP-positive astrocytosis in cortex and hippocampus by 65.2% and 51.6%, respectively, and Iba-1-positive microgliosis by 64.1% and 79.6%; pro-inflammatory cytokines IL-1β, IL-6, and TNF-α levels decreased by 41.7%, 50.1%, and 55.6%, respectively. Oxidative stress was mitigated, as evidenced by increased glutathione (GSH) levels, decreased oxidized glutathione (GSSG), an elevated GSH/GSSG ratio, and reduced ROS levels. Furthermore, W20/XD4-SPIONs reduced Aβ plaque burden and elevated synaptic protein levels of PSD-95 and synaptophysin, indicating synapse preservation. The design of W20/XD4-SPIONs uniquely combines targeted AβO recognition with enhanced microglial clearance mechanisms, demonstrating significant therapeutic benefits and potential diagnostic value in early-stage AD.
5.4. Biomimetic NP
Biomimetic nanoparticles, such as apolipoprotein-reconstituted high-density lipoprotein (rHDL) particles, have been engineered to mimic natural lipoproteins, enhancing BBB crossing and exhibiting high binding affinity to Aβ monomers and oligomers. For instance, ApoE3-rHDL nanoparticles have been shown to facilitate Aβ clearance by promoting microglial uptake and lysosomal degradation, reducing Aβ deposition and improving cognitive function in AD models.243 Similarly, donepezil-loaded ApoA-I rHDL nanoparticles simultaneously target Aβ clearance and inhibit acetylcholinesterase activity, offering a dual therapeutic strategy.244 A monosialotetrahexosylganglioside-modified reconstituted high-density lipoprotein nanoparticle (GM1-rHDL) was designed to bind Aβ with subnanomolar affinity (KD values of 2.13 × 10−10 M for Aβ1–42 monomer and 1.51 × 10−10 M for oligomer), significantly enhancing microglial uptake (increasing cellular uptake from 63.3 to 83.2 fluorescence units) and degradation of Aβ, and promoting its efflux across the blood–brain barrier (BBB) by 32.4% over controls.245 GM1-rHDL displayed increased brain biodistribution efficiency following intranasal administration, with AUCall values in the cortex and hippocampus 85% higher than rHDL. Furthermore, this nanostructure efficiently encapsulated the neuroprotective peptide NAP (encapsulation efficiency of 64.39%) to form the multifunctional platform RNAP-GM1-rHDL. In vitro, RNAP-GM1-rHDL significantly protected primary neurons from Aβ1–42 oligomer/glutamate-induced toxicity at femtomolar concentrations (10−14 M), outperforming GM1-rHDL alone in increasing neurite length and branch points. In AD model mice, intranasal administration of RNAP-GM1-rHDL reduced Aβ deposition by 66% compared to controls, ameliorated neuronal shrinkage observed in histological evaluations, and rescued memory deficits in the Morris water maze test, with no observable cytotoxicity or tissue toxicity. The GM1-rHDL nanoparticle functions by mimicking natural lipoproteins to enhance Aβ clearance via high-affinity binding, facilitating microglial degradation and BBB efflux, while concurrently delivering NAP to protect neurons, resulting in significant therapeutic benefits and safety in AD models. This work demonstrates the novel use of a biomimetic nanostructure as a safe and efficient multifunctional platform for combination therapy in Alzheimer's disease. However, further investigations are needed to assess the long-term safety and potential immunogenicity of GM1-rHDL nanoparticles in clinical settings.Additionally, nanoparticles coated with red blood cell (RBC) or macrophage membranes have been utilized to evade immune clearance and target inflammatory sites. Functionalization with BBB-penetrating ligands and mitochondrial-targeting moieties enables these nanoparticles to deliver therapeutic agents like curcumin directly to neuronal mitochondria, reducing oxidative stress and neuronal apoptosis.229 The incorporation of photothermal or photodynamic therapy using near-infrared irradiation further enhances BBB permeability and facilitates the depolymerization of existing Aβ fibrils. For example, nitrogen-doped carbon quantum dots encapsulated with macrophage membranes can chelate excess metal ions like Cu2+, inhibit Aβ aggregation, and ameliorate neuroinflammation.148 Similarly, red blood cell membrane-encapsulated carbon quantum dots and polydopamine nanoparticles target multiple AD pathological features, demonstrating improved cognitive function in AD models.144
Researchers designed a multifunctional nanocomposite (CQD–Ce–RBC) by growing cerium oxide (CeO2) nanocrystals in situ on red blood cell membranes (RBC) and encapsulating carbon quantum dots (CQDs), combining photothermal therapy (PTT) with antioxidant and metal-chelating properties to address multiple AD pathological targets.146 The RBC membrane enhances biocompatibility and prolongs circulation time, while CeO2 nanocrystals (∼5 nm) exhibit outstanding antioxidant activity by mimicking catalase and superoxide dismutase, scavenging ROS. Nitrogen-doped CQDs act as chelating agents for copper ions (Cu2+), preventing metal-ion-triggered Aβ aggregation, and their strong NIR absorption enables PTT to disrupt existing Aβ fibrils and temporarily open the BBB to improve drug delivery. In vitro experiments demonstrated that CQD–Ce–RBC significantly reduced ROS levels in SH-SY5Y neuronal cells and inhibited Aβ1–42 aggregation, with ROS levels reduced to 159.6% of control compared to 574.9% with Aβ treatment alone. In vivo, APP/PS1 transgenic mice treated with CQD–Ce–RBC combined with NIR irradiation (1.5 W cm−2, 5 min) every 48 hours for 6 weeks showed effective clearance of cerebral amyloid deposits and significant improvements in learning and memory, evidenced by reduced escape latency in the Morris Water Maze and increased central activity in the Open Field Test. Notably, levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 were markedly decreased, indicating reduced neuroinflammation.
In addition, a novel hybrid cell membrane-coated nanoparticle was developed to enhance BBB penetration and target neuroinflammatory lesions characteristic of AD pathology.246 The researchers hybridized membranes from platelets and chemokine (C–C motif) receptor 2 (CCR2)-overexpressing HEK293T cells to create liposomes capable of chemotactic migration toward regions with elevated chemokine ligand 2 (CCL2). Two drugs with different mechanisms, rapamycin (an autophagy enhancer) and 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU, a soluble epoxide hydrolase inhibitor), were co-loaded into these liposomes to achieve multitargeted therapy. In vitro, the dual–drug-loaded hybrid liposomes (TR@CPLs) significantly rescued cell viability in AD models more effectively than single-drug treatments. In 5 × FAD transgenic mice, intravenous administration of TR@CPLs three times a week for six weeks resulted in significant cognitive improvement, as demonstrated by behavioral tests such as the novel object recognition test, where recognition indices increased markedly compared to controls. Additionally, treated mice exhibited a substantial reduction in amyloid plaque deposition in the cortex and hippocampus, and decreased neuroinflammation, evidenced by reduced glial cell infiltration. Biodistribution studies showed that TR@CPLs effectively crossed the BBB, with fluorescence signals in the brain peaking at 12 hours post-injection and accumulated around microglia and astrocytes. Safety assessments, including histological examination of major organs, indicated no apparent abnormalities, suggesting good biocompatibility. The hybrid cell membrane-coated liposomes, designed by fusing platelet membranes with CCR2-overexpressing cell membranes, leveraged chemotactic targeting to neuroinflammatory sites, effectively delivering rapamycin and TPPU across the BBB and achieving significant therapeutic effects without observable toxicity.
Another study hypothesized that flavin mononucleotide (FMN), an intermediate of riboflavin metabolism, inhibits microglial riboflavin kinase (RFK) expression via regulation of lysine-specific methyltransferase 2B (KMT2B), thereby attenuating inflammation-associated cognitive impairment (Fig. 11).247 Researchers designed biomimetic microglial nanoparticles (MNPs@FMN) composed of FMN-encapsulated human serum albumin nanoparticles coated with microglial BV2 cell membranes to improve BBB penetration and microglial-targeted delivery efficiency. In vitro, FMN supplementation decreased RFK expression by approximately 51.2% (fold-change = 0.488, p = 0.0037) and reduced pro-inflammatory cytokines such as Il-1β, Il-6, and TNF-α in lipopolysaccharide (LPS)-treated primary microglia. In vivo, intravenous administration of MNPs@FMN in LPS-induced inflammatory mice and 5 × FAD Alzheimer's disease mice significantly ameliorated cognitive deficits, with a 4.688-fold increase in time spent in the target zone of the Morris water maze compared to the LPS-PBS group (p = 0.0063). MNPs@FMN restored synaptic plasticity, enhancing long-term potentiation amplitude by 29.1% (FC = 1.291, p = 0.0016) at 50–60 minutes post-induction, and reduced hippocampal expression of RFK and pro-inflammatory markers. The design of MNPs@FMN effectively delivers FMN to microglia, inhibits RFK via KMT2B regulation, and attenuates neuroinflammation without detectable pathological changes in major organs or alterations in serum biochemical indicators of liver or kidney function, demonstrating both safety and therapeutic efficacy.
 | ||
| Fig. 11 Schematic illustration of the preparation and mechanism of action of MNPs@FMN in memory protection. (A) Preparation process of biomimetic nanocarriers (MNPs@FMN). (B) Proposed mechanism whereby MNPs@FMN crosses the blood–brain barrier by binding to cell surface receptors on brain endothelial cells, accumulates in microglia, and releases flavin mononucleotide (FMN). The released FMN inhibits riboflavin kinase (RFK) via KMT2B, while RFK promotes the TNFR1/NF-κB signaling pathway. Ultimately, MNPs@FMN restores cognitive function by suppressing the inflammatory response.247 | ||
Biomimetic nanoparticles functionalized with BBB-targeting peptides have also been developed to enhance brain delivery of neuroprotective agents.146 For instance, RBC membrane-coated nanoparticles functionalized with the TGN peptide efficiently deliver curcumin to the brain, resulting in improved therapeutic outcomes in AD models.248 Erythrocyte membrane-modified upconversion nanoparticles loaded with curcumin enhance photodynamic therapy efficiency against Aβ aggregates under near-infrared irradiation.145 Moreover, macrophage membrane-coated molybdenum disulfide quantum dots address multiple AD pathological targets, including ROS scavenging and inhibition of Aβ aggregation.149 Prussian blue nanoparticles encapsulated with biomimetic membranes also chelate excess metal ions, scavenge reactive oxygen species, and ameliorate neuroinflammation, resulting in improved cognitive deficits in AD models.147
Moreover, macrophage (MA) membrane-coated solid lipid nanoparticles (SLNs) were engineered by integrating rabies virus glycoprotein (RVG29) and triphenylphosphine cation (TPP) onto the MA membrane surface, forming RVG/TPP-MASLNs, to deliver the antioxidant genistein (GS) directly to neuronal mitochondria.228 The hypothesis was that this biomimetic nanosystem could evade reticuloendothelial system (RES) clearance, cross the BBB, selectively target neurons, and accumulate in mitochondria to attenuate oxidative stress implicated in AD progression. In vitro experiments demonstrated that RVG/TPP-MASLNs-GS significantly reduced mitochondrial ROS levels in Aβ25–35-damaged HT22 neuronal cells, decreasing apoptosis rates from 50.27% to 14.35%. In vivo, APP/PS1 transgenic mice treated with RVG/TPP-MASLNs-GS showed improved cognitive function, with escape latency in the Morris water maze reduced compared to controls, and a notable reduction in hippocampal Aβ1–42 levels. The design leverages MA membranes for RES evasion, RVG29 for BBB penetration and neuronal targeting, and TPP for mitochondrial targeting, ensuring efficient and safe delivery of GS without inducing toxicity in major organs over 14 days. This work is significant as it presents a multifunctional biomimetic nanosystem capable of targeted delivery to neuronal mitochondria, demonstrating therapeutic efficacy against AD in vitro and in vivo. However, limitations include the need for further evaluation of long-term safety, potential immunogenic responses, and scalability for clinical translation.
These strategies have collectively demonstrated significant improvements in cognitive deficits and neuropathological features in various AD animal models. While these nanotherapeutic approaches offer promising avenues for AD treatment by addressing multiple pathological targets and enhancing brain delivery, further studies are essential to evaluate their long-term safety, pharmacokinetics, and clinical translatability.
5.5. Extracellular vesicles (EVs)
Lesion-recognizing nanoparticles were developed to synergistically deliver small interfering RNAs (siRNAs) targeting β-site amyloid precursor protein cleaving enzyme 1 (BACE1) and caspase-3.110 The nanoparticles consisted of rabies virus glycoprotein peptide (RVG29)-modified mesenchymal stem cell-derived EVs (REXO) as the shell and a ROS-responsive cationic polymer (BAP88) loaded with siRNAs as the core, forming particles approximately 155.5 nm in diameter with a near-neutral ζ potential of −5.78 mV. In vitro, the nanoparticles exhibited low cytotoxicity with cell viabilities above 85.8% in human nasal epithelial and SH-SY5Y neuronal cells at an N/P ratio of 5. After intranasal administration in 3 × Tg-AD mice, the nanoparticles effectively crossed the nasal mucosa and accumulated in the hippocampus, with Cy5-siRNA mass ratios reaching 19.6%. The RVG29 modification facilitated neuronal targeting and membrane fusion, enabling direct cytoplasmic entry and controlled siRNA release in high-ROS environments, leading to significant downregulation of BACE1 and caspase-3 expression. Treatment with these nanoparticles reduced Aβ42 levels, decreased the number of reactive astrocytes by a significant margin, and restored neuronal integrity, as evidenced by Nissl staining. Behavioral assessments demonstrated improved cognitive function, with mice showing a 50% reduction in escape latency during the Morris water maze test compared to untreated controls and spending significantly more time (p < 0.01) in the target quadrant. Importantly, the nanoparticles exhibited no observable toxicity in nasal mucosa, olfactory bulb, or brain tissues, as confirmed by hematoxylin and eosin staining.Recent advancements have also explored extracellular vesicle-based interventions to target pathogenic mechanisms in AD. In one study,249 researchers found that ultrasound stimulation of human astrocytes significantly increased EVrelease-approximately five-fold-which, when delivered to the brains of APP/PS1 transgenic mice via focused ultrasound-mediated blood–brain barrier opening, led to a notable reduction in amyloid plaques and Aβ levels without observable adverse effects. These EVs acted as nanocarriers facilitating Aβ clearance, offering a non-invasive therapeutic approach for AD. Another study250 investigated vesicle-like nanoparticles derived from garlic chives, which were shown to inhibit the NLRP3 inflammasome in primary macrophages and in mouse models. The active component, identified as the phospholipid 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), suppressed NLRP3-mediated inflammation, suggesting potential for treating neuroinflammation associated with AD. While both studies present promising nanoparticle-based strategies for AD treatment, further research is necessary to assess long-term effects and confirm efficacy in AD-specific neuroinflammatory contexts.
5.6. Lipid nanoparticle
Lipid nanoparticles (LNPs) have gained significant attention in the treatment of AD due to their ability to deliver therapeutic agents across the BBB with high efficiency and specificity. LNPs are nano-sized vesicles composed of lipid molecules, capable of encapsulating a variety of therapeutic substances such as RNA. Their biocompatibility and versatile delivery capabilities make them promising candidates for targeting the complex pathophysiological mechanisms of AD.Recent advancements have introduced innovative LNP-based strategies targeting different aspects of AD pathology. One such approach involves the use of LNPs for gene therapy targeting microglial cells to reduce neuroinflammation.127 Multiple lipid nanoparticle (LNP) formulations were evaluated for RNA delivery to microglia, leading to the identification of a lead microglia LNP (MG-LNP) that efficiently transfected human induced pluripotent stem cell-derived microglia-like cells (iMGLs) with minimal toxicity, achieving up to 92 ± 2% PU.1 transcriptional silencing at 500 ng mL−1 within 12 hours without significant cell death. The MG-LNP showed enhanced delivery efficiency to inflammatory iMGLs, indicating preferential uptake by activated microglia. In vivo experiments demonstrated that intraperitoneal injection of MG-LNP resulted in widespread reporter expression across multiple organs, while intracisternal injection into the cerebrospinal fluid led to preferential expression in the brain, with luminescence decreasing by 41 ± 12% and 58 ± 9.7% in the hippocampus and cortex, respectively, after four days. MG-LNP-mediated delivery of siRNA targeting PU.1 significantly reduced PU.1 levels in iMGLs and attenuated neuroinflammation in lipopolysaccharide (LPS)-induced neuroinflammatory mice and CK-p25 transgenic mice that model AD-associated chronic neuroinflammation. Specifically, repeated intrathecal administration reduced hippocampal PU.1-positive cells from 66 ± 10 to 32 ± 17 nuclei per field and decreased microglial activation markers, including IBA1 by 61%, C1Q by 62%, and GFAP by 39%. The design demonstrates a novel MG-LNP capable of delivering siRNA to microglia via intrathecal administration, effectively reducing neuroinflammation with minimal off-target effects, thereby providing a potent vehicle for neuroinflammation-directed gene therapies in AD. The novelty of this work lies in overcoming the challenge of transfecting microglia in vivo using LNPs, paving the way for anti-inflammatory RNA therapeutics targeting microglia.
Another strategy focuses on enhancing senolytic therapy by improving BBB penetration to selectively eliminate senescent cells in the brain. A study251 developed neurotransmitter-derived LNPs loaded with SSK1, a prodrug activated by β-galactosidase, which is upregulated in senescent cells. These SSK1-loaded nanoparticles (SSK1-NPs) demonstrated efficient BBB penetration and selectively induced apoptosis in senescent neuronal cells while sparing healthy cells. In aged AD mouse models, intravenous administration of SSK1-NPs significantly reduced expression of senescence-associated genes, decreased amyloid-beta accumulation, and improved cognitive function.
A third innovative method utilizes biomimetic lipid nanocomposites for a “Drug-Carrier” synergy therapy, simultaneously targeting amyloid-beta clearance and Tau phosphorylation pathways. Researchers designed a nanocomposite252 by grafting apolipoprotein A-I mimetic peptides fused with angiopep-2 onto a lipid nanoparticle loaded with methylene blue (MB) to inhibit Tau aggregation. The optimized nanoparticles effectively crossed the BBB via receptor-mediated transcytosis, targeted microglia for enhanced amyloid-beta clearance, and delivered MB to neurons to inhibit Tau phosphorylation. In AD mouse models, intravenous administration of this nanocomposite significantly alleviated AD symptoms, rescued neuron viability, and improved cognitive functions. This work presents a novel approach that concurrently addresses multiple pathological features of AD using a biomimetic nanocomposite.
6. Future perspectives and conclusions
Alzheimer's disease continues to pose a significant challenge due to its complex pathophysiology and the limitations of current therapeutic approaches, which largely offer symptomatic relief without altering disease progression. Nanoparticle-based delivery systems have emerged as a promising strategy to address these challenges by enhancing drug delivery across the BBB and enabling targeted therapy to affected neuronal tissues (Table 4).| Nanoparticle design | Function of nanoparticle | Reference |
|---|---|---|
| Macrophage membrane-coated solid lipid nanoparticles with RVG29 and TPP | Targeted delivery of antioxidants to neuronal mitochondria across the blood–brain barrier | 228 |
| Donepezil-loaded apolipoprotein A-I-reconstituted HDL (rHDL/Do) | Aβ-targeting clearance and acetylcholinesterase inhibition | 244 |
| Biomimetic microglial nanoparticle (MNPs@FMN) | Enhanced microglial-targeted delivery across the blood–brain barrier | 247 |
| RBC membrane-coated polycaprolactone (PCL) nanoparticles loaded with curcumin and functionalized with TGN peptide | Targeted delivery of curcumin across the blood–brain barrier for Alzheimer's disease therapy | 248 |
| Erythrocyte membrane-modified core–shell upconversion nanoparticle loaded with curcumin | Biomimetic nanobait to improve photodynamic therapy efficiency | 145 |
| RBC membrane-camouflaged human serum albumin nanoparticles bearing T807 and TPP | Targeted delivery of antioxidants to neuronal mitochondria | 229 |
| Hybrid cell membrane-coated liposomes with membranes from platelets and CCR2 cells | Targeted drug delivery across the blood–brain barrier to neuroinflammatory lesions | 246 |
| Design includes upconversion nanoparticles and a chelating agent to create a nanoprobe capable of detecting and capturing Cu(2+) ions | Detects and captures Cu(2+) ions and enables upconversion luminescence imaging | 150 |
| Mesoporous silica nanomaterials loaded with curcumin and IR780, grafted with cerium oxide nanoparticles and peptide K | Inhibiting β-amyloid aggregation and scavenging reactive oxygen species (ROS) | 151 |
| Hybrid nanodrug (AuM) composed of memantine attached via polymer linkers to a gold nanoparticle | Selective inhibition of extrasynaptic NMDARs and neuroprotection | 253 |
| Porous silicon nanoparticles functionalized with biotin-polyethylene glycol and loaded with ANA | Targeted delivery to AD brain and amyloid-beta plaque disaggregation | 155 |
| Custom-made peptide dendrimers conjugated to star-shaped and spherical gold nanostructures (H3/H6–AuNS/AuNP) | Delivery of neuroprotectants and protection of neurons | 160 |
| RB-loaded upconverting nanocomposites with rattle-structured organosilica shell on NaYF4:Yb, Er nanocrystals | NIR-responsive inhibitor of Aβ aggregation and suppressor of Aβ-induced cytotoxicity | 164 |
| EGCG-stabilized selenium nanoparticles coated with Tet-1 peptide (Tet-1-EGCG@Se) | Inhibits Aβ fibrillation and disaggregates Aβ fibrils | 167 |
| Small-sized Pd hydride (PdH) nanoparticles | High payload and sustained release of hydrogen | 170 |
| Res-selenium-peptide nanocomposite (TGN-Res@SeNPs) | Eliminates Aβ aggregate-induced neurotoxicity and mitigates gut microbiota imbalance | 171 |
| UCNP@SiO2@Cur/CQ | Controlled sequential drug release, Cu2+ chelation, and ROS removal | 232 |
| Mesoporous nano-selenium (MSe) release delivery system (MSe-Res/Fc-β-CD/Bor) based on borneol target and β-cyclodextrin nanovalves | Controlled release and targeted delivery across the blood–brain barrier | 176 |
| Magnetic nanoparticles conjugated with ΛA | Aβ clearance | 179 |
| Magnetic nanoparticles conjugated with D-TLKIVWC (7-DP) | Facilitate transport across the blood–brain barrier | 227 |
| H2O2 responsive controlled-release mesoporous silica nanoparticles (MSNs) | Controlled release of AD therapeutic metal chelator in response to H2O2 levels | 183 |
| Tg–CS/DMY@SeNPs | Inhibit Aβ aggregation and reduce inflammatory cytokines | 185 |
| Quercetin modified polysorbate 80-coated AuPd core–shell structure | Activate autophagy and promote amyloid-β clearance | 187 |
| Gold and platinum nanoparticles coated with multiple ligands | Increase binding affinity of Aβ-specific small molecules to inhibit Aβ peptide aggregation | 193 |
| Protoporphyrin IX (PX)-modified oxidized mesoporous carbon nanospheres (PX@OMCN@PEG(OP)@RVGs) | Inhibits tau phosphorylation and amyloid beta aggregation, enhances blood–brain barrier permeability | 194 |
| Congo red/Rutin-MNPs | Diagnosis and treatment of Alzheimer's disease | 235 |
| UCNP@C60-pep (upconversion nanoparticle and Aβ-target peptide KLVFF) | Near-infrared-switchable ROS producer and scavenger, Aβ-targeting, and imaging capabilities | 196 |
| Gold nanoparticle-capped mesoporous silica (MSN-AuNPs)-based H2O2-responsive controlled release system | Targeted delivery of metal chelator CQ and inhibition of Aβ aggregation | 197 |
| Curcumin and SPIO nanoparticles encapsulated by DSPE–PEG and modified with CRT and QSH peptides | Early diagnosis via MRI and therapeutic intervention by reducing β-amyloid plaque burden | 204 |
| Neurotransmitter-derived lipidoid (NT-lipidoid) nanoparticle loaded with cationic tau-targeting PROTAC via DNA-intercalation | Delivery of tau-targeting PROTAC across the blood–brain barrier to induce tau protein degradation | 254 |
| Pan@TRF@Liposome NPs | Modulating CRM1-mediated PKM2 nuclear translocation | 94 |
| Liposomal nanoparticles coupled with PEG encapsulating astaxanthin | Enhance water solubility of ATX and scavenge formaldehyde reducing amyloid-beta deposition | 95 |
| Curcumin-decorated nanoliposomes maintaining planar structure | Vectors for targeted delivery of diagnostic and therapeutic molecules | 255 |
| Lipid nanoparticles loaded with Aβ peptides and rapamycin | Effectively delivers rapamycin and Aβ peptides to dendritic cells | 256 |
| Liposomal nanodrug incorporating felodipine | Blood–brain barrier crossing drug delivery assisted by low-intensity pulse ultrasound | 93 |
| Liposomes and solid lipid nanoparticles functionalized with phosphatidic acid and cardiolipin | Targeted delivery of diagnostic and therapeutic molecules | 257 |
| Integrated ceria nanozymes into MOFs loaded with siSOX9 and RA | Promotes neuron differentiation and eliminates ROS | 113 |
| Fe-MIL-88B-NH2-NOTA-DMK6240/MB | Magnetic resonance imaging and inhibition of tau aggregation | 258 |
| Inhibitor-conjugated NIR laser-propelled Janus nanomotor (JNM-I) | Modulation of amyloid-β aggregation | 211 |
| Nanovehicles (nanoparticles-IgG4.1) loaded with imaging agents and therapeutic agents | Target cerebrovascular amyloid deposits for diagnostic imaging and drug delivery | 213 |
| Metformin-based supramolecular nanodrugs | Selective penetration of BBB and Aβ-responsive on-demand drug release | 259 |
| Traceable nano-biohybrid complexes loaded with CRISPR/Cas9 plasmids | Efficient delivery of CRISPR-chem drugs into brain lesions and accurate imaging | 115 |
| Stepwise metal-phenolic coordination of rhein and polydopamine to create K8@Fe–Rh/Pda nanoparticles | Inhibit Aβ aggregation, repair neuronal damage, promote mitochondrial biogenesis, and inhibit neuronal apoptosis | 216 |
| Biocompatible metal-phenolic network (MPN) with EGCG and Zn(II) on gold nanoparticles | Inhibits amyloid beta aggregation and toxicity, crosses blood–brain barrier | 217 |
| Carbon nitride dots (CNDs) and black carbon dots (B-CDs) conjugated with memantine hydrochloride (MH) | Delivery of memantine hydrochloride across the blood–brain barrier and inhibition of tau aggregation | 260 |
| Human serum albumin encapsulated quercetin (HSA@QC) nanoparticles | Natural phyto-antioxidant albumin nanoagent for treating advanced Alzheimer's disease | 238 |
| Nanoformulation of α-mangostin | Efficiently delivers α-mangostin to microglia to rejuvenate their clearance capacity | 261 |
| Poly(lactide-co-glycolide) (PLGA) and polyethylene glycol (PEG)-2000 based biodegradable nanoparticles | Encapsulation of anthocyanins to enhance bioavailability and stability | 99 |
| PEGylated poly(lactic acid) polymer conjugated with TGN and QSH peptides at a maleimide/peptide molar ratio of 3 (T3Q3-NP) | Enhanced and precise targeted delivery to amyloid plaque in the brains of AD model mice | 262 |
| Curcumin-encapsulated PLGA nanoparticles | Induce neurogenesis by promoting neural stem cell proliferation and neuronal differentiation | 263 |
| Clioquinol and donepezil co-encapsulated human serum albumin (HSA) nanoparticles modified with TAT and GM1 | Inhibit Aβ aggregation, regulate acetylcholine imbalance, and enhance brain delivery | 139 |
| LK7-conjugated poly(lactic-co-glycolic acid) nanoparticles | Inhibition of Aβ42 aggregation and reduction of cytotoxicity | 136 |
| PEGylated, biodegradable poly(alkyl cyanoacrylate) polymeric nanoparticles functionalized with CuAAC, rhodamine B dye, and targeting ligands | Drug delivery and specific targeting of Aβ(1–42) species in Alzheimer's disease | 134 |
| Targeted multimodal polyglutamate-based nanoconjugate with propargylamine, bisdemethoxycurcumin/genistein, and Angiopep-2 moiety | Neuroprotection, neurotrophic effects, BBB passage enhancement, reduction of β amyloid aggregates | 143 |
| Tween 80 coated polylactide-co-glycolide (PLGA) nanoparticles | Oral delivery of estradiol to the brain | 264 |
| Mitochondria-targeted polymeric nanoparticle system based on PLGA-b-PEG–TPP blended with PLGA-b-PEG–OH or PLGA–COOH | Efficient delivery of mitochondria-acting therapeutics to the mitochondrial matrix | 240 |
| Biodegradable PLGA nanoparticles loaded with memantine and surface-coated with polyethylene glycol | Targeted delivery across the blood–brain barrier with controlled release | 265 |
| PEG–PLA nanoparticles loaded with α-mangostin | Increase LDL receptor expression in microglia and improve clearance of amyloid beta | 266 |
| Chitosan/TPP nanoparticles loaded with Resveratrol and modified with TG peptide | Delivery of Resveratrol to the brain | 241 |
| PLGA–PEG skeleton loaded with fingolimod and modified with mannose | Oral brain-targeting delivery of fingolimod for Alzheimer's treatment | 267 |
| Negatively charged polymeric nanoparticles (NP10) | Inhibits primary nucleation of Aβ aggregation and enhances EGCG binding to Aβ | 137 |
| Hybrid peptide VLC (VHS + COG1410) conjugated to curcumin via phenylboronic ester bond | Targeted delivery to pericyte lesions and release upon ROS stimulation | 106 |
| Dual-drug loaded PEGylated PLGA nanoparticles (EGCG/AA NPs) | Increased stability and bioavailability of EGCG, enabling accumulation in major organs including the brain | 100 |
| PEGylated dendrigraft poly-L-lysines (DGLs) modified with Aleuria aurantia lectin (AAL) and β-amyploid (Aβ)-binding peptides (KLVFF) | Co-delivery of BACE1 siRNA and rapamycin into the brain | 126 |
| Poly(ethylene glycol)–poly(L-lactide) (PEG–PLA) nanoparticles | Improved biodistribution and facilitated Aβ clearance | 108 |
| Amorphous PDLLA-dextran bottlebrush with controlled graft density and side chain length forming micelles, vesicles, and compound vesicles | Codelivery of hydrophilic antioxidants (citric acid, vitamin C, gallic acid) | 109 |
| Endogenous apolipoprotein A-I and mimicking peptide 4F fused angiopep-2 grafted onto lipid nanocomposite with methylene blue | Crosses BBB, targets Aβ clearance, inhibits tau aggregation | 252 |
| SSK1-loaded neurotransmitter-derived lipid nanoparticles | Facilitates BBB penetration and delivery of SSK1 to eliminate senescent cells | 251 |
Future research should prioritize the development of nanoparticles with improved safety profiles. Utilizing biocompatible and biodegradable materials can reduce toxicity and immunogenicity, which are major barriers to clinical application. Surface modifications, such as coating with polyethylene glycol (PEGylation), can further enhance biocompatibility by minimizing unintended interactions with the immune system and prolonging circulation time. Enhancing targeting precision is another critical area for advancement. Functionalizing nanoparticles with ligands or antibodies specific to Alzheimer's disease biomarkers—such as amyloid-beta plaques and tau protein tangles—can significantly improve targeting efficiency. This specificity not only increases therapeutic efficacy but also minimizes off-target effects, thereby reducing potential side effects. Optimizing the pharmacokinetic properties of nanoparticles is essential to maximize therapeutic benefits. Adjusting parameters like size, shape, and surface charge influences their distribution, circulation time, and clearance rates. A thorough understanding of these factors allows for the design of nanoparticles with favorable absorption, distribution, metabolism, and excretion (ADME) properties, enhancing their overall effectiveness. In adiditon, the integration of theranostic nanoparticles offers a dual-function platform that combines therapeutic and diagnostic capabilities. By incorporating imaging agents, these nanoparticles enable real-time monitoring of drug delivery and treatment response, which supports the development of personalized treatment strategies. This approach not only enhances therapeutic outcomes but also aids in early detection and intervention, which are crucial for managing Alzheimer's disease.
One of the primary clinical translation challenges is the intricate nano-bio interactions within the human body.268 After administration, NPs encounter various biological molecules, leading to the formation of a protein corona that can alter their physicochemical properties. This modification may affect the NPs' ability to cross the BBB, target specific brain regions, and might trigger immune responses or unintended side effects. Additionally, the heterogeneity of AD pathology complicates the targeting of NPs, as the disease affects different brain regions and progresses differently among patients. Nanotoxicity is another concern impeding clinical translation. The long-term effects of NPs in the brain are not fully understood, and there is a risk of accumulation leading to neuroinflammation or cytotoxicity. Rigorous safety evaluations are required to assess the biocompatibility, biodegradability, and potential adverse effects of NPs. Regulatory agencies demand comprehensive toxicity studies, which can be time-consuming and costly, slowing down the development process.
Manufacturing challenges also play a role. Scaling up NP production while maintaining consistent quality, stability, and reproducibility is difficult. Complex synthesis methods may lead to batch-to-batch variations, affecting the efficacy and safety of the nanomedicine. Moreover, stringent regulatory standards necessitate robust quality control measures, adding to the complexity and cost of development. Regulatory challenges are significant in the context of nanomedicine for AD. Given the novel properties of NPs, existing regulatory frameworks may not be fully applicable. There is often a lack of standardized guidelines for evaluating nanomedicines, leading to uncertainties in the approval process. Regulatory agencies require detailed characterization of NPs, including their physicochemical properties, pharmacokinetics, and interactions with biological systems. Demonstrating the safety and efficacy of nanomedicines in clinical trials is complicated by the need for specialized assessment techniques and endpoints relevant to AD.
In conclusion, while nanomedicine holds great promise for advancing AD treatment, numerous clinical translation challenges exist. Collaboration among researchers, clinicians, and industry stakeholders is crucial for accelerating the translation of nanoparticle-based therapies from the laboratory to clinical practice. Rigorous preclinical and clinical studies are necessary to address challenges related to toxicity, targeting precision, and regulatory approval processes. By overcoming these hurdles, nanoparticle technologies hold significant promise for developing effective treatments that can alter the course of Alzheimer's disease.
In conclusion, nanoparticle-based delivery systems offer a transformative approach for the treatment and diagnosis of Alzheimer's disease. Their ability to enhance drug stability, bioavailability, and targeted delivery across the BBB—combined with the therapeutic potential of new drugs—offers a powerful strategy to combat this debilitating condition. Continued innovation and interdisciplinary collaboration are critical to realizing the full potential of these technologies, ultimately providing new hope for individuals affected by Alzheimer's disease.
Author contributions
Liqin Liu (investigation; writing – original draft; writing – review & editing); Haini He (investigation; writing – original draft); Bin Du and Yang He (funding acquisition; investigation; project administration; writing – original draft; writing – review & editing).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (No. GZC20241118), and the Fundamental Research Funds for the Central University (No. SCU2022D006). The authors have no acknowledgments to report.References
- M. Zvěřová, Clinical aspects of Alzheimer's disease, Clin. Biochem., 2019, 72, 3–6, DOI:10.1016/j.clinbiochem.2019.04.015.
- A. P. Skaria, The economic and societal burden of Alzheimer disease: managed care considerations, Am. J. Manag. Care, 2022, 28(10 Suppl), S188–S196, DOI:10.37765/ajmc.2022.89236.
- A. A. Rostagno, Pathogenesis of Alzheimer’s Disease, Int. J. Mol. Sci., 2023, 24(1), 107, DOI:10.3390/ijms24010107.
- J. Zhang, Y. Zhang, J. Wang, Y. Xia, J. Zhang and L. Chen, Recent advances in Alzheimer's disease: Mechanisms, clinical trials and new drug development strategies, Signal Transduct. Targeted Ther., 2024, 9(1), 211, DOI:10.1038/s41392-024-01911-3.
- S. Salloway and J. Cummings, Aducanumab, Amyloid Lowering, and Slowing of Alzheimer Disease, Neurology, 2021, 97(11), 543–544, DOI:10.1212/WNL.0000000000012451.
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, The antibody aducanumab reduces Aβ plaques in Alzheimer's disease, Nature, 2016, 537(7618), 50–56, DOI:10.1038/nature19323.
- S. Salloway, M. Farlow, E. McDade, D. B. Clifford, G. Wang, J. J. Llibre-Guerra, J. M. Hitchcock, S. L. Mills, A. M. Santacruz, A. J. Aschenbrenner, J. Hassenstab, T. L. S. Benzinger, B. A. Gordon, A. M. Fagan, K. A. Coalier, C. Cruchaga, A. A. Goate, R. J. Perrin, C. Xiong, Y. Li, J. C. Morris, B. J. Snider, C. Mummery, G. M. Surti, D. Hannequin, D. Wallon, S. B. Berman, J. J. Lah, I. Z. Jimenez-Velazquez, E. D. Roberson, D. van, H. Christopher, L. S. Honig, R. Sánchez-Valle, W. S. Brooks, S. Gauthier, D. R. Galasko, C. L. Masters, J. R. Brosch, G.-Y. R. Hsiung, S. Jayadev, M. Formaglio, M. Masellis, R. Clarnette, J. Pariente, B. Dubois, F. Pasquier, C. R. Jack, R. Koeppe, P. J. Snyder, P. S. Aisen, R. G. Thomas, S. M. Berry, B. A. Wendelberger, S. W. Andersen, K. C. Holdridge, M. A. Mintun, R. Yaari, J. R. Sims, M. Baudler, P. Delmar, R. S. Doody, P. Fontoura, C. Giacobino, G. A. Kerchner and R. J. Bateman, A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease, Signal Transduct. Targeted Ther., 2021, 27(7), 1187–1196, DOI:10.1038/s41591-021-01369-8.
- A. J. Espay, K. P. Kepp and K. Herrup, Lecanemab and Donanemab as Therapies for Alzheimer’s Disease: An Illustrated Perspective on the Data, eNeuro, 2024, 11(7), ENEURO.0319-23.2024, DOI:10.1523/ENEURO.0319-23.2024.
- R. I. Teleanu, M. D. Preda, A.-G. Niculescu, O. Vladâcenco, C. I. Radu, A. M. Grumezescu and D. M. Teleanu, Current Strategies to Enhance Delivery of Drugs across the Blood–Brain Barrier, Pharmaceutics, 2022, 14(5), 987, DOI:10.3390/pharmaceutics14050987.
- A. Chakraborty, N. M. de Wit, W. M. van der Flier and H. E. de Vries, The blood–brain barrier in Alzheimer's disease, Vasc. Pharmacol., 2017, 89, 12–18, DOI:10.1016/j.vph.2016.11.008.
- P. Poudel and S. Park, Recent Advances in the Treatment of Alzheimer’s Disease Using Nanoparticle-Based Drug Delivery Systems, Pharmaceutics, 2022, 14(4), 835, DOI:10.3390/pharmaceutics14040835.
- C.-T. Lu, Y.-Z. Zhao, H. L. Wong, J. Cai, L. Peng and X.-Q. Tian, Current approaches to enhance CNS delivery of drugs across the brain barriers, Int. J. Nanomed., 2014, 9, 2241–2257, DOI:10.2147/IJN.S61288.
- W. M. Pardridge, Blood–brain barrier delivery, Drug discovery today, 2007, 12(1–2), 54–61, DOI:10.1016/j.drudis.2006.10.013.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases, J. Controlled Release, 2016, 235, 34–47, DOI:10.1016/j.jconrel.2016.05.044.
- Y. Zhou, Z. Peng, E. S. Seven and R. M. Leblanc, Crossing the blood–brain barrier with nanoparticles, J. Controlled Release, 2018, 270, 290–303, DOI:10.1016/j.jconrel.2017.12.015.
- X. Li, J. Tsibouklis, T. Weng, B. Zhang, G. Yin, G. Feng, Y. Cui, I. N. Savina, L. I. Mikhalovska, S. R. Sandeman, C. A. Howel and S. V. Mikhalovsky, Nano carriers for drug transport across the blood–brain barrier, J. Drug Targeting, 2017, 25(1), 17–28, DOI:10.1080/1061186X.2016.1184272.
- J. K. Sahni, S. Doggui, J. Ali, S. Baboota, L. é Dao and C. Ramassamy, Neurotherapeutic applications of nanoparticles in Alzheimer's disease, J. Controlled Release, 2011, 152(2), 208–231, DOI:10.1016/j.jconrel.2010.11.033.
- A. Cano, P. Turowski, M. Ettcheto, J. T. Duskey, G. Tosi, E. Sánchez-López, M. L. García, A. Camins, E. B. Souto, A. Ruiz, M. Marquié and M. Boada, Nanomedicine-based technologies and novel biomarkers for the diagnosis and treatment of Alzheimer's disease: from current to future challenges, J. Nanobiotechnol., 2021, 19(1), 122, DOI:10.1186/s12951-021-00864-x.
- T. R. Hunter, L. E. Santos, F. Tovar-Moll, F. De and G. Fernanda, Alzheimer's disease biomarkers and their current use in clinical research and practice, Mol. Psychiatry, 2025, 30(1), 272–284, DOI:10.1038/s41380-024-02709-z.
- D. J. Selkoe and J. Hardy, The amyloid hypothesis of Alzheimer's disease at 25 years, EMBO Mol. Med., 2016, 8(6), 595–608, DOI:10.15252/emmm.201606210.
- R. J. O'Brien and P. C. Wong, Amyloid precursor protein processing and Alzheimer's disease, Annu. Rev. Neurosci., 2011, 34, 185–204, DOI:10.1146/annurev-neuro-061010-113613.
- B. Allinquant, C. Clamagirand and P. Marie-Claude, Role of cholesterol metabolism in the pathogenesis of Alzheimer's disease, Curr. Opin. Clin. Nutr. Metab. Care, 2014, 17(4), 319–323, DOI:10.1097/MCO.0000000000000069.
- S. Carmona, J. Hardy and R. Guerreiro, The genetic landscape of Alzheimer disease, Handb. Clin. Neurol., 2018, 148, 395–408, DOI:10.1016/B978-0-444-64076-5.00026-0.
- D. R. Thal, U. Rüb, M. Orantes and H. Braak, Phases of A beta-deposition in the human brain and its relevance for the development of AD, Neurology, 2002, 58(12), 1791–1800, DOI:10.1212/WNL.58.12.1791.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade, J. Alzheimer's Dis., 2018, 64(s1), S567–S610, DOI:10.3233/JAD-179941.
- L. Mucke, Neuroscience: Alzheimer's disease, Nature, 2009, 461(7266), 895–897, DOI:10.1038/461895a.
- C. Haass and D. J. Selkoe, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nature reviews, Mol. Cell Biol., 2007, 8(2), 101–112, DOI:10.1038/nrm2101.
- K. S. Kosik, C. L. Joachim and D. J. Selkoe, Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease, Proc. Natl. Acad. Sci. U. S. A., 1986, 83(11), 4044–4048, DOI:10.1073/pnas.83.11.4044.
- N. Nukina and Y. Ihara, One of the antigenic determinants of paired helical filaments is related to tau protein, J. Biochem., 1986, 99(5), 1541–1544, DOI:10.1093/oxfordjournals.jbchem.a135625.
- J. G. Wood, S. S. Mirra, N. J. Pollock and L. I. Binder, Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau), Proc. Natl. Acad. Sci. U. S. A., 1986, 83(11), 4040–4043, DOI:10.1073/pnas.83.11.4040.
- S. Dujardin and B. T. Hyman, Tau Prion-Like Propagation: State of the Art and Current Challenges, Adv. Exp. Med. Biol., 2019, 1184, 305–325, DOI:10.1007/978-981-32-9358-8_23.
- P. d'Errico and M. Meyer-Luehmann, Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer's Disease, Front. Aging Neurosci., 2020, 12, 265, DOI:10.3389/fnagi.2020.00265.
- S. Prokop, K. R. Miller, S. R. Labra, R. M. Pitkin, K. Hoxha, S. Narasimhan, L. Changolkar, A. Rosenbloom, V. M.-Y. Lee and J. Q. Trojanowski, Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer's disease patient brain samples, Acta Neuropathol., 2019, 138(4), 613–630, DOI:10.1007/s00401-019-02048-2.
- P. Barbier, O. Zejneli, M. Martinho, A. Lasorsa, V. Belle, C. Smet-Nocca, P. O. Tsvetkov, F. Devred and I. Landrieu, Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects, Front. Aging Neurosci., 2019, 11, 204, DOI:10.3389/fnagi.2019.00204.
- E. Y. Hayden and D. B. Teplow, Amyloid β-protein oligomers and Alzheimer's disease, Alzheimer's Res. Ther., 2013, 5(6), 60, DOI:10.1186/alzrt226.
- E. Tönnies and E. Trushina, Oxidative Stress, Synaptic Dysfunction, and Alzheimer's Disease, J. Alzheimer's Dis., 2017, 57(4), 1105–1121, DOI:10.3233/JAD-161088.
- W.-J. Huang, X. Zhang and W.-W. Chen, Role of oxidative stress in Alzheimer's disease, Biomed. Rep., 2016, 4(5), 519–522, DOI:10.3892/br.2016.630.
- B. V. Zlokovic, Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders, Nat. Rev. Neurosci., 2011, 12(12), 723–738, DOI:10.1038/nrn3114.
- M. Sochocka, E. S. Koutsouraki, K. Gasiorowski and J. Leszek, Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy, CNS Neurol. Disord. – Drug Targets, 2013, 12(6), 870–881, DOI:10.2174/18715273113129990072.
- Y.-C. Kao, P.-C. Ho, Y.-K. Tu, I.-M. Jou and K.-J. Tsai, Lipids and Alzheimer’s Disease, Int. J. Mol. Sci., 2020, 21(4), 1505, DOI:10.3390/ijms21041505.
- I. J. Martins, E. Hone, J. K. Foster, S. I. Sünram-Lea, A. Gnjec, S. J. Fuller, D. Nolan, S. E. Gandy and R. N. Martins, Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer's disease and cardiovascular disease, Mol. Psychiatry, 2006, 11(8), 721–736, DOI:10.1038/sj.mp.4001854.
- Z. Korade and A. K. Kenworthy, Lipid rafts, cholesterol, and the brain, Neuropharmacology, 2008, 55(8), 1265–1273, DOI:10.1016/j.neuropharm.2008.02.019.
- C.-C. Liu, C.-C. Liu, T. Kanekiyo, H. Xu and G. Bu, Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy, Nat. Rev. Neurol., 2013, 9(2), 106–118, DOI:10.1038/nrneurol.2012.263.
- C. M. Karch and A. M. Goate, Alzheimer's disease risk genes and mechanisms of disease pathogenesis, Biol. Psychiatry, 2015, 77(1), 43–51, DOI:10.1016/j.biopsych.2014.05.006.
- P. B. Verghese, J. M. Castellano and D. M. Holtzman, Apolipoprotein E in Alzheimer's disease and other neurological disorders, Lancet Neurol., 2011, 10(3), 241–252, DOI:10.1016/S1474-4422(10)70325-2.
- E. E. Tuppo and H. R. Arias, The role of inflammation in Alzheimer's disease, Int. J. Biochem. Cell Biol., 2005, 37(2), 289–305, DOI:10.1016/j.biocel.2004.07.009.
- L. Mezzaroba, D. F. Alfieri, C. Simão, A. Name, V. Reiche and E. Maria, The role of zinc, copper, manganese and iron in neurodegenerative diseases, Neurotoxicology, 2019, 74, 230–241, DOI:10.1016/j.neuro.2019.07.007.
- L. Wang, Y.-L. Yin, X.-Z. Liu, P. Shen, Y.-G. Zheng, X.-R. Lan, C.-B. Lu and J.-Z. Wang, Current understanding of metal ions in the pathogenesis of Alzheimer's disease, Transl. Neurodegener., 2020, 9, 10, DOI:10.1186/s40035-020-00189-z.
- D. H. Upton, C. Ung, S. M. George, M. Tsoli, M. Kavallaris and D. S. Ziegler, Challenges and opportunities to penetrate the blood–brain barrier for brain cancer therapy, Theranostics, 2022, 12(10), 4734–4752, DOI:10.7150/thno.69682.
- M. Segarra, M. R. Aburto and A. Acker-Palmer, Blood–Brain Barrier Dynamics to Maintain Brain Homeostasis, Trends Neurosci., 2021, 44(5), 393–405, DOI:10.1016/j.tins.2020.12.002.
- W. M. Pardridge, CNS drug design based on principles of blood–brain barrier transport, J. Neurochem., 1998, 70(5), 1781–1792, DOI:10.1046/j.1471-4159.1998.70051781.x.
- C. W. Fong, Permeability of the Blood–Brain Barrier: Molecular Mechanism of Transport of Drugs and Physiologically Important Compounds, J. Membr. Biol., 2015, 248(4), 651–669, DOI:10.1007/s00232-015-9778-9.
- A. Bhattacharya, D. K. Kaushik, B. M. Lozinski and V. W. Yong, Beyond barrier functions: Roles of pericytes in homeostasis and regulation of neuroinflammation, J. Neurosci. Res., 2020, 98(12), 2390–2405, DOI:10.1002/jnr.24715.
- E. G. Knox, M. R. Aburto, G. Clarke, J. F. Cryan and C. M. O'Driscoll, The blood–brain barrier in aging and neurodegeneration, Mol. Psychiatry, 2022, 27(6), 2659–2673, DOI:10.1038/s41380-022-01511-z.
- M. D. Sweeney, Z. Zhao, A. Montagne, A. R. Nelson and B. V. Zlokovic, Blood–Brain Barrier: From Physiology to Disease and Back, Physiol. Rev., 2019, 99(1), 21–78, DOI:10.1152/physrev.00050.2017.
- M. I. Teixeira, C. M. Lopes, M. H. Amaral and P. C. Costa, Current insights on lipid nanocarrier-assisted drug delivery in the treatment of neurodegenerative diseases, Eur. J. Pharm. Biopharm., 2020, 149, 192–217, DOI:10.1016/j.ejpb.2020.01.005.
- J. A. Endicott and V. Ling, The biochemistry of P-glycoprotein-mediated multidrug resistance, Annu. Rev. Biochem., 1989, 58, 137–171, DOI:10.1146/annurev.bi.58.070189.001033.
- M. F. Fromm, Importance of P-glycoprotein at blood-tissue barriers, Trends Pharmacol. Sci., 2004, 25(8), 423–429, DOI:10.1016/j.tips.2004.06.002.
- U. H. Langen, S. Ayloo and C. Gu, Development and Cell Biology of the Blood–Brain Barrier, Annu. Rev. Cell Dev. Biol., 2019, 35, 591–613, DOI:10.1146/annurev-cellbio-100617-062608.
- W. M. Pardridge, Drug and gene targeting to the brain with molecular Trojan horses, Nat. Rev. Drug Discov., 2002, 1(2), 131–139, DOI:10.1038/nrd725.
- B. Nabi, S. Rehman, S. Khan, S. Baboota and J. Ali, Ligand conjugation: An emerging platform for enhanced brain drug delivery, Brain Res. Bull., 2018, 142, 384–393, DOI:10.1016/j.brainresbull.2018.08.003.
- M. Choudhari, S. Hejmady, N. S. Ranendra, S. Damle, G. Singhvi, A. Alexander, P. Kesharwani and S. Kumar Dubey, Evolving new-age strategies to transport therapeutics across the blood-brain-barrier, Int. J. Pharm., 2021, 599, 120351, DOI:10.1016/j.ijpharm.2021.120351.
- Y. Anraku, H. Kuwahara, Y. Fukusato, A. Mizoguchi, T. Ishii, K. Nitta, Y. Matsumoto, K. Toh, K. Miyata, S. Uchida, K. Nishina, K. Osada, K. Itaka, N. Nishiyama, H. Mizusawa, T. Yamasoba, T. Yokota and K. Kataoka, Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain, Nat. Commun., 2017, 8(1), 1001, DOI:10.1038/s41467-017-00952-3.
- B. Qu, X. Li, M. Guan, X. Li, L. Hai and Y. Wu, Design, synthesis and biological evaluation of multivalent glucosides with high affinity as ligands for brain targeting liposomes, Eur. J. Med. Chem., 2014, 72, 110–118, DOI:10.1016/j.ejmech.2013.10.007.
- R. I. Henkin, Intranasal delivery to the brain, Nat. Biotechnol., 2011, 29(6), 480, DOI:10.1038/nbt.1866.
- Y. Liu, Y. Gong, W. Xie, A. Huang, X. Yuan, H. Zhou, X. Zhu, X. Chen, J. Liu, J. Liu and X. Qin, Microbubbles in combination with focused ultrasound for the delivery of quercetin-modified sulfur nanoparticles through the blood–brain barrier into the brain parenchyma and relief of endoplasmic reticulum stress to treat Alzheimer's disease, Nanoscale, 2020, 12(11), 6498–6511, 10.1039/C9NR09713A.
- D.-D. Li, Y.-H. Zhang, W. Zhang and P. Zhao, Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer's Disease, Front. Neurosci., 2019, 13, 472, DOI:10.3389/fnins.2019.00472.
- T. T. Nguyen, T. T. D. Nguyen, T. K. O. Nguyen, T. K. Vo and V. G. Vo, Advances in developing therapeutic strategies for Alzheimer's disease, Biomed. Pharmacother., 2021, 139, 111623, DOI:10.1016/j.biopha.2021.111623.
- C. H. van Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer and T. Iwatsubo, Lecanemab in Early Alzheimer's Disease, N. Engl. J. Med., 2023, 388(1), 9–21, DOI:10.1056/NEJMoa2212948.
- T. Naqvi, A. Abu, G. M. Hasan and M. I. Hassan, Targeting Tau Hyperphosphorylation via Kinase Inhibition: Strategy to Address Alzheimer's Disease, Curr. Top. Med. Chem., 2020, 20(12), 1059–1073, DOI:10.2174/1568026620666200106125910.
- C.-X. Gong and K. Iqbal, Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease, Curr. Med. Chem., 2008, 15(23), 2321–2328, DOI:10.2174/092986708785909111.
- H. Keren-Shaul, A. Spinrad, A. Weiner, O. Matcovitch-Natan, R. Dvir-Szternfeld, T. K. Ulland, E. David, K. Baruch, D. Lara-Astaiso, B. Toth, S. Itzkovitz, M. Colonna, M. Schwartz and I. Amit, A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease, Cell, 2017, 169(7), 1276–1290, DOI:10.1016/j.cell.2017.05.018.
- A. Y. Kim, S. Al Jerdi, R. MacDonald and C. R. Triggle, Alzheimer's disease and its treatment-yesterday, today, and tomorrow, Front. Pharmacol., 2024, 15, 1399121, DOI:10.3389/fphar.2024.1399121.
- H. Sugimoto, Y. Yamanishi, Y. Iimura and Y. Kawakami, Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors, Curr. Med. Chem., 2000, 7(3), 303–339, DOI:10.2174/0929867003375191.
- P. N. Tariot, M. R. Farlow, G. T. Grossberg, S. M. Graham, S. McDonald and I. Gergel, Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial, JAMA, 2004, 291(3), 317–324, DOI:10.1001/jama.291.3.317.
- K. Wojtunik-Kulesza, M. Rudkowska and A. Orzeł-Sajdłowska, Aducanumab-Hope or Disappointment for Alzheimer’s Disease, Int. J. Mol. Sci., 2023, 24(5), 4367, DOI:10.3390/ijms24054367.
- L. Jönsson, A. Wimo, R. Handels, G. Johansson, M. Boada, S. Engelborghs, L. Frölich, F. Jessen, P. G. Kehoe, M. Kramberger, A. de Mendonςa, P. J. Ousset, N. Scarmeas, P. J. Visser, G. Waldemar and B. Winblad, The affordability of lecanemab, an amyloid-targeting therapy for Alzheimer's disease: an EADC-EC viewpoint, Lancet Reg. Health Eur., 2023, 29, 100657, DOI:10.1016/j.lanepe.2023.100657.
- U. Kaur, J. Reddy, A. Tiwari, S. Chakrabarti and S. S. Chakrabarti, Lecanemab: More Questions Than Answers!, Clin. Drug Invest., 2024, 44(1), 1–10, DOI:10.1007/s40261-023-01331-1.
- W. A. Banks, Drug delivery to the brain in Alzheimer's disease: consideration of the blood–brain barrier, Adv. Drug Deliv. Rev., 2012, 64(7), 629–639, DOI:10.1016/j.addr.2011.12.005.
- A. R. Jones and E. V. Shusta, Blood–brain barrier transport of therapeutics via receptor-mediation, Pharm. Res., 2007, 24(9), 1759–1771, DOI:10.1007/s11095-007-9379-0.
- P. C. Pires and A. O. Santos, Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies, J. Controlled Release, 2018, 270, 89–100, DOI:10.1016/j.jconrel.2017.11.047.
- C. Ross, M. Taylor, N. Fullwood and D. Allsop, Liposome delivery systems for the treatment of Alzheimer's disease, Int. J. Nanomed., 2018, 13, 8507–8522, DOI:10.2147/IJN.S183117.
- M. Agrawal, T. Ajazuddin, K. Dulal, S. Saraf, S. Saraf, S. G. Antimisiaris, S. Mourtas, M. Hammarlund-Udenaes and A. Alexander, Recent advancements in liposomes targeting strategies to cross blood–brain barrier (BBB) for the treatment of Alzheimer's disease, J. Controlled Release, 2017, 260, 61–77, DOI:10.1016/j.jconrel.2017.05.019.
- G. Zhong, H. Long, T. Zhou, Y. Liu, J. Zhao, J. Han, X. Yang, Y. Yu, F. Chen and S. Shi, Blood–brain barrier Permeable nanoparticles for Alzheimer's disease treatment by selective mitophagy of microglia, Biomaterials, 2022, 288, 121690, DOI:10.1016/j.biomaterials.2022.121690.
- J. Ahmad, S. Akhter, M. Rizwanullah, M. A. Khan, L. Pigeon, R. T. Addo, N. H. Greig, P. Midoux, C. Pichon and M. A. Kamal, Nanotechnology Based Theranostic Approaches in Alzheimer's Disease Management: Current Status and Future Perspective, Curr. Alzheimer Res., 2017, 14(11), 1164–1181, DOI:10.2174/1567205014666170508121031.
- D. S. Knopman, H. Amieva, R. C. Petersen, G. Chételat, D. M. Holtzman, B. T. Hyman, R. A. Nixon and D. T. Jones, Alzheimer disease, Nat. Rev. Dis. Prim., 2021, 7(1), 33, DOI:10.1038/s41572-021-00269-y.
- M. P. Mattson, Pathways towards and away from Alzheimer's disease, Nature, 2004, 430(7000), 631–639, DOI:10.1038/nature02621.
- L. Ordóñez-Gutiérrez and F. Wandosell, Nanoliposomes as a Therapeutic Tool for Alzheimer's Disease, Front. Synaptic Neurosci., 2020, 12, 20, DOI:10.3389/fnsyn.2020.00020.
- D. B. Vieira and L. F. Gamarra, Getting into the brain: liposome-based strategies for effective drug delivery across the blood–brain barrier, Int. J. Nanomed., 2016, 11, 5381–5414, DOI:10.2147/IJN.S117210.
- C. Spuch and C. Navarro, Liposomes for Targeted Delivery of Active Agents against Neurodegenerative Diseases (Alzheimer's Disease and Parkinson's Disease), J. Drug Deliv., 2011, 2011, 469679, DOI:10.1155/2011/469679.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Liposomal Formulations in Clinical Use: An Updated Review, Pharmaceutics, 2017, 9(2), 12, DOI:10.3390/pharmaceutics9020012.
- M. Hasan, K. Elkhoury, C. J. F. Kahn, E. Arab-Tehrany and M. Linder, Preparation, Characterization, and Release Kinetics of Chitosan-Coated Nanoliposomes Encapsulating Curcumin in Simulated Environments, Molecules, 2019, 24(10), 2023, DOI:10.3390/molecules24102023.
- X. He, Y. Peng, S. Huang, Z. Xiao, G. Li, Z. Zuo, L. Zhang, X. Shuai, H. Zheng and X. Hu, Blood–Brain Barrier-Crossing Delivery of Felodipine Nanodrug Ameliorates Anxiety-Like Behavior and Cognitive Impairment in Alzheimer's Disease, Adv. Sci., 2024, 11(34), e2401731, DOI:10.1002/advs.202401731.
- Y. Chen, L. Huang, Z. Luo, D. Han, W. Luo, R. Wan, Y. Li, Y. Ge, W.-W. Lin, Y. Xie, M. Sun, Q. Wang, Z. Li, S. Chen, Y. Yang, B. Huang and Y. Xu, Pantothenate-encapsulated liposomes combined with exercise for effective inhibition of CRM1-mediated PKM2 translocation in Alzheimer's therapy, J. Controlled Release, 2024, 373, 336–357, DOI:10.1016/j.jconrel.2024.07.010.
- Z. Gu, H. Zhao, Y. Song, Y. Kou, W. Yang, Y. Li, X. Li, L. Ding, Z. Sun, J. Lin, Q. Wang, X. Li, X. Yang, X. Huang, C. Yang and Z. Tong, PEGylated-liposomal astaxanthin ameliorates Aβ neurotoxicity and Alzheimer-related phenotypes by scavenging formaldehyde, J. Controlled Release, 2024, 366, 783–797, DOI:10.1016/j.jconrel.2024.01.019.
- N. Filipczak, J. Pan, S. S. K. Yalamarty and V. P. Torchilin, Recent advancements in liposome technology, Adv. Drug Deliv. Rev., 2020, 156, 4–22, DOI:10.1016/j.addr.2020.06.022.
- R. Liu, J. Yang, L. Liu, Z. Lu, Z. Shi, W. Ji, J. Shen and X. Zhang, An "Amyloid-β Cleaner" for the Treatment of Alzheimer's Disease by Normalizing Microglial Dysfunction, Adv. Sci., 2020, 7(2), 1901555, DOI:10.1002/advs.201901555.
- K. Qian, X. Bao, Y. Li, P. Wang, Q. Guo, P. Yang, S. Xu, F. Yu, R. Meng, Y. Cheng, D. Sheng, J. Cao, M. Xu, J. Wu, T. Wang, Y. Wang, Q. Xie, W. Lu and Q. Zhang, Cholinergic Neuron Targeting Nanosystem Delivering Hybrid Peptide for Combinatorial Mitochondrial Therapy in Alzheimer's Disease, ACS Nano, 2022, 16(7), 11455–11472, DOI:10.1021/acsnano.2c05795.
- F. Ul Amin, S. A. Shah, H. Badshah, M. Khan and M. O. Kim, Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Aβ1-42-induced oxidative stress, J. Nanobiotechnol., 2017, 15(1), 12, DOI:10.1186/s12951-016-0227-4.
- A. Cano, M. Ettcheto, J.-H. Chang, E. Barroso, M. Espina, B. A. Kühne, M. Barenys, C. Auladell, J. Folch, E. B. Souto, A. Camins, P. Turowski and M. L. García, Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer's disease mice model, J. Controlled Release, 2019, 301, 62–75, DOI:10.1016/j.jconrel.2019.03.010.
- C. Ye, M. Cheng, L. Ma, T. Zhang, Z. Sun, C. Yu, J. Wang and Y. Dou, Oxytocin Nanogels Inhibit Innate Inflammatory Response for Early Intervention in Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2022, 14(19), 21822–21835, DOI:10.1021/acsami.2c00007.
- Q. Huang, C. Jiang, X. Xia, Y. Wang, C. Yan, X. Wang, T. Lei, X. Yang, W. Yang, G. Cheng and H. Gao, Pathological BBB Crossing Melanin-Like Nanoparticles as Metal-Ion Chelators and Neuroinflammation Regulators against Alzheimer's Disease, Research, 2023, 6, 0180, DOI:10.34133/research.0180.
- H. M. Gebril, A. Aryasomayajula, L. de, R. N. Mariana, K. E. Uhrich and P. V. Moghe, Nanotechnology for microglial targeting and inhibition of neuroinflammation underlying Alzheimer's pathology, Transl. Neurodegener., 2024, 13(1), 2, DOI:10.1186/s40035-023-00393-7.
- P. Liu, T. Zhang, Q. Chen, C. Li, Y. Chu, Q. Guo, Y. Zhang, W. Zhou, H. Chen, Z. Zhou, Y. Wang, Z. Zhao, Y. Luo, X. Li, H. Song, B. Su, C. Li, T. Sun and C. Jiang, Biomimetic Dendrimer-Peptide Conjugates for Early Multi-Target Therapy of Alzheimer's Disease by Inflammatory Microenvironment Modulation, Adv. Mater., 2021, 33(26), e2100746, DOI:10.1002/adma.202100746.
- Q. Guo, S. Xu, P. Yang, P. Wang, S. Lu, D. Sheng, K. Qian, J. Cao, W. Lu and Q. Zhang, A dual-ligand fusion peptide improves the brain-neuron targeting of nanocarriers in Alzheimer's disease mice, J. Controlled Release, 2020, 320, 347–362, DOI:10.1016/j.jconrel.2020.01.039.
- P. Yang, Y. Li, K. Qian, L. Zhou, Y. Cheng, J. Wu, M. Xu, T. Wang, X. Yang, Y. Mu, X. Liu and Q. Zhang, Precise Modulation of Pericyte Dysfunction by a Multifunctional Nanoprodrug to Ameliorate Alzheimer's Disease, ACS Nano, 2024, 18(22), 14348–14366, DOI:10.1021/acsnano.4c00480.
- Q. Luo, Y.-X. Lin, P.-P. Yang, Y. Wang, G.-B. Qi, Z.-Y. Qiao, L. Bing-Nan, K. Zhang, J.-P. Zhang, L. Wang and H. Wang, A self-destructive nanosweeper that captures and clears amyloid β-peptides, Nat. Commun., 2018, 9(1), 1802, DOI:10.1038/s41467-018-04255-z.
- L. Yao, X. Gu, Q. Song, X. Wang, M. Huang, M. Hu, L. Hou, T. Kang, J. Chen, H. Chen and X. Gao, Nanoformulated alpha-mangostin ameliorates Alzheimer's disease neuropathology by elevating LDLR expression and accelerating amyloid-beta clearance, J. Controlled Release, 2016, 226, 1–14, DOI:10.1016/j.jconrel.2016.01.055.
- F. Dai, X. Li, K. Lv, J. Wang and Y. Zhao, Combined Core Stability and Degradability of Nanomedicine via Amorphous PDLLA-Dextran Bottlebrush Copolymer for Alzheimer's Disease Combination Treatment, ACS Appl. Mater. Interfaces, 2023, 15(22), 26385–26397, DOI:10.1021/acsami.3c03174.
- J. Li, H. Peng, W. Zhang, M. Li, N. Wang, C. Peng, X. Zhang and Y. Li, Enhanced Nose-to-Brain Delivery of Combined Small Interfering RNAs Using Lesion-Recognizing Nanoparticles for the Synergistic Therapy of Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2023, 15(46), 53177–53188, DOI:10.1021/acsami.3c08756.
- Y. Wang, X. Wang, R. Xie, J. C. Burger, Y. Tong and S. Gong, Overcoming the Blood–Brain Barrier for Gene Therapy via Systemic Administration of GSH-Responsive Silica Nanocapsules, Adv. Mater., 2023, 35(6), e2208018, DOI:10.1002/adma.202208018.
- S. Gupta, V. Singh, S. Ganesh, N. K. Singhal and R. Sandhir, siRNA Mediated GSK3β Knockdown Targets Insulin Signaling Pathway and Rescues Alzheimer's Disease Pathology: Evidence from In Vitro and In Vivo Studies, ACS Appl. Mater. Interfaces, 2022, 14(1), 69–93, DOI:10.1021/acsami.1c15305.
- D. Yu, M. Ma, Z. Liu, Z. Pi, X. Du, J. Ren and X. Qu, MOF-encapsulated nanozyme enhanced siRNA combo: Control neural stem cell differentiation and ameliorate cognitive impairments in Alzheimer's disease model, Biomaterials, 2020, 255, 120160, DOI:10.1016/j.biomaterials.2020.120160.
- H. Park, J. Oh, G. Shim, B. Cho, Y. Chang, S. Kim, S. Baek, H. Kim, J. Shin, H. Choi, J. Yoo, J. Kim, W. Jun, M. Lee, C. J. Lengner, Y.-K. Oh and J. Kim, In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer's disease, Nat. Neurosci., 2019, 22(4), 524–528, DOI:10.1038/s41593-019-0352-0.
- J. Shen, Z. Lu, J. Wang, Q. Hao, W. Ji, Y. Wu, H. Peng, R. Zhao, J. Yang, Y. Li, Z. Shi and X. Zhang, Traceable Nano-Biohybrid Complexes by One-Step Synthesis as CRISPR-Chem Vectors for Neurodegenerative Diseases Synergistic Treatment, Adv. Mater., 2021, 33(27), e2101993, DOI:10.1002/adma.202101993.
- E. Samaridou, H. Walgrave, E. Salta, D. M. Álvarez, V. Castro-López, M. Loza and M. J. Alonso, Nose-to-brain delivery of enveloped RNA - cell permeating peptide nanocomplexes for the treatment of neurodegenerative diseases, Biomaterials, 2020, 230, 119657, DOI:10.1016/j.biomaterials.2019.119657.
- W. Li, X. Peng, X. Mei, M. Dong, Y. Li and H. Dong, Multifunctional DNA Tetrahedron for Alzheimer's Disease Mitochondria-Targeted Therapy by MicroRNA Regulation, ACS Appl. Mater. Interfaces, 2023, 15(19), 22977–22984, DOI:10.1021/acsami.3c03181.
- Q. Ouyang, K. Liu, Q. Zhu, H. Deng, Y. Le, W. Ouyang, X. Yan, W. Zhou and J. Tong, Brain-Penetration and Neuron-Targeting DNA Nanoflowers Co-Delivering miR-124 and Rutin for Synergistic Therapy of Alzheimer's Disease, Small, 2022, 18(14), e2107534, DOI:10.1002/smll.202107534.
- D. Huang, Y. Cao, X. Yang, Y. Liu, Y. Zhang, C. Li, G. Chen and Q. Wang, A Nanoformulation-Mediated Multifunctional Stem Cell Therapy with Improved Beta-Amyloid Clearance and Neural Regeneration for Alzheimer's Disease, Adv. Mater., 2021, 33(13), e2006357, DOI:10.1002/adma.202006357.
- Y. Liu, S. An, J. Li, Y. Kuang, X. He, Y. Guo, H. Ma, Y. Zhang, B. Ji and C. Jiang, Brain-targeted co-delivery of therapeutic gene and peptide by multifunctional nanoparticles in Alzheimer's disease mice, Biomaterials, 2016, 80, 33–45, DOI:10.1016/j.biomaterials.2015.11.060.
- M. Inoue, T. Higashi, Y. Hayashi, R. Onodera, K. Fujisawa, T. Taharabaru, R. Yokoyama, K. Ouchi, Y. Misumi, M. Ueda, Y. Inoue, M. Mizuguchi, T. Saito, T. C. Saido, Y. Ando, H. Arima, K. Motoyama and H. Jono, Multifunctional Therapeutic Cyclodextrin-Appended Dendrimer Complex for Treatment of Systemic and Localized Amyloidosis, ACS Appl. Mater. Interfaces, 2022, 14(36), 40599–40611, DOI:10.1021/acsami.2c09913.
- Y. Li, Y. Li, W. Ji, Z. Lu, L. Liu, Y. Shi, G. Ma and X. Zhang, Positively Charged Polyprodrug Amphiphiles with Enhanced Drug Loading and Reactive Oxygen Species-Responsive Release Ability for Traceable Synergistic Therapy, J. Am. Chem. Soc., 2018, 140(11), 4164–4171, DOI:10.1021/jacs.8b01641.
- P. Wang, X. Zheng, Q. Guo, P. Yang, X. Pang, K. Qian, W. Lu, Q. Zhang and X. Jiang, Systemic delivery of BACE1 siRNA through neuron-targeted nanocomplexes for treatment of Alzheimer's disease, J. Controlled Release, 2018, 279, 220–233, DOI:10.1016/j.jconrel.2018.04.034.
- L. L. Israel, T. Sun, O. Braubach, A. Cox, E. S. Shatalova, H.-M. Rashid, A. Galstyan, Z. Grodzinski, X. Y. Song, O. Chepurna, V. A. Ljubimov, A. Chiechi, S. Sharma, C. Phebus, Y. Wang, J. Y. Ljubimova, K. L. Black and E. Holler, β-Amyloid targeting nanodrug for neuron-specific delivery of nucleic acids in Alzheimer's disease mouse models, J. Controlled Release, 2023, 361, 636–658, DOI:10.1016/j.jconrel.2023.08.001.
- H. J. Shin, I. Kim, C. Soo, G. Seung, K. Lee, H. Park, J. Shin, D. Kim, J. Beom, Y. Y. Yi, D. P. Gupta, G. J. Song, W.-S. Chung, C. J. Lee and D. W. Kim, Rejuvenating aged microglia by p16ink4a-siRNA-loaded nanoparticles increases amyloid-β clearance in animal models of Alzheimer's disease, Mol. Neurodegener., 2024, 19(1), 25, DOI:10.1186/s13024-024-00715-x.
- X. Yang, W. Yang, X. Xia, T. Lei, Z. Yang, W. Jia, Y. Zhou, G. Cheng and H. Gao, Intranasal Delivery of BACE1 siRNA and Rapamycin by Dual Targets Modified Nanoparticles for Alzheimer's Disease Therapy, Small, 2022, 18(30), e2203182, DOI:10.1002/smll.202203182.
- W. T. Ralvenius, J. L. Andresen, M. M. Huston, J. Penney, J. M. Bonner, O. S. Fenton, R. Langer and L.-H. Tsai, Nanoparticle-Mediated Delivery of Anti-PU.1 siRNA via Localized Intracisternal Administration Reduces Neuroinflammation, Adv. Mater., 2024, 36(8), e2309225, DOI:10.1002/adma.202309225.
- S. U. Islam, A. Shehzad, M. B. Ahmed and Y. S. Lee, Intranasal Delivery of Nanoformulations: A Potential Way of Treatment for Neurological Disorders, Molecules, 2020, 25(8), 1929, DOI:10.3390/molecules25081929.
- A. Schambach, C. J. Buchholz, R. Torres-Ruiz, K. Cichutek, M. Morgan, I. Trapani and H. Büning, A new age of precision gene therapy, Lancet (London, England), 2024, 403(10426), 568–582, DOI:10.1016/S0140-6736(23)01952-9.
- B. Anand, Q. Wu, M. Nakhaei-Nejad, G. Karthivashan, L. Dorosh, S. Amidian, A. Dahal, X. Li, M. Stepanova, H. Wille, F. Giuliani and S. Kar, Significance of native PLGA nanoparticles in the treatment of Alzheimer's disease pathology, Bioact. Mater., 2022, 17, 506–525, DOI:10.1016/j.bioactmat.2022.05.030.
- P. S. Paul, J.-Y. Cho, Q. Wu, G. Karthivashan, E. Grabovac, H. Wille, M. Kulka and S. Kar, Unconjugated PLGA nanoparticles attenuate temperature-dependent β-amyloid aggregation and protect neurons against toxicity: implications for Alzheimer's disease pathology, J. Nanobiotechnol., 2022, 20(1), 67, DOI:10.1186/s12951-022-01269-0.
- M. Silva-Abreu, A. C. Calpena, P. Andrés-Benito, E. Aso, I. A. Romero, D. Roig-Carles, R. Gromnicova, M. Espina, I. Ferrer, M. L. García and D. Male, PPARγ agonist-loaded PLGA-PEG nanocarriers as a potential treatment for Alzheimer's disease: in vitro and in vivo studies, Int. J. Nanomed., 2018, 13, 5577–5590, DOI:10.2147/IJN.S171490.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, Inhibition of amyloid beta protein fibrillation by polymeric nanoparticles, J. Am. Chem. Soc., 2008, 130(46), 15437–15443, DOI:10.1021/ja8041806.
- B. Le Droumaguet, J. Nicolas, D. Brambilla, S. Mura, A. Maksimenko, L. De Kimpe, E. Salvati, C. Zona, C. Airoldi, M. Canovi, M. Gobbi, N. Magali, B. La Ferla, F. Nicotra, W. Scheper, O. Flores, M. Masserini, K. Andrieux and P. Couvreur, Versatile and efficient targeting using a single nanoparticulate platform: application to cancer and Alzheimer's disease, ACS Nano, 2012, 6(7), 5866–5879, DOI:10.1021/nn3004372.
- S. Fan, Y. Zheng, X. Liu, W. Fang, X. Chen, W. Liao, X. Jing, M. Lei, E. Tao, Q. Ma, X. Zhang, R. Guo and J. Liu, Curcumin-loaded PLGA-PEG nanoparticles conjugated with B6 peptide for potential use in Alzheimer's disease, Drug Deliv., 2018, 25(1), 1091–1102, DOI:10.1080/10717544.2018.1461955.
- N. Xiong, X.-Y. Dong, J. Zheng, F.-F. Liu and Y. Sun, Design of LVFFARK and LVFFARK-functionalized nanoparticles for inhibiting amyloid β-protein fibrillation and cytotoxicity, ACS Appl. Mater. Interfaces, 2015, 7(10), 5650–5662, DOI:10.1021/acsami.5b00915.
- H. Liu, L. Yu, X. Dong and Y. Sun, Synergistic effects of negatively charged hydrophobic nanoparticles and (-)-epigallocatechin-3-gallate on inhibiting amyloid β-protein aggregation, J. Colloid Interface Sci., 2017, 491, 305–312, DOI:10.1016/j.jcis.2016.12.038.
- L. Zhu, L. Xu, X. Wu, F. Deng, R. Ma, Y. Liu, F. Huang and L. Shi, Tau-Targeted Multifunctional Nanoinhibitor for Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2021, 13(20), 23328–23338, DOI:10.1021/acsami.1c00257.
- H. Yang, W. Mu, D. Wei, Y. Zhang, Y. Duan, J.-X. Gao, X.-Q. Gong, H.-J. Wang, X.-L. Wu, H. Tao and J. Chang, A Novel Targeted and High-Efficiency Nanosystem for Combinational Therapy for Alzheimer's Disease, Adv. Sci., 2020, 7(19), 1902906, DOI:10.1002/advs.201902906.
- C. Zhang, X. Zheng, X. Wan, X. Shao, Q. Liu, Z. Zhang and Q. Zhang, The potential use of H102 peptide-loaded dual-functional nanoparticles in the treatment of Alzheimer's disease, J. Controlled Release, 2014, 192, 317–324, DOI:10.1016/j.jconrel.2014.07.050.
- W.-B. Li, L.-L. Xu, S.-L. Wang, Y.-Y. Wang, Y.-C. Pan, L.-Q. Shi and D.-S. Guo, Co-Assembled Nanoparticles toward Multi-Target Combinational Therapy of Alzheimer's Disease by Making Full Use of Molecular Recognition and Self-Assembly, Adv. Mater., 2024, 36(28), e2401918, DOI:10.1002/adma.202401918.
- R. Zhang, Y. Li, B. Hu, Z. Lu, J. Zhang and X. Zhang, Traceable Nanoparticle Delivery of Small Interfering RNA and Retinoic Acid with Temporally Release Ability to Control Neural Stem Cell Differentiation for Alzheimer's Disease Therapy, Adv. Mater., 2016, 28(30), 6345–6352, DOI:10.1002/adma.201600554.
- A. Duro-Castano, C. Borrás, V. Herranz-Pérez, M. C. Blanco-Gandía, I. Conejos-Sánchez, A. Armiñán, C. Mas-Bargues, M. Inglés, J. Miñarro, M. Rodríguez-Arias, J. M. García-Verdugo, J. Viña and M. J. Vicent, Targeting Alzheimer’s disease with multimodal polypeptide-based nanoconjugates, Sci. Adv., 2021, 7(13), eabf9180, DOI:10.1126/sciadv.abf9180.
- J. Liu, M. Chi, L. Li, Y. Zhang and M. Xie, Erythrocyte membrane coated with nitrogen-doped quantum dots and polydopamine composite nano-system combined with photothermal treatment of Alzheimer's disease, J. Colloid Interface Sci., 2024, 663, 856–868, DOI:10.1016/j.jcis.2024.02.219.
- X. Wang, W. Zhang, L. Hou, W. Geng, J. Wang, Y. Kong, C. Liu, X. Zeng and D. Kong, A Biomimetic Upconversion Nanobait-Based Near Infrared Light Guided Photodynamic Therapy Alleviates Alzheimer's Disease by Inhibiting β-Amyloid Aggregation, Adv. Healthcare Mater., 2024, 13(9), e2303278, DOI:10.1002/adhm.202303278.
- M. Chi, J. Liu, L. Li, Y. Zhang and M. Xie, CeO2 In Situ Growth on Red Blood Cell Membranes: CQD Coating and Multipathway Alzheimer's Disease Therapy under NIR, ACS Appl. Mater. Interfaces, 2024, 16(28), 35898–35911, DOI:10.1021/acsami.4c02088.
- L. Li, Y. Xiong, Y. Zhang, Y. Yan, R. Zhao, F. Yang and M. Xie, Biofilm-camouflaged Prussian blue synergistic mitochondrial mass enhancement for Alzheimer's disease based on Cu2+ chelation and photothermal therapy, J. Controlled Release, 2024, 375, 269–284, DOI:10.1016/j.jconrel.2024.09.009.
- P. Ye, L. Li, X. Qi, M. Chi, J. Liu and M. Xie, Macrophage membrane-encapsulated nitrogen-doped carbon quantum dot nanosystem for targeted treatment of Alzheimer's disease: Regulating metal ion homeostasis and photothermal removal of β-amyloid, J. Colloid Interface Sci., 2023, 650(Pt B), 1749–1761, DOI:10.1016/j.jcis.2023.07.132.
- X. Qi, L. Li, P. Ye and M. Xie, Macrophage Membrane-Modified MoS2 Quantum Dots as a Nanodrug for Combined Multi-Targeting of Alzheimer's Disease, Adv. Healthcare Mater., 2024, 13(6), e2303211, DOI:10.1002/adhm.202303211.
- Z. Cui, W. Bu, W. Fan, J. Zhang, D. Ni, Y. Liu, J. Wang, J. Liu, Z. Yao and J. Shi, Sensitive imaging and effective capture of Cu(2+): Towards highly efficient theranostics of Alzheimer's disease, Biomaterials, 2016, 104, 158–167, DOI:10.1016/j.biomaterials.2016.06.056.
- C. Wang, X. Song, P. Li, S. Sun, J. Su, S. Liu and W. Wei, Multifunctional Nanocarrier for Synergistic Treatment of Alzheimer's Disease by Inhibiting β-Amyloid Aggregation and Scavenging Reactive Oxygen Species, ACS Appl. Mater. Interfaces, 2024, 16(21), 27127–27138, DOI:10.1021/acsami.4c02825.
- L. Zhang, K. Cao, J. Xie, X. Liang, H. Gong, Q. Luo and H. Luo, Aβ42 and ROS dual-targeted multifunctional nanocomposite for combination therapy of Alzheimer's disease, J. Nanobiotechnol., 2024, 22(1), 278, DOI:10.1186/s12951-024-02543-z.
- L. Xiao, D. Zhao, W.-H. Chan, M. M. F. Choi and H.-W. Li, Inhibition of beta 1-40 amyloid fibrillation with N-acetyl-L-cysteine capped quantum dots, Biomaterials, 2010, 31(1), 91–98, DOI:10.1016/j.biomaterials.2009.09.014.
- Q.-Y. Li, X. Yu, X. Li, L.-N. Bao, Y. Zhang, M.-J. Xie, M. Jiang, Y. Q. Wang, K. Huang and L. Xu, Silicon-Carbon Dots-Loaded Mesoporous Silica Nanocomposites (mSiO2@SiCDs): An Efficient Dual Inhibitor of Cu2+-Mediated Oxidative Stress and Aβ Aggregation for Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2023, 15(47), 54221–54233, DOI:10.1021/acsami.3c10053.
- J. Kim, H. Um, N. H. Kim and D. Kim, Potential Alzheimer's disease therapeutic nano-platform: Discovery of amyloid-beta plaque disaggregating agent and brain-targeted delivery system using porous silicon nanoparticles, Bioact. Mater., 2023, 24, 497–506, DOI:10.1016/j.bioactmat.2023.01.006.
- T. Yin, W. Xie, J. Sun, L. Yang and J. Liu, Penetratin Peptide-Functionalized Gold Nanostars: Enhanced BBB Permeability and NIR Photothermal Treatment of Alzheimer's Disease Using Ultralow Irradiance, ACS Appl. Mater. Interfaces, 2016, 8(30), 19291–19302, DOI:10.1021/acsami.6b05089.
- K. Hou, J. Zhao, H. Wang, B. Li, K. Li, X. Shi, K. Wan, J. Ai, J. Lv, D. Wang, Q. Huang, H. Wang, Q. Cao, S. Liu and Z. Tang, Chiral gold nanoparticles enantioselectively rescue memory deficits in a mouse model of Alzheimer's disease, Nat. Commun., 2020, 11(1), 4790, DOI:10.1038/s41467-020-18525-2.
- K. Ge, Y. Mu, M. Liu, Z. Bai, Z. Liu, D. Geng and F. Gao, Gold Nanorods with Spatial Separation of CeO2 Deposition for Plasmonic-Enhanced Antioxidant Stress and Photothermal Therapy of Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2022, 14(3), 3662–3674, DOI:10.1021/acsami.1c17861.
- M. Ma, N. Gao, Y. Sun, X. Du, J. Ren and X. Qu, Redox-Activated Near-Infrared-Responsive Polyoxometalates Used for Photothermal Treatment of Alzheimer's Disease, Adv. Healthcare Mater., 2018, 7(20), e1800320, DOI:10.1002/adhm.201800320.
- C. Morfill, S. Pankratova, P. Machado, N. K. Fernando, A. Regoutz, F. Talamona, A. Pinna, M. Klosowski, R. J. Wilkinson, R. A. Fleck, F. Xie, A. E. Porter and D. Kiryushko, Nanostars Carrying Multifunctional Neurotrophic Dendrimers Protect Neurons in Preclinical In Vitro Models of Neurodegenerative Disorders, ACS Appl. Mater. Interfaces, 2022, 14(42), 47445–47460, DOI:10.1021/acsami.2c14220.
- Y.-H. Liao, Y.-J. Chang, Y. Yoshiike, Y.-C. Chang and Y.-R. Chen, Negatively charged gold nanoparticles inhibit Alzheimer's amyloid-β fibrillization, induce fibril dissociation, and mitigate neurotoxicity, Small, 2012, 8(23), 3631–3639, DOI:10.1002/smll.201201068.
- X. Yin, H. Zhou, T. Cao, X. Yang, F. Meng, X. Dai, Y. Wang, S. Li, W. Zhai, Z. Yang, N. Chen and R. Zhou, Rational Design of Dual-Functionalized Gd@C82 Nanoparticles to Relieve Neuronal Cytotoxicity in Alzheimer's Disease via Inhibition of Aβ Aggregation, ACS Nano, 2024, 18(24), 15416–15431, DOI:10.1021/acsnano.3c08823.
- X. Guo, C. Li, J. Zhang, M. Sun, J. Xu, C. Xu, H. Kuang and L. Xu, Chiral nanoparticle-remodeled gut microbiota alleviates neurodegeneration via the gut-brain axis, Nat. Aging, 2023, 3(11), 1415–1429, DOI:10.1038/s43587-023-00516-9.
- S. Kuk, B. I. Lee, J. S. Lee and C. B. Park, Rattle-Structured Upconversion Nanoparticles for Near-IR-Induced Suppression of Alzheimer’s β-Amyloid Aggregation, Small, 2017, 13(11), 1603139, DOI:10.1002/smll.201603139.
- Z. Jia, X. Yuan, J.-A. Wei, X. Guo, Y. Gong, J. Li, H. Zhou, L. Zhang and J. Liu, A Functionalized Octahedral Palladium Nanozyme as a Radical Scavenger for Ameliorating Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2021, 13(42), 49602–49613, DOI:10.1021/acsami.1c06687.
- N. Andrikopoulos, Z. Song, X. Wan, A. M. Douek, I. Javed, C. Fu, Y. Xing, F. Xin, Y. Li, A. Kakinen, K. Koppel, R. Qiao, A. K. Whittaker, J. Kaslin, T. P. Davis, Y. Song, F. Ding and P. C. Ke, Inhibition of Amyloid Aggregation and Toxicity with Janus Iron Oxide Nanoparticles, Chem. Mater., 2021, 33(16), 6484–6500, DOI:10.1021/acs.chemmater.1c01947.
- J. Zhang, X. Zhou, Q. Yu, L. Yang, D. Sun, Y. Zhou and J. Liu, Epigallocatechin-3-gallate (EGCG)-stabilized selenium nanoparticles coated with Tet-1 peptide to reduce amyloid-β aggregation and cytotoxicity, ACS Appl. Mater. Interfaces, 2014, 6(11), 8475–8487, DOI:10.1021/am501341u.
- N. Xiong, Y. Zhao, X. Dong, J. Zheng and Y. Sun, Design of a Molecular Hybrid of Dual Peptide Inhibitors Coupled on AuNPs for Enhanced Inhibition of Amyloid β-Protein Aggregation and Cytotoxicity, Small, 2017, 13(13), 1601666, DOI:10.1002/smll.201601666.
- J. Zhang, S. Zhu, P. Jin, Y. Huang, Q. Dai, Q. Zhu, P. Wei, Z. Yang, L. Zhang, H. Liu, G. Xu, L. Chen, E. Gu, Y. Zhang, L. Wen and X. Liu, Graphene oxide improves postoperative cognitive dysfunction by maximally alleviating amyloid beta burden in mice, Theranostics, 2020, 10(26), 11908–11920, DOI:10.7150/thno.50616.
- L. Zhang, P. Zhao, C. Yue, Z. Jin, Q. Liu, X. Du and Q. He, Sustained release of bioactive hydrogen by Pd hydride nanoparticles overcomes Alzheimer's disease, Biomaterials, 2019, 197, 393–404, DOI:10.1016/j.biomaterials.2019.01.037.
- C. Li, N. Wang, G. Zheng and L. Yang, Oral Administration of Resveratrol-Selenium-Peptide Nanocomposites Alleviates Alzheimer's Disease-like Pathogenesis by Inhibiting Aβ Aggregation and Regulating Gut Microbiota, ACS Appl. Mater. Interfaces, 2021, 13(39), 46406–46420, DOI:10.1021/acsami.1c14818.
- Q. Han, X. Wang, X. Liu, Y. Zhang, S. Cai, C. Qi, C. Wang and R. Yang, MoO3-x nanodots with dual enzyme mimic activities as multifunctional modulators for amyloid assembly and neurotoxicity, J. Colloid Interface Sci., 2019, 539, 575–584, DOI:10.1016/j.jcis.2018.12.093.
- D. Liu, W. Li, X. Jiang, S. Bai, J. Liu, X. Liu, Y. Shi, Z. Kuai, W. Kong, R. Gao and Y. Shan, Using near-infrared enhanced thermozyme and scFv dual-conjugated Au nanorods for detection and targeted photothermal treatment of Alzheimer's disease, Theranostics, 2019, 9(8), 2268–2281, DOI:10.7150/thno.30649.
- J. Li, G. Gao, X. Tang, M. Yu, M. He and T. Sun, Isomeric Effect of Nano-Inhibitors on Aβ40 Fibrillation at The Nano-Bio Interface, ACS Appl. Mater. Interfaces, 2021, 13(4), 4894–4904, DOI:10.1021/acsami.0c21906.
- C. Ren, D. Li, Q. Zhou and X. Hu, Mitochondria-targeted TPP-MoS2 with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer's disease model, Biomaterials, 2020, 232, 119752, DOI:10.1016/j.biomaterials.2019.119752.
- J. Sun, C. Wei, Y. Liu, W. Xie, M. Xu, H. Zhou and J. Liu, Progressive release of mesoporous nano-selenium delivery system for the multi-channel synergistic treatment of Alzheimer's disease, Biomaterials, 2019, 197, 417–431, DOI:10.1016/j.biomaterials.2018.12.027.
- T. Yin, L. Yang, Y. Liu, X. Zhou, J. Sun and J. Liu, Sialic acid (SA)-modified selenium nanoparticles coated with a high blood–brain barrier permeability peptide-B6 peptide for potential use in Alzheimer's disease, Acta Biomater., 2015, 25, 172–183, DOI:10.1016/j.actbio.2015.06.035.
- C. N. Loynachan, G. Romero, M. G. Christiansen, R. Chen, R. Ellison, T. T. O'Malley, U. P. Froriep, D. M. Walsh and P. Anikeeva, Targeted Magnetic Nanoparticles for Remote Magnetothermal Disruption of Amyloid-β Aggregates, Adv. Healthcare Mater., 2015, 4(14), 2100–2109, DOI:10.1002/adhm.201500487.
- Z. Du, C. Liu, Z. Liu, H. Song, P. Scott, X. Du, J. Ren and X. Qu, In vivo visualization of enantioselective targeting of amyloid and improvement of cognitive function by clickable chiral metallohelices, Chem. Sci., 2023, 14(3), 506–513, 10.1039/d2sc05897a.
- R. Prades, S. Guerrero, E. Araya, C. Molina, E. Salas, E. Zurita, J. Selva, G. Egea, C. López-Iglesias, M. Teixidó, M. J. Kogan and E. Giralt, Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor, Biomaterials, 2012, 33(29), 7194–7205, DOI:10.1016/j.biomaterials.2012.06.063.
- X. Guo, Q. Lie, Y. Liu, Z. Jia, Y. Gong, X. Yuan and J. Liu, Multifunctional Selenium Quantum Dots for the Treatment of Alzheimer's Disease by Reducing Aβ-Neurotoxicity and Oxidative Stress and Alleviate Neuroinflammation, ACS Appl. Mater. Interfaces, 2021, 13(26), 30261–30273, DOI:10.1021/acsami.1c00690.
- E. Park, L. Y. Li, C. He, A. Z. Abbasi, T. Ahmed, W. D. Foltz, R. O'Flaherty, M. Zain, R. P. Bonin, A. M. Rauth, P. E. Fraser, J. T. Henderson and X. Y. Wu, Brain-Penetrating and Disease Site-Targeting Manganese Dioxide-Polymer-Lipid Hybrid Nanoparticles Remodel Microenvironment of Alzheimer's Disease by Regulating Multiple Pathological Pathways, Adv. Sci., 2023, 10(12), e2207238, DOI:10.1002/advs.202207238.
- J. Geng, M. Li, L. Wu, C. Chen and X. Qu, Mesoporous silica nanoparticle-based H2O2 responsive controlled-release system used for Alzheimer's disease treatment, Adv. Healthcare Mater., 2012, 1(3), 332–336, DOI:10.1002/adhm.201200067.
- M. Li, C. Xu, L. Wu, J. Ren, E. Wang and X. Qu, Self-assembled peptide-polyoxometalate hybrid nanospheres: two in one enhances targeted inhibition of amyloid β-peptide aggregation associated with Alzheimer's disease, Small, 2013, 9(20), 3455–3461, DOI:10.1002/smll.201202612.
- L. Yang, Y. Cui, H. Liang, Z. Li, N. Wang, Y. Wang and G. Zheng, Multifunctional Selenium Nanoparticles with Different Surface Modifications Ameliorate Neuroinflammation through the Gut Microbiota-NLRP3 Inflammasome-Brain Axis in APP/PS1 Mice, ACS Appl. Mater. Interfaces, 2022, 14(27), 30557–30570, DOI:10.1021/acsami.2c06283.
- J. Jang and C. B. Park, Magnetoelectric dissociation of Alzheimer's β-amyloid aggregates, Sci. Adv., 2022, 8(19), eabn1675, DOI:10.1126/sciadv.abn1675.
- Y. Liu, H. Zhou, T. Yin, Y. Gong, G. Yuan, L. Chen and J. Liu, Quercetin-modified gold-palladium nanoparticles as a potential autophagy inducer for the treatment of Alzheimer's disease, J. Colloid Interface Sci., 2019, 552, 388–400, DOI:10.1016/j.jcis.2019.05.066.
- B. G. Anand, Q. Wu, G. Karthivashan, K. P. Shejale, S. Amidian, H. Wille and S. Kar, Mimosine functionalized gold nanoparticles (Mimo-AuNPs) suppress β-amyloid aggregation and neuronal toxicity, Bioact. Mater., 2021, 6(12), 4491–4505, DOI:10.1016/j.bioactmat.2021.04.029.
- Y. Li, H. Tang, H. Zhu, A. Kakinen, D. Wang, N. Andrikopoulos, Y. Sun, A. Nandakumar, E. Kwak, T. P. Davis, D. T. Leong, F. Ding and P. C. Ke, Ultrasmall Molybdenum Disulfide Quantum Dots Cage Alzheimer's Amyloid Beta to Restore Membrane Fluidity, ACS Appl. Mater. Interfaces, 2021, 13(25), 29936–29948, DOI:10.1021/acsami.1c06478.
- M. J. Kogan, N. G. Bastus, R. Amigo, D. Grillo-Bosch, E. Araya, A. Turiel, A. Labarta, E. Giralt and V. F. Puntes, Nanoparticle-mediated local and remote manipulation of protein aggregation, Nano Lett., 2006, 6(1), 110–115, DOI:10.1021/nl0516862.
- N. Liu, X. Liang, C. Yang, S. Hu, Q. Luo and H. Luo, Dual-targeted magnetic mesoporous silica nanoparticles reduce brain amyloid-β burden via depolymerization and intestinal metabolism, Theranostics, 2022, 12(15), 6646–6664, DOI:10.7150/thno.76574.
- I. Javed, G. Peng, Y. Xing, T. Yu, M. Zhao, A. Kakinen, A. Faridi, C. L. Parish, F. Ding, T. P. Davis, P. C. Ke and S. Lin, Inhibition of amyloid beta toxicity in zebrafish with a chaperone-gold nanoparticle dual strategy, Nat. Commun., 2019, 10(1), 3780, DOI:10.1038/s41467-019-11762-0.
- C. Streich, L. Akkari, C. Decker, J. Bormann, C. Rehbock, A. Müller-Schiffmann, N. Felix Carlsson, L. Nagel-Steger, D. Willbold, B. Sacca, C. Korth, T. Schrader and S. Barcikowski, Characterizing the Effect of Multivalent Conjugates Composed of Aβ-Specific Ligands and Metal Nanoparticles on Neurotoxic Fibrillar Aggregation, ACS Nano, 2016, 10(8), 7582–7597, DOI:10.1021/acsnano.6b02627.
- M. Xu, H. Zhou, Y. Liu, J. Sun, W. Xie, P. Zhao and J. Liu, Ultrasound-Excited Protoporphyrin IX-Modified Multifunctional Nanoparticles as a Strong Inhibitor of Tau Phosphorylation and β-Amyloid Aggregation, ACS Appl. Mater. Interfaces, 2018, 10(39), 32965–32980, DOI:10.1021/acsami.8b08230.
- N. Liu, C. Yang, X. Liang, K. Cao, J. Xie, Q. Luo and H. Luo, Mesoporous silica nanoparticle-encapsulated Bifidobacterium attenuates brain Aβ burden and improves olfactory dysfunction of APP/PS1 mice by nasal delivery, J. Nanobiotechnol., 2022, 20(1), 439, DOI:10.1186/s12951-022-01642-z.
- Z. Du, N. Gao, X. Wang, J. Ren and X. Qu, Near-Infrared Switchable Fullerene-Based Synergy Therapy for Alzheimer's Disease, Small, 2018, e1801852, DOI:10.1002/smll.201801852.
- L. Yang, T. Yin, Y. Liu, J. Sun, Y. Zhou and J. Liu, Gold nanoparticle-capped mesoporous silica-based H2O2-responsive controlled release system for Alzheimer's disease treatment, Acta Biomater., 2016, 46, 177–190, DOI:10.1016/j.actbio.2016.09.010.
- Y.-J. Chang, Y.-H. Chien, C.-C. Chang, P.-N. Wang, Y.-R. Chen and Y.-C. Chang, Detection of Femtomolar Amyloid-β Peptides for Early-Stage Identification of Alzheimer's Amyloid-β Aggregation with Functionalized Gold Nanoparticles, ACS Appl. Mater. Interfaces, 2024, 16(3), 3819–3828, DOI:10.1021/acsami.3c12750.
- L. Xu, M. Guo, C.-T. Hung, X.-L. Shi, Y. Yuan, X. Zhang, R.-H. Jin, W. Li, Q. Dong and D. Zhao, Chiral Skeletons of Mesoporous Silica Nanospheres to Mitigate Alzheimer's β-Amyloid Aggregation, J. Am. Chem. Soc., 2023, 145(14), 7810–7819, DOI:10.1021/jacs.2c12214.
- Q. Han, S. Cai, L. Yang, X. Wang, C. Qi, R. Yang and C. Wang, Molybdenum Disulfide Nanoparticles as Multifunctional Inhibitors against Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2017, 9(25), 21116–21123, DOI:10.1021/acsami.7b03816.
- X.-G. Liu, L. Zhang, S. Lu, D.-Q. Liu, Y.-R. Huang, J. Zhu, W.-W. Zhou, Y. Xiao-Lin and R.-T. Liu, Superparamagnetic iron oxide nanoparticles conjugated with Aβ oligomer-specific scFv antibody and class A scavenger receptor activator show therapeutic potentials for Alzheimer's Disease, J. Nanobiotechnol., 2020, 18(1), 160, DOI:10.1186/s12951-020-00723-1.
- Y. Guan, M. Li, K. Dong, N. Gao, J. Ren, Y. Zheng and X. Qu, Ceria/POMs hybrid nanoparticles as a mimicking metallopeptidase for treatment of neurotoxicity of amyloid-β peptide, Biomaterials, 2016, 98, 92–102, DOI:10.1016/j.biomaterials.2016.05.005.
- H. Moorthy, L. P. Datta, S. Samanta and T. Govindaraju, Multifunctional Architectures of Cyclic Dipeptide Copolymers and Composites, and Modulation of Multifaceted Amyloid-β Toxicity, ACS Appl. Mater. Interfaces, 2022, 14(51), 56535–56547, DOI:10.1021/acsami.2c16336.
- Y. Ruan, Y. Xiong, W. Fang, Q. Yu, Y. Mai, Z. Cao, K. Wang, M. Lei, J. Xu, Y. Liu, X. Zhang, W. Liao and J. Liu, Highly sensitive Curcumin-conjugated nanotheranostic platform for detecting amyloid-beta plaques by magnetic resonance imaging and reversing cognitive deficits of Alzheimer's disease via NLRP3-inhibition, J. Nanobiotechnol., 2022, 20(1), 322, DOI:10.1186/s12951-022-01524-4.
- D. Sun, W. Zhang, Q. Yu, X. Chen, M. Xu, Y. Zhou and J. Liu, Chiral penicillamine-modified selenium nanoparticles enantioselectively inhibit metal-induced amyloid β aggregation for treating Alzheimer's disease, J. Colloid Interface Sci., 2017, 505, 1001–1010, DOI:10.1016/j.jcis.2017.06.083.
- H. Zhang, C. Hao, A. Qu, M. Sun, L. Xu, C. Xu and H. Kuang, Light-Induced Chiral Iron Copper Selenide Nanoparticles Prevent β-Amyloidopathy In Vivo, Angew. Chem., 2020, 59(18), 7131–7138, DOI:10.1002/anie.202002028.
- L. Liu, W. Liu, Y. Sun and X. Dong, Serum albumin-embedding copper nanoclusters inhibit Alzheimer's β-amyloid fibrillogenesis and neuroinflammation, J. Colloid Interface Sci., 2024, 672, 53–62, DOI:10.1016/j.jcis.2024.05.193.
- J. Wang, Y. Fan, Y. Tan, X. Zhao, Y. Zhang, C. Cheng and M. Yang, Porphyrinic Metal-Organic Framework PCN-224 Nanoparticles for Near-Infrared-Induced Attenuation of Aggregation and Neurotoxicity of Alzheimer's Amyloid-β Peptide, ACS Appl. Mater. Interfaces, 2018, 10(43), 36615–36621, DOI:10.1021/acsami.8b15452.
- J. Jang, Y. Jo and C. B. Park, Metal-Organic Framework-Derived Carbon as a Photoacoustic Modulator of Alzheimer's β-Amyloid Aggregate Structure, ACS Nano, 2022, 16(11), 18515–18525, DOI:10.1021/acsnano.2c06759.
- S. Xiao, D. Zhou, P. Luan, B. Gu, L. Feng, S. Fan, W. Liao, W. Fang, L. Yang, E. Tao, R. Guo and J. Liu, Graphene quantum dots conjugated neuroprotective peptide improve learning and memory capability, Biomaterials, 2016, 106, 98–110, DOI:10.1016/j.biomaterials.2016.08.021.
- W. Liu, W. Wang, X. Dong and Y. Sun, Near-Infrared Light-Powered Janus Nanomotor Significantly Facilitates Inhibition of Amyloid-β Fibrillogenesis, ACS Appl. Mater. Interfaces, 2020, 12(11), 12618–12628, DOI:10.1021/acsami.0c02342.
- W. Zhang, N. Smith, Y. Zhou, C. M. McGee, M. Bartoli, S. Fu, J. Chen, J. B. Domena, A. Joji, H. Burr, G. Lv, E. K. Cilingir, S. Bedendo, M. L. Claure, A. Tagliaferro, D. Eliezer, E. A. Veliz, F. Zhang, C. Wang and R. M. Leblanc, Carbon dots as dual inhibitors of tau and amyloid-beta aggregation for the treatment of Alzheimer's disease, Acta Biomater., 2024, 183, 341–355, DOI:10.1016/j.actbio.2024.06.001.
- K. M. Jaruszewski, G. L. Curran, S. K. Swaminathan, J. T. Rosenberg, S. C. Grant, S. Ramakrishnan, V. J. Lowe, J. F. Poduslo and K. K. Kandimalla, Multimodal nanoprobes to target cerebrovascular amyloid in Alzheimer's disease brain, Biomaterials, 2014, 35(6), 1967–1976, DOI:10.1016/j.biomaterials.2013.10.075.
- B. Shi, J. Zhao, Z. Xu, C. Chen, L. Xu, C. Xu, M. Sun and H. Kuang, Chiral Nanoparticles Force Neural Stem Cell Differentiation to Alleviate Alzheimer's Disease, Adv. Sci., 2022, 9(29), e2202475, DOI:10.1002/advs.202202475.
- X. Yin, H. Zhou, M. Zhang, J. Su, X. Wang, S. Li, Z. Yang, Z. Kang and R. Zhou, C3N nanodots inhibits Aβ peptides aggregation pathogenic path in Alzheimer's disease, Nat. Commun., 2023, 14(1), 5718, DOI:10.1038/s41467-023-41489-y.
- Z. Yin, Z. Zhang, D. Gao, G. Luo, T. Ma, Y. Wang, L. Lu and X. Gao, Stepwise Coordination-Driven Metal-Phenolic Nanoparticle as a Neuroprotection Enhancer for Alzheimer's Disease Therapy, ACS Appl. Mater. Interfaces, 2023, 15(1), 524–540, DOI:10.1021/acsami.2c18060.
- N. Andrikopoulos, Y. Li, A. Nandakumar, J. F. Quinn, T. P. Davis, F. Ding, N. Saikia and P. C. Ke, Zinc-Epigallocatechin-3-gallate Network-Coated Nanocomposites against the Pathogenesis of Amyloid-Beta, ACS Appl. Mater. Interfaces, 2023, 15(6), 7777–7792, DOI:10.1021/acsami.2c20334.
- H. Liu, X. Dong, F. Liu, J. Zheng and Y. Sun, Iminodiacetic acid-conjugated nanoparticles as a bifunctional modulator against Zn2+-mediated amyloid β-protein aggregation and cytotoxicity, J. Colloid Interface Sci., 2017, 505, 973–982, DOI:10.1016/j.jcis.2017.06.093.
- H. Derakhshankhah, M. J. Hajipour, E. Barzegari, A. Lotfabadi, M. Ferdousi, A. A. Saboury, E. P. Ng, M. Raoufi, H. Awala, S. Mintova, R. Dinarvand and M. Mahmoudi, Zeolite Nanoparticles Inhibit Aβ-Fibrinogen Interaction and Formation of a Consequent Abnormal Structural Clot, ACS Appl. Mater. Interfaces, 2016, 8(45), 30768–30779, DOI:10.1021/acsami.6b10941.
- M. Li, C. Zhao, J. Ren and X. Qu, Chiral Metallo-Supramolecular Complex Directed Enantioselective Self-Assembly of β-Sheet Breaker Peptide for Amyloid Inhibition, Small, 2015, 11(36), 4651–4655, DOI:10.1002/smll.201501329.
- B. Zhang, G. Gao, Z. Zhang, B. Cao and T. Sun, Ligand steric effects of nano-inhibitors on Aβ fibrillation at the nano-bio interfaces, Appl. Surf. Sci., 2023, 640, 158427, DOI:10.1016/j.apsusc.2023.158427.
- M. Nazıroğlu, S. Muhamad and L. Pecze, Nanoparticles as potential clinical therapeutic agents in Alzheimer's disease: focus on selenium nanoparticles, Expet Rev. Clin. Pharmacol., 2017, 10(7), 773–782, DOI:10.1080/17512433.2017.1324781.
- Y. Deng, G. Gao, L. Yu, Z. Zhang, B. Zhang, H. Li, X. Zhang, L. Shen and T. Sun, Engineering Core/Ligands Interfacial Anchors of Nanoparticles for Efficiently Inhibiting Both Aβ and Amylin Fibrillization, Small, 2024, 20(40), e2312046, DOI:10.1002/smll.202312046.
- G. Gao, T. Zhang, W. Zhang, Z. Luo, Z. Zhang, Z. Gu, L. Yu, Q. Mu and T. Sun, High efficiency and related mechanism of Au(RC) nanoclusters on disaggregating Aβ fibrils, J. Colloid Interface Sci., 2022, 621, 67–76, DOI:10.1016/j.jcis.2022.04.085.
- G. Gao, X. Liu, Z. Gu, Q. Mu, G. Zhu, T. Zhang, C. Zhang, L. Zhou, L. Shen and T. Sun, Engineering Nanointerfaces of Au25 Clusters for Chaperone-Mediated Peptide Amyloidosis, Nano Lett., 2022, 22(7), 2964–2970, DOI:10.1021/acs.nanolett.2c00149.
- S. Mourtas, A. N. Lazar, E. Markoutsa, C. Duyckaerts and S. G. Antimisiaris, Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease, Eur. J. Med. Chem., 2014, 80, 175–183, DOI:10.1016/j.ejmech.2014.04.050.
- K. Hou, H. Pan, H. Shahpasand-Kroner, C. Hu, R. Abskharon, P. Seidler, M. Mekkittikul, M. Balbirnie, C. Lantz, M. R. Sawaya, J. L. Dolinsky, M. Jones, X. Zuo, J. A. Loo, S. Frautschy, G. Cole and D. S. Eisenberg, D-peptide-magnetic nanoparticles fragment tau fibrils and rescue behavioral deficits in a mouse model of Alzheimer's disease, Sci. Adv., 2024, 10(18), eadl2991, DOI:10.1126/sciadv.adl2991.
- Y. Han, C. Gao, H. Wang, J. Sun, M. Liang, Y. Feng, Q. Liu, S. Fu, L. Cui, C. Gao, Y. Li, Y. Yang and B. Sun, Macrophage membrane-coated nanocarriers Co-Modified by RVG29 and TPP improve brain neuronal mitochondria-targeting and therapeutic efficacy in Alzheimer's disease mice, Bioact. Mater., 2021, 6(2), 529–542, DOI:10.1016/j.bioactmat.2020.08.017.
- C. Gao, Y. Wang, J. Sun, Y. Han, W. Gong, Y. Li, Y. Feng, H. Wang, M. Yang, Z. Li, Y. Yang and C. Gao, Neuronal mitochondria-targeted delivery of curcumin by biomimetic engineered nanosystems in Alzheimer's disease mice, Acta Biomater., 2020, 108, 285–299, DOI:10.1016/j.actbio.2020.03.029.
- J. M. Dowding, W. Song, K. Bossy, A. Karakoti, A. Kumar, A. Kim, B. Bossy, S. Seal, M. H. Ellisman, G. Perkins, W. T. Self and E. Bossy-Wetzel, Cerium oxide nanoparticles protect against Aβ-induced mitochondrial fragmentation and neuronal cell death, Cell Death Differ., 2014, 21(10), 1622–1632, DOI:10.1038/cdd.2014.72.
- Y. Zhang, Z. Wang, X. Li, L. Wang, M. Yin, L. Wang, N. Chen, C. Fan and H. Song, Dietary Iron Oxide Nanoparticles Delay Aging and Ameliorate Neurodegeneration in Drosophila, Adv. Mater., 2016, 28(7), 1387–1393, DOI:10.1002/adma.201503893.
- M. Ma, N. Gao, Y. Sun, J. Ren and X. Qu, A Near-Infrared Responsive Drug Sequential Release System for Better Eradicating Amyloid Aggregates, Small, 2017, 13(46), 1701817, DOI:10.1002/smll.201701817.
- H. J. Kwon, M.-Y. Cha, D. Kim, D. K. Kim, M. Soh, K. Shin, T. Hyeon and I. Mook-Jung, Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer's Disease, ACS Nano, 2016, 10(2), 2860–2870, DOI:10.1021/acsnano.5b08045.
- D. Kim, H. J. Kwon and T. Hyeon, Magnetite/Ceria Nanoparticle Assemblies for Extracorporeal Cleansing of Amyloid-β in Alzheimer's Disease, Adv. Mater., 2019, 31(19), e1807965, DOI:10.1002/adma.201807965.
- B. Hu, F. Dai, Z. Fan, G. Ma, Q. Tang and X. Zhang, Nanotheranostics: Congo Red/Rutin-MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer's Disease in APPswe/PS1dE9 Transgenic Mice, Adv. Mater., 2015, 27(37), 5499–5505, DOI:10.1002/adma.201502227.
- X. Cai, K. Zhang, X. Xie, X. Zhu, J. Feng, Z. Jin, H. Zhang, M. Tian and H. Chen, Self-assembly hollow manganese Prussian white nanocapsules attenuate Tau-related neuropathology and cognitive decline, Biomaterials, 2020, 231, 119678, DOI:10.1016/j.biomaterials.2019.119678.
- S. M. ElMorsy, D. A. Gutierrez, S. Valdez, J. Kumar, R. J. Aguilera, M. Noufal, H. Sarma, S. Chinnam and M. Narayan, Graphene acid quantum dots: A highly active multifunctional carbon nano material that intervene in the trajectory towards neurodegeneration, J. Colloid Interface Sci., 2024, 670, 357–363, DOI:10.1016/j.jcis.2024.05.072.
- Y. Dou, D. Zhao, F. Yang, Y. Tang and J. Chang, Natural Phyto-Antioxidant Albumin Nanoagents to Treat Advanced Alzheimer's Disease, ACS Appl. Mater. Interfaces, 2021, 13(26), 30373–30382, DOI:10.1021/acsami.1c07281.
- F. Mohebichamkhorami, M. Faizi, M. Mahmoudifard, A. Hajikarim-Hamedani, S. S. Mohseni, A. Heidari, Y. Ghane, M. Khoramjouy, M. Khayati, R. Ghasemi, H. Zali, S. Hosseinzadeh and E. Mostafavi, Microfluidic Synthesis of Ultrasmall Chitosan/Graphene Quantum Dots Particles for Intranasal Delivery in Alzheimer's Disease Treatment, Small, 2023, 19(40), e2207626, DOI:10.1002/smll.202207626.
- S. Marrache and S. Dhar, Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(40), 16288–16293, DOI:10.1073/pnas.1210096109.
- L. Yang, Y. Wang, Z. Li, X. Wu, J. Mei and G. Zheng, Brain targeted peptide-functionalized chitosan nanoparticles for resveratrol delivery: Impact on insulin resistance and gut microbiota in obesity-related Alzheimer's disease, Carbohydr. Polym., 2023, 310, 120714, DOI:10.1016/j.carbpol.2023.120714.
- A. Qu, M. Sun, J.-Y. Kim, L. Xu, C. Hao, W. Ma, X. Wu, X. Liu, H. Kuang, N. A. Kotov and C. Xu, Stimulation of neural stem cell differentiation by circularly polarized light transduced by chiral nanoassemblies, Nat. Biomed. Eng., 2021, 5(1), 103–113, DOI:10.1038/s41551-020-00634-4.
- Q. Song, M. Huang, L. Yao, X. Wang, X. Gu, J. Chen, J. Chen, J. Huang, Q. Hu, T. Kang, Z. Rong, H. Qi, G. Zheng, H. Chen and X. Gao, Lipoprotein-based nanoparticles rescue the memory loss of mice with Alzheimer's disease by accelerating the clearance of amyloid-beta, ACS Nano, 2014, 8(3), 2345–2359, DOI:10.1021/nn4058215.
- H. Zhang, Y. Zhao, M. Yu, Z. Zhao, P. Liu, H. Cheng, Y. Ji, Y. Jin, B. Sun, J. Zhou and Y. Ding, Reassembly of native components with donepezil to execute dual-missions in Alzheimer's disease therapy, J. Controlled Release, 2019, 296, 14–28, DOI:10.1016/j.jconrel.2019.01.008.
- M. Huang, M. Hu, Q. Song, H. Song, J. Huang, X. Gu, X. Wang, J. Chen, T. Kang, X. Feng, D. Jiang, G. Zheng, H. Chen and X. Gao, GM1-Modified Lipoprotein-like Nanoparticle: Multifunctional Nanoplatform for the Combination Therapy of Alzheimer's Disease, ACS Nano, 2015, 9(11), 10801–10816, DOI:10.1021/acsnano.5b03124.
- R.-R. Lin, L.-L. Jin, Y.-Y. Xue, Z.-S. Zhang, H.-F. Huang, D.-F. Chen, Q. Liu, Z.-W. Mao, Z.-Y. Wu and Q.-Q. Tao, Hybrid Membrane-Coated Nanoparticles for Precise Targeting and Synergistic Therapy in Alzheimer's Disease, Adv. Sci., 2024, 11(24), e2306675, DOI:10.1002/advs.202306675.
- M. Zhang, H. Chen, W. Zhang, Y. Liu, L. Ding, J. Gong, R. Ma, S. Zheng and Y. Zhang, Biomimetic Remodeling of Microglial Riboflavin Metabolism Ameliorates Cognitive Impairment by Modulating Neuroinflammation, Adv. Sci., 2023, 10(12), e2300180, DOI:10.1002/advs.202300180.
- J. Gu, C. Yan, S. Yin, H. Wu, C. Liu, A. Xue, X. Lei, N. Zhang and F. Geng, Erythrocyte membrane-coated nanocarriers modified by TGN for Alzheimer's disease, J. Controlled Release, 2024, 366, 448–459, DOI:10.1016/j.jconrel.2023.12.030.
- Z. Deng, J. Wang, Y. Xiao, F. Li, L. Niu, X. Liu, L. Meng and H. Zheng, Ultrasound-mediated augmented exosome release from astrocytes alleviates amyloid-β-induced neurotoxicity, Theranostics, 2021, 11(9), 4351–4362, DOI:10.7150/thno.52436.
- B. Liu, X. Li, H. Yu, X. Shi, Y. Zhou, S. Alvarez, M. J. Naldrett, S. D. Kachman, S.-H. Ro, X. Sun, S. Chung, L. Jing and J. Yu, Therapeutic potential of garlic chive-derived vesicle-like nanoparticles in NLRP3 inflammasome-mediated inflammatory diseases, Theranostics, 2021, 11(19), 9311–9330, DOI:10.7150/thno.60265.
- W. Ji, H. Zhou, W. Liang, W. Zhang, B. Gong, T. Yin, J. Chu, J. Zhuang, J. Zhang, Y. Luo, Y. Liu, J. Gao and Y. Yin, SSK1-Loaded Neurotransmitter-Derived Nanoparticles for Alzheimer's Disease Therapy via Clearance of Senescent Cells, Small, 2024, 20(26), e2308574, DOI:10.1002/smll.202308574.
- G. Han, K. Bai, X. Yang, C. Sun, Y. Ji, J. Zhou, H. Zhang and Y. Ding, Drug-Carrier Synergy Therapy for Amyloid-β Clearance and Inhibition of Tau Phosphorylation via Biomimetic Lipid Nanocomposite Assembly, Adv. Sci., 2022, 9(14), e2106072, DOI:10.1002/advs.202106072.
- A. Savchenko, G. B. Braun and E. Molokanova, Nanostructured Antagonist of Extrasynaptic NMDA Receptors, Nano Lett., 2016, 16(9), 5495–5502, DOI:10.1021/acs.nanolett.6b01988.
- B. Gong, W. Zhang, W. Cong, Y. Gu, W. Ji, T. Yin, H. Zhou, H. Hu, J. Zhuang, Y. Luo, Y. Liu, J. Gao and Y. Yin, Systemic Administration of Neurotransmitter-Derived Lipidoids-PROTACs-DNA Nanocomplex Promotes Tau Clearance and Cognitive Recovery for Alzheimer's Disease Therapy, Adv. Healthcare Mater., 2024, 13(32), e2400149, DOI:10.1002/adhm.202400149.
- S. Mourtas, M. Canovi, C. Zona, D. Aurilia, A. Niarakis, B. La Ferla, M. Salmona, F. Nicotra, M. Gobbi and S. G. Antimisiaris, Curcumin-decorated nanoliposomes with very high affinity for amyloid-β1-42 peptide, Biomaterials, 2011, 32(6), 1635–1645, DOI:10.1016/j.biomaterials.2010.10.027.
- M. Jung, S. Lee, S. Park, J. Hong, C. Kim, I. Cho, H. S. Sohn, K. Kim, I. W. Park, S. Yoon, S. Kwon, J. Shin, D. Lee, M. Kang, S. Go, S. Moon, Y. Chung, Y. S. Kim and B.-S. Kim, A Therapeutic Nanovaccine that Generates Anti-Amyloid Antibodies and Amyloid-specific Regulatory T Cells for Alzheimer's Disease, Adv. Mater., 2023, 35(3), e2207719, DOI:10.1002/adma.202207719.
- M. Gobbi, F. Re, M. Canovi, M. Beeg, M. Gregori, S. Sesana, S. Sonnino, D. Brogioli, C. Musicanti, P. Gasco, M. Salmona and M. E. Masserini, Lipid-based nanoparticles with high binding affinity for amyloid-beta1-42 peptide, Biomaterials, 2010, 31(25), 6519–6529, DOI:10.1016/j.biomaterials.2010.04.044.
- J. Zhao, F. Yin, L. Ji, C. Wang, C. Shi, X. Liu, H. Yang, X. Wang and L. Kong, Development of a Tau-Targeted Drug Delivery System Using a Multifunctional Nanoscale Metal-Organic Framework for Alzheimer's Disease Therapy, ACS Appl. Mater. Interfaces, 2020, 12(40), 44447–44458, DOI:10.1021/acsami.0c11064.
- Z. Fan, T. Ren, Y. Wang, H. Jin, D. Shi, X. Tan, D. Ge, Z. Hou, X. Jin and L. Yang, Aβ-responsive metformin-based supramolecular synergistic nanodrugs for Alzheimer's disease via enhancing microglial Aβ clearance, Biomaterials, 2022, 283, 121452, DOI:10.1016/j.biomaterials.2022.121452.
- W. Zhang, N. Kandel, Y. Zhou, N. Smith, C. L. B. Ferreira, P. Braulio, C. Miranda, L. Matteo, K. J. Mintz, C. Wang and R. M. Leblanc, Drug delivery of memantine with carbon dots for Alzheimer's disease: blood–brain barrier penetration and inhibition of tau aggregation, J. Colloid Interface Sci., 2022, 617, 20–31, DOI:10.1016/j.jcis.2022.02.124.
- D. Wang, X. Gu, X. Ma, J. Chen, Q. Zhang, Z. Yu, J. Li, M. Hu, X. Tan, Y. Tang, J. Xu, M. Xu, Q. Song, H. Song, G. Jiang, Z. Tang, X. Gao and H. Chen, Nanopolyphenol rejuvenates microglial surveillance of multiple misfolded proteins through metabolic reprogramming, Acta Pharm. Sin. B, 2023, 13(2), 834–851, DOI:10.1016/j.apsb.2022.07.014.
- C. Zhang, X. Wan, X. Zheng, X. Shao, Q. Liu, Q. Zhang and Y. Qian, Dual-functional nanoparticles targeting amyloid plaques in the brains of Alzheimer's disease mice, Biomaterials, 2014, 35(1), 456–465, DOI:10.1016/j.biomaterials.2013.09.063.
- S. K. Tiwari, S. Agarwal, B. Seth, A. Yadav, S. Nair, P. Bhatnagar, M. Karmakar, M. Kumari, L. K. S. Chauhan, P. Devendra Kumar, V. Srivastava, D. Singh, S. K. Gupta, A. Tripathi, R. K. Chaturvedi and K. C. Gupta, Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer's disease model via canonical Wnt/β-catenin pathway, ACS Nano, 2014, 8(1), 76–103, DOI:10.1021/nn405077y.
- G. Mittal, H. Carswell, R. Brett, S. Currie and M. N. V. R. Kumar, Development and evaluation of polymer nanoparticles for oral delivery of estradiol to rat brain in a model of Alzheimer's pathology, J. Controlled Release, 2011, 150(2), 220–228, DOI:10.1016/j.jconrel.2010.11.013.
- E. Sánchez-López, M. Ettcheto, M. A. Egea, M. Espina, A. Cano, A. C. Calpena, A. Camins, N. Carmona, A. M. Silva, E. B. Souto and M. L. García, Memantine loaded PLGA PEGylated nanoparticles for Alzheimer's disease: in vitro and in vivo characterization, J. Nanobiotechnol., 2018, 16(1), 32, DOI:10.1186/s12951-018-0356-z.
- Y. Tang, J. Gao, T. Wang, Q. Zhang, A. Wang, M. Huang, R. Yu, H. Chen and X. Gao, The effect of drug loading and multiple administration on the protein corona formation and brain delivery property of PEG-PLA nanoparticles, Acta Pharm. Sin. B, 2022, 12(4), 2043–2056, DOI:10.1016/j.apsb.2021.09.029.
- T. Lei, Z. Yang, C. Jiang, X. Wang, W. Yang, X. Yang, R. Xie, F. Tong, X. Xia, Q. Huang, Y. Du, Y. Huang and H. Gao, Mannose-Integrated Nanoparticle Hitchhike Glucose Transporter 1 Recycling to Overcome Various Barriers of Oral Delivery for Alzheimer's Disease Therapy, ACS Nano, 2024, 18(4), 3234–3250, DOI:10.1021/acsnano.3c09715.
- P. Zhang, Y. Xiao, X. Sun, X. Lin, S. Koo, A. V. Yaremenko, D. Qin, N. Kong, O. C. Farokhzad and W. Tao, Cancer nanomedicine toward clinical translation: Obstacles, opportunities, and future prospects, Med, 2023, 4(3), 147–167, DOI:10.1016/j.medj.2022.12.001.
| This journal is © The Royal Society of Chemistry 2025 |