
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of perovskite type oxide LaxCe1−xcoo3 in lignin depolymerization and mono-phenol preparation
Tao Zhuab,
Yujie Wang *a,
Miao Changa,
Jie Xua and
Xiaobin Haoa
*a,
Miao Changa,
Jie Xua and
Xiaobin Haoa
aSchool of Materials and Chemical Engineering, Chuzhou University, Chuzhou, Anhui 239000, China. E-mail: wangyj120@163.com
bSchool of Life Sciences, Anhui University, Hefei, Anhui 230601, China
First published on 17th January 2025
Abstract
This study successfully prepared LaxCe1−xCoO3 (x = 0.2, 0.4, 0.6, 0.8, 1) series perovskite oxide catalysts by co precipitation, and found that La0.6Ce0.4CoO3 can significantly improve the yield of bio-oil under specific conditions (lignin to catalyst mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, reaction temperature of 240 °C, time of 10 hours, using methanol and ethanol as solvents). Furthermore, NH3-TPD, GC-MS, and FTIR analyses revealed the thermal decomposition behavior of key bonding structures such as β-O-4 and C–O–C during the catalytic process, while generating various mono-phenolic products such as guaiacol and 2,6-dimethylphenol. In addition, studies have shown that the physicochemical properties of perovskite type oxide catalysts have a significant impact on the chemical properties of lignin oil. By increasing the acidity of the catalyst, not only can the yield of lignin oil and phenols be improved, but also the yield of carbon can be reduced. More importantly, the catalyst performed well in the test of lignin catalyzed hydrogenation to produce monophenols, significantly improving the conversion rate of lignin and the yield of various monophenols compared to noncatalytic processes.
:2, reaction temperature of 240 °C, time of 10 hours, using methanol and ethanol as solvents). Furthermore, NH3-TPD, GC-MS, and FTIR analyses revealed the thermal decomposition behavior of key bonding structures such as β-O-4 and C–O–C during the catalytic process, while generating various mono-phenolic products such as guaiacol and 2,6-dimethylphenol. In addition, studies have shown that the physicochemical properties of perovskite type oxide catalysts have a significant impact on the chemical properties of lignin oil. By increasing the acidity of the catalyst, not only can the yield of lignin oil and phenols be improved, but also the yield of carbon can be reduced. More importantly, the catalyst performed well in the test of lignin catalyzed hydrogenation to produce monophenols, significantly improving the conversion rate of lignin and the yield of various monophenols compared to noncatalytic processes.
1 Introduction
With the increasingly severe global energy crisis and environmental issues, developing efficient and environmentally friendly energy conversion and storage technologies has become an important topic in today's scientific research.1 Among numerous renewable biomass resources, lignin is the only nonpetroleum resource in nature that can provide renewable aromatic compounds. Its efficient utilization is of great significance for promoting the development of green chemistry and bioenergy.2 Lignin is a three-dimensional amorphous high molecular weight aromatic polymer mainly composed of three phenylpropane structural units, namely hydroxyphenyl, guaiacyl, and syringayl, connected by C–O–C and C–C bonds.3,4 However, due to the complex structure, strong stability, and large molecular size of lignin, its effective utilization has always been a challenge.In order to fully utilize lignin and make it profitable, we need to rely on depolymerization technology to extract the platform chemicals from lignin. At present, various thermochemical methods have been studied, which can depolymerize lignin into monomers and dimers, mainly alkaline hydrolysis,5 acid hydrolysis, pyrolysis,6 oxidation,7 and hydrogenolysis.8 Among them, hydrogenolysis is one of the effective methods for depolymerizing lignin into phenolic biooil, and the reaction conditions are mild. For hydrogenolysis, the source of hydrogen can be H2 molecules or hydrogen donating reagents. It is worth noting that in the process of lignin hydrolysis, alcohol solvents, especially methanol, ethanol, and isopropanol, are widely used due to their unique properties. These solvents exhibit high efficiency in dissolving lignin and possess potential hydrogen donor characteristics, making them particularly outstanding solvent choices in lignin hydrolysis processes.9 Barta et al.10 used supercritical methanol as a solvent and Cu doped porous metal oxides as catalysts, effectively promoting the depolymerization of lignin and converting it into high-value monomer compounds. Ma et al.11 used isopropanol as a solvent and hydrogen transfer promoter, and employed Ni/ZrP catalyst to hydrogenate lignin. The results showed that isopropanol was more effective in H2 generation and facilitated the hydrogenation deoxygenation of lignin derived phenolic compounds, inhibiting the re polymerization reaction between unstable intermediates and phenols.
However, the hydrogenation of lignin into high yield, high selectivity, and low-cost monophenols remains a challenge. To address these issues, various catalysts have been used to break the ether bonds of lignin, including homogeneous catalysts and heterogeneous metal catalysts. Metal heterogeneous catalysts have the advantages of selectivity, reusability, and no emission of pollutants, and are widely used in the catalytic cracking of lignin and lignin model compounds. Metal catalysts are generally dominated by precious metals such as ruthenium,12 platinum,13 palladium, etc. Although precious metals can provide many attractive properties, they are difficult to meet the needs of industrial scale due to their high price and limited quantity. Therefore, it is necessary to develop inexpensive catalysts with better catalytic effects. Some inexpensive metals, such as Ni,14 Fe,15 Cu,16 Co,17 etc., have been explored in some research reports.
Zhao et al.18 achieved selective hydrogenolysis of renewable lignin using low-cost catalyst Ni/MgO, achieving a lignin conversion rate of 93.4% and a phenolic monomer yield of 15.0% under optimal reaction conditions. Compared with pure MgO, the introduction of Ni can effectively improve the conversion rate of lignin and increase the yield of bio-oil. Co based catalysts also exhibit good performance in catalyzing hydrogenolysis. Recently, some researchers have turned to bimetallic catalysts because compared to single metal catalysts, bimetallic catalysts can form more catalytic centers through interactions between metal atoms. These catalytic centers can more effectively bind with reactants in the reaction, promoting the progress of the reaction.
Jiang et al.19 investigated the catalytic performance of Ni Pd bimetallic catalysts for lignin depolymerization. Among them, the Ni50Pd50/SBA-15 catalyst has the best depolymerization effect on corn stover lignin, with the highest total mono-phenolic yield (8.14%), which is 1.96 times that of the single metal catalyst Ni/SBA-15 and 1.44 times that of Pd/SBA-15. Therefore, developing an inexpensive bimetallic catalyst to catalyze the depolymerization of lignin is of great significance.
Perovskite materials, with their unique crystal structure and excellent physical and chemical properties, have demonstrated extraordinary application potential in many fields. In the field of energy conversion,20,21 perovskite materials have become a key component in systems such as solar cells and fuel cells due to their efficient energy conversion capabilities, playing a crucial role and greatly promoting the development and effective utilization of clean energy. In the field of materials science, the tunable optoelectronic properties and stability of perovskite materials make them ideal candidates for photodetectors and storage devices.22,23 In addition, in the field of catalysis, perovskite materials are widely used in catalytic oxidation,24,25 photocatalytic water splitting,26 and environmental pollutant degradation27 due to their abundant active sites and good stability, demonstrating excellent performance.
Lignin depolymerization, as a key step in biomass conversion, also requires catalytic materials with high activity and stability to break its complex molecular structure and achieve high selectivity and efficiency in conversion. The performance characteristics exhibited by perovskite materials in other fields, such as controllable active sites, high stability, and good adaptability to complex chemical reactions, are highly compatible with the core requirements of lignin depolymerization process.
Especially, the ability of perovskite materials to optimize catalytic performance by finely regulating their composition and structure makes them an ideal catalyst for lignin depolymerization. Therefore, inspired by the achievements of perovskite materials, introducing them into the field of lignin depolymerization for research is not only a natural extension of their application, but also an important exploration direction for efficient utilization of biomass resources. This cross disciplinary attempt will help reveal the potential of perovskite materials in biomass catalytic conversion, promoting the efficient conversion of biomass into high value-added chemicals and energy.
Among them, LaxCe1−xCoO3, as a new type of perovskite catalyst, not only inherits the high stability characteristics of traditional perovskite materials, but also achieves catalytic performance control by regulating the composition of A-site ions (such as La/Ce ratio), providing the possibility for efficient implementation of specific catalytic reactions. This unique tunability has increasingly attracted attention to the application research of LaxCe1−xCoO3 in fields such as catalytic oxidation and photocatalysis, and has achieved a series of remarkable results. However, despite the enormous potential of LaxCe1−xCoO3 in various catalytic fields, its research in the key biomass conversion field of lignin depolymerization is almost blank.
Lignin, as a renewable resource with abundant reserves in nature, its effective depolymerization and transformation are of great significance for the development of sustainable energy and chemicals. Given this, we have reason to believe that by preparing LaxCe1−xCoO3 catalysts with different La/Ce ratios and systematically studying their performance in catalyzing the depolymerization of sodium lignosulfonate, we aim to reveal the relationship between catalyst structure and performance, and provide new approaches and strategies for the efficient conversion and utilization of biomass resources. Specifically, this study will comprehensively evaluate the catalytic activity, selectivity, and stability of LaxCe1−xCoO3 catalyst in the sodium lignosulfonate depolymerization reaction through steps such as catalyst preparation, characterization, and performance testing. At the same time, combined with the structural analysis of catalysts, key scientific issues such as active sites and reaction mechanisms of catalysts are explored in depth, providing scientific basis for further optimizing catalyst performance and expanding its application scope. In summary, this study not only has significant academic value, but also has important practical significance for promoting the high-value utilization of lignin and promoting the sustainable development of biomass resources.
2 Experimental section
2.1 Catalyst preparation
LaxCe1−xCoO3 (x = 0.2, 0.4, 0.6, 0.8, 1) was prepared by co precipitation method, and a transparent solution was prepared by mixing stoichiometric amounts of lanthanum nitrate hexahydrate, cerium nitrate hexahydrate, and cobalt nitrate hexahydrate. Subsequently, under stirring in an 80 °C water bath, a precipitant sodium carbonate solution was added to adjust the pH value to 8.0–8.5, resulting in a purple flocculent mixture. Then precipitate the mixture overnight and filter under vacuum. The sample obtained by filtration was dried in a drying oven at 110 °C for 12 hours, and then calcined in a muffle furnace at 800 °C for 5 hours to obtain LaxCe1−xCoO3 (x = 0.2, 0.4, 0.6, 0.8, 1) perovskite samples.2.2 Catalyst characterization
The BSD-PS2 analyzer was used to measure the N2 adsorption desorption isotherm at N2 isothermal (196 °C), and the pore structure and specific surface area of the catalyst were determined. Calculate the specific surface area, pore size, and pore volume using BET equation and BJH model, respectively. The crystal phase analysis of the sample was carried out using a powder X-ray diffractometer (XRD) equipped with Cu Kα radiation (1¼ 0.154 nm), 40 kV, 30 mA, and a scanning range of 10–80. In order to observe the morphology and dispersion of surface elements, scanning electron microscopy (SEM) analysis and energy dispersive X-ray spectroscopy (EDS) plotting were used. The elemental states of metals were measured using X-ray photoelectron spectroscopy (XPS) experiments on a Thermo-Fisher Scientific K-A apparatus. The acidity of the catalyst was measured using a BELCAT-B instrument from Osaka, Japan, and the acidic sites under NH3 gas were determined using TCD.2.3 Depolymerization of lignin
The depolymerization experiment of lignin was conducted in a high-pressure reactor. Firstly, add 0.6 g of sodium lignosulfonate and 0.3 g of catalyst (with a mass ratio of lignin to catalyst of 2:1) into the reaction vessel. Then add 10 mL of isopropanol and 10 mL of ethanol. Finally, heat the reactor to the expected temperature (180–260 °C) and react for 6–14 hours. After the reaction is complete, cool the reactor to ambient temperature. The reaction product is divided into three parts: gas, liquid, and solid. Due to the relatively small gas production rate (less than 0.5 wt%), it can be ignored. Use filtration method for solid–liquid separation and wash the kettle body with anhydrous ethanol. The obtained liquid product is subjected to solvent removal under a rotary evaporator to obtain biooil. In order to recycle the used catalyst, the coke, catalyst, and unreacted lignin were washed with acetone and dimethyl sulfoxide solution, respectively. The mixed residual solid obtained after washing was dried and ground into powder, and then calcined in a muffle furnace. The obtained solid is the regenerated catalyst. Repeat all experiments three times and take the average to reduce errors.
2.4 Analysis of lignin conversion products
Gas chromatography-mass spectrometry (GC-MS, Agilent 7890A) and HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm) were used to detect and identify the components of bio-oil. The working conditions of GC-MS are: the initial temperature of the column temperature chamber is 80 °C, and then the temperature is raised to 200 °C at a rate of 10 °C min−1. The quantitative analysis of phenolic monomers after the reaction and the solvent analysis collected after the reaction were performed using flame ionization detection gas chromatography (GC-FID, Agilent 6890N). The chromatographic column is an HP-5 MS capillary column (30 m × 0.25 mm × 0.25 μm). The working conditions of GC-FID are as follows: maintain the column temperature at 70 °C for 2 minutes, then increase it to 200 °C at a rate of 10 °C min−1, and maintain it for another 2 minutes. The yields of biooil, monomers, and charcoal are determined by the following formula: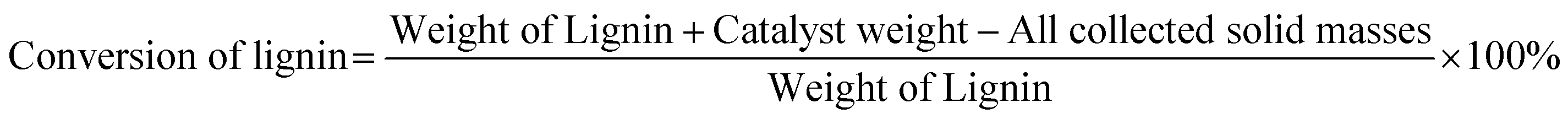



The changes in functional groups of lignin and product biooil were detected using the VERTEX 80V infrared spectroscopy (FT-IR) instrument from Bruker, Germany. Solid KBr was used for compression, with a sample to KBr mass ratio of 1/100 and a detection wavelength range of 4000–400 cm−1. Each spectrum was scanned 32 times.
Determine the elemental composition content of raw lignin and product biooil using 2400 II PE from the United States. Wrap the test sample in aluminum foil, set the O2 and He flow rates to 30 mL min−1 and 250 mL min−1, respectively, and burn at 1200 °C. The content of C, H, and N elements can be measured, and the content of O element can be obtained using the law of conservation of mass. And the Dulong formula can be used to calculate the high calorific value of related products: HHV (MJ/Kg) = 0.335 × C + 1.422 × H − 0.154 × O − 0.145 × N.
3 Results and discussion
3.1 Characterization of LaxCe1−xCoO3 catalysts
 | ||
| Fig. 1 XRD spectra of LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) series catalysts. | ||
The occurrence of this phenomenon may be due to the following reasons: in traditional perovskite structures, A-site ions are usually divalent or trivalent, while Ce3+ ions are more unstable and easily oxidized to Ce4+ ions. Therefore, in the catalyst, Ce mainly exists in the state of Ce4+. However, if all Ce4+ ions enter the structure of perovskite, it will lead to charge imbalance in the LaxCe1−xCoO3 catalyst. In order to compensate for this charge imbalance, some La and Ce ions did not participate in the formation of the perovskite structure, resulting in a corresponding number of B-site transition metal ions not entering the perovskite structure, but existing in the form of their respective oxides.
 | ||
| Fig. 2 SEM image of LaxCe1−x CoO3 catalysts ((A) x = 0, (B) x = 0.2, (C) x = 0.4, (D) x = 0.6, (E) x = 0.8). | ||
From the distribution of La element in Fig. 3, it can be seen that as the Ce content gradually increases, the concentration of La element gradually decreases, which is consistent with the expected design of La being partially replaced by Ce, further confirming the successful doping of Ce into the catalyst lattice. Specifically, in the La0.6Ce0.4CoO3 catalyst (Fig. 3C), La, Ce, O, and Co elements exhibit uniform distribution, indicating that all elements have good dispersibility within the catalyst. This good dispersibility may be a key factor in the superior catalytic performance of the La0.6Ce0.4CoO3 catalyst. It is worth noting that as the content of Ce element increases (as shown in Fig. 3D and E), the surface of the catalyst becomes rougher, which is attributed to the possible induction of uneven grain growth and the formation of irregular clusters by the addition of Ce element. In catalysts with lower Ce content, such as La0.6Ce0.4CoO3 (Fig. 3C), the appropriate introduction of Ce not only helps to improve the redox performance of the catalyst and promote the generation of active oxygen species, but also maintains the stability of the lattice structure, thereby gradually improving the catalytic performance.
 | ||
| Fig. 3 EDS spectrum of LaxCe1−x CoO3 ((A) x = 0, (B) x = 0.2, (C) x = 0.4, (D) x = 0.6, (E) x = 0.8). | ||
 | ||
| Fig. 4 The nitrogen adsorption desorption isotherms (A) and pore size distribution curves (B) of LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) series catalysts prepared. | ||
In mesoporous materials, due to the small pore size, N2 molecules are subject to certain limitations during the adsorption process, resulting in incomplete reversibility of the adsorption and desorption processes, thus forming hysteresis loops on the isotherm.
Table 1 presents the specific surface area, pore size, and pore volume results of LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) perovskite catalysts. Among them, the specific surface area of the catalyst was calculated by the Brunner–Emmett–Teller (BET) method, while the pore size and pore volume were calculated by the Barrett–Joyner–Halenda (BJH) desorption method. According to the table, the specific surface area of LaxCe1−xCoO3 catalyst is significantly larger than that of LaCoO3 catalyst (6.2792 m2 g−1), indicating that cerium doping may alter the surface structure and active sites of LaCoO3. But when the doping ratio of Ce is 0.4, the specific surface area of the catalyst (4.1991 m2 g−1) is smaller than that of undoped (6.2792 m2 g−1), which may be due to the fact that doping sacrifices some of the specific surface area. Based on the XRD pattern, we can observe that although there were no significant impurity peaks observed at a Ce doping level of 0.4, the slight changes in lattice parameters indicate that the lattice structure has been finetuned, which may lead to lattice distortion or differences in ion radius, further affecting the surface structure of the catalyst. Secondly, the observation results of SEM images show that when the Ce doping amount is 0.4, the surface of the catalyst particles becomes smoother and the pore structure between particles is reduced. This morphological change is consistent with the specific surface area data calculated by BET method, that is, the specific surface area of LaxCe1−xCoO3 catalyst is smaller when Ce doping is 0.4 than when undoped. The phenomenon of surface smoothing and reduction of pore structure may be due to surface reconstruction caused by Ce doping. The surface reconstruction caused by doping may reduce the roughness and pore structure, thereby reducing the specific surface area. Although surface reconstruction may create new conditions for improving catalytic performance, it seems that this reconstruction has not yet reached its optimal state in La0.6Ce0.4CoO3 catalyst, resulting in a failure to maintain a high level of specific surface area.
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| La0.2Ce0.8CoO3 | 6.9267 | 0.1349 | 77.9012 |
| La0.4Ce0.6CoO3 | 6.5720 | 0.0690 | 41.9963 |
| La0.6Ce0.4CoO3 | 4.1991 | 0.0671 | 63.9190 |
| La0.8Ce0.2CoO3 | 8.7927 | 0.1024 | 46.5841 |
| LaCoO3 | 6.2792 | 0.1186 | 75.5511 |
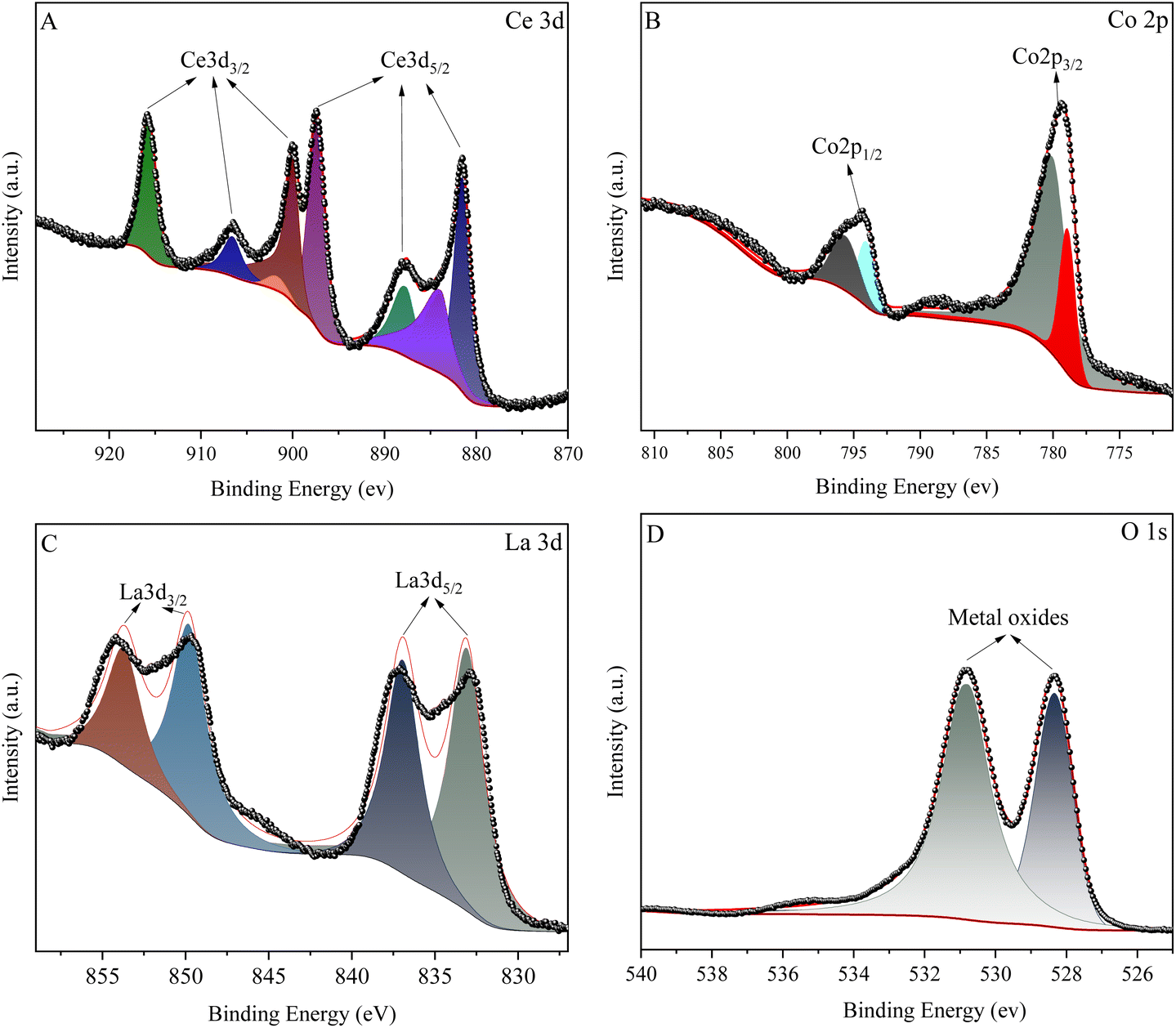 | ||
| Fig. 5 XPS spectrum of La0.6Ce0.4CoO3 catalyst. ((A) Ce 3d, (B) Co 2p, (C) La 3d, (D) O 1s). | ||
In the XPS spectrum of Ce3d (Fig. 5a), eight peaks were successfully fitted. Among them, the four peaks of 899.99 eV, 901.77 eV, 906.58 eV, and 915.74 eV belong to the Ce3d3/2 region, while the four peaks of 881.51 eV, 883.88 eV, 887.69 eV, and 897.39 eV belong to the Ce3d5/2 region. Specifically, the peaks at 883.88 eV and 906.58 eV correspond to the characteristics of Ce3+, while the remaining peaks represent Ce4+ with different electronic configuration states. This indicates that in La1−xCexCoO3, the Ce element has a variable valence, but mainly exists in the form of Ce4+, which is attributed to the good thermal stability of Ce4+at higher calcination temperatures.
For the XPS spectrum of Co2p (Fig. 5b), the Co2p spectrum is divided into two asymmetric parts: Co2p1/2 and Co2p3/2. In the Co2p3/2 region, peaks with binding energies of approximately 778.92 eV and 780.06 eV correspond to Co3+ and Co2+, respectively. The sub peaks at 796.65 eV and 794.07 eV are attributed to Co2+ and Co3+ in Co2p1/2, respectively. Therefore, we can confirm that the Co element in the sample exists in the form of Co2+ and Co3+. As Ce3+ replaces La3+, the relative concentration of Co2+ slightly increases. This is because when the doping of Ce3+ reaches saturation, some Ce3+ will transform into Ce4+ species. In order to maintain electronic neutrality, the valence state of some cobalt in the B site will change from Co3+ to Co2+. The increase in Co2+ content helps promote the formation of oxygen vacancies. Meanwhile, the presence of Ce also promotes the conversion of Co2+/Co3+ redox pairs, effectively improving the catalytic performance of the catalyst.
In Fig. 5c, the XPS spectrum of element La was successfully fitted as two 3d3/2 peaks (located at 849.79 eV and 853.79 eV) and two 3d5/2 peaks (located at 833.09 eV and 836.95 eV). In addition, the O 1s spectrum shown in Fig. 5d reveals two main oxygen species at 530.79 eV and 528.33 eV, corresponding to chemisorbed oxygen and surface lattice oxygen, respectively.
As shown in Fig. 6, the LaCoO3 catalyst exhibits broad NH3 desorption peaks at 473.3 °C, 645.0 °C, and 789.8 °C, indicating that the catalyst only contains strong acidic sites. After doping with Ce element, the NH3 adsorption peaks of La0.2Ce0.8CoO3 and La0.4Ce0.6CoO3 are single, which may indicate that the high proportion of Ce element doping changes the original distribution of acidic sites, resulting in the disappearance or merging of some acidic sites into one main acidic site.
 | ||
| Fig. 6 NH3-TPD curves of LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) series catalysts prepared. | ||
As the Ce doping ratio decreases, the strong acidic sites NH3 desorption peak of La0.8Ce0.2CoO3 and La0.6Ce0.4CoO3 catalysts gradually flattens, and more acidic sites of different intensities appear. This change indicates that moderate doping of Ce can increase the number of strong acidic sites in the catalyst, thereby enhancing its total acidity. This enhanced acidic site is beneficial for the activation and hydrogenolysis of C–O bonds by the catalyst, thereby improving the catalytic hydrogenolysis performance of La0.6Ce0.4CoO3.
3.2 Products yield
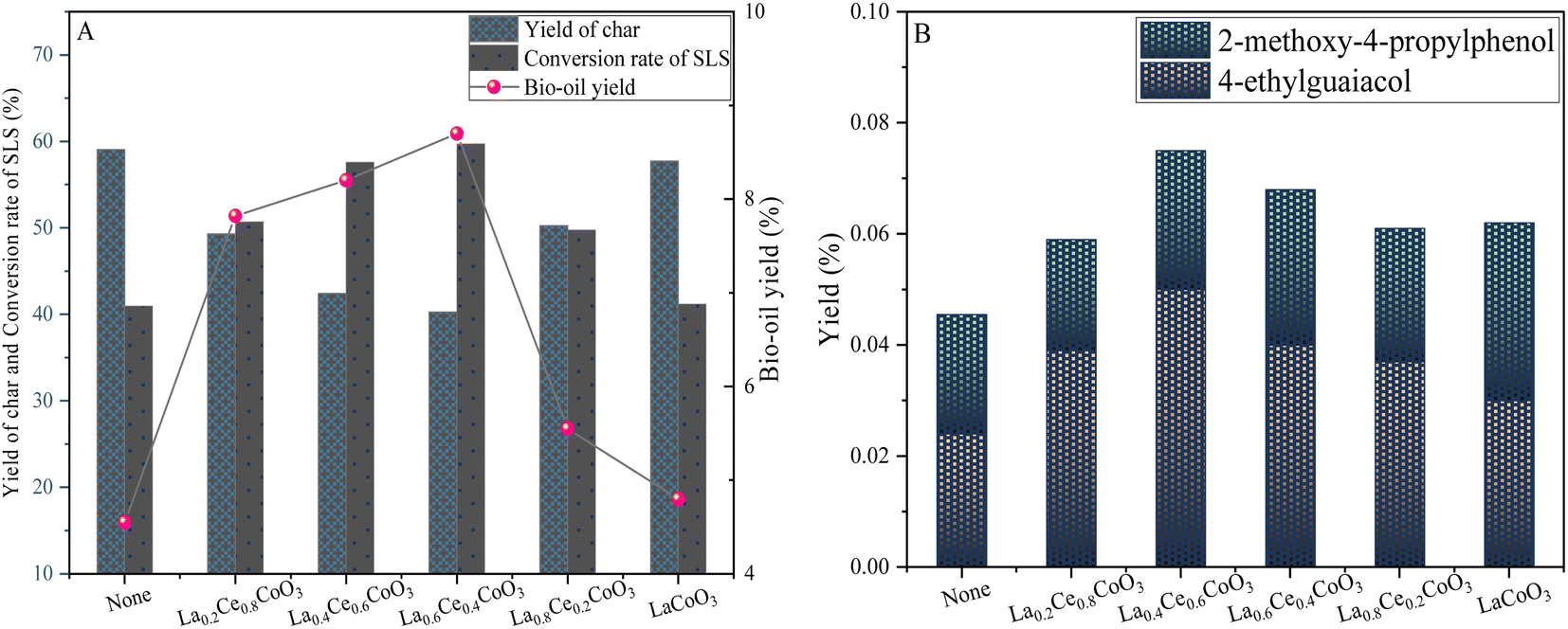 | ||
| Fig. 7 Yield plots of lignin sulfonate sodium depolymerization products (A) and phenolic products (B) for LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) series catalysts (0.6 g sodium lignosulfonate, 0.3 g catalyst, 240 °C, 1.1 MPaN2, 12 h, 10 mL anhydrous ethanol and 10 mL isopropanol solvent system). | ||
It is worth noting that all catalysts have some degree of improved product yield compared to non-catalytic experiments. With the increase of Ce doping from 0 to 0.4, the bio-oil yield increased significantly from 4.8 wt% to 8.7 wt%, and the yields of 4-ethylguaiacol and 2-methoxy-4-propylphenol were also significantly improved. At the same time, the conversion rate of lignin also increased from 41.16 wt% to 59.7 wt%. This increase may be attributed to the fact that the doping of Ce elements changed the crystal structure and surface properties of the LaCoO3 catalyst. We found that the specific surface area of the catalyst increased with the increase of Ce doping. For example, the specific surface area of the La0.4Ce0.6CoO3 catalyst is 6.5720 m2 g−1, which is increased compared to the undoped LaCoO3 (6.2792 m2 g−1), which is conducive to providing more active sites for the dispersion of the catalyst and the conversion of lignin (Table 1). However, the La0.8Ce0.2CoO3 catalyst has the largest specific surface area, but its depolymerization effect on lignin is not outstanding due to the low amount of Ce doping, relatively few active sites in the catalyst, and relatively limited redox ability. These results indicate that the change of Ce doping content is a key variable, but the effect on lignin catalytic depolymerization is nonlinear.
When the doping amount of Ce was increased to 0.4 to form La0.6Ce0.4CoO3 catalyst, although the specific surface area was the lowest among the LaxCe1−xCoO3 catalysts (x = 0.2, 0.4, 0.8, 1), the lignin conversion rate and bio-oil yield were the largest, indicating that although the specific surface area was an important factor affecting the catalytic effect in the LaxCe1−xCoO3 catalysts, it was not the only determinant. The activity of the catalyst is also affected by a combination of various factors such as acidity. Therefore, the reason why the La0.6Ce0.4CoO3 catalyst exhibits the best catalytic effect is that it achieves a relatively optimal equilibrium state in these aspects.
However, when the adulteration of Ce was further increased from 0.4 to 0.8, the yield of bio-oil decreased, from 8.7 wt% to 7.82 wt%, while the conversion rate of lignin also decreased from 59.7 wt% to 50.7 wt%. This phenomenon indicates that although the doping of too high Ce metal in La0.4Ce0.6CoO3 and La0.2Ce0.8CoO3 catalysts increases the active metal site, it also brings problems such as uneven metal distribution on the surface of the catalyst and excessive metal particle size, which may block the reaction site and lead to excessive hydrolysis of lignin.
Based on the above analysis, this study concluded that it is more appropriate to fix the doping amount of Ce metal in the catalyst at 0.6, which can not only ensure sufficient active sites, but also avoid the negative effects caused by excessive doping.
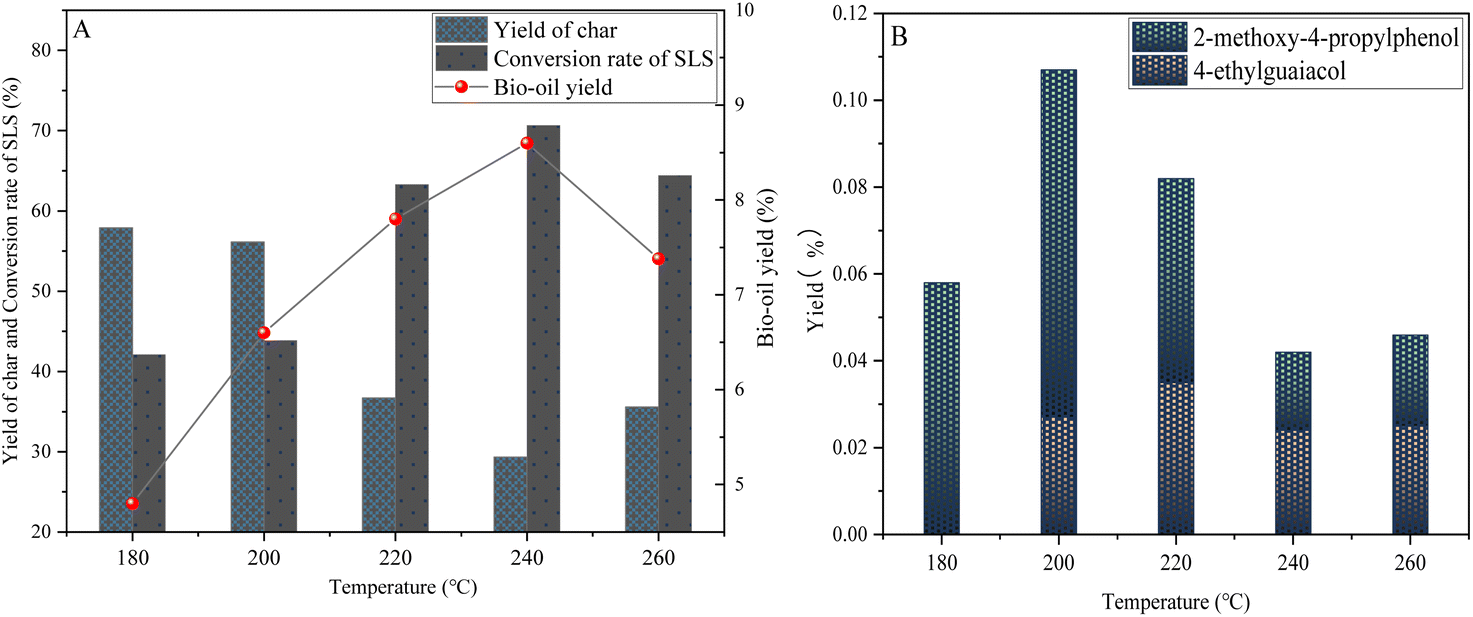 | ||
| Fig. 8 Yield of sodium lignosulfonate depolymerization products under temperature (A) and phenolic product yield (B) (0.6 g sodium lignosulfonate, 0.3 g catalyst, 1.1 MPaN2, 12 h, 10 mL anhydrous ethanol and 10 mL isopropanol solvent system). | ||
This study indicates that when at the initial temperature point (180 °C), the vibration inside the lignin molecule is slowed down, which makes it difficult for the otherwise stable chemical bonds (such as β-O-4 ether bonds, α-O-4 ether bonds, etc.) to obtain enough energy to break. Therefore, the depolymerization effect of lignin under low temperature conditions is not good, and it is difficult to effectively release small molecule lignin fragments, which limits the formation of important phenolic compounds (such as 4-ethylguaiacol in bio-oil) in the subsequent conversion process.
However, with the gradual increase of temperature (200–240 °C), the depolymerization effect of lignin showed significant changes. This change has a profound impact on the production efficiency of bio-oil and the amount of carbon by-products generated.
When the temperature rises to 200 °C, the vibration of lignin molecules begins to intensify, making it easier for previously stable chemical bonds (such as β-O-4, α-O-4 ether bonds, etc.) to break, releasing more active intermediates. These intermediates are effectively adsorbed under the action of mesoporous catalysts, thereby limiting their free movement, reducing the chance of repolymerization, increasing their contact frequency with the active metal sites of the catalyst, promoting bio-oil yield and lignin conversion, which peaks at 240 °C.
At 240 °C, this effect reaches its peak, at which point the bio-oil yield and lignin conversion rate both reach their highest points within their respective temperature ranges. At the same time, the yield of phenolic substances such as 4-ethylguaiacol also significantly increases. However, it is worth noting that when the temperature further increased to 260 °C, the situation reversed. Although higher temperatures should theoretically be more conducive to the breaking of chemical bonds, at this time, the intermediate reaction activity is too strong, irregular collisions between molecules intensify, and condensation is prone to occur, ultimately forming a large amount of coke. Therefore, the yield of bio-oil decreased to 7.38 wt%, while the yield of coke sharply increased to 35.58 wt%.
Based on the study of temperature in the lignin depolymerization reaction and energy-saving considerations, 240 °C was identified as the ideal reaction temperature, and the yields of phenolic substances such as 4-ethylguaiacol and 2-methoxy-4-propyllignin have been significantly improved in this temperature range.
 | ||
| Fig. 9 Time dependent graph of lignin sulfonate sodium depolymerization product yield (A) and phenolic product yield graph (B) (0.6 g sodium lignosulfonate, 0.3 g catalyst, 1.1 MPaN2, 240 °C, 10 mL anhydrous ethanol and 10 mL isopropanol solvent system). | ||
However, when the reaction time was further extended to 12 hours, although the lignin conversion rate continued to increase to 70.65 wt%, the bio-oil yield unexpectedly decreased to 6.86 wt%, this phenomenon may be due to the inactivation of the active site as the reaction continued, and the active site was covered by the depolymerization product.
The effective specific surface area decreases and the catalytic efficiency decreases. At the same time, the coke yield also decreased slightly to 29.3 wt%, but this does not mean that all the depolymerization products are effectively converted to bio-oil, but some of the products may have formed more complex coke structures through condensation reactions.
The data in Fig. 9(B) confirms this trend: during the process of extending the reaction time from 6 hours to 10 hours, the yield of 4-ethylguaiacol significantly increases, indicating that the catalyst has a high selective catalytic ability for the cleavage of specific chemical bonds (such as ethyl and methoxy groups on the aromatic ring) within this time range. However, the yields of 4-ethylguaiacol and 2-methoxy-4-propylphenol decreased slightly at 12 and 14 hours, which may be related to changes in acidity. Excessive reaction time may lead to uneven acidity distribution on the surface of the catalyst, weaken its catalytic effect on the target phenolic compound, and at the same time promote the polycondensation reaction between the depolymerization products, thereby reducing the content of target phenols.
In summary, although prolonging the reaction time can improve the yield of mono-phenolic compounds within a certain range, too long reaction time will lead to polycondensation reaction of the depolymerization products due to factors such as excessive molecular cleavage, thermodynamic equilibrium shift, reduction of specific surface area and change of acidity, resulting in coke production and reduction of phenolic content. Therefore, considering the depolymerization effect and energy consumption, 10 hours was considered to be an ideal reaction time for the depolymerization effect of lignin.
As shown in Fig. 10(A), when the mixed solvent of methanol and ethanol is used, the bio-oil yield is significantly increased to 13.05 wt%, which is much higher than that of single solvent methanol (8.61 wt%) or ethanol (11.28 wt%), and the carbon yield is also relatively low. This finding shows that the mixing of alcohol solvents as a good hydrogen donor can effectively enhance hydrogen transport and promote the hydrolysis of lignin.
 | ||
| Fig. 10 Yield plots of solvent induced depolymerization products of sodium lignosulfonate (A) and phenolic products (B) (0.6 g sodium lignosulfonate, 0.3 g catalyst, 1.1 MPaN2, 10 h, 240 °C). | ||
Further analysis showed that the alcohol solvent not only served as the reaction medium, but also provided a stability mechanism for the lignin depolymerization process through its unique chemical properties. Specifically, the hydroxyl group of the alcohol solvent forms hydrogen bonds or other weak interactions with the active sites on the lignin fragment, stabilizing the depolymerized fragment that is susceptible to reaggregation or degradation (as described in Fig. 10(B)). This stabilization effect significantly reduces ineffective reaggregation reactions, increasing the purity and yield of the target product.
In particular, the blended solvents exhibit superior properties over the individual solvents. The mixing of alcohols such as methanol, ethanol and isopropanol optimizes the physicochemical properties of the solvent, such as solubility, polarity and hydrogen bond formation capacity, through the synergistic effect between the components. This optimization not only promotes the efficient transport of hydrogen, but also effectively inhibits the occurrence of adverse side reactions, so that the yield of target products such as 4-ethylguaiacol and 2-methoxy-4-propyllignanl is much higher than that of other solvent systems.
In conclusion, the alcohol solvents and their hybrid systems significantly improved the yield of biooil and the selectivity of target products by providing a stable chemical environment, promoting the transfer and utilization of hydrogen, and inhibiting the repolymerization of depolymerization products during lignin-catalyzed hydrogenolysis. This discovery not only provides a new strategy for the efficient conversion of lignin, but also opens up new avenues for the production of biofuels and chemicals.
Compared with ref. 28 and 29 its production process is simpler and does not require complex loading processes. Only a single catalyst is needed to achieve efficient catalysis, and it exhibits unique catalytic advantages under similar lignin depolymerization conditions.
The yield of its EA soluble product is as high as 13.05 wt%, which is at a relatively high level among similar catalysts (ref. 28 and 30), fully demonstrating the excellent catalytic activity of the catalyst. What is particularly outstanding is that the catalyst has excellent selectivity for the main product 4-ethylguaiacol, which has high industrial application value. In addition, the catalytic system can achieve efficient conversion under relatively mild reaction conditions (lower temperature and pressure), which not only reduces energy consumption but also provides convenience for subsequent industrial scaling up. In summary, the La0.6Ce0.4CoO3 catalyst has significant advantages in yield, product selectivity, and reaction conditions, making it an efficient and economical mild condition catalyst. In the future, by further optimizing reaction time and exploring more suitable reaction systems, its comprehensive performance is expected to be further improved (Table 2).
| Material | Catalytic | Reaction condition | Product | EA soluble product yield (wt%) | References |
|---|---|---|---|---|---|
| Cork lignin | V–Cu/ZrO2 | 150 °C, 5MPaO2, 10 min | Vanillin | 9 | 30 |
| Organic solvent lignin | 5%Pt–1%Ni/HTC | 200 °C, 1.5 h, 2.0 MPaH2 | Phenol, 2-methoxyphenol | 18 | 28 |
| Sodium lignosulfonate | La0.6Ce0.4CoO3 | 240 °C, 10 h, 1.1 MPaN2 | 4-Ethyl guaiacol | 13.05 | This chapter's work |
| Lignin | CoO/m-SEP | 260 °C, 4 h, 4.0 MPaH2 | Phenol | 8.12 | 31 |
| Alkali lignin | 10Ni10Fe/MgSiO3 | 300 °C, 1 h, 1.0 MPaN2 | Catechins, guaiacol, phenol | 14.29 | 29 |
 | ||
| Fig. 11 Test of catalytic depolymerization of lignin using recycled catalyst (A), phenolic product yield (B) and XRD analysis (C) (0.6 g sodium lignosulfonate, 0.3 g La0.6Ce0.4CoO3, 1.1 MPaN2, 10 h, 240 °C, 10 mL methanol, 10 mL ethanol). | ||
Fig. 11(C) shows the XRD patterns of fresh catalyst and old catalyst recovered after 3 cycles. It can be seen that the characteristic peak intensity of CeO2 is significantly reduced at 2θ = 28.5°, 32.9°, and 47.5°, while only an extremely weak characteristic peak of La2O is detected at 2θ = 58.92°, indicating that the metal elements on the catalyst surface have partially detached during multiple lignin depolymerization processes, causing changes in its crystal structure, resulting in surface roughness, element loss, and limiting the diffusion and adsorption ability of reactant molecules on the catalyst surface.
Despite these unfavorable factors, the results of the third cycle experiment (lignin conversion rate: 54.3 wt%, product oil yield: 11.66 wt%) were still far better than those of the experiment without catalyst (lignin conversion rate: 40.96 wt%, product oil yield: 4.55 wt%). After 3 cycles, the catalyst still maintains a certain level of catalytic activity. Therefore, the La0.6Ce0.4CoO3 catalyst exhibits good stability and recyclability in the process of lignin catalyzed depolymerization.
3.3 Characterization of bio-oils
 | ||
| Fig. 12 GC-MS analysis of the depolymerization products of sodium lignosulfonate by La0.6Ce0.4CoO3 (0.6 g sodium lignosulfonate, 0.3 g catalyst, 1.1 MPaN2, 240 °C, 10 h, 10 mL ethanol and 10 mL methanol solvent system). | ||
In addition to phenolic compounds, a small amount of fatty ester compounds such as ethyl dodecanoate are also generated during the depolymerization process, which may originate from triglyceride impurities in lignin raw materials. By optimizing the reaction conditions and using La0.6Ce0.4CoO3 catalyst for lignin depolymerization reaction, the total yield of monomer compounds was significantly improved, far higher than the yield under no catalyst conditions. This discovery not only demonstrates the high efficiency of La0.6Ce0.4CoO3 catalyst in lignin depolymerization reaction, but also opens up new avenues for high-value utilization of lignin. These phenolic compounds have broad application prospects in chemical, pharmaceutical and other fields, providing strong technical support for the deep development of lignin resources.
 | ||
| Fig. 13 FT-IR spectra of sodium lignosulfonate and lignin oil. | ||
The appearance of these absorption peaks also indirectly confirms the structure of the polymer products and indicates that after catalytic action, lignin will be efficiently converted into guaiacol and its derivatives. In addition, stretching vibration peaks of other specific functional groups were also observed. For example, the absorption peaks appearing near 3400 cm−1, 1600 cm−1, 1514 cm−1, and 800 cm−1 are caused by the stretching vibrations of phenolic hydroxyl, aliphatic hydroxyl, and aromatic ring outer plane C–H.
The –OH vibration signal of bio-oil is significantly enhanced, and the peak range of the signal is the widest, indicating abundant hydrogen bonds within the bio-oil molecules. In addition, during the process of lignin depolymerization to generate bio-oil, although chemical bonds are broken and recombined, some major functional groups such as methyl, methoxy, hydroxyl, etc. still remain in the bio-oil. These functional groups are relatively stable during the pyrolysis process and are not easily completely destroyed or converted. In addition, the similarity between the infrared spectra of the two is high, which means that the extraction process of bio-oil is more effective and can better preserve the effective components in lignin.
| Sample | Element content, (wt%) | |||||||
|---|---|---|---|---|---|---|---|---|
| C | H | O | N | S | O/C | H/C | HHV (MJ kg−1) | |
| Lignin | 34.17 | 4.19 | 54.08 | 0.10 | 7.46 | 1.58 | 0.13 | 9.06 |
| Lignin oil | 45.56 | 7.24 | 44.1 | 0.19 | 2.91 | 0.96 | 0.16 | 18.74 |
 | ||
| Fig. 14 1H NMR spectra of La0.6Ce0.4CoO3 depolymerized bio-oil (A) and none catalyst bio-oil 1H NMR spectra (B) (0.6 g sodium lignosulfonate, 1.1 MPaN2, 240 °C, 10 h, 10 mL ethanol and 10 mL methanol solvent system). | ||
Based on the above results, the sample in Fig. 14(A) is rich in phenolic hydroxyl groups and methylated phenolic compounds, and also has many oxidation sites, which is consistent with the results of FTIR and GC-MS.
In contrast, the phenol content in Fig. 14(B) is lower, and the aromatic hydrogen signal is weakened, which may be attributed to the higher degree of methoxy substitution, leading to an enhanced shielding effect of the aromatic ring. These differences reflect that the catalyst (La0.6Ce0.4CoO3) is more effective in depolymerizing lignin.
3.4 Lignin depolymerization mechanism
GC-MS analysis results based on EA soluble products and relevant literature reports. We have made a reasonable speculation on the depolymerization mechanism of sodium lignosulfonate (SLS) under the action of perovskite oxide catalyst. Perovskite oxide catalysts have the characteristic of releasing adsorbed oxygen and lattice oxygen at high temperatures, and supplementing lattice oxygen during the regeneration process of perovskite.32 During the pyrolysis of lignin, the adsorbed oxygen and lattice oxygen released by perovskite at high temperatures oxidize Cα–OH in SLS to Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, thereby reducing the bond energy of Cα–Cβ bonds, lowering the energy threshold for bond cleavage, and promoting the decomposition of lignin structure.33,34 During this process, alcohols (methanol and ethanol) play a crucial role as in situ hydrogen donors. They generate highly reactive free radicals such as hydrogen atoms, methoxy groups (–OCH3), and ethoxy groups (–OC2H5) through decomposition reactions.35 These free radicals not only promote the catalytic hydrogenation process, but also selectively act on the C–O bonds of lignin aryl ether adsorbed and activated by oxygen vacancies in perovskite catalysts, especially the β-O-4 and α-O-4 bonds, leading to effective cleavage of these chemical bonds. As the depolymerization reaction progresses, these free radicals generated by alcohol decomposition undergo structural rearrangement under the combined influence of catalytic activity at the active sites of the catalyst and thermal effects, further promoting the cleavage of Cα–Cβ bonds. This series of reactions ultimately formed highly reactive phenolic intermediates such as 3,5-diethyl-4-hydroxyacetophenone and ferulic acid, providing an important foundation for subsequent conversion and utilization. In addition, ethanol can stabilize phenolic intermediates through O-alkylation and C-alkylation, effectively inhibit the re polymerization reaction, avoid the formation of carbon residue, and improve monomer yield.36 Subsequently, we emphasized the important role of catalysts in the further cracking process of lignin.
O, thereby reducing the bond energy of Cα–Cβ bonds, lowering the energy threshold for bond cleavage, and promoting the decomposition of lignin structure.33,34 During this process, alcohols (methanol and ethanol) play a crucial role as in situ hydrogen donors. They generate highly reactive free radicals such as hydrogen atoms, methoxy groups (–OCH3), and ethoxy groups (–OC2H5) through decomposition reactions.35 These free radicals not only promote the catalytic hydrogenation process, but also selectively act on the C–O bonds of lignin aryl ether adsorbed and activated by oxygen vacancies in perovskite catalysts, especially the β-O-4 and α-O-4 bonds, leading to effective cleavage of these chemical bonds. As the depolymerization reaction progresses, these free radicals generated by alcohol decomposition undergo structural rearrangement under the combined influence of catalytic activity at the active sites of the catalyst and thermal effects, further promoting the cleavage of Cα–Cβ bonds. This series of reactions ultimately formed highly reactive phenolic intermediates such as 3,5-diethyl-4-hydroxyacetophenone and ferulic acid, providing an important foundation for subsequent conversion and utilization. In addition, ethanol can stabilize phenolic intermediates through O-alkylation and C-alkylation, effectively inhibit the re polymerization reaction, avoid the formation of carbon residue, and improve monomer yield.36 Subsequently, we emphasized the important role of catalysts in the further cracking process of lignin.
The valence state cycling of Ce4+/Ce3+ of cerium (Ce) element in the catalyst (La0.6Ce0.4CoO3) promotes the formation of oxygen vacancies on the surface of CeO2, effectively inhibits coking during the catalytic process, improves the selectivity of target phenolic compounds, and further decomposes aromatic intermediates into mono-phenolic compounds such as cresol and 2,6-dimethylphenol.37
Finally, we summarized the key role of the La0.6Ce0.4CoO3 catalyst in this reaction system. Its highly dispersed active sites promote the release of active hydrogen, while the abundant mesoporous structure and oxygen vacancies significantly improve the adsorption and activation performance of the catalyst for lignin molecular fragments. Together, these properties make the catalyst excellent for promoting lignin depolymerization, improving biooil yield, and high selectivity for specific compounds (Fig. 15).
 | ||
| Fig. 15 Possible pathways of SLS catalytic depolymerization to monomeric phenols. | ||
3.5 DFT simulation calculation
In order to further elucidate the possible reaction process of lignin cracking catalyzed by La0.6Ce0.4CoO3, DFT simulation calculations were conducted on the hydrogenolysis reaction of lignin on La0.6Ce0.4CoO3. As shown in Fig. 16, all spin calculations were performed within the framework of density functional theory (DFT) using the projection enhanced plane wave method, as implemented in the Vienna ab initio simulation package (VASP).38,39 The general gradient approximation proposed by Perdew, Burke, and Ernzerhof was chosen as the Exchange Correlation Potential (GGA-PBE).40,41 The cutoff energy of plane waves is set to 400 eV. In the iterative solution of the Kohn Sham equation, the energy standard is set to 1 × 10−6 eV. The van der Waals interaction was incorporated into Grimme's DFT-D3 method.42 The (110) surface of LaCoO3 is represented as LaCoO3 catalyst, and the substitution of one La atom with Ce atom is represented as Ce doped LaCoO3 catalyst. Apply a vacuum thickness of 15 Å along the z-direction to avoid periodic interactions. A 2 × 2 × 1 Monkhorst Pack grid was used in K-sampling. A lignin molecular model was constructed and optimized in 15 × 15 × 15 Å cubic cells with a single k-point. | ||
| Fig. 16 Side and top views of DFT calculation reaction model of lignin on LaxCe1−x CoO3/LaCoO3. | ||
In the undoped LaCoO3 system, lignin molecules tend to adsorb onto the catalyst surface in a tilted manner through the O end, forming O–Co bonds with bond lengths of 0.214 nm and 0.206 nm, respectively. Although this adsorption mode exhibits relatively high adsorption energy, the large interaction angle between molecules and catalyst surfaces limits the effective contact area between lignin and catalyst, thereby affecting adsorption stability. In contrast, Ce doped LaCoO3 catalyst promotes parallel adsorption of lignin molecules with C and O atoms on the surface, forming C–Co bonds (0.216 nm) and O–Ce bonds (0.228 nm). This parallel adsorption mode significantly increases the contact area between molecules and catalysts, not only improving adsorption stability, but also reducing the total energy of the system, which is beneficial for lignin depolymerization reaction. The introduction of Ce adds additional active sites to the catalytic system, such as Ce–O bonds. Meanwhile, the variable valence state of Ce3+/Ce4+ and the formation of oxygen vacancies significantly improve the electronic conductivity and bond activation efficiency of the catalyst. In summary, the cerium doped LaCoO3 catalyst not only significantly reduces the activation energy of lignin depolymerization, but also exhibits excellent catalytic performance. This provides new research directions and technical support for the efficient conversion of lignin.
4 Conclusions
In the catalytic depolymerization of lignin to produce monophenols using LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) perovskite oxide catalysts prepared by co precipitation method, the lignin conversion rate and carbon yield are closely related to the doping amount of metal Ce. Under the optimal reaction conditions, the catalytic conversion rate of lignin can reach 59.4%. Compare with non-catalytic processes, catalytic processes significantly improve the conversion rate of lignin and the yield of bio-oil. The results showed that the perovskite catalyst LaxCe1−xCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) also exhibited significant activity and structural stability in the lignin catalyzed depolymerization process, making it an efficient multiphase catalyst in lignin catalysis.Data availability
The data supporting this article can be found in the following websites (published papers). (1) https://www.sciencedirect.com/science/article/abs/pii/S0016236121029604 (2) https://www.nature.com/articles/s41929-017-0007-z (3) https://www.sciencedirect.com/science/article/pii/S2468025721000261 (4) https://pubs.acs.org/doi/10.1021/acssuschemeng.9b05041.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by Anhui Provincial Science and Technology Key R&D Program (2022I07020035) and Science and Technology Project of Chuzhou (2022ZD002).References
- Y. Q. Yang, L. P. Xiao, W. Z. Xiao, X. Y. Li, Q. Wang. and R. C. Sun, Nitrogen-doped carbon anchored ruthenium nanoparticles for biofuel upgrade, Fuel, 2022, 314, 123100 CrossRef CAS.
- Z. H. Sun, G. Bottari, A. Afanasenko, C. A. S. Marc, J. Peter and B. Katalin, Complete lignocellulose conversion with integrated catalyst recycling yielding valuable aromatics and fuels, Nat. Catal., 2018, 17, 82–92 CrossRef.
- W. L. Ciaran, P. C. J. Kamer, C. S. Lancefield and P. J. Deuss, An Introduction to Model Compounds of Lignin Linking Motifs; Synthesis and Selection Considerations for Reactivity Studies, ChemSusChem, 2020, 13, 4238–4255 CrossRef PubMed.
- C. F. Zhang and F. Wang, Catalytic Lignin Depolymerization to Aromatic Chemicals, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: an interplay of lignocellulose fractionation Depolymerization, and upgrading, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- Y. Y. Fan, C. Liu, X. C. Kong, Y. Han, M. Lei and R. Xiao, A new perspective on polyethylene-promoted lignin pyrolysis with mass transfer and radical explanation, Green Energy Environ., 2021, 7, 1318–1326 CrossRef.
- S. S. Chen, Q. Q. Lu, W. Y. Han, P. X. Yan, H. L. Wang and W. B. Zhu, Insights into the oxidation–reduction strategy for lignin conversion to high-value aromatics, Fuel, 2021, 283, 119–172 Search PubMed.
- B. Biswas, A. Kumar, B. B. Krishna and T. Bhaskar, Effects of solid base catalysts on depolymerization of alkali lignin for the production of phenolic monomer compounds, Renewable Energy, 2021, 175, 270–280 CrossRef CAS.
- X. D. Liu, Z. C. Jiang, S. S. Feng, H. Z, J. M. Li and C. W. Hu, Catalytic depolymerization of organosolv lignin to phenolic monomers and low molecular weight oligomers, Fuel, 2019, 244, 247–257 CrossRef CAS.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol, Green Chem., 2010, 12, 1640–1647 RSC.
- H. W. Ma, H. W. L, W. J. Zhao, L. X. Li, S. J. Liu, J. X. Long and X. H. Li, Selective depolymerization of lignin catalyzed by nickel supported on zirconium phosphate, Green Chem., 2019, 21, 658–668 RSC.
- R. Y. Shu, R. X. Li, B. Q. Lin, B. W. Luo and Z. P. Tian, High dispersed Ru/SiO2-ZrO2 catalyst prepared by polyol reduction method and its catalytic applications in the hydrodeoxygenation of phenolic compounds and pyrolysis lignin-oil, Fuel, 2020, 265, 117–127 CrossRef.
- C. Zhu., S. Y. Ding, H. Hojo and H. Einaga, Controlling Diphenyl Ether Hydrogenolysis Selectivity by Tuning the Pt Support and H-Donors under Mild Conditions, ACS Catal., 2021, 11, 12661–12672 CrossRef CAS.
- B. P. Álvarez and R. Martin, Ni-Catalyzed Reduction of Inert C−O Bonds: A New Strategy for Using Aryl Ethers as Easily Removable Directing Groups, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef PubMed.
- G. S. Qi, E. Joseph and T. Ralph, Selective Catalytic Reduction of NO with NH3 over Fe-ZSM-5 Catalysts Prepared by Sublimation of FeCl3 at Different Temperatures, Catal. Lett., 2004, 226, 120–128 CrossRef CAS.
- S. Jeong, S. Yang and D. H. Kim, Depolymerization of Protobind lignin to produce monoaromatic compounds over Cu/ZSM-5 catalyst in supercritical ethanol, Mol. Catal., 2017, 442, 140–146 CrossRef CAS.
- G. G. Hao, H. Y. Liu, Z. B. Chang, K. C. Song, X. Yang., H. Ma and W. G. Wang, Catalytic depolymerization of the dealkaline lignin over Co–Mo–S catalysts in supercritical ethanol, Biomass Bioenergy, 2022, 157, 106–118 CrossRef.
- W. G. Zhao, X. Li, H. W. Li, X. L. Zheng, H. W. Ma, G. X. Long and X. H. Li, Selective Hydrogenolysis of Lignin Catalyzed by the Cost-Effective Ni Metal Supported on Alkaline MgO, ACS Sustainable Chem. Eng., 2019, 7, 19750–19760 CrossRef CAS.
- B. X. Jiang, G. Hu, Y. H. Qiao, X. X. Jiang and P. Lu, Depolymerization of lignin over Ni-Pd bimetallic catalyst using isopropanol as in situ hydrogen source, Energy Fuels, 2019, 33, 8786–8793 CrossRef CAS.
- T. Todorov, O. Gunawan, S. Jay Chey and D. B. Mitzi, Progress towards applications of perovskite solar cells, Thin Solid Films, 2011, 519, 7378–7381 CrossRef CAS.
- M. Shellaiah and K. W. Sun, Review on Sensing Applications of Perovskite Nanomaterials, Chemosensors, 2020, 8, 149–172 CrossRef.
- N. Sun., F. G. Jin, X. L. Liu, X. W. Liu, G. X. Li, Y. Shen, F. Wang, X. Y. Chu, Z. Wu, G. H. Li and X. L. Lv, In Situ Co-exsolution of Metal Nanoparticle-Decorated Double Perovskites as Anode Materials for Solid Oxide Fuel Cells, ACS Appl. Energy Mater., 2021, 4, 7992–8002 CrossRef CAS.
- H. B. Deng, L. Lin., Y. Sun, C. S. Pang, J. P. Zhuang, P. K. Ouyang, J. J. Li and S. J. Liu, Activity and Stability of Perovskite-Type Oxide LaCoO3 Catalyst in Lignin Catalytic Wet Oxidation to Aromatic Aldehydes Process, Energy Fuels, 2009, 23, 19–24 CrossRef CAS.
- H. B. Deng, L. Lin., Y. Sun., C. S. Pang, J. P. Zhuang, P. K. Ouyang, Z. J. Li and S. J. Liu, Perovskite-type Oxide LaMnO3: An Efficient and Recyclable Heterogeneous Catalyst for the Wet Aerobic Oxidation of Lignin to Aromatic Aldehydes, Catal. Lett., 2008, 126, 106–111 CrossRef CAS.
- J. H. Zhang, H. B. Deng and L. Lin, Wet Aerobic Oxidation of Lignin into Aromatic Aldehydes Catalysed by a Perovskite-type Oxide: LaFe1-xCuxO3 (x=0, 0.1, 0.2), Molecules, 2009, 14, 27–47 Search PubMed.
- H. B. Deng, L. Lin and S. J. Liu, Catalysis of Cu-Doped Co-Based Perovskite-Type Oxide in Wet Oxidation of Lignin To Produce Aromatic Aldehydes, Energy Fuels, 2010, 24, 4797–4802 CrossRef CAS.
- Y. X. Li, J. P. Zhu, Z. J. Zhang and Y. S. Qu, Preparation of Syringaldehyde from Lignin by Catalytic Oxidation of Perovskite-Type Oxides, ACS Omega, 2020, 5, 2107–2113 CrossRef CAS PubMed.
- R. Süss, G. Aufischer, L. Zeilerbauer, B. Kamm, G. Meissner, H. Spod and C. Paulik, Depolymerization of organosolv lignin by supported Pt metal catalysts, Catal. Commun., 2022, 170, 450–467 CrossRef.
- A. Laobuthee, A. Khankhuean, P. Panith, C. Veranitisagul and N. Laosiripojana, Ni–Fe Cocatalysts on Magnesium Silicate Supports for the Depolymerization of Kraft Lignin, ACS Omega, 2023, 8, 8675–8682 CrossRef CAS PubMed.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, S. Bjelić, C. P. Hulteberg and A. Riisager, Oxidative Depolymerization of Kraft Lignin to Aromatics Over Bimetallic V–Cu/ZrO2 Catalysts, Top. Catal., 2023, 66, 1369–1380 CrossRef CAS.
- M. Q. Chen, Y. Cao, Y. S. Wang, Z. L. Yang, Q. Wang, Q. Q. Sun and J. Wang, Depolymerization of lignin over CoO/m-SEP catalyst under supercritical methanol, J. Renewable Sustainable Energy, 2019, 11, 5670–5688 Search PubMed.
- K. Zhao, F. He., Z. Huang., G. Q. Wei, A. Q. Zheng, H. B. Li and Z. L. Zhao, Perovskite-type oxides LaFe1−xCoxO3 for chemical looping steam methane reforming to syngas and hydrogen co-production, Appl. Energy, 2016, 168, 193–203 CrossRef CAS.
- L. D. M. S. Borel, T. S. Lira, J. A. Ribeiro, C. H. Ataíde and M. A. S. Barrozo, Pyrolysis of brewer's spent grain: Kinetic study and products identification, Ind. Crops Prod., 2018, 121, 388–395 CrossRef CAS.
- V. Nair and R. Vinu, Production of guaiacols via catalytic fast pyrolysis of alkali lignin using titania, zirconia and ceria, J. Anal. Appl. Pyrolysis, 2016, 119, 31–39 CrossRef CAS.
- M. Kleinert, J. R. Gasson and T. Barth, Optimizing solvolysis conditions for integrated Depolymerization and hydrodeoxygenation of lignin to produce liquid biofuel, J. Anal. Appl. Pyrolysis, 2009, 85, 108–117 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Catalytic Depolymerization of Lignin in Supercritical Ethanol, ChemSusChem, 2015, 7, 293–307 Search PubMed.
- L. J. Lei, Y. H. Wang, Z. X. Zhang, J. H. An and F. Wang, Transformations of Biomass, Its Derivatives, and Downstream Chemicals over Ceria Catalysts, ACS Catal., 2020, 10, 8788–8814 CrossRef CAS.
- G. G. Kresse. and J. J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse a. and J. Furthmüller b, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- J. P. Perdew., K. Burke. and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. A. White. and D. M. Bird-Implementation, of Gradient-Corrected Exchange-Correlation Potentials in Car-Parrinello Total-Energy-Calculations, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 4954–4957 CrossRef CAS PubMed.
- S. G. J. Antony., S. Ehrlich. and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 104–154 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |