DOI:
10.1039/D4RA07788A
(Review Article)
RSC Adv., 2025,
15, 14558-14586
Recent advances in the application of β-ketonitriles as multifunctional intermediates in organic chemistry
Received
1st November 2024
, Accepted 28th April 2025
First published on 6th May 2025
Abstract
This review discusses the diverse applications of β-ketonitriles in organic synthesis, highlighting protocols that generate various compounds, including cyclic hydrocarbons, aromatic compounds, heterocycles, spirocycles, and fused heterocycles. These compounds serve as valuable building blocks for biologically active scaffolds like chromenes, quinolines, and natural products. It provides an overview of 69 research articles published from 2014 to 2023, focusing on reactions involving benzoyl acetonitrile derivatives and other β-ketonitriles. The methodologies include cascade, domino, and sequential reactions facilitated by different catalysts, showcasing the versatility of β-ketonitriles in organic chemistry.
1. Introduction
β-Ketonitriles are used as soluble molecules in water and are used in the synthesis of organic compounds, pharmacology, pure chemistry, and applied chemistry.1
These compounds are used as precursors for the production of anti-cancer,2 anti-inflammatory,3 and antimalarial drugs,4 and anti-HIV agents.5 Additionally, β-ketonitriles have been studied extensively in medicinal chemistry due to their potential therapeutic applications. For example, some derivatives have shown promising activity against cancer cells, while others have been studied for their potential as anti-inflammatory agents or as inhibitors of enzymes involved in various disease pathways.6 Ongoing research in this area may lead to the development of new drugs with improved efficacy and reduced side effects. Various methods have been presented for synthesizing β-ketonitriles.7 Kiyokawa et al. completely summarized the synthesis of these valuable compounds in a review.8 One well-known β-ketonitrile is benzoylacetonitrile, utilized as a starting material for synthesizing various heterocyclic compounds. It can undergo different reactions to produce various types of heterocycles such as pyridines,9 pyrimidines,10 and pyrazoles.11 One common reaction is the condensation of benzoylacetonitrile with aldehydes or ketones to form pyridine. This reaction is known as the Hantzsch synthesis and involves the formation of a dihydropyridine intermediate, which is then oxidized to the pyridine product.9 Benzoylacetonitrile can also undergo cyclization reactions with different reagents to form various heterocyclic compounds. For example, a reaction with hydrazine derivatives can lead to the formation of pyrazoles,11 while a reaction with urea or thiourea can lead to the formation of pyrimidines.10 In addition, benzoylacetonitrile can be used as a starting material for the synthesis of benzonitriles.12 Overall, benzoylacetonitrile is a versatile starting material for synthesizing a wide range of heterocyclic compounds with potential applications in pharmaceuticals and agrochemicals.13 In 2013, Bakr F. and colleagues successfully synthesized various heterocycles such as pyran, pyridazine, pyrimidine, pyrazine, and triazine compounds using benzoylacetonitrile as a precursor.14 They conducted a review of the use of benzoyl acetonitrile from 1985 to 2013. In the last decade, many efforts have been made in this field, leading to numerous interesting results. Benzoylacetonitrile was used as a precursor in several multicomponent and one-pot reactions that led to the construction of functionalized cyclic molecules. In addition to some common carbocyclic and heterocyclic compounds, other unusual compounds, such as propellanes, carbazoles, quinoxalines, benzofuropyrroles, N-fused bicyclic systems, and other polycyclic heterocycles, were also reported. To provide a better understanding of the topic, this review is mainly organized according to different product structures.
2. Acyclic compounds
2.1. α-Ketoesters
α-Ketoesters are valuable in the synthesis of drugs and organic compounds because they are good precursors for α-hydroxy esters, chiral compounds, and heterocycles. Due to their versatile applications, several methods have been developed for their synthesis.15 In this review, we will discuss two optimized methods that use β-ketonitriles. Xie and their colleagues report a facile and straightforward approach to obtaining α-ketoesters and α-ketoamides 3 from β-ketonitriles 1, phenyliodine(III) diacetate (PIDA) 2, and alcohols and amines under refluxing conditions (Scheme 1).16
 |
| | Scheme 1 Synthesis of α-ketoesters 3 with phenyliodine(III) diacetate. | |
The following year, Cheng Guo et al. reported a new method for esterification that does not require transition metals or photocatalysts. This process involves using visible light to facilitate the decyanation of aryl ketonitriles 1 in the presence of dioxygen and alcohols 4 at room temperature, forming α-ketoesters 5 (53–93% yield). This reaction enables C–H bond functionalization, C–C σ-bond cleavage, and dioxygen activation to be achieved in a single step, cleavage, and dioxygen activation to be achieved in a single step (Scheme 2).17
 |
| | Scheme 2 Reactions of β-ketonitriles and alcohols for constructing α-ketoesters 5. | |
2.2. 3,5-Diketoesters
In 2016, Rao and colleagues successfully developed a simple and convenient method for synthesizing various 3,5-dioxopentanoates 7 (3,5-diketo esters) and 3-amino-5-oxopent-3-enoates 8. This is achieved through a zinc-mediated condensation of β-ketonitriles 1 (also known as α-cyano ketones) with ethyl bromoacetate 6. By using an acidic workup, the reaction yields 3,5-dioxopentanoates (55–81%), while a basic workup yields ethyl (3Z)-3-amino-5-oxo-5-pent-3-enoates (62–82%). This method allows for direct and regioselective synthesis of 3,5-dioxopentanoates or the corresponding enamines (Scheme 3).18 Further investigation of this method has shown that the keto and nitrile functional groups are in a geminal relationship, leading to enolization and the possibility of the Blaise process.
 |
| | Scheme 3 Synthesis of 3,5-dioxopentanoates 7 (3,5-diketo esters) and 3-amino-5-oxopent-3-enoates 8. | |
3. Carbocyclic compounds
3.1. Aliphatic carboxylic compounds
Gao et al. have developed a green cascade approach for the preparation of a variety of diastereoselective polysubstituted cyclopentene derivatives through metal-free oxidative [2 + 1 + 1 + 1] annulation of aldehydes 9 and β-ketonitriles 1. According to their research, different groups at the para-position of the aromatic aldehyde ring were better tolerated than the substituents at the meta-position, resulting in the corresponding para-substituted diphenylcyclopentene products in higher yields compared to the meta-substituted analogs. To confirm the application of this method, they selected certain substrates such as 2-naphthaldehyde, cyclohexane-carboxaldehyde, and 3-thiophenecarboxaldehyde instead of the aromatic aldehyde ring. Except for cyclohexane, other substitutions produced the desired product in moderate yields with high stereoselectivities (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr). Other carbonyl acetonitrile substrates such as benzoyl acetonitrile, furanylacetonitrile, and phenyl nitrile acetate, also showed good reactivity in this reaction. The mechanism of this method involves a four-step cascade reaction involving air oxidation and Michael addition to obtain the final product (Scheme 4).19
:5 dr). Other carbonyl acetonitrile substrates such as benzoyl acetonitrile, furanylacetonitrile, and phenyl nitrile acetate, also showed good reactivity in this reaction. The mechanism of this method involves a four-step cascade reaction involving air oxidation and Michael addition to obtain the final product (Scheme 4).19
 |
| | Scheme 4 Synthesis of diastereoselective polysubstituted cyclopentenes 10. | |
3.2. Aromatic carboxylic compounds
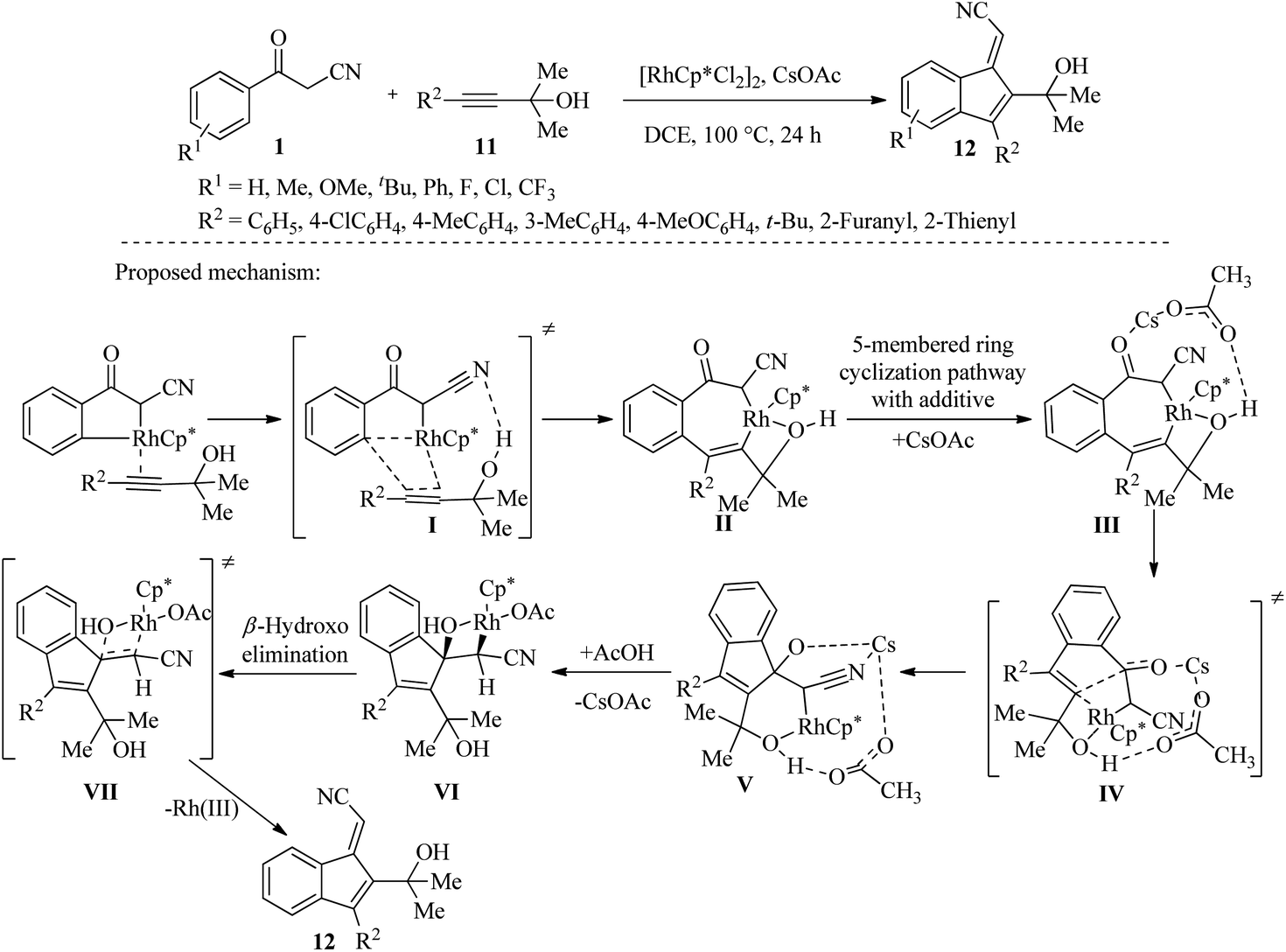
3.2.1. Synthesis of functionalized benzofulvenes. In 2019, Zhang and colleagues carried out a Rh(III)-catalyzed synthesis of functionalized benzofulvenes (40–83%) 12 using benzoyl acetonitrile 1 and propargyl alcohols 11. The researchers used density functional theory (DFT) calculations to perform mechanical modeling and found that the hydroxyl group and CsOAc played crucial roles in mediating the 5-membered ring cyclization. These two components formed a thermodynamically stable Rh(III) intermediate that was essential for the reaction (Scheme 5).20
 |
| | Scheme 5 Synthesis of functionalized benzofulvenes 12. | |
3.2.2. Synthesis of substituted naphthalenes. An efficient and practical method for synthesizing cyano-substituted naphthalene derivatives is described by reactions of β-ketonitriles 1 and 1,2-bis(halomethyl)benzenes 13 including a rearrangement aromatization of benzo[c]oxepine in present Cs2CO3 in DMSO at 40 °C. The mechanism of this method involves the C-alkylation of compound 13 provided intermediate I, which appeared in two tautomeric forms: I and I′. The subsequent O-alkylation of enolic formed I and presented as the seven-membered ring II. Meanwhile, the competing C-alkylation yielded the five-membered ring 15 as a by-product. Finally, rearrangement of I followed by aromatization in the presence of Cs2CO3 afforded the desired naphthalene-based product 14 (Scheme 6).21
 |
| | Scheme 6 Synthesis of substituted naphthalenes 14. | |
3.2.3. Synthesis of substituted phenanthrene-1-carboxylates. Shu-Jiang Tu et al. have developed a mild reaction condition to prepare a wide range of phenanthrene-1-carboxylates. The method involves a metal-free [2 + 2] cycloaddition/ring expansion sequence of yne-allenone esters 16 with β-keto nitriles 1, using 1.0 equivalent of Cs2CO3 as a base promoter. The reaction is carried out in 1,4-dioxane at room temperature for 2.5 hours under air conditions. The final product, phenanthrene-1-carboxylates 17, is obtained with a 56–97% yield (Scheme 7).22
 |
| | Scheme 7 Synthesis of substituted phenanthrene-1-carboxylates 17. | |
4. Heterocyclic compounds
4.1. Five-membered rings
4.1.1. Five-membered rings with one hetero atom.
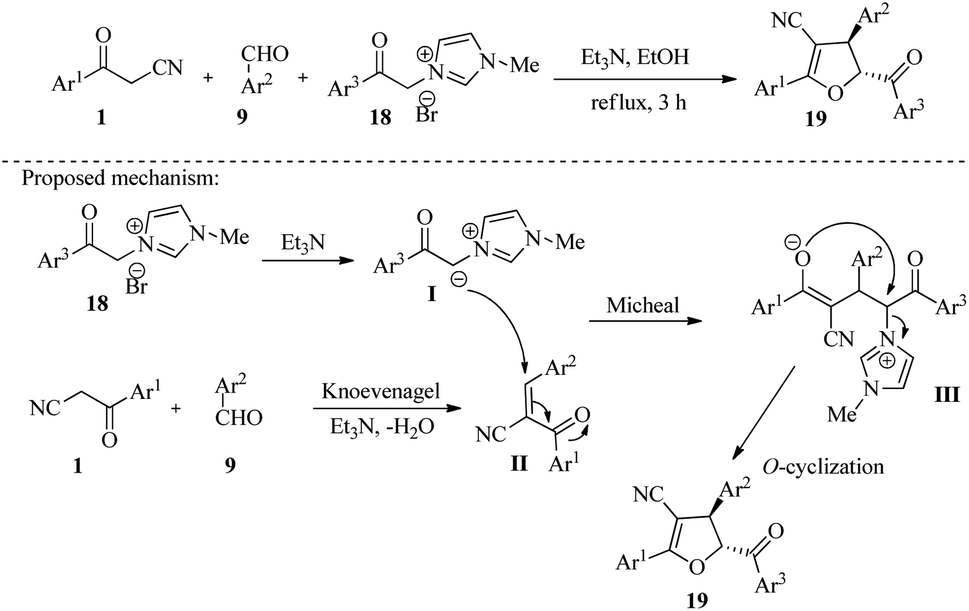
4.1.1.1. Polysubstituted furans, dihydrofurans, furo[2,3-b]furans. In 2021, Sathiyamoorthi and colleagues developed a new chemical reaction that produces highly substituted dihydrofuran derivatives 19 in 90–97% yield. The reaction involves a one-pot three-component tandem strategy that uses aroyl acetonitriles 1, aromatic aldehydes 9, and 1-methyl-3-(2-oxo-2-phenylethyl)-1H-imidazol-3-ium bromides 18. The reaction mechanism follows a convergence condensation-Michael addition-O-cyclization process, which results in the formation of three new bonds (two C–C and one C–O) to produce the final product (Scheme 8).23
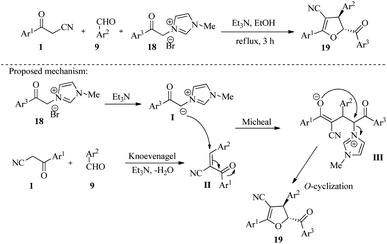 |
| | Scheme 8 Synthesis of multi-substituted 4,5-dihydrofurans 19. | |
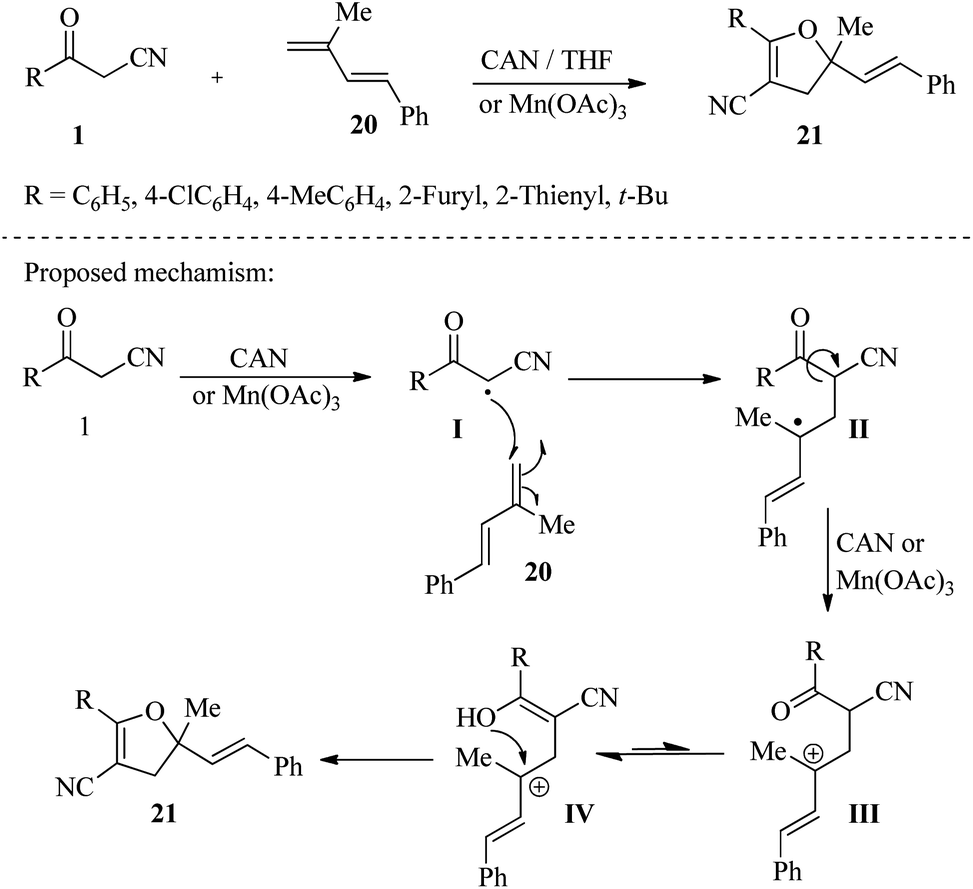
In a different study, Hocaoglu and colleagues utilized the radical addition of β-ketonitriles 1 to conjugated dienes 20 to produce 5-ethenyl-4,5-dihydrofuran-3-carbonitriles 21 in yields ranging from low to good. In this method, Mn(OAc)3 and CAN were used as radical oxidants in the reactions. Based on these results, the authors concluded that Mn(OAc)3 is effective in reactions of 3-oxopropanenitriles with dienes that contain a thiophen-2-yl group. Conversely, CAN is more efficient with dienes that do not contain thiophen-2-yl (Scheme 9).24 Wang and their team have reported on a solvent/base-switchable platform that allows for the selective conversion of β-ketonitriles 1 to multi-substituted 2,3-dihydrofuran-2-carbonitriles 22 and 4,5-dihydrofuran-3-carbonitriles 23 without the use of metals. By simply changing the solvent and reaction base, the same oxidant (TBHP) can produce the desired products through two distinct pathways. Through mechanistic exploration, this method has shown that the reaction of β-ketonitriles under non-polar and polar solvents leads to unexpected rearrangement of the –CN group migration and hydroxylation, respectively, resulting in different products of hydrofurans (Scheme 10).25
 |
| | Scheme 9 Synthesis of 5-ethenyl-4,5-dihydrofuran-3-carbonitriles 21. | |
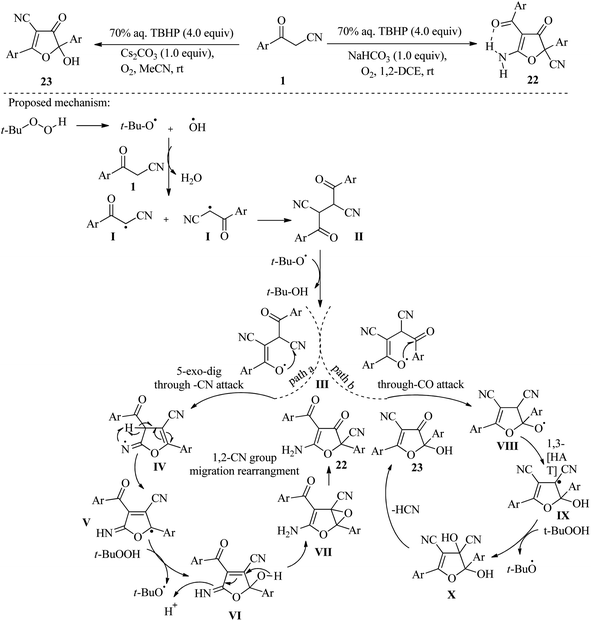 |
| | Scheme 10 Synthesis of multi-substituted 2,3-dihydrofuran-2-carbonitriles 22 and 4,5-dihydrofuran-3-carbonitriles 23. | |
In 2016, Feng and coworkers obtained regio-diastereo and enantioselective 2,3-dihydro-furans 25 in 93–92% yield from the reaction of substituted β-ketonitriles 1 with bromonitrostyrenes 24 in toluene solvent at 60 °C for 72 h by using achiral N,N′-dioxide as organocatalyst. In the mechanical exploration of this reaction, bromonitrostyrenes are first added to substituted β-ketonitrile from Michael's process (C–C bond formation). Then, by adjusting the reaction conditions, the O-alkylation path is preferred to the C-alkylation path. With this strategy, the desired chiral 2,3-dihydrofurans 25 were obtained in up to 95% yield with 95:5 dr and 93% ee (Scheme 11).26
 |
| | Scheme 11 Synthesis of 2,3-dihydrofurans 25. | |
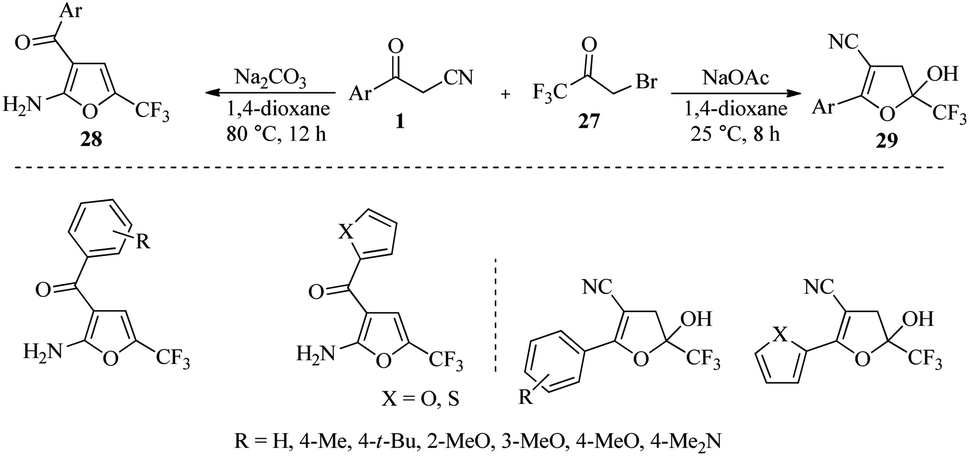
In a different research, Weng et al. discovered that by using two different bases, Na2CO3 and NaOAc, in the reaction of β-ketonitriles 1 with 3-bromo-1,1,1-trifluoroacetone 27, it becomes possible to selectively synthesize two trifluoromethylated furans 28 and dihydrofuranols 29 without using any metals (Scheme 12).27
 |
| | Scheme 12 Synthesis of trifluoromethylated furans 28 and dihydrofuranols 29. | |
Zhang and colleagues have developed a new method for synthesizing chiral bicyclic dihydrofurans 31 with two vicinal carbon stereocenters. The method involves a Pd-catalyzed asymmetric allylic substitution cascade reaction of allylic dicarbonates 30 with β-ketonitriles 1. The products are obtained in high yields and with up to 97% ee (Scheme 13).28
 |
| | Scheme 13 Synthesis of bicyclic dihydrofurans 31. | |
The following year, Xinwei He et al., synthesized a series of polysubstituted furans 33 by the reactions of propargylamines 32 with β-ketonitriles 1 in the presence of Na2CO3 under heating at 90 °C for 24 h. Further exploration of the mechanism of this reaction indicates that it proceeds through a sequence of steps. First, there is a 1,4-conjugate addition of β-ketonitriles 1 to propargylamines 32, followed by a 5-exo-dig annulation/isomerization to form the fully substituted furans 33 (Scheme 14).29
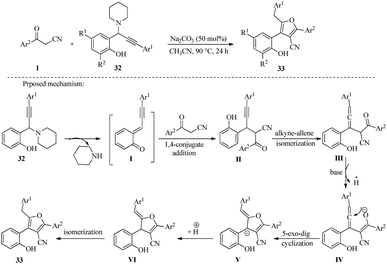 |
| | Scheme 14 Synthesis of polysubstituted furans 33. | |
In 2019, Jeong and coworkers synthesized highly substituted furan-linked biheteroaryls 36 by a three-component cascade reaction of aryl glyoxals 34, isonitriles 35, and aroyl or heteroaryl β-ketonitriles 1 in the presence of p-TsOH and EtOH under reflux condition for 4 h. The reaction sequence involves a Knoevenagel condensation of aryl glyoxals 34 with β-ketonitriles 1 followed by an isocyanide 35 insertion via formal [4 + 1] cycloaddition, and then a rapid [1,3]-H shift to afford novel bi-heterocycles with unique decorations (Scheme 15).30
 |
| | Scheme 15 Synthesized highly substituted furan-linked biheteroaryls 36. | |
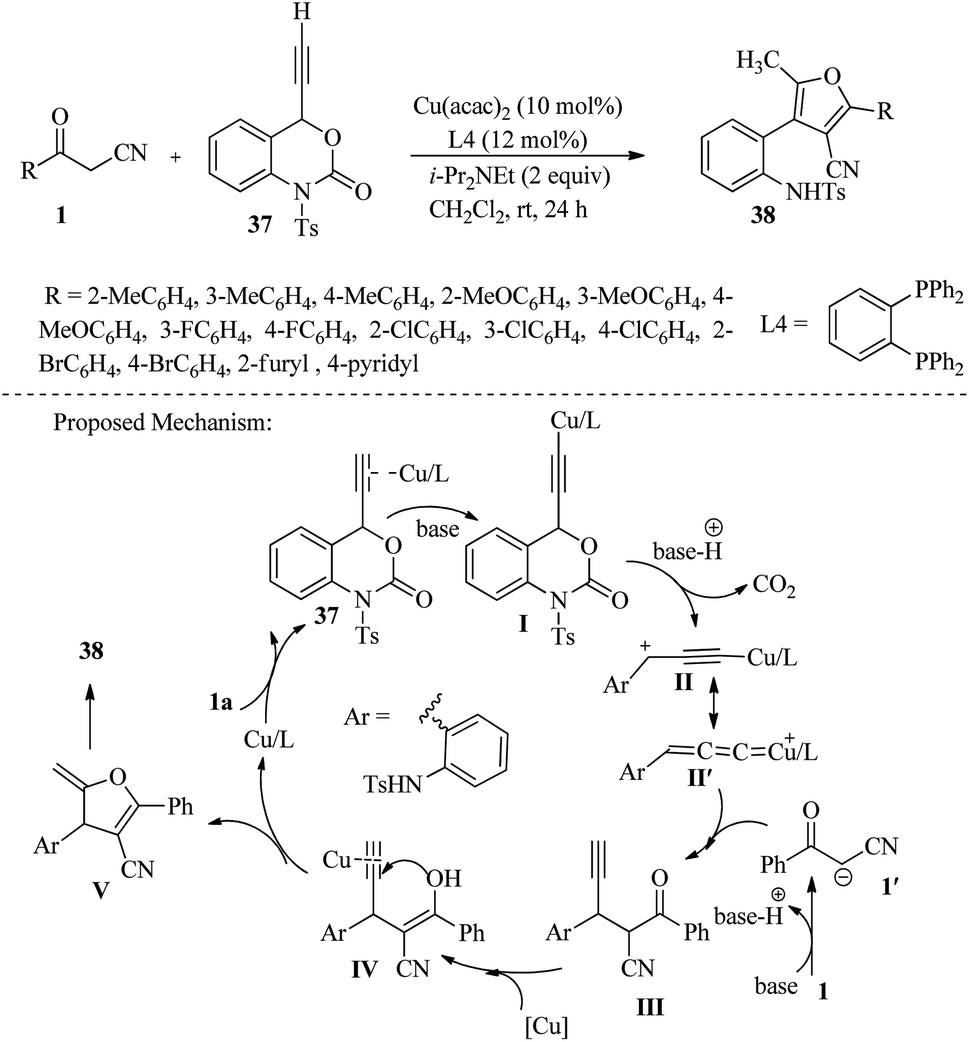
In 2020, Zhong and colleagues achieved moderate to high yields of tetrasubstituted furan derivatives 38 through [3 + 2] annulation of ethynyl benzoxazinanones 37 with β-ketonitriles 1. This was done in the presence of Cu(acac)2 as a catalyst and i-Pr2NEt in CH2Cl2 at room temperature over 24 hours. It was found that intermediate copper allenylidines acted as a C2 synthon during the cycloaddition reaction (Scheme 16).31
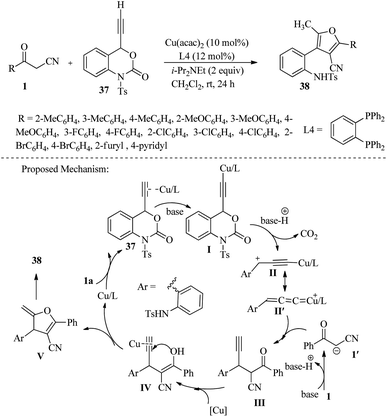 |
| | Scheme 16 Synthesis of tetrasubstituted furan 38. | |
In 2021, Wan et al. reported the synthesis of substituted-furo[2,3-b]furans 40 by oxidizing malononitrile 39 to 2-hydroxy-malononitrile I, which can then react with β-ketonitriles 1 in CH3CN in the presence of 0.5 equiv. of benzoic acid at room temperature for 12 h. In the mechanistic exploration of this reaction, it was hypothesized that SeO2 oxidizes malononitrile to 2-hydroxy-malononitrile, which can then react with β-ketonitriles 1 and during the two processes of Michael addition and intramolecular cyclization, it provides target compounds with 27–56% efficiency. The authors have highlighted that the synthesized compounds exhibit interesting photoluminescence properties in both solution and solid-state (Scheme 17).32
 |
| | Scheme 17 Synthesis of substituted-furo[2,3-b]furans 40. | |
In a recent study, Gnanasambandam and his colleagues synthesized heterocyclic furo(2,3-b)furan derivatives 43 with an efficiency rate of 60–80%. They achieved this through a three-component reaction that involved methylglyoxal 41, benzothiazole acetonitrile 42, and β-ketonitriles 1, with the use of DABCO (1,4-diazabicyclo[2.2.2]octane) as a base and EtOH/H2O solvent mixture at room temperature for two hours. The reaction involved Knoevenagel condensation, Michael addition, intramolecular cyclization, and tautomerization processes (Scheme 18).33
 |
| | Scheme 18 Synthesis of heterocyclic furo(2,3-b)furan derivatives 43. | |
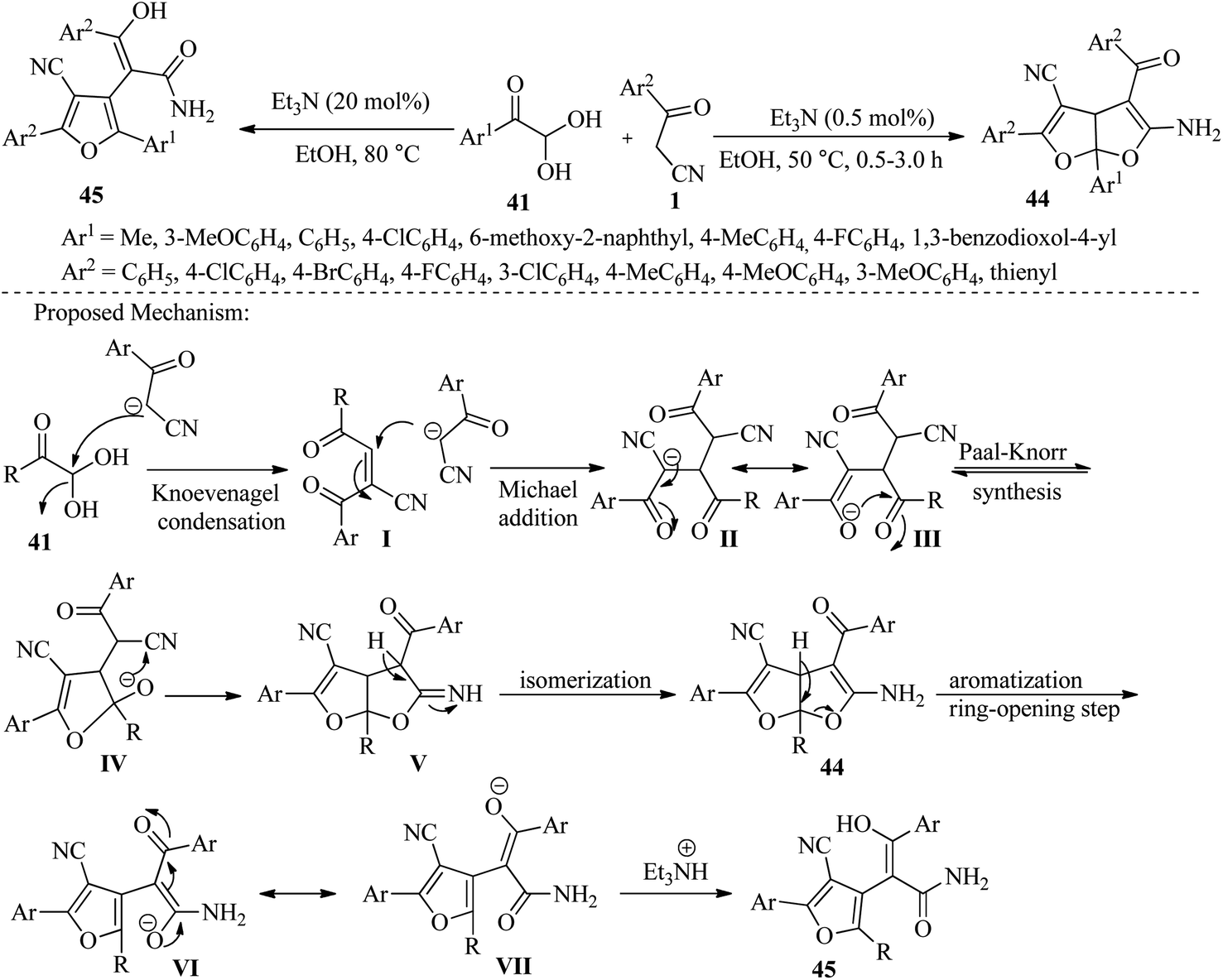
Followed by Jeong et al. have disclosed a highly efficient one-step protocol for constructing highly functionalized dihydrofuro[2,3-b]furans 44 and substituted phenyl furan 3-hydroxy-3-phenylacrylamides 45. This is achieved through a three-component cascade reaction between aromatic or aliphatic glyoxals 41, β-ketonitriles 1, and two different equivalent amounts of triethylamine. The reaction can be performed in two different conditions: at room temperature or under reflux, with excellent yields obtained in both cases. The protocol involves a Knoevenagel and Michael adduct via Paal–Knorr cyclization with aromatic or aliphatic glyoxal and β-ketonitriles under mild heating conditions. The final product can be easily obtained through a simple filtration method (Scheme 19).34
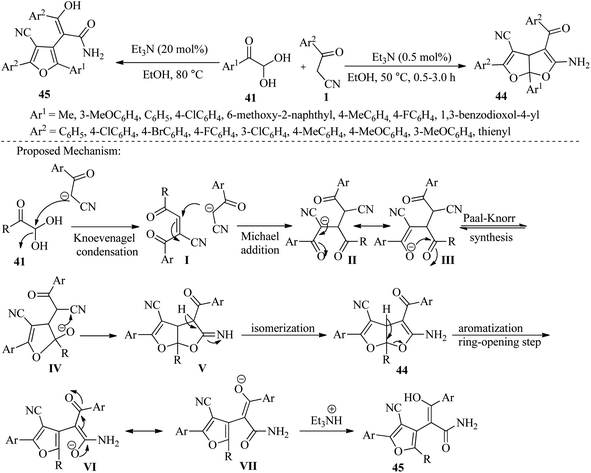 |
| | Scheme 19 Synthesis of functionalized dihydrofuro[2,3-b]furans 44 and substituted phenyl furan 3-hydroxy-3-phenylacrylamides 45. | |
Also, Fang and their team have successfully developed a novel method to create a large variety of cyclic molecules, such as dihydrofurofurans 47 dihydrocyclopentafuranols 48 and, by performing regioselectivity-switchable reactions between alkynyl α-diketones 46 and β-ketonitriles 1. This approach provides a platform for generating a broad range of structurally diverse heterocycles with new dihydrofuran-cyclopentenone skeletons and dihydrofurofurans.
The study also revealed that base and solvent play a critical role in modulating the regioselectivity of this reaction (Scheme 20).35
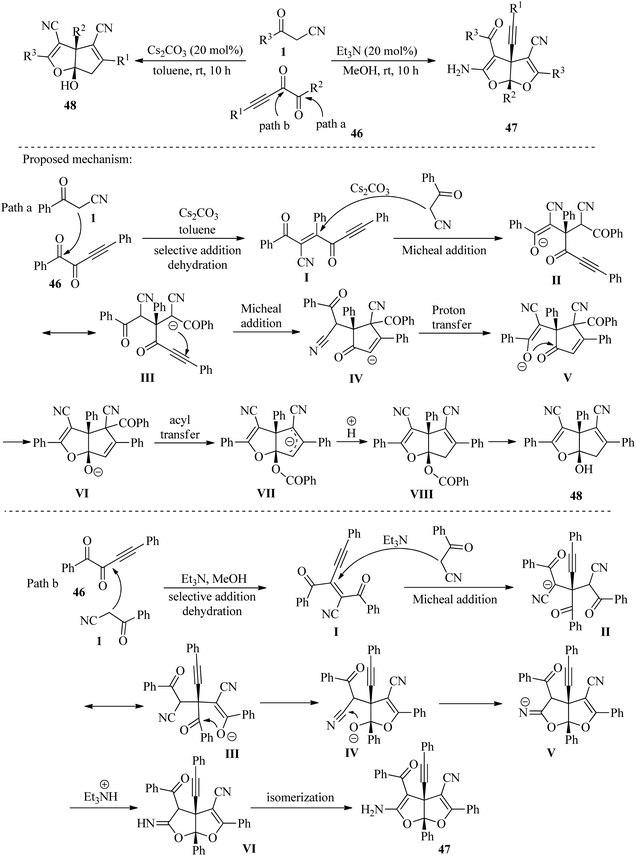 |
| | Scheme 20 Synthesis of dihydrofurofurans 47 and dihydrocyclopentafuranols 48. | |
 |
| | Scheme 21 Synthesis of functionalized 3-cyanopyrroles 51. | |
4.1.2. Five-membered rings with two hetero atom.
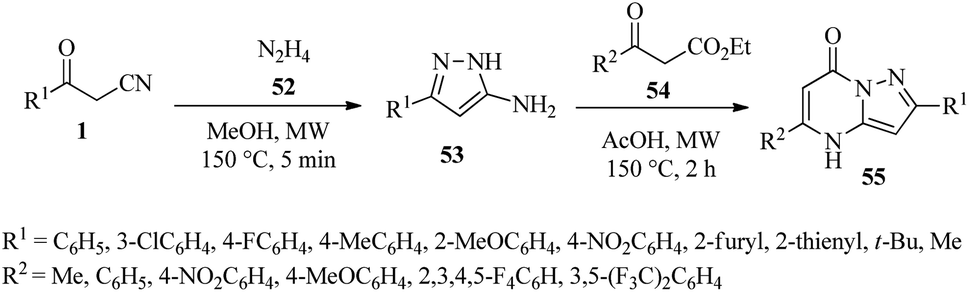
4.1.2.1. Synthesis of 5-aminopyrazole and pyrazolo[1,5-a]pyrimidinones. One of the important applications of β-ketonitriles derivatives is their use as intermediates for the synthesis of 5-amino pyrazoles. To prepare 5-amino pyrazoles, β-ketonitrile 1 is heated with hydrazine in various ways, either in a solvent or with the help of microwaves.37 Reports indicate that 5-amino pyrazoles play an important role in the synthesis of nitrogen-containing heterocyclic compounds, including pyrazolopyrimidinones. For example, Kelada et al. reacted the derivatives of β-ketonitriles 1 (0.9 mmol, 1.0 equiv.), hydrazine 52 (1.2 mmol, 1.3 equiv.), MeOH (1 mL) under microwave conditions (100 W, 150 °C) for 5 min have reported the synthesis of 5-amino pyrazoles 53. After obtaining this intermediate, they continued this process by adding AcOH (0.5 mmol, 0.6 equiv.) and ketoester derivatives 54 (0.9 mmol, 1.0 equiv.) under microwave conditions (100 W, 150 °C) for 2 h and various heterocycles of pyrazolopyrimidinones 55 have been synthesized (Scheme 22).38
 |
| | Scheme 22 Synthesis of 5-amino pyrazoles 53 and pyrazolo[1,5-a]pyrimidinones 55. | |
In a separate study, Liu and colleagues used NIS to accomplish a quasi-three-component reaction of β-ketonitriles 1 and aryl sulfonyl hydrazides 56 in ethanol solvent under reflux conditions, yielding 3-aryl-4-(arylthio)-1H-pyrazol-5-amine derivatives 59 and 3-phenyl-1-(phenylsulfonyl)-4-(phenylthio)-1H-pyrazol-5-amines 60. In the mechanistic investigation of this method, it was discovered that the process involves a sequence of cyclization, sulfonylation, and removal of aryl sulfonyl in the presence of NIS (Scheme 23).39
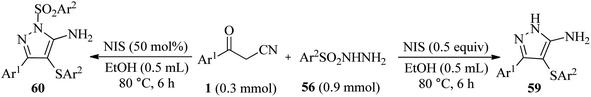 |
| | Scheme 23 Synthesis of 3-aryl-4-(arylthio)-1H-pyrazol-5-amine derivatives 59 and 3-phenyl-1-(phenylsulfonyl)-4-(phenylthio)-1H-pyrazol-5-amines 60. | |
 |
| | Scheme 24 Synthesis of phenylselanyl-1H-1,2,3-triazole-4-carbonitriles 62. | |
Kachanov et al. produced 1,3-oxaselenole heterocycles 63 and 64 in good yield with a cyano group using aroyl acetonitrile 1 and selenium(IV) oxide. The resulting products were found to react with ammonia, hydrazine, or primary amines, during which an aryl rearrangement was observed (Scheme 25).41
 |
| | Scheme 25 Synthesis of 1,3-oxaselenoles 63 and 64. | |
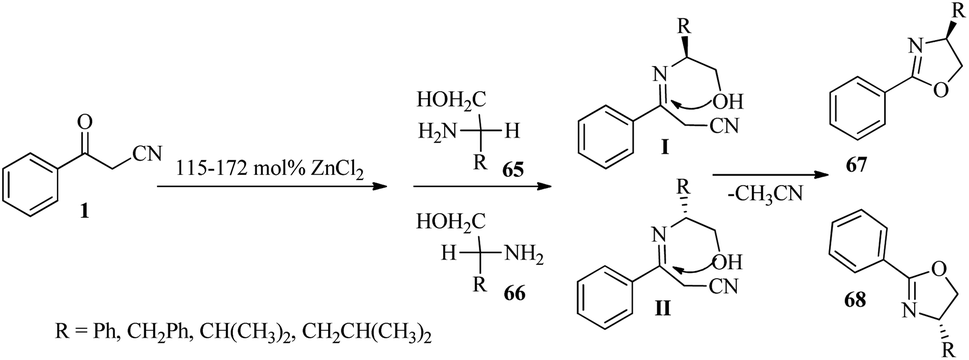
In another study, a series of 2-oxazolines (67 and 68) were produced using a simple one-pot method under inert and non-moisture conditions from β-ketonitrile 1 and β-amino alcohols 65 and 66 with 115–172 mol% ZnCl2 (Scheme 26).42
 |
| | Scheme 26 Synthesis of 2-oxazolines 67 and 68. | |
An effective oxidative system, which does not require the use of metals, has been developed using TBHP and AIBN. This system has allowed for the successful synthesis of substituted 2-aminothioazoles 70 by reacting β-ketonitrile 1 with thiourea 69. Mechanistic studies have shown that the reaction proceeds through the formation of a C–S bond via a radical process, followed by the formation of a C–N bond through an intramolecular condensation reaction (Scheme 27).43
 |
| | Scheme 27 Synthesis of substituted 2-aminothioazoles 70. | |
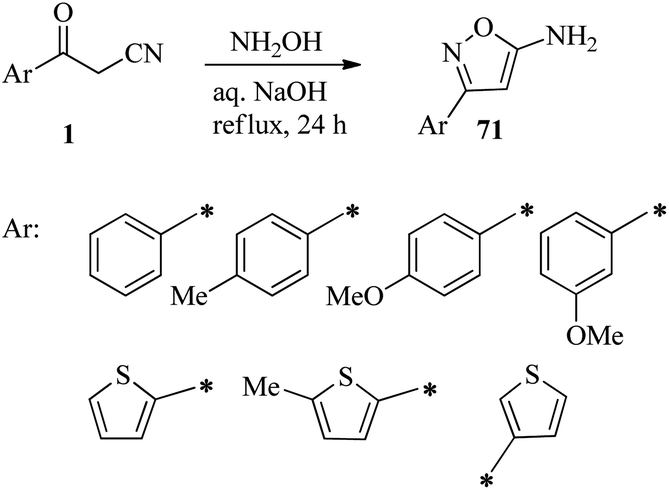
Krasavin et al. have prepared a set of isoxazole-5-amines 71 by reacting readily available β-ketonitriles 1 with hydroxylamine in 15% aqueous NaOH solution at reflux for 14 hours (Scheme 28).44
 |
| | Scheme 28 Synthesis of substituted isoxazole-5-amines 71. | |
4.2. Six-membered rings
4.2.1. Six-membered rings with one heteroatom.
4.2.1.1. Synthesis of dihydropyrans and pyrans. In 2014, a highly efficient synthesis of 3,4-dihydropyrans 75 was reported by catalyzing L-diphenylprolinol trimethylsilyl ether 73 through the Michael addition of β-ketonitriles 1 to β-unsaturated aldehyde derivatives 72.The desired compounds were acquired with exceptional yields (up to 91%) and enantioselectivities (up to 98% ee) (Scheme 29).45
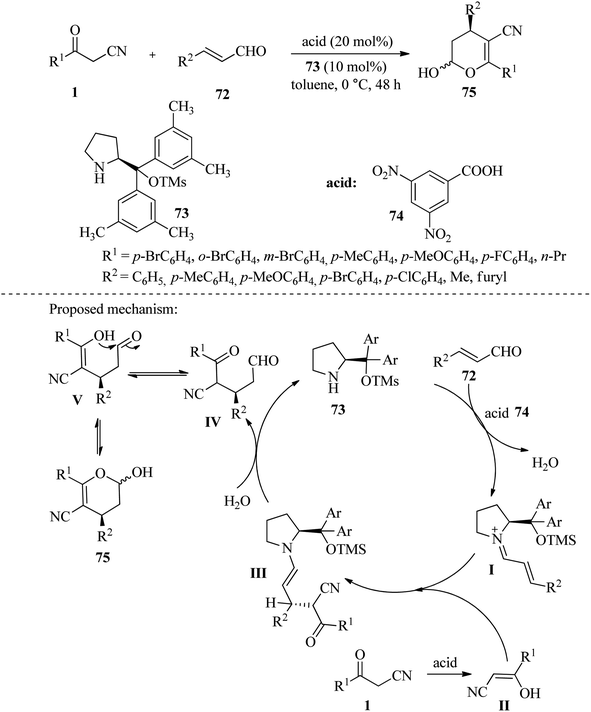 |
| | Scheme 29 Synthesis of 3,4-dihydropyrans 75. | |
In 2016, Tong et al. found that by using 6′-deoxy-6′-[(L)-N,N-(2,2′-oxydiethyl)-valine amido]quinine 77 as the catalyst, the formation of 4H-pyrans 78 through (3 + 3) annulations of β′-acetoxy allenoates 76 with β-ketonitriles 1 can occur quickly and with excellent enantioselectivity. Catalyst 77 has three functions, including Lewis base (quinuclidine N), H-bond donor (amide NH), and Brønsted base (morpholine N), each playing crucial roles in the chemo- and enantio-selectivity for the construction of 4H-pyrans 78 (Scheme 30).46
 |
| | Scheme 30 Synthesis of 4H-pyran derivatives 78 by using 6′-deoxy-6′-[(L)-N,N-(2,2′-oxydiethyl)-valine amido]quinine 77 as a catalyst. | |
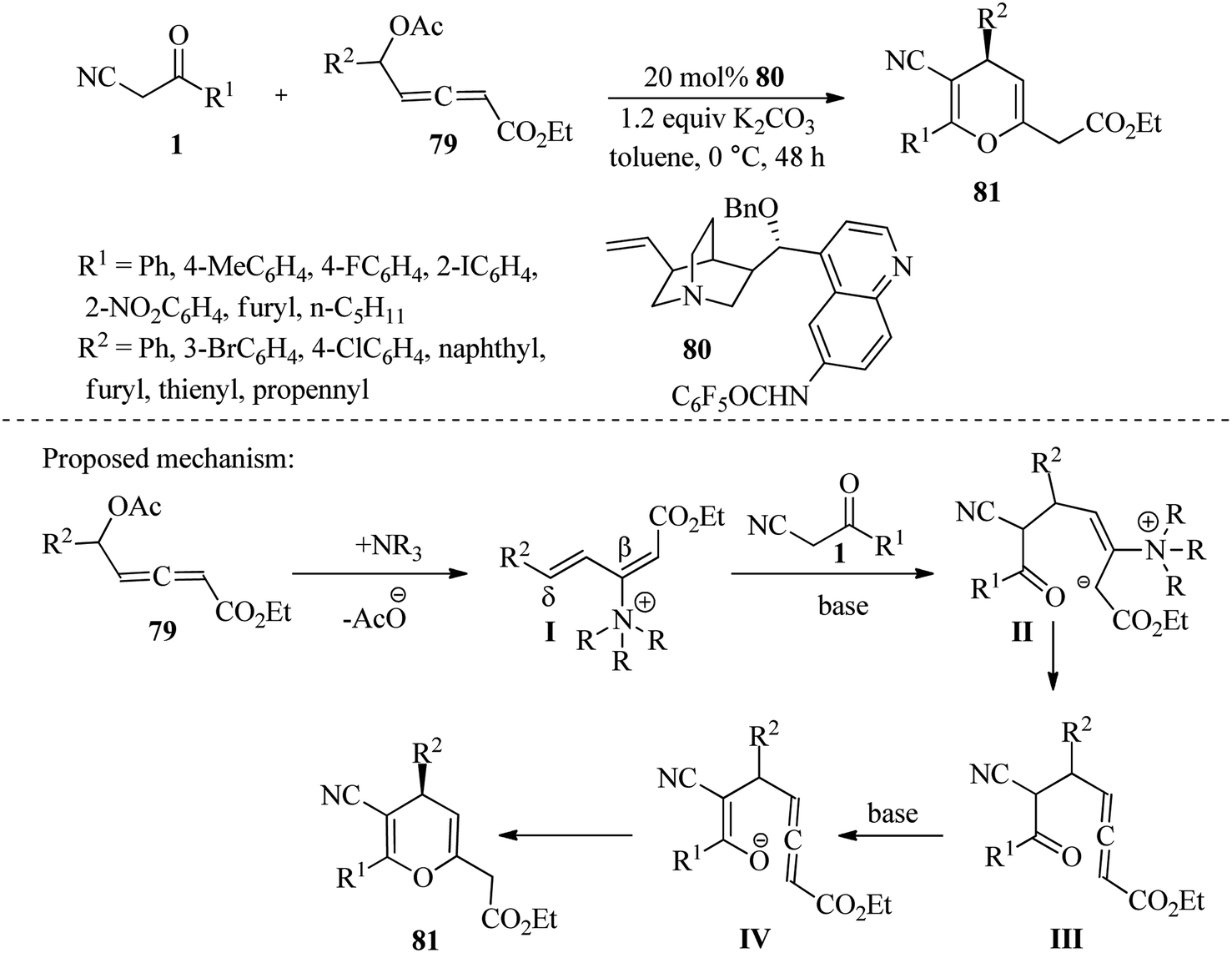
The following year, Zhou et al. discovered that using 6′-deoxy-6′ perfluorobenzamido-quinine 80 as a catalyst can result in the quick formation of 4H-pyrans 81 through [3 + 3] annulations of δ-acetoxy allenoates 79 and β-ketonitriles 1 with excellent enantioselectivity. The researchers found that the amide NH of 80 plays a crucial role as an H-bond donor, facilitating the formation of cationic intermediate I and increasing the electrophilicity of its δ-position (Scheme 31).47
 |
| | Scheme 31 Synthesis of 4H-pyrans 81 by using 6′-deoxy-6′ perfluorobenzamido-quinine 80 as a catalyst. | |
In 2022, Zhang et al. realized that using Cu(OAc)2 as the catalyst can result in the quick formation of polysubstituted 4H-pyran derivatives 84 with a quaternary CF3-containing center through (3 + 3) annulations of alkynyl ketimines 82 and β-ketonitriles 1 with excellent yields (86–99%) and good enantioselectivities (71–78% ee). The researchers found that the reaction involves a process with a base-catalyzed or chiral thiourea-catalyzed Mannich-type reaction followed by a highly regioselective copper-catalyzed ring-closing reaction on the alkynyl moiety in a 6-endo-dig fashion (Scheme 32).48
 |
| | Scheme 32 Synthesis of 4H-pyran derivatives 84 with a quaternary CF3-containing center. | |
In 2019, the same group synthesized derivatives of 2-amino-4H-pyran-3,5-dicarbonitrile 85 through a three-component reaction. This reaction involved aldehydes 9, malononitrile 39, and β-ketonitriles 1 and was carried out in a mixture of ethanol and water at room temperature. The resulting products had a wide range of functional groups and were obtained in high yields (Scheme 33).49
 |
| | Scheme 33 Synthesis of 2-amino-4H-pyran-3,5-dicarbonitrile derivatives 85. | |
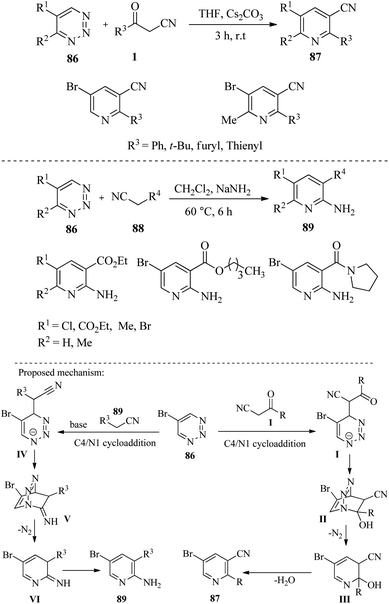 |
| | Scheme 34 Synthesis of highly substituted pyridines 87 and 89. | |
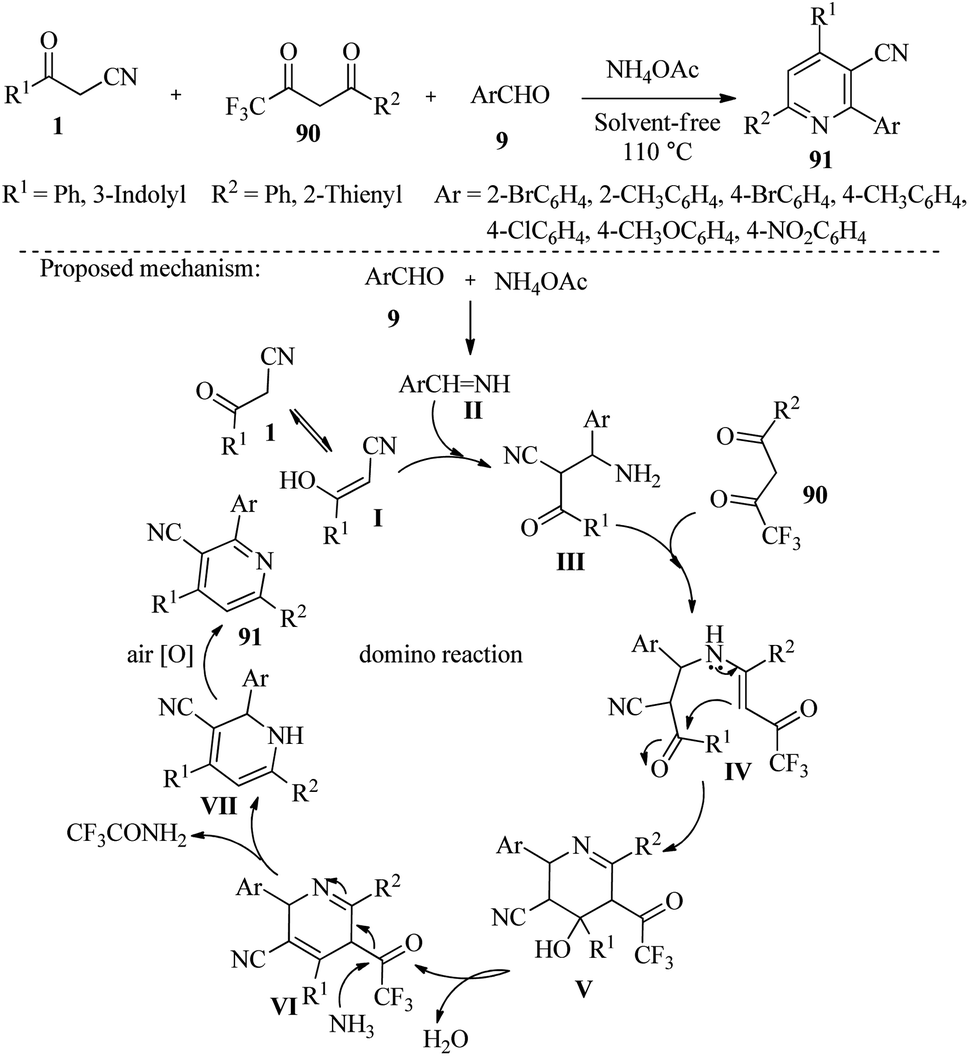
Bharkavi et al. reported a series of new compounds called 2,6-diaryl-4-(1H-indol-3-yl)-3-cyanopyridines 91. These compounds were produced in good yields through the domino reactions of β-ketonitriles 1, 4,4,4-trifluoro-1-phenylbutane-1,3-dione 90, and aromatic aldehydes 9 in the presence of ammonium acetate without the use of solvents. This process creates two C–C and two C–N bonds, leading to the formation of a six-membered ring in a single operation (Scheme 35).51
 |
| | Scheme 35 Synthesis of 2,6-diaryl-4-(1H-indol-3-yl)-3-cyanopyridines 91. | |
4.2.2. Six-membered rings with two heteroatom.
4.2.2.1. Synthesis of pyrimidines. In 2014, Siddiqui et al. applied the Friedlander reaction of guanidines, aldehydes, and cyano ketones under solvent-free conditions at 85 °C in the presence of chitosan as a green catalyst to get 2-aminopyrimidine carbonitriles 93.In the mechanistic study of this reaction, it is explained that the hydroxyl group of chitosan activates the carbonyl group of aldehyde 9 at first, which then undergoes condensation with guanidine 92 leading to the formation of intermediate I. The amine group present on the surface of chitosan facilitates the formation of III. The final step involves the addition of intermediates I and III to form a new intermediate IV, which undergoes intramolecular cyclization followed by aromatization V to produce the desired product 93 in good yield (Scheme 36).52
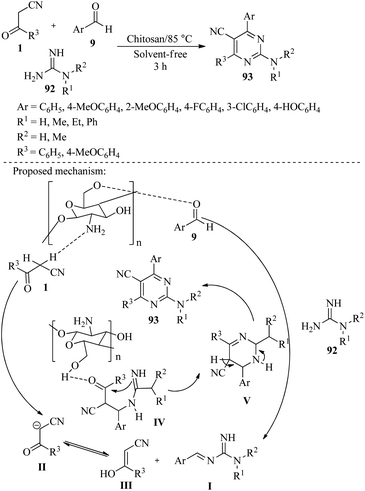 |
| | Scheme 36 Synthesis of 2-aminopyrimidine carbonitriles 93. | |
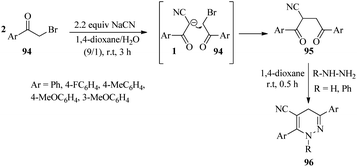 |
| | Scheme 37 Synthesis of dihydropyridazines 96. | |
4.3. Synthesis of quinoline and isoquinoline derivatives
Palanimuthu and his colleagues have developed a simple and efficient method for synthesizing new, densely functionalized tetrahydroquinoline derivatives. This is achieved through the aza-Diels–Alder reaction between β-ketonitriles 1 aromatic aldehydes 9, and 2-alkenyl amines 97. The reaction uses a catalytic amount of TEA and takes place under very mild conditions. The resulting tetrahydroquinoline isomers exo 98 and endo 99 are synthesized with excellent diastereoselectivity and high yields (Scheme 38).54
 |
| | Scheme 38 Synthesis of tetrahydroquinoline isomers exo 98 and endo 99. | |
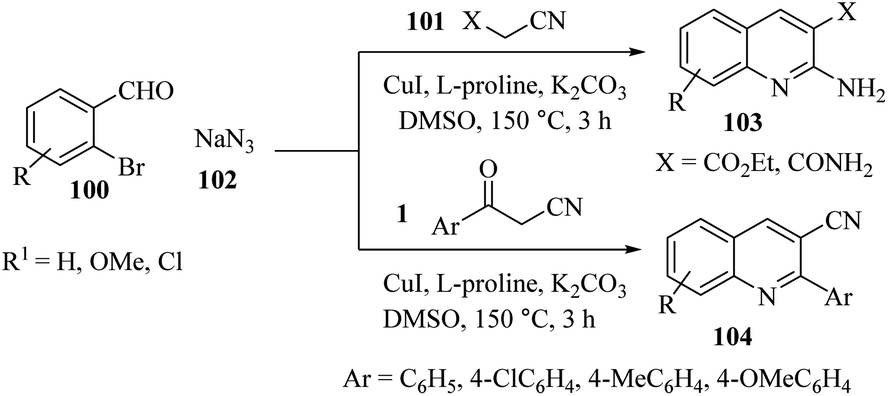
In 2016, Dhiman used copper as a catalyst for the three-component domino reactions of 2-bromobenzaldehydes 100, active methylene nitriles (1 and 101), and sodium azide 102. The reactions were performed in DMSO under reflux at 150 °C in the presence of L-proline and K2CO3 leading to the synthesis of 2-aminoquinolines 103 and 2-arylquinoline-3 carbonitriles 104. Mechanistic exploration of this reaction shows that the formation of substituted quinolines involves Knoevenagel condensation of ortho-bromobenzaldehyde with active methylene nitriles followed by copper-catalyzed reductive amination and intramolecular cyclization (Scheme 39).55
 |
| | Scheme 39 Synthesis of 2-aminoquinolines 103 and 2-arylquinoline-3 carbonitriles 104. | |
Recently, our group reported an efficient synthesis of functionalized pyrano[2,3-b]quinoline and benzo[h]pyrano[2,3-b]quinoline derivatives 107a and 107b by using 2-chloroquinoline-3-carbaldehyde or 2-chlorobenzo[h]quinoline-3-carbaldehyd 105a and 105b, 1-aryl-2-(1,1,1-triphenyl-λ5-phosphanylidene)ethan-1-one (Wittig reagent) 106 and β-ketonitriles 1 in EtOH under reflux at 80 °C in the presence of Et3N in good to excellent yields. The mechanical of this reaction involves the two processes of C–C bond formation (Michael addition) and intramolecular cyclization (by attacking the oxygen atom of active methylene compounds) (Scheme 40).56
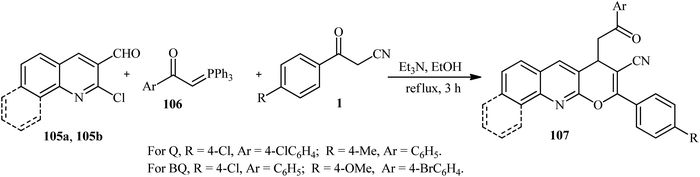 |
| | Scheme 40 Synthesis of functionalized pyrano[2,3-b]quinoline and benzo[h]pyrano[2,3-b]quinoline derivatives. | |
Hussain et al. have developed an efficient method to produce 3-cyano-4-quinolone derivatives 109. This is achieved through decarboxylative cyclization of isatoic anhydrides 108 with β-ketonitriles 1 using DABCO as a non-toxic, eco-friendly, and inexpensive reagent under microwave conditions. The reaction is performed in CH3CN under reflux at 80 °C for 30 minutes. This approach provides an easy pathway for making this class of compounds in good to high yield from readily available starting materials in short reaction times (Scheme 41).57
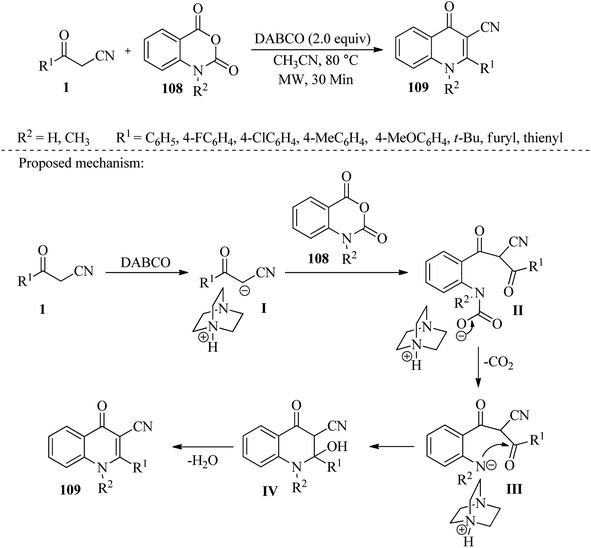 |
| | Scheme 41 Synthesis of 3-cyano-4-quinolone derivatives 109. | |
Yang et al. conducted a study on synthesizing 3-trifluoromethyl-isoquinolines 111, which are important compounds in biology, using an Rh(III)-catalyzed annulation of β-ketonitriles 1 and CF3-substituted imidoyl sulfonium ylides (TFISYs) 110. The reaction was conducted in THF solvent with NaOAc at 60 °C for 24 hours. The transformation involved removing a C–H bond and an unusual C–C bond from benzoyl acetonitrile, while also removing a molecule of dimethyl sulfoxide (DMSO) and acetonitrile (CH3CN). This reaction has several advantages, including low catalyst loadings, mild conditions, wide substrate scope, high efficiency, and scalability (Scheme 42).58
 |
| | Scheme 42 Synthesis of 3-trifluoromethyl-isoquinolines 111. | |
4.4. Synthesis of chromene derivatives
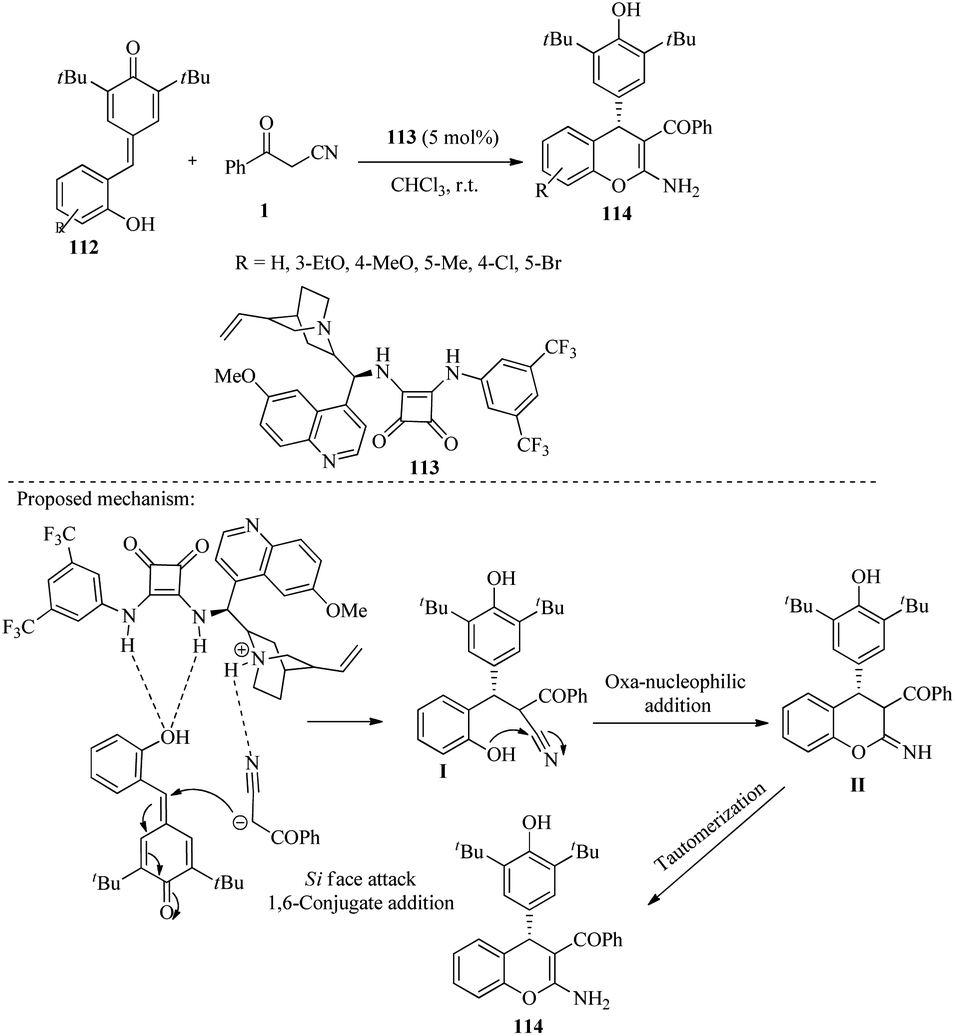
Duan et al. have developed an efficient method to produce 2-amino-4-aryl-4H-chromenes 114. This is achieved through reactions of ortho-hydroxyl-substituted p-QMs 112 with β-ketonitriles 1, using asymmetric organocatalytic chiral 113 at mild conditions in CHCl3 solvent. The process involves 1,6-conjugate addition/annulation/tautomerization (Scheme 43).59
 |
| | Scheme 43 Synthesis of 2-amino-4-aryl-4H-chromenes 114. | |
In 2018, He et al. developed a concise and efficient method for synthesizing functionalized 2-aryl-4H-chromenes 116 using propargylamines 115 and β-ketonitriles 1 in the presence of FeCl3 as a catalyst. This method is environmentally friendly and utilizes CH3CN solvent for 24 hours under reflux conditions. The reaction features a highly efficient tandem sequence of 1,4-conjugate addition, 6-endo-dig cyclization, and oxidation. This protocol accommodates various functional groups, rendering it a practical and effective method for synthesizing 2-aryl-4H-chromene skeletons 116 (Scheme 44).60
 |
| | Scheme 44 Synthesis of functionalized 2-aryl-4H-chromenes 116. | |
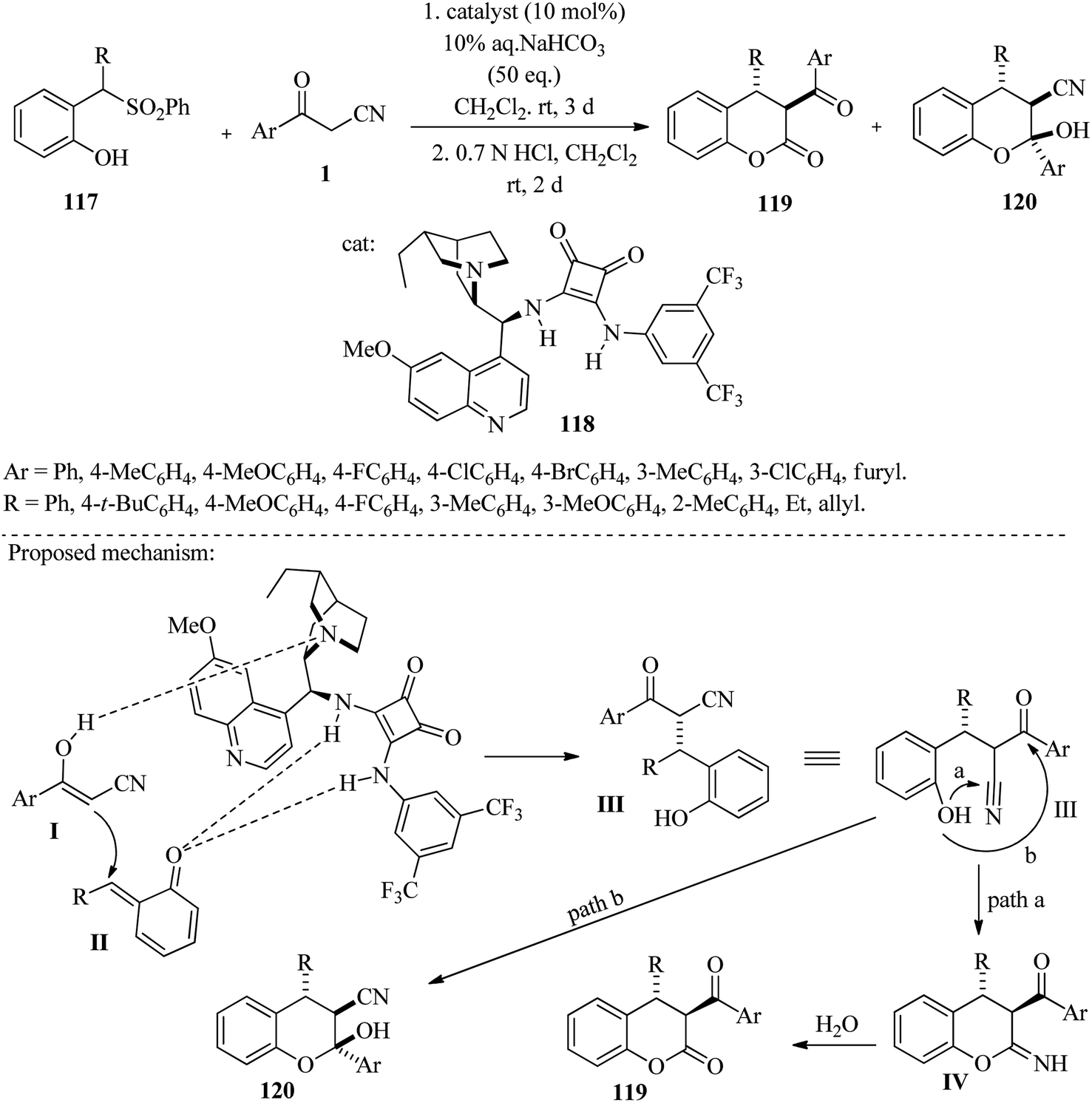
The Pan group recently developed an organocatalytic method for the synthesis of chiral 3,4-dihydrocoumarins 119 and tetrasubstituted chromas 120. This process involves Michael addition and intramolecular cyclization of β-ketonitriles 1 and 2-sulfonyl methyl phenols 117. The reaction takes place in dichloromethane with sodium bicarbonate at room temperature. In the reaction mechanism, the amino group of catalyst 118 activates the carbonyl groups and acts as a Lewis acid to facilitate the reaction (Scheme 45).61
 |
| | Scheme 45 Synthesis of chiral 3,4-dihydrocoumarins 119 and tetrasubstituted chromas 120. | |
Jiang and his team conducted research on the domino reaction of two molecules of β-ketonitriles 1 and one molecule of 2-aryl-3-nitro chromene 121, in the presence of triethylamine. They performed this reaction in tetrahydrofuran solvent to synthesize various derivatives of dihydrofuro[2,3-c]chromenes 122, and by using polar protic solvents such as ethanol and methanol, to synthesize chromeno[3,4-b] substituted pyridines 123. These products were obtained under mild conditions. One of the most significant advantages of this process is the wide range of products produced with satisfactory efficiency (Scheme 46).62
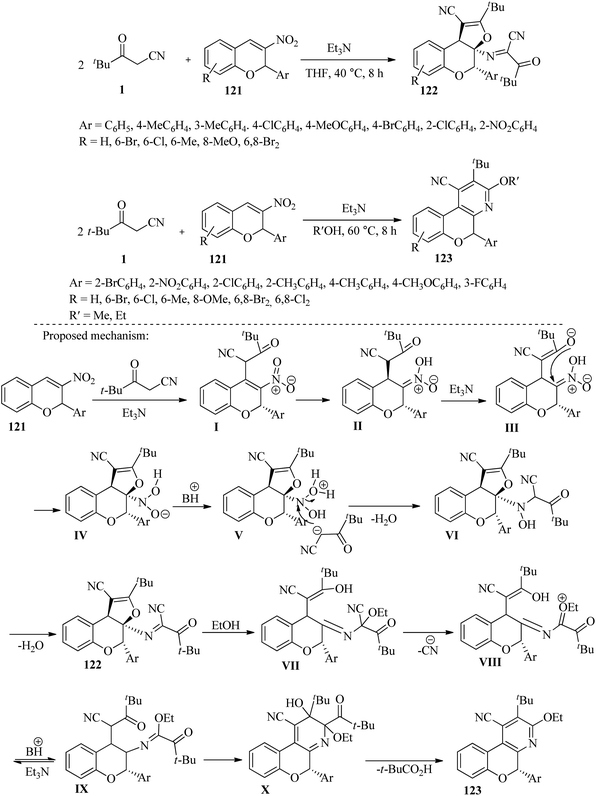 |
| | Scheme 46 Synthesis of dihydrofuro[2,3-c]chromene 122 and chromeno[3,4-b] substituted pyridines 123. | |
Also, our research group a general and efficient method for the chemoselective synthesis of benzo[c]chromen-6-ones 125 has been developed. The reactions were accomplished in the presence of Et3N in EtOH under reflux conditions to afford functionalized benzo[c]chromen-6-ones 125 in 70–91% yield. The mechanism of this reaction includes a base-promoted nucleophilic substitution/deprotonation/intramolecular aldol condensation/carboxylic acid or alkyl hydrogen carbonate elimination/aromatization reaction of β-ketonitriles 1 and α,β-unsaturated coumarins 124 (Scheme 47).63
 |
| | Scheme 47 Synthesis of benzo[c]chromen-6-ones 125. | |
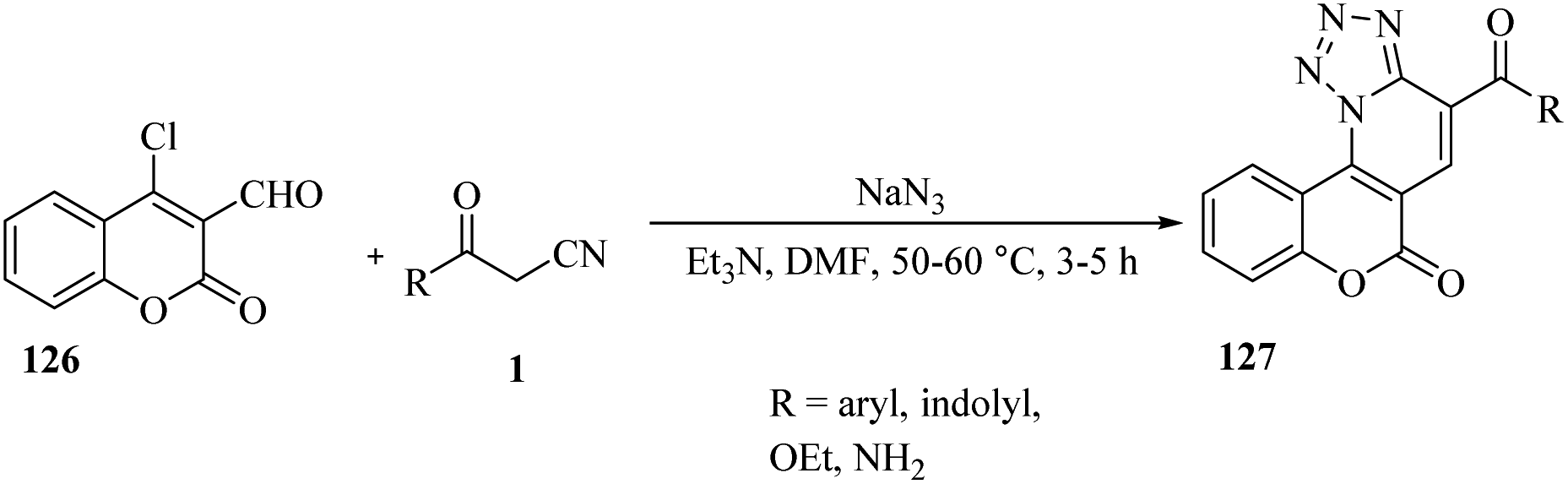
Bhuyan and colleagues have developed an effective and versatile method for the selective synthesis of tetrazole-fused pyrido[3,2-c]coumarin derivatives 127.
This is achieved through a one-pot three-component reaction of 4-chloro-3-formylcoumarin 126 via intramolecular 1,3-cycloaddition reaction of azides to β-ketonitriles 1. The reactions were carried out in DMF under mild conditions with the addition of one drop of Et3N. The resulting products were obtained with yields ranging between 75-86% (Scheme 48).64
 |
| | Scheme 48 Synthesis of tetrazole-fused pyrido[3,2-c]coumarin derivatives 127. | |
In another study Alizadeh and Rostampoor developed a method for synthesizing dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-ones 128 through a three-component reaction involving α,β-unsaturated coumarins 124, β-ketonitriles 1, and hydrazine hydrate 52. This process includes 1,4-addition and aza-Michael addition, forming two carbon–nitrogen (C–N) bonds. The reactions were accomplished in the presence of Et3N in EtOH under reflux conditions to afford products up to 77–92% yield (Scheme 49).65
 |
| | Scheme 49 Synthesis of dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-ones 128. | |
Choudhury and his team developed a concise method using molecular iodine to synthesize dihydrochromeno[4,3-b]pyrazolo[4,3-e]pyridin-6(7H)-ones 130 through a four-component reaction involving aromatic aldehydes 9, 4-hydroxycoumarin 129, β-ketonitriles 1, and hydrazine hydrate 52 in EtOH under reflux conditions. The synthesis process involves Knoevenagel condensation, Michael addition, and intramolecular condensation (Scheme 50).66
 |
| | Scheme 50 Synthesis of dihydrochromeno[4,3-b]pyrazolo[4,3-e]pyridin-6(7H)-ones 130. | |
4.5. Synthesis of spiro compounds
Baradarani and his team developed a simple and concise method catalyzed by DMAP for synthesizing a series of spiro[4H-pyran-3,3′-oxindoles] 132. This was achieved through a three-component reaction of isatin (5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-1,2-dione) 131 with various β-ketonitriles 1 and malononitrile derivatives 39 in ethanol under reflux. The synthesis process involves Knoevenagel condensation, Michael addition, and intramolecular condensation (Scheme 51).67
 |
| | Scheme 51 Synthesis of spiro[4H-pyran-3,3′-oxindoles] 132. | |
Wu and colleagues developed a method using just 2 mol% of a chiral organocatalyst 134, producing chiral spiro[4H-pyran-oxindole] derivatives 135 with 97–99% yields and 76–97% enantioselectivities. This is achieved through a one-pot reaction of β-ketonitriles 1 and isatylidene malononitriles 133 in CH2Cl2 at −10 °C and adding 1 mol% morpholine. The addition of 2% mol of tertiary amine catalyst 134, which contains many hydrogen bond donors, activates the carbonyl β-ketonitrile 1 group. This activation promotes the Michael addition process, resulting in the formation of spirooxindole through intramolecular cyclization (Scheme 52).68
 |
| | Scheme 52 Synthesis of chiral spiro[4H-pyran-oxindole] derivatives 135. | |
4.6. Synthesis of propellanes
In 2017, Yavari and his team revealed a method for synthesizing dioxopropanes 138 and 139 that is highly chemoselective and regioselective. The process involves reacting adducts of acenaphthoquinolidene malonitriles 136 (and ninhydrylidene malonitriles 137) with β-ketonitriles 1 in 70% aqueous MeOH at room temperature. The reaction mechanism involves three processes: Michael addition and two molecular cyclization processes that form one C–C bond and two C–O bonds (Scheme 53).69
 |
| | Scheme 53 Synthesis of dioxopropanes 138 and 139. | |
Jeong and his team developed a simple and concise method catalyzed by DBU for synthesizing new substituted heterocyclic[4.3.3]propellane 141. This was done through a one-pot reaction of a-diketones 140 with variously substituted β-ketonitriles 1 in MetOH under reflux conditions for 8–12 h. The synthesis process involves Knoevenagel, Michael, and intramolecular/Paal–Knorr cyclization (Scheme 54).70
 |
| | Scheme 54 Synthesis of substituted heterocyclic[4.3.3]propellane 141. | |
In 2022, the same group synthesized fused (epoxyetheno)indeno-furans 143 by reacting acenaphthoquinolidene 142 and β-ketonitriles 1. The reaction was carried out in the presence of 30 mol% morpholine in DCM under mild conditions for 3 hours. The reaction mechanism is similar to Scheme 54 and involves three processes: Knoevenagel, Michael, and intramolecular/Paal–Knorr cyclization (Scheme 55).71
 |
| | Scheme 55 Synthesis of fused (epoxyetheno)indeno-furans 143. | |
4.7. Nitrogen-containing heterocycles
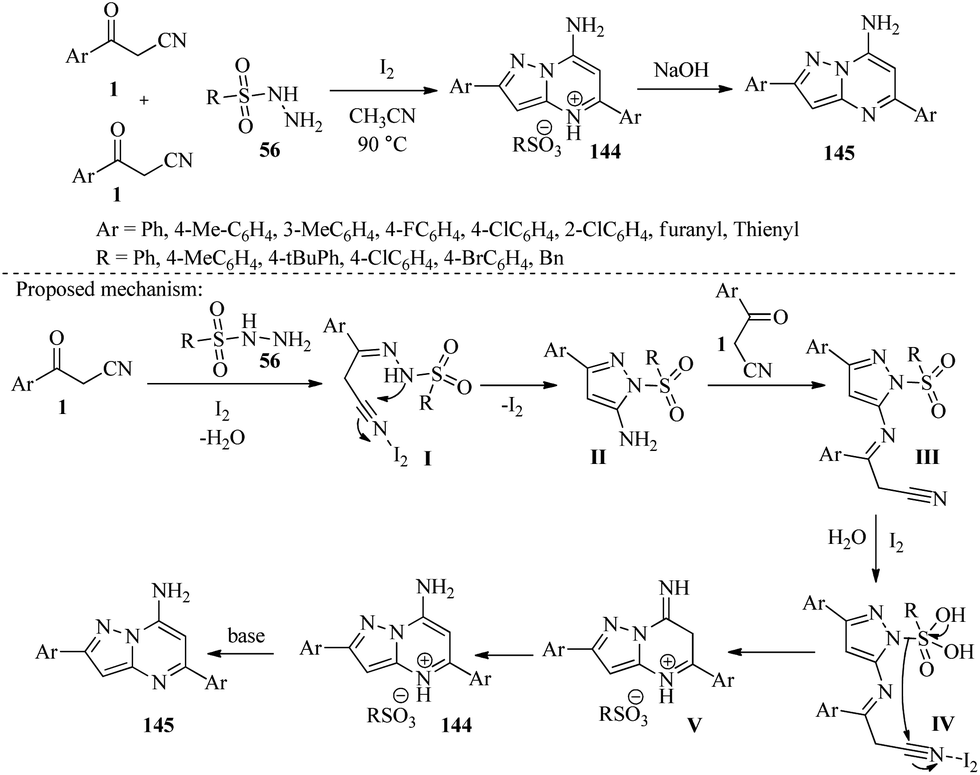
4.7.1. Bicyclic system. Sun and colleagues developed an iodine-catalyzed three-component bi-cyclization of β-ketonitriles 1 with sulfonylhydrazides 56 to synthesize a range of functionalized pyrazolo[1,5-a]pyrimidine-4-iome sulfonates 144 in a highly regioselective manner. These sulfonates can be quantitatively converted into densely functionalized pyrazolo[1,5-a]pyrimidines 145 in the presence of bases (Scheme 56).72
 |
| | Scheme 56 Synthesis of functionalized pyrazolo[1,5-a]pyrimidine-4-iome sulfonates 144 and pyrazolo[1,5-a]pyrimidines 145. | |
In 2021, Kim and his colleagues developed a reaction between β-ketonitriles 1 and N-substituted pyrrole-2-carboxaldehyde 146 in CH3CN, in the presence of piperidinium acetate, which allows selective access to 5-acylindolizine-7-carbonitrile 147 through a Knoevenagel condensation–intramolecular aldol cyclization sequence (Scheme 57).73
 |
| | Scheme 57 Synthesis of 5-acylindolizine-7-carbonitrile 147. | |
In a recent study, Khosropour and his colleagues developed a novel catalyst named 3-(propylthio)propane-1-sulfonic acid-functionalized MCM-41 (PTPSA@MCM-41) 148 which was found to be effective and reusable. This catalyst was used for the multicomponent synthesis of dihydro-1H-pyrazolo-[3,4-b]pyridines 149 and 1H-pyrazolo[3,4-b]pyridines 150 through the reaction of aldehydes 9, β-ketonitriles 1, and 1H-pyrazol-5-amines 53 at 80 °C under solvent-free conditions. One significant advantage of this catalytic system is its high catalytic activity and reusability, mild reaction conditions, simple operation, and benign environmental impact (Scheme 58).74
 |
| | Scheme 58 Synthesis of dihydro-1H-pyrazolo-[3,4-b]pyridines 149 and 1H-pyrazolo-[3,4-b]pyridines. | |
A selective protocol has been developed for the synthesis of quinoxaline derivatives 152 under the same reaction conditions simply from the reaction of phenylenediamine 151 with various derivatives of β-ketonitriles 1 in the presence of visible light in THF solvent and room temperature. This is a novel protocol that accommodates various β-ketonitriles 1 and binary aromatic amines via visible light-induced electron transfer and oxidative coupling. This metal-free method works at room temperature with various substrates and does not require extra oxidants. It typically gives moderate to good yields (Scheme 59).75
 |
| | Scheme 59 Synthesis of synthesis of quinoxaline derivatives 152. | |
4.7.2. Tricyclic system. A team of researchers led by Behbehan et al. has developed a simple and efficient method for synthesizing benzo[4,5]imidazo[1,2-b]pyridazines 154 using intramolecular SNAr. They used either H2O/AcONa or AcOH/AcONa as the reaction medium and microwave irradiation as an energy source. The entire process involves only one step, which is the reaction between 3-oxo-2-arylhydrazonopropanals 153 and β-ketonitriles 1 such as (3-oxo-3-phenylpropionitrile, 3-oxo-3-hetarylpropionitrile, ethyl cyanoacetate, and 2-cyanoacetamide). The final compounds were produced with an overall yield of 89% to 99% (Scheme 60).76
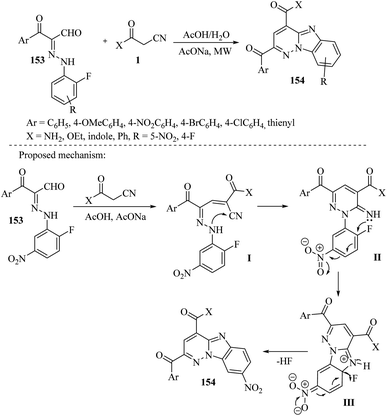 |
| | Scheme 60 Synthesis of benzo[4,5]imidazo[1,2-b]pyridazines derivatives 154. | |
Singh and his team synthesized a series of pyrazolo[1,5-c]quinazolines 159 that function as inhibitors for EGFR. They used a highly efficient multicomponent route involving a palladium-catalyzed four-component one-pot tandem reaction.
In this one-pot process, they first formed azomethine 158 by reacting azidobenzaldehyde derivatives 155, isocyanides 156, and arylsulfonylhydrazides 157 in the presence of Pd(OAc)2 (7.5 mol%) in the toluene solvent. Then, they added β-ketonitriles 1 to the reaction mixture in the presence of DABCO, which resulted in pyrazolo[1,5-c]quinazoline formation. The target compounds were screened against MDA-MB-231, A549, and H1299 cancer cell lines (Scheme 61).77
 |
| | Scheme 61 Synthesis of pyrazolo[1,5-c]quinazolines derivatives 159. | |
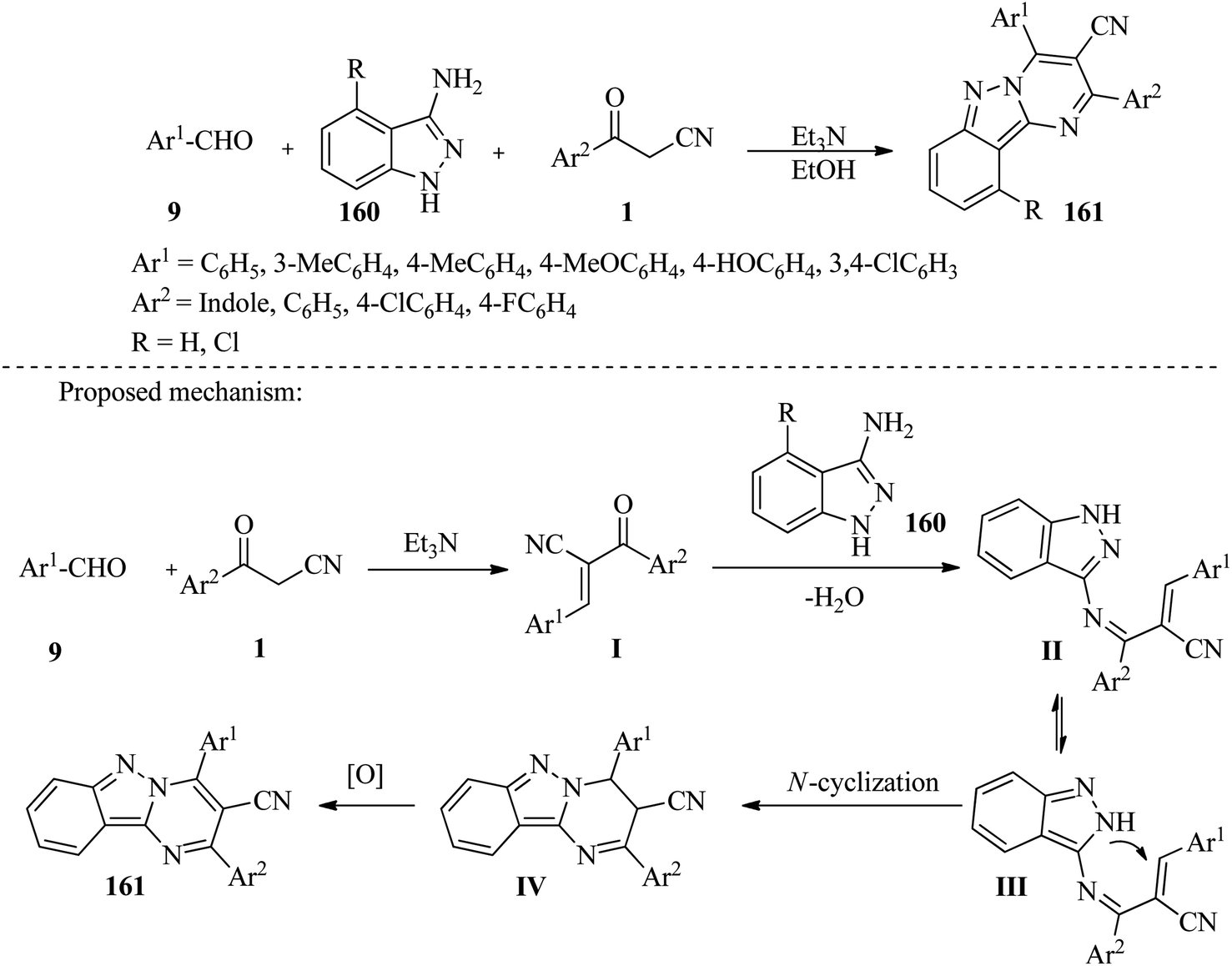
In 2017, Rong and co-workers developed a simple and efficient method for the synthesis of pyrimido[1,2-b]indazol-3-carbonitrile derivatives 161 from aromatic aldehydes 9, 1H-indazol-3-amine (4-chloro-1H-indazol-3-amine) 160 and β-ketonitriles 1 (3-(1H-indol-3-yl)-3-oxopropanenitrile or 3-oxo-3-arylpro-panenitrile) under metal-free conditions. This was a very successful technique for making pyrimido[1,2-b]indazole compounds 161 using only ethanol and triethylamine in a typical laboratory setting. This process has many advantages, including simple operation, high efficiency, easy isolation, and a wide range of substrates (Scheme 62).78
 |
| | Scheme 62 Synthesis of pyrimido[1,2-b]indazol-3-carbonitrile derivatives 161. | |
4.7.3. Tetracyclic system. New N-fused heterocyclic compounds (pyrazolo[5′,1′:2,3]pyrimido[6,1-a]isoindol derivatives) 164 have been synthesized through a simple one-pot four-component reaction involving hydrazine hydrate 52, 3-oxoalkanonitriles 1,2-phthaldehydic acid 162, and various CH-acids 163 in ethanol, using 4-toluenesulfonic acid as a catalyst. Researchers found the most important advantage of this reaction is the formation of all rings during the reaction time and the use of 4-toluenesulfonic acid as an available and cheap catalyst and ethanol as a green organic solvent (Scheme 63).79
 |
| | Scheme 63 Synthesis of pyrazolo[5′,1′:2,3]pyrimido[6,1-a]isoindol derivatives 164. | |
4.8. Polycyclic system
Yang et al. have developed an efficient method for synthesizing polysubstituted cyanocarbazoles 167. The reaction can be carried out in two steps using Cs2CO3 and FeBr2 (5 mol%) as catalysts. The process involves reacting indolyl alkynyl ketones 165 with β-ketonitriles 1 in the presence of nitrogen gas and DMSO solvent under reflux conditions. The transformation involves Cs2CO3-promoted C–C σ-bond activation of α β-ketonitriles, followed by Fe-catalyzed selective C–H and/or C–C bond activations. This selective C–C σ-bond insertion and C–H activation could have broad implications for discovering new routes in organic chemistry that lead to advanced molecular scaffolds (Scheme 64).80
 |
| | Scheme 64 Synthesis of polysubstituted cyanocarbazoles 167. | |
In another study, Jeong et al. have developed a one-pot coupling method that involves three components: 2-hydroxybenzaldehydes 168, β-ketonitriles 1, and isonitriles 156. This method results in the construction of a new tricyclic 2-phenyl-1H-benzofuro[2,3-b]pyrrole ring 169. The reaction sequence begins with a Knoevenagel condensation of 2-hydroxybenzaldehydes 168 with β-ketonitriles 1, followed by the nucleophilic addition of the divalent isocyanic carbon 156. This reaction produces a reactive nitrilium carbon that can be easily trapped by a nearby phenolic group of 2-hydroxybenzaldehydes 168, yielding diverse benzofuro[2,3-b]pyrroles 169 in a single step (Scheme 65).81
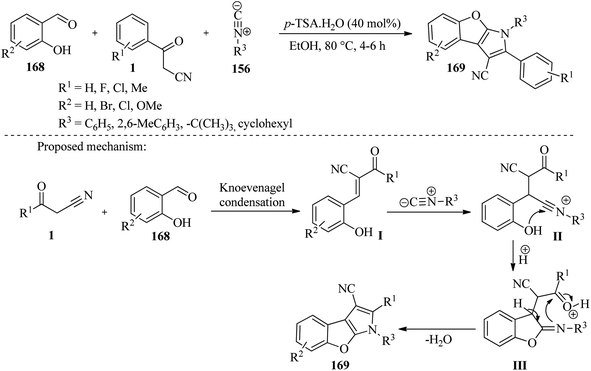 |
| | Scheme 65 Synthesis of polysubstituted benzofuro[2,3-b]pyrroles 169. | |
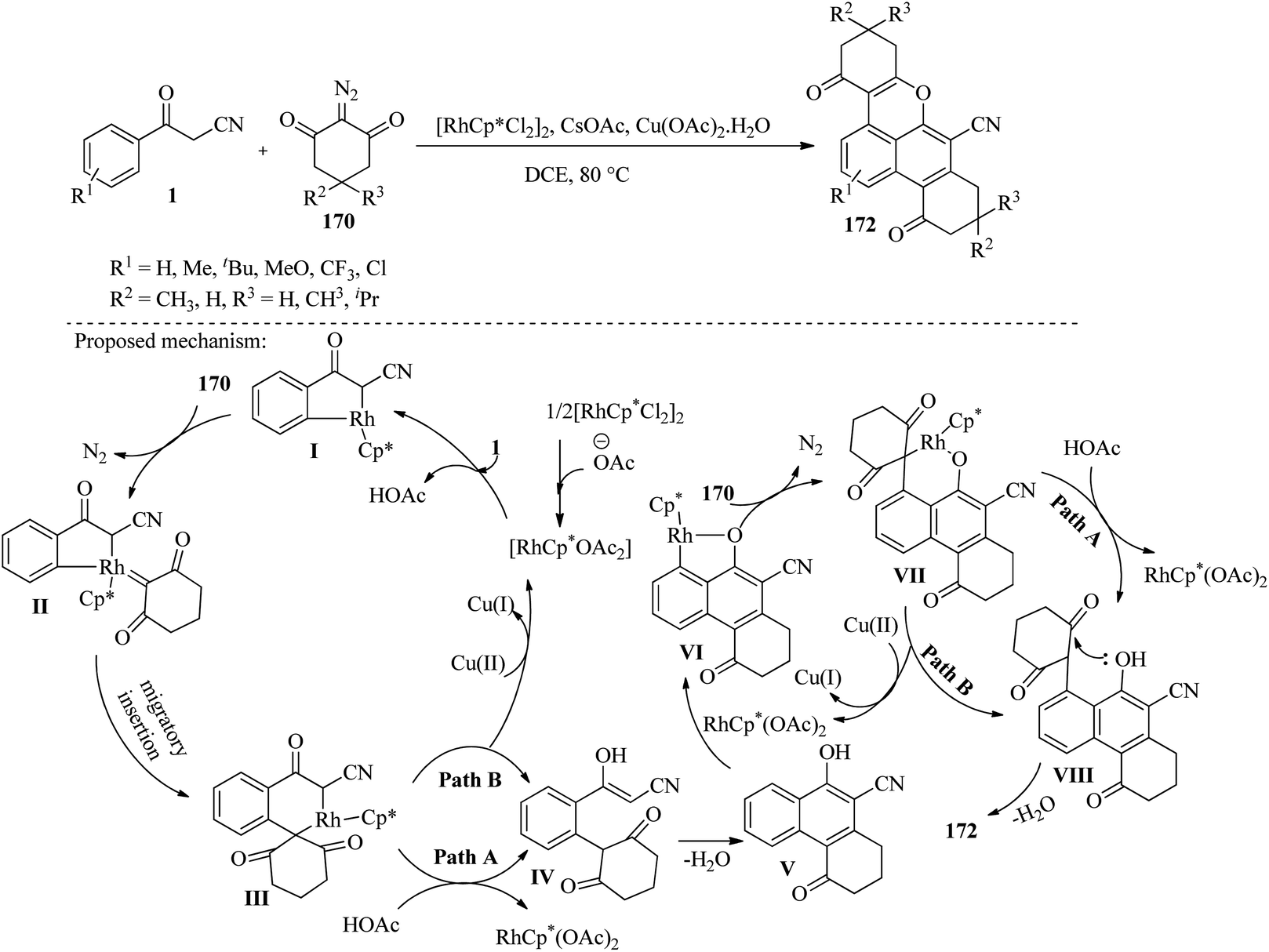
According to Wang et al., the use of an iridium catalyst facilitates the cascade annulation reactions of β-ketonitriles 1 with diazo compounds 170 and 171, leading to the formation of substituted naphtho[1,8-bc]pyrans 172 and 173. The reactions involve sequential cleavage of C(sp2)–H/C(sp3)–H and C(sp2)–H/O–H bonds, resulting in the production of different types of naphtho[1,8-bc]pyrans 172 and 173 depending on whether cyclic or open-chain diazo compounds are used. The researchers found that the reactions yield most products in moderate to good amounts and work well with a wide range of substrates (Scheme 66).82
 |
| | Scheme 66 Synthesis of polysubstituted naphtho[1,8-bc]pyrans 172 and 173. | |
Continuing research in this field, Zhang and colleagues have developed an efficient approach for synthesizing functionalized naphtho[1,8-bc]pyrans 172 through Rh(III)-catalyzed cascade reactions of β-ketonitriles 1 with cyclic 2-diazo-1,3-dicarbonyl compounds 170. The formation of the title compounds involves a cascade process that goes through two steps. The process begins with the cleavage of C(sp2)AH/C(sp3)AH bonds, followed by metalation and carbenoid insertion of 170 with I, resulting in intramolecular annulation to yield substituted 1-naphthol V as a key intermediate. In the second step, the in situ formed 1-naphthol intermediate V undergoes C(sp2)AH/OAH bonds cleavage, metalation, carbenoid insertion with 170, and an intramolecular cyclization to give the naphtho[1,8-bc]pyran product. According to the authors, this new method for synthesizing naphtho[1,8-bc]pyran derivatives 172 offers several advantages over previously reported methods, including a simple operating method, readily available substrates, high efficiency, and excellent atom economy (Scheme 67).83
 |
| | Scheme 67 Synthesis of polysubstituted naphtho[1,8-bc]pyrans 172. | |
In a recent report, Wang and colleagues demonstrated the successful application of rhodium(III) catalysis in the synthesis of naphthols 175 and 2,3-dihydronaphtho[1,8-bc]pyrans 176.
The process is involved the cascade activation of β-ketonitriles 1 and annulation with sulfoxonium ylides 174. This study shows that this method is very efficient, selective, and versatile, and a wide range of β-ketonitrile 1 and sulfoxonium ylide 174 have been successfully employed. The redox-neutral conditions and broad substrate scope make this approach suitable for the synthesis of complex structures that are otherwise challenging to access (Scheme 68).84
 |
| | Scheme 68 Synthesis of naphthols 175 and 2,3-dihydronaphtho[1,8-bc]pyrans 176. | |
Bai et al. have reported the development of three-component reactions that involve 4-hydroxyindole 177, aldehydes 9, and β-ketonitriles 1 for the synthesis of 3,4-fused tricyclic indoles 178. These reactions utilize either potassium fluoride or diethylamine as a catalyst, offering straightforward access to 3,4-fused tricyclic indoles with good to excellent yields (Scheme 69).85
 |
| | Scheme 69 Synthesis of 3,4-fused tricyclic indoles 178. | |
5. Conclusion
This review summarizes the last decade of advancements in using β-ketonitriles as vinylogous nucleophiles for synthesizing biologically active scaffolds such as functionalized coumarins, quinolines, five and six-membered heterocycles, spiro and polycyclic heterocycles, propellanes and other useful cyclic and acyclic compounds, as well as natural products.
The dual reactivity of β-ketonitriles, serving as both electrophiles from the CN moiety and nucleophiles from hydroxyl and CH groups, enables the design of novel tandem reactions. Their high reactivity and ability to coordinate with catalysts through hydrogen bonding and acid-base interactions make them valuable substrates in synthetic organic chemistry, particularly for synthesizing chiral scaffolds.
Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Research Council of Tarbiat Modares University for its support of this work.
Notes and references
- M. H. Elnagdi, M. R. H. Elmoghayar and G. E. H. Elgemeie, Synthesis, 1984, 1984, 1–26 CrossRef.
- N. R. Mohamed, N. Y. Khaireldin, A. F. Fahmy and A. A. El-Sayed, Pharma Chem., 2010, 2, 400 CAS.
- R. E. Khidre, B. F. Abdel-Wahab and F. A.-R. Badria, Lett. Drug Des. Discovery, 2011, 8, 640 CrossRef CAS.
- B. Zarranz, A. Jaso, I. Aldana, A. Monge, S. Maurel, E. Deharo, V. Jullian and M. Sauvain, Chem. Abstr., 2006, 144, 266586 Search PubMed.
- F. Al-Omran, A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2001, 57, 10163 CrossRef CAS.
-
(a) R. E. Khidre and B. F. Abdelwahab, Turk. J. Chem., 2013, 37, 685–711 CrossRef CAS;
(b) M. H. Elnagdi, M. R. H. Elmoghayar and G. E. H. Elgemeie, Synthesis, 1984, 1984, 1–26 CrossRef;
(c) C. R. Hauser and C. J. Eby, J. Am. Chem. Soc., 1957, 79, 728–731 CrossRef CAS.
- I. Yavari, S. Shaabanzadeh and S. Sheikhi, ChemistrySelect, 2020, 5, 564–568 CrossRef CAS.
- K. Kiyokawa, T. Nagata and S. Minakata, Synthesis, 2018, 50, 485–498 CrossRef CAS.
- J. M. Robinson, M. Ahmed, N. J. Alaniz, T. R. Boyles, C. D. Brasher, K. A. Floyd and C. D. Wright, J. Heterocycl. Chem., 1998, 35, 65–69 CrossRef CAS.
- S. V. Ryabukhin, A. S. Plaskon, E. N. Ostapchuk, D. M. Volochnyuk, O. V. Shishkin, A. N. Shivanyuk and A. A. Tolmachev, Org. Lett., 2007, 9, 4215–4218 CrossRef CAS PubMed.
- P. S. N. Reddy and P. Reddy, Heterocycl. Commun., 2004, 10, 163–166 CAS.
- Q. Jia and J. Wang, Org. Lett., 2016, 18, 2212–2215 CrossRef CAS PubMed.
- R. E. Khidre and B. F. Abdel-Wahab, Curr. Org. Chem., 2013, 17, 430–445 CrossRef CAS.
- B. F. Abdel-Wahab, M. F. El-Mansy and R. E. Khidre, J. Iran. Chem. Soc., 2013, 10, 1085–1102 CrossRef CAS.
-
(a) A. Raghunadh, S. B. Meruva, N. A. Kumar, G. S. Kumar, L. V. Rao and U. S. Kumar, Synthesis, 2012, 44, 283–289 CAS;
(b) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed;
(c) A. Nagaki, D. Ichinari and J. I. Yoshida, Chem. Commun., 2013, 49, 3242–3244 RSC.
- Y. Xie, J. Liu, Y. Huang and L. Yao, Tetrahedron Lett., 2015, 56, 3793–3795 CrossRef CAS.
- C. Xu, N. N. Zhang, X. J. Li, Y. Q. Ge, P. H. Diao and C. Guo, Synlett, 2018, 29, 1065–1070 CrossRef CAS.
- H. S. P. Rao and N. Muthanna, Synlett, 2016, 27, 2014–2018 CrossRef CAS.
- N. Gao, X. Liu, X. Yuan, B. Hu, P. Jiang, J. Lin and Y. Jin, J. Org. Chem., 2022, 87, 10298–10308 CrossRef CAS.
- X. Song, B. N. Do Doan, X. Zhang, R. Lee and X. Fan, Org. Lett., 2020, 22, 46–51 CrossRef CAS.
- J. Wang, J. Xiang, M. Wang, J. Guan and A. Wu, Tetrahedron, 2014, 70, 1412–1417 CrossRef CAS.
- H. K. Sha, T. Xu, F. Liu, B. Z. Tang, W. J. Hao, S. J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10415–10418 RSC.
- S. Sathiyamoorthi, A. I. Almansour, S. K. Raju, A. Natarajan and R. R. Kumar, Synth. Commun., 2021, 51, 234–244 CrossRef CAS.
- B. Hocaoglu and M. Yilmaz, Synth. Commun., 2019, 49, 1938–1946 CrossRef CAS.
- B. S. Gore, C. Y. Kuo, A. M. Garkhedkar, Y. L. Chang and J. J. Wang, Org. Lett., 2020, 22, 6160–6165 CrossRef CAS.
- J. Feng, X. Yuan, W. Luo, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2016, 22, 15650–15653 CrossRef CAS PubMed.
- J. Wang, S. Chen, W. Wu, S. Wen and Z. Weng, J. Org. Chem., 2019, 84, 15685–15696 CrossRef CAS PubMed.
- K. Xu, H. Liu, Y. Hou, J. Shen, D. Liu and W. Zhang, Chem. Commun., 2019, 55, 13295–13298 RSC.
- Q. Li, X. He, J. Tao, M. Xie, H. Wang, R. Li and Y. Shang, Adv. Synth. Catal., 2019, 361, 1874–1886 CrossRef CAS.
- S. G. Balwe, J. S. Kim, Y. I. Kim and Y. T. Jeong, Tetrahedron, 2019, 75, 797–807 CrossRef CAS.
- D. Zhong, F. Jiang, S. Shen, L. Wang, W. Wang, Y. Wu and H. Guo, Adv. Synth. Catal., 2020, 362, 5026–5030 CrossRef CAS.
- J. Zhou, M. Huang, X. Zhu and Y. Wan, Chin. Chem. Lett., 2021, 32, 445–448 CrossRef CAS.
- L. Palanivel and V. Gnanasambandam, Org. Biomol. Chem., 2020, 18, 3082–3092 RSC.
- M. B. Yadav and Y. T. Jeong, Org. Biomol. Chem., 2021, 19, 7409–7419 RSC.
- S. Nagaraju, S. Liu, J. Liu, S. Yang, R. Liu, Z. Chen and X. Fang, Org. Lett., 2019, 21, 10075–10080 CrossRef CAS.
- M. Xia, Z. Moussa and Z. Judeh, Molecules, 2022, 27, 5285 CrossRef CAS PubMed.
- A. Shaabani, M. T. Nazeri and R. Afshari, Mol. Diversity, 2019, 23, 751–807 CrossRef CAS PubMed.
-
(a) M. Kelada, J. M. Walsh, R. W. Devine, P. McArdle and J. C. Stephens, Beilstein J. Org. Chem., 2018, 14, 1222–1228 CrossRef CAS PubMed;
(b) Z. He, C. Charleton, R. W. Devine, M. Kelada, J. M. Walsh, G. E. Conway and J. F. Curtin, Eur. J. Med. Chem., 2021, 224, 113736–113747 CrossRef CAS.
- Y. Wei, P. Liu, Y. Liu, J. He, X. Li, S. Li and J. Zhao, Org. Biomol. Chem., 2021, 19, 3932–3939 RSC.
- L. Savegnago, M. do Sacramento, L. M. Brod, M. G. Fronza, N. Seus, E. J. Lenardao and D. Alves, RSC Adv., 2016, 6, 8021–8031 RSC.
- A. V. Kachanov, A. V. Zamaraev, A. V. Gerasimenko, K. V. Maslov, O. Y. Slabko and V. A. Kaminskii, Synlett, 2018, 29, 2035–2038 CrossRef CAS.
- M. Luo, J. C. Zhang and H. Yin, J. Chem. Sci., 2015, 127, 163–166 CrossRef CAS.
- J. Sun, H. Ge, X. Zhen, X. An, G. Zhang, D. Zhang-Negrerie and K. Zhao, Tetrahedron, 2018, 74, 2107–2114 CrossRef CAS.
- M. Krasavin, M. Korsakov, Z. Zvonaryova, E. Semyonychev, T. Tuccinardi, S. Kalinin and C. T. Supuran, Bioorg. Med. Chem., 2017, 25, 1914–1925 CrossRef CAS PubMed.
- Z. Niu, X. He and Y. Shang, Tetrahedron: Asymmetry, 2014, 25, 796–801 CrossRef CAS.
- C. Ni and X. Tong, J. Am. Chem. Soc., 2016, 138, 7872–7875 CrossRef CAS.
- W. Zhou, C. Ni, J. Chen, D. Wang and X. Tong, Org. Lett., 2017, 19, 1890–1893 CrossRef CAS PubMed.
- C. Sheng, Z. Ling, T. Ahmad, F. Xie and W. Zhang, Chem.–Eur. J., 2022, 28, e202200128, DOI:10.1002/chem.202200128.
- M. Zhang, M. N. Chen, J. M. Li, N. Liu and Z. H. Zhang, ACS Comb. Sci., 2019, 21, 685–691 CrossRef CAS PubMed.
- Y. Zhang, H. Luo, Q. Lu, Q. An, Y. Li, S. Li and B. Li, Chin. Chem. Lett., 2021, 32, 393–396 CrossRef CAS.
- C. Bharkavi, P. Gunasekaran, S. V. Kumar, M. Sakthi and S. Perumal, Tetrahedron Lett., 2014, 55, 5486–5490 CrossRef CAS.
- I. R. Siddiqui, P. Rai and A. Srivastava, New J. Chem., 2014, 38, 3791–3795 RSC.
- C. K. Chan, Y. L. Chan, Y. L. Tsai and M. Y. Chang, J. Org. Chem., 2016, 81, 8112–8120 CrossRef CAS PubMed.
- A. Palanimuthu, C. Chen and G. H. Lee, Tetrahedron Lett., 2021, 72, 153063 CrossRef CAS.
- S. Dhiman, H. K. Saini, N. K. Nandwana, D. Kumar and A. Kumar, RSC Adv., 2016, 6, 23987–23994 RSC.
- A. Alizadeh and A. Rostampoor, J. Iran. Chem. Soc., 2022, 19, 1239–1249 CrossRef CAS.
- M. S. Rao and S. Hussain, Tetrahedron Lett., 2021, 76, 153187 CrossRef CAS.
- Z. Yang, P. Li, Z. Chen and X. F. Wu, J. Catal., 2023, 427, 115098 CrossRef CAS.
- C. Duan, L. Ye, W. Xu, X. Li, F. Chen, Z. Zhao and X. Li, Chin. Chem. Lett., 2018, 29, 1273–1276 CrossRef CAS.
- X. He, H. Wang, X. Cai, Q. Li, J. Tao and Y. Shang, Org. Biomol. Chem., 2018, 16, 7191–7202 RSC.
- C. Gharui, C. Parida and S. C. Pan, J. Org. Chem., 2021, 86, 13071–13081 CrossRef CAS PubMed.
- W. Jiang, J. Sun, R. Z. Liu and C. G. Yan, Org. Biomol. Chem., 2018, 16, 5816–5822 RSC.
- A. Alizadeh and B. Farajpour, ChemistrySelect, 2021, 6, 6150–6153 CrossRef CAS.
- P. Borah, P. S. Naidu and P. J. Bhuyan, Tetrahedron Lett., 2012, 53, 5034–5037 CrossRef CAS.
- A. Alizadeh and A. Rostampoor, ChemistrySelect, 2022, 7, DOI:10.1002/slct.202200299.
- S. Pal, M. N. Khan, S. Karamthulla and L. H. Choudhury, RSC Adv., 2013, 3, 15705–15711 RSC.
- P. Zafari, B. E. Saatluo, A. Rashidi, M. M. Baradarani and J. A. Jouleb, Arkivoc, 2021, 9–16 Search PubMed.
- J. Xie, W. L. Xing, F. Sha and X. Y. Wu, Eur. J. Org Chem., 2016, 2016, 3983–3992 CrossRef CAS.
- I. Yavari, A. Khajeh-Khezri, S. Bahemmat and M. R. Halvagar, Synlett, 2017, 28, 1785–1788 CrossRef CAS.
- M. B. Yadav, S. R. Bhosle and Y. T. Jeong, Tetrahedron Lett., 2022, 98, 153815 CrossRef CAS.
- M. B. Yadav, S. S. Vagh and Y. T. Jeong, Eur. J. Org Chem., 2022, 2022, e202101534 CrossRef CAS.
- J. Sun, J. K. Qiu, B. Jiang, W. J. Hao, C. Guo and S. J. Tu, J. Org. Chem., 2016, 81, 3321–3328 CrossRef CAS PubMed.
- S. Kim, J. H. Lee, S. H. Yoon and I. Kim, Org. Biomol. Chem., 2021, 19, 5806–5817 RSC.
- S. Safaei, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and V. Mirkhani, J. Iran. Chem. Soc., 2017, 14, 1583–1589 CrossRef CAS.
- C. Zhou, P. Diao, X. Li, Y. Ge and C. Guo, Chin. Chem. Lett., 2019, 30, 371–374 CrossRef CAS.
- H. Behbehani and H. M. Ibrahim, RSC Adv., 2015, 5, 89226–89237 RSC.
- A. J. Ansari, G. Joshi, U. P. Yadav, A. K. Maurya, V. K. Agnihotri, S. Kalra and D. M. Sawant, Bioorg. Chem., 2019, 93, 103314 CrossRef PubMed.
- L. Li, H. Xu, L. Dai, J. Xi, L. Gao and L. Rong, Tetrahedron, 2017, 73, 5358–5365 CrossRef CAS.
- M. Alizadeh-Kouzehrash and A. Rahmati, Tetrahedron, 2020, 76, 130923 CrossRef CAS.
- Y. Yang, J. Huang, H. Tan, L. Kong, M. Wang, Y. Yuan and Y. Li, Org. Biomol. Chem., 2019, 17, 958–965 RSC.
- S. G. Balwe and Y. T. Jeong, New J. Chem., 2020, 44, 3632–3636 RSC.
- K. Yan, B. Li and B. Wang, Adv. Synth. Catal., 2018, 360, 2272–2279 CrossRef CAS.
- B. Zhang, B. Li, C. Guo, X. Zhang and X. Fan, Tetrahedron Lett., 2018, 59, 3094–3099 CrossRef CAS.
- P. Hu, Y. Zhang, Y. Xu, S. Yang, B. Liu and X. Li, Org. Lett., 2018, 20, 2160–2163 CrossRef CAS PubMed.
- R. Bai, J. Yang, L. Min, C. Liu, F. Wu and Y. Gu, Tetrahedron, 2016, 72, 2170–2177 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*