
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in pyrazolo[1,5-a]pyrimidines: synthesis and their role as protein kinase inhibitors in cancer treatment
Terungwa H. Iorkula*a,
Osasere Jude-Kelly Osayawea,
Daniel A. Odogwua,
Latifat Oluwatobi Ganiyua,
Emmanuel Faderinb,
Raymond Femi Awoyemic,
Busayo Odunayo Akodub,
Ikhazuagbe Hilary Ifijen *d,
Omowunmi Rebecca Aworindee,
Peter Agyemange and
Odo Lovelyn Onyinyechif
*d,
Omowunmi Rebecca Aworindee,
Peter Agyemange and
Odo Lovelyn Onyinyechif
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah, USA. E-mail: terungwa@chem.byu.edu
bDepartment of Pharmaceutical Sciences, Southern Illinois University, 1Harirpin Dr, Edwardsville, IL 62026, USA
cDepartment of Chemistry, Mississippi State University, MS 39762, USA
dDepartment of Research Outreach, Rubber Research Institute of Nigeria, Benini City, Nigeria. E-mail: larylans4u@yahoo.com
eDepartment of Chemistry, Michigan Technological University, 1400 Townsend Dr, Houghton, MI 49931, USA
fFaculty of Pharmaceutical Sciences, University of Nigeria, Nsukka, Nigeria
First published on 5th February 2025
Abstract
Pyrazolo[1,5-a]pyrimidines are a notable class of heterocyclic compounds with potent protein kinase inhibitor (PKI) activity, playing a critical role in targeted cancer therapy. Protein kinases, key regulators in cellular signalling, are frequently disrupted in cancers, making them important targets for small-molecule inhibitors. This review explores recent advances in pyrazolo[1,5-a]pyrimidine synthesis and their application as PKIs, with emphasis on inhibiting kinases such as CK2, EGFR, B-Raf, MEK, PDE4, BCL6, DRAK1, CDK1 and CDK2, Pim-1, among others. Several synthetic strategies have been developed for the efficient synthesis of pyrazolo[1,5-a]pyrimidines, including cyclization, condensation, three-component reactions, microwave-assisted methods, and green chemistry approaches. Palladium-catalyzed cross-coupling and click chemistry have enabled the introduction of diverse functional groups, enhancing the biological activity and structural diversity of these compounds. Structure–activity relationship (SAR) studies highlight the influence of substituent patterns on their pharmacological properties. Pyrazolo[1,5-a]pyrimidines act as ATP-competitive and allosteric inhibitors of protein kinases, with EGFR-targeting derivatives showing promise in non-small cell lung cancer (NSCLC) treatment. Their inhibitory effects on B-Raf and MEK kinases are particularly relevant in melanoma. Biological evaluations, including in vitro and in vivo studies, have demonstrated their cytotoxicity, kinase selectivity, and antiproliferative effects. Despite these advances, challenges such as drug resistance, off-target effects, and toxicity persist. Future research will focus on optimizing synthetic approaches, improving drug selectivity, and enhancing bioavailability to increase clinical efficacy.
1 Introduction
1.1 Background on protein kinase inhibitors (PKIs) in cancer treatment
Cancer remains one of the leading causes of mortality worldwide, with an estimated 10 million deaths in 2020 alone. The complex nature of cancer, characterized by uncontrolled cell proliferation, invasion, and metastasis, presents significant challenges in developing effective treatments.1–5 Among the various therapeutic strategies, targeted therapy has emerged as a highly promising approach, focusing on the selective inhibition of molecular pathways crucial for cancer cell survival and growth. Protein kinase inhibitors (PKIs) have become central to this strategy, offering a mechanism to block aberrant signaling pathways that drive oncogenesis.6–9Protein kinases are enzymes that regulate a wide array of cellular processes, including cell growth, differentiation, apoptosis, and metabolism, by transferring a phosphate group from ATP to specific substrates. Disruption of kinase activity, often due to genetic mutations or overexpression, is a hallmark of many cancers. As a result, kinases represent attractive targets for therapeutic intervention, with PKIs designed to specifically inhibit the activity of these enzymes, thereby disrupting the signalling pathways essential for cancer cell proliferation and survival.10–12 Eqn (1) shows the phosphorylation process, where adenosine triphosphate (ATP) donates a phosphate group to a protein through the activity of kinase or phosphatase enzymes. This reaction results in the formation of adenosine diphosphate (ADP) and a phosphorylated protein.
The phosphorylation process depicted in eqn (1) is a prime example of a post-translational modification (PTM), a critical mechanism for regulating protein activity in cellular pathways. These modifications, such as the addition or removal of a phosphate group, play pivotal roles in controlling signalling cascades that maintain cellular homeostasis. In cancer, however, disruption of these processes can lead to aberrant activation of oncogenic pathways, contributing to uncontrolled cell growth and survival.13,14 To better understand the role of phosphorylation as a PTM in these pathways, it is important to examine its broader implications and how disruptions in this process can drive cancer progression.
Post-translational modifications (PTMs) are chemical changes made to proteins after they are synthesized, significantly expanding the functional diversity of the proteome. One of the most critical PTMs is phosphorylation, a process where a phosphate group is added to a protein by an enzyme called a kinase or removed by a phosphatase. Phosphorylation plays a central role in regulating various cellular processes, including signal transduction, metabolism, and cell cycle progression. In the context of the described pathway, phosphorylation modifies a protein's activity, often serving as an on/off switch that influences its function, stability, or ability to interact with other molecules. For instance, a phosphorylated protein can become activated to perform a specific role in a signalling cascade or deactivated to terminate the signal.13,14
In oncogenic pathways, dysregulation of phosphorylation is a frequent contributor to disease progression. Overactive protein kinases or defective phosphatases can lead to uncontrolled signalling, driving abnormal cell growth, division, and survival—key hallmarks of cancer. For example, hyperphosphorylation caused by excessive kinase activity can continuously activate pathways like MAPK/ERK or PI3K/Akt, which are often implicated in cancer. These modifications are directly linked to oncogenesis, as they can amplify signals that promote proliferation or inhibit apoptosis (programmed cell death). Understanding the relationship between PTMs, such as phosphorylation, and oncogenic pathways is essential for developing targeted cancer therapies, including kinase inhibitors designed to block abnormal phosphorylation. Thus, phosphorylation serves as a prime example of how PTMs regulate both normal cellular functions and pathological processes.15–20
| Protein + ATP → ATP + phophorylated protein | (1) |
The success of PKIs in cancer treatment is exemplified by drugs such as imatinib, erlotinib, and vemurafenib, which target specific kinases implicated in chronic myeloid leukemia, non-small cell lung cancer, and melanoma, respectively.13,14 These inhibitors have not only transformed the treatment landscape for these cancers but have also validated the kinase inhibition approach as a viable and effective strategy in oncology. Despite these successes, the development of new PKIs remains a critical area of research, driven by the need to overcome resistance mechanisms, improve selectivity, and target additional kinases implicated in other cancers.15–17
One of the most promising classes of compounds in this regard is pyrazolo[1,5-a] pyrimidines. These heterocyclic structures have garnered significant attention due to their versatile chemistry and potent biological activity. Pyrazolo[1,5-a]pyrimidines have been identified as effective inhibitors of various protein kinases, including those that are not yet effectively targeted by existing therapies.18–20 Their ability to modulate kinase activity through different mechanisms, combined with their potential for structural modification, makes them ideal candidates for the development of next-generation PKIs in cancer treatment.21–23
This review explored the advances in the synthesis of pyrazolo[1,5-a] pyrimidines and their emerging role as potent protein kinase inhibitors. Key synthetic methodologies that enabled the development of these compounds were discussed, their structure–activity relationships analyzed, and their therapeutic potential in targeting critical kinases involved in cancer highlighted. By examining these aspects, the review provided a comprehensive overview of the current state of pyrazolo[1,5-a] pyrimidine research and its implications for the future of targeted cancer therapy.
2 Overview of pyrazolo[1,5-a] pyrimidine compounds
Pyrazolo[1,5-a]pyrimidine compounds represent an important class of heterocyclic molecules that have attracted significant attention in medicinal chemistry due to their wide range of biological activities and potential therapeutic applications. These compounds exhibit diverse pharmacological properties, including anticancer, antiviral, anti-inflammatory, and kinase inhibitory activities, making them promising candidates for drug development.21,22The versatility of pyrazolo[1,5-a]pyrimidines is further highlighted by the existence of several isomeric forms, each characterized by a distinct arrangement of nitrogen atoms within the bicyclic structure. These isomers include pyrazolo[5,1-b]pyrimidine, pyrazolo[5,1-c]pyrimidine, pyrazolo[3,4-d]pyrimidine, and pyrazolo[4,3-d]pyrimidine. Fig. 1 displays the isomeric forms of pyrazolo[1,5-a]pyrimidine. The structural differences between these isomers can lead to variations in their chemical reactivity, biological activity, and interaction with molecular targets, thereby expanding their utility in the design of novel therapeutic agents. This structural diversity allows medicinal chemists to explore different binding modes and mechanisms of action, enhancing the potential for discovering highly selective and potent drugs.23–26
 | ||
| Fig. 1 Selected isomers of pyrazolo[1,5-a]pyrimidine. | ||
These compounds are characterized by their unique structural framework, which consists of a fused bicyclic system incorporating a pyrazole ring and a pyrimidine ring.24–27 The pyrazole ring, a five-membered heterocycle containing two adjacent nitrogen atoms, is fused at the 1,2-positions with the six-membered pyrimidine ring, which contains two nitrogen atoms at the 1 and 3 positions This fused bicyclic core structure forms the basis for the wide-ranging chemical diversity and biological activity observed in pyrazolo[1,5-a] pyrimidine derivatives.28,29
The structural features of pyrazolo[1,5-a] pyrimidines are integral to their pharmacological properties. The fused ring system provides a rigid, planar framework that is highly amenable to chemical modifications at various positions on the rings. Substitutions at different positions of the pyrazolo[1,5-a] pyrimidine scaffold can significantly influence the electronic properties, lipophilicity, and overall molecular conformation of the compounds, thereby affecting their interaction with biological targets.30–32 For instance, modifications at the 3-, 5-, and 7 (pyrimidine ring) positions or at the 4- and 6- (pyrazole ring) positions have been shown to enhance binding affinity to specific protein targets, such as kinases, through hydrogen bonding, hydrophobic interactions, and π–π stacking. The ability to introduce a wide variety of substituents, including alkyl, aryl, amino, and halogen groups, at these positions allows for the fine-tuning of the compounds' pharmacokinetic and pharmacodynamic properties, making them highly versatile in drug design.33–36
The historical development of pyrazolo[1,5-a] pyrimidines dates back to the mid-20th century when they were first synthesized as part of efforts to explore the chemical and biological properties of fused heterocyclic systems. Early studies primarily focused on their synthesis and chemical reactivity, with researchers investigating various routes to construct the fused ring system, including cyclization reactions of appropriate precursors. Over time, the pharmacological potential of pyrazolo[1,5-a] pyrimidines became evident as these compounds were found to exhibit a broad spectrum of biological activities, including anti-inflammatory, antiviral, and anticancer properties.37–39
The significance of pyrazolo[1,5-a] pyrimidines in medicinal chemistry was further solidified in the 1980s and 1990s when these compounds were identified as potent inhibitors of various enzymes, particularly protein kinases. Protein kinases, which play crucial roles in cell signalling pathways, became prominent drug targets for the treatment of cancer and other diseases characterized by disrupted cell growth and proliferation. The unique structural features of pyrazolo[1,5-a] pyrimidines, including their ability to mimic ATP and interact with the ATP-binding pocket of kinases, positioned them as attractive candidates for the development of kinase inhibitors. This led to an explosion of research into the synthesis of novel pyrazolo[1,5-a] pyrimidine derivatives with enhanced selectivity and potency against specific kinase targets.40–42
In the ensuing decades, numerous pyrazolo[1,5-a]pyrimidines-based kinase inhibitors were developed, some of which have progressed to clinical trials and even received regulatory approval for the treatment of various cancers (Fig. 2). These developments underscored the therapeutic potential of pyrazolo[1,5-a]pyrimidines and cemented their status as a valuable scaffold in the design of targeted therapies. The continued interest in pyrazolo[1,5-a]pyrimidines is driven by their structural versatility, which allows for the exploration of new chemical spaces and the development of compounds with improved efficacy, reduced toxicity, and favourable pharmacokinetic profiles.43–45
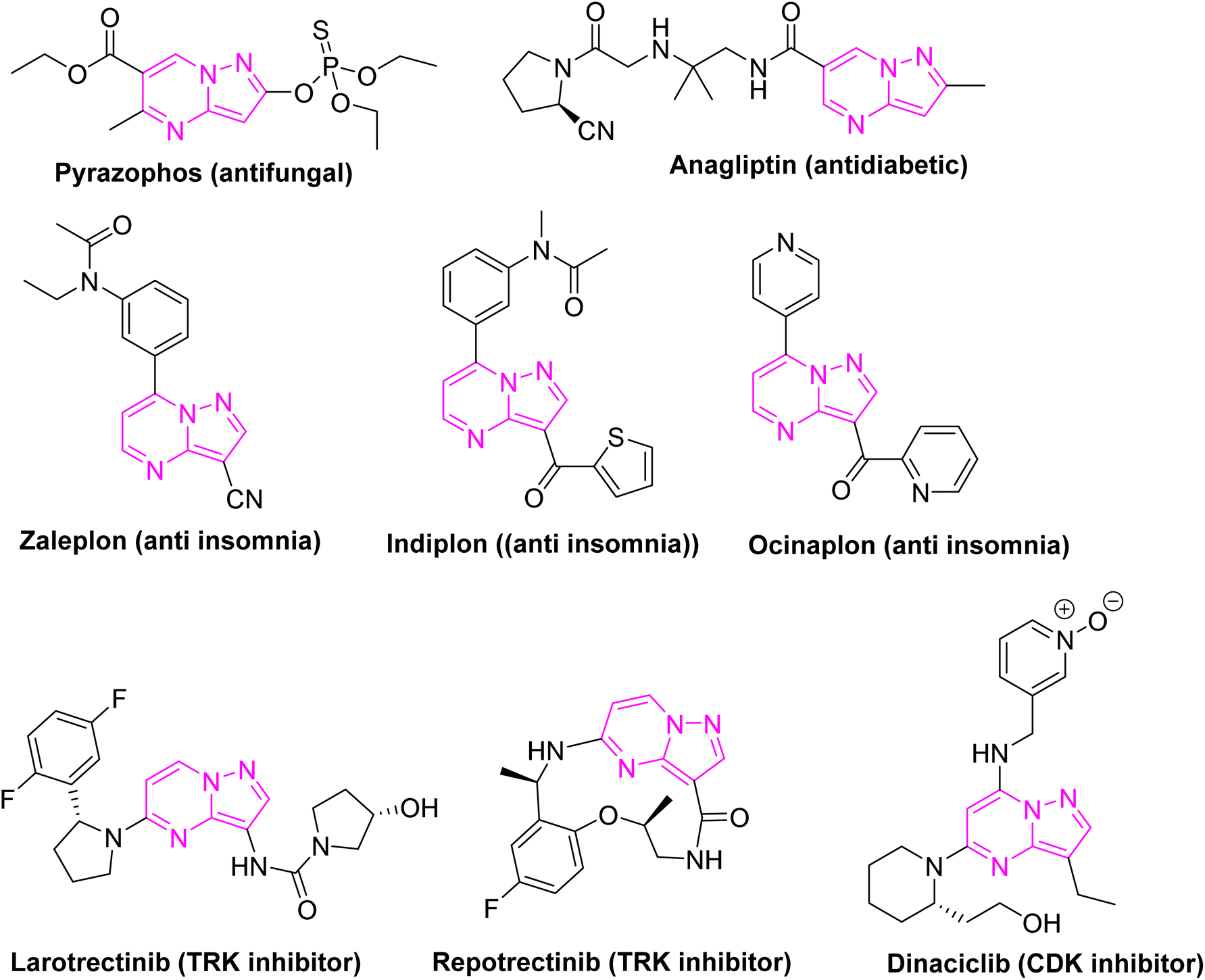 | ||
| Fig. 2 Marketed drugs with pyrazolo[1,5-a]pyrimidine skeleton. | ||
Pyrazolo[1,5-a]pyrimidines have evolved from relatively obscure chemical entities to cornerstone compounds in medicinal chemistry, particularly in the field of oncology. Their structural features not only provide a robust foundation for drug development but also offer extensive opportunities for the discovery of new therapeutics targeting a wide range of diseases. The historical journey of pyrazolo[1,5-a]pyrimidines reflect their growing importance in the design and development of next-generation kinase inhibitors and other therapeutic agents, making them a focal point of ongoing research in drug discovery.
3 Synthetic strategies for pyrazolo[1,5-a]pyrimidines
The synthesis of pyrazolo[1,5-a]pyrimidines has garnered significant attention due to their broad spectrum of biological activities, particularly their role as protein kinase inhibitors in cancer therapy. The fused bicyclic structure of pyrazolo[1,5-a]pyrimidines, combining a five-membered pyrazole ring with a six-membered pyrimidine ring, offers a versatile scaffold for chemical modifications, making them attractive targets for synthetic organic chemistry. Several synthetic strategies have been developed over the years to construct these compounds, each offering distinct advantages depending on the desired substitution pattern and the specific application.42–45 Fig. 3 presents a schematic representation of the synthetic routes utilized to produce pyrazolo[1,5-a]pyrimidines through a cyclization strategy.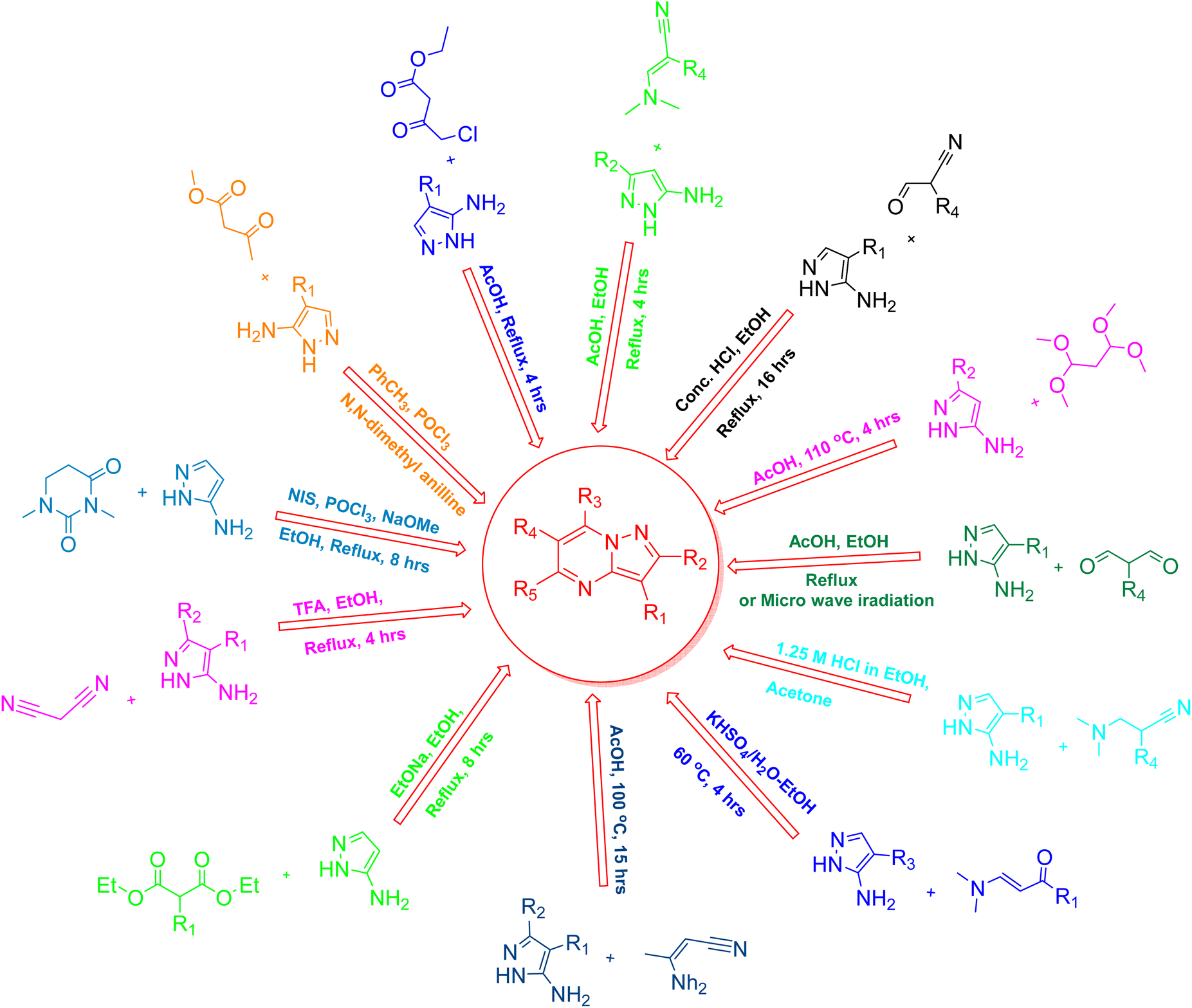 | ||
| Fig. 3 Schematic representation of synthetic routes to afford pyrazolo[1,5-a]pyrimidines by cyclisation strategy. | ||
3.1 Cyclization approaches
The synthesis of pyrazolo[1,5-a]pyrimidines through cyclization strategies remains a widely adopted approach due to its efficiency in constructing the fused bicyclic system. This process typically starts with the formation of the pyrazole ring, which is usually accomplished via the condensation of hydrazine derivatives and carbonyl-containing compounds. Common reactions involve hydrazines or hydrazones reacting with α,β-unsaturated carbonyl compounds, leading to the formation of substituted pyrazoles. For instance, the reaction of hydrazines with β-ketoesters or β-diketones is frequently employed to generate the pyrazole ring.46–50Once the pyrazole ring is established, the subsequent step in cyclization focuses on constructing the pyrimidine ring. This is often achieved through various methodologies, such as using carbodiimides or isocyanates, which facilitate the formation of the pyrimidine ring by reacting with functional groups already present on the pyrazole ring.51,52 Another common approach involves the reaction of pyrazole derivatives with aldehydes or ketones, where reaction conditions, such as acid or base catalysts, aid in forming the pyrimidine ring through imine or oxime intermediates.53,54 Alternatively, amides or ureas can be used to provide the nitrogen atoms required for the pyrimidine ring, with specific conditions such as elevated temperatures or catalytic systems driving the reaction to completion.55
One of the key advantages of the cyclization strategy is its flexibility, allowing the introduction of various substituents on both the pyrazole and pyrimidine rings. This adaptability is achieved by modifying reaction conditions or employing different reagents, which enable the incorporation of diverse functional groups at specific positions on the fused ring system.56,57 This versatility is crucial for tailoring the physicochemical properties and biological activities of the resulting pyrazolo[1,5-a]pyrimidines, which is particularly valuable in drug development. Moreover, these cyclization methods can be adapted to achieve regioselective synthesis, offering precise control over the placement of substituents. For instance, the choice of substituents on the pyrazole ring can influence the reactivity and selectivity of the cyclization process, leading to distinct substitution patterns on the pyrimidine ring.58–60
Overall, cyclization strategies for synthesizing pyrazolo[1,5-a]pyrimidines offer a versatile and adaptable approach, enabling the construction of these compounds with precise control over their structure and functionality. These methods are highly significant for developing pyrazolo[1,5-a]pyrimidine-based pharmaceuticals and other bioactive molecules.
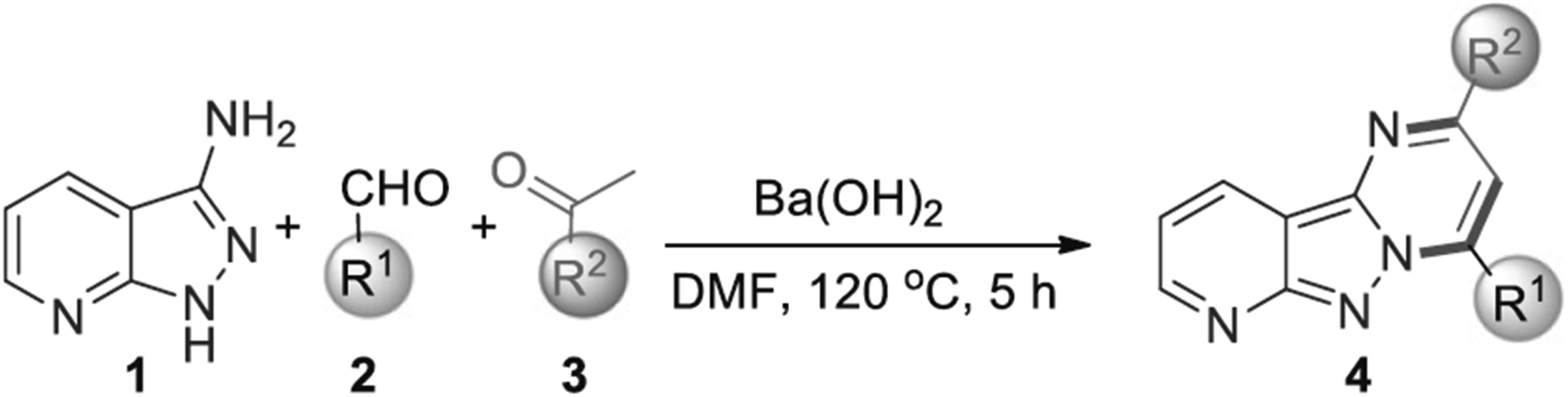
In the study by Sikdar et al. (2023), a one-pot cyclization methodology was developed to synthesize 3-halo-pyrazolo[1,5-a]pyrimidine derivatives through a reaction involving amino pyrazoles, enaminones (or chalcone), and sodium halides.46 This approach utilizes a cyclization strategy to efficiently form the pyrazolo[1,5-a]pyrimidine core structure, followed by oxidative halogenation to introduce halogen atoms into the final compound.
The cyclization process, depicted in Scheme 1, begins with a cyclocondensation reaction between amino groups of the pyrazoles and enaminones (or chalcone) in the presence of potassium persulfate (K2S2O8). This reaction establishes the pyrazole ring. Subsequent oxidative halogenation, using sodium halides and K2S2O8, introduces halogens at the 3-position of the pyrazolo[1,5-a]pyrimidine structure. This method was noted for its efficiency and versatility, as it employs readily available reagents and operates under mild conditions.
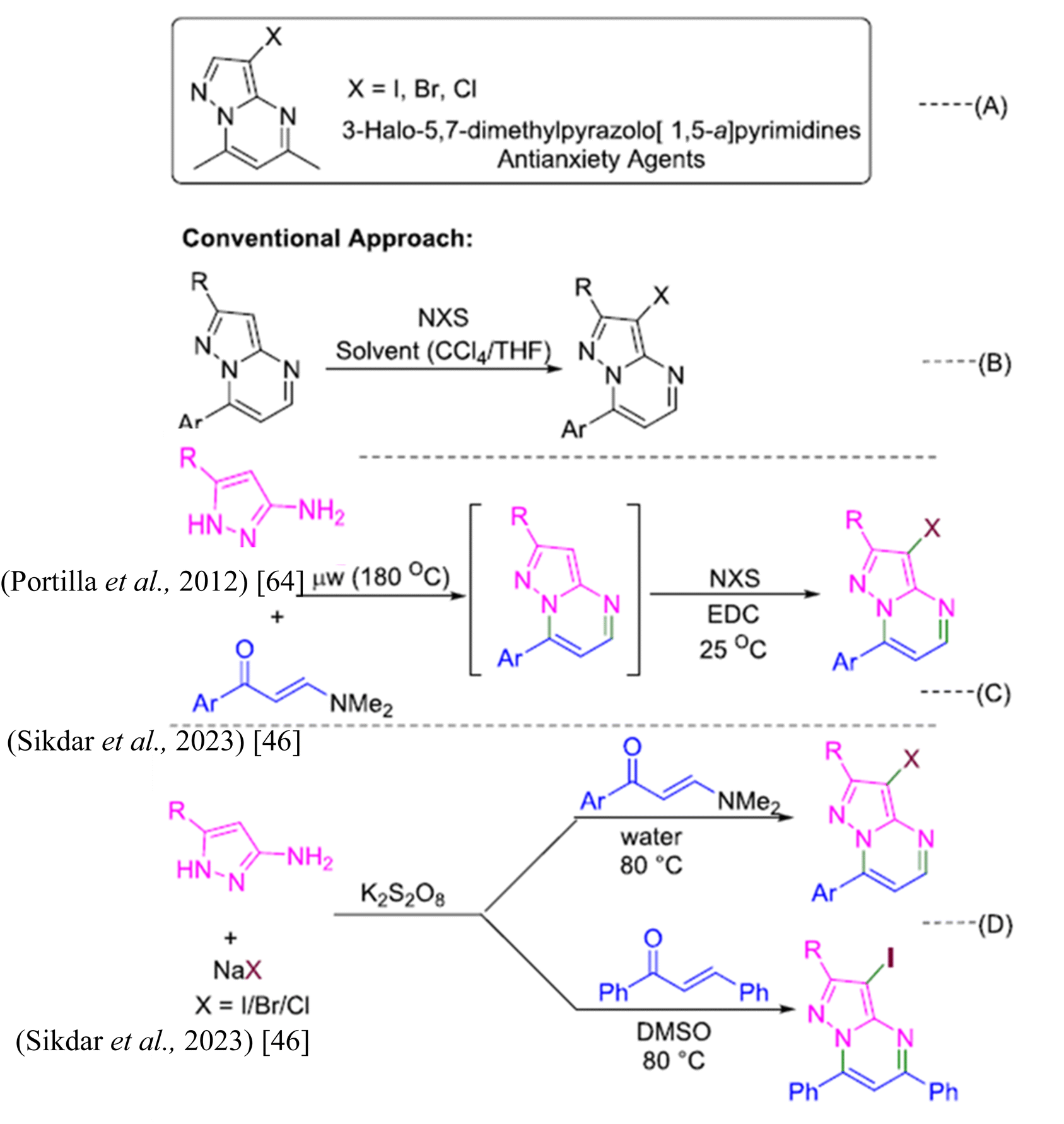 | ||
| Scheme 1 (a–c) Strategies toward the synthesis of halogenated pyrazolo[1,5-a]pyrimidines.46 | ||
The synthesized compounds exhibited several notable properties. For instance, the reaction conditions were optimized to enhance the yield of 3-iodo-pyrazolo[1,5-a]pyrimidine derivatives. The study found that using a combination of sodium iodide (NaI) and K2S2O8 in water was highly effective, achieving nearly quantitative yields of the desired products. This demonstrates the method's high efficiency and suitability for large-scale synthesis. The use of water as a solvent was particularly advantageous, as it supported the efficient formation of the pyrazolo[1,5-a]pyrimidine core and facilitated the oxidative halogenation process.
The substrate scope was explored extensively, revealing that various aryl enaminones, including those with electron-donating and electron-withdrawing groups, afforded 3-iodo-pyrazolo[1,5-a]pyrimidine derivatives with high to excellent yields. This broad functional group tolerance indicates that the methodology is highly adaptable. The study also demonstrated that enaminones with naphthalenyl moieties and heteroaryl enaminones yielded 3-iodinated pyrazolo[1,5-a]pyrimidines efficiently.
Scheme 1 illustrates the synthesis of 3-halo-pyrazolo[1,5-a]pyrimidine derivatives using different halogen sources and conditions, emphasizing the effectiveness of the cyclization approach. Furthermore, the methodology was extended to the synthesis of 3-bromo- and 3-chloro-pyrazolo[1,5-a]pyrimidines using sodium bromide (NaBr) and sodium chloride (NaCl), respectively. While the yields of brominated derivatives were high, albeit with longer reaction times, chlorinated derivatives were produced with moderate yields. This flexibility in halogenation further illustrates the utility of the method.
The study also addressed the formation of pyrazolo[1,5-a]pyrimidine derivatives using chalcone. Although water was not suitable for this transformation, dimethyl sulfoxide (DMSO) was found to be an effective solvent, allowing for successful synthesis of 3-iodo-pyrazolo[1,5-a]pyrimidines. The cyclization approach was demonstrated to be regioselective, with mono-iodination occurring primarily at the 3-position of the pyrazolo[1,5-a]pyrimidine ring.
In general, the cyclization approach developed by Sikdar et al.46 is characterized by its simplicity, efficiency, and wide applicability. The synthesized 3-halo-pyrazolo[1,5-a]pyrimidines exhibit high yields and excellent functional group tolerance, making this method a valuable addition to the synthetic chemist's toolkit for constructing complex heterocyclic compounds.
In the study by Portilla et al. (2012), the synthesis of pyrazolo[1,5-a]pyrimidines are explored through a cyclization reaction involving 3-substituted-5-amino-1H-pyrazoles and different cyclic β-dicarbonyl compounds, leading to the formation of cyclopentapyrazolo[1,5-a]pyrimidines.61 The authors reported that the reaction between 3-substituted-5-amino-1H-pyrazoles and either 2-acetylcyclopentanone or 2-ethoxycarbonylcyclopentanone resulted in the regioselective formation of cyclopentapyrazolo[1,5-a]pyrimidines in good yields, highlighting the influence of the β-dicarbonyl compounds on controlling the reaction pathway (Scheme 2). Notably, both fusion and microwave irradiation methods provided faster reaction times and higher yields compared to traditional reflux methods in ethanol, emphasizing the efficiency of solvent-free conditions.
 | ||
| Scheme 2 Synthesis of cyclopentapyrazolo[1,5-a]pyrimidines 12 and 13. | ||
The study also delves into the reaction of 3-substituted-5-amino-1H-pyrazoles with 2-acetylbutyrolactone, which led to the formation of 6-(2-hydroxyethyl)pyrazolo[1,5-a]pyrimidin-7(4H)-ones (Scheme 3). Interestingly, this reaction involved the opening of the butyrolactone ring as a final step in the cyclization, distinguishing it from the reactions involving cyclic β-dicarbonyl compounds. Despite attempts to replicate the solvent-free synthesis approach, the reaction with butyrolactone did not yield satisfactory results under these conditions.
 | ||
| Scheme 3 Synthesis of 6-(2-hydroxyethyl)pyrazolo[1,5-a]pyrimidinone 20. | ||
In the context of structure elucidation, NMR and solid-state studies were critical in confirming the regioselectivity of the synthesized compounds. The 1H and 13C NMR spectra provided clear evidence of the cyclization occurring through the nitrogen of the pyrazolic ring rather than the carbon, ruling out the formation of alternative regioisomeric products such as cyclopentapyrazolo[3,4-b]pyridines (Scheme 2). Moreover, X-ray diffraction analysis of some derivatives further corroborated the structural assignments, particularly in distinguishing between angular and linear isomers.
The mechanistic insights gained from the study, particularly the role of β-dicarbonyl compounds in directing the regiochemistry, were further demonstrated through the reaction with 2-acetylbutyrolactone. The formation of intermediate (3Z)-3-{1-[(5-R-1H-pyrazol-3-yl)amino]ethylidene}-4,5-dihydrofuranone (Scheme 4) supports the proposed reaction pathway, where the condensation between the aminopyrazole and the carbonyl group of the lactone occurs prior to ring opening and cyclization.
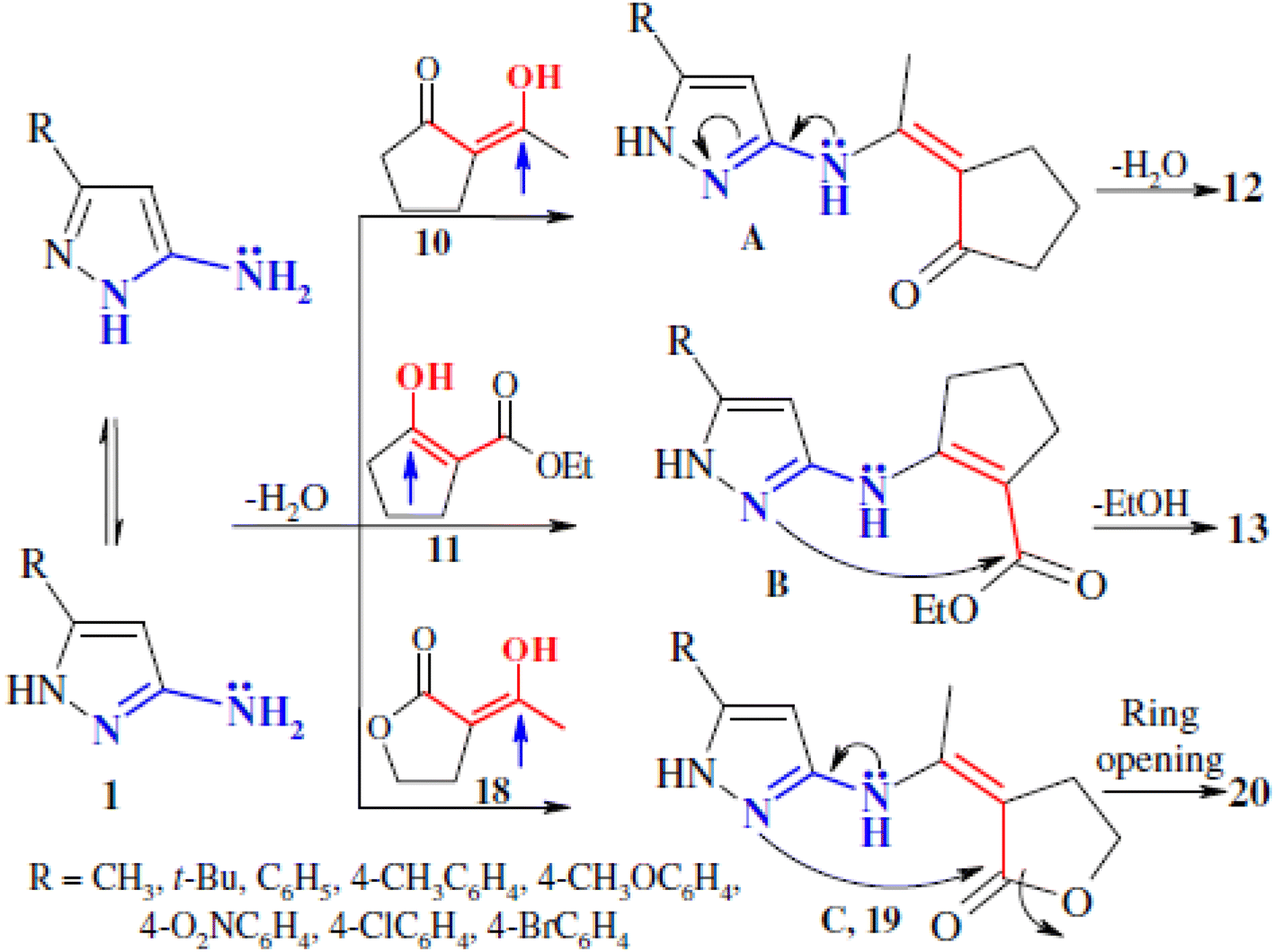 | ||
| Scheme 4 Mechanism for the formation of compounds 12, 13 and 20. | ||
These findings underscore the unique pathways in the synthesis of pyrazolo[1,5-a]pyrimidines and highlight the crucial role of the β-dicarbonyl moieties in influencing the regioselectivity and final products, as demonstrated in Schemes 3–5.
 | ||
| Scheme 5 The reaction of 5-aminopyrazole derivatives 1 with benzylidene malononitrile derivatives 2.62 | ||
The synthesis of pyrazolo[1,5-a]pyrimidines through cyclization strategies is a notable focus in the study by Castillo et al. (2016), where a microwave-assisted approach was developed to regioselectively synthesize functionalized 6-(aryldiazenyl)pyrazolo[1,5-a]pyrimidin-7-amines. This method involves the cyclization of 3-oxo-2-(2-arylhydrazinylidene) butanenitriles with 5-amino-1H-pyrazoles under solvent-free conditions.49 The microwave-assisted cyclization process was key to achieving high yields and purity of the desired products in a short reaction time.
In the initial phase of the study, the researchers optimized the reaction conditions by exploring different solvents and heating methods. Conventional heating in solvents like ethanol and toluene proved ineffective, while the use of high-boiling solvents such as DMSO and DMF resulted in moderate yields. However, the breakthrough came when microwave irradiation was employed under solvent-free conditions. This approach significantly enhanced the reaction efficiency, producing the target pyrazolo[1,5-a]pyrimidin-7-amines in near-quantitative yields with a reaction time as short as four minutes.
The cyclization strategy employed in this study is distinguished by its operational simplicity, high atom economy, and eco-compatibility. The use of microwave irradiation not only accelerated the cyclization process but also minimized the need for chromatographic purification, making it a sustainable and practical method for synthesizing these heterocyclic compounds. The resultant pyrazolo[1,5-a]pyrimidines exhibited high regioselectivity, and their structures were confirmed by NMR spectroscopy. The study also highlights that the synthesized compounds have potential as intermediates in the development of biologically active N-fused heteroaromatic systems, demonstrating the versatility and utility of the cyclization method developed by Castillo et al. (2016).49
The study by Moustafa et al. (2022) explores the synthesis of various pyrazolo[1,5-a]pyrimidine derivatives through cyclization reactions, focusing on the reaction of N-(5-amino-4-cyano-1H-pyrazole-3-yl)-benzamide (compound 1) with different electrophiles.62 The authors highlight how the interaction with these electrophiles leads to distinct isomeric derivatives, with cyclization being a key step in forming the pyrazolo[1,5-a]pyrimidine scaffold.
One of the pivotal findings discussed in the paper is the reaction of compound 1 with benzylidene malononitrile (compound 2a) under microwave irradiation (120 °C for 20 minutes). This reaction selectively yields the 7-aminopyrazolo[1,5-a]pyrimidine derivative (compound 4a), rather than its isomeric form, 5-aminopyrazolo[1,5-a]pyrimidine (compound 3a). The formation of 4a was confirmed using mass spectrometry and IR spectra, with peaks indicating the presence of the amino group, cyano groups, and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. Additionally, 1H NMR and 13C NMR spectra further supported the structure, with the proton assignments being consistent with the expected anisotropic effects of the pyrazole ring nitrogen. Single-crystal X-ray diffraction provided further unambiguous structural confirmation.
N bond. Additionally, 1H NMR and 13C NMR spectra further supported the structure, with the proton assignments being consistent with the expected anisotropic effects of the pyrazole ring nitrogen. Single-crystal X-ray diffraction provided further unambiguous structural confirmation.
The scope of the reaction was extended by reacting compound 1 with several substituted cinnamoyl derivatives (2b–f) under similar microwave conditions. This resulted in the formation of 7-aminopyrazolo[1,5-a]pyrimidine derivatives (4b–f), which displayed consistent NMR and mass spectral characteristics with compound 4a. For instance, the structure of compound 4d was also confirmed using single-crystal X-ray diffraction, and these results were encapsulated in Scheme 5, providing a visual representation of the reaction pathways leading to the formation of the different derivatives.
Additionally, the study contrasts these findings with earlier work, which often reported the formation of 5-aminopyrazolo[1,5-a]pyrimidines. The selectivity observed in the current study suggests that microwave irradiation plays a crucial role in determining the regioselectivity of the cyclization. For example, earlier reports indicated that reactions involving N-(5-amino-4-cyano-1H-pyrazole-3-yl)-benzamide (1) with cinnamoyl derivatives under different conditions typically yielded 5-amino isomers, while in the present study, 7-amino isomers dominate.
Furthermore, Moustafa et al. explore the reaction of compound 1 with (E)-3-(dimethylamino)-1-arylprop-2-en-1-one derivatives (5a–f). This reaction, similarly, conducted under microwave conditions, results in the formation of 7-arylpyrazolo[1,5-a]pyrimidines (7a–f). Analytical data, including mass spectrometry and NMR spectra, ruled out the possible formation of the 5-arylpyrazolo[1,5-a]pyrimidine isomers (6a), further highlighting the selectivity for 7-aryl products in this reaction sequence. These transformations were summarized in Schemes 6 and 7, visually representing the selective cyclization pathways observed under controlled microwave heating.
 | ||
| Scheme 6 The reaction of 5-aminopyrazole derivatives 1 with enaminone derivatives 5.62 | ||
 | ||
| Scheme 7 Mechanism of the formation of 4af and 7a–d.62 | ||
Overall, Moustafa et al. demonstrate that the regioselectivity in the cyclization of N-(5-amino-4-cyano-1H-pyrazole-3-yl)-benzamide with various electrophiles can be significantly modulated by employing microwave-assisted reactions. The study provides a robust foundation for understanding the factors influencing the selective formation of pyrazolo[1,5-a]pyrimidines over other possible isomeric forms.
In 2013, Abdelhamid & Gomha explored the synthesis of a diverse array of pyrazolo[1,5-a]pyrimidine derivatives through an innovative cyclization process. Their work primarily focused on the reaction of sodium 3-oxo-3-(1-phenyl-1H-pyrazol-4-yl)prop-1-en-1-olate (compound 2) with various heterocyclic amines, including cyanoacetamide, cyanothioacetamide, and 2-cyanoacetohydrazide. This key step, depicted in Scheme 8, allowed for the formation of a series of fused heterocyclic structures, such as pyrazolo[1,5-a]pyrimidines (compounds 5a–d), which displayed a remarkable diversity in their chemical architecture.
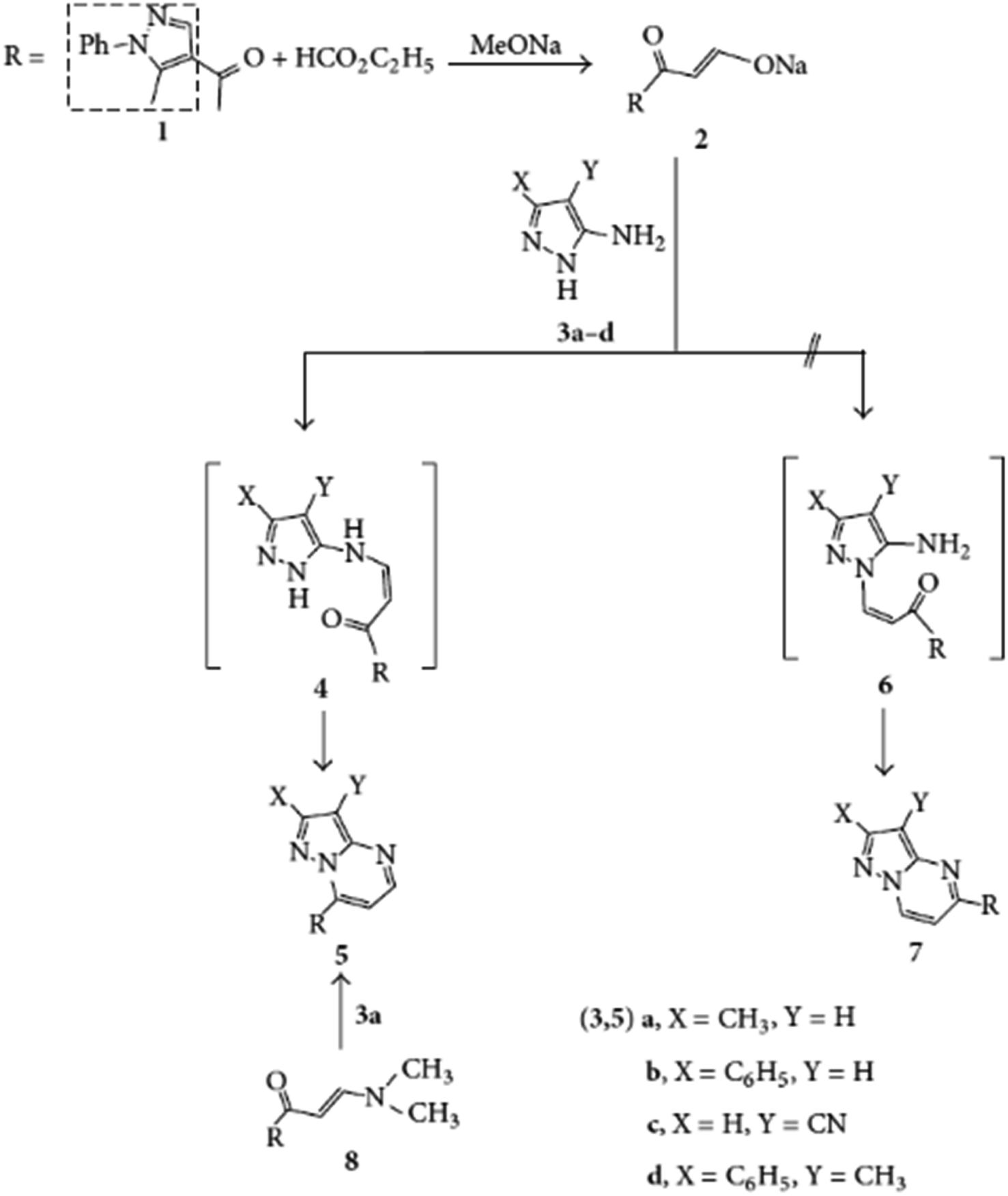 | ||
| Scheme 8 Synthesis of pyrazolo[1,5-a]pyrimidines (5a–d).63 | ||
Further exploration of these reactions, outlined in Scheme 9, led to the synthesis of other complex heterocycles, such as pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine (compound 9), benzo[4,5]imidazo[1,2-a]pyrimidine (compound 10), and [1,2,4]triazolo[1,5-a]pyrimidine (compound 11). The authors employed a variety of heterocyclic amines and utilized a stepwise synthetic approach to ensure high yields and structural diversity, which they meticulously confirmed using techniques such as elemental analysis, IR, NMR, and mass spectrometry.
 | ||
| Scheme 9 Synthesis of pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine (9), benzoimidazo[1,2-a]pyrimidine (10), triazolo[4,3-a]pyrimidine (11), and pyrimidinones (12–14).63 | ||
In addition to the pyrazolo[1,5-a]pyrimidines, Abdelhamid & Gomha also reported the synthesis of novel pyridine derivatives (compounds 12–14), as shown in Scheme 10. Notably, the transformation of pyridinethione (compound 13) through its reaction with α-halo ketones and α-halo esters resulted in the formation of thieno[2,3-b]pyridine derivatives (compounds 15–18). These reactions demonstrated the versatility of their methodology, providing a pathway for the efficient synthesis of sulfur-containing fused heterocycles, which hold potential for various biomedical applications.
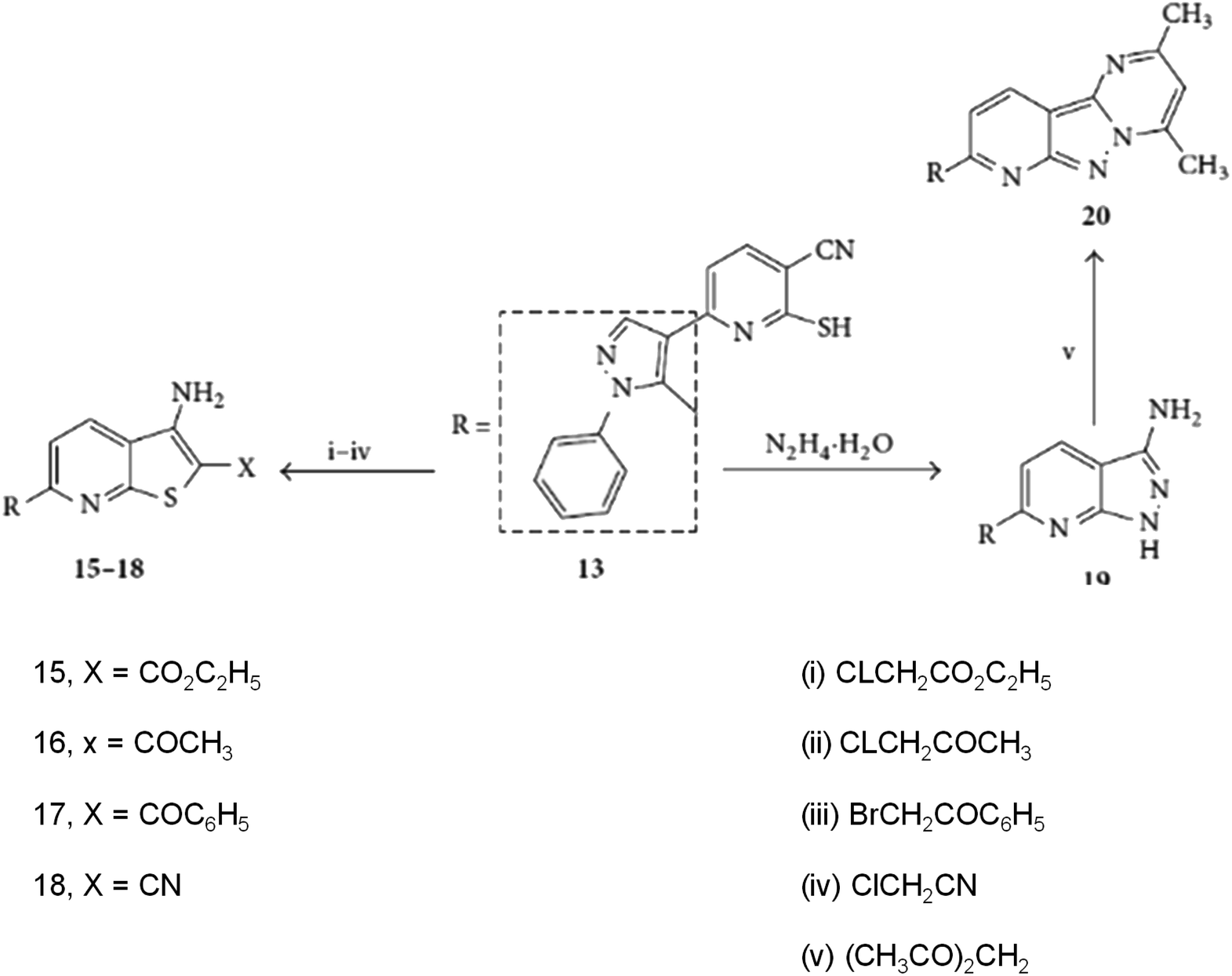 | ||
| Scheme 10 Synthesis of thieno[2,3-b]pyridines (15–18) and pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine (20).63 | ||
The study presented these alternative synthetic routes (illustrated in Schemes 8–10) to emphasize the flexibility and adaptability of their approach in constructing a wide range of fused heterocyclic systems. Abdelhamid & Gomha's work stands out for its ability to produce a diverse array of biologically relevant compounds, with potential applications in drug discovery for treating conditions such as bacterial and fungal infections, as well as cancer. Their findings contribute to the growing body of research focused on the medicinal chemistry of pyrazolo[1,5-a]pyrimidine-based compounds, positioning these heterocyclic structures as promising candidates for further pharmacological investigation.
3.2 Condensation reactions
A frequently employed strategy for the synthesis of pyrazolo[1,5-a]pyrimidines is the condensation of 5-aminopyrazoles with β-dicarbonyl compounds or their equivalents. In this method, the 5-aminopyrazole acts as a nucleophile, attacking the carbonyl carbon of the β-dicarbonyl compound, followed by cyclization to form the pyrimidine ring. This reaction typically proceeds under acidic or basic conditions and can be enhanced by the use of catalysts such as Lewis's acids or bases, which facilitate the formation of the fused ring system. The reaction mechanism involves the initial nucleophilic addition of the amino group to the electrophilic carbonyl center, followed by cyclization and dehydration to generate the desired pyrazolo[1,5-a]pyrimidine core.61,64The choice of β-dicarbonyl compound is crucial, as it directly influences the substitution pattern on the pyrimidine ring, enabling the fine-tuning of the electronic and steric properties of the final product.64 By selecting different β-dicarbonyl compounds, chemists can introduce a variety of functional groups at specific positions, which can be beneficial for optimizing the biological activity of these compounds. This method provides a straightforward approach to access a range of pyrazolo[1,5-a]pyrimidines with diverse substitution patterns, making it a valuable strategy for medicinal chemistry applications.61
Furthermore, the use of β-dicarbonyl equivalents, such as esters or nitriles, expands the scope of this method, allowing for the synthesis of more structurally diverse derivatives. The reaction conditions, such as temperature, pH, and the nature of the catalyst, can be adjusted to optimize yields and selectivity, providing flexibility in the synthesis of pyrazolo[1,5-a]pyrimidine derivatives with tailored properties. However, some challenges may arise from the reactivity of certain β-dicarbonyl compounds, requiring careful optimization of the reaction conditions to prevent side reactions or unwanted by-products.65,66
For example, Poursattar et al. (2015) reported a straightforward and efficient method for synthesizing novel pyrazolo[1,5-a]pyrimidine analogues through the condensation of 1,3-diketones or keto esters with substituted 5-aminopyrazoles in the presence of sulfuric acid (H2SO4), using acetic acid (AcOH) as the solvent.67 This synthetic approach involved the reaction of 5-amino-3-arylamino-1H-pyrazole-4-carbonitriles (1a–f) and ethyl 5-amino-3-arylamino-1H-pyrazole-4-carboxylate (2a,b) with various β-dicarbonyl compounds, such as pentane-2,4-dione, ethyl acetoacetate, ethyl isobutyrylacetate, and ethyl butyrylacetate (3a–d), to produce the corresponding pyrazolo[1,5-a]pyrimidine derivatives (4a–m) in high yields ranging from 87% to 95%, as illustrated in Scheme 11.
 | ||
| Scheme 11 Synthesis of pyrazolo[1,5-a]pyrimidine derivatives (4a–i, 4l–m, 4j, 4k).67 | ||
The methodology utilized in this study was notable for its simplicity and the high yield of the desired products, making it a valuable addition to the synthetic chemistry of heterocyclic compounds. The reaction conditions were optimized to ensure the efficient formation of the pyrazolo[1,5-a]pyrimidine core, which is a privileged scaffold in medicinal chemistry due to its potential biological activities.
Thirteen examples of the conversion of 5-amino-3-arylamino-1H-pyrazole-4-carbonitriles (1a–f) and ethyl 5-amino-3-arylamino-1H-pyrazole-4-carboxylate (2a,b) to the corresponding 4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives (4a–m) were successfully demonstrated, with the reaction times, melting points, and yields of the synthesized compounds. The high yields and mild reaction conditions emphasize the efficiency and practicality of this synthetic route for preparing a wide range of pyrazolo[1,5-a]pyrimidine derivatives, which could be explored further for their potential applications in various fields.
3.3 Three-component reactions
Another well-established method involves the three-component reaction of 3-amino-1H-pyrazoles with aldehydes and activated methylene compounds, such as malononitrile or ethyl cyanoacetate. This one-pot reaction proceeds via the formation of an imine intermediate, followed by nucleophilic attack by the activated methylene compound and subsequent cyclization to yield the pyrazolo[1,5-a]pyrimidine core.68–70 This strategy is particularly advantageous for the rapid synthesis of diverse pyrazolo[1,5-a]pyrimidine derivatives, as it allows for the simultaneous introduction of substituents on both the pyrazole and pyrimidine rings in a single step. Additionally, the use of different aldehydes and activated methylene compounds provides access to a wide variety of substitution patterns, making this method highly versatile for structure–activity relationship (SAR) studies.69One of the primary benefits of this method is its efficiency and simplicity. By combining all reactants in one step, it reduces the need for multiple reaction phases and purification steps, ultimately saving time and resources. The reaction generally proceeds under mild conditions, minimizing the risk of degradation or side reactions that could complicate product isolation. Moreover, this approach allows for easy tuning of the final product's properties by simply varying the aldehyde or activated methylene compound, which is especially useful in medicinal chemistry for exploring a range of biological activities.71,72
However, there are some limitations to this approach. Although it offers structural diversity, the reaction may be sensitive to steric and electronic effects of the reactants, which could influence the yields and selectivity of the products. In some cases, optimizing conditions to favor cyclization over other potential side reactions may be necessary. Despite these challenges, the versatility and expediency of this method make it a powerful tool for the synthesis of pyrazolo[1,5-a]pyrimidines.
Hoang et al. (2018) developed an efficient three-component strategy for the Rh(III)-catalyzed annulation of 3-aminopyrazoles, aldehydes, and sulfoxonium ylides, yielding diverse pyrazolo[1,5-a]pyrimidines.73 This reaction, performed under microwave heating with short reaction times, closely aligns with the general mechanism of the three-component reaction of 3-amino-1H-pyrazoles with aldehydes and activated methylene compounds, such as malononitrile or ethyl cyanoacetate. In both cases, the process begins with the formation of an imine intermediate, followed by nucleophilic attack and cyclization, ultimately leading to the pyrazolo[1,5-a]pyrimidine core.
In the study, Hoang et al. optimized the reaction conditions by exploring various parameters, focusing on the coupling of benzaldehyde, aminopyrazole, and sulfoxonium ylide to synthesize pyrazolopyrimidine.73 It was determined that the best yields were obtained using a cationic Rh(III) catalyst with KOAc, pivalic acid, and 3 Å molecular sieves as additives in dioxane under microwave conditions at 150 °C. Notably, the reaction tolerates a wide range of substituted aminopyrazoles and aromatic aldehydes, including those with electron-withdrawing, electron-donating, and basic nitrogen functionalities, among others.
The versatility of the aldehyde scope is evident, as benzaldehydes with electron-withdrawing and electron-donating groups at various positions, along with heteroaromatic aldehydes such as pyridinecar. For instance, the incorporation of 4-pyridinecarboxaldehyde resulted in a 58% yield of the product, illustrating the method's tolerance to unhindered basic heterocyclic nitrogens. The reaction also accommodates various functional groups, including ester, carboxylic acid, and acidic secondary anilide, as highlighted in Scheme 12.
 | ||
| Scheme 12 Aldehyde scope for three-component synthesis of pyrazolopyrimidines 8a standard conditions with 5 (0.60 mmol), 6a (0.30 mmol), and 7a (0.45 mmol) a0.2 M.73 | ||
The study further explored the scope of sulfoxonium ylides. A variety of sulfoxonium ylides, including those with aryl, heteroaryl, and alkyl substituents, were employed, leading to good yields of the corresponding pyrazolopyrimidine products. This highlights the method's ability to regioselectively introduce aromatic substituents with diverse electronic properties. For instance, the reaction of electron-rich and electron-poor aryl-substituted ylides efficiently yielded the corresponding pyrazolopyrimidines in high yields, as seen in Scheme 13.
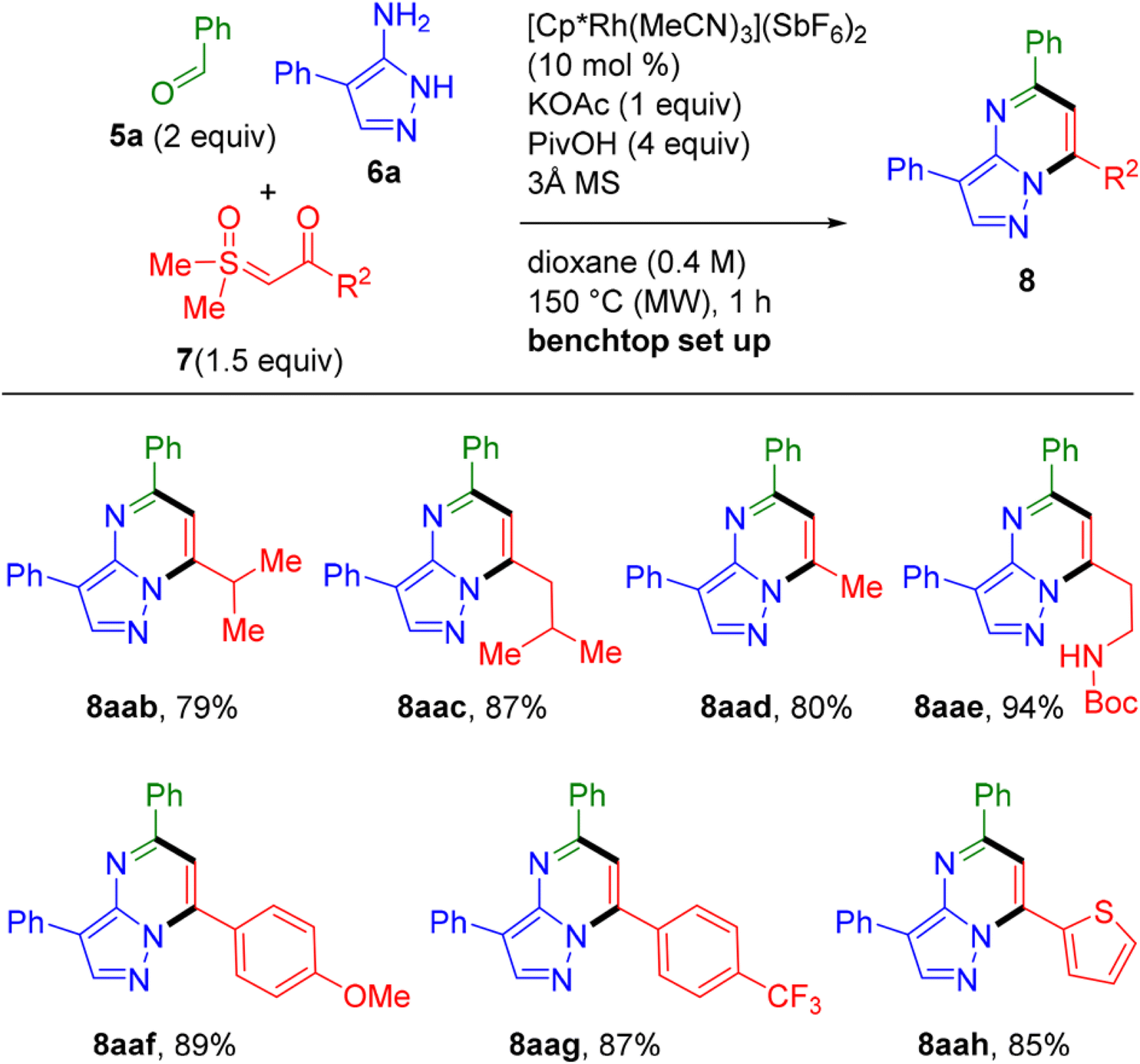 | ||
| Scheme 13 Sulfoxonium ylide scope for three-component synthesis of pyrazolopyrimidines.73 | ||
Overall, Hoang et al.'s approach offers a robust and versatile method for synthesizing pyrazolo[1,5-a]pyrimidines, expanding the toolbox for creating this important heterocyclic scaffold.73 The study's findings align well with traditional three-component reactions involving 3-amino-1H-pyrazoles, demonstrating the effectiveness of the Rh(III)-catalyzed process in producing structurally diverse and functionally rich pyrazolopyrimidines.
3.4 Microwave-assisted synthesis
Microwave-assisted synthesis is highly valued for its capacity to dramatically shorten reaction times while boosting yields, efficiency, and environmental sustainability. By delivering rapid and uniform heating, microwave irradiation increases the reactivity of starting materials, promoting faster molecular interactions and accelerating cyclization reactions. This technique is especially advantageous in multicomponent reactions, where multiple substrates react in a single step to form complex heterocyclic structures.74–76For instance, the microwave-assisted three-component reaction of 3-amino-1H-pyrazoles, aldehydes, and β-dicarbonyl compounds has been a successful and widely adopted approach for synthesizing pyrazolo[1,5-a]pyrimidines. Under microwave conditions, this reaction proceeds within minutes and typically yields products in high purity. Compared to conventional heating methods, which often require several hours of refluxing and can lead to complex workups and side reactions, microwave irradiation streamlines the process, significantly reducing reaction times and enhancing overall efficiency.77,78 This accelerated method also minimizes the formation of by-products, resulting in cleaner reactions and simpler product isolation. Moreover, microwave-assisted synthesis is advantageous for optimizing green chemistry principles. It often requires less solvent or can be performed under solvent-free conditions, contributing to environmentally friendly reaction processes. The energy efficiency of microwave systems further supports sustainability by reducing energy consumption compared to traditional heating techniques.79,80
In summary, microwave-assisted synthesis has become a valuable tool in the preparation of pyrazolo[1,5-a]pyrimidines, offering a practical route to these bioactive heterocycles with enhanced speed, higher yields, and reduced environmental impact. As a result, it represents a promising approach for both academic and industrial applications, particularly in the development of new pharmaceuticals and biologically active compounds.
In their 2018 study, Fahim et al. focus on the microwave-assisted synthesis of novel fused heterocyclic compounds, particularly pyrazolo[1,5-a]pyrimidines.81 The application of microwave irradiation in this context significantly enhances the reactivity of the starting materials, which facilitates rapid cyclization reactions. The approach not only accelerates reaction times but also improves the yield, making it a highly efficient method for synthesizing these complex heterocycles. Schemes 14 and 15 from the study demonstrate the regioselective synthesis of heterocyclic derivatives, starting from 2-cyano-N-cyclohexylacetamide (3) and various nitrogen nucleophiles.
 | ||
| Scheme 14 Synthesis of 2-cyano-N-cyclohexylacetamide (3), followed by its reaction with aromatic aldehyde derivatives and the subsequent reaction of acrylamide derivatives with hydrazine derivatives.81 | ||
 | ||
| Scheme 15 Reaction of acrylamide 5 with 2-amino benzimidazole, amino pyrazole, and aminotriazole.81 | ||
In the first step of the process, cyclohexylamine (1) reacts with ethyl cyanoacetate (2) under microwave irradiation to yield 2-cyano-N-cyclohexylacetamide (3). This intermediate serves as a key precursor in the synthesis of a wide array of fused heterocyclic derivatives, as depicted in Scheme 14. The versatility of compound 3 is showcased by its subsequent reaction with aldehydes 4a–f, producing the corresponding 2-cyano-N-cyclohexyl-3-acrylamide derivatives (5a–f). The high efficiency of the microwave-assisted method is further evident in the conversion of these acrylamides into pyrazole derivatives 7a and 7b, using hydrazine derivatives. The cyclization mechanism leading to these pyrazoles is facilitated by the uniform heating and enhanced reactivity provided by microwave irradiation.
Scheme 15 highlights additional transformations, where compound 5b reacts with various nucleophilic reagents to yield different fused heterocycles, such as pyrimido[1,2-a]benzimidazole (10), pyrazolo[1,5-a]pyrimidine (12), and triazolo[1,5-a]pyrimidine (14). Notably, microwave-assisted synthesis allows these reactions to occur in a short time frame, yielding high-purity products. The IR, 1H NMR, and 13C NMR spectra provided confirm the successful synthesis of these compounds, displaying characteristic absorption bands and chemical shifts that align with their proposed structures. The study effectively demonstrates how microwave irradiation can be utilized to streamline the synthesis of complex heterocyclic scaffolds, making it a valuable tool in the development of novel compounds with potential applications in pharmaceuticals and materials science.
Fahim et al.'s investigation provides an important contribution to the growing body of research on microwave-assisted synthesis, showing its advantages in generating a variety of nitrogen-containing heterocycles.81 This method not only improves reaction efficiency but also enhances product selectivity, providing a sustainable pathway for heterocyclic compound production.
Hoang et al. (2018) developed a three-component strategy using Rh(III)-catalyzed annulation for the efficient synthesis of pyrazolo[1,5-a]pyrimidines. The reaction involves 3-aminopyrazoles, aldehydes, and sulfoxonium ylides under microwave heating, yielding diverse products.82 The method, which operates under straightforward benchtop conditions, showcases versatility with various aromatic and heteroaromatic aldehydes, tolerating electron-withdrawing, electron-donating, basic nitrogen, halides, and acidic functionalities. The authors also introduced ester and methoxy groups directly onto the pyrimidine ring using ethyl glyoxylate and trimethyl orthoformate, respectively.
A detailed optimization study was conducted using benzaldehyde, aminopyrazole, and sulfoxonium ylide, showing that a cationic Rh(III) catalyst with KOAc and PivOH in dioxane at 150 °C yielded the best results. Substitutions at different positions on benzaldehyde (para, meta, ortho) were tolerated, although ortho-substituted products showed a moderate reduction in yield. The reaction was also successful with functional groups such as esters, carboxylic acids, and secondary anilides, as well as with heterocyclic aldehydes like pyridinecarboxaldehydes, furan, thiophene, and pyrazolecarboxaldehydes.
The sulfoxonium ylides scope was equally broad, incorporating aryl, alkyl, and heteroaryl groups. The method demonstrated regioselective introduction of substituents, and different aminopyrazoles also yielded good results. Disubstituted and electron-deficient aminopyrazoles worked well, though 2-aminoimidazoles were ineffective. This efficient protocol is applicable for synthesizing pyrazolopyrimidines with potential pharmaceutical relevance.
The study by Shekarrao et al. (2014) demonstrates an efficient palladium-catalyzed microwave-assisted synthesis of pyrazole-fused heterocycles, specifically focusing on pyrazolo[3,4-b]pyridines, pyrazolo[3,4-b]quinolines, pyrazolo[1,5-a]pyrimidines, and pyrazolo[1,5-a]quinazolines.83 This approach stands out due to its solvent-free conditions and the use of microwave irradiation, which significantly accelerates the reaction while improving yields. The method employed β-halovinyl/aryl aldehydes and 3-aminopyrazoles or 5-aminopyrazoles as starting materials, with a palladium catalyst facilitating the reaction.
The reaction of β-bromovinylaldehyde (1a) with 5-aminopyrazole (2a) in the presence of 2.5 mol% PdCl2 and triphenylphosphine (PPh3) as a ligand, under traditional thermal conditions, initially yielded compound 3a in a modest 15% yield after 24 hours (Scheme 16). However, microwave irradiation significantly enhanced both the reaction speed and yield. Under optimized microwave conditions (700 W, 120 °C, 14 bar, solvent-free), the yield of compound 3a increased to 81%, reducing the reaction time to just 15 minutes. This is a dramatic improvement compared to the conventional heating approach.
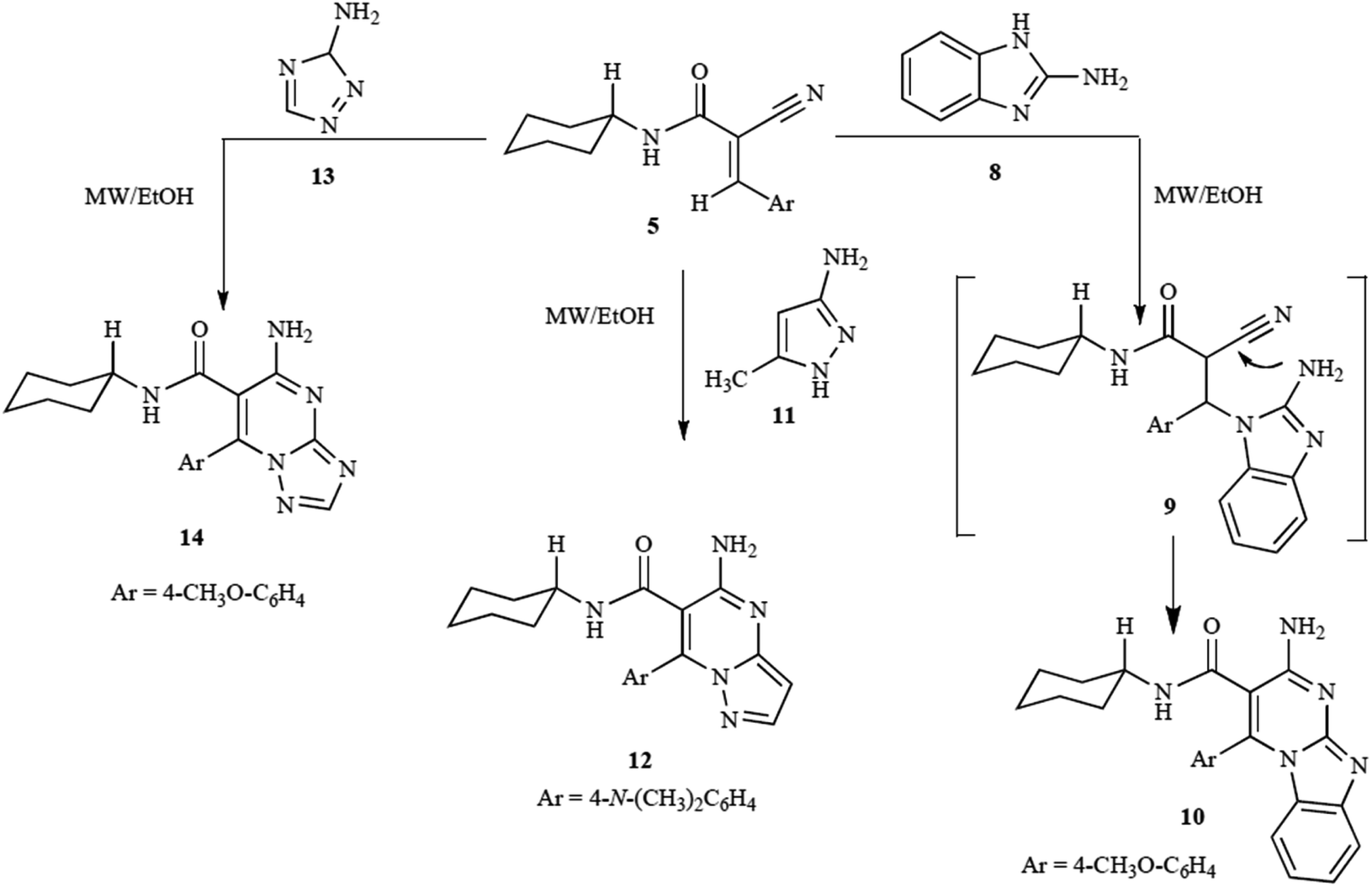 | ||
| Scheme 16 Synthesis of pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a]quinazolines.82 | ||
The optimization of the reaction further explored the role of catalyst loading, solvent choice, and microwave power. It was found that solvent-free conditions yielded the best results, as DMF and DMSO solvents provided lower yields of 3a (22% and 15%, respectively). Catalyst loading adjustments showed that increasing the amount of Pd(OAc)2 to 5 mol% did not improve the yield, while decreasing it to 1.5 mol% resulted in a reduction of the product yield to 64%. Notably, microwave power also influenced the reaction outcome, with 500 W and 800 W resulting in slightly lower yields of 3a (52% and 77%, respectively), as compared to the optimized power setting of 700 W.
The reaction also produced an imine derivative (4a) as a byproduct under certain conditions, particularly in the absence of a catalyst or at suboptimal microwave power settings. This highlights the sensitivity of the reaction to precise microwave and catalyst conditions, emphasizing the efficiency of the optimized method.
The scope of the reaction was evaluated by using various β-halovinyl/aryl aldehydes and aminopyrazoles, which resulted in good yields (67–80%) of pyrazolo[3,4-b]pyridines and related heterocycles. The versatility of this approach was further demonstrated by applying it to steroidal β-bromovinyl aldehydes, yielding steroidal pyrazolo[3,4-b]pyridines (3i–m) with similarly high efficiency. Additionally, 2-bromobenzaldehyde derivatives bearing electron-donating or electron-withdrawing groups were successfully employed to synthesize pyrazolo[3,4-b]quinolines in 68–76% yields.
This study highlights the advantages of microwave-assisted synthesis, particularly its ability to reduce reaction times and increase yields in the formation of biologically significant pyrazole-fused heterocycles.
The study by Bassoude et al. (2016) presents an efficient one-pot, two-step method for synthesizing 7-substituted pyrazolo[1,5-a]pyrimidines using a palladium-catalyzed direct C–H arylation followed by a saponification–decarboxylation reaction.84 In this study, the authors highlight the advantages of their method over traditional Negishi coupling reactions, including shorter reaction times, higher yields, and the use of commercially available, less toxic reagents.
The initial stage of the study focused on optimizing the direct C–H arylation of 7-ethoxycarbonylmethyl-5-methyl-2-phenylpyrazolo[1,5-a]pyrimidine (1). The authors tested several reaction parameters such as ligands, bases, and reaction temperatures to improve the yield of the desired arylated product. After optimizing these conditions, they found that using a combination of Pd(OAc)2, DavePhos, and Cs2CO3 under microwave irradiation provided the best results, reducing the reaction time significantly and yielding the desired product in 81% (Scheme 17).
 | ||
| Scheme 17 Comparison between the literature method and this study's synthesis.84 | ||
To further improve the efficiency of their synthetic approach, the authors investigated conditions for the saponification–decarboxylation of the arylated intermediate. By using microwave irradiation, they were able to shorten the reaction time to 20 minutes while achieving an 87% yield of the final product. Encouraged by these results, the researchers developed a sequential one-pot process that combined both the C–H arylation and the saponification–decarboxylation steps, leading to a high overall yield of the 7-substituted pyrazolo[1,5-a]pyrimidine product.
The study demonstrates the versatility of this one-pot method, as it was successfully applied to a range of aryl halides with different electronic properties, yielding a variety of substituted pyrazolo[1,5-a]pyrimidines. Electron-rich and electron-poor aryl halides were well tolerated, though ortho-substituents led to slightly lower yields due to steric hindrance. Nonetheless, the method was effective across a broad scope of substrates, with yields ranging from 48% to 80%.
Overall, this study highlights the advantages of microwave-assisted synthesis in the preparation of heterocyclic compounds, particularly in accelerating reaction times and improving yields. The one-pot approach developed by Bassoude et al.84 provides a practical and efficient route to 7-substituted pyrazolo[1,5-a]pyrimidines, which are valuable scaffolds in medicinal chemistry. Their method represents a significant improvement over traditional techniques, offering a streamlined synthesis that is both time- and cost-efficient.
3.5 Transition metal-catalyzed reactions
In recent years, transition metal-catalyzed reactions have significantly broadened the synthetic toolkit for constructing pyrazolo[1,5-a]pyrimidines, complementing traditional cyclization methods. Among these, palladium-catalyzed cross-coupling reactions, such as the Suzuki–Miyaura and Sonogashira couplings, have gained prominence. These methodologies have been effectively employed to introduce aryl or alkynyl groups at specific positions on the pyrazolo[1,5-a]pyrimidine core, thus facilitating structural modification and diversification.85–87 By leveraging these reactions, chemists can explore previously inaccessible chemical spaces and develop new analogs with potentially enhanced biological properties.88,89Moreover, the application of other transition metals, including copper and gold, has opened further avenues for synthesizing pyrazolo[1,5-a]pyrimidines. Notably, these metals have been used in processes such as C–H activation and annulation reactions. These methodologies enable the direct construction of the fused ring system from simpler starting materials, offering a more efficient route to complex molecular architectures.90,91 This strategic use of transition metal catalysis continues to evolve, offering new possibilities for the design and synthesis of bioactive pyrazolo[1,5-a]pyrimidine derivatives.
The study by Krishnammagari & Jeong (2018) presents an efficient protocol for the synthesis of novel aza-fused polysubstituted pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine derivatives, utilizing a transition metal-free approach.92 This method employs readily available starting materials: an aromatic aldehyde, acetophenone, and 1H-pyrazolo[3,4-b]pyridin-3-amine, with Ba(OH)2 as a base under reflux conditions, facilitating the formation of the desired products through a sequential Aldol reaction, imine formation, intramolecular N-cyclization, and auto-oxidation. The process, outlined in Scheme 18, involves the formation of new C–C and C–N bonds, making it a versatile and practical approach with broad functional group tolerance.
 | ||
| Scheme 18 Synthesis of polysubstituted pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine derivatives.92 | ||
In the optimization of the reaction, 1H-pyrazolo[3,4-b]pyridin-3-amine, 4-ethoxybenzaldehyde, and acetophenone were selected as model substrates for the synthesis of compound 4b. When the reaction was conducted in ethanol without a base, no product was obtained, demonstrating the necessity of a base for the cyclization process. However, the introduction of NaOMe as a base led to a modest 27% yield of 4b, as identified through 1H NMR, 13C NMR, and mass spectrometry. This outcome prompted further investigation into the use of both organic and inorganic bases, with Ba(OH)2 ultimately being identified as the most effective, yielding 72% of the desired product in ethanol. The use of high-boiling solvents such as DMF and DMSO significantly improved the yields, with DMF providing the highest yield of 92% at 120 °C.
This method showed remarkable versatility across a wide range of substrates, including benzaldehydes with electron-donating and electron-withdrawing substituents. The reaction's functional group tolerance was highlighted by the successful synthesis of a series of highly functionalized pyrimidofused derivatives. Electron-donating groups like methoxy (OMe) and methyl (Me), as well as electron-withdrawing groups such as halides (F, Cl, Br), were well-tolerated under the reaction conditions. The reaction mechanism proceeds through an initial aldol condensation between acetophenones and aldehydes, followed by the formation of a β-hydroxy keto intermediate (A), which undergoes dehydration to form an α,β-unsaturated keto intermediate (B). This intermediate then couples with an amine to form an imine (C), which cyclizes intramolecularly and undergoes auto-oxidation to yield the final products.
The success of this methodology lies in its simplicity, efficiency, and ability to provide a wide range of pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine derivatives in good to excellent yields, making it a valuable tool for the synthesis of complex heterocyclic compounds under mild conditions.
Ren et al. (2021) developed a one-step synthesis method for diversely substituted pyrazolo[1,5-a]pyrimidines using saturated ketones and 3-aminopyrazoles.93 The transformation involves the in situ generation of α,β-unsaturated ketones through a radical process, which then undergoes [3 + 3] annulation with 3-aminopyrazoles in a single reaction vessel. A key feature of this method is the dual C(sp3)–H bond functionalization of inactive ketones, which is necessary for forming the target compounds.
The significance of this work lies in its ability to provide functionalized pyrazolo[1,5-a]pyrimidines with potential antitumor activity, starting from readily available substrates. The process offers an efficient and practical approach to constructing these biologically relevant heterocycles, making it valuable for drug discovery and development. Mechanistic studies of the reaction confirm that the radical formation of α,β-unsaturated ketones is crucial for the success of the subsequent annulation step.
Kotla et al. (2017) developed a palladium-catalyzed intramolecular dehydrogenative coupling reaction for the synthesis of fused imidazo[1,2-a]pyrimidines and pyrazolo[1,5-a]pyrimidines, which are biologically important heterocyclic compounds.94 The process begins with the reaction of 1H-benzo[d]imidazole-2-amine and 2-phenylacetaldehyde, initially yielding a low product yield of less than 10%. However, by optimizing the conditions—such as selecting PdCl2 as the palladium catalyst, using K2CO3 as the base, and toluene as the solvent—yields increased to 80%.
The study further evaluated other palladium salts, solvents, and bases, confirming that PdCl2 with K2CO3 in toluene at 80 °C provided optimal results. The protocol was extended to synthesize various imidazo[1,2-a]pyrimidine derivatives, with electron-donating substituents yielding higher amounts compared to electron-withdrawing ones. Additionally, the method was applicable to the synthesis of fused pyrazolo[1,5-a]pyrimidines using single-ring heterocycles, producing good to excellent yields.
A plausible reaction mechanism was proposed, where the key step involves the formation of a seven-membered palladium cycle and a 1,2-palladium migration, followed by reductive elimination and Wacker–Tsuji type oxidation. This method offers a simple and efficient approach to synthesizing a wide range of substituted pyrimidines under mild conditions, with potential utility in constructing complex derivatives.
3.6 Green chemistry approaches
Recent advances in green chemistry have significantly impacted the synthesis of pyrazolo[1,5-a]pyrimidines, with an increasing focus on adopting environmentally friendly and sustainable methodologies. One of the key strategies involves the use of water as a solvent, which is both abundant and non-toxic. Water-based reactions not only reduce the reliance on harmful organic solvents but also often lead to improved reaction rates and selectivities due to the unique properties of water as a solvent. Additionally, this approach aligns with the principles of green chemistry by minimizing hazardous waste and reducing the environmental footprint associated with traditional solvent systems.95,96The employment of reusable catalysts is another critical advancement in this field. Transition metal catalysts, enzyme-based systems, and even metal–organic frameworks (MOFs) have been developed to catalyze pyrazolo[1,5-a]pyrimidine formation with the advantage of being recycled and reused over multiple reaction cycles. This reduces the consumption of expensive or rare catalytic materials and lowers the overall production costs. Furthermore, reusable catalysts help in reducing the generation of toxic by-products, contributing to more sustainable manufacturing processes.97,98
Solvent-free reactions, which eliminate the need for solvent altogether, represent another innovative green chemistry approach. These reactions typically involve the use of solid-state techniques, microwave irradiation, or mechanochemical methods to drive the synthesis of pyrazolo[1,5-a]pyrimidines. Such solvent-free protocols not only minimize solvent waste but also offer increased atom economy and can often be performed under milder conditions, thus enhancing energy efficiency.83,99–103
Collectively, these green chemistry approaches not only aim to reduce the use of hazardous reagents and solvents but also enhance the overall efficiency, yield, and cost-effectiveness of pyrazolo[1,5-a]pyrimidine synthesis. As a result, they are becoming particularly attractive for large-scale production in the pharmaceutical and chemical industries, where sustainable practices are increasingly prioritized to meet environmental regulations and reduce production costs. The integration of these green methodologies represents a significant step forward in the field of synthetic chemistry, promoting more responsible and sustainable manufacturing processes for biologically important compounds like pyrazolo[1,5-a]pyrimidines.
Konda et al. (2022)104 and Metwally et al. (2022)105 both explore environmentally friendly approaches to the synthesis of pyrazolo[1,5-a]pyrimidines, aiming to minimize environmental impact while developing compounds with significant antimicrobial potential.
In Konda et al.'s study, the researchers aimed to design a highly efficient synthesis of thienyl pyrazolo[1,5-a]pyrimidines using polyethylene glycol-400 (PEG-400) as a green solvent.104 PEG-400, a biodegradable and non-toxic solvent, was chosen for its eco-friendly properties, ease of recovery, and reusability without a loss of efficacy. The reaction involved condensing 4-(4′-chloro-phenylazo)-5-amino pyrazole with chalcones (α,β-unsaturated carbonyl compounds) in the presence of NaOH as a base. This method led to excellent product yields in shorter reaction times, illustrating both the efficiency and sustainability of the process. Notably, this approach avoided the use of hazardous solvents and expensive catalysts, contributing to its green chemistry credentials. The newly synthesized thienyl pyrazolo[1,5-a]pyrimidines were tested for their antimicrobial properties, showing that substitutions with hydroxyl and halo groups on the thienyl moiety significantly enhanced their antibacterial and antifungal activity, highlighting their potential for medical applications.
Metwally et al. (2022), on the other hand, explored biocatalysis as a green synthetic strategy. The researchers used pepsin, a natural enzyme, to catalyze the multicomponent reaction of 4-formylphenyl benzoates, malononitrile, and pyrazolones to synthesize pyrano[2,3-c]pyrazoles and pyrazolo[1,5-a]pyrimidines.105 This reaction was carried out by grinding the reactants in a mortar at room temperature for 1.5–2 hours, offering a solvent-free and energy-efficient alternative to traditional synthetic methods. Additionally, the methodology was extended to prepare new pyrazolo[1,5-a]pyrimidines by using 1,3-cyclic diketones and 4-arylazo-5-aminopyrazoles, yielding the desired products in good to excellent quantities. The synthesized compounds were tested against multiple bacterial strains, and certain derivatives, such as compounds 14a and 14f, exhibited moderate antibacterial activity, particularly against Klebsiella pneumoniae and Staphylococcus aureus, with inhibition zones of 29.6–30.3 mm and MIC values of 125–250 μg mL−1.
Both studies highlight the advantages of green chemistry in the synthesis of bioactive compounds. Konda et al. utilized PEG-400, a recoverable solvent, to streamline the process, while Metwally et al. demonstrated the effectiveness of biocatalysis in a solvent-free environment. These approaches not only reduced environmental harm but also improved the efficiency and cost-effectiveness of the synthesis, making them viable for larger-scale production. The antimicrobial results from both studies are promising, with structural differences in the synthesized compounds (such as the thienyl moiety in Konda et al. and cyclic diketones in Metwally et al.) influencing the observed biological activities. Together, these works illustrate the growing trend toward sustainable synthetic methodologies in pharmaceutical chemistry, demonstrating how innovative green chemistry techniques can enhance both environmental sustainability and the therapeutic potential of new chemical entities.
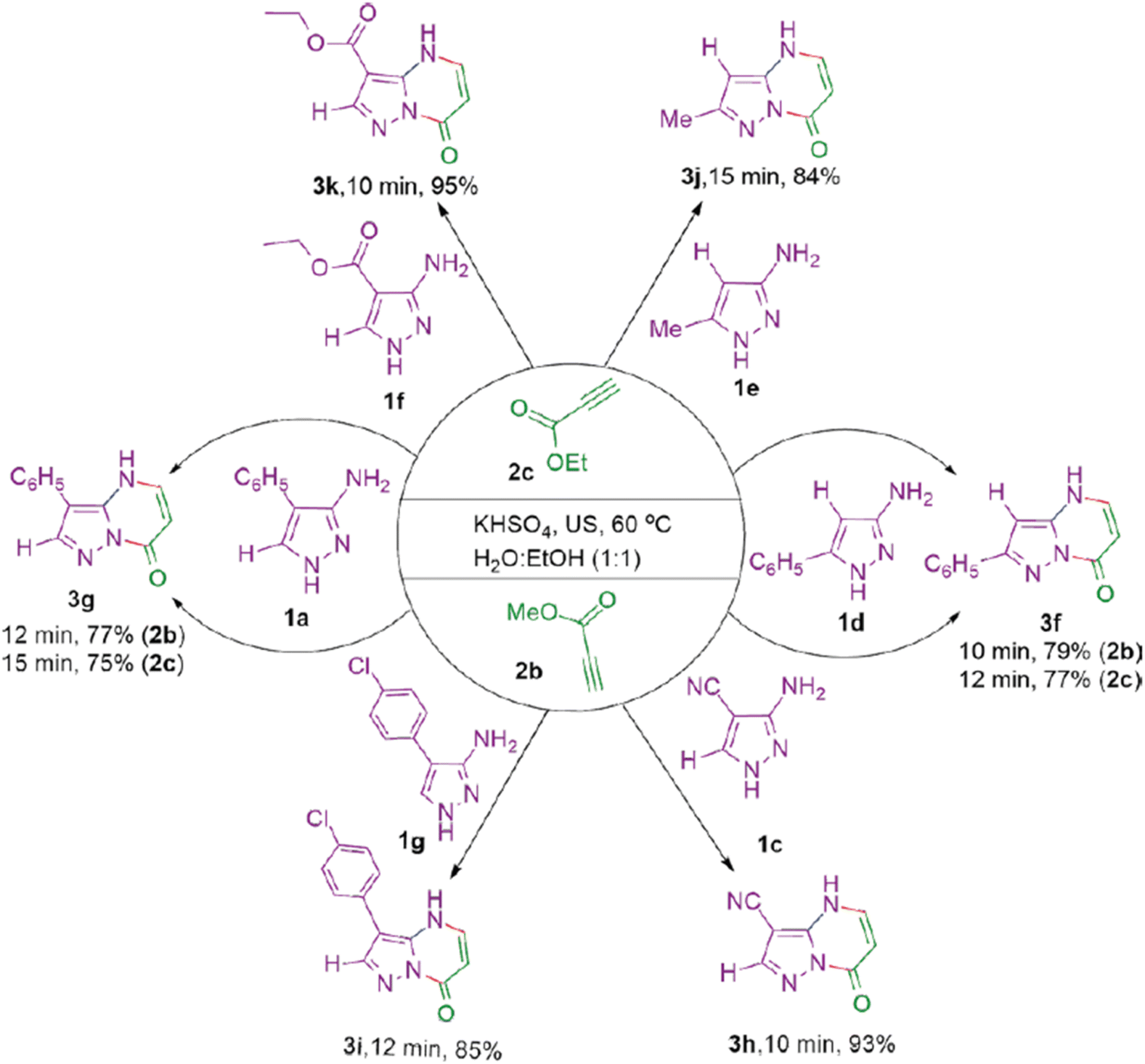
Das et al. (2024) present a study that aligns well with the principles of green chemistry, focusing on the ultrasonic irradiation-assisted synthesis of pyrazolo[1,5-a]pyrimidines in an aqueous ethanol medium.106 This method reflects recent trends in sustainable synthetic strategies, where the use of eco-friendly solvents and energy-efficient techniques plays a central role. By employing water–ethanol mixtures as the reaction medium, the study minimizes the reliance on organic solvents that are often hazardous and environmentally detrimental. Ultrasonic irradiation further reduces the environmental footprint by accelerating reaction rates, thereby lowering the energy requirements traditionally associated with such chemical processes.
The synthesis involves the reaction of 3-aminopyrazoles with alkynes like dimethyl acetylenedicarboxylate (DMAD), methyl propiolate, and ethyl propiolate under ultrasound (US) irradiation, facilitated by potassium bisulfate (KHSO4) as a catalyst. The use of KHSO4, a relatively benign catalyst, adds to the green chemistry aspect of the work, as it avoids toxic or heavy metal catalysts that can pose environmental hazards. Scheme 19 illustrates the reaction of 3-amino-4-phenylpyrazole (1a) with DMAD (2a) in ethanol–water, which yields the corresponding pyrazolo[1,5-a]pyrimidine product (3a) in high yield and purity. The reaction conditions, optimized through sonication at 60 °C, emphasize efficiency, reducing reaction times to a few minutes.
 | ||
| Scheme 19 Optimization of reaction conditions.106 | ||
The expansion of the method to other aminopyrazoles (Scheme 20) and alkynes (Scheme 21) underscores the versatility and scalability of this green synthetic approach. The reactions proceed smoothly, yielding products in high yields (84–95%) within short timeframes, further demonstrating the cost-effectiveness of this technique. The spectral data (1H NMR, 13C NMR, FT-IR, and MS) for the synthesized compounds confirm the structures, and the simplicity of product isolation (via filtration) enhances the overall sustainability of the method.
 | ||
| Scheme 20 Synthesis of 2 or/and 3-substituted methyl 7-oxo-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylate.106 | ||
 | ||
| Scheme 21 Synthesis of a 2 or/and 3-substituted pyrazolo[1,5-a]pyrimidine-7(4H)-one.106 | ||
This work exemplifies how advancements in green chemistry, particularly the use of water-based systems and ultrasonic irradiation, can significantly reduce the environmental impact of synthetic organic chemistry. The method not only offers a cleaner alternative to traditional approaches but also improves the efficiency and scalability of pyrazolo[1,5-a]pyrimidine synthesis, making it suitable for large-scale production in pharmaceutical contexts.
In general, the synthesis of pyrazolo[1,5-a]pyrimidines have evolved significantly over the years, with a variety of synthetic strategies available to construct these biologically important compounds. From traditional cyclization reactions to modern microwave-assisted methods and transition metal-catalyzed couplings, each approach offers unique advantages that can be tailored to the specific needs of the desired application. The continued development of new synthetic methodologies, particularly those aligned with green chemistry principles, will undoubtedly expand the chemical space of pyrazolo[1,5-a]pyrimidines and enhance their potential as therapeutic agents in the treatment of cancer and other diseases.
4 Structural diversification and functionalization
The pyrazolo[1,5-a]pyrimidine core is a versatile scaffold for drug discovery, with its heterocyclic structure allowing for extensive functionalization to optimize pharmacological properties. This section explores how different substitution patterns affect biological activity and examines the synthetic strategies employed to introduce diverse functional groups, while maintaining functional group tolerance.4.1 Substitution patterns and their effects on biological activity
Position 3: substituents at position 3 of the pyrazole ring, such as alkyl, aryl, and heteroaryl groups, have been found to enhance lipophilicity, leading to improved membrane permeability and, consequently, enhanced bioavailability. For instance, aryl groups introduced here often facilitate stronger hydrophobic interactions with target proteins, improving binding affinity.109
The study by Roux et al. (2016) investigates a new series of pyrazolo[1,5-a]pyrimidine derivatives, with the aim of developing potent phosphodiesterase-4 (PDE4) inhibitors for the treatment of inflammatory diseases, such as dry eye. PDE4 inhibitors block the hydrolysis of cyclic adenosine monophosphate (cAMP), which plays a key role in regulating inflammation.110 While two PDE4 inhibitors—Roflumilast and Apremilast—are already approved for COPD and psoriasis, there remains a demand for novel scaffolds with improved pharmacological profiles. The study draws on the structure of GSK256066, a potent PDE4 inhibitor (IC50 = 0.01 nM), and utilizes a rescaffolding approach to design new derivatives with enhanced activity and potential therapeutic benefits.
Compound 1 was identified as the starting point, showing moderate PDE4 inhibition (IC50 = 165 nM) but no cellular activity. Optimizations at key positions of the pyrazolo[1,5-a]pyrimidine scaffold led to the synthesis of compound 10, which demonstrated a significant improvement in activity (IC50 = 0.7 nM), as well as a marked increase in cellular potency. The study leverages a structure-based approach, using modeling studies (PDB structure 3GWT) to guide modifications at positions 2, 3, 6, and 7 of the scaffolds, which were essential for enhancing both potency and selectivity. The introduction of methyl sulfone at position 2 in compound 5 was pivotal, yielding a 6-fold improvement in activity compared to compound 1, as well as observable cellular activity for the first time (IC50 s = 1820 nM and 1500 nM for compounds 5 and 6, respectively).
Position 3 was a critical focus of optimization. As discussed, introducing substituents at this position—such as alkyl, aryl, and heteroaryl groups—enhanced lipophilicity, improving membrane permeability and bioavailability. This is exemplified in Scheme 22, where the synthesis of compound 7 involved the introduction of a 3,5-dichlorophenyl group, which improved IC50 to 16 nM, although cellular activity remained absent. Larger hydrophobic groups, such as isopropyl in compounds 12 and 13, also increased activity by 3- to 5-fold compared to earlier compounds, confirming that a hydrophobic pocket near position 3 played a role in enhancing binding affinity to PDE4 through stronger hydrophobic interactions.
 | ||
| Scheme 22 (i) 3-Amino-4-alkyl pyrazole, ethyl (ethoxymethylene)cyanoacetate (1 equiv.), AcOH, reflux, 4 h, yield: 60%; (ii) NaOH (10 equiv.), EtOH, reflux, 4 h, yield: 62%; (iii) ArI (3 equiv.), CuI (1 equiv.), proline (1 equiv.), K2CO3 (3 equiv.), DMF, microwave, 110 °C, 2 h, yield: 5–35%; (iv) 1-hydroxybenzotriazole (HOBt, 3 equiv.), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 3 equiv.), R3NH2 (excess), DMF, room temperature, 2 h, yield: 8–33%.110 | ||
Further structural modifications are outlined in Scheme 23, where the preparation of methyl sulfone-containing derivatives required a new synthetic route. Compounds 5 and 6 exhibited improved potencies due to the introduction of this functional group, illustrating the value of cyclization strategies in enhancing molecular interactions with the active site. The synthesis pathway involved cyclizing the primary amide of pyrazolo[1,5-a]pyrimidine to afford compounds with a 200-fold increase in potency, as observed with compounds 15 and 16.
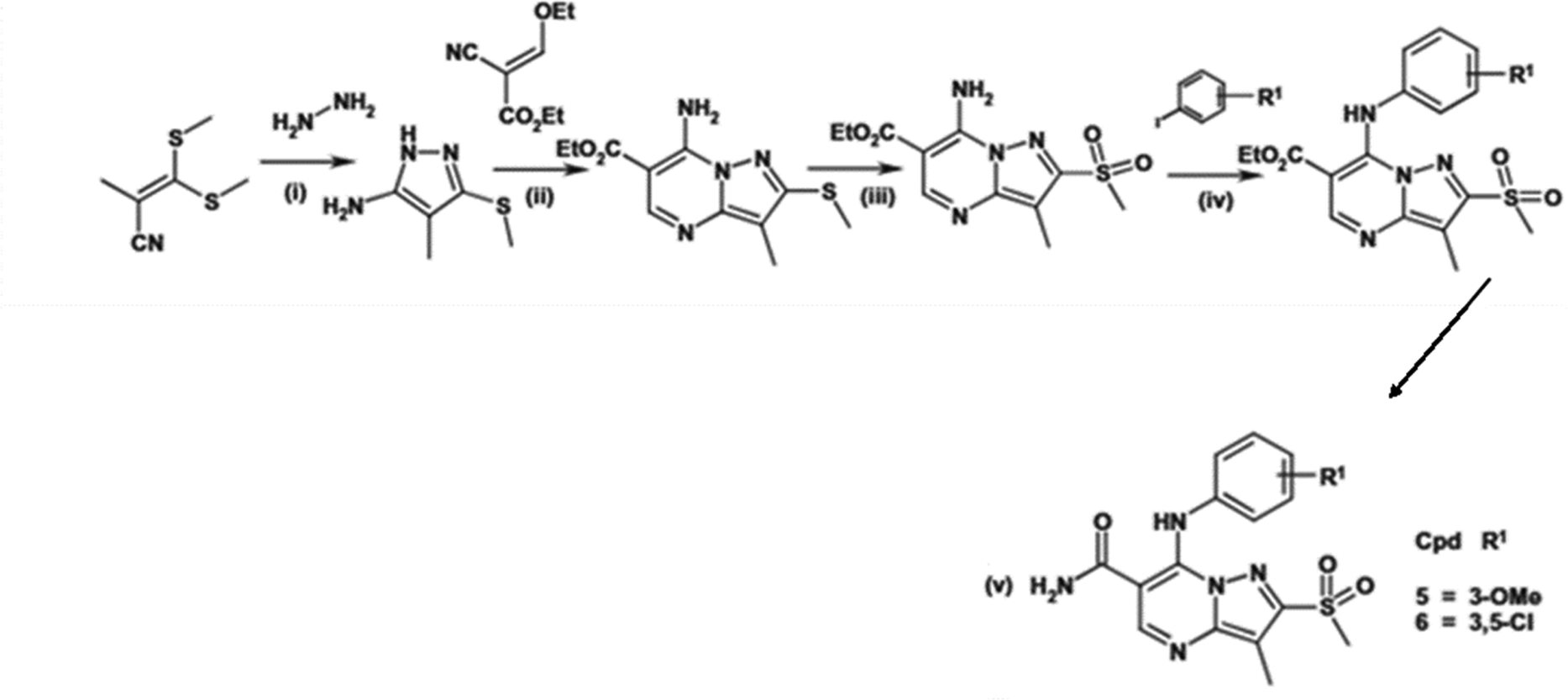 | ||
| Scheme 23 (i) Hydrazine hydrate (10 equiv.), ethanol, reflux, 72 h, yield: 36%; (ii) ethyl (ethoxymethylene)cyanoacetate (1 equiv.), AcOH, reflux, 1 h, yield: 43%; (iii) hydrogen peroxide (5 equiv.), AcOH, 70 °C, 16 h, yield: 80%; (iv) ArI (3 equiv.), CuI (1 equiv.), proline (0.7 equiv.), K2CO3 (3 equiv.), DMF, microwave, 110 °C, 2 h, yield: 34%; (v) ammonium hydroxide (excess), THF, 50 °C, 48 h, yield: 14–15%.110 | ||
This systematic optimization of substituents at key positions of the pyrazolo[1,5-a]pyrimidine scaffold demonstrates how minor structural changes can significantly impact PDE4 inhibition and cellular activity. The incorporation of bulky groups at position 3, as well as the introduction of methyl sulfone moieties at position 2, played critical roles in enhancing bioavailability and binding affinity, leading to highly potent inhibitors like compound 10 with sub-nanomolar activity (IC50 = 0.7 nM). The study provides valuable insights into structure–activity relationships that guide future PDE4 inhibitor development.
Positions 5 and 7: these positions on the pyrazole ring are frequently modified with electron-donating or electron-withdrawing groups, which affect the molecule's electronic distribution. Electron-withdrawing groups such as halogens can increase the compound's metabolic stability and potency, while electron-donating groups, such as methoxy, may enhance the binding interactions with specific enzymes or receptors.
In the study by Roux et al.110 (2016), the optimization of pyrazolo[1,5-a]pyrimidine derivatives as phosphodiesterase-4 (PDE4) inhibitors illustrates the crucial role of structural modifications at positions 5 and 7 on the pyrazole ring.111 Through systematic structure–activity relationship (SAR) studies, the research demonstrates how electron-donating and electron-withdrawing groups at these positions significantly influence the pharmacological activity, binding affinity, and metabolic stability of the compounds.
Position 5 modifications: SAR investigations reveal that electron-withdrawing groups at position 5, such as halogens, contribute to increased metabolic stability and improved potency of the pyrazolo[1,5-a]pyrimidine derivatives.112 For example, the incorporation of halogen substituents like chlorine or fluorine enhances the electron-deficient nature of the pyrazole ring, leading to better interaction with PDE4's active site. This finding aligns with previous studies on kinase inhibitors, where halogen atoms at specific positions were shown to increase binding affinity by stabilizing the inhibitor within the ATP-binding pocket. The halogens at position 5 likely increase the overall metabolic stability of the compounds by making them less susceptible to oxidative degradation, a common issue in drug metabolism.113–115
In this study, the chloro and bromo substituents on position 5 enhanced the PDE4 inhibitory activity, leading to compounds with higher potency. Compound 4, which includes a chloro group at position 5, exhibited sub-nanomolar inhibition of PDE4, with an IC50 in the low nanomolar range. This demonstrates that halogenation at this position optimizes electronic distribution in the molecule, favoring stronger binding interactions with target enzymes while also improving the compound's pharmacokinetic profile116–120
Position 7 modifications: position 7 is another site frequently modified with electron-donating groups, such as methoxy (–OCH3) or amino groups, which can enhance binding interactions with specific receptors or enzymes. In the context of the PDE4 inhibitors developed in this study, electron-donating groups at position 7 play a pivotal role in enhancing the binding affinity to PDE4 by engaging in hydrogen bonding or electrostatic interactions with amino acid residues in the active site.121–125
For instance, methoxy groups, known for their electron-donating properties, can enhance the compound's interaction with key residues in the binding pocket by increasing the electron density on the adjacent atoms of the pyrazole ring. This increased electron density can strengthen the binding interactions with polar or charged amino acid residues, which is crucial for maximizing the inhibitory effect.126,127 In some derivatives explored by Roux et al. methoxy substitution at position 7 was associated with improved cellular potency, likely due to enhanced receptor binding and better overall drug–receptor interactions. Additionally, the methoxy groups can influence the conformational flexibility of the molecule, potentially allowing it to adopt more favorable binding orientations within the PDE4 active site. This flexibility, combined with enhanced electronic interactions, contributes to the compound's ability to inhibit PDE4 more effectively, translating into improved anti-inflammatory activity.110,128–130
Dual effects of modifications at positions 5 and 7: the combination of electron-withdrawing and electron-donating groups at positions 5 and 7 provides a balanced approach to optimizing both binding interactions and metabolic stability. For instance, halogen atoms at position 5 improve the compound's resistance to metabolic degradation, while electron-donating groups at position 7 enhance the binding affinity with PDE4 by strengthening electrostatic and hydrogen bonding interactions.131–133
The synergistic effects of these modifications are evident in compounds 10, which exhibited exceptional potency as a PDE4 inhibitor with an IC50 of 0.7 nM. The presence of a halogen at position 5 and a methoxy group at position 7 allowed this compound to achieve optimal binding within the PDE4 active site, demonstrating how SAR-guided modifications at these positions can significantly enhance both potency and drug-like properties.134–136
In summary, modifications at positions 5 and 7 of the pyrazolo[1,5-a]pyrimidine scaffold play a critical role in fine-tuning the electronic properties, binding affinity, and metabolic stability of PDE4 inhibitors. Electron-withdrawing groups, such as halogens, at position 5 improve metabolic stability and enhance potency, while electron-donating groups, such as methoxy, at position 7 strengthen binding interactions with the enzyme's active site. This systematic approach to SAR analysis not only optimizes drug efficacy but also informs the broader design of pyrazolo[1,5-a]pyrimidine-based therapeutics targeting various enzymes and receptors.
Fraley et al. (2002) conducted a detailed study on the optimization of 3,6-disubstituted pyrazolo[1,5-a]pyrimidine derivatives to improve the pharmacokinetics and cellular activity of Kinase Insert Domain Receptor (KDR) kinase inhibitors.137 Their efforts aimed to enhance the physical properties of these compounds, which are essential in angiogenesis inhibition—a process highly relevant in diseases such as cancer, rheumatoid arthritis, and diabetic retinopathy. The researchers began by making modifications to the 6-aryl ring, incorporating solubilizing functionalities, including basic side chains and 4-pyridinonyl groups, to address solubility and polarity issues. The importance of pyrimidine ring modifications is clearly demonstrated in this study, as adjustments to positions 4 and 6 significantly influenced both the physical properties and the potency of the inhibitors.
A key aspect of Fraley et al.'s approach involved the introduction of basic amines to the 6-aryl ring via ether linkages.137 This modification led to the synthesis of analogs that were more potent in biochemical assays and had increased cellular activity, as demonstrated by compounds such as 4a, which showed enhanced potency in comparison to the initial lead compounds. The structure–activity relationships (SARs) derived from these compounds revealed that the addition of basic side chains improved solubility and potency, a trend that correlates with previously established studies on pyrimidine ring modifications (Scheme 24). For example, substitution at the 6-position with polar groups, such as amino or hydroxyl moieties, has been shown to improve interaction with biological targets, which is consistent with the work on kinase inhibitors discussed by Fraley et al.137
 | ||
| Scheme 24 Synthesis of compounds 5a and 5b.137 | ||
Further, the addition of the 4-pyridinonyl substituent to the 6-position of the pyrazolo[1,5-a]pyrimidine core (Scheme 25) resulted in compounds with significantly improved pharmacokinetics. Compounds 5a–c exhibited better aqueous solubility and reduced lipophilicity, which translated into increased cellular potency, as shown in Table 1, which outlines the KDR kinase and cellular activity of these compounds.137 This enhancement aligns with Fraley et al.'s earlier findings and supports the broader understanding of how alterations to the pyrimidine ring, particularly at the 4 and 6 positions, can influence the overall polarity and hydrogen bonding capacity of the molecule. Moreover, these modifications, as discussed in the context of angiogenesis, demonstrate how the strategic functionalization of heterocyclic rings can directly improve therapeutic outcomes in cancer treatment by targeting vascular endothelial growth factor receptor (VEGFR) pathways.63
 | ||
| Scheme 25 Synthesis of compound 5c.137 | ||
|
|||||
|---|---|---|---|---|---|
| Compound | R | 3-Ar/Het | KDR IC50 (nM) | ECMA IC50 (nM) | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
| 5a |  |
 |
14 | 70 | 2.04 |
| 5b |  |
 |
13 | 80 | 2.0 |
| 5c |  |
 |
7 | 20 | 1.6 |

4.2 Structure–activity relationship (SAR) studies
Systematic structure–activity relationship (SAR) studies have played a pivotal role in revealing how the structural modifications of pyrazolo[1,5-a]pyrimidines influence their biological activities. These studies allow researchers to gain insights into the specific chemical features that optimize pharmacological efficacy.138 For example, SAR investigations into pyrazolo[1,5-a]pyrimidine-based kinase inhibitors have revealed that the incorporation of small hydrophobic groups at position 3 significantly enhances binding to the ATP pockets of kinases. This feature results in more potent kinase inhibition, which is a valuable attribute for anticancer agents targeting disrupted kinase pathways. Similarly, studies on antimicrobial derivatives of pyrazolo[1,5-a]pyrimidines have shown that electron-withdrawing substituents at position 5 generally improve antibacterial potency. This likely occurs due to better penetration of bacterial cell walls, underscoring the impact of functional group manipulation in drug design.139,140In a specific instance, Novikova et al. (2023) explored the reduction of pyrazolo[1,5-a]pyrimidine derivatives using complex hydrides to form tetrahydropyrazolo[1,5-a]pyrimidines (THPPs).107 Their research demonstrated that the pyrimidine ring preferentially reduces over the pyrazole ring, producing stereoisomers that are key to medicinal chemistry. The study highlights the synthesis of syn- and anti-isomers, confirming through nuclear magnetic resonance (NMR) spectroscopy that both configurations can be obtained depending on the conditions employed. This finding is particularly noteworthy because it expands the structural diversity of pyrazolo[1,5-a]pyrimidines, offering more options for fine-tuning biological activities in drug development.
The synthetic methodology used by Novikova et al. (2023) (depicted in Scheme 26) involved a three-step process starting from ethyl 2-cyanoacetate.107 The final product, ethyl 5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxylate, was obtained in high yield and subsequently subjected to reduction. The results of this reduction are illustrated in Scheme 27, where the reaction with sodium borohydride (NaBH4) led to a complex mixture of products, including the desired tetrahydropyrazolo[1,5-a]pyrimidines. Notably, the stereochemical outcomes favoured the syn-isomer over the anti-isomer in a 7:1 ratio when methanol was used as the solvent. However, by altering the reaction conditions—specifically, using tetrabutylammonium borohydride in an aprotic solvent such as chloroform—the proportion of anti-isomer increased to 1:1.
 | ||
| Scheme 26 Synthesis of ethyl 5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxylate. Reaction conditions: (a) dimethylformamidine dimethyl acetal, 70 °C, 6 hours, yield 90%; (b) hydrazine hydrate, ethanol/water, 90 °C, 4 hours, yield 82%; (c) acetylacetone, acetic acid/ethanol, 100 °C, 6 hours, yield 80%.107 | ||
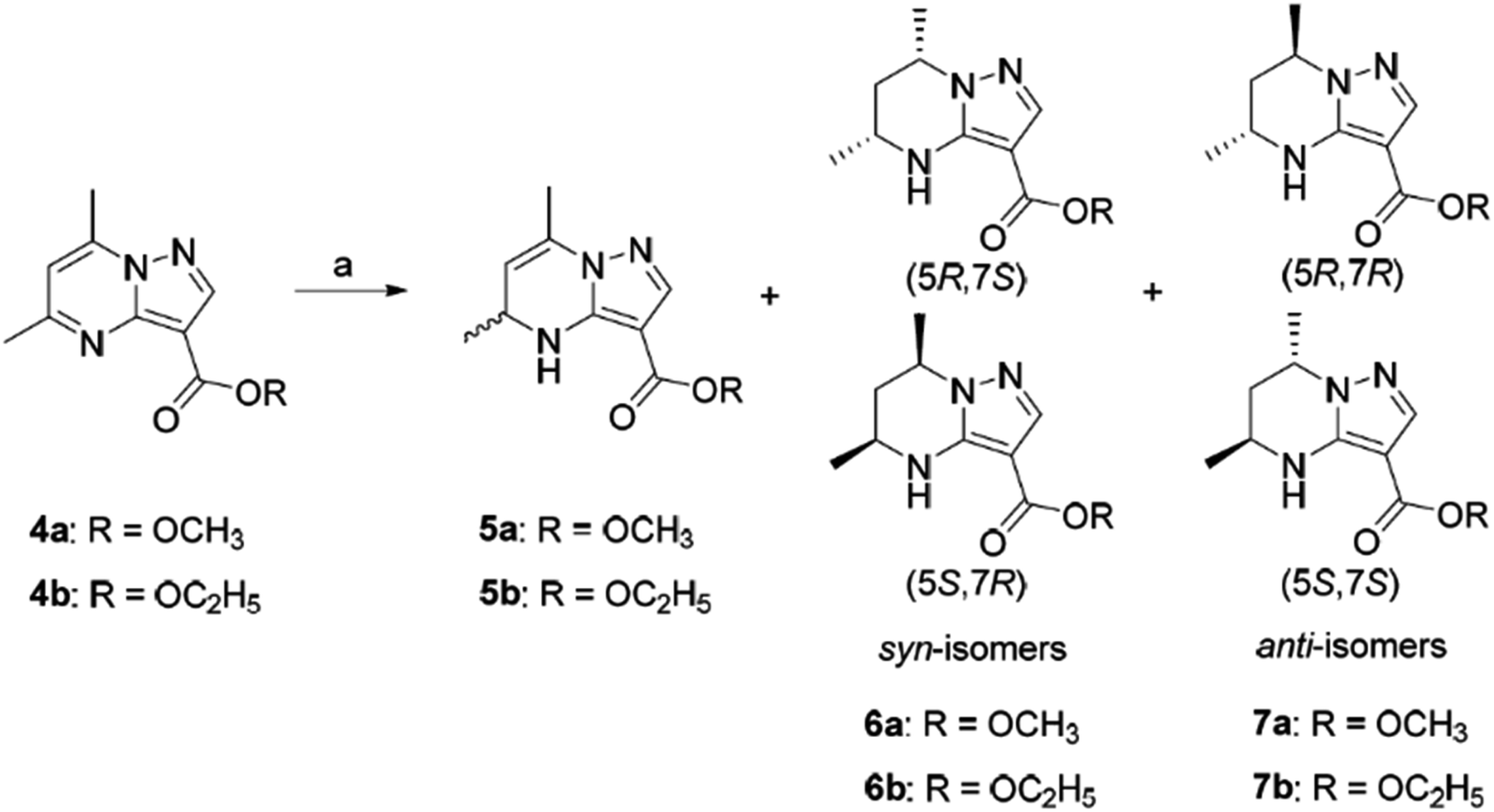 | ||
| Scheme 27 Dearomatization of 5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxylate. Reagents and conditions: (a) NaBH4, ROH, RONa, RCH3, and C2H5.107 | ||
This work illustrates the flexibility and potential of pyrazolo[1,5-a]pyrimidines as scaffolds for designing biologically active molecules. The ability to control stereochemistry, as demonstrated in the formation of syn- and anti-isomers, is crucial for optimizing interactions with biological targets. For instance, the lability of the anti-isomeric tetrahydropyrazolo[1,5-a]pyrimidines allow these molecules to adjust to the active sites of various targets, making them highly adaptable for therapeutic applications. This adaptability is supported by computational studies that confirmed the conformational flexibility of the anti-isomers. The computational data provided insight into the energy states and geometric parameters of the synthesized compounds, which is important for understanding their interaction dynamics within biological systems.
Overall, the systematic exploration of pyrazolo[1,5-a]pyrimidine derivatives through SAR and synthetic modifications, as seen in the work by Novikova et al. and others, emphasizes the versatility of this scaffold in drug discovery. The ability to fine-tune properties such as potency, selectivity, and conformational stability positions pyrazolo[1,5-a]pyrimidines as promising candidates for developing new therapeutics across a range of disease areas.
Elbakry et al. (2023) explored the synthesis and biological evaluation of new pyrazolo[1,5-a]pyrimidine derivatives 5a–c, focusing on their anticancer activity and inhibition of CDK2.141 The authors employed systematic structure–activity relationship (SAR) studies to elucidate how chemical modifications to the pyrazolo[1,5-a]pyrimidine scaffold impact biological efficacy. Previous SAR analyses on kinase inhibitors have demonstrated that small hydrophobic groups at position 3 of pyrazolo[1,5-a]pyrimidines enhance ATP pocket binding, while electron-withdrawing groups at position 5 improve antibacterial activity. In line with this, Elbakry et al. sought to determine how similar modifications would affect the antiproliferative activities of their newly synthesized compounds.
Scheme 28 outlines the synthetic route leading to the pyrazolo[1,5-a]pyrimidine derivatives 5a–c. Starting from 3,5-diaminopyrazole derivatives (3), a nucleophilic addition to enaminones (4a–c) followed by the elimination of the dimethylamine group resulted in intermediates 7 and 8. The subsequent intramolecular cyclization of intermediate 8 led to the formation of intermediate 9, which then underwent aromatization via water elimination to afford the final products, 5a–c. The regioselectivity of this reaction and the formation of the isomers were confirmed by 1H-NMR analysis and spectral data, as shown in Scheme 29, which details the reaction mechanism.
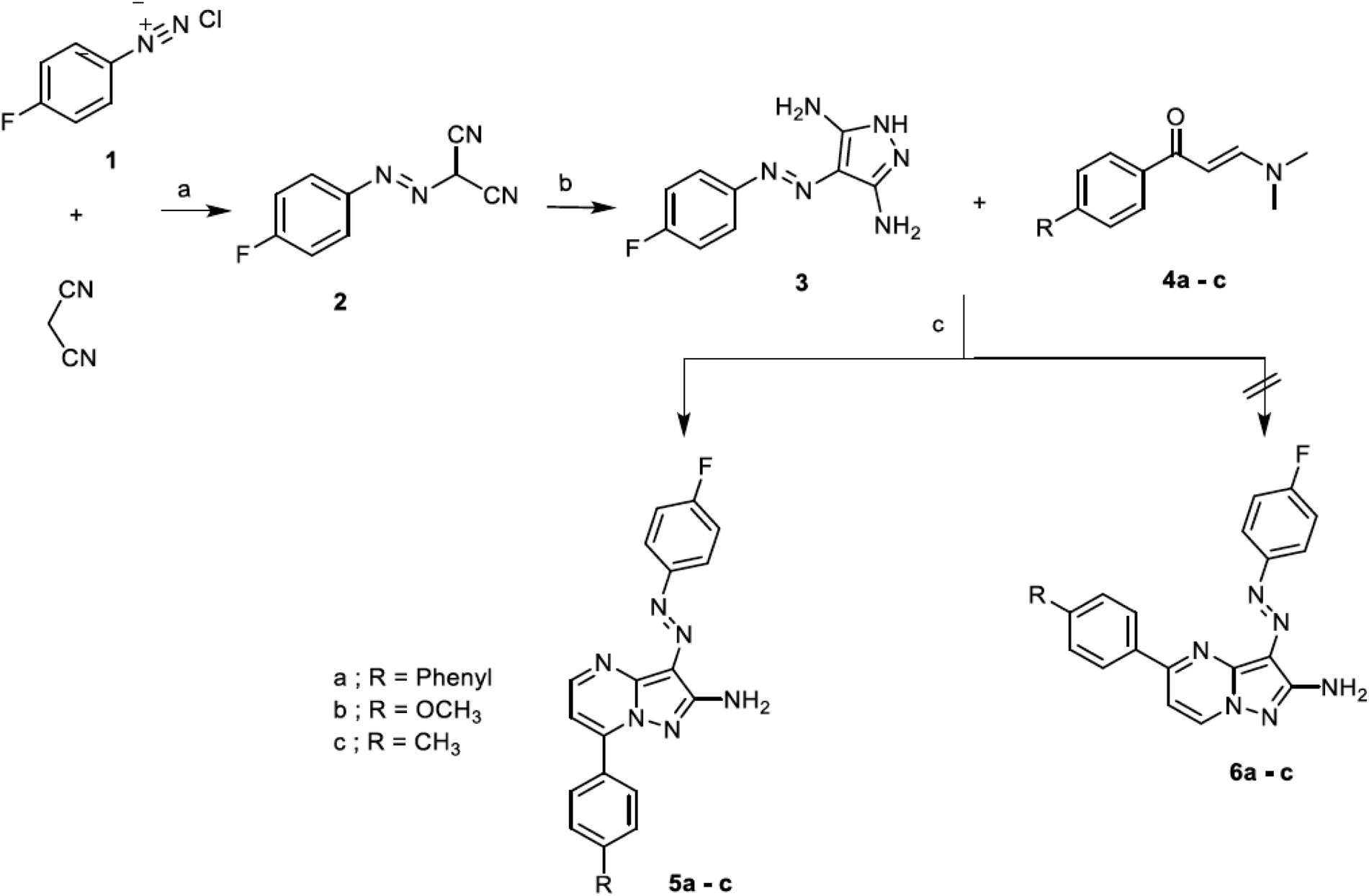 | ||
| Scheme 28 The preparation of 3-((4-fluorophenyl)diazenyl)-7-(4-substituted phenyl)pyrazolo[1,5-a]pyrimidin-2-amine 5a–c. Reagents and condition: (a) sodium acetate/EtOH, stirring for 30 min/cooling. (b) NH2NH2·H2O/EtOH, reflux 6 h. (c) GL acetic acid, reflux 7 h. | ||
 | ||
| Scheme 29 The suggested reaction mechanism for the formation of pyrazolopyrimidines 5a–c.141 | ||
The biological evaluation of these compounds revealed their promising cytotoxic activity against three human cancer cell lines—MCF-7 (breast cancer), HCT-116 (colon cancer), and HepG-2 (liver cancer). Among the three compounds, 5b (with a methoxy group) demonstrated the most potent activity against HCT-116, with an IC50 value of 8.64 μM, which is comparable to that of the reference drug doxorubicin (IC50 = 5.49 μM). In contrast, compounds 5a (with a phenyl group) and 5c (with a methyl group) exhibited moderate activity, further underscoring the significance of functional group modifications on biological outcomes. The SAR results confirmed that the methoxy substitution in 5b played a key role in enhancing its anticancer efficacy.
Docking studies provided additional insights into the molecular interactions between the pyrazolo[1,5-a]pyrimidine derivatives and CDK2. The docking simulations, conducted using the MOE-2014 software, revealed that compounds 5a–c bind with high affinity to the ATP binding site of CDK2, with binding energies of −12.8, −11.54, and −11.59 kcal mol−1, respectively. Critical interactions were observed between the NH2 groups of the pyrazolo[1,5-a]pyrimidines and Leu83 in the hinge region of CDK2, as well as Pi-alkyl interactions with Ile10 and Pi-anion interactions with Asp86 in the cases of 5a and 5b. These molecular interactions suggest that the pyrazolo[1,5-a]pyrimidine scaffold is well-suited for CDK inhibition, making it a valuable target for anticancer drug development.
The ADME (absorption, distribution, metabolism, and excretion) profile of the compounds was evaluated, showing that all derivatives adhered to Lipinski's rule of five, with the exception of 5a, which had a higher MlogP value. Additionally, the predicted absorption rates were favourable, with all compounds displaying appropriate cell membrane permeability. The analysis also suggested that compounds 5a and 5b would not cross the blood–brain barrier, minimizing potential central nervous system side effects.
In general, the study by Elbakry et al.141 (2023) demonstrates the successful synthesis of new pyrazolo[1,5-a]pyrimidine derivatives and their potent anticancer activity, particularly against HCT-116. The research highlights the importance of SAR studies in optimizing pharmacological efficacy through strategic chemical modifications. The docking studies further validate the potential of these compounds as CDK2 inhibitors, supporting their development as anticancer agents.
4.3 Functional group tolerance in synthesis
4.3.1.1 Palladium-catalyzed cross-coupling. Palladium-catalyzed cross-coupling reactions, such as the Suzuki–Miyaura and Heck reactions, are extensively employed to introduce aryl, alkyl, and vinyl groups at various positions on the pyrazole ring. These reactions are especially valuable for constructing carbon–carbon (C–C) bonds, which are crucial for synthesizing pyrazole derivatives with a wide range of steric and electronic properties.142,143 The ability of these reactions to efficiently form C–C bonds under mild conditions, often with high yields and selectivity, has made them indispensable in the design of complex organic molecules.
The Suzuki–Miyaura reaction, for instance, typically involves the coupling of aryl or vinyl boronic acids with aryl halides in the presence of a palladium catalyst and a base. This method allows for the introduction of a wide array of substituents onto the pyrazole ring, enabling fine-tuning of the molecule's electronic properties, which is critical for optimizing biological activity. Similarly, the Heck reaction facilitates the coupling of alkenes with aryl halides or vinyl halides, providing access to vinyl-substituted pyrazole derivatives. These types of modifications can significantly alter the pharmacokinetics and receptor interactions of pyrazole-based compounds, making them more suitable for therapeutic applications.144
The versatility of palladium-catalyzed cross-coupling reactions has been demonstrated in numerous studies, where they are employed to introduce diverse functional groups onto various heterocyclic frameworks, including pyrazoles, imidazoles, and other biologically relevant molecules. This versatility is largely due to palladium's unique ability to facilitate both oxidative addition and reductive elimination processes, which are essential steps in cross-coupling reactions.145,146 These reactions have revolutionized the synthesis of heterocyclic compounds, particularly in medicinal chemistry, where they allow for the rapid assembly of complex molecular scaffolds with specific functional group arrangements.
In addition to their use in small-scale laboratory settings, these palladium-catalyzed reactions are also highly suitable for large-scale industrial applications. Their efficiency, coupled with the growing availability of palladium catalysts, has led to widespread adoption in pharmaceutical synthesis, agrochemicals, and materials science.147–151 Moreover, advances in green chemistry have begun to influence the development of these cross-coupling reactions, with efforts focusing on minimizing the use of hazardous solvents and reagents, further enhancing the environmental sustainability of these valuable synthetic tools.
By enabling the precise introduction of functional groups onto pyrazole rings and other heterocycles, palladium-catalyzed cross-coupling reactions continue to play a pivotal role in the development of novel bioactive compounds and functional materials, showcasing their enduring importance in modern organic synthesis.
The work of Kotla et al. (2017) illustrates the utility of palladium-catalyzed reactions in organic synthesis.94 In their study, they optimized a reaction involving 1H-benzo[d]imidazole-2-amine and 2-phenylacetaldehyde to produce phenyl(3-phenylbenzo[4,5]imidazo[1,2-a]pyrimidin-2-yl)methanone using a palladium catalyst. Initially, other metal salts such as CuCl2, Fe, Zn, and Sn were tested, but PdCl2 proved to be the most effective, yielding the desired product in 80%. This significant yield improvement highlights the unique catalytic properties of palladium in facilitating C–C bond formation.
The development and optimization of these palladium-catalyzed reactions allow for the introduction of various substituents on heterocycles, significantly impacting the steric and electronic properties of the resultant compounds. The introduction of aryl or alkyl groups via Suzuki–Miyaura cross-coupling, for instance, is a widely applied strategy for modifying the electronic nature of pyrazole rings, as well as other heterocycles, which are frequently used as pharmacophores in medicinal chemistry. Similarly, Heck coupling allows for the vinylation of these rings, adding further versatility to synthetic approaches.
Kotla et al. further explored the scope of their developed reaction by synthesizing a series of imidazo[1,2-a]pyrimidine derivatives.94 The results of their experiments demonstrated that starting materials with electron-donating groups yielded higher amounts of the desired products compared to those with electron-withdrawing groups, which aligns with the general trends observed in cross-coupling chemistry. This trend underscores the role of electronic factors in these reactions, as electron-rich substrates often enhance the coupling efficiency by stabilizing intermediate palladium species.
Beyond merely introducing functional groups, palladium-catalyzed cross-coupling reactions also enable the fine-tuning of the physicochemical properties of target molecules. This fine-tuning is particularly evident in the synthesis of kinase inhibitors, as demonstrated by Kotla et al.'s study. By employing Suzuki cross-coupling to install pyridinonyl or thienyl groups, they were able to enhance both the polarity and solubility of KDR kinase inhibitors, which in turn led to improved pharmacokinetic profiles. Such strategic modifications underscore the importance of palladium-catalyzed cross-coupling in drug development, where both efficacy and bioavailability need to be optimized.
In general, palladium-catalyzed cross-coupling reactions are indispensable for modern synthetic chemistry, enabling the formation of C–C bonds with precision and versatility. As shown in studies like that of Kotla et al., these reactions are not only useful for synthesizing complex heterocyclic compounds but also for tailoring their physicochemical and biological properties, making them crucial tools in pharmaceutical and material sciences.
In the study by Kotla et al. (2017), the reaction of 1H-benzo[d]imidazole-2-amine with phenylacetaldehyde in the presence of various metal salts demonstrated the superiority of palladium catalysts, specifically PdCl2, which afforded significantly higher yields than other metal salts like CuCl2 or FeCl2. This highlights palladium's remarkable efficacy in promoting cross-coupling reactions, not just in pyrazole systems but across a range of heterocyclic compounds.94 The optimization of reaction conditions, including base and solvent selection, further reinforced the importance of fine-tuning reaction parameters in achieving optimal yields. This study underlined the practical utility of palladium in heterocycle synthesis, particularly when generating structurally complex compounds such as phenyl-substituted imidazo[1,2-a]pyrimidines, a structure relevant to biological and pharmacological applications.
Hsiao et al. (2022) further expanded the scope of palladium-catalyzed C–H bond activation by introducing a direct cross-dehydrogenative coupling approach to synthesize 3,3′-bipyrazolo[1,5-a]pyridine derivatives.152 This method demonstrated excellent functional group tolerance and efficiency, with yields up to 94%. The mechanistic insights gained through kinetic isotope effect experiments and density functional theory calculations offered a deeper understanding of the palladium-catalyzed C–H activation process. The subsequent derivatizations of these bipyrazolo[1,5-a]pyridine products, including palladium-mediated ortho C–H activation followed by iodine-induced chlorination, enabled further functionalization, enhancing π-conjugation and twisting conformations. These derivatives hold potential in the development of organic luminescent materials, showcasing the broad applicability of palladium-catalyzed cross-coupling in the design of advanced materials.
Nguyen et al. (2021) provided another illustration of palladium's versatility in oxidative C–H/C–H cross-coupling reactions, specifically involving pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a]pyridines.153 This regioselective process, enabled by Pd(OAc)2 and AgOAc without the need for directing groups, exemplifies the direct functionalization of heteroarenes, expanding the toolkit available for constructing complex molecular architectures. The coupling with five-membered heteroarenes like thiophenes, thiazoles, and furans demonstrated the wide applicability of this method in constructing heterocyclic frameworks. Such transformations are valuable for the synthesis of biologically active molecules and advanced materials, where the precise incorporation of heteroaryl units is often necessary.
Overall, palladium-catalyzed cross-coupling reactions continue to play a pivotal role in modern organic synthesis, enabling the efficient and selective formation of C–C bonds in a wide array of heterocyclic systems. The work by Kotla, Hsiao, and Nguyen et al. demonstrates not only the robustness of these reactions but also their adaptability to different substrates and reaction conditions, paving the way for future innovations in both synthetic methodologies and material development.
Nucleophilic substitution and halogenation: another common functionalization approach involves the halogenation of specific positions on the pyrazolo[1,5-a]pyrimidine core, followed by nucleophilic substitution reactions. This allows for the introduction of amino, thiol, or hydroxyl groups, which can dramatically alter the molecule's solubility and binding interactions. Halogenation followed by substitution has been particularly useful in creating pyrazolo[1,5-a]pyrimidine derivatives with enhanced antimicrobial and anti-inflammatory properties.
4.3.1.2 Click chemistry. Click chemistry, especially copper-catalyzed azide–alkyne cycloaddition (CuAAC), has emerged as an efficient method to functionalize pyrazolo[1,5-a]pyrimidines. This technique enables the rapid attachment of bioactive moieties such as peptides, carbohydrates, and fluorescent tags, facilitating the development of multifunctional compounds for imaging, targeted drug delivery, and diagnostic applications.154
The research on the inhibition of the protein–protein interaction between B-cell lymphoma 6 (BCL6) and corepressors has led to the identification of a pyrazolo[1,5-a]pyrimidine series as potential BCL6 inhibitors.154 Click chemistry has become an essential method for functionalizing pyrazolo[1,5-a]pyrimidines, which can facilitate the attachment of bioactive moieties like peptides and fluorescent tags. This approach has enabled the rapid development of multifunctional compounds aimed at enhancing diagnostic and therapeutic applications such as imaging and targeted drug delivery. Scheme 30 highlights the functionalization of pyrazolo[1,5-a]pyrimidines via click chemistry, showcasing its ability to fine-tune compounds for better biological interactions.154 Using structure-based drug design, the binding affinity of these compounds was improved significantly by displacing crystallographic water and enhancing ligand–protein interactions. These optimizations led to increased bioactivity and selectivity, particularly against off-targets like CK2.24,25
 | ||
| Scheme 30 Synthesis of acyclic pyrazolo[1,5-a]pyrimidines.154 aReagents and conditions: (a) c-PrNH2, EtOH, 99%; (b) Pd/C, H2, EtOAc, 53%; (c) c-PrNH2 or PhNH2 or 6-amino-3,4-dihydroquinolin-2(1H)-one, DIPEA, NMP, rt-100 °C, then pyrrolidine-R6, μw, 65–130 °C, 25–64%; (d) 6-amino-3,4-dihydroquinolin-2(1H)-one, EtOH, 80 °C, 91%; (e) (S) or (R)-prolinol, DIPEA, NMP, 80 °C, 65–67%.154 | ||
The introduction of macrocyclization further improved the affinity and kinetics of the BCL6 inhibitors. In acyclic compounds 8, free rotation of the pyrrolidine allows for multiple conformations of the chiral alcohol group. Upon macrocyclization, such as in compound 11, this rotation is restricted, leading to a more stable bioactive conformation, enhancing binding affinity.155,156 Notably, the (5S)-methanol substitution in macrocyclic compounds 21 led to a six-fold increase in affinity due to favourable hydrogen bonding interactions, displacing structural water as predicted by solvent analysis.157
Click chemistry played a pivotal role in designing these BCL6 inhibitors, allowing for the precise positioning of functional groups, such as the (5S)-methanol in the macrocyclic structures, which stabilized ligand–protein interactions. This chemical strategy was instrumental in optimizing the off-rate kinetics and increasing the residence time of the inhibitors, which are critical parameters for achieving sustained therapeutic effects in cellular models.158–160 However, despite these advancements, the observed antiproliferative effects across various diffuse large B-cell lymphoma (DLBCL) lines were weak, and the link between BCL6 potency and cancer cell proliferation remains unclear, leaving the therapeutic hypothesis of BCL6 inhibition in DLBCL unresolved.154
The research underlines the value of integrating click chemistry into drug development processes to rapidly generate multifunctional compounds. Scheme 30 serves as a clear reference point for how bioactive moieties can be effectively incorporated into pyrazolo[1,5-a]pyrimidine structures, advancing both the selectivity and functionality of BCL6 inhibitors.154
Jismy et al. (2018) presented a streamlined approach to synthesizing trifluoromethylated 3,5-disubstituted pyrazolo[1,5-a]pyrimidines, utilizing principles inspired by click chemistry, a concept designed for efficiency, modularity, and high regioselectivity.161 Click chemistry is prized for its simplicity in generating target compounds with minimal byproducts. In this study, Jismy and colleagues demonstrate a two-step synthetic process that reflects the core tenets of click chemistry, specifically in its focus on high-yield reactions and the formation of heterocycles.
The first step involves a one-pot reaction between 3-aminopyrazoles and ethyl 4,4,4-trifluorobut-2-ynoate, producing 7-trifluoromethylated pyrazolo[1,5-a]pyrimidin-5-ones. This step aligns well with click chemistry due to its efficiency and the use of readily available materials, yielding high regioselectivity. Without the need for complex protecting group strategies, the reaction proceeds cleanly, forming heterocyclic frameworks in a straightforward manner. Following this, the core pyrazolo[1,5-a]pyrimidin-5-one intermediates undergo C–O bond activation, catalyzed by a phosphonium salt, which facilitates subsequent Suzuki–Miyaura cross-coupling reactions. These cross-couplings introduce various aryl groups, enabling the creation of 5-arylated pyrazolo[1,5-a]pyrimidines in high yields.
This synthetic route demonstrates the modularity that defines click chemistry, as a wide variety of boronic acids can be employed in the cross-coupling step. The reaction tolerates both electron-rich and electron-deficient substituents, allowing for significant functional diversity. Moreover, the use of microwave-assisted conditions further enhances the reaction's efficiency, demonstrating how this methodology adheres to click chemistry's principles by optimizing reaction time and yield under mild conditions. This approach, confirmed by single-crystal X-ray crystallography, ensures that the desired regioisomers are obtained with excellent selectivity.
The trifluoromethylated pyrazolo[1,5-a]pyrimidines synthesized through this methodology exhibit interesting properties, particularly in the context of their bioactivity. For instance, compound 4k shows promise as an inhibitor of FUBP1, a target with relevance in cancer biology. The incorporation of fluorine atoms imparts unique electronic and lipophilic characteristics to the molecules, which can improve metabolic stability, membrane permeability, and binding affinity in biological systems. Additionally, the observed structural variations from the introduction of different aryl groups offer an opportunity to fine-tune these properties for specific applications, especially in medicinal chemistry.
Further functionalization is demonstrated through a one-pot Sonogashira coupling for C-5 alkynylation of the pyrazolo[1,5-a]pyrimidine core. This reaction, known for its efficiency and use of mild copper-catalyzed conditions, produces C–C bonds cleanly and without extensive purification steps, in keeping with click chemistry's goal of minimizing side reactions and maximizing yields. The trifluoromethyl group plays a crucial role in modifying the physicochemical properties of the final compounds, influencing their reactivity, stability, and potential biological activity. Fluorinated molecules are often sought after in drug discovery due to their improved pharmacokinetic profiles, and the ease of introducing fluorinated motifs via this synthetic approach is particularly advantageous.
In summary, the work by Jismy et al.161 exemplifies the application of click chemistry principles in the synthesis of trifluoromethylated pyrazolo[1,5-a]pyrimidines, emphasizing modularity, efficiency, and regioselectivity. The synthetic pathway offers flexibility in functional group incorporation, yielding biologically relevant compounds with desirable properties, including potential as pharmaceutical agents. The observed properties, such as bioactivity and enhanced stability conferred by the fluorine atoms, highlight the practical advantages of this approach, making it a valuable tool for advancing the design of novel molecules in drug discovery and other fields.
5 Mechanisms of action as protein kinase inhibitors
5.1 Overview of kinase inhibition
Kinase inhibitors encompass a wide range of substances that target and inhibit the activity of one or more kinases—enzymes that mediate the transfer of phosphate groups, a critical step in phosphorylation, which is essential for cellular signaling.162 Phosphorylation regulates numerous physiological processes, including cell growth, differentiation, survival, and division (see Fig. 1 above). Therefore, by inhibiting these kinases, kinase inhibitors can modulate key signaling pathways involved in cellular homeostasis. These pathways are particularly important in the context of cancer, where kinase activity is frequently disrupted, leading to uncontrolled cellular proliferation and tumor progression.163Kinase inhibitors hold significant therapeutic potential in oncology, as they specifically target aberrant kinases responsible for the malignancy of cancer cells. By disrupting the signaling cascades that cancer cells rely on for survival and division, these inhibitors can effectively impede tumor growth.164,165 For instance, specific kinase inhibitors have been designed to target receptor tyrosine kinases (RTKs) or serine/threonine kinases, which are often overactive or mutated in cancers such as breast, lung, and colorectal carcinomas. Among the various classes of kinase inhibitors, pyrazolo[1,5-a]pyrimidines have garnered considerable attention due to their potent inhibitory effects on kinases closely associated with cancer and other diseases.165 These compounds act by binding to the ATP-binding pocket of kinases, thereby preventing the transfer of phosphate groups that drive signal transduction pathways critical for cancer cell survival and proliferation. Ongoing research into pyrazolo[1,5-a]pyrimidines continue to expand their therapeutic applications, as structural modifications to the core scaffold have been shown to enhance both their selectivity and efficacy against specific kinases implicated in malignancies.23,166,167
As a result, pyrazolo[1,5-a]pyrimidine derivatives not only exhibit promising anticancer properties but also offer potential therapeutic options for other kinase-related diseases, making them a valuable addition to the arsenal of kinase inhibitors under investigation for drug development.
Once bound to the kinase, many pyrazolo[1,5-a]pyrimidines form stable complexes, forcing the enzyme into an inactive conformation. This prevents ATP binding and halts phosphorylation events critical for cell signaling, especially in cases where kinase mutations or overexpression drive cancerous cell proliferation and tumor growth. The inhibition of aberrant kinase activity through these compounds is particularly relevant in oncology, where disrupted kinases contribute to uncontrolled cell growth, making them key therapeutic targets.149–152
In addition to inhibiting the ATP-binding pocket, pyrazolo[1,5-a]pyrimidines may modulate kinase activity through other mechanisms, such as exploiting the viscoelastic properties of kinases. Some of these compounds can chelate to inactive conformations of kinases, locking them in an inactive state and preventing their activation and subsequent phosphorylation of downstream substrates.19,168 This dual ability to target multiple kinases, through both ATP competition and stabilization of inactive conformations, has positioned pyrazolo[1,5-a]pyrimidines as a highly desirable class of drugs, especially in the development of anticancer therapies. Their ability to selectively inhibit overactive or mutated kinases makes them promising candidates in precision oncology, where personalized treatment strategies are increasingly focused on targeting specific molecular drivers of cancer.
One particularly relevant application of pyrazolo[1,5-a]pyrimidine derivatives is in the inhibition of kinases associated with cancer, where disruption of these enzymes often contributes to uncontrolled cell growth. For example, Hanke (2022) explored the development of a pyrazolo[1,5-a]pyrimidine-based inhibitor targeting aerine/ihreonine kinase 17A (DRAK1), a member of the death-associated protein kinase (DAPK) family.169 DRAK1 is part of the so-called “dark kinome”, meaning its cellular functions and roles in diseases are not well understood. However, recent findings have implicated DRAK1 in the progression of glioblastoma multiforme (GBM) and other cancers, sparking interest in developing selective inhibitors for this target.
Through structure-guided optimization of a pyrazolo[1,5-a]pyrimidine-based macrocyclic scaffold, the researchers developed CK156 (34), a compound with high in vitro potency (KD = 21 nM) and selectivity, as confirmed by kinome-wide screens. The crystal structure of CK156 (34) revealed that it acts as a type I inhibitor, which means it binds to the active conformation of the kinase, blocking ATP binding and preventing phosphorylation of downstream substrates. This inhibition could theoretically halt the oncogenic signaling that drives tumor growth in GBM and other cancers.
However, despite the high potency of CK156 (34) in biochemical assays, the compound demonstrated limited efficacy in inhibiting the growth of glioma cells in both 2D and 3D cultures, with significant effects only observed at low micromolar concentrations. This suggests a potential discrepancy between genetic knockdown studies of DRAK1 and pharmacological inhibition by CK156 (34). Such differences could be due to compensatory mechanisms within the cell, the influence of the cellular microenvironment, or the inability of the inhibitor to fully recapitulate the effects of genetic ablation.
The mechanism of action for pyrazolo[1,5-a]pyrimidines is not solely limited to ATP competition. These inhibitors may also lock kinases into inactive conformations, stabilizing their inactive state and further preventing phosphorylation events.19,168 This dual mode of inhibition, both by competing for ATP and by stabilizing the inactive form of kinases, makes pyrazolo[1,5-a]pyrimidines versatile tools in cancer treatment. Their ability to selectively inhibit overactive or mutated kinases enhances their appeal in precision oncology, where targeted therapies are designed to disrupt specific molecular drivers of cancer.
In the case of DRAK1, CK156 (34) represents a significant step forward in developing selective inhibitors for this relatively unexplored kinase. Although the in vitro potency of the compound is promising, further optimization may be required to improve its cellular efficacy and therapeutic potential in cancer models. The findings from Hanke (2022) also highlight the importance of understanding the mechanistic nuances of kinase inhibition, particularly when translating biochemical potency into meaningful therapeutic outcomes.169
The study by Williamson et al. (2005) explores the protein structure-guided design of pyrazolo[1,5-a]pyrimidines, focusing on their role as inhibitors of cyclin-dependent kinase 2 (CDK2).170 CDK2 plays a central role in regulating the mammalian cell cycle, making it a critical target in cancer therapy. These pyrazolo[1,5-a]pyrimidine derivatives exhibit significant potency and selectivity for CDK2 and CDK1, which are both involved in cell cycle progression. By interfering with the ATP-binding pocket of CDK2, these compounds prevent the enzyme from catalyzing the phosphorylation of its substrates, thus halting cell division, particularly in tumor cells.
One key aspect of the mechanistic action of these compounds involves mimicking ATP, similar to other pyrazolo[1,5-a]pyrimidine-based kinase inhibitors. By binding to the ATP-binding site, these compounds compete with ATP and block kinase activity, effectively inhibiting cell proliferation. Structural analysis via X-ray crystallography revealed the binding interactions between pyrazolo[1,5-a]pyrimidines and CDK2. Notably, a “donor–acceptor–donor” interaction motif was identified, which involves hydrogen bonding with the backbone of residues like Leu83 and Glu81 in CDK2. These interactions lock the enzyme into an inactive conformation, akin to the way pyrazolo[1,5-a]pyrimidines inhibit other kinases by stabilizing inactive forms.
Interestingly, further structure–activity relationship (SAR) studies showed that modifications at specific positions on the pyrazolopyrimidine scaffold, such as the 3-, 5-, and 7-positions, significantly influenced the potency and selectivity of the inhibitors. For instance, the introduction of an isopropyl group at the 3-position enhanced CDK2 potency while reducing activity against glycogen synthase kinase-3β (GSK-3β), which is therapeutically antagonistic to CDK2. Such specificity, achieved by fine-tuning the interactions with key active site residues, underscores the potential of pyrazolo[1,5-a]pyrimidines as selective kinase inhibitors.
The study also revealed that these inhibitors could selectively downregulate CDK2-mediated phosphorylation of retinoblastoma protein in cancer cells, leading to cell cycle arrest in the G2/M phase. This finding aligns with the expected mode of action for dual CDK1/CDK2 inhibitors, which further illustrates their relevance in disrupting tumor cell growth.
In general, the study by Williamson et al.170 contributes to the broader understanding of how pyrazolo[1,5-a]pyrimidines can be fine-tuned to selectively target specific kinases like CDK2 and CDK1. By exploiting their ability to bind the ATP pocket and stabilize inactive enzyme conformations, these compounds hold significant promise in cancer treatment, where disrupted kinase activity is a major driver of disease progression.
Degorce et al. (2016) focused on improving the cellular lipophilic ligand efficiency (LLE) of a series of 2-anilino-pyrimidine IGF-1R kinase inhibitors, aiming to discover novel compounds with enhanced physicochemical properties.171 By substituting the imidazo[1,2-a]pyridine group in their previously reported inhibitor with a pyrazolo[1,5-a]pyridine, they achieved improved IGF-1R cellular potency. This shift in the chemical structure was a critical factor in enhancing the potency and selectivity of the compounds, specifically against the IGF-1R kinase, while minimizing undesirable activity against CDKs and other kinases.
The study introduced amino-pyrazole derivatives and identified specific substitutions, such as the 3-methoxy and 3-methyl analogues, that helped restore kinase selectivity while maintaining reasonable IGF-1R activity. These modifications were essential as previous analogues suffered from a lack of selectivity, particularly showing activity against CDK1, CDK2, CDK7, and CDK9, which posed toxicity concerns. By adjusting the chemical structure, they managed to balance cellular potency and lipophilicity, improving cell LLE without negatively impacting hERG ion channel activity, a known toxicity risk in drug development.
Crystallographic studies further elucidated the mechanistic basis of improved selectivity, revealing how specific methyl substitutions on the pyrazole ring induced conformational changes in the binding of the inhibitors to the kinase domain of IGF-1R. This structural insight was crucial for designing compounds with enhanced selectivity, particularly dimethyl pyrazole analogues, which showed significant improvements in selectivity ratios against CDK2. Moreover, these compounds exhibited fewer off-target kinase hits in broader kinase panels, demonstrating their potential for improved efficacy and safety profiles in kinase-targeted therapies.
Overall, the work by Degorce et al. aligns with the broader theme of optimizing kinase inhibitors through structural modifications, a principle seen in the development of pyrazolo[1,5-a]pyrimidines as discussed in kinase inhibition studies.171 These modifications underscore the importance of balancing potency, selectivity, and physicochemical properties in the design of next-generation kinase inhibitors for targeted cancer therapies.
5.1.2.1 ATP-competitive inhibitors. ATP-competitive inhibitors exert their function by binding to the ATP-binding cleft of kinases, directly competing with ATP for this critical binding site. Since ATP is a ubiquitous molecule present in all cells and tissues, a major challenge associated with these inhibitors is achieving selectivity for the targeted kinase, while avoiding inhibition of other kinases that also rely on ATP for their enzymatic activity.174,175 This selectivity is crucial for minimizing off-target effects, as kinases play central roles in various signalling pathways across different cellular contexts.
Pyrazolo[1,5-a] pyrimidines are a class of small molecules that are thought to function primarily as ATP competitors, meaning they occupy the ATP-binding site of the kinase and thus prevent the kinase from phosphorylating its substrates. By binding in this manner, they attenuate downstream signaling pathways that depend on the phosphorylation events catalyzed by the kinase.176,177 This type of inhibition is typically reversible, meaning that the inhibitory effect persists only as long as the inhibitor remains bound to the kinase. Once the inhibitor dissociates from the kinase, or is degraded or replaced, normal kinase activity can resume. This reversible nature of ATP-competitive inhibitors can offer flexibility in therapeutic applications, but also poses challenges in maintaining sustained inhibition, particularly in rapidly proliferating cells or dynamic signaling environments. Thus, achieving both potent and selective inhibition is critical to the success of ATP-competitive inhibitors in therapeutic settings.178,179
5.1.2.2 Allosteric inhibitors. Allosteric inhibitors differ significantly from ATP-competitive inhibitors in that they bind to a different location on the kinase, typically at the enzyme's inactive site rather than the ATP-binding site. By binding to these allosteric sites, these inhibitors induce conformational changes in the kinase that alter the shape and structure of the enzyme's active site, ultimately affecting its catalytic activity.180,181 This mechanism of inhibition often involves stabilizing the kinase in an inactive conformation or obstructing ATP binding indirectly, thus preventing phosphorylation of the kinase's substrates without directly competing with ATP.182
One of the key advantages of allosteric inhibitors is their higher specificity. The allosteric site is usually unique to a particular kinase or a subset of kinases, making this form of inhibition less likely to interfere with other kinases that share similar ATP-binding sites. Consequently, allosteric inhibitors can offer more selective therapeutic targeting, minimizing off-target effects that are commonly associated with ATP-competitive inhibitors.183,184 While pyrazolo[1,5-a] pyrimidines are traditionally categorized as ATP-competitive inhibitors, there have been modifications to the core structure of these molecules that have endowed them with the potential to act as allosteric inhibitors. Structural changes in the base template of pyrazolo[1,5-a] pyrimidines have been described in the literature, allowing them to engage allosteric sites and modulate kinase activity in a non-ATP-competitive manner. This dual mechanism of action adds versatility to pyrazolo[1,5-a] pyrimidines, providing opportunities to develop inhibitors with enhanced selectivity and efficacy.185,186
5.2 Pyrazolo[1,5-a] pyrimidines as inhibitors of specific kinases
One of the key advantages of the pyrazolo[1,5-a]pyrimidine structure is its capacity to selectively inhibit specific kinases, particularly those that play critical roles in disease-related pathways such as cancer. Kinases like the epidermal growth factor receptor (EGFR) have been central to advancements in cancer therapy, with EGFR inhibitors showing more precise and effective outcomes compared to traditional chemotherapy. This selectivity reduces off-target effects and enhances the overall efficacy of treatment.165,168CK2 inhibitors, like silmitasertib, have entered clinical trials for various cancers, yet they face challenges with off-target effects. The need for a more selective CK2 inhibitor became evident, prompting Krämer et al. (2020) to explore the pyrazolo[1,5-a]pyrimidine scaffold to develop such inhibitors.195 Their study focused on optimizing this scaffold, ultimately leading to the development of IC20 (31), a highly potent CK2 inhibitor with a dissociation constant (KD) of 12 nM. IC20 (31) exhibited excellent selectivity for CK2, as evidenced by X-ray crystallography revealing a type-I binding mode. This structural characteristic of the pyrazolo[1,5-a]pyrimidine scaffold is crucial as it provides a strong interaction with CK2's ATP-binding site, particularly through its carboxylic acid moiety, which is essential for potency. However, this feature also presented a limitation in cellular contexts, as the compound's IC50 in cellular assays dropped to low micromolar levels. Despite this, IC20 (31) remains a valuable tool compound for studying CK2's role in diseases (Fig. 4).
 | ||
| Fig. 4 Binding affinity of IC20 (31, Panel A) and IC19 (32, Panel B) determined by isothermal titration calorimetry reveals two-digit nanomolar binding affinity of these compounds towards CK2. Minor differences are identifiable in their overall Gibbs free energy, as well as in their enthalpic and entropic contribution to CK2 binding.195 | ||
In their efforts to develop CK2 inhibitors, the researchers made several modifications to the pyrazolo[1,5-a]pyrimidine structure. For example, they introduced phenyl moieties at the 3-position and an ether linker at the 5-position of the pyrimidine ring. These structural modifications were synthesized using a sequence of reactions, including the Mitsunobu reaction for macrocyclization (Fig. 5). The resulting macrocyclic compounds and their acyclic derivatives were evaluated for their kinase inhibition profiles. Among the tested compounds, IC20 (31) and its macrocyclic derivative, IC19 (32), showed the highest selectivity for CK2. Importantly, the carboxylic acid group and a BOC group on the 5-position amine were critical for maintaining the binding affinity and selectivity of these inhibitors.
 | ||
| Fig. 5 Selectivity and cellular potency of acyclic and macrocyclic pyrazolo[1,5-a]pyrimidines. (A) IC19 (32) and IC20 (31) were screened in a KinomeScan by DiscoverX (now belonging to Eurofins) against a panel of 469 kinases (including disease relevant mutants). Red circles identify potential kinases that are affected by these compounds. (B) Cellular potency of pyrazolo [1,5-a]pyrimidines on CK2a/CK2a0 determined by NanoBRET™ in HEK293T cells. The respective IC50 is indicated in the figure legend. (C) Potency of pyrazolo [1,5-a]pyrimidines on CK2a/CK2a0 determined by NanoBRET™ after permeabilizing the cells. The respective IC50 is indicated in the figure legend (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).195 | ||
To assess the selectivity of the synthesized compounds, the study screened them against a panel of 48 kinases, including known CK2 off-targets. This screen highlighted the selectivity of IC20 (31) and IC19 (32), both of which demonstrated strong binding to CK2 while minimizing interactions with other kinases. The study also confirmed the binding mode of these inhibitors through crystallographic studies, which revealed that both IC20 (31) and IC19 (32) bind to the ATP-binding pocket of CK2 in a type-I fashion, forming critical interactions with the kinase back pocket residues (Fig. 4). Specifically, the carboxylic acid group of IC20 (31) forms polar interactions with conserved residues in CK2, including a salt bridge with lysine K68 and a hydrogen bond with glutamate E81. This interaction network, along with additional interactions formed by the BOC group, explains the potency and selectivity of IC20 (31) as a CK2 inhibitor.
The importance of structural features like the carboxylic acid group is further emphasized by the observed loss of activity when this group is replaced with a methyl ester, highlighting the critical role of these functional groups in maintaining CK2 binding. Moreover, the structural analysis revealed that CK2a and CK2a′ possess a unique DWG motif (instead of the more common DFG motif found in other kinases), further contributing to the selectivity of these inhibitors. The BOC group in IC20 (31) also plays a significant role by stabilizing the binding interaction through hydrogen bonding with the hinge region of CK2 (Fig. 5). These findings provide insight into the structure–activity relationships (SAR) of pyrazolo[1,5-a]pyrimidines as CK2 inhibitors, which could guide the future design of more potent and selective inhibitors for cancer therapy.
The optimization of pyrazolo[1,5-a]pyrimidines as specific kinase inhibitors, particularly targeting B-Raf, demonstrates the scaffold's versatility in achieving selectivity and potency. Gopalsamy et al. (2009) focused on the strategic introduction of functional groups at the 2-position of the scaffold, aiming to exploit key interactions within the ATP-binding site of kinases.196 The incorporation of heterocyclic groups like 4-pyridyl was particularly effective, as it enabled a direct hydrogen bond with the hinge region residue Cys531, significantly enhancing kinase inhibition (IC50 of 0.032 μM for B-Raf).
This SAR-driven approach not only improved enzyme potency but also allowed fine-tuning of the selectivity profile across kinases, addressing common issues of cross-reactivity seen with other kinase inhibitors. The structural modifications, specifically targeting the hinge region, underscore the importance of scaffold rigidity and spatial arrangement in kinase binding pockets, offering insights into how pyrazolo[1,5-a]pyrimidines can be optimized for other kinases beyond B-Raf. The design and successful binding of compound 9 highlight the potential for this scaffold to serve as a modular framework in developing selective inhibitors for various kinases critical in oncogenic signalling pathways.
The inhibition of EGFR through specific kinase inhibitors has emerged as an effective treatment strategy, especially for patients with EGFR-dependent cancers. These inhibitors work by selectively blocking the aberrant signaling pathways that cancer cells rely on for their proliferation.201 Pyrazolo[1,5-a]pyrimidines, in particular, have shown promise in targeting EGFR, effectively competing with ATP for binding to the kinase's active site. This is especially important in cases where EGFR is mutated and remains constitutively active, promoting cancer progression.45,202 By occluding the ATP-binding site, pyrazolo[1,5-a]pyrimidine compounds prevent the phosphorylation events that are necessary for EGFR's kinase activity. This inhibition disrupts the signalling cascades that drive cancer cell growth, proliferation, and survival.203,204 As a result, these compounds provide a targeted approach to treat EGFR-dependent cancers, offering a more precise and less toxic alternative to conventional chemotherapy. Their ability to block EGFR in its mutated and active state highlights the potential of pyrazolo[1,5-a]pyrimidines as a significant therapeutic option for cancers characterized by disrupted EGFR signalling.205
Dickerson et al. (2024) explored recent advances in targeted cancer therapy, particularly focusing on gefitinib, the first EGFR tyrosine kinase inhibitor (TKI).206 Gefitinib, part of the anilinoquinazoline class, targets the tyrosine kinase domain of EGFR and demonstrates high binding specificity, with an IC50 of 0.033 μM efficacy in EGFR-dependent tumors. Its 60% bioavailability and selective targeting further substantiate its therapeutic potential. As an ATP-competitive inhibitor, gefitinib obstructs receptor phosphorylation by mimicking ATP structurally, with the nitrogen in the 4-anilinoquinazoline core playing a pivotal role in the interaction. However, the morpholine moiety in gefitinib exhibits suboptimal binding, which may contribute to its limited efficacy over time. The structural interaction of gefitinib with the EGFR tyrosine kinase pocket is visualized in Fig. 6, showing how it occupies the ATP-binding site.206
 | ||
| Fig. 6 Diagram showing gefitinib within the EGFR-TK pocket.206 | ||
This inhibition mechanism underlies its clinical success in treating non-small cell lung cancer (NSCLC). A phase III clinical trial sponsored by AstraZeneca in 2006 demonstrated that gefitinib provided improved outcomes in NSCLC patients, with a progression-free survival (PFS) of 10.8 months compared to 5.4 months in the carboplatin group, and an overall survival (OS) of 30.5 months versus 23.6 months. Moreover, gefitinib exhibited fewer toxicities, including diarrhea and rash, than traditional chemotherapy agents like paclitaxel and carboplatin, though the study duration was only six weeks.207–210
Gefitinib was first approved in 2003 as a targeted therapy for NSCLC, but its effectiveness was soon challenged by resistance, particularly due to the T790M mutation in about 50% of patients within 9–15 months of treatment. This led to the development of second-generation TKIs such as afatinib, which incorporates a covalent bond linking the TKI to the C797 residue of EGFR. Clinical trials have shown that afatinib prolongs treatment duration before disease progression, with a PFS of 13.7 months compared to 11.5 months for gefitinib. Despite this improvement, resistance remains a critical issue, necessitating the design of third-generation TKIs.211–213
The key characteristics, mechanisms of action, and clinical outcomes of gefitinib and afatinib are compared in Table 2 (Dickerson et al., 2024). This comparison highlights the ongoing evolution of targeted therapies to overcome resistance mechanisms, particularly in EGFR-reliant cancers like NSCLC.
| Parameter | Gefitinib (first-generation TKI) | Afatinib (second-generation TKI) |
|---|---|---|
| Classification | Small molecule within the anilinoquinazoline class | Anilinoquinazoline core with additional features |
| Mechanism of action | EGFR inhibitor, targets TK domain | Irreversible EGFR inhibition, targets mutation sites |
| Core interaction | Binds T790 residue via hydrogen bonding with Met793 | Covalent bond with C797; hydrogen bond with Met793 |
| Binding potency | IC50 = 0.033 μM against EGFR | Enhanced potency, multiple interactions with EGFR |
| Resistance | T790M mutation impairs binding | Partially overcomes T790M; other mutations still pose challenges |
| Clinical efficacy | PFS = 10.8 months, OS = 30.5 months | Longer treatment duration compared to gefitinib, PFS = 13.7 months |
| Safety profile | Common side effects: diarrhea, rash, elevated ALT | Higher risk of toxicity than gefitinib, but improved efficacy |
| Trial comparisons | Compared with carboplatin (traditional chemotherapy) | Compared with erlotinib (first-gen TKI) and cisplatin (chemotherapy) |
| Notable findings | Reduced toxicity compared to carboplatin | More effective against T790M mutation but with increased off-target toxicity |
| FDA approval | 2003 (marketed as Iressa®) | 2013 (marketed as Gilotrif®) |
Dickerson et al. (2024) emphasize that while gefitinib and other TKIs have significantly advanced targeted cancer therapies, ongoing efforts are essential to address treatment resistance and enhance overall treatment efficacy.206
5.2.2.1 Role in non-small cell lung cancer (NSCLC) treatment. Non-small cell lung carcinoma (NSCLC) is the most common type of lung cancer and is frequently associated with mutations in the epidermal growth factor receptor (EGFR) gene. These mutations cause an upregulation of the EGFR signaling pathway, promoting uncontrolled cell growth and division.207–209 The discovery of EGFR mutations in a subset of NSCLC patients revolutionized treatment by shifting the focus to EGFR-targeted therapies, which have proven superior to traditional chemotherapy. Current treatments for NSCLC often involve EGFR inhibitors, including pyrazolo[1,5-a] pyrimidine-based compounds, which have shown significant efficacy in these patients. Individuals with EGFR mutations tend to respond well to these inhibitors, as their tumors shrink, and they experience longer progression-free survival.210,211
One of the key advantages of EGFR inhibitors is their selectivity for cancer cells with EGFR mutations, resulting in minimal effects on healthy cells and fewer side effects compared to chemotherapy. This precision makes EGFR inhibitors a recommended treatment option for NSCLC patients harbouring EGFR mutations.212,213
Pyrazolo[1,5-a] pyrimidines have been particularly effective in targeting both wild-type EGFR and its mutated forms. These compounds inhibit not only wild-type EGFR but also many EGFR mutations that have developed resistance to first-generation EGFR inhibitors like erlotinib and gefitinib. One notable mutation, T790M, frequently arises in patients treated with first-generation EGFR inhibitors, rendering these drugs ineffective. However, pyrazolo[1,5-a] pyrimidines have demonstrated activity against this resistant EGFR form, offering renewed hope for patients with drug-resistant NSCLC.214–216
In addition, Ho et al. (2022) explored the significance of baseline plasma EGFR mutation status in guiding NSCLC treatment.217 Their study highlighted the value of plasma EGFR mutation detection in managing NSCLC, emphasizing the importance of combining both tissue and plasma analyses for a more accurate assessment. The research analyzed EGFR mutation status in paired tumor and plasma samples from 137 advanced NSCLC patients before and after EGFR-TKI treatment. Plasma EGFR-activating mutations were detected in 65% (89/137) of patients at baseline, which aligns with previous studies, although variability in detection rates across studies is often due to differences in assay sensitivity and patient disease stages.
By providing insights into plasma-based EGFR mutation detection, the study underscores the growing role of non-invasive testing in personalizing NSCLC treatments. The ability of pyrazolo[1,5-a] pyrimidine-based inhibitors to target both EGFR mutations and drug-resistant variants offers a promising avenue for enhancing treatment outcomes in this challenging cancer type. Table 3 show the detection rate of baseline EGFR-activating mutations in plasma samples217
| Mutation type | Number of patients detected | Percentage detected (%) |
|---|---|---|
| EGFR-activating mutation | 89/137 | 65.0 |
| EGFR T790M mutation | 28/137 | 20.4 |
Ho et al. (2022), found that patients with detectable EGFR-activating mutations in plasma had a shorter progression-free survival (PFS) and overall survival (OS), suggesting that the absence of these mutations might indicate a lower systemic tumor burden.217 This reinforces the prognostic value of baseline plasma EGFR mutation testing for guiding therapy. A key result was the detection of EGFR T790M mutation in 20.4% (28/137) of EGFR-mutated, treatment-naïve tumors, a finding made possible by the use of the ultra-sensitive ddPCR platform. The study noted that EGFR T790M was often present as a minor subpopulation and that its presence at baseline was not associated with acquired resistance at disease progression. The findings highlight the challenges of relying solely on tissue biopsy for T790M detection, as the intratumor heterogeneity can contribute to sampling bias, leading to inconsistent detection rates. The concordance rate for T790M mutation between paired tissue and plasma samples at disease progression was only 71.4%, indicating that neither tissue nor plasma testing alone is sufficient for guiding subsequent third-generation EGFR-TKI treatment. This finding emphasizes the need for combined or repeated testing to ensure appropriate patient selection for osimertinib therapy.
Ho et al. (2022), also observed that pre-treatment EGFR T790M was significantly associated with brain metastasis in patients treated with first- or second-generation EGFR-TKIs.217 These results suggest a potential benefit of targeting pre-existing T790M clones to prevent CNS progression in NSCLC. However, the study found no significant difference in overall survival between patients with and without pre-treatment T790M mutations, highlighting variability in prognostic outcomes depending on assay methodologies and treatment regimens used. The study underscores the importance of liquid biopsy for accurate assessment of EGFR mutations in NSCLC, offering a promising approach for personalized treatment. The results support the use of ddPCR for EGFR mutation detection as a complementary tool to tissue biopsy for the precision management of NSCLC. Further research with larger cohorts and standardized methods is necessary to validate these findings and improve clinical practice. Accurately identifying tumors that harbor EGFR-activating as well as T790M resistance mutations is critical for the precision management of NSCLC, and this study provides valuable insights for optimizing treatment strategies.
5.2.2.2 Studies of pyrazolo[1,5-a] pyrimidines as EGFR inhibitors. Pyrazolo[1,5-a]pyrimidines have garnered significant attention in recent years due to their potential as anticancer agents, specifically as inhibitors of the epidermal growth factor receptor (EGFR). EGFR is a well-known target in cancer therapy, as its overexpression or mutation is associated with the progression of several cancers, including lung and breast cancers. Pyrazolo[1,5-a]pyrimidine derivatives, with their unique N-heterocyclic structure, exhibit strong kinase inhibition, making them promising candidates for targeting EGFR-driven tumor growth.218–220
Recent studies have focused on synthesizing and evaluating various pyrazolo[1,5-a]pyrimidine compounds for their EGFR inhibitory activity. These compounds typically function by binding to the ATP-binding site of the EGFR kinase, thereby preventing autophosphorylation and downstream signalling required for tumour cell proliferation. Molecular docking studies have confirmed the favourable interactions between these derivatives and EGFR's active site, reinforcing their potential as dual EGFR/HER2 inhibitors, which may provide broader anticancer efficacy. Recent studies highlight the potential of pyrazolo[1,5-a]pyrimidine derivatives as potent EGFR inhibitors, focusing on their ability to act as anticancer agents through kinase inhibition and apoptotic induction.220–222
Sivaiah et al. (2023) explored the synthesis of pyrazolo[1,5-a]pyrimidine derivatives (6a–o) for their potential anticancer activities, targeting EGFR and HER2 pathways.223 Structural confirmation was achieved through elemental analysis and spectroscopy, with several compounds showing significant cytotoxicity against cancer cell lines, particularly HepG2, MCF-7, and HCT116. Notably, compounds 6a and 6b exhibited potent EGFR and HER2 inhibition, with IC50 values of 0.163 μM (EGFR) and 0.116 μM (HER2) for 6a, and 0.126 μM (EGFR) and 0.083 μM (HER2) for 6b. Molecular docking further demonstrated these compounds' ability to strongly bind to EGFR and HER2, making them promising candidates for dual kinase inhibition in cancer therapy.
Othman et al. (2021) synthesized pyrazolo[1,5-a]pyrimidine derivatives with C5 substitutions and evaluated them for antiproliferative activity against cancer cell lines such as A549, MDA-MB-231, and DU-145.224 Compound 6h, in particular, showed significant efficacy against MDA-MB-231 with an IC50 of 2.6 μM. Mechanistic studies revealed these compounds' ability to arrest the cell cycle at the subG1 phase and inhibit the EGFR/STAT3 signaling pathway, critical for cell survival and apoptosis. The observed upregulation of apoptotic proteins (p53, p21, Bax) and downregulation of anti-apoptotic proteins (Bcl-2, procaspase-9) confirmed the compounds' apoptotic activity.
Byeon et al. (2022) focused on overcoming resistance in EGFR-mutant NSCLC, where mutations such as L858R/T790M and L858R/T790M/C797S lead to resistance against third-generation TKIs like osimertinib.225 Their study introduced compound A, a fourth-generation allosteric inhibitor that selectively targets L858R activating mutations while sparing wild-type EGFR. Compound A demonstrated nanomolar potency and showed significant antitumor activity in mouse models resistant to osimertinib. Its selective inhibition of mutated EGFR, including C797S, positions compound A as a promising candidate for future clinical development in treating EGFR-mutant NSCLC. In general, pyrazolo[1,5-a]pyrimidine derivatives show great promise as potent EGFR inhibitors, with studies emphasizing their kinase inhibition, apoptotic activity, and potential to overcome drug resistance in cancer therapy.
Pyrazolo[1,5-a]pyrimidines have emerged as promising scaffolds in the design of potent inhibitors for the MAPK pathway, particularly targeting B-Raf and MEK kinases. These heterocyclic compounds, characterized by their fused ring structure, offer unique chemical properties that enable selective inhibition of kinase activity. Structurally versatile, pyrazolo[1,5-a]pyrimidines allow for modification at various positions, facilitating the design of molecules with high specificity and potency against mutant forms of B-Raf and MEK.23,25,165
Recent studies have demonstrated that pyrazolo[1,5-a]pyrimidine derivatives can effectively bind to the ATP-binding site of these kinases, preventing their phosphorylation activity and halting downstream signaling. This blockade of the MAPK pathway inhibits cancer cell proliferation and induces apoptosis, making pyrazolo[1,5-a]pyrimidines valuable candidates for therapeutic development. Moreover, the ability of these compounds to target multiple nodes within the pathway, such as dual inhibition of B-Raf and MEK, enhances their therapeutic potential by reducing the likelihood of resistance development—a common challenge in kinase-targeted therapies MEK.23,25,165
The study by Gopalsamy et al. (2009) focuses on the development of pyrazolo[1,5-a]pyrimidines as potent B-Raf kinase inhibitors, specifically targeting the V600E mutation implicated in various cancers.196 This mutation induces constitutive activation of the B-Raf isoform, promoting uncontrolled cell proliferation through the ERK signaling pathway. The study highlights the potential of B-Raf inhibitors in treating conditions like melanoma and colorectal cancer. Through high-throughput screening (HTS), Gopalsamy et al. identified pyrazolo[1,5-a]pyrimidine-3-carboxylate as a lead compound, which showed promising activity in B-Raf inhibition.
The optimization of the lead compound was driven by structure–activity relationship (SAR) studies, which focused on modifying the ester moiety and substituents at the 2-position of the pyrazolopyrimidine scaffold. This approach aimed to enhance binding interactions within the ATP pocket of B-Raf. Initially, the ester group was identified as metabolically unstable, leading the researchers to explore amide replacements. These modifications resulted in the discovery of compounds with increased stability and potency, including those bearing water-solubilizing functionalities. Of particular note, the replacement of the ester with amides led to enhanced microsomal stability and stronger inhibitory effects.
To further optimize the interaction between the pyrazolo[1,5-a]pyrimidine scaffold and the B-Raf protein, Gopalsamy et al. focused on incorporating polar groups in the 2-position to form hydrogen bonds with the kinase's hinge region, specifically targeting Cys531. Their SAR analysis revealed that the introduction of heterocyclic groups, such as the 4-pyridyl substituent, significantly improved enzyme inhibition. The predicted binding model showed that the nitrogen atom of the 4-pyridyl group was well-positioned to form a hydrogen bond with Cys531, a key interaction that contributed to the enhanced potency of these analogs. In contrast, other substituted phenyl rings at this position led to a substantial loss of activity.
In addition to B-Raf inhibition, the study also explored the ability of these pyrazolo[1,5-a]pyrimidine derivatives to inhibit the proliferation of tumor cell lines. The increased potency of the 4-pyridyl analog (compound 9) in enzyme inhibition translated to improved cellular growth inhibition, making it a strong candidate for further development as a cancer therapeutic. Further profiling of compound 9 against various tumor cell lines confirmed its efficacy, highlighting the potential of this compound as a lead in the design of targeted cancer therapies.
The study also noted the importance of flexibility in the linker region connecting the core scaffold to the substituents. Urea analogs, in place of amides, maintained key interactions with Glu500 and Asp593, essential for binding. Additionally, polar groups introduced near the pyridyl moiety were well tolerated, as these groups were ideally positioned to reach solvent-exposed regions of the binding pocket, enhancing the drug's solubility and efficacy.
In summary, Gopalsamy et al. provide a detailed exploration of pyrazolo[1,5-a]pyrimidines as B-Raf inhibitors, emphasizing the significance of strategic modifications to the scaffold in improving enzyme inhibition and cellular potency.196 Their work underscores the potential of these compounds in cancer therapy, particularly in targeting the RAS-RAF-MEK pathway. The SAR-driven optimization process demonstrates the value of structure-based drug design in developing highly potent and selective kinase inhibitors.
Ren et al. (2021) reported the discovery of a novel series of ATP-competitive B-Raf inhibitors based on the pyrazolo[1,5-a]pyrimidine scaffold, designed to selectively target the B-RafV600E mutation.93 This mutation, commonly found in melanoma, leads to constitutive activation of the MAPK pathway, driving cancer progression. The lead compound, 17, demonstrated potent and selective inhibition of B-RafV600E, excellent cellular activity, and favourable physicochemical properties, making it a promising candidate for further development.
The study began with earlier lead compounds like 1, which exhibited excellent B-Raf inhibition but had solubility issues due to dimer formation in the crystal lattice. A structural redesign, aimed at disrupting this dimerization, led to compound 17, which improved solubility by lowering the melting point and reducing crystal lattice energy, while maintaining strong inhibitory activity. By introducing alkoxy and heteroaryl substituents, the authors optimized binding at the ATP cleft, leveraging hydrophobic contacts with residues like Trp531 and Ser535/536. The key outcome was the identification of compound 17, a methoxypyrazolopyrimidine, which exhibited a 68 nM IC50 for pERK activity—seven times more potent than the unsubstituted lead. This compound also showed improved aqueous solubility and favorable pharmacokinetic properties, making it an attractive candidate for further preclinical evaluation.
Kolathur et al. (2024) offer a comprehensive analysis of B-Raf mutations, focusing specifically on the V600E mutation and its significant role in melanoma development.230 The study underscores that this mutation is a primary driver of skin cancer, with more than 50% of melanoma cases exhibiting the B-Raf mutation, and V600E being the most common variant. This mutation results in the persistent activation of the B-Raf protein and its related signaling pathways in the MAPK cascade, facilitating unchecked cell proliferation and tumor formation.
Although B-Raf inhibitors such as vemurafenib and dabrafenib have been approved to target these mutations, the study highlights the challenge of resistance. A notable percentage of patients with B-Raf V600E mutations (15–20%) exhibit inadequate responses to these treatments. This resistance underscores the necessity for alternative therapeutic strategies, as solely targeting the B-Raf and MEK pathways may not yield sufficient efficacy for all melanoma patients.
Table 4 of the study provides a summary of the major findings, detailing the mechanisms responsible for resistance to B-Raf-targeted therapies and highlighting the complexities involved in melanoma treatment. The insights from this study are vital for understanding why additional treatments and combination therapies are essential to overcome resistance and enhance outcomes for melanoma patients. The findings reveal a complex interplay between mutations and therapeutic resistance in melanoma, highlighting the need for further investigation into these factors to improve survival rates. For instance, while primary driver mutations can be targeted by existing drugs, additional mutations within the same pathway may reactivate downstream signaling, enabling cancer progression. Moreover, previous studies on MEK inhibitors, such as trametinib, in combination with B-Raf inhibitors have demonstrated that MEK inhibitors tend to be more effective and better tolerated than monotherapy. Nevertheless, the authors caution that acquired resistance continues to challenge treatment efficacy, necessitating further exploration of novel approaches and combination therapies.
| Major findings | Description |
|---|---|
| Mutation prevalence | Over 50% of melanoma cases harbor B-Raf mutations, primarily V600E, which is implicated in tumor progression |
| Drug resistance | Resistance mechanisms include reactivation of the MAPK pathway due to mutations in MEK1/2 and dimerization of B-Raf, leading to treatment failure |
| B-Raf inhibitor efficacy | Vemurafenib and dabrafenib exhibit limited efficacy in certain patients; combination therapies with MEK inhibitors yield improved outcomes |
| Signaling pathway adaptation | Tumor cells adapt through alterations in signaling pathways, notably the PI3K/AKT pathway, in response to B-Raf inhibition, complicating treatment strategies |
| Dimerization mechanisms | Dimerization of mutated BRAF proteins, including splice variants, results in bypassing inhibition, emphasizing the need for dual-targeted therapies |
In summary, Kolathur et al. (2024) synthesize current knowledge regarding genetics, B-Raf inhibitors, and resistance mechanisms in melanoma, offering valuable insights and recommendations for the next generation of therapeutic strategies that could potentially improve patient prognosis in this intricate disease.
6 Biological evaluation and pharmacological properties
Pyrazolo[1,5-a]pyrimidines have emerged as a promising class of compounds with significant biological and pharmacological potential, particularly as protein kinase inhibitors in cancer treatment. These heterocyclic compounds are structurally characterized by a fused pyrazole and pyrimidine ring system, which allows for diverse chemical modifications. This structural versatility enables pyrazolo[1,5-a]pyrimidines to effectively target various protein kinases that are disrupted in cancer cells. Protein kinases are enzymes that play critical roles in controlling cellular processes such as proliferation, differentiation, and apoptosis, which are often altered in cancerous cells. The aberrant activation or overexpression of specific kinases drives tumor growth and progression, making them important therapeutic targets in oncology.The pyrazolo[1,5-a]pyrimidine scaffold has shown great affinity for the ATP-binding pockets of protein kinases. Many of these compounds act by competing with ATP, thereby inhibiting the kinase's ability to phosphorylate downstream substrates. This inhibition disrupts critical signalling pathways, leading to the suppression of tumor cell proliferation, induction of apoptosis, and in some cases, inhibition of angiogenesis. Several members of this compound class have been investigated for their ability to inhibit kinases such as the epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), Bcr–Abl, and cyclin-dependent kinases (CDKs), all of which are implicated in different types of cancer.18,165,166,231
EGFR is a receptor tyrosine kinase that is often mutated or overexpressed in cancers such as non-small cell lung cancer, glioblastoma, and colorectal cancer. The activation of EGFR triggers several downstream pathways that promote cancer cell survival and proliferation, including the RAS-RAF-MEK-ERK and PI3K-AKT-mTOR pathways. Pyrazolo[1,5-a]pyrimidines have demonstrated potent inhibitory activity against both wild-type and mutant EGFR, making them promising agents in targeting EGFR-driven cancers. In particular, these compounds have shown efficacy against tumors harboring the T790M mutation, which confers resistance to first-generation EGFR inhibitors. This ability to overcome drug resistance is a key advantage of pyrazolo[1,5-a]pyrimidines in cancer therapy.230,232
Another important target for pyrazolo[1,5-a]pyrimidine-based inhibitors is VEGFR, a kinase involved in tumor angiogenesis. Angiogenesis is the process by which tumors form new blood vessels to supply themselves with oxygen and nutrients, facilitating their growth and metastatic spread. By inhibiting VEGFR, pyrazolo[1,5-a]pyrimidines can disrupt this process, effectively “starving” the tumor and slowing its progression. This anti-angiogenic effect is particularly important in cancers that rely heavily on vascularization, such as renal cell carcinoma and certain types of breast cancer.233–237
In chronic myeloid leukemia (CML), the Bcr–Abl fusion protein, produced by the Philadelphia chromosome translocation, acts as a constitutively active tyrosine kinase that drives uncontrolled cell proliferation. Pyrazolo[1,5-a]pyrimidines have been developed to inhibit Bcr–Abl activity, providing a therapeutic option for patients with CML, including those who have developed resistance to first-line treatments like imatinib. These compounds not only target the wild-type form of Bcr–Abl but also its drug-resistant mutants, offering a broader spectrum of activity and reducing the likelihood of relapse.238–241
In addition to receptor tyrosine kinases, pyrazolo[1,5-a]pyrimidines have been shown to inhibit serine/threonine kinases such as CDKs. CDKs are key regulators of the cell cycle, and their disruption is a hallmark of many cancers. By inhibiting CDKs, pyrazolo[1,5-a]pyrimidines can induce cell cycle arrest, particularly at the G1/S transition, thereby preventing cancer cells from proliferating. This makes CDK inhibitors highly effective in cancers characterized by rapid cell division, such as hormone receptor-positive breast cancer.44,242,243
The pharmacological properties of pyrazolo[1,5-a]pyrimidines extend beyond their kinase inhibitory activity. These compounds have been shown to possess favorable pharmacokinetic profiles, including good bioavailability, metabolic stability, and the ability to cross biological barriers such as the blood–brain barrier. These properties are crucial for achieving therapeutic concentrations at the tumor site while minimizing off-target effects. Furthermore, pyrazolo[1,5-a]pyrimidines have demonstrated a low toxicity profile in preclinical studies, making them suitable candidates for further development as anticancer agents.244
One of the major challenges in cancer therapy is the development of drug resistance, often caused by secondary mutations in the target kinase or activation of alternative signaling pathways. Pyrazolo[1,5-a]pyrimidines offer a potential solution to this problem due to their ability to target multiple kinases simultaneously or to inhibit both wild-type and mutant forms of a kinase. This multi-targeted approach not only reduces the likelihood of resistance but also enhances the overall efficacy of treatment by simultaneously disrupting several oncogenic pathways.245,246 In addition, the use of pyrazolo[1,5-a]pyrimidines in combination with other cancer therapies, such as chemotherapy, radiation, or immunotherapy, has shown synergistic effects. For example, combining kinase inhibitors with immune checkpoint inhibitors can enhance the immune system's ability to recognize and destroy cancer cells, while inhibiting kinases involved in DNA repair can sensitize tumors to DNA-damaging agents.247
In a nutshell, pyrazolo[1,5-a]pyrimidines represent a highly versatile and potent class of protein kinase inhibitors with significant potential in cancer treatment. Their ability to target key oncogenic kinases, overcome drug resistance, and synergize with existing therapies makes them promising candidates for the development of next-generation cancer therapeutics. As research into the structure–activity relationships and pharmacological properties of pyrazolo[1,5-a]pyrimidines continue, it is likely that more effective and personalized treatment options will emerge, offering new hope for patients with cancer.
6.1 In vitro studies
For example, cell line assays with non-small cell lung cancer (NSCLC) models, particularly those with mutations in the epidermal growth factor receptor (EGFR), have demonstrated that pyrazolo[1,5-a]pyrimidines can effectively inhibit tumor cell proliferation by targeting EGFR's kinase activity. In recent studies exploring the efficacy of pyrazolo[1,5-a]pyrimidines in non-small cell lung cancer (NSCLC) models, particularly those harboring mutations in the epidermal growth factor receptor (EGFR), a significant focus has been on their ability to inhibit tumor cell proliferation by targeting kinase activity.201 One of the prominent targets in this area of research is the RET (REarranged during Transfection) kinase, which has emerged as an oncogenic driver in multiple cancer types, including lung adenocarcinoma. Mathison et al.165 (2020) investigated the potential of selective inhibitors designed to target RET kinase gain-of-function aberrations, leading to the identification of the pyrazolo[1,5-a]pyrimidine compound WF-47-JS03 (1), which exhibited remarkable selectivity against RET while sparing the KDR (Kinase insert Domain Receptor) kinase.
In cell line assays involving Ba/F3 cells transfected with various oncogenic kinase fusions, including the KIF5B-RET fusion, compound 1 showed potent RET inhibition. This was particularly relevant to NSCLC models, as targeting the RET fusion is a crucial therapeutic strategy in tumors driven by such mutations. These findings underscore the potential of pyrazolo[1,5-a]pyrimidines to serve as effective inhibitors of kinase-driven tumorigenesis in cancers like NSCLC.
The study also included a structure–activity relationship (SAR) investigation that led to significant improvements in both the potency and selectivity of the compound. One key modification was at the C5 position of the pyrazolo[1,5-a]pyrimidine core, where changing the ether linkage to a nitrogen-based linkage enhanced RET selectivity. Fig. 7 illustrated the dose-dependent tumor regression observed in a KIF5B-RET-driven xenograft model when mice were treated with 1 at 10, 30, and 60 mg kg−1 once daily. The 10 mg per kg dose was well-tolerated and led to substantial tumor regression without causing the severe lung toxicity observed at higher doses. This highlights the delicate balance between efficacy and toxicity when using pyrazolo[1,5-a]pyrimidine-based RET inhibitors, which is consistent with the narrow therapeutic window often associated with these scaffolds.109
 | ||
| Fig. 7 RET inhibitor compound 1 (WF-47-JS03) significantly inhibits tumor growth in RIE KIF5B-RET xenograft mice and is well tolerated at 1, 3, and 10 mg kg−1 in the 10 days study.165 | ||
Another aspect of Mathison et al.'s (2020) work focused on the in vivo pharmacokinetics and pharmacodynamics of compound 1.165 The study demonstrated that the compound had good oral bioavailability and significant brain penetration, a desirable trait for treating cancers with central nervous system involvement. The ability of compound 1 to inhibit tumor growth effectively, particularly at a dose of 10 mg kg−1, was corroborated by Fig. 8, which showcases the long-term tumor abatement following a 21 days oral dosing regimen in a disease-relevant LC-2 tumor tissue model. The sustained efficacy of the treatment even after cessation of dosing suggests a potential for long-lasting therapeutic benefits in RET-driven cancers.
 | ||
| Fig. 8 Treatment of LC-2/ad tumor-bearing mice with 1 (WF-47-JS03) for 21 days is well-tolerated and leads to significant regression at 10 mg kg−1.165 | ||
The findings from Mathison et al. (2020) offer compelling evidence that pyrazolo[1,5-a]pyrimidines, specifically compound 1, can serve as effective RET kinase inhibitors, providing a promising therapeutic strategy for NSCLC and other RET-driven malignancies.165 The study's results demonstrated the compound's ability to induce tumor regression while also highlighting the challenges associated with toxicity at higher doses. The work sets the stage for further refinement of these compounds to optimize their therapeutic potential in cancer treatment.
In breast cancer cell lines overexpressing human epidermal growth factor receptor 2 (HER2), these compounds have shown promising results in disrupting HER2-mediated signaling pathways, leading to reduced cancer cell growth and increased apoptosis. In leukemia models, particularly chronic myeloid leukemia (CML) cells expressing the Bcr–Abl fusion protein, pyrazolo[1,5-a]pyrimidines have exhibited the ability to inhibit Bcr–Abl activity and induce cell death even in drug-resistant variants, highlighting their therapeutic potential.238
In addition to their effects on cancer cell lines, the kinase selectivity profiling of pyrazolo[1,5-a]pyrimidines provided essential data regarding their specificity for different kinase targets. Protein kinases are part of large and diverse families, with each kinase playing a role in various cellular processes. Therefore, achieving selectivity is critical for minimizing off-target effects and reducing potential toxicity in normal cells. Advances in kinase selectivity profiling allow researchers to screen pyrazolo[1,5-a]pyrimidines against a broad panel of kinases to identify their primary targets and off-target interactions.252–255
High-throughput screening techniques, such as kinase assays using recombinant enzymes, have become invaluable in this profiling. By measuring the inhibition of kinase activity across multiple targets, researchers can identify the most potent kinases for which pyrazolo[1,5-a]pyrimidines have high affinity. These assays typically measure the compound's ability to compete with ATP for binding at the kinase's active site, providing a quantitative measure of inhibition (IC50 values) for each target. Advances in selectivity profiling have shown that certain pyrazolo[1,5-a]pyrimidines exhibit a high degree of specificity for kinases such as EGFR, VEGFR, and Bcr–Abl, making them highly attractive for targeted therapies in cancers driven by these kinases.168
Kurz et al. (2022) contributed to the growing body of research surrounding serine/threonine kinase 17A (DRAK1), part of the death-associated protein kinase (DAPK) family. DRAK1 is relatively underexplored, classified within the “dark kinome”, with limited knowledge of its cellular functions and role in pathophysiological processes.168
In the study, the researchers aimed to optimize a pyrazolo[1,5-a]pyrimidine-based macrocyclic scaffold to develop potent and selective DRAK1 inhibitors. High-throughput screening techniques, such as kinase assays using recombinant enzymes, were pivotal in profiling potential inhibitors. These assays allowed the team to measure the inhibition of kinase activity across multiple targets, quantifying the compound's ability to compete with ATP for binding at the kinase's active site. Through this method, the team could determine the IC50 values, which provide a quantitative measure of inhibition for each target.
Advances in selectivity profiling played a significant role in this study, highlighting the pyrazolo[1,5-a]pyrimidines' specificity for kinases, particularly the engineered compound CK156 (34). This compound emerged from structure-guided optimization, exhibiting high in vitro potency with a dissociation constant (KD) of 21 nM, reflecting its strong binding affinity for DRAK1. Kinome-wide screens confirmed its selectivity, which is a key element in drug discovery, as highly selective inhibitors reduce off-target effects, thereby improving the potential for targeted therapies in cancers like glioblastoma.
The study further revealed the structural basis for CK156 (34)'s activity, as crystallographic analyses confirmed that it functions as a type I inhibitor, binding to the active conformation of the kinase. This mode of action differentiates it from type II inhibitors, which target inactive kinase conformations, and could influence its therapeutic applications.
Despite the promising in vitro data, the translation to cellular models presented a challenge. Contrary to genetic knockdown studies of DRAK1, CK156 (34) only inhibited the growth of glioma cells in 2D and 3D cultures at low micromolar concentrations. This discrepancy suggests that while CK156 (34) is a potent DRAK1 inhibitor in biochemical assays, its cellular efficacy may be limited by factors such as drug uptake, stability, or potential compensatory mechanisms in glioma cells. These findings underline the complexity of translating kinase inhibition data from in vitro settings to more physiologically relevant models and highlight the importance of continued optimization and testing in drug development pipelines. This study exemplifies the utility of high-throughput kinase screening techniques in identifying and optimizing selective inhibitors within challenging targets like DRAK1. It also underscores the importance of combining biochemical data with cellular models to fully understand the therapeutic potential and limitations of these compounds in cancer treatment.
Further insights into selectivity are gained through cell-based kinase assays, where the effect of pyrazolo[1,5-a]pyrimidines on kinase signaling pathways is assessed in a more complex biological context. In these assays, the phosphorylation status of downstream substrates is measured to determine whether the compound effectively blocks kinase activity within living cells. This approach is particularly useful for studying kinases involved in signaling networks, such as the PI3K-AKT-mTOR and MAPK pathways, which are often activated in cancer cells. By examining the effects of pyrazolo[1,5-a]pyrimidines on these pathways, researchers can better understand their mode of action and potential for therapeutic intervention.
The study by Xu et al. (2015) presents an extensive exploration into the development of potent and selective Pim-1 kinase inhibitors using pyrazolo[1,5-a]pyrimidine-based compounds.256 These compounds were identified through a combination of virtual screening and lead optimization strategies aimed at improving both the potency and selectivity of the inhibitors for therapeutic applications, especially in the context of cancer. Pim-1 kinase, along with its family members Pim-2 and Pim-3, plays a critical role in various oncogenic processes, such as cancer cell survival, cytokine signaling, and tumorigenesis, making it an attractive target for therapeutic intervention. Notably, Pim-1 does not require upstream phosphorylation for activation, making its kinase activity primarily regulated by transcriptional and translational control, which is relevant for cancer progression and cell proliferation.
Xu et al. began their investigation by identifying an initial hit compound that demonstrated moderate inhibition of Pim-1 kinase, with an IC50 value of 52 μM.256 This compound, part of the pyrazolo[1,5-a]pyrimidine chemotype, provided a scaffold for further chemical modifications aimed at enhancing its potency while avoiding the undesirable effects observed with earlier generations of Pim-1 inhibitors, such as SGI-1776. SGI-1776, while potent, was known to exhibit significant off-target effects, including hERG (human Ether-à-go-go-Related Gene) channel inhibition, which can lead to cardiotoxicity.
To improve selectivity and minimize side effects, Xu et al. focused on combining elements of SGI-1776 with the pyrazolo[1,5-a]pyrimidine core structure. This led to the generation of compound 1, which exhibited an IC50 value of 45 nM against Pim-1 kinase, representing a significant improvement over the initial hit. Further structural modifications were systematically made to optimize the physicochemical properties of the inhibitors, ensuring both potency and selectivity.
The study utilized both biochemical kinase assays and cell-based assays to assess the efficacy of the compounds. In the biochemical assays, the pyrazolo[1,5-a]pyrimidine compounds strongly inhibited both Pim-1 and Flt-3 kinases, the latter of which is also implicated in oncogenic processes, particularly in hematologic cancers. These results are consistent with Table 5, which presents the hERG IC50 and Flt-3 IC50 values for selected compounds. For instance, compound 1 demonstrated an hERG IC50 value of 1.9 μM, indicating low cardiotoxic risk, while the Flt-3 inhibition data remained undetermined (ND) for this compound, suggesting a potential focus on Pim-1 selectivity in initial assays.
| Comp. no. | hERG IC50, μM | Flt-3 IC50, nM |
|---|---|---|
| 1 | 1.9 | ND |
| SGI-1776 | <1.0 | ND |
| 9 | >30 | 157 |
| 9a | >30 | 53 |
| 11a | >30 | 271 |
| 11b | >30 | 125 |
In cell-based kinase assays, these pyrazolo[1,5-a]pyrimidine compounds effectively inhibited the phosphorylation of downstream targets, such as the BAD protein, which is involved in apoptotic signalling. This inhibition was shown to suppress 2D colony formation in a clonogenic survival assay, demonstrating that the cellular activity was mediated through Pim-1 inhibition. This ability to block Pim-1-mediated signalling in living cells adds a layer of complexity to the compound's mode of action, revealing its potential for therapeutic intervention in cancers driven by Pim-1.
Further insights into the selectivity of these inhibitors were provided by a kinome-wide panel, which showed that the lead compounds were highly selective for Pim-1, with little off-target activity across 119 oncogenic kinases. This selective profile, combined with the improved safety over previous inhibitors like SGI-1776, makes these pyrazolo[1,5-a]pyrimidine compounds attractive candidates for future drug development. Importantly, the absence of significant hERG inhibition at 30 μM concentrations suggests a favourable therapeutic window, reducing the risk of cardiotoxicity commonly associated with kinase inhibitors.
In summary, the study by Xu et al. demonstrates the successful lead optimization of pyrazolo[1,5-a]pyrimidine compounds as selective Pim-1 kinase inhibitors with potential applications in cancer therapy. By leveraging both biochemical and cell-based assays, the researchers were able to gain valuable insights into the compounds' selectivity and mechanism of action, particularly in blocking oncogenic signalling pathways. The findings suggest that these inhibitors, with their improved potency and safety profiles, hold promise for future development as targeted cancer therapies.
The 2012 study by Hanan et al. investigates the development of selective Jak2 inhibitors as potential treatments for chronic myeloproliferative neoplasms, focusing on the optimization of pyrazolo[1,5-a]pyrimidine compounds.257 Fig. 9 in the study presents the co-crystal structure of compound 8 within the Jak2 kinase domain. This structural analysis reveals that the N1-nitrogen of the pyrazolopyrimidine core forms a hydrogen bond with the backbone NH of Leu932. Additionally, a potential weak nonclassical hydrogen bond is noted between the C–H \moiety at C-7 of the pyrazolopyrimidine core and the backbone carbonyl of Glu930, with a distance of 3.4 Å. Both the amide carbonyl and the pyrazole N2-nitrogen of compound 8 are observed to form hydrogen bonds with water molecules, and the terminal phenyl ring occupies the hydrophobic sugar region of the ATP binding site. Fig. 10 illustrates the in vivo efficacy of compound 7j in a SCID mouse SET2 xenograft model, which is dependent on Jak2 for growth. At an oral dose of 100 mg kg−1, compound 7j demonstrates a time-dependent knockdown of pSTAT5, a downstream target of Jak2, indicating effective inhibition of Jak2 activity in this model.
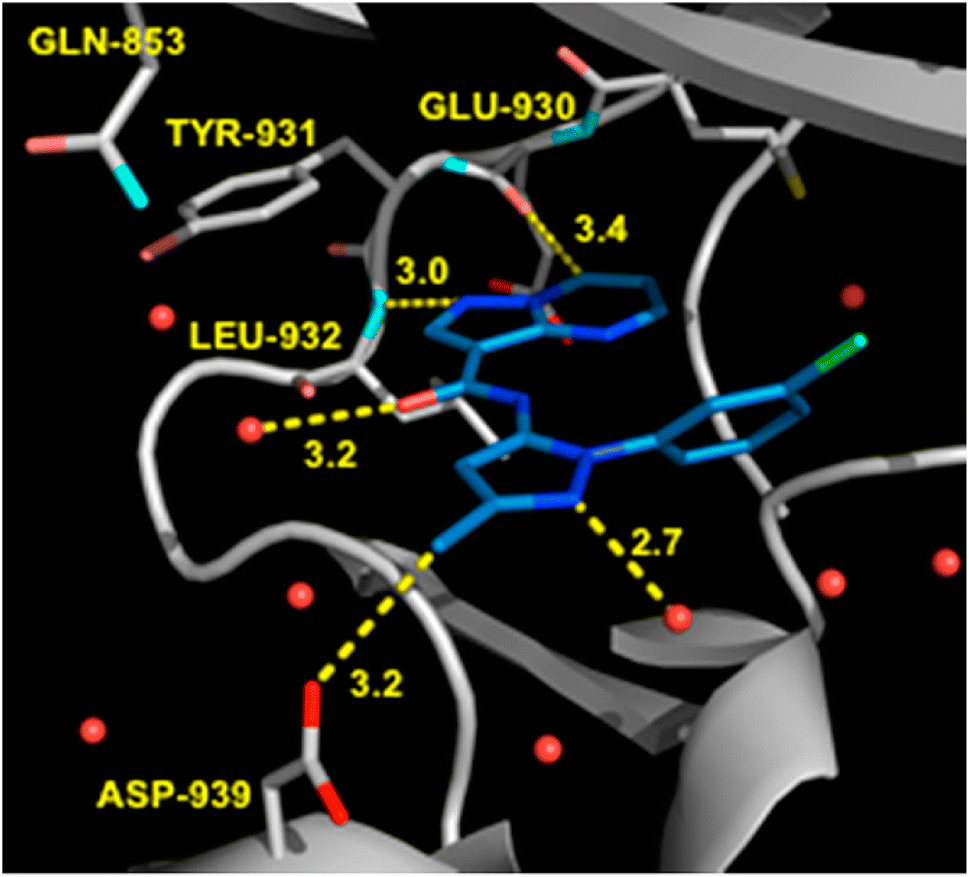 | ||
| Fig. 9 Co-crystal structure (2.3 Å) of compound 8 in the active site of the Jak2 kinase domain with the P-loop removed to allow a better view of the key active site interactions. Dashed lines indicate close contacts between ligand and protein with distances labelled in Å.257 | ||
 | ||
| Fig. 10 PK/PD study of 7j at an oral dose of 100 mg kg−1 in a SET2 xenograft model.257 | ||
Scheme 31 outlines the synthetic route employed to prepare the pyrazolo[1,5-a]pyrimidine compounds investigated in this study. The synthesis begins with the reaction of potassium ethyl malonate with carbonyldiimidazole (CDI) and magnesium chloride in tetrahydrofuran (THF) at 50 °C. Subsequent steps involve the use of various reagents and conditions, including diethyl carbonate, sodium hydride, and dimethylformamide (DMF), to construct the desired pyrazolo[1,5-a]pyrimidine scaffold. The final compounds are obtained through purification processes such as recrystallization or chromatography.
 | ||
| Scheme 31 General synthetic procedure for 5-amino-pyrazolo[1,5-a]pyrimidine-3-carboxamidesa257 aReaction conditions: (a) 21% EtONa, EtOH, 95%; (b) HOAc, H2O, 85%; (c) N-iodosuccinimide, DMF, 86%; (d) CO (1 atm), Pd(OAc)2, Et3N, MeOH; (e) 5% aq. LiOH, 68% (2 steps); (f) POCl3, DIPEA, 87%; (g) H2NR, DIPEA, DCM, 52–92%; (h) NH3, iPrOH, DIPEA, μW 120 °C, 15 min, 9–80%; (i) H2NCH3, H2O, μW 100 °C, 15 min, 62%.257 | ||
One of the key challenges in developing kinase inhibitors is achieving a balance between selectivity and potency. Highly selective compounds are less likely to cause off-target effects, but they may also be less effective in treating cancers with complex signaling networks. On the other hand, broader-spectrum inhibitors may target multiple kinases involved in cancer progression but at the cost of increased toxicity. In vitro kinase profiling helps researchers to fine-tune the selectivity of pyrazolo[1,5-a]pyrimidines, optimizing their efficacy while minimizing potential side effects.18
Overall, in vitro studies of cell line assays and kinase selectivity profiling have significantly advanced our understanding of pyrazolo[1,5-a]pyrimidines as kinase inhibitors in cancer treatment. These studies provide critical data on the efficacy, selectivity, and potential therapeutic applications of these compounds, paving the way for their continued development as targeted cancer therapies. As new methods in in vitro testing continue to evolve, the future of pyrazolo[1,5-a]pyrimidine-based therapies look promising, with the potential to offer more effective and personalized treatments for patients with various types of cancer.
6.2 Cytotoxicity and antiproliferative effects of pyrazolo[1,5-a]pyrimidines
Pyrazolo[1,5-a]pyrimidines represent a promising class of compounds with significant potential in medicinal chemistry, particularly in the development of anticancer agents. Their unique structural features allow for diverse biological activities, making them targets for investigations into their cytotoxic and antiproliferative effects on cancer cells. This write-up explores the mechanisms underlying the cytotoxicity of pyrazolo[1,5-a]pyrimidines and their antiproliferative properties, highlighting relevant studies and potential therapeutic applications.244Additionally, pyrazolo[1,5-a]pyrimidines may induce cytotoxic effects through the generation of reactive oxygen species (ROS), which can trigger apoptosis (programmed cell death). Studies have shown that certain pyrazolo[1,5-a]pyrimidine derivatives increase ROS levels, leading to oxidative stress and subsequent cell death in cancer cells. This mechanism highlights the dual nature of pyrazolo[1,5-a]pyrimidines in targeting both specific molecular pathways and broader cellular stress responses.18,258
6.2.1.1 Antiproliferative activity. The antiproliferative effects of pyrazolo[1,5-a]pyrimidines have been widely explored across multiple cancer types, with preclinical studies highlighting their promise as potent anticancer agents. These compounds have shown remarkable activity against various cancer cell lines, including breast, prostate, and leukemia, underscoring their broad therapeutic potential. The versatility of the pyrazolo[1,5-a]pyrimidine scaffold makes it an attractive target for drug development in oncology.19,212
One of the key aspects driving this interest is the structure–activity relationship (SAR) analysis, which has shed light on how specific structural modifications can significantly enhance the potency and selectivity of these compounds. SAR studies have demonstrated that the introduction of functional groups at particular positions on the pyrazolo[1,5-a]pyrimidine core can increase the compounds' ability to selectively target cancer cells while minimizing their effects on healthy cells. This is crucial for developing anticancer agents with fewer side effects, as the goal is to maximize efficacy against cancerous tissues while reducing toxicity to normal tissues.258,259
In addition to improved selectivity, some pyrazolo[1,5-a]pyrimidine derivatives have been shown to exert their antiproliferative effects through mechanisms such as cell cycle arrest and apoptosis induction. For example, derivatives that inhibit proteins involved in cancer cell survival and proliferation, such as cyclin-dependent kinases (CDKs) and Bcl-2, have demonstrated significant cytotoxicity against tumor cells. Moreover, these compounds can be further optimized through combination therapies, where they are used alongside other chemotherapeutic agents to achieve synergistic effects. This combined approach has the potential to enhance the overall therapeutic outcome by targeting multiple pathways involved in cancer progression.260,261
The development of pyrazolo[1,5-a]pyrimidine derivatives as anticancer agents remain a promising avenue in oncology, with ongoing research focused on refining their molecular structure to enhance selectivity, potency, and safety. Their ability to selectively inhibit cancer cell proliferation while sparing healthy cells positions them as valuable candidates for further investigation in cancer therapy.261,262
Kamal et al. (2016) synthesized and evaluated a series of anilinonicotinyl-linked pyrazolo[1,5-a]pyrimidine conjugates (6a–x) for their cytotoxicity against breast cancer (MCF-7) cells.244 Among these, compounds 6a and 6c exhibited significant antiproliferative activity, along with the ability to arrest the cell cycle at the G2/M phase. This arrest is critical as it prevents cancer cells from proliferating by halting their division. The study also demonstrated that these compounds exerted their effects by modulating key proteins involved in cell cycle regulation, including cyclin D1, Bcl-2, and survivin, which are often overexpressed in cancer cells to promote unchecked growth (Fig. 11). By inhibiting these proteins, the pyrazolo[1,5-a]pyrimidine derivatives effectively reduce cancer cell viability.
 | ||
| Fig. 11 (a–d) SiHa, HeLa (cervical cancer), MCF-7 (breast cancer), IMR-32 (neuroblastoma) were studied at 4 lM of conjugates for 24 h using MTT assay.244 | ||
Another crucial finding from this study is the downregulation of estrogen receptor alpha (ERα) activity by compounds 6a and 6c in estrogen-positive breast cancer cells. ERα plays a significant role in driving the growth of hormone-dependent breast cancer, and its inhibition is a desirable target in the treatment of such cancers. Estrogen-positive breast cancers account for more than 70% of all breast cancer cases, and ERα has been identified as a prognostic marker that also predicts the response to endocrine therapy. The inhibition of ERα by pyrazolo[1,5-a]pyrimidine conjugates provide a dual benefit by not only inhibiting cell proliferation directly but also modulating hormone receptor signaling pathways that fuel the cancer's progression (Fig. 12).
 | ||
| Fig. 12 The cell cycle analysis was conducted in MCF-7 cells treated with 6a, 6c at concentrations of 4, 8, 16, 32 μM for 48 h. Here doxorubicin (Doxo), the anthracycline antibiotic as well as one of most effective anticancer agents used to treat against breast cancer was used as positive control. Conjugates induced G2/M cell cycle arrest. Increased concentration of drug caused apoptosis as indicated by percentage of cells in G0 phase.244 | ||
Fused heterocyclic structures, such as pyrazolo[1,5-a]pyrimidines, are also known for their broad pharmacological activities, which include antibacterial, antifungal, antitumor, and anti-inflammatory properties. Their structural similarity to purines enables them to act as ATP-competitive inhibitors for various kinase enzymes, which are critical in cancer progression. For example, pyrazolo[1,5-a]pyrimidines have been shown to inhibit cyclin-dependent kinases (CDKs), checkpoint kinase 1 (CHK1), and mammalian target of rapamycin (mTOR), among others. These enzymes play pivotal roles in regulating cell cycle progression and survival, making them attractive targets for anticancer therapy.
In addition to their monotherapeutic potential, pyrazolo[1,5-a]pyrimidines have demonstrated synergistic effects when used in combination with other chemotherapeutic agents. Such combination therapies can lead to enhanced antiproliferative effects, potentially reducing the required doses of each drug and minimizing toxicity. Moreover, SAR studies indicate that specific functional groups, such as those at the 2- or 7-position of the pyrazolo[1,5-a]pyrimidine ring, can further improve selectivity for cancer cells, increasing their therapeutic index and reducing collateral damage to normal cells.
In summary, the continued exploration of pyrazolo[1,5-a]pyrimidine derivatives as anticancer agents hold significant promise, especially when considering the modifications that can be made to optimize their antiproliferative effects. Their ability to target both cell cycle regulators and hormone receptors, such as ERα, makes them particularly valuable in the treatment of breast cancer. Further studies, particularly those focused on combination therapies and in vivo efficacy, will be essential in translating these preclinical findings into clinical applications.
The cytotoxicity and antiproliferative effects of pyrazolo[1,5-a]pyrimidines underscore their potential as therapeutic agents in cancer treatment. Their ability to inhibit critical signaling pathways and induce oxidative stress positions them as versatile compounds in the fight against cancer. Ongoing research into the structural optimization of these compounds will likely yield novel derivatives with improved efficacy and selectivity. Ultimately, the continued exploration of pyrazolo[1,5-a]pyrimidines in cancer therapy may lead to the development of effective and targeted treatments for various malignancies, paving the way for improved patient outcomes.
In the study conducted by Attia et al. (2019), the antiproliferative activity of newly synthesized pyrazolo[1,5-a]pyrimidine compounds were evaluated against a variety of cancer cell lines, including Huh-7 (liver cancer), HeLa (cervical cancer), MCF-7 (breast cancer), and MDA-MB231 (triple-negative breast cancer) cell lines.263 Several of the compounds demonstrated notable antiproliferative effects, particularly compounds 11f, 16b, and 11i.
Compound 11f, which contains a 4-chlorophenyl substitution, showed moderate antiproliferative activity against the Huh-7 cell line with an IC50 value of 6.3 μM, compared to the reference drug doxorubicin (IC50 = 3.2 μM). The chloro substitution on the phenyl ring contributes positively to the compound's cytotoxic potential, as supported by the structure–activity relationship (SAR) analysis, which highlights that 11f's antiproliferative activity is stronger compared to its fluoro (11e, IC50 = 75.2 μM) and bromo (11g, IC50 = 88.4 μM) analogs. However, when tested against other cell lines, such as MDA-MB231, compound 11f exhibited much lower antiproliferative activity, with an IC50 of 74.25 μM, indicating its selective action on liver cancer cells.
On the other hand, compound 16b, which features a 4-methoxyphenyl group, demonstrated potent antiproliferative activity, particularly against HeLa cells, with an IC50 value of 7.8 μM, slightly better than that of doxorubicin (IC50 = 8.1 μM). The introduction of the methoxy group appears to enhance the compound's effectiveness, as evidenced by its superior performance compared to its unsubstituted counterpart, compound 16a (IC50 = 15.5 μM). Furthermore, compound 16b also showed promising results against MDA-MB231 cells with an IC50 value of 5.74 μM. SAR analysis emphasizes that methoxy substitution on the aryl ring plays a crucial role in increasing the antiproliferative efficacy of compound 16b.
Among the synthesized compounds, 11i, which incorporates a naphthyl ring, exhibited the most remarkable antiproliferative activity. Against the MCF-7 cell line, compound 11i achieved an IC50 of 3.0 μM, significantly outperforming doxorubicin (IC50 = 5.9 μM). Additionally, 11i demonstrated potent antiproliferative activity against MDA-MB231 cells with an IC50 of 4.32 μM. The higher lipophilicity of the naphthyl substitution in compound 11i appears to contribute to its enhanced cytotoxicity, particularly against breast cancer cell lines. The SAR analysis also indicates that replacing the phenyl ring with a more hydrophobic naphthyl group substantially boosts the activity of compound 11i, especially in MCF-7 and MDA-MB231 cells.
Flow cytometry analysis was conducted to examine the impact of compounds 11f, 11i, and 16b on cell cycle distribution. Treatment of MCF-7 cells with compound 11i resulted in a marked reduction in the G1 phase, with significant arrest in both the S and G2/M phases (Fig. 13). This indicates that compound 11i interferes with the progression of the cell cycle, ultimately inhibiting proliferation. Similarly, treatment of MDA-MB231 cells with compound 11i induced a significant G1 phase arrest, accompanied by a reduction in the S phase (Fig. 14). In HeLa cells, compound 16b treatment led to a reduction in the G1 phase and significant arrest in the G2/M phase. Interestingly, compound 11f showed no notable impact on the cell cycle of Huh-7 cells, suggesting that its antiproliferative mechanism may differ or be less related to cell cycle modulation.
 | ||
| Fig. 13 Cell cycle analysis in MCF-7 cells. Cells were subjected to cell cycle analysis using flow cytometry. Representative histogram of the gated cells in the G0/G1, S, and G2/M phases for (a): control; (b): compound 11i; (c): PTX. (d): quantitative analysis of distribution or proportion of the cells in each phase was performed from at least 10000 cells per sample. Each bar represents mean ± SEM of the data obtained from three independent experiments. *p < 0.05 vs. control.263 | ||
 | ||
| Fig. 14 Cell cycle analysis in MDA-Mb-231 cells. Cells were subjected to cell cycle analysis using flow cytometry. Representative histogram of the gated cells in the G0/G1, S, and G2/M phases for (a): control; (b): compound 11i. (c): quantitative analysis of distribution or proportion of the cells in each phase was performed from at least 10000 cells per sample. Each bar represents mean ± SEM of the data obtained from three independent experiments. *p < 0.05 vs. control.263 | ||
These findings highlight the selective and potent antiproliferative activities of certain pyrazolo[1,5-a]pyrimidine derivatives, with compounds11i and 16b standing out as promising candidates for further development in cancer therapy, particularly for breast cancer and cervical cancer. The SAR analysis provides valuable insights into how specific chemical modifications can enhance or reduce cytotoxic activity, paving the way for the design of more effective antiproliferative agents.
6.3 In vivo evaluation of the anti-cancer efficacy of pyrazolo[1,5-a]pyrimidines
In vivo studies play a crucial role in determining the therapeutic potential of small-molecule inhibitors such as pyrazolo[1,5-a]pyrimidines in cancer treatment. While in vitro studies offer initial insights into the biological activity of these compounds, in vivo evaluations provide a more comprehensive understanding of their pharmacokinetics, bioavailability, toxicity, and overall efficacy in living organisms. The anti-cancer potential of pyrazolo[1,5-a]pyrimidines has been evaluated in various animal models, particularly in targeting key oncogenic pathways such as the Ras-Raf-MEK-ERK signaling pathway, which is often disrupted in cancers.264,265Pyrazolo[1,5-a]pyrimidines have shown promising results in vivo, particularly in cancers driven by mutations in the B-Raf and MEK kinases, such as melanoma, colorectal, and thyroid cancers. By inhibiting these kinases, pyrazolo[1,5-a]pyrimidines prevent the phosphorylation of downstream targets in the MAPK pathway, effectively halting cancer cell proliferation and inducing apoptosis. These in vivo studies are essential for assessing the selectivity of these inhibitors, ensuring they target cancerous cells without causing significant damage to normal tissues, thereby minimizing off-target effects.266,267
For example, in mouse xenograft models, pyrazolo[1,5-a]pyrimidine derivatives have demonstrated significant tumor regression and prolonged survival when administered systemically. In these models, the compounds have shown strong anti-tumor activity against both primary tumors and metastases, particularly in cancers that harbor the B-Raf V600E mutation. This mutation, found in a significant percentage of melanomas and other cancers, leads to constitutive activation of the MAPK pathway, and pyrazolo[1,5-a]pyrimidines have been effective in inhibiting this aberrant signalling.268,269
Additionally, in vivo studies have explored the pharmacokinetics of pyrazolo[1,5-a]pyrimidines, determining their absorption, distribution, metabolism, and excretion (ADME) profiles. These factors are critical for understanding how the compounds behave in a complex biological system and ensuring their therapeutic efficacy. The stability of pyrazolo[1,5-a]pyrimidines in the bloodstream, their ability to penetrate tissues, and their clearance rates are vital parameters that influence their overall anti-cancer potential.270 Toxicity assessments in vivo are also an essential component of these studies. Pyrazolo[1,5-a]pyrimidines must exhibit an acceptable safety profile, with limited adverse effects at therapeutic doses. Early in vivo toxicity studies have demonstrated that some derivatives of pyrazolo[1,5-a]pyrimidines can be well tolerated, with manageable side effects. However, further optimization may be necessary to improve their therapeutic window and reduce potential toxicity.26,44
In summary, in vivo evaluations of pyrazolo[1,5-a]pyrimidines have provided critical data supporting their potential as effective anti-cancer agents. These studies highlight the compounds' ability to selectively target cancer-driving kinases, reduce tumor growth, and offer promising therapeutic outcomes in animal models. However, ongoing research is required to further refine these compounds and move towards clinical trials in humans.
For example, in El Sayed et al. (2018) focused heavily on in vitro assays and molecular modelling to evaluate the anti-cancer potential of pyrazolo[1,5-a]pyrimidines and pyrido[2,3-d]pyrimidines.271 These compounds were primarily tested for their tyrosine kinase inhibitory activity, targeting EGFR kinase and their cytotoxicity against cancer cell lines.
The study's main approach involved in vitro assays, including the NCI 60 cell line panel across various cancer types (leukemia, breast, renal, etc.), which provided essential preliminary data for the therapeutic potential of these compounds. Key findings from these in vitro tests indicated that specific compounds, like 9b and 6f, were particularly effective, showing inhibitory effects on MCF-7 breast cancer cells and renal cell lines. The most active compound, 9b, demonstrated an EGFR inhibition of 81.72% at 25 nM and an IC50 of 8.4 nM, comparable to Sorafenib, a well-known kinase inhibitor.
Although in vivo animal models were not used directly in this study, the strong EGFR inhibitory activity of compounds like 9b suggests their potential for further in vivo evaluations. Typically, such promising in vitro results are followed by xenograft models in mice to assess pharmacokinetics, toxicity, and antitumor efficacy in living organisms, which could be a next step for this research. The study provides a solid foundation for future preclinical investigations, where in vivo animal models would be essential for confirming the efficacy of these compounds in real-world cancer therapies.
In the study by Kosugi et al. (2012), the in vivo evaluation of pyrazolo[1,5-a]pyrimidine derivatives were conducted to assess their potential as inhibitors of MAPKAP-K2 (Mitogen-Activated Protein Kinase-Activated Protein Kinase 2), a kinase implicated in inflammatory diseases.272 The in vivo experiments were crucial for determining the pharmacokinetics, efficacy, and overall therapeutic potential of these compounds.
The research explored the anti-inflammatory properties of key pyrazolo[1,5-a]pyrimidine derivatives, including compound (S)-44, which had shown promising in vitro results. Compound (S)-44 was selected for further in vivo studies due to its strong MAPKAP-K2 inhibition (IC50 < 100 nM) and superior selectivity over CDK2 (Cyclin-Dependent Kinase 2) (Fig. 15). In a model of acute inflammation, compound (S)-44 demonstrated potent efficacy, effectively reducing paw edema in rats following carrageenan injection. This significant anti-inflammatory activity correlated well with the inhibition of MAPKAP-K2, as compound (S)-44 was able to modulate the downstream effects of this kinase, particularly its role in regulating pro-inflammatory cytokines. This effect was evident when compound (S)-44 decreased TNF-α (Tumor Necrosis Factor-alpha) levels in plasma, a key mediator of inflammation. Fig. 16 illustrates the reduction in inflammatory markers observed in these in vivo models, providing a clear link between kinase inhibition and anti-inflammatory outcomes.
 | ||
| Fig. 15 MAPKAP-K2 inhibitors demonstrating in vivo efficacy.272 | ||
 | ||
| Fig. 16 Predicted binding mode of compound (1) in the MAPKAP-K2 homology model. Equivalent residue numbers in CDK2 are shown in parentheses.272 | ||
Additionally, the pharmacokinetic profile of (S)-44 was assessed. The compound exhibited favorable properties, including adequate bioavailability and a half-life that supported its potential use as an orally administered anti-inflammatory agent. These results indicated that (S)-44 could maintain effective concentrations in systemic circulation, which was essential for sustained kinase inhibition and therapeutic action.
Fig. 17 highlights the structure–activity relationship (SAR) findings from the in vivo studies, showing that specific modifications at the 5- and 7-positions of the pyrazolo[1,5-a]pyrimidine core played a pivotal role in enhancing both the potency and selectivity of these compounds. The para-substitution on the 7-phenyl group (compound 64) led to improved binding in the MAPKAP-K2 pocket, contributing to its enhanced anti-inflammatory effects in vivo.
 | ||
| Fig. 17 The effect of compound (S)-44 on serum TNF-α concentration in mouse endotoxin shock model. Statistical analysis was carried out with the Dunnet's multiple comparison test, with P < 0.05 considered as significant. *P < 0.05 and ***P < 0.001 (vs. control).272 | ||
Overall, the in vivo studies provided strong evidence that pyrazolo[1,5-a]pyrimidine derivatives, particularly (S)-44, could effectively inhibit MAPKAP-K2, reduce inflammation, and show promise as potential therapeutic agents for inflammatory diseases. The combination of potent kinase inhibition, selectivity, and favorable pharmacokinetics laid the groundwork for further preclinical and clinical investigations.
The study by Fouda et al. (2019) highlighted the synthesis of 2-arylazomalononitrile derivatives, which are precursors to pyrazolo[1,5-a]pyrimidine analogs. These compounds, synthesized via diazotization of aniline derivatives followed by coupling with malononitrile, exhibited significant anticancer activity.273 The in vitro cytotoxicity of these compounds was assessed against several human cancer cell lines, including MCF-7 (breast cancer), HePG2 (liver cancer), and HCT 116 (colon cancer). The IC50 values, which represent the concentration of the compound required to inhibit 50% of cell viability, were measured to compare the effectiveness of the tested compounds across different tumor cell lines. As shown in Fig. 18, the tested compounds displayed varying degrees of cytotoxicity depending on the cell line, with some compounds showing higher potency against specific cancer types.
 | ||
| Fig. 18 The differences of IC50% of new tested chemicals compounds effects on different tumor cells (MCF-7, HePG2, and HCT 116 cells).273 | ||
The differences in IC50 values among the different tumor cells suggest that the anticancer activity of pyrazolo[1,5-a]pyrimidine derivatives is influenced by the molecular characteristics of each cancer type. For instance, the MCF-7 cells were found to be more resistant to certain compounds compared to HCT 116 or HePG2 cells, highlighting the need for tailored therapeutic strategies. Additionally, these findings emphasize the potential of pyrazolo[1,5-a]pyrimidines as selective inhibitors, offering an advantage over non-specific cytotoxic agents currently used in chemotherapy. The detailed analysis of structure–activity relationships in the study further supports the rational design of more potent derivatives with improved selectivity for cancer cells.
In the study by Kamal et al. (2013), a novel series of pyrazolo[1,5-a]pyrimidine derivatives linked to 2-aminobenzothiazole (compounds 6a–t) were synthesized and tested for their anticancer potential.244 Two standout compounds from this series, referred to as 6m and 6p, exhibited particularly significant anticancer activity. These compounds are pyrazolo[1,5-a]pyrimidine derivatives that possess specific structural features responsible for their enhanced cytotoxic effects against various human cancer cell lines.
Compound 6m is characterized by the presence of a 3,4,5-trimethoxy substitution on the phenyl ring attached to the pyrazolo[1,5-a]pyrimidine core, while compound 6p features a 4-fluoro substitution on this same phenyl ring. These specific substitutions were found to be critical in enhancing the compounds' overall anticancer activity. The study showed that both 6m and 6p exhibited potent cytotoxic effects, with IC50 values ranging from 1.94 to 7.07 μM, depending on the type of cancer cell line being treated. For instance, 6p demonstrated a particularly low GI50 value of 0.7 μM against lung cancer cell lines, which is indicative of its strong antiproliferative effect.
Both 6m and 6p were observed to induce G2/M cell cycle arrest, which prevents cancer cells from progressing through the cell division process. This was confirmed through flow cytometry analysis in lung cancer cells (A549), showing that at a concentration of 2 μM, 6m and 6p led to an accumulation of 52% and 64% of cells in the G2/M phase, respectively (Fig. 19). These effects are thought to be due to the downregulation of Cdk1 (cyclin-dependent kinase 1), a key enzyme involved in the G2/M transition, as indicated by western blot analysis.
 | ||
| Fig. 19 Flow cytometric analysis of compounds 6p, 6m and roscovitine in A-549 lung cancer cell line. (A) control cells, (B) roscovitine (2 μM), (C) 6m (1 μM), (D) 6m (2 μM), (E) 6p (1 μM) and (F) 6p (2 μM).244 | ||
In addition to cell cycle arrest, both compounds triggered caspase-3-dependent apoptosis, a programmed cell death process, in the treated cancer cells. This was demonstrated by the detection of nuclear fragmentation and chromatin condensation through Hoechst staining, key indicators of apoptosis. DNA fragmentation assays further confirmed the apoptotic effects of these compounds, underscoring their ability to induce cancer cell death effectively.
Overall, the study demonstrated that compounds like6m and 6p—bearing the pyrazolo[1,5-a]pyrimidine core with 2-aminobenzothiazole linkage—hold substantial promise as anticancer agents. Their ability to induce both cell cycle arrest and apoptosis suggests their potential for further development in cancer therapy, particularly given their potency across different cancer cell lines (Fig. 20). The structure–activity relationship (SAR) analysis revealed that the substitution patterns on the phenyl ring of the pyrazolo[1,5-a]pyrimidine unit play a crucial role in determining the compounds' efficacy, with 6m and 6p emerging as the most promising candidates.
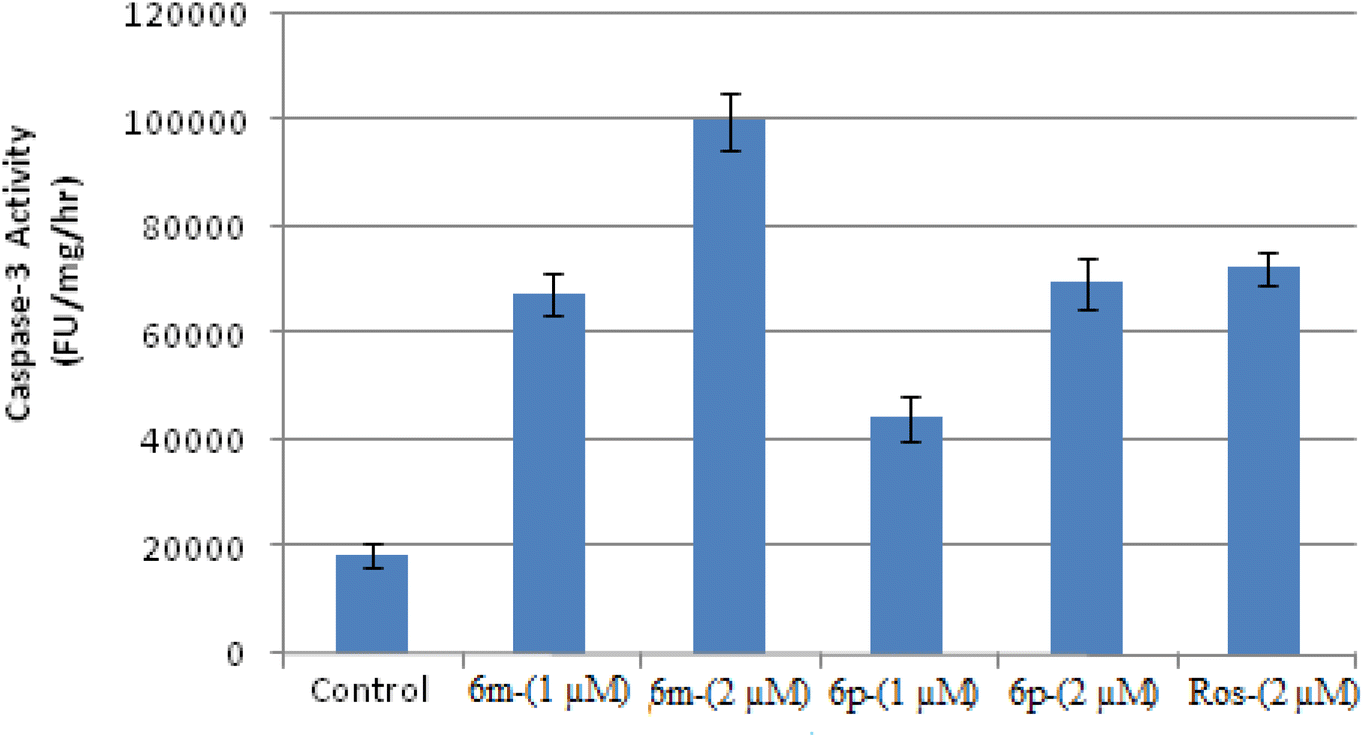 | ||
| Fig. 20 Effect of compounds 6p and 6m on caspase-3 activity: A-549 cells were treated with compounds 6p and 6m at 1 and 2 μM concentrations for 48 h. Roscovitine is used as a positive control. Values indicated are the mean ± SD of two different experiments performed in triplicates.244 | ||
Liu et al. (2016) describe a scaffold-hopping exercise that initiates with imidazo[1,2-a]pyrazines and explores pyrazolo[1,5-a][1,3,5]triazines, ultimately leading to the identification of pyrazolo[1,5-a]pyrimidines as a novel class of potent TTK inhibitors.274–276 This research provided significant structural insights through X-ray crystallography of a representative compound, which indicated a 1(1)/2 type inhibition mechanism. The findings suggest that by incorporating polar moieties in hydrophobic regions, researchers could enhance the physicochemical properties of these compounds while retaining their inhibitory potency.
The synthesis of pyrazolo[1,5-a]pyrimidines begin with 3-bromo-5,7-dichloro-pyrazolo[1,5-a]pyrimidine and proceeds through a series of steps, including nucleophilic aromatic substitution, protection of nitrogen, and Suzuki–Miyaura coupling. These synthetic routes have allowed the development of a range of pyrazolo[1,5-a]pyrimidines (designated as compounds 7 to 24) with varying potencies. In comparative analyses, the new pyrazolo[1,5-a]pyrimidines demonstrated comparable potency to their pyrazolotriazine counterparts, with improved cell activity and oral bioavailability. However, some lead compounds were found to be dissolution-limited, hindering their plasma levels from reaching maximum tolerated doses.
To address bioavailability, researchers initiated a lead optimization program focusing on modifying physicochemical properties without compromising potency. By introducing polar and basic solubilizing elements into solvent-accessible regions, they identified compounds with enhanced oral exposure. Notably, compound 10 achieved significant cancer cell growth inhibition and demonstrated dose-dependent plasma concentration increases, establishing a maximum tolerated dose of 6.5 mg kg−1 in preclinical studies.
A detailed examination of TTK binding interactions through co-crystallization revealed unique structural features, including hydrogen bonding patterns with key residues in the TTK enzyme. Such insights guide the development of compounds that maintain their potency while being optimized for better oral bioavailability. Further exploration involved modifying the compounds with hydroxyl functions and weak bases, leading to additional potent TTK inhibitors that showed promising activity against cancer cell lines. Scheme 32 illustrates the scaffold hopping of TTK inhibitors.
 | ||
| Scheme 32 TTK inhibitor scaffold hopping.274 | ||
Ultimately, four potent pyrazolo[1,5-a]pyrimidine compounds were selected for further evaluation against a panel of human kinases and tested in xenograft models. Among these, compound 24 emerged as a standout candidate due to its excellent in vitro potency (TTK IC50 = 0.1 nM) and selective inhibition profile, with minimal interaction with cytochrome P450 enzymes. The compound demonstrated effective growth inhibition across various cancer cell lines, positioning it as a strong candidate for further preclinical evaluation and potential clinical application in cancer therapy. Overall, the work highlights the therapeutic promise of pyrazolo[1,5-a]pyrimidines as selective anticancer agents, with ongoing studies aimed at optimizing their pharmacokinetic and safety profiles for clinical use.274
Overexpression of P-glycoprotein (P-gp) and other ATP-binding cassette (ABC) transporters is a major mechanism through which MDR manifests, as these proteins effectively pump chemotherapeutic agents out of the cells, thereby reducing their intracellular concentrations and efficacy. This is particularly problematic in the treatment of central nervous system (CNS) tumors, where the blood–brain barrier restricts drug delivery.166 Research conducted by Fallacara et al. (2019) demonstrated that a specific set of pyrazolo[3,4-d]pyrimidines, which were developed as novel Src tyrosine kinase inhibitors, exhibited significant activity against CNS tumors in vivo.166 Their findings indicated that these compounds could enhance the intracellular accumulation of fluorescent substrate Rho 123, which suggests a potential to overcome the limitations imposed by P-gp and improve the effectiveness of traditional chemotherapeutics such as paclitaxel in P-gp overexpressing cancer cells.
Furthermore, the therapeutic potential of pyrazolo[1,5-a]pyrimidines extend beyond their role as P-gp inhibitors. According to Arias-Gómez et al.260 (2021), these derivatives represent a vast family of N-heterocyclic compounds that have garnered significant interest not only in medicinal chemistry but also in material science due to their promising photophysical properties. The exploration of diverse synthesis pathways and post-functionalization strategies for these compounds has enhanced their structural diversity and application potential. By improving the functional characteristics of pyrazolo[1,5-a]pyrimidines, researchers hope to design drugs that are more effective against cancer. This includes an emphasis on their anticancer activity and their ability to inhibit specific enzymes related to cancer progression. The ongoing advances in the synthesis and functionalization of pyrazolo[1,5-a]pyrimidines underscore their potential to lead to novel and rational drug designs, ultimately contributing to improved outcomes in cancer treatment strategies. Overall, the incorporation of pyrazolo[1,5-a]pyrimidines into therapeutic regimens may offer new avenues for combating drug-resistant cancers, particularly in challenging cases such as glioblastoma.
7 Challenges and future directions
The development of pyrazolo[1,5-a]pyrimidines as potent protein kinase inhibitors has garnered significant attention in cancer research due to their potential to interfere with key cellular signaling pathways involved in tumorigenesis. Protein kinases are essential for regulating various cellular functions, including growth, survival, and differentiation. Their disruption in cancer leads to uncontrolled cell proliferation and survival, making them attractive therapeutic targets.277 Pyrazolo[1,5-a]pyrimidines have shown promise in inhibiting specific kinases, thus blocking cancer progression. However, despite these promising applications, several challenges remain in their synthesis, biological specificity, and clinical utility, necessitating further research and innovation to enhance their efficacy and safety in cancer therapy.18,19The synthesis of pyrazolo[1,5-a]pyrimidines is often complex and involves multiple steps that can present significant hurdles, particularly in achieving high yields and selectivity. Traditional synthetic routes typically rely on cyclocondensation reactions or transition-metal-catalyzed processes, both of which can involve harsh reaction conditions and yield low amounts of product. Additionally, synthesizing pyrazolo[1,5-a]pyrimidine derivatives with specific functional groups that are crucial for enhancing their kinase inhibition properties poses a considerable challenge.248,278,279 While advances in organic synthesis, such as microwave-assisted reactions and flow chemistry, have provided new avenues for more efficient production, these methods still require optimization for large-scale applications. Scalability is another critical issue; developing industrial-scale processes that are both cost-effective and environmentally sustainable remains a significant bottleneck in the production of these compounds. Improving the synthesis process to make it more efficient and adaptable to large-scale production is crucial for advancing pyrazolo[1,5-a]pyrimidines to clinical use.266,280–282
One of the key challenges in developing pyrazolo[1,5-a]pyrimidines as cancer therapeutics lies in achieving specificity for their kinase targets. Kinase inhibitors must selectively block the activity of a specific kinase without affecting others, a difficult task given the highly conserved nature of the ATP-binding site across the kinase family. Off-target effects, where the drug inadvertently inhibits other kinases, can lead to toxicities and unwanted side effects, thus limiting the clinical applicability of these inhibitors. Achieving the necessary selectivity to avoid such toxicities remains an ongoing challenge in the design of pyrazolo[1,5-a]pyrimidines.23,43
Resistance to kinase inhibitors also poses a significant hurdle. Cancer cells can develop resistance through various mechanisms, including mutations in the kinase target or activation of compensatory pathways that bypass the blocked kinase. This resistance reduces the effectiveness of the inhibitors over time, necessitating the development of next-generation pyrazolo[1,5-a]pyrimidine derivatives that can overcome these resistance mechanisms. In addition, some kinases perform important functions independent of their kinase activity, such as acting as scaffolds for protein–protein interactions. Inhibiting only the catalytic activity of these kinases may not fully disrupt their oncogenic role, further complicating the design of effective inhibitors.168,271
Beyond issues of specificity and resistance, the pharmacokinetics and delivery of pyrazolo[1,5-a]pyrimidines represent additional challenges. Many of these compounds suffer from poor solubility, which limits their bioavailability when administered orally. Additionally, rapid metabolism and clearance from the body reduce the amount of drug available to exert therapeutic effects, particularly in the tumor microenvironment. Enhancing the solubility, stability, and bioavailability of these molecules is critical to improving their overall efficacy in cancer treatment.45,137 Another challenge lies in ensuring that pyrazolo[1,5-a]pyrimidines are selectively delivered to tumor tissues while sparing healthy cells. Current formulations may not always achieve optimal biodistribution, leading to suboptimal therapeutic outcomes and increased toxicity. Developing advanced drug delivery systems, such as nanoparticles or liposomes, may help to improve the selective targeting of tumors and enhance the therapeutic index of pyrazolo[1,5-a]pyrimidines. In cancers affecting the central nervous system, like glioblastoma, these inhibitors must also cross the blood–brain barrier, a significant obstacle for most small-molecule drugs. Innovative delivery methods such as intranasal administration or convection-enhanced delivery may provide solutions to this problem.45,137
The future of pyrazolo[1,5-a]pyrimidines in cancer treatment lies in rational drug design, where advances in computational chemistry and structural biology can be leveraged to create more selective and potent kinase inhibitors. By studying the crystal structures of kinase–inhibitor complexes, researchers can design inhibitors that fit more precisely into the ATP-binding pockets of target kinases, reducing off-target effects and enhancing efficacy. Structure-based drug design can also help optimize the pharmacokinetic properties of these compounds, improving their absorption, distribution, metabolism, and excretion. Additionally, targeting multiple kinases simultaneously with multi-target kinase inhibitors may offer a more effective approach to cancer treatment. Cancer cells often rely on more than one signalling pathway for survival and blocking multiple pathways at once could provide a more comprehensive therapeutic strategy.154–157
Combination therapies are another promising direction for pyrazolo[1,5-a]pyrimidines. Pairing these kinase inhibitors with other cancer treatments, such as immune checkpoint inhibitors or chemotherapy, could enhance their effectiveness.200 For instance, combining kinase inhibitors with immunotherapy could potentiate the immune system's ability to attack tumors, while using them alongside chemotherapy may sensitize cancer cells to treatment. However, these combination strategies must be carefully developed through clinical trials to identify the most effective regimens and minimize adverse effects.283–286
As the field of personalized medicine continues to grow, the potential of pyrazolo[1,5-a]pyrimidines could be further realized through the identification of biomarkers that predict which patients are most likely to respond to these inhibitors. By tailoring treatments based on the specific genetic and molecular characteristics of an individual's cancer, clinicians could maximize therapeutic efficacy while minimizing unnecessary treatments and side effects. Advances in genomics and proteomics will be key to identifying these biomarkers and selecting the most appropriate kinase inhibitor for each patient's cancer subtype.287,288
While significant progress has been made in preclinical studies, the translation of pyrazolo[1,5-a]pyrimidine-based kinase inhibitors into clinical trials has been slow. Addressing safety, efficacy, and tolerability concerns in early-phase trials will be crucial for advancing these compounds through the clinical pipeline. Regulatory approvals from agencies such as the FDA and EMA will depend on comprehensive data demonstrating the therapeutic potential, pharmacokinetics, and toxicity profiles of these compounds.289
Pyrazolo[1,5-a]pyrimidines represent a promising class of protein kinase inhibitors with significant potential in cancer therapy. However, challenges related to their synthesis, specificity, pharmacokinetics, and clinical development remain. Overcoming these hurdles will require continued research, innovation in drug design and delivery, and rigorous clinical testing. With advances in technology and a deeper understanding of cancer biology, pyrazolo[1,5-a]pyrimidines could become powerful tools in the fight against cancer, offering more effective and personalized treatment options for patients.
8 Conclusion
Pyrazolo[1,5-a]pyrimidines represent a highly versatile and promising class of compounds in the field of targeted cancer therapy, particularly as potent protein kinase inhibitors (PKIs). Their ability to selectively inhibit key oncogenic kinases such as as CK2, EGFR, B-Raf, MEK, PDE4, BCL6, DRAK1, CDK1 and CDK2, Pim-1, among others, highlights their potential as therapeutic agents in the treatment of various malignancies, including non-small cell lung cancer (NSCLC) and melanoma. Advances in synthetic methodologies, such as cyclization, microwave-assisted synthesis, and palladium-catalyzed cross-coupling, have enabled the structural diversification and functionalization of these molecules, paving the way for more effective and selective kinase inhibitors. Structure–activity relationship (SAR) studies have revealed the critical role of specific substitution patterns in enhancing both pharmacological properties and kinase selectivity. Despite these achievements, several challenges remain. Resistance mechanisms, off-target effects, and suboptimal pharmacokinetics continue to impede the clinical success of many kinase inhibitors, including pyrazolo[1,5-a]pyrimidines. Overcoming these limitations will require a more in-depth understanding of the molecular basis of kinase inhibition and the development of next-generation inhibitors with improved specificity, reduced toxicity, and enhanced bioavailability.Moving forward, the integration of green chemistry principles, novel synthetic approaches, and precision medicinal strategies could accelerate the translation of pyrazolo[1,5-a]pyrimidines from preclinical models to clinical applications. Continued research in this area holds significant promise for advancing cancer treatment, particularly in overcoming drug resistance and improving patient outcomes with highly targeted kinase inhibition.
Data availability
This study is a review and does not include primary data. All data supporting the findings and conclusions of this review are drawn from previously published works, which are appropriately cited within the manuscript. The references for these works are included in the article.Author contributions
Terungwa H. Iorkula contributed to the conceptualization, drafting, and revision of the manuscript, as well as provided essential resources. Osasere Jude-Kelly Osayawe, Daniel A. Odogwu, Latifat Oluwatobi, Emmanuel Faderin, Omowunmi Rebecca Aworinde, Peter Agyemang, Odo Lovelyn Onyinyechi, Raymond Femi Awoyemi, and Busayo Odunayo Akodu were involved in revising the manuscript and contributing resources. Ikhazuagbe Hilary Ifijen prepared the initial draft and also participated in the manuscript's revision.Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.References
- M. Maliki, I. H. Ifijen and E. U. Ikhuoria, et al.). Copper nanoparticles and their oxides: optical, anticancer, and antibacterial properties, Int. Nano Lett., 2022, 12(4), 379–398, DOI:10.1007/s40089-022-00380-2.
- I. H. Ifijen, M. Maliki and B. Anegbe, Synthesis, photocatalytic degradation, and antibacterial properties of selenium or silver doped zinc oxide nanoparticles: a detailed review, OpenNano, 2022, 8, 100082, DOI:10.1016/j.onano.2022.100082.
- I. H. Ifijen and M. Maliki, A comprehensive review on the synthesis and photothermal cancer therapy of titanium nitride nanostructures, Inorg. Nano-Met. Chem., 2022, 53(4), 366–387, DOI:10.1080/24701556.2022.2068596.
- A. Efunnuga, A. Efunnuga and A. P. Onivefu, et al.). Nanomedicine advancements: vanadium oxide nanoparticles as a game-changer in antimicrobial and anticancer therapies, J. Bionanoscience, 2024, 3715–3756, DOI:10.1007/s12668-024-01566-y.
- S. I. Omonmhenle and I. H. Ifijen, Advancements in layered double hydroxide-based chemotherapeutic nanosystems for cancer treatment, J. Appl. Sci. Environ. Manage., 2023, 27(4), 815–821, DOI:10.4314/jasem.v27i4.24.
- K. Bhullar, N. Lagarón, E. McGowan, I. Parmar, A. Jha, B. Hubbard and H. Rupasinghe, Kinase-targeted cancer therapies: progress, challenges and future directions, Mol. Cancer, 2018, 17 DOI:10.1186/s12943-018-0804-2.
- L. Kondapalli, K. Soltani and M. Lacouture, The promise of molecular targeted therapies: protein kinase inhibitors in the treatment of cutaneous malignancies, J. Am. Acad. Dermatol., 2005, 53(2), 291–302, DOI:10.1016/J.JAAD.2005.02.011.
- C. Pottier, M. Fresnais, M. Gilon, G. Jerusalem, R. Longuespée and N. Sounni, Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy, Cancers, 2020, 12, 731, DOI:10.3390/cancers12030731.
- Y. Ling, J. Liu, J. Qian, C. Meng, J. Guo, W. Gao, B. Xiong, C. Ling and Y. Zhang, Recent Advances in Multi-target Drugs Targeting Protein Kinases and Histone Deacetylases in Cancer Therapy, Curr. Med. Chem., 2020, 7264–7288, DOI:10.2174/0929867327666200102115720.
- T. Otto and P. Sicinski, Cell cycle proteins as promising targets in cancer therapy, Nat. Rev. Cancer, 2017, 17, 93–115, DOI:10.1038/nrc.2016.138.
- K. Bhullar, N. Lagarón, E. McGowan, I. Parmar, A. Jha, B. Hubbard and H. Rupasinghe, Kinase-targeted cancer therapies: progress, challenges and future directions, Mol. Cancer, 2018, 17 DOI:10.1186/s12943-018-0804-2.
- C. Chu and T. Lee, Targeting protein kinases in cancer stem cells, Essays Biochem., 2022, 399–412, DOI:10.1042/EBC20220002.
- M. Heinrich, Abstract ED01-03: targeted therapy with protein kinase inhibitors Lessons learned from the last 20 years, Mol. Cancer Ther., 2019, 18, 03, DOI:10.1158/1535-7163.TARG-19-ED01-03.
- S. Ghione, N. Mabrouk, C. Paul, A. Bettaieb and S. Plenchette, Protein kinase inhibitor-based cancer therapies: considering the potential of nitric oxide (NO) to improve cancer treatment, Biochem. Pharmacol., 2020, 113855, DOI:10.1016/j.bcp.2020.113855.
- A. Passaro, P. Jänne, T. Mok and S. Peters, Overcoming therapy resistance in EGFR-mutant lung cancer, Nat. Cancer, 2021, 2, 377–391, DOI:10.1038/s43018-021-00195-8.
- D. Camidge, W. Pao and L. Sequist, Acquired resistance to TKIs in solid tumours: learning from lung cancer, Nat. Rev. Clin. Oncol., 2014, 11, 473–481, DOI:10.1038/nrclinonc.2014.104.
- A. Desai and C. Lovly, Strategies to overcome resistance to ALK inhibitors in non-small cell lung cancer: a narrative review, Transl. Lung Cancer Res., 2023, 12, 615–628, DOI:10.21037/tlcr-22-708.
- D. Heathcote, H. Patel, S. Kroll, P. Hazel, M. Periyasamy, M. Alikian, S. Kanneganti, A. Jogalekar, B. Scheiper, M. Barbazanges, A. Blum, J. Brackow, A. Siwicka, R. Pace, M. Fuchter, J. Snyder, D. Liotta, P. Freemont, E. Aboagye, R. Coombes, A. Barrett and S. Ali, A novel pyrazolo[1,5-a]pyrimidine is a potent inhibitor of cyclin-dependent protein kinases 1, 2, and 9, which demonstrates antitumor effects in human tumor xenografts following oral administration, J. Med. Chem., 2010, 53(24), 8508–8522, DOI:10.1021/jm100732t.
- R. Jorda, L. Havlícek, I. McNae, M. Walkinshaw, J. Voller, A. Šturc, J. Navrátilová, M. Kuzma, M. Mistrik, J. Bartek, M. Strnad and V. Kryštof, Pyrazolo[4,3-d]pyrimidine bioisostere of roscovitine: evaluation of a novel selective inhibitor of cyclin-dependent kinases with antiproliferative activity, J. Med. Chem., 2011, 54(8), 2980–2993, DOI:10.1021/jm200064p.
- C. Greco, R. Catania, D. Balacco, V. Taresco, F. Musumeci, C. Alexander, A. Huett and S. Schenone, Synthesis and Antibacterial Evaluation of New Pyrazolo[3,4-d]pyrimidines Kinase Inhibitors, Molecules, 2020, 25, 5354, DOI:10.3390/molecules25225354.
- G. Vignaroli, G. Iovenitti, C. Zamperini, F. Coniglio, P. Calandro, A. Molinari, A. Fallacara, A. Sartucci, A. Calgani, D. Colecchia, A. Mancini, C. Festuccia, E. Dreassi, M. Valoti, F. Musumeci, M. Chiariello, A. Angelucci, M. Botta and S. Schenone, Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model, J. Med. Chem., 2017, 60(14), 6305–6320, DOI:10.1021/acs.jmedchem.7b00637.
- M. Nešović, A. Rankov, A. Podolski-Renić, I. Nikolic, G. Tasic, A. Mancini, S. Schenone, M. Pešić and J. Dinić, Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, Inhibit Focal Adhesion Kinase and Suppress Human Glioblastoma Invasion In Vitro and In Vivo, Cancers, 2020, 12, 1570, DOI:10.3390/cancers12061570.
- I. Nassar, M. Aal, W. El-Sayed, M. Shahin, E. Elsakka, M. Mokhtar, M. Hegazy, M. Hagras, A. Mandour and N. Ismail, Discovery of pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives as novel CDK2 inhibitors: synthesis, biological and molecular modeling investigations, RSC Adv., 2022, 12, 14865–14882, 10.1039/d2ra01968j.
- M. Hammouda, H. Gaffer and K. Elattar, Insights into the medicinal chemistry of heterocycles integrated with a pyrazolo[1,5-a]pyrimidine scaffold, RSC Med. Chem., 2022, 13(10), 1150–1196, 10.1039/d2md00192f.
- K. Abdellatif and R. Bakr, New advances in synthesis and clinical aspects of pyrazolo[3,4-d]pyrimidine scaffolds, Bioorg. Chem., 2018, 78, 341–357, DOI:10.1016/j.bioorg.2018.03.032.
- A. Naglah, A. Askar, A. Hassan, T. Khatab, M. Al-Omar and M. Bhat, Biological Evaluation and Molecular Docking within Silico Physicochemical, Pharmacokinetic and Toxicity Prediction of Pyrazolo[1,5-a]pyrimidines, Molecules, 2020, 25, 1431, DOI:10.3390/molecules25061431.
- S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. Hampannavar, N. Thapliyal and V. Palakollu, An insight on synthetic and medicinal aspects of pyrazolo[1,5-a]pyrimidine scaffold, Eur. J. Med. Chem., 2017, 126, 298–352, DOI:10.1016/j.ejmech.2016.11.019.
- A. Hassan, N. Morsy, W. Aboulthana and A. Ragab, In vitro enzymatic evaluation of some pyrazolo[1,5-a]pyrimidine derivatives: design, synthesis, antioxidant, anti-diabetic, anti-Alzheimer, and anti-arthritic activities with molecular modeling simulation, Drug Dev. Res., 2022, 84, 3–24, DOI:10.1002/ddr.22008.
- L. Crocetti, G. Guerrini, F. Melani, C. Vergelli and M. Giovannoni, 4,5-Dihydro-5-Oxo-Pyrazolo[1,5-a]Thieno[2,3-c]Pyrimidine: A Novel Scaffold Containing Thiophene Ring. Chemical Reactivity and In Silico Studies to Predict the Profile to GABAA Receptor Subtype, Molecules, 2023, 28, 3054, DOI:10.3390/molecules28073054.
- A. Al-Azmi, Pyrazolo[1,5-a]pyrimidines: A Close Look into their Synthesis and Applications, Curr. Org. Chem., 2019, 721–743, DOI:10.2174/1385272823666190410145238.
- D. Compton, S. Sheng, K. Carlson, N. Rebacz, I. Lee, B. Katzenellenbogen and J. Katzenellenbogen, Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity, J. Med. Chem., 2004, 47(24), 5872–5893, DOI:10.1021/JM049631K.
- A. Abdallah and G. Elgemeie, New Route to the Synthesis of Novel Pyrazolo[1,5-a]pyrimidines and Evaluation of their Antimicrobial Activity as RNA Polymerase Inhibitors, Med. Chem., 2022, 926–948, DOI:10.2174/1573406418666220302092414.
- J. Lim, M. Altman, J. Baker, J. Brubaker, H. Chen, Y. Chen, T. Fischmann, C. Gibeau, M. Kleinschek, E. Leccese, C. Lesburg, J. Maclean, L. Moy, E. Mulrooney, J. Presland, L. Rakhilina, G. Smith, D. Steinhuebel and R. Yang, Discovery of 5-Amino-N-(1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide Inhibitors of IRAK4, ACS Med. Chem. Lett., 2015, 6(6), 683–688, DOI:10.1021/acsmedchemlett.5b00107.
- L. Squarcialupi, M. Falsini, D. Catarzi, F. Varano, M. Betti, K. Varani, F. Vincenzi, D. Ben, C. Lambertucci, R. Volpini and V. Colotta, Exploring the 2- and 5-positions of the pyrazolo[4,3-d]pyrimidin-7-amino scaffold to target human A1 and A2A adenosine receptors, Bioorg. Med. Chem., 2016, 24(12), 2794–2808, DOI:10.1016/j.bmc.2016.04.048.
- F. Varano, D. Catarzi, M. Falsini, D. Ben, M. Buccioni, G. Marucci, R. Volpini and V. Colotta, Novel human adenosine receptor antagonists based on the 7-amino-thiazolo[5,4-d]pyrimidine scaffold. Structural investigations at the 2-, 5- and 7-positions to enhance affinity and tune selectivity, Bioorg. Med. Chem. Lett., 2019, 29(4), 563–569, DOI:10.1016/j.bmcl.2018.12.062.
- D. Kishore, C. Balakumar, A. Rao, P. Roy and K. Roy, QSAR of adenosine receptor antagonists: exploring physicochemical requirements for binding of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives with human adenosine A(3) receptor subtype, Bioorg. Med. Chem. Lett., 2011, 21(2), 818–823, DOI:10.1016/j.bmcl.2010.11.094.
- S. Degorce, S. Boyd, J. Curwen, R. Ducray, C. Halsall, C. Jones, F. Lach, E. Lenz, M. Pass, S. Pass and C. Trigwell, Discovery of a Potent, Selective, Orally Bioavailable, and Efficacious Novel 2-(Pyrazol-4-ylamino)-pyrimidine Inhibitor of the Insulin-like Growth Factor-1 Receptor (IGF-1R), J. Med. Chem., 2016, 59(10), 4859–4866, DOI:10.1021/acs.jmedchem.6b00203.
- Y. Koizumi, Y. Tanaka, T. Matsumura, Y. Kadoh, H. Miyoshi, M. Hongu, K. Takedomi, J. Kotera, T. Sasaki, H. Taniguchi, Y. Watanabe, M. Takakuwa, K. Kojima, N. Baba, I. Nakamura and E. Kawanishi, Discovery of a pyrazolo[1,5-a]pyrimidine derivative (MT-3014) as a highly selective PDE10A inhibitor via core structure transformation from the stilbene moiety, Bioorg. Med. Chem., 2019, 3440–3450, DOI:10.1016/j.bmc.2019.06.021.
- S. Sheikhi-Mohammareh, F. Oroojalian, H. Beyzaei, M. Moghaddam-manesh, A. Salimi, F. Azizollahi and A. Shiri, Domino protocol for the synthesis of diversely functionalized derivatives of a novel fused pentacyclic antioxidant/anticancer fluorescent scaffold: pyrazolo[5'',1'':2',3']pyrimido[4',5':5,6][1,4]thiazino[2,3-b]quinoxaline, Talanta, 2023, 262, 124723, DOI:10.2139/ssrn.4369510.
- M. Stypik, M. Zagozda, S. Michałek, B. Dymek, D. Zdżalik-Bielecka, M. Dziachan, N. Orłowska, P. Gunerka, P. Turowski, J. Hucz-Kalitowska, A. Stańczak, P. Stanczak, K. Mulewski, D. Smuga, F. Stefaniak, L. Gurba-Bryśkiewicz, A. Leniak, Z. Ochal, M. Mach, K. Dzwonek, M. Lamparska-Przybysz, K. Dubiel and M. Wieczorek, Design, Synthesis, and Development of pyrazolo[1,5-a]pyrimidine Derivatives as a Novel Series of Selective PI3Kδ Inhibitors: Part I—Indole Derivatives, Pharmaceuticals, 2022, 15, 949, DOI:10.3390/ph15080949.
- N. Ismail, G. Ali, D. Ibrahim and A. Elmetwali, Medicinal attributes of pyrazolo[1,5-a]pyrimidine based scaffold derivatives targeting kinases as anticancer agents, Futur. J. Pharm. Sci., 2016, 2, 60–70, DOI:10.1016/J.FJPS.2016.08.004.
- L. Vymětalová, L. Havlícek, A. Šturc, Z. Skrášková, R. Jorda, T. Pospíšil, M. Strnad and V. Kryštof, 5-Substituted 3-isopropyl-7-[4-(2-pyridyl)benzyl]amino-1(2)H-pyrazolo[4,3-d]pyrimidines with anti-proliferative activity as potent and selective inhibitors of cyclin-dependent kinases, Eur. J. Med. Chem., 2016, 110, 291–301, DOI:10.1016/j.ejmech.2016.01.011.
- C. Tintori, A. Fallacara, M. Radi, C. Zamperini, E. Dreassi, E. Crespan, G. Maga, S. Schenone, F. Musumeci, C. Brullo, A. Richters, F. Gasparrini, A. Angelucci, C. Festuccia, S. Monache, D. Rauh and M. Botta, Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma, J. Med. Chem., 2015, 58(1), 347–361, DOI:10.1021/jm5013159.
- S. Almehmadi, A. Alsaedi, M. Harras and T. Farghaly, Synthesis of a new series of pyrazolo[1,5-a]pyrimidines as CDK2 inhibitors and anti-leukemia, Bioorg. Chem., 2021, 117, 105431, DOI:10.1016/j.bioorg.2021.105431.
- A. Gaber, A. El-Morsy, F. Sherbiny, A. Bayoumi, K. El-Gamal, K. El-Adl, A. Al-karmalawy, R. Eldin, M. Saleh and H. Abulkhair, Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: Synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies, Arch. Pharmazie, 2021, e2100258, DOI:10.1002/ardp.202100258.
- P. Sikdar, T. Choudhuri, S. Paul, S. Das and A. Bagdi, K2S2O8-Promoted Consecutive Tandem Cyclization/Oxidative Halogenation: Access to 3-Halo-Pyrazolo[1,5-a]pyrimidines, ACS Omega, 2023, 8, 23851–23859, DOI:10.1021/acsomega.3c02270.
- H. Ibrahim, H. Behbehani and N. Mostafa, Scalable Sonochemical Synthetic Strategy for Pyrazolo[1,5-a]pyridine Derivatives: First Catalyst-Free Concerted [3 + 2] Cycloaddition of Alkyne and Alkene Derivatives to 2-Imino-1H-pyridin-1-amines, ACS Omega, 2019, 4, 7182–7193, DOI:10.1021/ACSOMEGA.9B00562.
- C. Ravi, S. Samanta, D. Mohan, N. Reddy and S. Adimurthy, Synthesis of Functionalized Pyrazolo[1,5-a]pyridines: [3+2] Cycloaddition of N-Aminopyridines and α,β-Unsaturated Carbonyl Compounds/Alkenes at Room Temperature, Synthesis, 2017, 49, 2513–2522, DOI:10.1055/s-0036-1588753.
- J. Castillo, D. Estupiñán, M. Nogueras, J. Cobo and J. Portilla, 6-(Aryldiazenyl)pyrazolo[1,5-a]pyrimidines as Strategic Intermediates for the Synthesis of Pyrazolo[5,1-b]purines, J. Org. Chem., 2016, 81(24), 12364–12373, DOI:10.1021/ACS.JOC.6B02431.
- H. Behbehani and H. Ibrahim, Synthetic Strategy for Pyrazolo[1,5-a]pyridine and Pyrido[1,2-b]indazole Derivatives through AcOH and O2-Promoted Cross-dehydrogenative Coupling Reactions between 1,3-Dicarbonyl Compounds and N-Amino-2-iminopyridines, ACS Omega, 2019, 4, 15289–15303, DOI:10.1021/acsomega.9b02430.
- X. Chu, B. Cheng, Y. Zhang, D. Ge, Z. Shen and T. Loh, Copper-catalyzed three-component cyclization of amidines, styrenes, and fluoroalkyl halides for the synthesis of modular fluoroalkylated pyrimidines, Chem. Commun., 2018, 54(21), 2615–2618, 10.1039/c7cc09571f.
- T. Wang, J. Xiong, W. Wang, R. Li, X. Tang and F. Xiong, Tunable regioselective synthesis of pyrazolo[3,4-d]pyrimidine derivatives via aza-Wittig cyclization and dimroth-type rearrangement, RSC Adv., 2015, 5, 19830–19837, 10.1039/C4RA15777J.
- G. Deng, F. Wang, S. Lu and B. Cheng, Synthesis of Pyrano[3,2-c]pyrazol-7(1H)-one Derivatives by Tandem Cyclization of 2-Diazo-3,5-dioxo-6-ynoates (Ynones), Org. Lett., 2015, 17(19), 4651–4653, DOI:10.1021/acs.orglett.5b02369.
- S. Basceken, S. Kaya and M. Balci, Intramolecular Gold-Catalyzed and NaH-Supported Cyclization Reactions of N-Propargyl Indole Derivatives with Pyrazole and Pyrrole Rings: Synthesis of Pyrazolodiazepinoindole, Pyrazolopyrazinoindole, and Pyrrolopyrazinoindole, J. Org. Chem., 2015, 80(24), 12552–12561, DOI:10.1021/acs.joc.5b02419.
- S. Basceken and M. Balci, Design of pyrazolo-pyrrolo-pyrazines and pyrazolo-pyrrolo-diazepines via AuCl3-catalyzed and NaH-supported cyclization of N-propargyl pyrazoles, J. Org. Chem., 2015, 80(8), 3806–3814, DOI:10.1021/acs.joc.5b00034.
- L. Dai, K. Mao, Z. Pan and L. Rong, Green metal-free synthesis of spiro-fused 3,4′-pyrazolo[4′,3′:5,6]pyrido[2,3-d]pyrimidine derivatives via deamination cyclization reactions in aqueous medium, Res. Chem. Intermed., 2018, 45, 769–788, DOI:10.1007/s11164-018-3642-3.
- A. Götzinger, F. Theßeling, C. Hoppe and T. Müller, One-Pot Coupling-Coupling-Cyclocondensation Synthesis of Fluorescent Pyrazoles, J. Org. Chem., 2016, 81(21), 10328–10338, DOI:10.1021/ACS.JOC.6B01326.
- R. Belaroussi, A. Bouakher, M. Marchivie, S. Massip, C. Jarry, A. Hakmaoui, G. Guillaumet, S. Routier and M. Akssira, Convenient Synthesis of New N-3-Substituted Pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidine-2,4(1H,3H)-dione Derivatives, Synthesis, 2013, 45, 2557–2566, DOI:10.1055/S-0033-1338514.
- N. Petek, B. Štefane, M. Novinec and J. Svete, Synthesis and biological evaluation of 7-(aminoalkyl)pyrazolo[1,5-a]pyrimidine derivatives as cathepsin K inhibitors, Bioorg. Chem., 2019, 84, 226–238, DOI:10.1016/j.bioorg.2018.11.029.
- M. Ezzati, J. Khalafy, A. Marjani and R. Prager, The catalyst-free syntheses of pyrazolo[3,4-b]quinolin-5-one and pyrazolo[4′,3′:5,6]pyrido[2,3-d]pyrimidin-5,7-dione derivatives by one-pot, three-component reactions, Tetrahedron, 2017, 73, 6587–6596, DOI:10.1016/J.TET.2017.10.004.
- J. Portilla, J. Quiroga, M. Nogueras and J. Cobo, Regioselective synthesis of fused pyrazolo[1,5-a]pyrimidines by reaction of 5-amino-1H-pyrazoles and β-dicarbonyl compounds containing five-membered rings, Tetrahedron, 2012, 68, 988–994, DOI:10.1016/J.TET.2011.12.001.
- M. S. Moustafa, A. M. Nour-Eldeen, S. M. Al-Mousawi, A. Abd El-Hameed, M. Magdy and K. U. Sadek, Regioselectivity in the reaction of 5-amino-3-anilino-1H-pyrazole-4-carbonitrile with cinnamonitriles and enaminones: Synthesis of functionally substituted pyrazolo[1,5-a]pyrimidine derivatives, Green Process. Synth., 2022, 11(1), 116–128, DOI:10.1515/gps-2022-0009.
- A. O. Abdelhamid and S. M. Gomha, Synthesis of new pyrazolo[1,5-a]pyrimidine, triazolo[4,3-a]pyrimidine derivatives, and thieno[2,3-b]pyridine derivatives from sodium 3-(5-methyl-1-phenyl-1H-pyrazol-4-yl)-3-oxoprop-1-en-1-olate, J. Chem., 2013, 2013, 327095, DOI:10.1155/2013/327095.
- V. Kokorekin, S. Neverov, V. Kuzina and V. Petrosyan, A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines, Molecules, 2020, 25, 4169, DOI:10.3390/molecules25184169.
- Y. Wu, H. Li, L. Liu, D. Wang, H. Yang and Y. Chen, Efficient Construction of Pyrazolo[1,5-a]pyrimidine Scaffold and its Exploration as a New Heterocyclic Fluorescent Platform, J. Fluoresc., 2008, 18, 357–363, DOI:10.1007/s10895-007-0275-0.
- O. Stepaniuk, V. Matviienko, I. Kondratov, I. Vitruk and A. Tolmachev, Synthesis of New Pyrazolo[1,5-a]pyrimidines by Reaction of β,γ-Unsaturated γ-Alkoxy-α-keto Esters with N-Unsubstituted 5-Aminopyrazoles, Synthesis, 2013, 45, 925–930, DOI:10.1055/S-0032-1318329.
- A. Poursattar, J. Khalafy, F. Salami and M. Ezzati, The synthesis of new pyrazolo[1,5-a]pyrimidine derivatives, Arch. Org. Chem., 2015, 2015, 277–286, DOI:10.3998/ark.5550190.p009.062.
- C. Messaoudi, B. Jismy, J. Jacquemin, H. Allouchi, H. M'Rabet and M. Abarbri, Stepwise synthesis of 2,6-difunctionalized ethyl pyrazolo[1,5-a]pyrimidine-3-carboxylate via site-selective cross-coupling reactions: experimental and computational studies, Org. Biomol. Chem., 2022, 9684–9697, 10.1039/d2ob01760a.
- S. Krishnammagari, B. Cho, J. Kim and Y. Jeong, An efficient and solvent-free one-pot multi-component synthesis of novel highly substituted pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine-3-carbonitrile derivatives catalyzed by tetramethylguanidine, Synth. Commun., 2018, 48, 2663–2674, DOI:10.1080/00397911.2018.1514053.
- A. Rahmati, One-pot synthesis of 2-alkyl-7-amino-5-aryl-pyrazolo[1,5-a]pyrimidine-6-carbonitriles via a domino three-component condensation-oxidation reaction, C. R. Chim., 2012, 15, 647–652, DOI:10.1016/J.CRCI.2012.06.006.
- F. Alizadeh-bami and H. Mehrabi, Green Synthesis of Novel [1,3,4]Thiadiazolo[3,2-a]Pyrimidines via Three-Component Reaction of 5-Amino-1,3,4-Thiadiazole-2-Thiol, Aromatic Aldehydes, and Meldrum's Acid, Polycyclic Aromat. Compd., 2022, 43, 7343–7354, DOI:10.1080/10406638.2022.2150654.
- W. Hao, P. Zhou, F. Wu, B. Jiang, S. Tu and G. Li, New Three-Component Bicyclization Leading to Densely Functionalized Pyrazolo[3,4-d]thiazolo[3,2-a]pyrimidines, Eur. J. Org Chem., 2016, 11, 1968–1971, DOI:10.1002/ejoc.201600163.
- G. Hoang, A. Streit and J. Ellman, Three-Component Coupling of Aldehydes, Aminopyrazoles, and Sulfoxonium Ylides via Rhodium(III)-Catalyzed Imidoyl C-H Activation: Synthesis of Pyrazolo[1,5- a]pyrimidines, J. Org. Chem., 2018, 83(24), 15347–15360, DOI:10.1021/acs.joc.8b02606.
- S. Gulati, S. John and N. Shankaraiah, Microwave-assisted multicomponent reactions in heterocyclic chemistry and mechanistic aspects, Beilstein J. Org. Chem., 2021, 17, 819–865, DOI:10.3762/bjoc.17.71.
- M. Driowya, A. Saber, H. Marzag, L. Demange, R. Benhida and K. Bougrin, Microwave-Assisted Synthesis of Bioactive Six-Membered Heterocycles and Their Fused Analogues, Molecules, 2016, 21, 492, DOI:10.3390/molecules21040492.
- T. Damera, R. Pagadala, S. Rana and S. Jonnalagadda, A Concise Review of Multicomponent Reactions Using Novel Heterogeneous Catalysts under Microwave Irradiation, Catalysts, 2023, 1034, DOI:10.3390/catal13071034.
- M. Driowya, A. Saber, H. Marzag, L. Demange, K. Bougrin and R. Benhida, Microwave-Assisted Syntheses of Bioactive Seven-Membered, Macro-Sized Heterocycles and Their Fused Derivatives, Molecules, 2016, 21, 1032, DOI:10.3390/molecules21081032.
- B. Banerjee, Recent developments on ultrasound-assisted one-pot multicomponent synthesis of biologically relevant heterocycles, Ultrason. Sonochem., 2017, 35, 15–35, DOI:10.1016/j.ultsonch.2016.10.010.
- J. Ng, E. Tiekink and A. Dolzhenko, Three-component microwave-assisted synthesis of 3,5-disubstituted pyrazolo[3,4-d]pyrimidin-4-ones, RSC Adv., 2022, 12, 8323–8332, 10.1039/d2ra00980c.
- E. Polo, K. Ferrer-Pertuz, J. Trilleras, J. Quiroga and M. Gutiérrez, Microwave-assisted one-pot synthesis in water of carbonylpyrazolo[3,4-b]pyridine derivatives catalyzed by InCl3 and sonochemical assisted condensation with aldehydes to obtain new chalcone derivatives containing the pyrazolopyridinic moiety, RSC Adv., 2017, 7, 50044–50055, 10.1039/C7RA10127A.
- A. M. Fahim, A. M. Farag, M. R. Shaaban and E. A. Ragab, Microwave-assisted synthesis of pyrazolo[1,5-a]pyrimidine, triazolo[1,5-a]pyrimidine, pyrimido[1,2-a]benzimidazole, triazolo[5,1-c][1,2,4]triazine and imidazo[2,1-c][1,2,4]triazine, Curr. Microwave Chem., 2018, 5(2), 111–119, DOI:10.2174/2213335605666180425144009.
- G. Hoang, A. Streit and J. Ellman, Three-Component Coupling of Aldehydes, Aminopyrazoles, and Sulfoxonium Ylides via Rhodium(III)-Catalyzed Imidoyl C-H Activation: Synthesis of Pyrazolo[1,5- a]pyrimidines, J. Org. Chem., 2018, 83(24), 15347–15360, DOI:10.1021/acs.joc.8b02606.
- K. Shekarrao, P. Kaishap, V. Saddanapu, A. Addlagatta, S. Gogoi and R. Boruah, Microwave-assisted palladium mediated efficient synthesis of pyrazolo[3,4-b]pyridines, pyrazolo[3,4-b]quinolines, pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a]quinazolines, RSC Adv., 2014, 4, 24001–24006, 10.1039/C4RA02865A.
- I. Bassoude, Z. Tber, E. Essassi, G. Guillaumet and S. Berteina-Raboin, A one-pot process for the microwave-assisted synthesis of 7-substituted pyrazolo[1,5-a]pyrimidine, RSC Adv., 2016, 6, 3301–3306, 10.1039/C5RA23417D.
- M. Balkenhohl, B. Salgues, T. Hirai, K. Karaghiosoff and P. Knochel, Regioselective Metalation and Functionalization of the Pyrazolo[1,5- a]pyridine Scaffold Using Mg- and Zn-TMP Bases, Org. Lett., 2018, 20(10), 3114–3118, DOI:10.1021/acs.orglett.8b01204.
- J. Sun, J. Qiu, B. Jiang, W. Hao, C. Guo and S. Tu, I2-Catalyzed Multicomponent Reactions for Accessing Densely Functionalized Pyrazolo[1,5-a]pyrimidines and Their Disulphenylated Derivatives, J. Org. Chem., 2016, 81(8), 3321–3328, DOI:10.1021/acs.joc.6b00332.
- C. Wiethan, S. Franceschini, H. Bonacorso and M. Stradiotto, Synthesis of pyrazolo[1,5-a]quinoxalin-4(5H)-ones via one-pot amidation/N-arylation reactions under transition metal-free conditions, Org. Biomol. Chem., 2016, 14(37), 8721–8727, 10.1039/C6OB01407K.
- S. Paul, S. Das, T. Choudhuri, P. Sikdar and A. Bagdi, Visible-Light-Induced Regioselective C-H Sulfenylation of Pyrazolo[1,5-a]pyrimidines via Cross-Dehydrogenative Coupling, J. Org. Chem., 2023, 4187–4198, DOI:10.1021/acs.joc.2c02665.
- T. Choudhuri, S. Paul, S. Das, D. Pathak and A. Bagdi, Visible-Light-Mediated Regioselective C3-H Selenylation of Pyrazolo[1,5-a]pyrimidines Using Erythrosine B as Photocatalyst, J. Org. Chem., 2023, 8992–9003, DOI:10.1021/acs.joc.3c00720.
- P. Kaswan, K. Pericherla, D. Purohit and A. Kumar, Synthesis of 5,7-diarylpyrazolo[1,5-a]pyrimidines via KOH mediated tandem reaction of 1H-pyrazol-3-amines and chalcones, Tetrahedron Lett., 2015, 56, 549–553, DOI:10.1016/J.TETLET.2014.11.121.
- N. Metwally, T. Koraa and S. Sanad, Green one-pot synthesis and in vitro antibacterial screening of pyrano[2,3-c]pyrazoles, 4H-chromenes and pyrazolo[1,5-a]pyrimidines using biocatalyzed pepsin, Synth. Commun., 2022, 52, 1139–1154, DOI:10.1080/00397911.2022.2074301.
- S. Krishnammagari and Y. Jeong, An Efficient and Transition Metal-Free Base-Promoted Multi-Component Synthesis of Aza-Fused Polysubstituted Pyrido[2′,3′:3,4]Pyrazolo[1,5-a]Pyrimidine Derivatives, Polycyclic Aromat. Compd., 2018, 40, 1068–1083, DOI:10.1080/10406638.2018.1526808.
- J. Ren, S. Ding, X. Li, R. Bi and Q. Zhao, An Approach for the Synthesis of Pyrazolo[1,5-a]pyrimidines via Cu(II)-Catalyzed [3+3] Annulation of Saturated Ketones with Aminopyrazoles, J. Org. Chem., 2021, 12762–12771, DOI:10.1021/acs.joc.1c01343.
- S. Kotla, J. Vandavasi, J. Wang, K. Parang and R. Tiwari, Palladium-Catalyzed Intramolecular Cross-Dehydrogenative Coupling: Synthesis of Fused Imidazo[1,2-a]pyrimidines and Pyrazolo[1,5-a]pyrimidines, ACS Omega, 2017, 2, 11–19, DOI:10.1021/acsomega.6b00417.
- M. Bakherad, A. Keivanloo, M. Gholizadeh, R. Doosti and M. Javanmardi, Using magnetized water as a solvent for a green, catalyst-free, and efficient protocol for the synthesis of pyrano[2,3-c]pyrazoles and pyrano[4′,3′:5,6]pyrazolo [2,3-d]pyrimidines, Res. Chem. Intermed., 2017, 43, 1013–1029, DOI:10.1007/s11164-016-2680-y.
- M. Daraie and M. Heravi, Molecular diversity of four–component synthesis of pyrazole based pyrido[2,3-d]pyrimidine-diones in water: a green synthesis, Arkivoc, 2016, 2016, 328–338, DOI:10.3998/ARK.5550190.P009.603.
- S. Krishnammagari and Y. Jeong, An efficient and green synthesis of novel highly functionalized nitrogen-fused pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine derivatives using recyclable choline hydroxide, Res. Chem. Intermed., 2018, 44, 7311–7329, DOI:10.1007/s11164-018-3558-y.
- X. Zhang, Y. Song, L. Gao, X. Guo and X. Fan, Highly facile and regio-selective synthesis of pyrazolo[1,5-a]pyrimidines via reactions of 1,2-allenic ketones with aminopyrazoles, Org. Biomol. Chem., 2014, 12(13), 2099–2107, 10.1039/c3ob42445f.
- A. Davoodnia, M. Roshani, S. Malaeke and M. Bakavoli, A rapid synthesis of isoxazolo[5,4-d]pyrimidin-4(5H)-ones under microwave irradiation with solid acid catalysis in solvent-free conditions, Chin. Chem. Lett., 2008, 19, 525–528, DOI:10.1016/J.CCLET.2008.01.037.
- D. Sureja and K. Vadalia, Microwave assisted, solvent-free synthesis and in-vitro antimicrobial screening of some novel pyrazolo[3,4-d]pyrimidin-4(5H)-one derivatives, Beni-Suef Univ. J. Basic Appl. Sci., 2017, 6, 33–38, DOI:10.1016/J.BJBAS.2016.12.006.
- A. Oliveira-Campos, A. Sivasubramanian, L. Rodrigues, J. Seijas, M. Vázquez-Tato, F. Peixoto, C. Abreu, H. Cidade, A. Oliveira and M. Pinto, Substituted Pyrazolo[3,4-d]pyrimidines: Microwave-Assisted, Solvent-Free Synthesis and Biological Evaluation, Helv. Chim. Acta, 2008, 91, 1336–1345, DOI:10.1002/HLCA.200890145.
- R. Rao, B. Mm, B. Maiti, R. Thakuria and K. Chanda, Efficient Access to Imidazo[1,2-a]pyridines/pyrazines/pyrimidines via Catalyst-Free Annulation Reaction under Microwave Irradiation in Green Solvent, ACS Comb. Sci., 2018, 20(3), 164–171, DOI:10.1021/acscombsci.7b00173.
- L. Ming, W. Shuwen, W. Lirong, Y. Huazheng and Z. Xiuli, A convenient, rapid, and highly selective method for synthesis of new pyrazolo[1,5-a]pyrimidines via the reaction of enaminones and 5-amino-1H-pyrazoles under microwave irradiation, J. Heterocycl. Chem., 2005, 42, 925–930, DOI:10.1002/JHET.5570420526.
- S. Konda, S. Chobe, A. Gosar, B. Hote and G. Mandawad, Polyethylene glycol-400 Prompted an Efficient Synthesis of Thienyl Pyrazolo[1,5-a] pyrimidines as Microbial Inhibitors, Curr. Org. Synth., 2022, 693–701, DOI:10.2174/1570179419666220304160938.
- N. Metwally, T. Koraa and S. Sanad, Green one-pot synthesis and in vitro antibacterial screening of pyrano[2,3-c]pyrazoles, 4H-chromenes and pyrazolo[1,5-a]pyrimidines using biocatalyzed pepsin, Synth. Commun., 2022, 52, 1139–1154, DOI:10.1080/00397911.2022.2074301.
- S. Das, T. R. A. Sangma, L. B. Marpna and J. N. Vishwakarma, A green synthetic strategy for pyrazolo[1,5-a]pyrimidin-7(4H)-one derivatives by the reaction of aminopyrazoles and symmetric/non-symmetric alkynes assisted by KHSO4 in aqueous media, Periodica Polytech., Chem. Eng., 2024, 68(3), 287–296, DOI:10.3311/PPch.23688.
- D. Novikova, A. Mustafa, T. Grigoreva, S. Vorona, S. Selivanov and V. Tribulovich, NMR-Verified Dearomatization of 5,7-Substituted Pyrazolo[1,5-a]pyrimidines, Molecules, 2023, 28, 6584, DOI:10.3390/molecules28186584.
- H. Sutherland, P. Choi, G. Lu, A. Giddens, A. Tong, S. Franzblau, C. Cooper, B. Palmer and W. Denny, Synthesis and Structure–Activity Relationships for the Anti-Mycobacterial Activity of 3-Phenyl-N-(Pyridin-2-ylmethyl)Pyrazolo[1,5-a]Pyrimidin-7-Amines, Pharmaceuticals, 2022, 15, 1125, DOI:10.3390/ph15091125.
- A. Ivachtchenko, E. Golovina, M. Kadieva, V. Kysil, O. Mitkin, S. Tkachenko and I. Okun, Synthesis and structure-activity relationship (SAR) of (5,7-disubstituted 3-phenylsulfonyl-pyrazolo[1,5-a]pyrimidin-2-yl)-methylamines as potent serotonin 5-HT(6) receptor (5-HT(6)R) antagonists, J. Med. Chem., 2011, 54(23), 8161–8173, DOI:10.1021/jm201079g.
- J. Roux, C. Leriche, P. Chamiot-Clerc, J. Feutrill, F. Halley, D. Papin, N. Derimay, C. Mugler, C. Grépin and L. Schio, Preparation and optimization of pyrazolo[1,5-a]pyrimidines as new potent PDE4 inhibitors, Bioorg. Med. Chem. Lett., 2016, 26(2), 454–459, DOI:10.1016/j.bmcl.2015.11.093.
- S. Sukanya, T. Venkatesh, S. Rao and M. Joy, Efficient L-Proline catalyzed synthesis of some new (4-substituted-phenyl)-1,5-dihydro-2H-pyrimido[4,5-d][1,3]thiazolo[3,2a]-pyrimidine-2,4(3H)-diones bearing thiazolopyrimidine derivatives and evaluation of their pharmacological activities, J. Mol. Struct., 2022, 1247, 131324, DOI:10.1016/J.MOLSTRUC.2021.131324.
- F. Stefanello, J. Vieira, J. Araújo, V. Souza, C. Frizzo, M. Martins, N. Zanatta, B. Iglesias and H. Bonacorso, Solution and Solid-State Optical Properties of Trifluoromethylated 5-(Alkyl/aryl/heteroaryl)-2-methyl-pyrazolo[1,5-a]pyrimidine System, Photochem, 2022, 345–357, DOI:10.3390/photochem2020024.
- A. Oyebamiji, S. Akintelu, K. Odelade, B. Adetuyi, E. Akintayo, C. Akintayo, B. Semire and J. Babalola, Electron Withdrawing Group Effect On Biological Activities Of Pyrimidine Hybrids As Potential Anti-Matrix Metalloproteinase-7, Quím. Nova, 2023 DOI:10.21577/0100-4042.20230055.
- S. Sanad, M. Ahmed, A. Mekky and Z. Abdallah, Regioselective synthesis and theoretical calculations of Bis(pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidines) linked to benzofuran units via piperazine spacer: A DFT, MM2, and MMFF94 study, J. Mol. Struct., 2021, 1243, 130802, DOI:10.1016/J.MOLSTRUC.2021.130802.
- P. Baraldi, B. Cacciari, G. Spalluto, M. Villatoro, C. Zocchi, S. Dionisotti and E. Ongini, Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives: potent and selective A(2A) adenosine antagonists, J. Med. Chem., 1996, 39(5), 1164–1171, DOI:10.1021/JM950746L.
- P. Baraldi, B. Cacciari, G. Spalluto, M. Villatoro, C. Zocchi, S. Dionisotti and E. Ongini, Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives: potent and selective A(2A) adenosine antagonists, J. Med. Chem., 1996, 39(5), 1164–1171, DOI:10.1021/JM950746L.
- A. Bhat, R. Dongre, F. Almalki, M. Berredjem, M. Aissaoui, R. Touzani, T. Hadda and M. Akhter, Synthesis, biological activity and POM/DFT/docking analyses of annulated pyrano[2,3-d]pyrimidine derivatives: Identification of antibacterial and antitumor pharmacophore sites, Bioorg. Chem., 2020, 104480, DOI:10.1016/j.bioorg.2020.104480.
- B. Jismy, A. Tikad, M. Akssira, G. Guillaumet and M. Abarbri, Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions, Molecules, 2020, 25, 2062, DOI:10.3390/molecules25092062.
- A. Ivachtchenko, E. Golovina, M. Kadieva, A. Koryakova, O. Mitkin, S. Tkachenko, V. Kysil and I. Okun, 2-Substituted 5,6-dimethyl-3-phenylsulfonyl-pyrazolo[1,5-a]pyrimidines: new series of highly potent and specific serotonin 5-HT6 receptor antagonists, Eur. J. Med. Chem., 2011, 46(4), 1189–1197, DOI:10.1016/j.ejmech.2011.01.038.
- A. Ivachtchenko, E. Golovina, M. Kadieva, V. Kysil, O. Mitkin, A. Vorobiev and I. Okun, Antagonists of 5-HT6 receptors. Substituted 3-(phenylsulfonyl)pyrazolo[1,5-a]pyrido[3,4-e]pyrimidines and 3-(phenylsulfonyl)pyrazolo[1,5-a]pyrido[4,3-d]pyrimidines-Synthesis and 'structure-activity' relationship, Bioorg. Med. Chem. Lett., 2012, 22(13), 4273–4280, DOI:10.1016/j.bmcl.2012.05.036.
- R. Friesen, Y. Ducharme, R. Ball, M. Blouin, L. Boulet, B. Côté, R. Frenette, M. Girard, D. Guay, Z. Huang, T. Jones, F. Laliberté, J. Lynch, J. Mancini, E. Martins, P. Masson, E. Muise, D. Pon, P. Siegl, A. Styhler, N. Tsou, M. Turner, R. Young and Y. Girard, Optimization of a tertiary alcohol series of phosphodiesterase-4 (PDE4) inhibitors: structure-activity relationship related to PDE4 inhibition and human ether-a-go-go related gene potassium channel binding affinity, J. Med. Chem., 2003, 46(12), 2413–2426, DOI:10.1021/JM0204542.
- J. O'Donnell and H. Zhang, Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4), Trends Pharmacol. Sci., 2004, 25(3), 158–163, DOI:10.1016/J.TIPS.2004.01.003.
- X. Feng, H. Wang, M. Ye, X. Xu, Y. Xu, W. Yang, H. Zhang, G. Song and H. Ke, Identification of a PDE4-Specific Pocket for the Design of Selective Inhibitors, Biochemistry, 2018, 57(30), 4518–4525, DOI:10.1021/acs.biochem.8b00336.
- H. Zhang, Y. Zhao, Y. Huang, C. Deng, A. Hopper, M. Vivo, G. Rose and J. O'Donnell, Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats, Psychopharmacology, 2006, 186, 209–217, DOI:10.1007/s00213-006-0369-4.
- V. Lê, H. Hung, N. Ha, C. Le, P. Minh and D. Lam, Natural Phosphodiesterase-4 Inhibitors with Potential Anti-Inflammatory Activities from Millettia dielsiana, Molecules, 2023, 28, 7253, DOI:10.3390/molecules28217253.
- D. Hu, D. Zhao, M. He, H. Jin, Y. Tang, L. Zhang, G. Song and Z. Cui, Synthesis and bioactivity of 3,5-dimethylpyrazole derivatives as potential PDE4 inhibitors, Bioorg. Med. Chem. Lett., 2018, 28(19), 3276–3280, DOI:10.1016/j.bmcl.2018.03.031.
- M. Mey, A. Hatzelmann, G. Klink, I. Laan, G. Sterk, U. Thibaut, W. Ulrich and H. Timmerman, Novel selective PDE4 inhibitors. 2. Synthesis and structure-activity relationships of 4-aryl-substituted cis-tetra- and cis-hexahydrophthalazinones, J. Med. Chem., 2001, 44(16), 2523–2535, DOI:10.1021/JM010838C.
- Y. Li, D. Hu, D. Zhao, X. Liu, H. Jin, G. Song, Z. Cui and L. Zhang, Design, synthesis and biological evaluation of 2,4-disubstituted oxazole derivatives as potential PDE4 inhibitors, Bioorg. Med. Chem., 2017, 25(6), 1852–1859, DOI:10.1016/j.bmc.2017.01.047.
- R. Allcock, H. Blakli, Z. Jiang, K. Johnston, K. Morgan, G. Rosair, K. Iwase, Y. Kohno and D. Adams, Phosphodiesterase inhibitors. Part 1: Synthesis and structure-activity relationships of pyrazolopyridine-pyridazinone PDE inhibitors developed from ibudilast, Bioorg. Med. Chem. Lett., 2011, 21(11), 3307–3312, DOI:10.1016/j.bmcl.2011.04.021.
- K. Ochiai, S. Takita, A. Kojima, T. Eiraku, K. Iwase, T. Kishi, A. Ohinata, Y. Yageta, T. Yasue, D. Adams and Y. Kohno, Phosphodiesterase inhibitors. Part 5: hybrid PDE3/4 inhibitors as dual bronchorelaxant/anti-inflammatory agents for inhaled administration, Bioorg. Med. Chem. Lett., 2013, 23(1), 375–381, DOI:10.1016/j.bmcl.2012.08.121.
- D. Paes, M. Schepers, B. Rombaut, D. Hove, T. Vanmierlo and J. Prickaerts, The Molecular Biology of Phosphodiesterase 4 Enzymes as Pharmacological Targets: An Interplay of Isoforms, Conformational States, and Inhibitors, Pharmacol. Rev., 2021, 73, 1016–1049, DOI:10.1124/pharmrev.120.000273.
- X. Li, S. Vadrevu, A. Dunlop, J. Day, N. Advant, J. Tröger, E. Klussmann, E. Jaffrey, R. Hay, D. Adams, M. Houslay and G. Baillie, Selective SUMO modification of cAMP-specific phosphodiesterase-4D5 (PDE4D5) regulates the functional consequences of phosphorylation by PKA and ERK, Biochem. J., 2010, 428(1), 55–65, DOI:10.1042/BJ20091672.
- A. Jankowska, A. Świerczek, G. Chłoń-Rzepa, M. Pawłowski and E. Wyska, PDE7-Selective and Dual Inhibitors: Advances in Chemical and Biological Research, Curr. Med. Chem., 2017, 24(7), 673–700, DOI:10.2174/0929867324666170116125159.
- W. Richter, L. Unciuleac, T. Hermsdorf, T. Kronbach and D. Dettmer, Identification of inhibitor binding sites of the cAMP-specific phosphodiesterase 4, Cell. Signal., 2001, 13(4), 287–297, DOI:10.1016/S0898-6568(01)00150-4.
- A. Grewal, N. Sharma, S. Singh and S. Arora, In Silico Designing of Novel Thiazolidine-2-one Derivatives as Dual PDE4/7 Inhibitors for Inflammatory Disorders, J. Pharm. Technol. Res. Manag., 2019, 149–162, DOI:10.15415/jptrm.2017.52010.
- J. Day, B. Lindsay, T. Riddell, Z. Jiang, R. Allcock, A. Abraham, S. Sookup, F. Christian, J. Bogum, E. Martin, R. Rae, D. Anthony, G. Rosair, D. Houslay, E. Huston, G. Baillie, E. Klussmann, M. Houslay and D. Adams, Elucidation of a structural basis for the inhibitor-driven, p62 (SQSTM1)-dependent intracellular redistribution of cAMP phosphodiesterase-4A4 (PDE4A4), J. Med. Chem., 2011, 54(9), 3331–3347, DOI:10.1021/jm200070e.
- M. Fraley, R. Rubino, W. Hoffman, S. Hambaugh, K. Arrington, R. Hungate, M. Bilodeau, A. Tebben, R. Rutledge, R. Kendall, R. McFall, W. Huckle, K. Coll and K. Thomas, Optimization of a pyrazolo[1,5-a]pyrimidine class of KDR kinase inhibitors: improvements in physical properties enhance cellular activity and pharmacokinetics, Bioorg. Med. Chem. Lett., 2002, 12(24), 3537–3541, DOI:10.1016/S0960-894X(02)00827-2.
- G. Auzzi, F. Bruni, L. Cecchi, A. Costanzo, L. Vettori, R. Pirisino, M. Corrias, G. Ignesti, G. Banchelli and L. Raimondi, 2-Phenylpyrazolo[1,5-a]pyrimidin-7-ones. A new class of nonsteroidal antiinflammatory drugs devoid of ulcerogenic activity, J. Med. Chem., 1983, 26(12), 1706–1709, DOI:10.1021/JM00366A009.
- R. Jorda, L. Havlícek, M. Peřina, V. Vojáčková, T. Pospíšil, S. Djukic, J. Škerlová, J. Grúz, N. Renešová, P. Klener, P. Řezáčová, M. Strnad and V. Kryštof, 3,5,7-Substituted Pyrazolo[4,3-d]Pyrimidine Inhibitors of Cyclin-Dependent Kinases and Cyclin K Degraders, J. Med. Chem., 2022, 8881–8896, DOI:10.1021/acs.jmedchem.1c02184.
- A. Farag and A. Fahim, Synthesis, biological evaluation and DFT calculation of novel pyrazole and pyrimidine derivatives, J. Mol. Struct., 2019, 304–314, DOI:10.1016/J.MOLSTRUC.2018.11.008.
- O. Elbakry, M. Harras, M. Elsebaei, A. Mehany and H. El-Sehrawi, Development of pyrazolo[1,5-a]pyrimidine derivatives: Synthesis, anticancer activity and docking study, Int. J. Med. Pharmaceut. Sci., 2023 DOI:10.21608/aijpms.2023.203637.1207.
- V. Gembus, J. Bonfanti, O. Querolle, P. Jubault, V. Levacher and C. Hoarau, Palladium catalyzed one-pot sequential Suzuki cross-coupling-direct C-H functionalization of imidazo[1,2-a]pyrazines, Org. Lett., 2012, 14(23), 6012–6015, DOI:10.1021/ol3029059.
- M. Andrus and C. Song, Palladium-imidazolium carbene catalyzed aryl, vinyl, and alkyl Suzuki-Miyaura cross coupling, Org. Lett., 2001, 3(23), 3761–3764, DOI:10.1021/OL016724C.
- J. Li, X. Zhang, Y. Yao, Y. Gao, W. Yang and W. Zhao, Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling of Oxygen-Substituted Allylboronates with Aryl/Vinyl (Pseudo)Halides, J. Org. Chem., 2022, 6951–6959, DOI:10.1021/acs.joc.2c00634.
- H. Xu, K. Ekoue-Kovi and C. Wolf, Palladium-phosphinous acid-catalyzed cross-coupling of aryl and acyl halides with aryl-, alkyl-, and vinylzinc reagents, J. Org. Chem., 2008, 73(19), 7638–7650, DOI:10.1021/jo801445y.
- K. Kang, J. Kim, A. Lee, W. Kim and H. Kim, Palladium-Catalyzed Dehydrative Cross-Coupling of Allylic Alcohols and N-Heterocycles Promoted by a Bicyclic Bridgehead Phosphoramidite Ligand and an Acid Additive, Org. Lett., 2016, 18(3), 616–619, DOI:10.1021/acs.orglett.6b00001.
- J. Kerhervé, C. Botuha and J. Dubois, New asymmetric synthesis of protein farnesyltransferase inhibitors via palladium-catalyzed cross-coupling reactions of 2-iodo-imidazoles, Org. Biomol. Chem., 2009, 7(10), 2214–2222, 10.1039/b902601k.
- Z. Liu, N. Dong, M. Xu, Z. Sun and T. Tu, Mild Negishi cross-coupling reactions catalyzed by acenaphthoimidazolylidene palladium complexes at low catalyst loadings, J. Org. Chem., 2013, 78(15), 7436–7444, DOI:10.1021/jo400803s.
- C. So and F. Kwong, Palladium-catalyzed cross-coupling reactions of aryl mesylates, Chem. Soc. Rev., 2011, 40(10), 4963–4972, 10.1039/c1cs15114b.
- R. Kotipalli, A. Nagireddy and M. Reddy, Palladium-catalyzed cyclizative cross coupling of ynone oximes with 2-haloaryl N-acrylamides for isoxazolyl indoline bis-heterocycles, Org. Biomol. Chem., 2022, 2609–2614, 10.1039/d2ob00065b.
- Q. Liu, B. He, P. Qian and L. Shao, N-Heterocyclic carbene-palladium(ii)-1-methylimidazole complex catalyzed direct C-H bond arylation of imidazo[1,2-a]pyridines with aryl chlorides, Org. Biomol. Chem., 2017, 15(5), 1151–1154, 10.1039/c6ob02704k.
- P. Hsiao, R. Chang, A. Sue, J. Chu, G. Liao, Y. Lee and J. Huang, Synthesis and Mechanistic Investigation of Bipyrazolo[1,5-a]pyridines via Palladium-Catalyzed Cross-Dehydrogenative Coupling of Pyrazolo[1,5-a]pyridines, J. Org. Chem., 2022, 9851–9863, DOI:10.1021/acs.joc.2c00895.
- T. Nguyen, L. Poli and A. Garrison, Palladium-catalyzed oxidative C-H/C-H cross-coupling of pyrazolo[1,5-a]azines with five-membered heteroarenes, Chem. Commun., 2021, 827–830, 10.1039/d1cc06337e.
- W. McCoull, Discovery of Pyrazolo[1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors, J. Med. Chem., 2017, 60(10), 4386–4402, DOI:10.1021/acs.jmedchem.7b00359.
- Y. Xing, W. Guo, M. Wu, J. Xie, D. Huang, P. Hu, M. Zhou, L. Zhang, Q. Zhang, P. Wang, X. Wang, G. Wang, H. Wu, C. Zhou, Y. Chen, M. Liu, Z. Yi and Z. Sun, An orally available small molecule BCL6 inhibitor effectively suppresses diffuse large B cell lymphoma cells growth in vitro and in vivo, Cancer Lett., 2021 DOI:10.21203/rs.3.rs-820765/v1.
- H. Gu, J. He, Y. Li, D. Mi, T. Guan, W. Guo, B. Liu and Y. Chen, B-cell Lymphoma 6 Inhibitors: Current Advances and Prospects of Drug Development for Diffuse Large B-cell Lymphomas, J. Med. Chem., 2022, 15559–15583, DOI:10.1021/acs.jmedchem.2c01433.
- M. Cárdenas, W. Yu, W. Béguelin, M. Teater, H. Geng, R. Goldstein, E. Oswald, K. Hatzi, S. Yang, J. Cohen, R. Shaknovich, K. Vanommeslaeghe, H. Cheng, D. Liang, H. Cho, J. Abbott, W. Tam, W. Du, J. Leonard, O. Elemento, L. Cerchietti, T. Cierpicki, F. Xue, A. MacKerell and A. Melnick, Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma, J. Clin. Investig., 2016, 126(9), 3351–3362, DOI:10.1172/JCI85795.
- L. Cerchietti, K. Hatzi, E. Caldas-Lopes, S. Yang, M. Figueroa, R. Morin, M. Hirst, L. Mendez, R. Shaknovich, P. Cole, K. Bhalla, R. Gascoyne, M. Marra, G. Chiosis and A. Melnick, BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy, J. Clin. Investig., 2010, 120(12), 4569–4582, DOI:10.1172/JCI42869.
- Y. Ai, L. Hwang, A. MacKerell, A. Melnick and F. Xue, Progress toward B-Cell Lymphoma 6 BTB Domain Inhibitors for the Treatment of Diffuse Large B-Cell Lymphoma and Beyond, J. Med. Chem., 2021, 4333–4358, DOI:10.1021/acs.jmedchem.0c01686.
- W. Guo, Y. Xing, Q. Zhang, J. Xie, D. Huang, H. Gu, P. He, M. Zhou, S. Xu, X. Pang, M. Liu, Z. Yi and Y. Chen, Synthesis and Biological Evaluation of B-Cell Lymphoma 6 Inhibitors of N-phenyl-4-pyrimidinamine Derivatives Bearing Potent Activities against Tumor Growth, J. Med. Chem., 2020, 676–695, DOI:10.1021/acs.jmedchem.9b01618.
- B. Jismy, A. Tikad, M. Akssira, G. Guillaumet and M. Abarbri, Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions, Molecules, 2020, 25, 2062, DOI:10.3390/molecules25092062.
- G. Kurşunluoğlu, D. Erdoğan, E. Çağatay, E. Atalay, S. Guler, Y. Gungor and H. Kayali, The Role of Kinase Inhibitors in Cancer Therapies, Biochemistry, 2021 DOI:10.5772/intechopen.99070.
- N. Cohen, T. Kim and R. DeMatteo, Principles of Kinase Inhibitor Therapy for Solid Tumors, Ann. Surg., 2017, 265, 311–319, DOI:10.1097/SLA.0000000000001740.
- S. Whittaker, A. Mallinger, P. Workman and P. Clarke, Inhibitors of cyclin-dependent kinases as cancer therapeutics, Pharmacol. Ther., 2017, 173, 83–105, DOI:10.1016/j.pharmthera.2017.02.008.
- C. Mathison, D. Chianelli, P. Rucker, J. Nelson, J. Roland, Z. Huang, Y. Yang, J. Jiang, Y. Xie, R. Epple, B. Bursulaya, C. Lee, M. Gao, J. Shaffer, S. Briones, Y. Sarkisova, A. Galkin, L. Li, N. Li, C. Li, S. Hua, S. Kasibhatla, J. Kinyamu-Akunda, R. Kikkawa, V. Molteni and J. Tellew, Efficacy and Tolerability of Pyrazolo[1,5-a]pyrimidine RET Kinase Inhibitors for the Treatment of Lung Adenocarcinoma, ACS Med. Chem. Lett., 2020, 11(4), 558–565, DOI:10.1021/acsmedchemlett.0c00015.
- A. Fallacara, C. Zamperini, A. Podolski-Renić, J. Dinić, T. Stankovic, M. Stepanović, A. Mancini, E. Rango, G. Iovenitti, A. Molinari, F. Bugli, M. Sanguinetti, R. Torelli, M. Martini, L. Maccari, M. Valoti, E. Dreassi, M. Botta, M. Pešić and S. Schenone, A New Strategy for Glioblastoma Treatment: In Vitro and In Vivo Preclinical Characterization of Si306, a Pyrazolo[3,4-d]Pyrimidine Dual Src/P-Glycoprotein Inhibitor, Cancers, 2019, 11, 848, DOI:10.3390/cancers11060848.
- C. Greco, V. Taresco, A. Pearce, C. Vasey, S. Smith, R. Rahman, C. Alexander, R. Cavanagh, F. Musumeci and S. Schenone, Development of Pyrazolo[3,4-d]pyrimidine Kinase Inhibitors as Potential Clinical Candidates for Glioblastoma Multiforme, ACS Med. Chem. Lett., 2020, 11(5), 657–663, DOI:10.1021/acsmedchemlett.9b00530.
- C. Kurz, F. Preuss, A. Tjaden, M. Cusack, J. Amrhein, D. Chatterjee, S. Mathea, L. Berger, B. Berger, A. Krämer, M. Weller, T. Weiss, S. Müller, S. Knapp and T. Hanke, Illuminating the Dark: Highly Selective Inhibition of Serine/Threonine Kinase 17A with Pyrazolo[1,5-a]pyrimidine-Based Macrocycles, J. Med. Chem., 2022, 7799–7817, DOI:10.1021/acs.jmedchem.2c00173.
- T. Hanke, S. Mathea, J. Woortman, E. Salah, B.-T. Berger, A. Tumber, R. Kashima, A. Hata, B. Kuster, S. Müller and S. Knapp, Development and characterization of type I, type II, and type III LIM-kinase chemical probes, J. Med. Chem., 2022, 65(19), 13264–13287, DOI:10.1021/acs.jmedchem.2c01106.
- D. Williamson, M. Parratt, J. Bower, J. Moore, C. Richardson, P. Dokurno, A. Cansfield, G. Francis, R. Hebdon, R. Howes, P. Jackson, A. Lockie, J. Murray, C. Nunns, J. Powles, A. Robertson, A. Surgenor and C. Torrance, Structure-guided design of pyrazolo[1,5-a]pyrimidines as inhibitors of human cyclin-dependent kinase 2, Bioorg. Med. Chem. Lett., 2005, 15(4), 863–867, DOI:10.1016/J.BMCL.2004.12.073.
- S. Degorce, S. Boyd, J. Curwen, R. Ducray, C. Halsall, C. Jones, F. Lach, E. Lenz, M. Pass, S. Pass and C. Trigwell, Discovery of a Potent, Selective, Orally Bioavailable, and Efficacious Novel 2-(Pyrazol-4-ylamino)-pyrimidine Inhibitor of the Insulin-like Growth Factor-1 Receptor (IGF-1R), J. Med. Chem., 2016, 59(10), 4859–4866, DOI:10.1021/acs.jmedchem.6b00203.
- R. Roskoski, Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes, Pharmacol. Res., 2016, 103, 26–48, DOI:10.1016/j.phrs.2015.10.021.
- I. Abdelbaky, H. Tayara and K. Chong, Prediction of kinase inhibitors binding modes with machine learning and reduced descriptor sets, Sci. Rep., 2021, 11 DOI:10.1038/s41598-020-80758-4.
- F. Miljković, R. Rodríguez-Pérez and J. Bajorath, Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes, J. Med. Chem., 2020, 8738–8748, DOI:10.1021/acs.jmedchem.9b00867.
- J. Rabuck, S. Hyung, K. Ko, C. Fox, M. Soellner and B. Ruotolo, Activation state-selective kinase inhibitor assay based on ion mobility-mass spectrometry, Anal. Chem., 2013, 85(15), 6995–7002, DOI:10.1021/ac4012655.
- S. Harrison, K. Das, F. Karim, D. Maclean and D. Mendel, Non-ATP-competitive kinase inhibitors – enhancing selectivity through new inhibition strategies, Expet Opin. Drug Discov., 2008, 3, 761–774, DOI:10.1517/17460441.3.7.761.
- S. Chakraborty, T. Inukai, L. Fang, M. Golkowski and D. Maly, Targeting Dynamic ATP-Binding Site Features Allows Discrimination between Highly Homologous Protein Kinases, ACS Chem. Biol., 2019, 1249–1259, DOI:10.1021/acschembio.9b00214.
- Q. Liu, S. Kirubakaran, W. Hur, M. Niepel, K. Westover, C. Thoreen, J. Wang, J. Ni, M. Patricelli, K. Vogel, S. Riddle, D. Waller, R. Traynor, T. Sanda, Z. Zhao, S. Kang, J. Zhao, A. Look, P. Sorger, D. Sabatini and N. Gray, Kinome-wide Selectivity Profiling of ATP-competitive Mammalian Target of Rapamycin (mTOR) Inhibitors and Characterization of Their Binding Kinetics*, J. Biol. Chem., 2012, 287, 9742–9752, DOI:10.1074/jbc.M111.304485.
- M. Cherry and D. Williams, Recent kinase and kinase inhibitor X-ray structures: mechanisms of inhibition and selectivity insights, Curr. Med. Chem., 2004, 11(6), 663–673, DOI:10.2174/0929867043455792.
- P. Brear, D. Ball, K. Stott, S. D'Arcy and M. Hyvönen, Proposed Allosteric Inhibitors Bind to the ATP Site of CK2α, J. Med. Chem., 2020, 12786–12798, DOI:10.1021/acs.jmedchem.0c01173.
- C. Olivieri, G. Li, Y. Wang, M. S, C. Walker, J. Kim, C. Camilloni, A. Simone, M. Vendruscolo, D. Bernlohr, S. Taylor and G. Veglia, ATP-competitive inhibitors modulate the substrate binding cooperativity of a kinase by altering its conformational entropy, Sci. Adv., 2022, 8 DOI:10.1126/sciadv.abo0696.
- A. Zorba, V. Nguyen, A. Koide, M. Hoemberger, Y. Zheng, S. Kutter, C. Kim, S. Koide and D. Kern, Allosteric modulation of a human protein kinase with monobodies, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13937–13942, DOI:10.1073/pnas.1906024116.
- K. Yamada, J. Zhang, X. Xie, J. Reinhardt, A. Xie, D. Lasala, D. Kohls, D. Yowe, D. Burdick, H. Yoshisue, H. Wakai, I. Schmidt, J. Gunawan, K. Yasoshima, Q. Yue, M. Kato, M. Mogi, N. Idamakanti, N. Kreder, P. Drueckes, P. Pandey, T. Kawanami, W. Huang, Y. Yagi, Z. Deng and H. Park, Discovery and Characterization of Allosteric WNK Kinase Inhibitors, ACS Chem. Biol., 2016, 11(12), 3338–3346, DOI:10.1021/ACSCHEMBIO.6B00511.
- L. Skóra and W. Jahnke, 19F-NMR-Based Dual-Site Reporter Assay for the Discovery and Distinction of Catalytic and Allosteric Kinase Inhibitors, ACS Med. Chem. Lett., 2017, 8(6), 632–635, DOI:10.1021/acsmedchemlett.7b00084.
- P. Wu, M. Clausen and T. Nielsen, Allosteric small-molecule kinase inhibitors, Pharmacol. Therapeut., 2015, 156, 59–68, DOI:10.1016/j.pharmthera.2015.10.002.
- H. Feldman, M. Tong, L. Wang, R. Meza-Acevedo, T. Gobillot, I. Lebedev, M. Gliedt, S. Hari, A. Mitra, B. Backes, F. Papa, M. Seeliger and D. Maly, Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands, ACS Chem. Biol., 2016, 11(8), 2195–2205, DOI:10.1021/acschembio.5b00940.
- Ø. Bruserud and H. Reikvam, Casein Kinase 2 (CK2): A Possible Therapeutic Target in Acute Myeloid Leukemia, Cancers, 2023, 15, 3711, DOI:10.3390/cancers15143711.
- Y. Chen, Y. Wang, J. Wang, Z. Zhou, S. Cao and J. Zhang, Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects, J. Med. Chem., 2023, 2257–2281, DOI:10.1021/acs.jmedchem.2c01523.
- G. Cozza and L. Pinna, Casein kinases as potential therapeutic targets, Expert Opin. Ther. Targets, 2016, 20, 319–340, DOI:10.1517/14728222.2016.1091883.
- H. Chon, K. Bae, Y. Lee and J. Kim, The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies, Front. Pharmacol, 2015, 6 DOI:10.3389/fphar.2015.00070.
- E. Silva-Pavez and J. Tapia, Protein Kinase CK2 in Cancer Energetics, Front. Oncol., 2020, 10 DOI:10.3389/fonc.2020.00893.
- M. Licciardello and P. Workman, A New Chemical Probe Challenges the Broad Cancer Essentiality of CK2, Trends Pharmacol. Sci., 2021, 313–315, DOI:10.1016/j.tips.2021.02.002.
- C. Gowda, M. Sachdev, S. Muthusami, M. Kapadia, L. Petrovic-Dovat, M. Hartman, Y. Ding, C. Song, J. Payne, B. Tan and S. Dovat, Casein Kinase II (CK2) as a Therapeutic Target for Hematological Malignancies, Curr. Pharmaceut. Des., 2016, 23(1), 95–107, DOI:10.2174/1381612822666161006154311.
- A. Abdel-Magid, Inhibition of CK2: An Attractive Therapeutic Target for Cancer Treatment, ACS Med. Chem. Lett., 2013, 4(12), 1131–1132, DOI:10.1021/ml400410p.
- A. Krämer, C. Kurz, B. Berger, I. Celik, A. Tjaden, F. Greco, S. Knapp and T. Hanke, Optimization of pyrazolo[1,5-a]pyrimidines lead to the identification of a highly selective casein kinase 2 inhibitor, Eur. J. Med. Chem., 2020, 208, 112770, DOI:10.1016/j.ejmech.2020.112770.
- A. Gopalsamy, G. Ciszewski, Y. Hu, F. Lee, L. Feldberg, E. Frommer, S. Kim, K. Collins, D. Wojciechowicz and R. Mallon, Identification of pyrazolo[1,5-a]pyrimidine-3-carboxylates as B-Raf kinase inhibitors, Bioorg. Med. Chem. Lett., 2009, 19(10), 2735–2738, DOI:10.1016/j.bmcl.2009.03.129.
- C. Madeddu, C. Donisi, N. Liscia, E. Lai, M. Scartozzi and A. Macciò, EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment, Int. J. Mol. Sci., 2022, 23, 6489, DOI:10.3390/ijms23126489.
- P. Hsu, D. Jablons, C. Yang and L. You, Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC), Int. J. Mol. Sci., 2019, 20, 3821, DOI:10.3390/ijms20153821.
- J. Greenhalgh, A. Boland, V. Bates, F. Vecchio, Y. Dundar, M. Chaplin and J. Green, First-line treatment of advanced epidermal growth factor receptor (EGFR) mutation positive non-squamous non-small cell lung cancer, Cochrane Database Syst. Rev., 2021, 3, CD010383, DOI:10.1002/14651858.CD010383.pub3.
- F. Agustoni, K. Suda, H. Yu, S. Ren, C. Rivard, K. Ellison, C. Caldwell, L. Rozeboom, K. Brovsky and F. Hirsch, EGFR-directed monoclonal antibodies in combination with chemotherapy for treatment of non-small-cell lung cancer: an updated review of clinical trials and new perspectives in biomarkers analysis, Cancer Treat Rev., 2019, 72, 15–27, DOI:10.1016/j.ctrv.2018.08.002.
- M. Abdelgawad, R. Bakr, O. Alkhoja and W. Mohamed, Design, synthesis and antitumor activity of novel pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors, Bioorg. Chem., 2016, 66, 88–96, DOI:10.1016/j.bioorg.2016.03.011.
- A. Ayati, S. Moghimi, M. Toolabi and A. Foroumadi, Pyrimidine-based EGFR TK inhibitors in targeted cancer therapy, Eur. J. Med. Chem., 2021, 221, 113523, DOI:10.1016/j.ejmech.2021.113523.
- H. Cheng, S. Nair, B. Murray, C. Almaden, S. Bailey, S. Baxi, D. Behenna, S. Cho-Schultz, D. Dalvie, D. Dinh, M. Edwards, J. Feng, R. Ferre, K. Gajiwala, M. Hemkens, A. Jackson-Fisher, M. Jalaie, T. Johnson, R. Kania, S. Kephart, J. Lafontaine, B. Lunney, K. Liu, Z. Liu, J. Matthews, A. Nagata, S. Niessen, M. Ornelas, S. Orr, M. Pairish, S. Planken, S. Ren, D. Richter, K. Ryan, N. Sach, H. Shen, T. Smeal, J. Solowiej, S. Sutton, K. Tran, E. Tseng, W. Vernier, M. Walls, S. Wang, S. Weinrich, S. Xin, H. Xu, M. Yin, M. Zientek, R. Zhou and J. Kath, Discovery of 1-{(3R,4R)-3-[({5-Chloro-2-[(1-methyl-1H-pyrazol-4-yl)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}oxy)methyl]-4-methoxypyrrolidin-1-yl}prop-2-en-1-one (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants, J. Med. Chem., 2016, 59(5), 2005–2024, DOI:10.1021/acs.jmedchem.5b01633.
- E. Tricker, C. Xu, S. Uddin, M. Capelletti, D. Ercan, A. Ogino, C. Pratilas, N. Rosen, N. Gray, K. Wong and P. Jänne, Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer, Cancer Discov., 2015, 5(9), 960–971, DOI:10.1158/2159-8290.CD-15-0063.
- E. Sobh, M. Dahab, E. Elkaeed, A. Alsfouk, I. Ibrahim, A. Metwaly and I. Eissa, Discovery of new thieno[2,3-d]pyrimidines as EGFR tyrosine kinase inhibitors for cancer treatment, Future Med. Chem., 2023, 1167–1184, DOI:10.4155/fmc-2023-0086.
- H. Dickerson, A. Diab and O. Al Musaimi, Epidermal growth factor receptor tyrosine kinase inhibitors in cancer: Current use and future prospects, Int. J. Mol. Sci., 2024, 25(18), 10008, DOI:10.3390/ijms251810008.
- Z. Wang, N. Yan, H. Sheng, Y. Xiao, J. Sun and C. Cao, Single-cell Transcriptomic Analysis Reveals an Immunosuppressive Network Between POSTN CAFs and ACKR1 ECs in TKI-resistant Lung Cancer, Cancer Genomics Proteomics, 2024, 21, 65–78, DOI:10.21873/cgp.20430.
- S. Moser, L. Apter, J. Solomon, G. Chodick, M. Wollner and N. Siegelmann-Danieli, Time on Treatment and Survival Outcomes for Patients Treated with First-line Osimertinib vs. Other Tyrosine Kinase Inhibitors, for EGFR Mutation-positive Metastatic Non-small Cell Lung Cancer: Real-world Experience Data, Anticancer Res., 2024, 44, 257–265, DOI:10.21873/anticanres.16809.
- J. Sun, R. Hu, M. Han, Y. Tan, M. Xie, S. Gao and J. Hu, Mechanisms underlying therapeutic resistance of tyrosine kinase inhibitors in chronic myeloid leukemia, Int. J. Biol. Sci., 2024, 20, 175–181, DOI:10.7150/ijbs.86305.
- W. Zhou, A. Lim, M. Edderkaoui, A. Osipov, H. Wu, Q. Wang and S. Pandol, Role of YAP Signaling in Regulation of Programmed Cell Death and Drug Resistance in Cancer, Int. J. Biol. Sci., 2024, 20, 15–28, DOI:10.7150/ijbs.83586.
- C. Babiloni-López, N. Fritz, R. Ramírez-Campillo and J. Colado, Water-Based Exercise in Patients with Nonspecific Chronic Low-Back Pain: A Systematic Review with Meta-Analysis, J. Strength Cond. Res., 2024, 38, 206–219, DOI:10.1519/JSC.0000000000004635.
- J. Lee, J. Choi, K. Lee, M. Lee, J. Ku, D. Oh, Y. Shin, D. Kim, I. Cho, W. Paik, J. Ryu, Y. Kim, S. Lee and S. Lee, Antiproliferative Activity of Piceamycin by Regulating Alpha-Actinin-4 in Gemcitabine-Resistant Pancreatic Cancer Cells, Biomol. Ther., 2024, 32(1), 123–135, DOI:10.4062/biomolther.2023.109.
- S. Lee, S. Joo, J. Lee, A. Kwak, K. Kim, S. Cho, G. Yoon, Y. Choi, J. Park and J. Shim, Licochalcone C Inhibits the Growth of Human Colorectal Cancer HCT116 Cells Resistant to Oxaliplatin, Biomol. Ther., 2024, 32(1), 104–114, DOI:10.4062/biomolther.2023.167.
- M. Fawwaz, K. Mishiro, B. Purwono, R. Nishii and K. Ogawa, Synthesis and evaluation of a rociletinib analog as prospective imaging double mutation L858R/T790M in non-small cell lung cancer, J. Pharm. Pharmacogn. Res., 2024, 231–242, DOI:10.56499/jppres23.1743_12.2.231.
- D. Udovicic-Gagula, N. Džananović and M. Dorić, Prevalence of epidermal growth factor receptor (EGFR) mutations and correlation with histological patterns in lung adenocarcinoma in patients from Bosnia and Herzegovina, Med. Glas., 2024, 21(1), 8–15, DOI:10.17392/1627-23.
- J. Nulali, K. Zhang, M. Long, Y. Wan, Y. Liu, Q. Zhang, L. Yang, J. Hao, L. Yang and H. Song, ALYREF-mediated RNA 5-Methylcytosine modification Promotes Hepatocellular Carcinoma Progression Via Stabilizing EGFR mRNA and pSTAT3 activation, Int. J. Biol. Sci., 2024, 20, 331–346, DOI:10.7150/ijbs.82316.
- H. Ho, F. Wang, C. Chiang, C. Tsai, C. Chiu and T. Chou, Dynamic assessment of tissue and plasma EGFR-activating and T790M mutations with droplet digital PCR assays for monitoring response and resistance in non-small cell lung cancers treated with EGFR-TKIs, Int. J. Mol. Sci., 2022, 23(19), 11353, DOI:10.3390/ijms231911353.
- J. Wu, Y. Chen, R. Li, Y. Guan, M. Chen, H. Yin, X. Yang, M. Jin, B. Huang, X. Ding, J. Yang, Z. Wang, Y. He, Q. Wang, J. Luo, P. Wang, Z. Mao, M. Huen, Z. Lou, J. Yuan and F. Gong, Synergistic anticancer effect by targeting CDK2 and EGFR-ERK signaling, J. Cell Biol., 2024, 223, 1, DOI:10.1083/jcb.202203005.
- G. Rady, M. Deeb, M. Sarg, A. Shalaby and A. Helwa, Imidazo- and pyrazolopyrimidine scaffolds as anticancer agents, Int. J. Med. Pharmaceut. Sci., 2024, 107–143, DOI:10.21608/aijpms.2023.191090.1194.
- W. Xuzhang, T. Lu, W. Jin, Y. Yu, Z. Li, L. Shen, X. Niu, X. Ai, L. Xia and S. Lu, Cisplatin-induced Pyroptosis Enhances the Efficacy of PD-L1 Inhibitor in Small-Cell Lung Cancer via GSDME/IL12/CD4Tem Axis, Int. J. Biol. Sci., 2024, 537–553, DOI:10.7150/ijbs.89080.
- N. Devi, V. Singh and A. Pathania, Synthesis of Pyrazolo-azepinone Derivatives via Morita-Baylis-Hillman Chemistry as Potent Antimicrobial Agents, New J. Chem., 2024, 2608–2623, 10.1039/d3nj04939f.
- Y. El-Ossaily, N. Alanazi, I. Althobaiti, H. Altaleb, N. Al-Muailkel, M. El-Sayed, M. Hussein, I. Ahmed, M. Alanazi, A. Fawzy, S. Abdel-Raheem and M. Tolba, Multicomponent approach to the synthesis and spectral characterization of some 3,5-pyrazolididione derivatives and evaluation as anti-inflammatory agents, Curr. Chem. Lett., 2024, 127–140, DOI:10.5267/j.ccl.2023.8.003.
- G. Sivaiah, R. Raveesha, S. B. Benaka Prasad, K. Y. Kumar, M. S. Raghu, F. Alharethy, M. K. Prashanth and B. H. Jeon, Synthesis, anticancer activity, and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives as EGFR/HER2 dual kinase inhibitors, J. Mol. Struct., 2023, 1289, 135877, DOI:10.1016/j.molstruc.2023.135877.
- I. Othman, Z. Alamshany, N. Tashkandi, M. Gad-Elkareem, M. Anwar and E. Nossier, New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: Synthesis, anticancer, antimicrobial evaluation and computational studies, Bioorg. Chem., 2021, 114, 105078, DOI:10.1016/j.bioorg.2021.105078.
- Y. Byeon, S. Jung, D. Yoon, S. Kang, J. Park, H. Jo, S. Choi, S. Park, S. Lee, Y. Tae, T. Kim, S. Cho, S. Kim, D. Ko, D. Kim and D. Kim, Abstract 5477: Compound A, a fourth-generation allosteric inhibitor, a potent and highly selective EGFR with L858R activating and C797S resistance mutations for the treatment of NSCLC, Cancer Res., 2022, 5477, DOI:10.1158/1538-7445.am2022-5477.
- Y. Dong, K. Tamari, M. Kishigami, S. Katsuki, K. Minami, S. Tatekawa, S. Shimizu, M. Koizumi and K. Ogawa, Irradiated Cell-derived Exosomes Enhance Cell Proliferation and Radioresistance via the MAPK/Erk Pathway, Cancer Genomics Proteomics, 2024, 21, 12–17, DOI:10.21873/cgp.20425.
- M. Piao, P. Fernando, K. Kang, P. Fernando, H. Herath, Y. Kim and J. Hyun, Rosmarinic Acid Inhibits Ultraviolet B-Mediated Oxidative Damage via the AKT/ERK-NRF2-GSH Pathway In Vitro and In Vivo, Biomol. Ther., 2024, 32(1), 84–93, DOI:10.4062/biomolther.2023.179.
- C. Fébrissy, M. Adlanmerini, C. Péqueux, F. Boudou, M. Buscato, A. Gargaros, S. Gilardi-Bresson, K. Boriak, H. Laurell, C. Fontaine, B. Katzenellenbogen, J. Katzenellenbogen, J. Guillermet-Guibert, J. Arnal, R. Metivier and F. Lenfant, Reprogramming of endothelial gene expression by tamoxifen inhibits angiogenesis and ERα-negative tumor growth, Theranostics, 2024, 14, 249–264, DOI:10.7150/thno.87306.
- M. Wu, J. Xie, Y. Xing, L. Zhang, H. Chen, B. Tang, M. Zhou, S. Lv, D. Huang, S. Jian, C. Zhou, M. Liu, W. Guo, Y. Chen and Z. Yi, Selectively targeting BCL6 using a small molecule inhibitor is a potential therapeutic strategy for ovarian cancer, Int. J. Biol. Sci., 2024, 486–501, DOI:10.7150/ijbs.86303.
- K. K. Kolathur, R. Nag, P. V. Shenoy, Y. Malik, S. M. Varanasi, R. S. Angom and D. Mukhopadhyay, Molecular Susceptibility and Treatment Challenges in Melanoma, Cells, 2024, 13(16), 1383, DOI:10.3390/cells13161383.
- G. Nitulescu, G. Stancov, O. Seremet, G. Nitulescu, D. Mihai, C. Duta-Bratu, S. Barbuceanu and O. Olaru, The Importance of the Pyrazole Scaffold in the Design of Protein Kinases Inhibitors as Targeted Anticancer Therapies, Molecules, 2023, 28, 5359, DOI:10.3390/molecules28145359.
- S. Planken, D. Behenna, S. Nair, T. Johnson, A. Nagata, C. Almaden, S. Bailey, T. Ballard, L. Bernier, H. Cheng, S. Cho-Schultz, D. Dalvie, J. Deal, D. Dinh, M. Edwards, R. Ferre, K. Gajiwala, M. Hemkens, R. Kania, J. Kath, J. Matthews, B. Murray, S. Niessen, S. Orr, M. Pairish, N. Sach, H. Shen, M. Shi, J. Solowiej, K. Tran, E. Tseng, P. Vicini, Y. Wang, S. Weinrich, R. Zhou, M. Zientek, L. Liu, Y. Luo, S. Xin, C. Zhang and J. Lafontaine, Discovery of N-((3R,4R)-4-Fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidine-3-yl)acrylamide (PF-06747775) through Structure-Based Drug Design: A High Affinity Irreversible Inhibitor Targeting Oncogenic EGFR Mutants with Selectivity over Wild-Type EGFR, J. Med. Chem., 2017, 60(7), 3002–3019, DOI:10.1021/acs.jmedchem.6b01894.
- D. Cross, S. Ashton, S. Ghiorghiu, C. Eberlein, A. N. Caroline, P. Spitzler, J. Orme, M. Finlay, A. W. Richard, M. Mellor, G. Hughes, A. Rahi, V. Jacobs, M. B. Red, E. Ichihara, J. Sun, H. Jin, P. Ballard, K. Al-Kadhimi, R. Rowlinson, T. Klinowska, G. Richmond, M. Cantarini, D. Kim, M. Ranson and W. Pao, AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discov., 2014, 4(9), 1046–1061, DOI:10.1158/2159-8290.CD-14-0337.
- W. Zhou, X. Liu, Z. Tu, L. Zhang, X. Ku, F. Bai, Z. Zhao, Y. Xu, K. Ding and H. Li, Discovery of pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R mutant, J. Med. Chem., 2013, 56(20), 7821–7837, DOI:10.1021/jm401045n.
- M. Günther, J. Lategahn, M. Juchum, E. Döring, M. Keul, J. Engel, H. Tumbrink, D. Rauh and S. Laufer, Trisubstituted Pyridinylimidazoles as Potent Inhibitors of the Clinically Resistant L858R/T790M/C797S EGFR Mutant: Targeting of Both Hydrophobic Regions and the Phosphate Binding Site, J. Med. Chem., 2017, 60(13), 5613–5637, DOI:10.1021/acs.jmedchem.7b00316.
- H. Tanaka, H. Sakagami, N. Kaneko, S. Konagai, H. Yamamoto, T. Matsuya, M. Yuri, Y. Yamanaka, M. Mori, M. Takeuchi, H. Koshio, M. Hirano and S. Kuromitsu, Mutant-Selective Irreversible EGFR Inhibitor, Naquotinib, Inhibits Tumor Growth in NSCLC Models with EGFR-Activating Mutations, T790M Mutation, and AXL Overexpression, Mol. Cancer Ther., 2019, 18, 1366–1373, DOI:10.1158/1535-7163.MCT-18-0976.
- K. Su, H. Chen, K. Li, M. Kuo, J. Yang, W. Chan, B. Ho, G. Chang, J. Shih, S. Yu and P. Yang, Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer, J. Clin. Oncol., 2012, 30(4), 433–440, DOI:10.1200/JCO.2011.38.3224.
- F. Manetti, A. Pucci, M. Magnani, G. Locatelli, C. Brullo, A. Naldini, S. Schenone, G. Maga, F. Carraro and M. Botta, Inhibition of Bcr-Abl Phosphorylation and Induction of Apoptosis by Pyrazolo[3,4-d]pyrimidines in Human Leukemia Cells, ChemMedChem, 2007, 2, 343–353, DOI:10.1002/cmdc.200600214.
- S. Wong, J. McLaughlin, D. Cheng, C. Zhang, K. Shokat and O. Witte, Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(50), 17456–17461, DOI:10.1073/PNAS.0407061101.
- M. Santucci, V. Corradi, M. Mancini, F. Manetti, M. Radi, S. Schenone and M. Botta, C6-Unsubstituted Pyrazolo[3,4-d]pyrimidines Are Dual Src/Abl Inhibitors Effective against Imatinib Mesylate Resistant Chronic Myeloid Leukemia Cell Lines, ChemMedChem, 2009, 4, 118–126, DOI:10.1002/cmdc.200800320.
- L. Hu, Y. Zheng, Z. Li, Y. Wang, Y. Lv, X. Qin and C. Zeng, Design, synthesis, and biological activity of phenyl-pyrazole derivatives as BCR-ABL kinase inhibitors, Bioorg. Med. Chem., 2015, 23(13), 3147–3152, DOI:10.1016/j.bmc.2015.04.083.
- A. Mandour, I. Nassar, M. Aal, M. Shahin, W. El-Sayed, M. Hegazy, A. Yehia, A. Ismail, M. Hagras, E. Elkaeed, H. Refaat and N. Ismail, Synthesis, biological evaluation, and in silico studies of new CDK2 inhibitors based on pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine scaffold with apoptotic activity, J. Enzyme Inhib. Med. Chem., 2022, 37, 1957–1973, DOI:10.1080/14756366.2022.2086866.
- B. Fanta, J. Lenjisa, T. Teo, L. Kou, L. Mekonnen, Y. Yang, S. Basnet, R. Hassankhani, M. Sykes, M. Yu and S. Wang, Discovery of N,4-Di(1H-pyrazol-4-yl)pyrimidin-2-amine-Derived CDK2 Inhibitors as Potential Anticancer Agents: Design, Synthesis, and Evaluation, Molecules, 2023, 28, 2951, DOI:10.3390/molecules28072951.
- A. Kamal, J. Tamboli, V. Nayak, S. Adil, M. Vishnuvardhan and S. Ramakrishna, Synthesis of pyrazolo[1,5-a]pyrimidine linked aminobenzothiazole conjugates as potential anticancer agents, Bioorg. Med. Chem. Lett., 2013, 23(11), 3208–3215, DOI:10.1016/j.bmcl.2013.03.129.
- A. Podolski-Renić, J. Dinić, T. Stankovic, I. Tsakovska, I. Pajeva, T. Tuccinardi, L. Botta, S. Schenone and M. Pešić, New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors, Cancers, 2021, 13, 5308, DOI:10.3390/cancers13215308.
- E. Lorthiois, M. Gerspacher, K. Beyer, A. Vaupel, C. Leblanc, R. Stringer, A. Weiss, R. Wilcken, D. Guthy, A. Lingel, C. Bomio-Confaglia, R. Machauer, P. Rigollier, J. Ottl, D. Arz, P. Bernet, G. Desjonqueres, S. Dussauge, M. Kazic-Legueux, M. Lozac'h, C. Mura, M. Sorge, M. Todorov, N. Warin, F. Zink, H. Voshol, F. Zécri, R. Sedrani, N. Ostermann, S. Brachmann and S. Cotesta, JDQ443, a Structurally Novel, Pyrazole-Based, Covalent Inhibitor of KRASG12C for the Treatment of Solid Tumors, J. Med. Chem., 2022, 16173–16203, DOI:10.1021/acs.jmedchem.2c01438.
- C. Karthikeyan, R. Malla, C. Ashby, H. Amawi, K. Abbott, J. Moore, J. Chen, C. Balch, C. Lee, P. Flannery, P. Trivedi, J. Faridi, S. Pondugula and A. Tiwari, Pyrimido[1′′,2′′:1,5]pyrazolo[3,4-b]quinolines: Novel compounds that reverse ABCG2-mediated resistance in cancer cells, Cancer Lett., 2016, 376(1), 118–126, DOI:10.1016/j.canlet.2016.03.030.
- J. Singleton, R. Dass, N. Neubert, R. Smith, Z. Webber, M. Hansen and M. Peterson, Synthesis and biological evaluation of novel pyrazolo[1,5-a]pyrimidines: Discovery of a selective inhibitor of JAK1 JH2 pseudokinase and VPS34, Bioorg. Med. Chem. Lett., 2019, 126813, DOI:10.1016/j.bmcl.2019.126813.
- R. Pal, G. Teli, S. Sengupta, L. Maji and G. Matada, An outlook of docking analysis and structure-activity relationship of pyrimidine-based analogues as EGFR inhibitors against non-small cell lung cancer (NSCLC), J. Biomol. Struct. Dynam., 2023, 9795–9811, DOI:10.1080/07391102.2023.2252082.
- S. Al-Muntaser, A. Al-karmalawy, A. El-Naggar, A. Ali, N. El-Sattar and E. Abbass, Novel 4-thiophenyl-pyrazole, pyridine, and pyrimidine derivatives as potential antitumor candidates targeting both EGFR and VEGFR-2; design, synthesis, biological evaluations, and in silico studies, RSC Adv., 2023, 13, 12184–12203, 10.1039/d3ra00416c.
- J. Gozgit, T. Chen, T. Clackson and V. Rivera, Abstract 2084: Ponatinib is a highly potent inhibitor of activated variants of RET found in MTC and NSCLC, Cancer Res., 2013, 73, 2084, DOI:10.1158/1538-7445.AM2013-2084.
- M. Gao, L. Duan, J. Luo, L. Zhang, X. Lu, Y. Zhang, Z. Zhang, Z. Tu, Y. Xu, X. Ren and K. Ding, Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors, J. Med. Chem., 2013, 56(8), 3281–3295, DOI:10.1021/jm301824k.
- P. Traxler, G. Bold, J. Frei, M. Lang, N. Lydon, H. Mett, E. Buchdunger, T. Meyer, M. Mueller and P. Furet, Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines, J. Med. Chem., 1997, 40(22), 3601–3616, DOI:10.1021/JM970124V.
- A. Wagner, M. Teufl, L. Gold, M. Lehner, C. Obinger, P. Sykacek and M. Traxlmayr, PhosphoFlowSeq - A high-throughput kinase activity assay for screening drug resistance mutations in EGFR, J. Mol. Biol., 2021, 167210, DOI:10.1016/j.jmb.2021.167210.
- F. Sherbiny, A. Bayoumi, A. El-Morsy, M. Sobhy and M. Hagras, Design, Synthesis, biological Evaluation, and molecular docking studies of novel Pyrazolo[3,4-d]Pyrimidine derivative scaffolds as potent EGFR inhibitors and cell apoptosis inducers, Bioorg. Chem., 2021, 116, 105325, DOI:10.1016/j.bioorg.2021.105325.
- Y. Xu, B. Brenning, S. Kultgen, J. Foulks, A. Clifford, S. Lai, A. Chan, S. Merx, M. Mccullar, S. Kanner and K. Ho, Synthesis and Biological Evaluation of Pyrazolo[1,5-a]pyrimidine Compounds as Potent and Selective Pim-1 Inhibitors, ACS Med. Chem. Lett., 2015, 6(1), 63–67, DOI:10.1021/ml500300c.
- E. Hanan, A. Abbema, K. Barrett, W. Blair, J. Blaney, C. Chang, C. Eigenbrot, S. Flynn, P. Gibbons, C. Hurley, J. Kenny, J. Kulagowski, L. Lee, S. Magnuson, C. Morris, J. Murray, R. Pastor, T. Rawson, M. Siu, M. Ultsch, A. Zhou, D. Sampath and J. Lyssikatos, Discovery of potent and selective pyrazolopyrimidine janus kinase 2 inhibitors, J. Med. Chem., 2012, 55(22), 10090–10107, DOI:10.1021/jm3012239.
- E. Husseiny, Synthesis, cytotoxicity of some pyrazoles and pyrazolo[1,5-a]pyrimidines bearing benzothiazole moiety and investigation of their mechanism of action, Bioorg. Chem., 2020, 102, 104053, DOI:10.1016/j.bioorg.2020.104053.
- S. Ioannidis, M. Lamb, T. Wang, L. Almeida, M. Block, A. Davies, B. Peng, M. Su, H. Zhang, E. Hoffmann, C. Rivard, I. Green, T. Howard, H. Pollard, J. Read, M. Alimzhanov, G. Bebernitz, K. Bell, M. Ye, D. Huszar and M. Zinda, Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway, J. Med. Chem., 2011, 54(1), 262–276, DOI:10.1021/jm1011319.
- A. Arias-Gómez, A. Godoy and J. Portilla, Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses as an Antitumor Scaffold, Molecules, 2021, 26, 2708, DOI:10.3390/molecules26092708.
- M. Hammouda, H. Gaffer and K. Elattar, Insights into the medicinal chemistry of heterocycles integrated with a pyrazolo[1,5-a]pyrimidine scaffold, RSC Med. Chem., 2022, 13(10), 1150–1196, 10.1039/d2md00192f.
- A. Abdallah and G. Elgemeie, Design, synthesis, docking, and antimicrobial evaluation of some novel pyrazolo[1,5-a] pyrimidines and their corresponding cycloalkane ring-fused derivatives as purine analogs, Drug Des. Dev. Ther., 2018, 12, 1785–1798, DOI:10.2147/DDDT.S159310.
- M. Attia, E. Elrazaz, S. El-Emam, A. Taher, H. Abdel-Aziz and K. Abouzid, Synthesis and in-vitro anti-proliferative evaluation of some pyrazolo[1,5-a]pyrimidines as novel larotrectinib analogs, Bioorg. Chem., 2019, 103458, DOI:10.1016/j.bioorg.2019.103458.
- R. Reddy, N. Sharadha and A. Kumari, Base-mediated [3 + 2]-cycloannulation strategy for the synthesis of pyrazolo[1,5-a]pyridine derivatives using (E)-β-iodovinyl sulfones, Org. Biomol. Chem., 2022, 4331–4337, 10.1039/d2ob00499b.
- A. Ballesteros-Casallas, M. Paulino, P. Vidossich, C. Melo, E. Jiménez, J.-C. Castillo, J. Portilla and G. P. Miscione, Synthesis of 2,7-diarylpyrazolo[1,5-a]pyrimidine derivatives with antitumor activity: Theoretical identification of targets, Eur. J. Med. Chem. Rep., 2022, 4, 100028, DOI:10.1016/j.ejmcr.2022.100028.
- S. Aranzazu, A. Tigreros, A. Arias-Gómez, J. Zapata-Rivera and J. Portilla, BF3-Mediated Acetylation of Pyrazolo[1,5-a]pyrimidines and Other π-Excedent (N-Hetero)arenes, J. Org. Chem., 2022, 9839–9850, DOI:10.1021/acs.joc.2c00881.
- C. N. Pillai and J. Chellapan, Effect of protonation and hydrogen bonding on 2,4,6-substituted pyrimidine and its salt complex-experimental and theoretical evidence, J. Mol. Model., 2014, 20(3), 2139, DOI:10.1007/s00894-014-2139-2.
- J. Castillo, A. Tigreros and J. Portilla, 3-Formylpyrazolo[1,5-a]pyrimidines as Key Intermediates for the Preparation of Functional Fluorophores, J. Org. Chem., 2018, 83(18), 10887–10897, DOI:10.1021/acs.joc.8b01571.
- G. Nitulescu, G. Stancov, O. Seremet, G. Nitulescu, D. Mihai, C. Duta-Bratu, S. Barbuceanu and O. Olaru, The Importance of the Pyrazole Scaffold in the Design of Protein Kinases Inhibitors as Targeted Anticancer Therapies, Molecules, 2023, 28, 5359, DOI:10.3390/molecules28145359.
- A. Kamal, S. Faazil, S. Hussaini, M. Ramaiah, M. Balakrishna, N. Patel, S. Pushpavalli and M. Pal-Bhadra, Synthesis and mechanistic aspects of 2-anilinonicotinyl-pyrazolo[1,5-a]pyrimidine conjugates that regulate cell proliferation in MCF-7 cells via estrogen signaling, Bioorg. Med. Chem. Lett., 2016, 26(8), 2077–2083, DOI:10.1016/j.bmcl.2016.02.072.
- M. T. El Sayed, H. Hussein, N. Elebiary, G. Hassan, S. Elmessery, A. Elsheakh, M. Nayel and H. Abdel-Aziz, Tyrosine kinase inhibition effects of novel Pyrazolo[1,5-a]pyrimidines and Pyrido[2,3-d]pyrimidines ligand: Synthesis, biological screening and molecular modeling studies, Bioorg. Chem., 2018, 78, 312–323, DOI:10.1016/j.bioorg.2018.03.009.
- T. Kosugi, D. Mitchell, A. Fujino, M. Imai, M. Kambe, S. Kobayashi, H. Makino, Y. Matsueda, Y. Oue, K. Komatsu, K. Imaizumi, Y. Sakai, S. Sugiura, O. Takenouchi, G. Unoki, Y. Yamakoshi, V. Cunliffe, J. Frearson, R. Gordon, C. Harris, H. Kalloo-Hosein, J. Le, G. Patel, D. Simpson, B. Sherborne, P. Thomas, N. Suzuki, M. Takimoto-Kamimura and K. Kataoka, Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAP-K2) as an antiinflammatory target: discovery and in vivo activity of selective pyrazolo[1,5-a]pyrimidine inhibitors using a focused library and structure-based optimization approach, J. Med. Chem., 2012, 55(15), 6700–6715, DOI:10.1021/jm300411k.
- A. Fouda, H. Abbas, E. Ahmed, A. Shati, M. Alfaifi and S. Elbehairi, Synthesis, In Vitro Antimicrobial and Cytotoxic Activities of Some New Pyrazolo[1,5-a]pyrimidine Derivatives, Molecules, 2019, 24, 1080, DOI:10.3390/molecules24061080.
- Y. Liu, R. Laufer, N. Patel, G. Ng, P. Sampson, S. Li, Y. Lang, M. Feher, R. Brokx, I. Beletskaya, R. Hodgson, O. Plotnikova, D. Awrey, W. Qiu, N. Chirgadze, J. Mason, X. Wei, D. Lin, Y. Che, R. Kiarash, G. Fletcher, T. Mak, M. Bray and H. Pauls, Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent, ACS Med. Chem. Lett., 2016, 7(7), 671–675, DOI:10.1021/acsmedchemlett.5b00485.
- N. Metwally, M. Mohamed and E. Ragb, Design, synthesis, anticancer evaluation, molecular docking and cell cycle analysis of 3-methyl-4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives as potent histone lysine demethylases (KDM) inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102929, DOI:10.1016/j.bioorg.2019.102929.
- C. Bagul, G. K. Rao, V. K. K. Makani, J. R. Tamboli, M. Pal-Bhadra and A. Kamal, Synthesis and biological evaluation of chalcone-linked pyrazolo[1,5-a]pyrimidines as potential anticancer agents, MedChemComm, 2017, 8(9), 1810–1816, 10.1039/C7MD00193B.
- A. Gopalsamy, G. Ciszewski, M. Shi, D. Berger, Y. Hu, F. Lee, L. Feldberg, E. Frommer, S. Kim, K. Collins, D. Wojciechowicz and R. Mallon, Hit to lead optimization of pyrazolo[1,5-a]pyrimidines as B-Raf kinase inhibitors, Bioorg. Med. Chem. Lett., 2009, 19(24), 6890–6892, DOI:10.1016/j.bmcl.2009.10.074.
- T. H. Iorkula, B. A. Tolman, J. D. Singleton and M. A. Peterson, An efficient microwave-assisted copper-catalyzed C-3 amination of 3-bromopyrazolo[1,5-a]pyrimidine, Tetrahedron Lett., 2023, 118, 154393, DOI:10.1016/j.tetlet.2023.154393.
- J. D. Singleton, R. Dass, N. R. Neubert, R. M. Smith, Z. Webber, M. D. H. Hansen and M. A. Peterson, Synthesis and biological evaluation of novel pyrazolo[1,5-a]pyrimidines: Discovery of a selective inhibitor of JAK1 JH2 pseudokinase and VPS34, Bioorg. Med. Chem. Lett., 2020, 30(2), 126813, DOI:10.1016/j.bmcl.2019.126813.
- A. T. Mahajan, Shivani, A. K. Datusalia, C. Coluccini, P. Coghi and S. Chaudhary, Pyrazolo[1,5-a]pyrimidine as a prominent framework for tropomyosin receptor kinase (Trk) inhibitors—Synthetic strategies and SAR insights, Molecules, 2024, 29(15), 3560, DOI:10.3390/molecules29153560.
- T. H. Iorkula, B. A. Tolman, S. R. Burt and M. A. Peterson, An efficient synthesis of C-6 aminated 3-bromoimidazo[1,2-b]pyridazines, Synth. Commun., 2023, 54(2), 121–132, DOI:10.1080/00397911.2023.2284350.
- A. T. Aborode, I. A. Onifade, M. M. Olorunshola, G. O. Adenikinju, I. J. Aruorivwooghene, A. C. Femi, O. O. Jude-Kelly, A. Osinuga, E. A. Omojowolo, A. F. Adeoye, S. Olapade, I. O. Adelakun, O. D. Moyinoluwa, O. M. Adeyemo, G. Y. Scott, R. A. Ogbonna, E. A. Fajemisin, O. Ehtasham, S. Toluwalashe, A. A. Bakre and T. H. Iorkula, et al.). Biochemical mechanisms and molecular interactions of vitamins in cancer therapy, Cancer. Pathog. Ther., 2024, 3–15, DOI:10.1016/j.cpt.2024.05.001.
- M. Bravo, A. Burgos-Molina, M. García-Aranda, M. Redondo and T. Téllez, Unlocking New Avenues in Breast Cancer Treatment: The Synergy of Kinase Inhibitors and Immunotherapy, Cancers, 2023, 15, 5499, DOI:10.3390/cancers15235499.
- B. Melosky, R. Juergens, V. Hirsh, D. Mcleod, N. Leighl, M. Tsao, P. Card and Q. Chu, Amplifying Outcomes: Checkpoint Inhibitor Combinations in First-Line Non-Small Cell Lung Cancer, Oncol., 2019, 64–77, DOI:10.1634/theoncologist.2019-0027.
- F. Esteva, V. Hubbard-Lucey, J. Tang and L. Pusztai, Immunotherapy and targeted therapy combinations in metastatic breast cancer, Lancet Oncol., 2019, 20(3), e175–e186, DOI:10.1016/S1470-2045(19)30026-9.
- F. Pessoa, C. Machado, E. Silva, L. Pantoja, R. Ribeiro, M. Moraes, M. Filho, R. Montenegro, A. Khayat and C. Moreira-Nunes, Solid Tumors and Kinase Inhibition: Management and Therapy Efficacy Evolution, Int. J. Mol. Sci., 2022, 23, 3830, DOI:10.3390/ijms23073830.
- A. Damont, V. Médran-Navarrete, F. Cacheux, B. Kuhnast, G. Pottier, N. Bernards, F. Marguet, F. Puech, R. Boisgard and F. Dollé, Novel Pyrazolo[1,5-a]pyrimidines as Translocator Protein 18 kDa (TSPO) Ligands: Synthesis, in Vitro Biological Evaluation, [(18)F]-Labeling, and in Vivo Neuroinflammation PET Images, J. Med. Chem., 2015, 58(18), 7449–7464, DOI:10.1021/acs.jmedchem.5b00932.
- L. He, Q. Qi, X. Wang, Y. Li, Y. Zhu, X. Wang and L. Xu, Synthesis of two novel pyrazolo[1,5-a]pyrimidine compounds with antibacterial activity and biophysical insights into their interactions with plasma protein, Bioorg. Chem., 2020, 99, 103833, DOI:10.1016/j.bioorg.2020.103833.
- M. Labroli, K. Paruch, M. Dwyer, C. Alvarez, K. Keertikar, C. Poker, R. Rossman, J. Duca, T. Fischmann, V. Madison, D. Parry, N. Davis, W. Seghezzi, D. Wiswell and T. Guzi, Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: a template-based approach-part 2, Bioorg. Med. Chem. Lett., 2011, 21(1), 471–474, DOI:10.1016/j.bmcl.2010.10.114.
| This journal is © The Royal Society of Chemistry 2025 |