
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of acrylamide-based covalent inhibitors of SARS-CoV-2 (SCoV-2) Nsp15 using high-throughput screening and machine learning†
Teena Bajaj‡
a,
Babak Mosavati‡bc,
Lydia H. Zhangd,
Mohammad S. Parsa e,
Huanchen Wangf,
Evan M. Kerekg,
Xueying Liangh,
Seyed Amir Tabatabaei Dakhilii,
Eddie Wehrij,
Silin Guok,
Rushil N. Desaic,
Lauren M. Orrk,
Mohammad R. K. Mofradl,
Julia Schaletzkyjm,
John R. Ussheri,
Xufang Dengh,
Robin Stanleyf,
Basil P. Hubbardg,
Daniel K. Nomurak and
Niren Murthy*h
e,
Huanchen Wangf,
Evan M. Kerekg,
Xueying Liangh,
Seyed Amir Tabatabaei Dakhilii,
Eddie Wehrij,
Silin Guok,
Rushil N. Desaic,
Lauren M. Orrk,
Mohammad R. K. Mofradl,
Julia Schaletzkyjm,
John R. Ussheri,
Xufang Dengh,
Robin Stanleyf,
Basil P. Hubbardg,
Daniel K. Nomurak and
Niren Murthy*h
aGraduate Program of Comparative Biochemistry, University of California, Berkeley, Berkeley, CA, USA. E-mail: bajajtiya@berkeley.edu
bInnovative Genomics Institute, University of California, Berkeley, Berkeley, CA, USA. E-mail: bmosavati@berkeley.edu
cDepartment of Bioengineering, University of California, Berkeley, Berkeley, CA, USA
dGraduate Program of Molecular Toxicology, University of California, Berkeley, Berkeley, CA, USA
eDepartment of Applied Science and Technology, University of California, Berkeley, CA, USA
fSignal Transduction Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, North Carolina, USA
gDepartment of Pharmacology, Li Ka Shing Institute of Virology, University of Alberta, Edmonton, Alberta, Canada
hDepartment of Physiological Sciences, College of Veterinary Medicine, Oklahoma Center for Respiratory and Infectious Disease, Oklahoma State University, Oklahoma, USA. E-mail: nmurthy@berkeley.edu
iFaculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada
jThe Henry Wheeler Center for Emerging and Neglected Diseases, University of California, Berkeley, Berkeley, CA, USA
kDepartment of Chemistry, University of California, Berkeley, Berkeley, CA, USA
lDepartment of Mechanical Engineering, University of California, Berkeley, CA, USA
mThe Molecular Therapeutics Initiative, University of California, Berkeley, 344 Li Ka Shing, Berkeley, CA, USA
First published on 3rd April 2025
Abstract
Non-structural protein 15 (Nsp15) is a SARS-CoV-2 (SCoV-2) endoribonuclease and is a promising target for drug development because of its essential role in evading the host immune system. However, developing inhibitors against Nsp15 has been challenging due to its structural complexity and large RNA binding surface. In this report, we screened a 2640 acrylamide-based compound library against Nsp15 and identified 10 fragments that reacted with cysteine residues on Nsp15 and inhibited its endoribonuclease activity with IC50s less than 5 μM. These compounds had several attractive properties, such as low molecular weight (180–300 g mol−1), log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P <3, zero violations to Lipinski's rules, and no apparent pan-assay interference (PAINs) properties. In addition, based on this data as a training set, we developed an artificial intelligence (AI) model that accelerated the hit to lead process and had a 73% accuracy for predicting new acrylamide-based Nsp15 inhibitors. Collectively, these results demonstrate that acrylamide fragments have great potential for developing Nsp15 inhibitors.
P <3, zero violations to Lipinski's rules, and no apparent pan-assay interference (PAINs) properties. In addition, based on this data as a training set, we developed an artificial intelligence (AI) model that accelerated the hit to lead process and had a 73% accuracy for predicting new acrylamide-based Nsp15 inhibitors. Collectively, these results demonstrate that acrylamide fragments have great potential for developing Nsp15 inhibitors.
Introduction
The emergence and rapid spread of the SARS-CoV-2 (SCoV-2) virus has stimulated the need for new drugs. Hundreds of drug discovery campaigns have been run to target essential proteins from SCoV-2 virus.1,2 For example, targeting RNA dependent RNA polymerase (RdRp)3 and main protease (Mpro)4,5 has led to the discovery of several promising antiviral drugs. However, alternative drugs that target other crucial proteins from SCoV-2 are still needed to respond to strain evolution and resistance development.6,7 The non-structural protein 15 (Nsp15) is a promising therapeutic target for drug development against SCoV-2 because its inhibition results in the upregulation of interferons and protects against viral infections via multiple pathways.8,9Nsp15 cleaves viral RNA and suppresses host sensors that recognize viral RNA and induce the production of interferons.10 Inhibition of Nsp15 activates the production of interferons and prevents the spread of viral infection through paracrine signaling pathways, consequently the activation of Nsp15 in a few cells can have global effects on anti-immunity.11 For example, infection of lung-derived epithelial cell lines and primary nasal epithelial air–liquid interface (ALI) cultures with mutant Nsp15 SCoV-2 virus caused an increased secretion of interferons and attenuated viral replication significantly.12 In another instance, infection of mutant Nsp15 MHV coronavirus into mouse bone marrow-derived macrophages resulted in an early and robust induction of interferon leading to rapid cell death.8 This suggests that viruses with mutant Nsp15 cannot infect mice effectively, due to their activation of the host immune response.8 Additionally, Nsp15 is also an evolutionarily conserved protein,13 with a possibility of discovering inhibitors efficacious against other coronaviruses. Though Nsp15 has great potential as a drug target, it is less explored in terms of drug discovery due to the challenge of drugging its very large binding interface.14 Only a handful of compounds have been identified that can inhibit Nsp15.15–17
Targeted covalent inhibition could be a promising route to drug classically “undruggable” proteins, such as Nsp15. SCoV-2 Nsp15 has several free cysteines (five cysteines: Cys103, Cys117, Cys291, Cys293 and Cys334) that play a role in subunit oligomerization and interactions with the RNA substrate that can potentially be targeted by covalent drugs. There is evidence that alkylation or other types of covalent modification of these cysteines by covalent drugs might have the potential to inhibit Nsp15 activity.16 Irreversible covalent modification of the cysteine near the active site is likely to be implicated in the mechanism of inhibition of Nsp15 through these compounds. However, electrophile libraries of covalent inhibitors have never been investigated before for identifying Nsp15 inhibitors.
In this report, we screened an acrylamide-based electrophile library containing 2640 compounds against Nsp15 and identified several fragments that inhibited Nsp15 with IC50s less than 5 μM, which had specificity for Nsp15 over other cysteine containing proteins. We selected an acrylamide library for screening because of their high selectivity for thiol nucleophiles and moderate reactivity to thiols at physiological pH. The identified fragments have promising predicted pharmacological properties and follow Lipinski's rule of five and are easy to synthesize. Mass spectrometry experiments showed that one of the ten compounds we identified modified the cysteine next to the Nsp15 active site (residue Cys293). Building on this, we used our experimental data to develop an innovative artificial intelligence platform that can predict potential inhibitors of Nsp15 and demonstrated that it has high prediction accuracy of ∼80%. Thus, this work both identifies a new chemical scaffold for the development of future drugs targeting SCoV-2 via Nsp15 and illustrates a novel approach to expedite the discovery and optimization of lead hits in a faster, and more economical manner. In conclusion, we demonstrate acrylamide-based Nsp15 inhibitors are interesting lead compounds for future drug discovery campaigns against coronaviruses.
Results and discussion
Rationale for selecting an acrylamide library to identify covalent inhibitors against SCoV-2 Nsp15
To discover covalent inhibitors that could sustain engagement efficiently with minimal off-target reactivity, we targeted the most nucleophilic residues present on the Nsp15 protein, those being cysteine residues.18 Typically, the most used electrophile building block employed in covalent inhibitors targeting cysteines are Michael acceptors, such as acrylamides.19 Acrylamides have been widely used as electrophiles in irreversible covalent inhibitors for many proteins bearing non-catalytic cysteines. For example, afatinib,20 ibrutinib,21 and AMG-510 (ref. 22) are acrylamide-based inhibitors of EGFR, BTK, and K-RasG12C, respectively.23 Here, we used an electrophile library containing 2640 acrylamide compounds from Enamine.Acrylamide-based compounds are covalent inhibitors against SCoV-2 Nsp15
Hexameric Nsp15 was recombinantly expressed and purified from bacterial cells using talon and size exclusion chromatography.13 We utilized two parameters (binding and inhibiting Nsp15) to discover acrylamide-based covalent inhibitors from high-throughput screening (HTS) (Fig. 1A). First, we used an activity-based protein profiling (ABPP) probe, cysteine-reactive tetramethylrhodamine-5-iodoacetamide dihydroiodide (IA-Rho)24 in a competitive manner to screen the acrylamide library to facilitate the discovery of covalent ligands against SCoV-2 Nsp15. The presence of cysteine-reactive compounds was expected to correspond with the disappearance of the IA-Rho-labeled Nsp15 band which can be visualized via gel electrophoresis for detection of Rho. We optimized the Nsp15 and IA-Rho concentrations to 0.25 μg and 0.5 μM, respectively. The negative control consisted of Nsp15, IA-Rho and DMSO. We initiated high throughput screening at a final concentration of 40 μM. A concentration of 40 μM was selected for the screening because Nsp15 is an undruggable target and would likely require high concentrations of compounds to identify inhibitors. We screened 2640 acrylamide-based compounds at a final concentration of 40 μM and the compounds that led to disappearance of the Nsp15 band were selected, followed by their confirmation with repurchased compounds. Repurchased compounds refer to the hits repurchased from ChemDiv as single compounds. Promising hits were repurchased to validate their activity further characterize them. The preliminary screening of the acrylamide library identified 829 initial hits that reacted with Nsp15 via its cysteines, corresponding to a hit rate of 31.4%.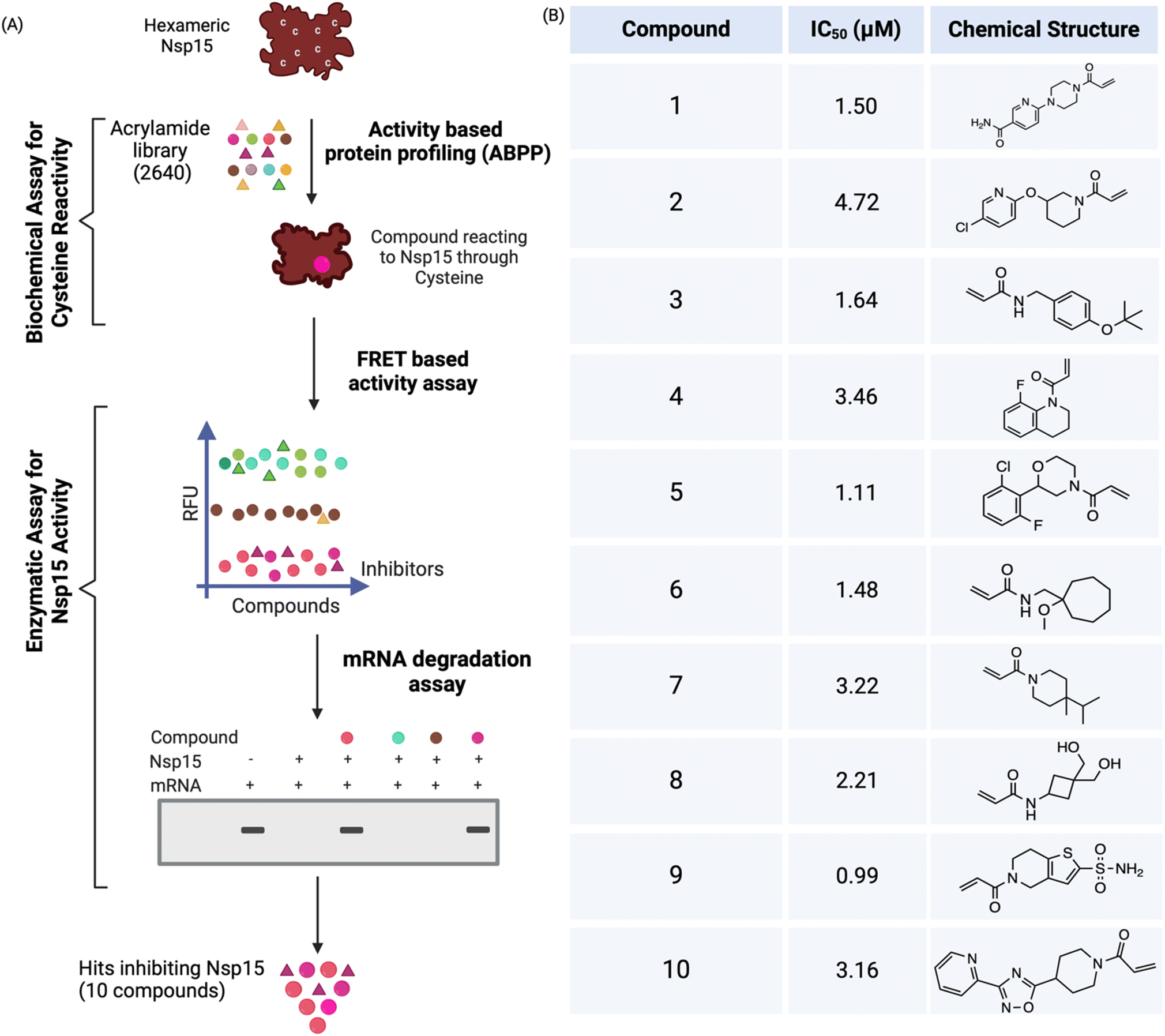 | ||
| Fig. 1 High-throughput screening (HTS) of acrylamide library consisting of 2640 compounds against Nsp15 identified ten inhibitors with sub-micromolar IC50s. (A) Hexameric Nsp15 was screened against the acrylamide library using three HTS assays including activity-based protein profiling (to find cysteine binders) and FRET and mRNA degradation assay (to find inhibitors) at the concentration of 40 μM; (B) ten potent covalent inhibitors of Nsp15 that have IC50s less than 5 μM were identified, and their chemical structures are shown. | ||
To further characterize and validate the potential binders as Nsp15 inhibitors, we used a fluorescence-based HTS assay that uses a DNA–RNA hybrid oligomer (5′FAM-dArUdAdA-TAMRA3′) with FRET pairs on the ends.25,26 As Nsp15 preferentially cleaves uridylates (rU),25 the endonuclease cleavage of this substrate determines the specific cleavage by Nsp15. Endonuclease cleavage of the oligomer by Nsp15 was quantified by measuring the fluorescence after exciting at 485 nm and measuring the emission at 535 nm. The optimized concentrations, Nsp15 (5 nM) and substrate (1 μM) showed a significant difference (>5-fold) between negative (in absence of Nsp15) and positive control (in presence of Nsp15) and had a Z′ calculated as >0.5. The dataset was normalized with negative and positive control and percentage inhibition was calculated.25,26 The screening of initial hits using this FRET assay resulted in the identification of 408 compounds (a hit rate of 15.4%) that inhibited endonuclease activity of Nsp15. To rule out the false positives, an orthogonal assay was performed to find out if these compounds could prevent mRNA degradation by Nsp15. The mRNA degradation assay confirmed several hits inhibiting 100% of Nsp15 activity and reduced the collection above to 308 (a confirmed hit rate of 11.6%, higher than 5% hit rate shown by fragment-based drug-discovery approach27). To narrow down the hit number for potent covalent inhibitors, all three assays were repeated at a second concentration of 10 μM. This further reduced the count to 60 compounds (a hit rate of 2%). Sixty compounds were repurchased and validated using all three assays at a concentration of 40 and 10 μM, and this resulted in the identification of 15 covalent inhibitors against Nsp15. Finally, a dose response fluorescent-based assay that used an RNA substrate13 was performed to validate and select potent inhibitors. This assay validated 10 compounds with IC50s less than 5 μM (Fig. 1B and S1†).
Acrylamide based Nsp15 inhibitors are non-toxic
We assessed the thiol reactivity of the top electrophile hits by incubating with reduced Ellman's reagent (5,5-dithio-bis-2-nitrobenzoic acid (DTNB)), and followed the absorbance of TNB2− at 412 nm wavelength for up to 5 hours28 (Fig. 2A). To measure the kinetic constants and evaluate the intrinsic reactivity of these acrylamide-based Nsp15 inhibitors towards thiols, we fitted the data to a second-order reaction rate equation and extrapolated the kinetic constant for the alkylation by the acrylamide-based inhibitors. All compounds showed an excellent fit to the kinetic model of one-phase exponential decay (R2 > 0.9). The kinetic rate constant (k) for Nsp15 inhibitors ranged from 1.5–4 M−1 s−1 (Fig. S2†).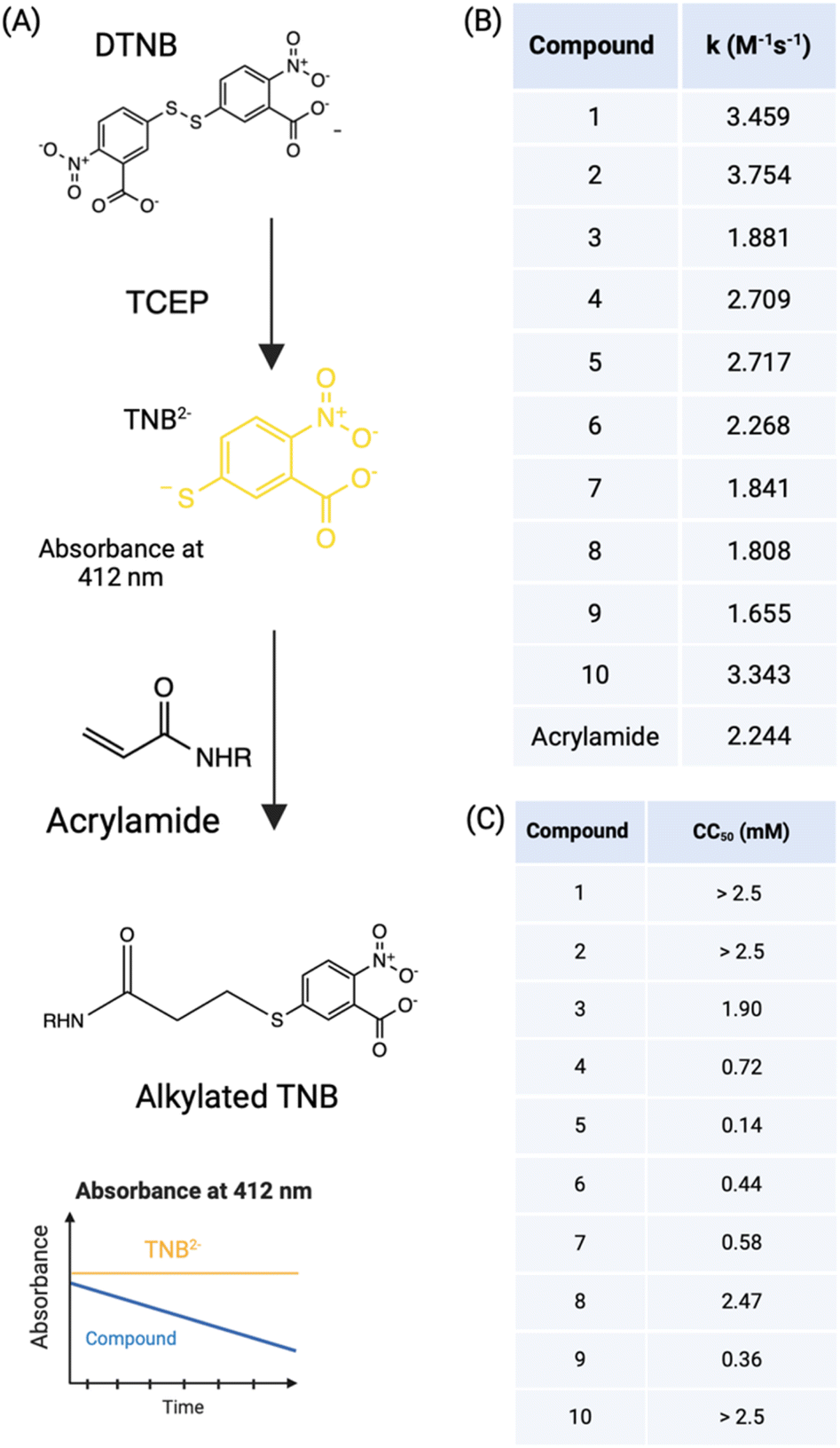 | ||
| Fig. 2 Acrylamide based inhibitors have moderate non-specific reactivity with thiols and are non-toxic to mammalian cells. (A) Scheme for determining thiol reactivity of Nsp15 covalent inhibitors (200 μM) using Ellman's reagent; (B) the kinetic rate constant of alkylation of TNB2− by acrylamide-based inhibitors (k) (M−1 s−1) was calculated and compared with acrylamide; (C) a cell viability assay utilizing the dye resazurin was performed with different concentrations (0–0.5 mM) of inhibitors and revealed the compounds to be non-toxic at the doses tested. The cytotoxic concentration 50% (CC50) (mM) is shown in the table for Caco-2 cells. | ||
Next, we assessed drug toxicity, which is a key parameter in clinical pharmacology and routinely performed during preclinical screening of drug candidates.29 To assess the drug response and toxicity, a resazurin assay was used to analyze the cell viability30 in response to Nsp15 inhibitor compound treatment. A range of concentrations (0.03–0.5 mM) of inhibitors was tested on Caco-2 cells (in vitro model of the intestinal epithelial cells), and cell viability was measured and the CC50 was calculated (Fig. 2C). At the lower concentrations (0.3–0.125 mM), most of the compounds (except compound 5) showed 100% cell viability, suggesting no toxicity at the tested concentrations. At the higher concentration (0.25–1 mM), compounds 1, 2, 3, 8 and 10 show cell viability greater than 85%, while the rest showed less than 40% viability, indicative of some negative effects on cell viability.
Acrylamide-based inhibitors show specificity and are active towards Nsp15 from other coronaviruses
Nsp15 is an evolutionary conserved protein and considered as a genetic marker for nidoviruses. Conservation of Nsp15 across species suggests that SCoV-2 Nsp15 inhibitors might be also used to target Nsp15 from other coronaviruses. SCoV-2 Nsp15 shares sequence identity with other coronavirus species such as SCoV-1, and Middle East respiratory syndrome coronavirus (MERS-CoV), with corresponding percentages of 88.44% and 51.47%, respectively (Fig. S3A†).16 We wondered if these ten compounds could also inhibit Nsp15 from SCoV-1 and MERS-CoV. To examine this, we tested the inhibitory activity of these compounds against Nsp15 from SCoV-2, SCoV-1 and MERS-CoV using a fluorescent based endonuclease assay. We observed that compounds inhibited SCoV-1 and MERS-CoV Nsp15 to varying extents. A dose response assay was performed to determine the IC50 values of these compounds against Nsp15 from SCoV-2, SCoV-1 and MERS-CoV (Fig. 3A and S3B). Since several inhibitors were able to work on other viral variants, we assert that these acrylamide compounds could serve as useful initial hits for development into second-generation compounds against other coronaviruses. | ||
| Fig. 3 Acrylamide based inhibitors show specificity towards Nsp15 from coronaviruses. (A) A dose response assay determining IC50s of ten inhibitors against Nsp15 from SCoV-2, SCoV-1 and MERS-CoV demonstrated that these inhibitors could also be utilized as initial hits for other coronaviruses; (B) effect of inhibitors on unrelated proteins, RNase A and SIRT1 show their specificity towards Nsp15. Results are expressed as percent activity relative to the DMSO control and were normalized based on quenching effects of the compounds in the respective assays using control substrates. Mean ± SD is shown (n = 3 independent experiments). | ||
To examine the broad specificity of the compounds, we also tested these ten compounds against a distantly related RNA endonuclease, RNase A that shares a similar catalytic mechanism with Nsp15. We observed that none of the inhibitors inhibited RNase A activity, suggesting the inhibitors are not acting through the conserved catalytic triad, as expected (RNase A and Nsp15 share the catalytic triad). We also tested these compounds against an unrelated enzyme SIRT1, that has been implicated for modifications (transnitrosation, glutathionylation) of cysteine residue as a mechanism of its physiological inhibition. As expected, and hypothesized, none of the compounds inhibited SIRT1 enzymatic activity, suggesting the nucleophilic cysteine in SIRT1 was not being alkylated by acrylamide based Nsp15 inhibitors. The statistical analysis, t-test of DMSO with each compound (compound 1–10) resulted in a p-value greater than 0.05 (p > 0.05), suggesting no significant difference in enzymatic activity. Together, these results demonstrate that these acrylamide-based compounds are relatively specific inhibitors that act on Nsp15 from various coronaviruses (Fig. 3B).
Ability of acrylamide-based inhibitors to inhibit Nsp15 in cells
To assess the inhibitory effect of these in vitro Nsp15 inhibitors in a cellular environment, we utilized a live virus infection assay based on a genetic ablation of Nsp15 activity resulting in a higher production of IFN-β during Nsp15 mutant virus infection.8 With a catalytic-inactive mutant (H234A) of Nsp15 as a positive control, we evaluated ten compounds in the Caco2-AT culture system. All the compound-treated cells did not produce more IFN-β compared to the untreated wild-type (WT) group, and some even produced less than the WT group. We found that the viral nucleocapsid (N) gene levels of all tested samples were comparable, indicating that all the cells were successfully infected (Fig. S4†). These results suggest that these ten compounds show no significant inhibitory effect on Nsp15 activity during SCoV-2 infection in the Caco2-AT test system. The lack of inhibitory effect might be due to their relative low potency (high IC50) of the compounds.Nsp15 covalent inhibitors are predicted to have favorable drug-like properties
Estimation of the pharmacokinetic profile of a drug candidate is a crucial aspect in drug development that includes parameters like its absorption, distribution, metabolism, and excretion (ADME).31 In this report, we carried out theoretical prediction of ADME parameters of the inhibitors using SwissADME,32,33 a free and readily accessible web tool. We predicted the physiochemical properties, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of these small molecules by importing their 2D structures into a webpage interface using the canonical simplified molecular input line entry system (SMILES) format.The ability of a drug to move across the membranes for transportation throughout the body is highly dependent on its physiochemical properties.34,35 All the Nsp15 inhibitors identified had optimal values for their physiochemical properties, indicating they should have good oral bioavailability, suggesting them as promising drug candidates36 (Fig. S5A†). SwissADME can also predict gastrointestinal (GI) absorption and blood–brain barrier (BBB) penetration,37 two pharmacokinetic behaviors associated with lipophilicity and polarity of the molecules. While all acrylamide based Nsp15 inhibitors were predicted to have a high level of GI absorption, six out of ten inhibitors showed a probability of crossing the BBB. Interestingly, all the inhibitors were deemed to be non-substrates of P-glycoprotein (P-gp), suggesting that they are unlikely to be effluxed from cells.
Drug-likeness is an essential aspect of drug development that evaluates the potential of a molecule to become an oral drug.38 These ten inhibitors followed Lipinski's rule of five,39 with zero violations and a bioavailability score of 0.55, displaying good bioavailability and demonstrating a similarity to other successfully developed oral drug candidates (drug-likeness).40 Importantly, the Nsp15 inhibitors identified herein did not show any PAINs alerts, as indicated by a score of zero. The ease with which these compounds can be synthesized is another positive consideration for their use as lead compounds (Fig. S5B†). To validate these results, we confirmed the drug likeness of these ten compounds using ADMETlab2.0.41
Compound 10 modifies a cysteine in the C-terminal domain of Nsp15
To determine the cysteine residue(s) that mediate the covalent interaction between Nsp15 and the compounds, we performed tandem mass spectrometry (MS/MS) analysis of protein–compound adducts subjected to trypsin digestion. Tandem mass spectrometry revealed that compound 10 is covalently reacting with Cys293. Cys293 is present in the C-terminal domain of Nsp15, next to the catalytic core of Nsp15 harboring endoribonuclease activity.42 Cys293 has been also been implicated in interaction with the drug Favipiravir through van der Waals interactions in molecular docking simulations43 (Fig. 4A).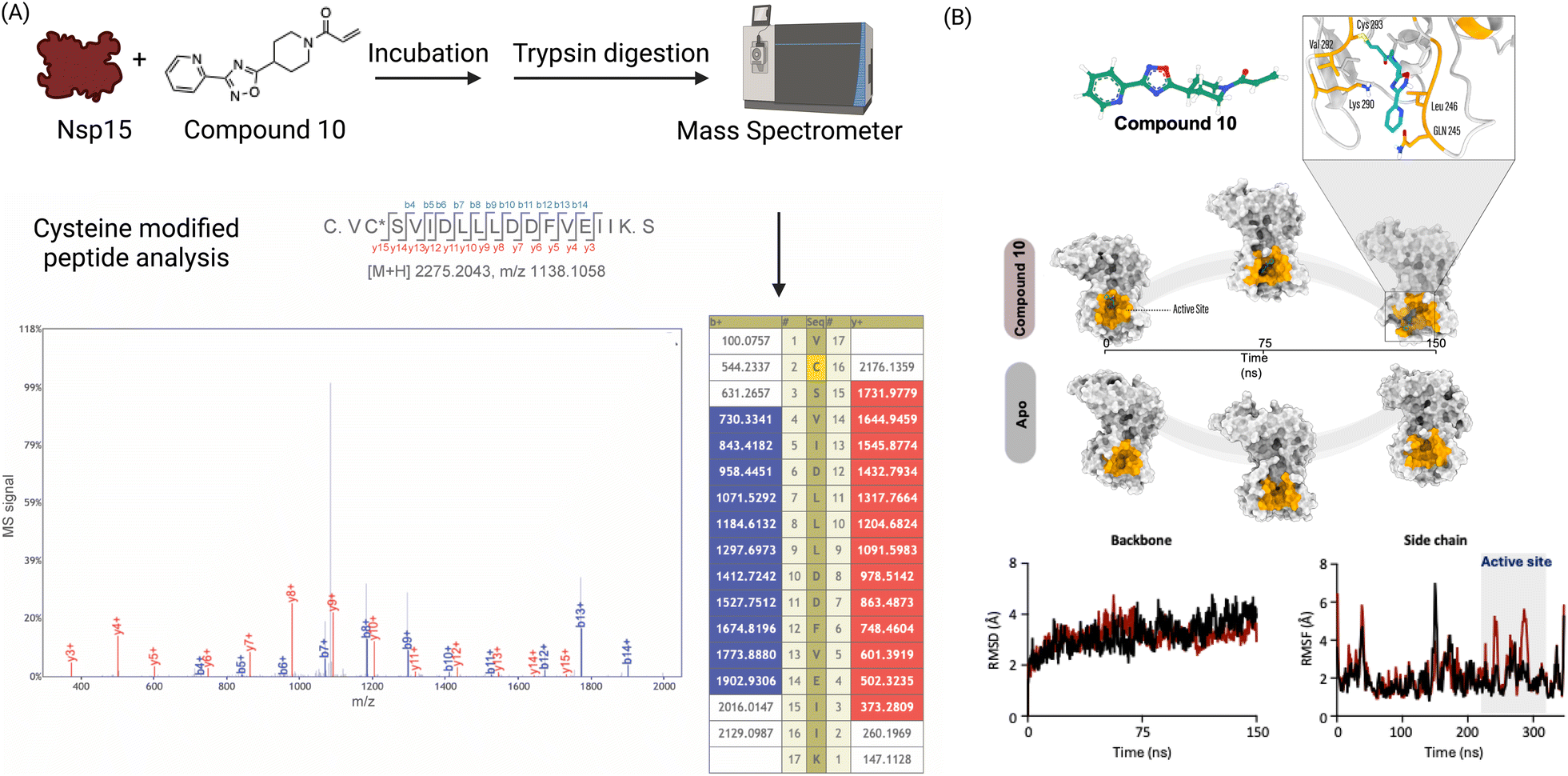 | ||
| Fig. 4 Compound 10 modifies Cys293 in Nsp15 and distorts the active site in the C-terminal domain of Nsp15. (A) Compound 10–Nsp15 was subjected to trypsin digestion followed by LC-mass spectrometry and analyzed by shotgun analysis. A peptide with Cys293 was found to be modified with compound 10; (B) structural analysis from the MD simulations of 150 ns of compound 10–Nsp15 complex in comparison to apo Nsp15 revealed that the irreversible binding of compound 10 to Nsp15 significantly distorts the active site. | ||
To assess the structural dynamics of Nsp15 following the irreversible covalent reaction of compound 10 to Cys293, we conducted molecular dynamics (MD) simulations of Nsp15 in its apo form and in complex with compound 10 (covalently bound to Cys293). Structural analysis from the MD simulations revealed that the irreversible binding of compound 10 to Nsp15 significantly distorts the active site (Fig. 4B). Although there is no observable change in the overall backbone of Nsp15, the side chain fluctuations in the compound 10-bound complex are altered, indicating a distortion of the binding site.
Development of an AI model to support Nsp15 hit-to-lead optimization
During drug discovery, turning an early stage hit molecule into a nanomolar-range lead molecule often requires numerous iterations, and even then, it carries a significant chance of failure. Therefore, to expedite this process, we utilized the power of AI to identify distinguishing characteristics between the successful and unsuccessful Nsp15 inhibitors in our library, an endeavor that would be impossible to be achieved manually. To the best of our knowledge, we are the first group to leverage AI for the screening of SCoV-2 Nsp15 inhibitors. Thus, we utilized our experimental HTS data to train sophisticated AI models to streamline the hit-to lead discovery process and make it less laborious and expensive (Fig. 5A). Our AI-driven methodology demonstrated a marked improvement in prediction accuracy and has the potential to reduce false positives and negatives. | ||
| Fig. 5 Development of an AI model to predict Nsp15 inhibitors and accelerate the process of hit to lead identification. (A) The results from the high throughput screening assay against Nsp15 were used to develop an AI model that could virtually screen thousands of compounds to predict inhibitors and non-inhibitors against Nsp15; (B) the AI model was developed by utilizing the chemical properties, integrating ChemBERTa, and machine learning techniques to predict Nsp15 inhibitors. The integrated model was tested on 50 unlabeled compounds, and its ability to predict inhibitors was determined. The model showed 73% accuracy in distinguish inhibitor from non-inhibitors; (C) representation of the test predictions made by the integrated AI model. The rest of the compounds are shown in Fig. S6B.† | ||
Artificial intelligence requires a vast training dataset comprising millions of data points; however, it is next to impossible to generate an experimental dataset this large in drug discovery. Effective training of AI models cannot be achieved with smaller dataset. Therefore, to overcome this challenge, we explored several strategies, including fine-tuning large language models, applying prompt engineering for ChatGPT-based predictions, and combining embeddings from language models with traditional machine learning techniques. We used various AI models, including Random Forest (RF), SVM, Random Forest (RF), Decision Tree (DT), Gradient Boosting (GB), Logistic Regression (LR), Naïve Bayes (NB), KNN, C4.5, Convolutional Neural Networks (CNNs), Recurrent Neural Networks (RNNs), LLMs models such as GPT-3.5-turbo, and GPT-4-turbo. Each of these models was optimized and fine-tuned to enhance accuracy and precision. Among these, the Gradient Boosting model demonstrated the best performance, as shown in Fig. S6A.†
We extensively experimented with different algorithm parameters and input features to improve model accuracy. For the machine learning models, we utilized a grid search approach to find the best hyperparameters for each model. We evaluated various LLM models for inhibitor prediction. After extensive testing and optimization of the models, we developed our model, which integrates Gradient Boosting and LLM. This model combines features and embedding vectors from the LLMs to train a new model for predicting inhibitors from a pool of chemical structures (Fig. 5B).
We measured the model's performance using four main metrics: recall, precision, F1 score, and efficiency. Recall checks how well the model finds all the correct results, which is important to avoid missing key compounds. Precision looks at how many of the positive predictions are actually correct, helping reduce false alarms. The F1 score combines recall and precision into one measure to give a balanced view of performance, especially when the data is uneven. Efficiency measures how quickly and easily the model makes predictions, which is important for real-time or large-scale use. Together, these metrics show how well the model works and where it can improve. The efficiency for all the individual models was greater than 50%, suggesting that our proposed pipeline is effective in dealing with small datasets for inhibitor prediction. This integrated approach significantly improved prediction accuracy, showing a precision value of 0.73 and an F1 score of 0.73, further indicating its high accuracy.
To validate our model, we tested it on an existing Nsp15 inhibitor data set, recently published.44 Our model predicted 9 out of 12 inhibitors listed in this recently published paper correctly, demonstrating its high accuracy. After validation, we employed our Integrated AI model to test 50 unlabeled compounds. This model predicted the unlabeled inhibitors with an accuracy of 73%. All the correct and incorrect predictions made on these 50 test compounds are presented in Fig. 5C and S6B.† Moving forward, we anticipate that this experimental-led AI driven experimental discovery platform will identify new potent lead compounds in a cheap and efficient manner. Moreover, the platform we present here can also be adapted for the discovery of inhibitors for other high-value target proteins.
Experimental
Materials and methods
:100 in 2XYT media to an OD of 1.0 (A600). SCoV-2 Nsp15 protein was expressed with 0.2 M isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 hours at 37 °C. The culture was harvested and resuspended in lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5% glycerol) supplemented with EDTA-free protease inhibitor tablets. The cells were sonicated, and lysate was clarified at 13000×g for 30 minutes at 4 °C. The supernatant was loaded on Talon HP column (Cytiva) at the speed of 1 mL min−1. The non-specific protein was removed by washing the column with lysis buffer supplemented with 10 mM imidazole. Nsp15 was eluted with high imidazole buffer (lysis buffer supplemented with 250 mM imidazole). The eluted fraction of Nsp15 were pooled, concentrated, and dialyzed against SEC buffer (20 mM HEPES pH 7.3, 150 mM NaCl, 5 mM MnCl2, 5 mM beta-mercaptoethanol (β-me)). The protein was stored in −80 °C until further use.SCoV-1 and MERS-CoV Nsp15 variants were purified as previously described.16 Briefly, BL21(DE3) pLYsS cells were transformed with pet28B+ plasmids encoding for these Nsp15 variants. Starter cultures were grown overnight at 37 °C in terrific broth (TB) in the presence of 100 μg mL−1 kanamycin. Larger cultures of TB-kanamycin were inoculated with starter culture and grown to an OD 600 nm of 0.6. Cultures were then cooled at 4 °C for 30 minutes before induction with 1 mM IPTG at 16 °C for 20 hours. Cells were then pelleted at 6000×g for 20 minutes at 4 °C and resuspended in lysis buffer (20 mM Tris pH 8.0, 150 mM NaCl, 5 mM imidazole, 0.1% Triton X-100, 1 mg mL−1 lysozyme) supplemented with EDTA-free Roche Complete Ultra protease inhibitor tablet (Sigma), 1 mM PMSF and 1 mM β-me. The lysate was incubated on ice for 30 minutes before sonication with a Branson Digital Sonifier at 25% amplitude (15 seconds on, 1 minute off) for 10 pulses. Debris was pelleted via centrifugation at 20000×g and the clarified lysate was incubated with Ni-NTA beads (Qiagen) at 4 °C for 4 hours with gentle rotation. The beads were washed in 20 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole, 0.01% Triton X-100, and 1 mM β-me. The proteins were eluted by incubating the beads with 10–250 mM imidazole. Fractions were analyzed for purity via SDS-PAGE and staining with Coomassie Brilliant Blue R-250 (BioRad). Pooled fractions were concentrated with a Pierce Protein concentrator 10K (Thermofisher). The buffer was exchanged during concentration with 20 mM HEPES pH 7.5, 150 mM NaCl, 0.1 mM DTT, and 10% glycerol. Concentrated protein was aliquoted and stored at −80 °C until usage. Protein concentration was measured using the DTT-resistant Pierce 660 nM Protein BCA Assay kit (Thermofisher).
μL buffer A (20 mM HEPES pH 7.2, 100 mM NaCl, 5 mM MnCl2) for 30 minutes. The final concentration of DMSO was 3.3%. Then, 20 μL of 0.5 μM RNA substrate (5′6-FAM-AAAUAA-3′6-TAMRA, GenScript) in a buffer A was added to the protein–compound complex and fluorescent intensity was measured at excitation/emission wavelength of 485/528 every 5 min for 120 minutes. The IC50 values were calculated based on the final concentrations of the compounds at the 45- or 90-minutes time points using GraphPad Prism.Specificity assay
The following SCoV-2 strain/isolate was obtained through BEI Resources, NIAID, NIH: Washington strain 1 (WA1) (NR-52281). A recombinant virus expressing catalytic-inactive Nsp15 (Nsp15mut) was generated using an infection clone as described here.47 These viruses were propagated once with Vero-AT cells to obtain large viral stocks and were titrated with Vero-AT cells.
Mass spectrometry
000 at 200 m/z; scan range, 350−1600 m/z. The top 20 most-abundant ions were subjected to collision-induced dissociation with a normalized collision energy of 35%, activation q 0.25, and precursor isolation width 2 m/z. Dynamic exclusion was enabled with a repeat count of 1, a repeat duration of 30 seconds, and an exclusion duration of 20 seconds. RAW files were analysed using PEAKS (Bioinformatics Solution Inc.) with the following parameters: semi-specific cleavage specificity at the C-terminal site of R and K, allowing for 5 missed cleavages, precursor mass tolerance of 15 ppm, and fragment ion mass tolerance of 0.5 daltons. Methionine oxidation was set as variable modifications and cysteine carbamidomethylation was set as a fixed modification. Peptide hits were filtered using a 5% FDR. Proteins with at least 2 unique peptides were filtered with a 5% FDR. Label free quantitation (LFQ) was performed using PEAKS quantitation module and default parameters with the following exceptions: top 2 peptides for each protein with a min of 10XE4 abundance was used and the TIC was used for all normalization including technical replicates.P (MolLogP), number of atoms, number of bonds, number of rings, rotatable bond counts, hydrogen bond donors, hydrogen bond acceptors, number of stereocenters, and topological polar surface area (TPSA). These features were normalized prior to their integration into the dataset. The dataset comprised 1920 entries, including 257 molecules identified as Nsp15 inhibitors (positive hits), 1613 molecules with no inhibitory action against Nsp15 (negative hits). The dataset was divided into training, validation, and testing sets with a 70%, 15%, 15% split: specifically, 70% (1309 compounds) for training, 15% (280 compounds) for validation, and 15% (280 compounds) for testing, and 50 molecules reserved for validation purposes as unlabeled compounds.ChemBERTa-based models
P, and other relevant descriptors. The combined embeddings and features were passed through a dropout layer to mitigate overfitting risks and subsequently fed into a dense layer for final predictions. This integration aimed to leverage both chemical structure information and specific molecular properties to improve prediction accuracy.Example prompt for GPT models:
“FC(F)(F)CC1CN(CCO1)C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CC ≫ YES O)CC ≫ YES |
| CCC(O)N1CCCCC1C2CCCO2 ≫ NO |
Based on the prior examples, for all of the following, predict whether they inhibit or not:
| CC1CC2CCCCC2N1C(O)CC≫ |
| CCC(O)N1CCN(CC1)S(O)(O)CC2CCON2≫” |
Including the Nsp15 protein sequence provided contextual biochemical information potentially enhancing prediction accuracy. For a comprehensive analysis, we engineered prompts that combined chemical structures (SMILES strings) with detailed quantitative features:
“The following are the drug information of molecules that do or do not inhibit the Nsp 15 protein of Covid-19 virus.
Each drug information is presented in one line in the following order: smiles strings, molecular weight, logP, number of atoms, number of bonds, number of rings, rotatable bonds count, hydrogen bond donors, hydrogen bond acceptors, number of stereocenters, Topological Polar Surface Area (TPSA).
| CNC(O)CC1CCN(CC1)C(O)CC, 210.277, 0.5471, 15, 15, 1, 3, 1, 2, 0, 49.41 ≫ 1 |
| CCC(O)N1CCSCC1C#N, 182.248, 0.63998, 12, 12, 1, 1, 0, 3, 1, 44.1 ≫ 0 |
Based on the prior examples, for all the following, predict whether they inhibit or not. Only output the smiles strings and your predictions, nothing else:
| CCC(O)NCC(O)N1CCC2CCCCC2C1, 244.294, 0.8735, 18, 19, 2, 3, 1, 2, 0, 49.41≫ |
| CN(CC(O)N1CCCC1)C(O)CC, 196.25, 0.2532, 14, 14, 1, 3, 0, 2, 0, 40.62≫” |
This prompt format provided extensive data for each molecule, allowing the GPT model to generate predictions based on both structural and physical properties.
| Recall = TP/TP + FN | (1) |
| Precision = TP/TP + FP | (2) |
| F1 score = 2 (precision × recall)/(precision + recall) | (3) |
| Accuracy = TP + TN/TP + TN + FP + FN | (4) |
The AUC value was considered as the indicator of classification model accuracy. Precision measures the accuracy of positive predictions made by a classifier. Recall, also known as sensitivity or true positive rate, measures the ability of a classifier to correctly identify all positive instances in the dataset. The F1 score is the harmonic mean of precision and recall, providing a single metric that balances both precision and recall.
Conclusions
Inhibition of Nsp15 has the potential to greatly improve the treatment of SCoV-2. Nsp15 is a crucial endoribonuclease present in all coronaviruses that aids viruses in evading the host immune response during viral infection. Nsp15 suppresses the production of interferons by infected cells by cleaving viral RNA. Down-regulating the production of interferons by SCoV-2 infected could have synergistic effects with inhibiting viral replication by preventing neighboring cells from being infected with viruses. Despite its potential, developing inhibitors against Nsp15 has been challenging due to its structural complexity and large binding interface. HTS against SCoV-2 Nsp15 have yielded a few inhibitors, however these compounds were frequently promiscuous hits or non-potent.Cysteine reactive acrylamide compounds have had great success as covalent inhibitors of proteins, especially in transforming “undruggable” proteins to druggable. Here, we took this opportunity to screen a never-been-explored acrylamide-based library against Nsp15. Acrylamide-containing drugs show prolonged on-target residence time due to irreversible cysteine engagement. We screened a 2640 acrylamide-based electrophile library and identified ten cysteine reactive inhibitors against Nsp15 with IC50s in the low micromolar range (less than 5 μM). These compounds are non-toxic in mammalian cells. These acrylamide-based inhibitors are specific to Nsp15 and can potentially be utilized as initial hits for targeting other coronaviruses. In conclusion, we present acrylamide-based fragments as new covalent inhibitors of Nsp15 enzymes from various coronaviruses and present a new AI-driven pipeline based on these results for the rapid and cheap identification of future lead compounds.
Data availability
The data and code used in this study are available online at https://github.com/babakmosavati/AI-Powered-Platform-Drug-Discovery.Author contributions
Conceptualization, N. M., D. K. N., B. P. H., R. S., X. D., J. R. U., J. S., M. R. K. M.; methodology, T. B., B. M., L. H. Z., E. M. K., H. W., X. L., S. A. T. D., E. W., S. G., R. N. D., M. S. P., J. S.; software, B. M., T. B., L. H. Z., E. M. K., H. W., X. L., S. A. T. D., M. S. P., E. W.; validation, B. M., T. B., N. M.; formal analysis, B. M., T. B., L. H. Z., E. M. K., H. W., X. L., S. A. T. D., E. W.; investigation, T. B., B. M. and N. M.; resources, N. M., D. K. N., B. P. H., R. S., X. D., J. S.; data curation, N. M. and B. M., T. B.; writing—original draft preparation, T. B., B. M., S. G., N. M.; writing—review and editing, N. M., T. B., B. M., J. S., B. P. H., R. S., X. D., L. H. Z.; visualization, T. B., B. M.; supervision, N. M., J. S.; project administration, N. M.; funding acquisition, N. M., J. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Basil P. Hubbard, Xufang Deng for providing critical suggestions for the manuscript. We would also acknowledge Robert Maxwell from QB3 Mass spectrometry facility for mass spectrometry experiments. The BSL-3 live virus work was supported by the Oklahoma Center for the Advancement of Science and Technology (OCAST) grant HR23096 to Xufang Deng. Niren Murthy would like to acknowledge NIH grants UG3NS115599, R33 and R61DA048444-01, RO1EB029320-01A1, RO1MH125979-01, and funding from the BAKAR Spark award, the Cystic Fibrosis Foundation, and the Innovative Genomics Institute. Julia Schaletzky, and Eddie Wehri were supported by the Henry Wheeler Center for Emerging and Neglected Diseases and through Fastgrants. Robin Stanley would like to acknowledge the in part support by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (1ZIAES103340).References
- Y. Zhu, J. Li and Z. Pang, Asian J. Pharm. Sci., 2021, 16, 4–23 Search PubMed.
- C. Pozzi, A. Vanet, V. Francesconi, L. Tagliazucchi, G. Tassone, A. Venturelli, F. Spyrakis, M. Mazzorana, M. P. Costi and M. Tonelli, J. Med. Chem., 2023, 66, 3664–3702 CrossRef CAS PubMed.
- I. Pauly, A. Kumar Singh, A. Kumar, Y. Singh, S. Thareja, M. A. Kamal, A. Verma and P. Kumar, Curr. Pharm. Des., 2022, 28, 3677–3705 CrossRef CAS PubMed.
- S. M. R. Hashemian, A. Sheida, M. Taghizadieh, M. Y. Memar, M. R. Hamblin, H. Bannazadeh Baghi, J. Sadri Nahand, Z. Asemi and H. Mirzaei, Biomed. Pharmacother., 2023, 162, 114367 CrossRef CAS PubMed.
- J. Liu, X. Pan, S. Zhang, M. Li, K. Ma, C. Fan, Y. Lv, X. Guan, Y. Yang and X. Ye, Lancet Reg. Health West. Pac., 2023, 33, 100694 Search PubMed.
- Y. Duan, H. Zhou, X. Liu, S. Iketani, M. Lin, X. Zhang, Q. Bian, H. Wang, H. Sun, S. J. Hong, B. Culbertson, H. Mohri, M. I. Luck, Y. Zhu, X. Liu, Y. Lu, X. Yang, K. Yang, Y. Sabo, A. Chavez, S. P. Goff, Z. Rao, D. D. Ho and H. Yang, Nature, 2023, 622, 376–382 CrossRef CAS PubMed.
- S. A. Moghadasi, E. Heilmann, A. M. Khalil, C. Nnabuife, F. L. Kearns, C. Ye, S. N. Moraes, F. Costacurta, M. A. Esler, H. Aihara, D. von Laer, L. Martinez-Sobrido, T. Palzkill, R. E. Amaro and R. S. Harris, Sci. Adv., 2023, 9, eade8778 CrossRef CAS PubMed.
- X. Deng, M. Hackbart, R. C. Mettelman, A. O'Brien, A. M. Mielech, G. Yi, C. C. Kao and S. C. Baker, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4251–E4260 CAS.
- E. Kindler, C. Gil-Cruz, J. Spanier, Y. Li, J. Wilhelm, H. H. Rabouw, R. Zust, M. Hwang, P. V'Kovski, H. Stalder, S. Marti, M. Habjan, L. Cervantes-Barragan, R. Elliot, N. Karl, C. Gaughan, F. J. van Kuppeveld, R. H. Silverman, M. Keller, B. Ludewig, C. C. Bergmann, J. Ziebuhr, S. R. Weiss, U. Kalinke and V. Thiel, PLoS Pathog., 2017, 13, e1006195 CrossRef PubMed.
- D. Zhang, L. Ji, X. Chen, Y. He, Y. Sun, L. Ji, T. Zhang, Q. Shen, X. Wang, Y. Wang, S. Yang, W. Zhang and C. Zhou, iScience, 2023, 26, 107705 CrossRef CAS PubMed.
- C. K. Yuen, J. Y. Lam, W. M. Wong, L. F. Mak, X. Wang, H. Chu, J. P. Cai, D. Y. Jin, K. K. To, J. F. Chan, K. Y. Yuen and K. H. Kok, Emerging Microbes Infect., 2020, 9, 1418–1428 CrossRef CAS PubMed.
- C. J. Otter, N. Bracci, N. A. Parenti, C. Ye, L. H. Tan, A. Asthana, J. J. Pfannenstiel, N. Jackson, A. R. Fehr, R. H. Silverman, N. A. Cohen, L. Martinez-Sobrido and S. R. Weiss, bioRxiv, 2023, preprint, DOI:10.1101/2023.11.15.566945.
- M. C. Pillon, M. N. Frazier, L. B. Dillard, J. G. Williams, S. Kocaman, J. M. Krahn, L. Perera, C. K. Hayne, J. Gordon, Z. D. Stewart, M. Sobhany, L. J. Deterding, A. L. Hsu, V. P. Dandey, M. J. Borgnia and R. E. Stanley, Nat. Commun., 2021, 12, 636 CrossRef CAS PubMed.
- M. N. Frazier, I. M. Wilson, J. M. Krahn, K. J. Butay, L. B. Dillard, M. J. Borgnia and R. E. Stanley, Nucleic Acids Res., 2022, 50, 8290–8301 CrossRef CAS PubMed.
- Y. Kim, J. Wower, N. Maltseva, C. Chang, R. Jedrzejczak, M. Wilamowski, S. Kang, V. Nicolaescu, G. Randall, K. Michalska and A. Joachimiak, Commun. Biol., 2021, 4, 193 CrossRef CAS PubMed.
- J. Chen, R. A. Farraj, D. Limonta, S. A. Tabatabaei Dakhili, E. M. Kerek, A. Bhattacharya, F. M. Reformat, O. M. Mabrouk, B. Brigant, T. A. Pfeifer, M. T. McDermott, J. R. Ussher, T. C. Hobman, J. N. M. Glover and B. P. Hubbard, J. Biol. Chem., 2023, 299, 105341 CrossRef CAS PubMed.
- J. Ortiz-Alcantara, K. Bhardwaj, S. Palaninathan, M. Frieman, R. S. Baric and C. C. Kao, Virus Adapt. Treat., 2010, 2, 125–133 CAS.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- P. Ábrányi-Balogh, L. Petri, T. Imre, P. Szijj, A. Scarpino, M. Hrast, A. Mitrović, U. P. Fonovič, K. Németh, H. Barreteau, D. I. Roper, K. Horváti, G. G. Ferenczy, J. Kos, J. Ilaš, S. Gobec and G. M. Keserű, Eur. J. Med. Chem., 2018, 160, 94–107 CrossRef PubMed.
- A. O. Walter, R. T. T. Sjin, H. J. Haringsma, K. Ohashi, J. Sun, K. Lee, A. Dubrovskiy, M. Labenski, Z. Zhu and Z. Wang, Cancer Discovery, 2013, 3, 1404–1415 CrossRef CAS PubMed.
- M. S. Davids and J. R. Brown, Future Oncol., 2014, 10, 957–967 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn and G. Goodman, J. Med. Chem., 2019, 63, 52–65 CrossRef PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- S. Wang, Y. Tian, M. Wang, M. Wang, G. B. Sun and X. B. Sun, Front. Pharmacol, 2018, 9, 353 CrossRef PubMed.
- K. Bhardwaj, J. Sun, A. Holzenburg, L. A. Guarino and C. C. Kao, J. Mol. Biol., 2006, 361, 243–256 CrossRef CAS PubMed.
- R. Choi, M. Zhou, R. Shek, J. W. Wilson, L. Tillery, J. K. Craig, I. A. Salukhe, S. E. Hickson, N. Kumar, R. M. James, G. W. Buchko, R. Wu, S. Huff, T. T. Nguyen, B. L. Hurst, S. Cherry, L. K. Barrett, J. L. Hyde and W. C. Van Voorhis, PLoS One, 2021, 16, e0250019 CrossRef CAS PubMed.
- S. G. Kathman and A. V. Statsyuk, MedChemComm, 2016, 7, 576–585 RSC.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- S. Parasuraman, J. Pharmacol. Pharmacother., 2011, 2, 74–79 CrossRef CAS PubMed.
- R. C. Borra, M. A. Lotufo, S. M. Gagioti, M. Barros Fde and P. M. Andrade, Braz. Oral Res., 2009, 23, 255–262 CrossRef PubMed.
- A. Reichel and P. Lienau, Handb. Exp. Pharmacol., 2016, 232, 235–260 CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- B. Bakchi, A. D. Krishna, E. Sreecharan, V. B. J. Ganesh, M. Niharika, S. Maharshi, S. B. Puttagunta, D. K. Sigalapalli, R. R. Bhandare and A. B. Shaik, J. Mol. Struct., 2022, 1259, 132712 CrossRef CAS.
- S. B. Bunally, C. N. Luscombe and R. J. Young, SLAS Discovery, 2019, 24, 791–801 CrossRef CAS PubMed.
- A. T. Garcia-Sosa, U. Maran and C. Hetenyi, Curr. Med. Chem., 2012, 19, 1646–1662 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- F. Protti Í, D. R. Rodrigues, S. K. Fonseca, R. J. Alves, R. B. de Oliveira and V. G. Maltarollo, ChemMedChem, 2021, 16, 1446–1456 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- Y. C. Martin, J. Med. Chem., 2005, 48, 3164–3170 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, W5–w14 CrossRef CAS PubMed.
- J. Chen, R. Abou Farraj, D. Limonta, S. A. T. Dakhili, E. M. Kerek, A. Bhattacharya, F. M. Reformat, O. M. Mabrouk, B. Brigant and T. A. Pfeifer, J. Biol. Chem., 2023, 299(11), 105341 CrossRef CAS PubMed.
- E. Şahin, A. Karanfil, M. Çol Ayvaz and E. Şahin, J. Mol. Struct., 2021, 1248, 131357 CrossRef.
- B. Van Loy, A. Stevaert and L. Naesens, Antiviral Res., 2024, 228, 105921 CrossRef CAS PubMed.
- J. Han, B. P. Hubbard, J. Lee, C. Montagna, H. W. Lee, D. A. Sinclair and Y. Suh, Cell Cycle, 2013, 12, 263–270 CrossRef CAS PubMed.
- D. Y. Chen, J. Turcinovic, S. Feng, D. J. Kenney, C. V. Chin, M. C. Choudhary, H. L. Conway, M. Semaan, B. J. Close, A. H. Tavares, S. Seitz, N. Khan, S. Kapell, N. A. Crossland, J. Z. Li, F. Douam, S. C. Baker, J. H. Connor and M. Saeed, iScience, 2023, 26, 106634 CrossRef CAS PubMed.
- Y. Hu, E. M. Lewandowski, H. Tan, X. Zhang, R. T. Morgan, X. Zhang, L. M. C. Jacobs, S. G. Butler, M. V. Gongora, J. Choy, X. Deng, Y. Chen and J. Wang, ACS Cent. Sci., 2023, 9, 1658–1669 CrossRef CAS PubMed.
- T. Xu, J. D. Venable, S. K. Park, D. Cociorva, B. Lu, L. Liao, J. Wohlschlegel, J. Hewel and J. Yates, Mol. Cell. Proteomics, 2006, 5, S174 Search PubMed.
- T. Xu, S. K. Park, J. D. Venable, J. A. Wohlschlegel, J. K. Diedrich, D. Cociorva, B. Lu, L. Liao, J. Hewel, X. Han, C. C. L. Wong, B. Fonslow, C. Delahunty, Y. Gao, H. Shah and J. R. Yates 3rd, J. Proteomics, 2015, 129, 16–24 CrossRef CAS PubMed.
- D. L. Tabb, W. H. McDonald and J. R. Yates, J. Proteome Res., 2002, 21–26 CrossRef CAS PubMed.
- D. L. Tabb, W. H. McDonald and J. R. Yates 3rd, J. Proteome Res., 2002, 1, 21–26 CrossRef CAS PubMed.
- S. K. Park, J. D. Venable, T. Xu and J. R. Yates, Nat. Methods, 2008, 5, 319–322 CrossRef CAS PubMed.
- S. K. R. Park, A. Aslanian, D. B. McClatchy, X. Han, H. Shah, M. Singh, N. Rauniyar, J. J. Moresco, A. F. M. Pinto, J. K. Diedrich, C. Delahunty and J. R. Yates III, Bioinformatics, 2014, 30, 2208–2209 CrossRef CAS PubMed.
- C. R. Søndergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef PubMed.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2019, 15, 1863–1874 CrossRef CAS PubMed.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, e4792 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
- T. R. Lane, F. Urbina, X. Zhang, M. Fye, J. Gerlach, S. H. Wright and S. Ekins, Mol. Pharm., 2022, 19, 4320–4332 CrossRef CAS PubMed.
- G. Tu, T. Fu, G. Zheng, B. Xu, R. Gou, D. Luo, P. Wang and W. Xue, J. Chem. Inf. Model., 2024, 64, 1433–1455 CrossRef CAS PubMed.
- B. Dudas and M. A. Miteva, Trends Pharmacol. Sci., 2024, 45, 39–55 CrossRef CAS PubMed.
- F. Wong, E. J. Zheng, J. A. Valeri, N. M. Donghia, M. N. Anahtar, S. Omori, A. Li, A. Cubillos-Ruiz, A. Krishnan, W. Jin, A. L. Manson, J. Friedrichs, R. Helbig, B. Hajian, D. K. Fiejtek, F. F. Wagner, H. H. Soutter, A. M. Earl, J. M. Stokes, L. D. Renner and J. J. Collins, Nature, 2024, 626, 177–185 CrossRef CAS PubMed.
- J. P. Vert, Nat. Biotechnol., 2023, 41, 750–751 CrossRef CAS PubMed.
- G. Turon, J. Hlozek, J. G. Woodland, A. Kumar, K. Chibale and M. Duran-Frigola, Nat. Commun., 2023, 14, 5736 CrossRef CAS PubMed.
- F. Grisoni, Curr. Opin. Struct. Biol., 2023, 79, 102527 CrossRef CAS PubMed.
- A. Tropsha, O. Isayev, A. Varnek, G. Schneider and A. Cherkasov, Nat. Rev. Drug Discovery, 2024, 23, 141–155 CrossRef PubMed.
- M. Ballarotto, S. Willems, T. Stiller, F. Nawa, J. A. Marschner, F. Grisoni and D. Merk, J. Med. Chem., 2023, 66, 8170–8177 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06955b |
| ‡ Equal contribution by authors. |
| This journal is © The Royal Society of Chemistry 2025 |