
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of quinoidal (di-)anionic coupled polymethine-oxonol dyes and their aromatic counterparts†
Benjamin
Mourot‡
 a,
Carmelo
Naim‡
b,
Olivier
Siri
a,
Denis
Jacquemin
*bc and
Simon
Pascal
*ab
a,
Carmelo
Naim‡
b,
Olivier
Siri
a,
Denis
Jacquemin
*bc and
Simon
Pascal
*ab
aAix Marseille Univ, CNRS UMR 7325, Centre Interdisciplinaire de Nanoscience de Marseille (CINaM), Campus de Luminy, case 913, Marseille cedex 09 13288, France. E-mail: simon.pascal@cnrs.fr
bNantes Université, CEISAM UMR 6230, CNRS, Nantes F-44000, France. E-mail: Denis.Jacquemin@univ-nantes.fr
cInstitut Universitaire de France (IUF), Paris F-75005, France
First published on 30th June 2025
Abstract
We report the synthesis of a new family of chromophores based on benzothiazole, indandione, and tricyanofuran moieties, forming aromatic neutral precursors or negatively charged quinoidal counterparts. Upon deprotonation, these dyes undergo marked bathochromic shifts, leading to highly delocalized coupled polymethine structures, particularly in their moderatly antiaromatic dianionic forms. The evolution of optical properties was systematically studied through pH-dependent absorption measurements and supported by TD-DFT and CC2 calculations. Distinct behaviors were identified across the series: ESIPT in benzothiazole dyes, photoacidity in indandione derivatives, and spirocyclization in tricyanofuran-containing dyes. These findings provide insights into structure–property relationships in anionic polymethine systems.
Introduction
Organic dyes capable of absorbing in the near-infrared (NIR) region are highly sought for modern applications, particularly in bio-imaging and molecular electronics.1–5 Among NIR chromophores, polymethines are one of the most common families used for accessing such wavelengths, owing to their remarkable optical properties, which stem from the delocalization of an even number of π-electrons along an unsaturated chain composed of an odd number of sp2-hybridized carbon atoms. Symmetrical polymethines are typically synthesized by Knoevenagel condensations, in which two electron-donating (D) or electron-withdrawing (A) moieties react with an unsaturated dialdehyde. The resulting polymethine bridge enables charge delocalization between the terminal units, giving rise to different classes of polymethine dyes.6–9Depending on the nature of the end groups, polymethines can be cationic (D–π–D, e.g. cyanines), neutral (D–π–A, e.g., merocyanines) or anionic (A–π–A, e.g., oxonols). While (mero)cyanines are well-documented in the literature,6,7,10,11 and can readily achieve NIR absorption with straightforward synthesis,12–14 negatively charged polymethines remain relatively scarce. These dyes typically incorporate electron-withdrawing end groups such as benzothiazole (A),15 1,3-indandione (B),16–18 or tricyanofuran (C),19 which are among the most common building blocks in negatively charged polymethines (Fig. 1). Rigidified anionic polymethines have also been designed introducing dipyrromethene,20 dioxaborine,21,22 or a central cyclohexyl ring linking tricyanofurans (D), further redshifting their optical properties towards the NIR region.23 Notably, negatively charged polymethine dyes have been explored in various applications, including light harvesting in solar cells,24,25 nonlinear optics,26–29 two-photon fluorescence bio-imaging,30 and halochromic sensing.31
 | ||
| Fig. 1 Optical properties of previously reported anionic polymethines featuring the different end groups. | ||
In their heuristic 1966 study, Dähne and Leupold proposed the concept of coupled polymethines, in which two polymethine subunits are linked by one or more σ bonds. This coupling results in the hybridization of the key molecular orbitals, reducing the HOMO–LUMO gap, thereby inducing a bathochromic shift in the absorption spectrum.32 The validity of this principle was demonstrated in the same study through a comparison between the simple trimethine oxonol (Ox3), which exhibits an absorption maximum at 267 nm, and its coupled counterpart (DiOx3), synthesized from 2,5-dihydroxybenzoquinone in basic media, which absorbs at 496 nm in aqueous sodium hydroxide (Fig. 2).32 Despite their intriguing optical properties, such coupled polymethines, and particularly negatively charged ones remain a relatively unexplored family of dyes.33
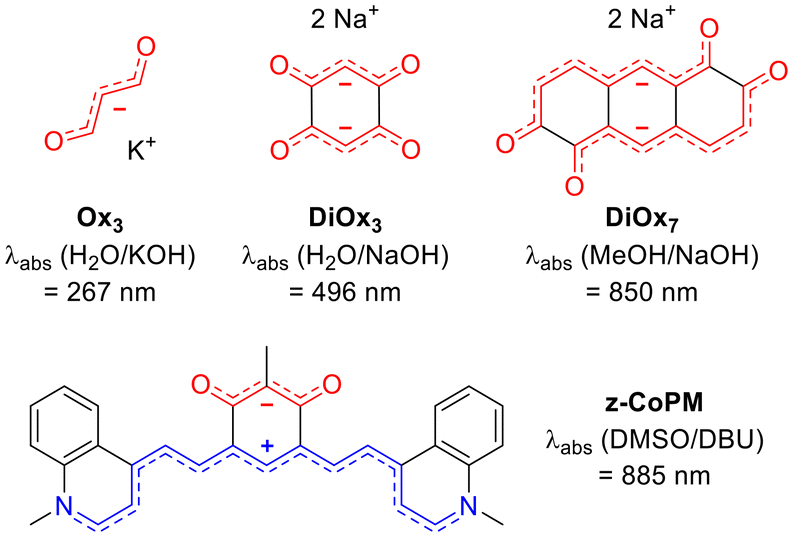 | ||
| Fig. 2 Dianionic and zwitterionic coupled oxonol dyes and their key optical properties. | ||
To date, a few dicationic coupled polymethines have been reported. These include the dicationic octamethyl-tetraaminobenzene studied by Staab and co-workers in 1986,34,35 acidochromic diaminobenzoquinonediiimine derivatives described by some of us,36–38 as well as the more recently reported dicationic hexaaminophenazine, which absorbs in the NIR region.39 Zwitterionic coupled polymethines, including a cationic and an anionic cyanine, have also been investigated, particularly derivatives of tetraazapentacene,40,41 hexaazaanthracene,42–44 or diaminobenzoquinonediimine incorporating electron-withdrawing groups on the nitrogen atoms, leading to intramolecular charge separation and unique solvatochromic properties.45–47 Additional examples include zwitterionic aminobenzoquinonemonoinimine derivatives,48,49 as well as the recently explored zwitterionic coupled heptamethine–oxonol systems (e.g., z-CoPM, Fig. 2), which exhibited dual acidochromic behavior in both solution and thin films.50
In contrast, dianionic coupled polymethines remain extremely rare. Beyond the previously mentioned DiOx3 in Dähne's seminal study, only a handful of examples have been described. Horner and Weber synthesized a derivative of 1,7-dihydoxy-naphthalene-2,6-dione, though its optical properties in basic media were not discussed in their work.51 Boldt reported a NIR-absorbing 2,6-dihydroxy-anthracene-1,5-dione dianion (DiOx7, Fig. 2) in 1964,52 and Khodorkovsky observed a bis-indantetraone derivative in 1994.53 The structural resemblance between DiOx3 and z-CoPM motivated us to further investigate dianionic coupled polymethines using an analogous synthetic strategy. This approach aims to explore how increasing negative charge influences the stabilization, optical redshift, and photophysical properties of these systems, offering a meaningful comparison to more commonly studied zwitterionic and dicationic analogues.
In this work, we report the synthesis of novel mono- and bis-adducts of benzothiazole (1a, 1b), 1,3-indandione (2a, 2b), and tricyanofuran (3a, 3b) chromophores (Fig. 3). These dyes were successfully isolated and characterized, exhibiting dual acidochromic switching properties. Starting from neutral aromatic compounds (CoPM·2H), deprotonation leads to the formation of highly delocalized and redshifted anionic (CoPM−·H) and dianionic (CoPM2−) species with quinoidal six-membered core. Additionally, the neutral dyes 1b and 2b were found to be emissive, with remarkably large Stokes shifts attributed to excited-state intramolecular proton transfer (ESIPT) in 1b and photoacid emission in 2b. Moreover, compound 3b revealed to undergo halochromic switching in basic medium, generating a spirocyclic isomer.
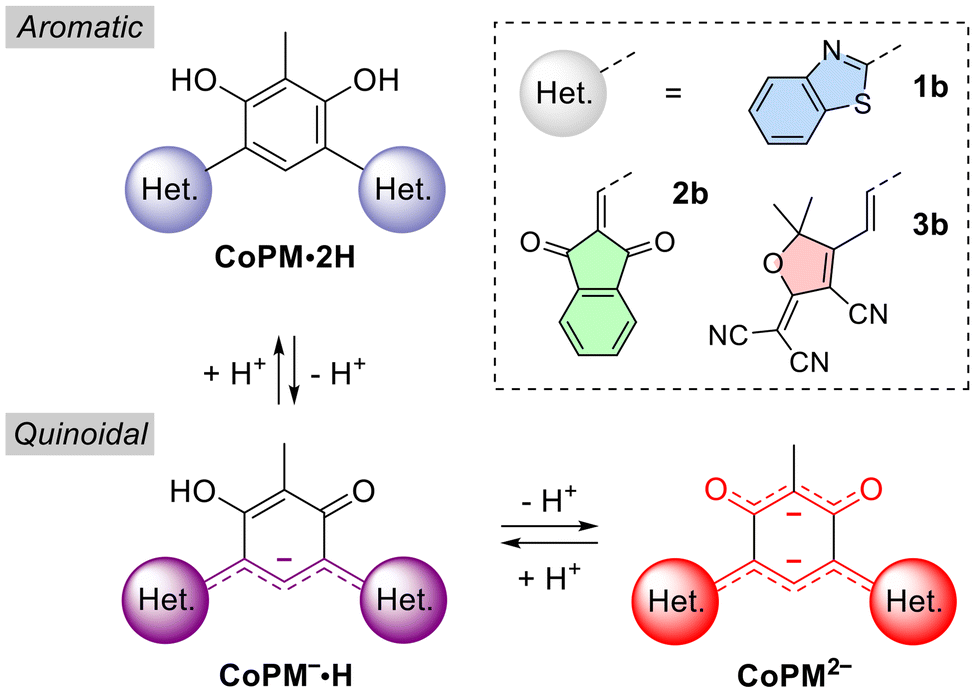 | ||
| Fig. 3 Protonation states of the compounds investigated. | ||
Results and discussion
Synthesis
The dialdehyde building block 4, synthesized through electrophilic formylation of 2-methylresorcinol,50 was used in condensation reactions with 2-aminothiophenol, 1,3-indandione or tricyanofuran 5 (Scheme 1). Initially, condensation of 4 with one equivalent of 2-aminothiophenol led to the precipitation of the pure mono-benzothiazole derivative 1a·2H in 70% yield. Under harsher conditions, i.e., boiling dimethylsulfoxide (DMSO), the reaction afforded the bis-benzothiazole 1b·2H with 81% yield. | ||
| Scheme 1 Synthesis of chromophores 1–3 under their neutral or dianionic forms. Conditions: (i) 2-aminothiophenol, EtOH, 25 °C, 16 h; (ii) 2-aminothiophenol, DMSO, reflux, 30 min; (iii) 1,3-indandione, EtOH, pyridine, reflux, 16 h; (iv) 1,3-indandione, 2,6-lutidine, 95 °C, 16 h; (v) 5, MeOH, pyridine, reflux, 64 h; (vi) 5, 2,6-lutidine, 95 °C, 16 h. | ||
Condensation of 4 with one equivalent of 1,3-indandione in boiling ethanol produced an orange precipitate identified as mono-condensed species 2a·2H, isolated in 53% yield after filtration. Its poor solubility suggested the need of a different solvent to facilitate a second condensation. Consequently, heating 4 with two equivalents of 1,3-indandione in 2,6-lutidine afforded the bis-indandione derivative 2b·2H with a rather modest 28% yield after precipitation in water.
Similarly, reacting 4 with one equivalent of tricyanofuran 5 at room temperature led to the precipitation of the pure mono-condensed adduct 3a·2H with 33% yield. Attempts to obtain the corresponding bis-condensed product 3b·2H using two equivalents of 5 in boiling ethanol were unsuccessful, yielding only 3a·2H with a higher 66% yield. Switching to boiling methanol further improved the yield of 3a·2H to 84%. However, heating 4 with two equivalents of 5 in 2,6-lutidine at 95 °C successfully afforded the bis-condensed product 3b2−, isolated in 56% yield after precipitation in diethyl ether. The dianionic structure of 3b2− was confirmed by 1H NMR, which showed signals corresponding to two equivalents of 2,6-lutidinium cations (see Fig. S24, ESI†), and HRMS analysis in methanol, which detected both dianionic (z = 2) and anionic (z = 1) species, indicating partial protonation in this protic solvent (refer to Fig. S35, ESI†).
Notably, solubilizing compounds 1a·2H, 1b·2H, 2a·2H, 2b·2H, or 3a·2H in polar solvents (e.g., methanol), lead to remarkable color changes, prompting further investigation into the acidochromic and solvatochromic properties of these dyes and the impact of their protonation states on photophysical properties.
Photophysical properties
To investigate the optical properties of the synthesized chromophores, their electronic absorption spectra were recorded in DMSO. Acidification and basification were achieved using 0.1 M trifluoroacetic acid (TFA) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), respectively. The corresponding spectra (Fig. 4) reveal significant variations of the absorption properties depending on the solution conditions: starting from neutral species in acidic media, spontaneous deprotonation occurs in DMSO, which can be further promoted under basic conditions. | ||
| Fig. 4 UV-vis-NIR electronic absorption spectra of mono-condensed (top row) and bis-condensed dyes (bottom row) dissolved at ca. 2 × 10−5 M in acidic (0.1 M TFA, black line), neutral (blue line) or basic (0.1 M DBU, red line) DMSO (with corresponding pictures of the solutions presented in this order from left to right). Below each experimental UV-vis-NIR electronic absorption spectra are presented the corresponding theoretical vibrationally-resolved absorption computed for each relevant protonation state (dashed lines). | ||
The absorption solvatochromism of the six dyes was examined in dichloromethane (DCM), methanol, and DMSO (Fig. S43–S45, ESI†). In these solvents, absorption profiles are primarily influenced by the acidic (e.g., DCM), or basic (e.g., DMSO) nature of the medium, which determines the predominance of either the neutral or deprotonated forms. To gain deeper insight into the protonation states of the species in solution, experimental absorption data were compared with the results of theoretical simulations. The computed spectra were obtained from vibrationally-resolved TD-DFT calculations using the cLR2 solvation model (DMSO),54 from which the wavelengths corresponding to the various peaks were extracted. Additional methodological details can be found in the ESI.† In general, at the selected level of theory, a blueshift with respect to experimental results of approximately 30 to 100 nm is expected for cyanine compounds when using TD-DFT.55 Improved estimates of absorption energies using CC2 corrections for the neutral and monoanionic species, are reported in Table S1 (ESI).† However, as outlined in the ESI,† such corrections could not be applied to the dianionic forms due to difficulties in reliably identifying the corresponding excited states. Theoretical calculations also provide a qualitative estimate of the molar absorptivity (ε), which reflects the absorption intensity. Although the absolute intensities are significantly overestimated by theory in the present case, the relative trends between peaks are well reproduced. In short, the compiled data (Table 1) provide a basis for correlating experimental observations with theoretical predictions, aiding identifying the dominant protonation states in different conditions.
| Experimental | Theoretical | ||||
|---|---|---|---|---|---|
| Dyes | Conditionsa | λ expmax [nm] (ε [M−1 cm−1]) | Species |
λ
theoabs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [nm] (ε [M−1 cm−1]) b [nm] (ε [M−1 cm−1]) |
NICS(0) [ppm] |
| a Pure DMSO or mixture of DMSO + 0.1 M TFA or DBU. b Theoretical absorption maxima and absorptivity coefficients determined in methanol. c Computed on the most stable isomer. sh: shoulder. | |||||
| 1a | DMSO/TFA | 355 (14700) |
1a·2H | 306 (33400) |
−7.16 |
| 282 (28000) |
264 (44800) |
||||
| DMSO | 436 (26000) |
1a−·H | 358 (31100)c |
−2.48c | |
| 374 (14900) |
311 (26200)c |
||||
| DMSO/DBU | 438 (26400) |
1a2− | 413 (34700) |
0.33 | |
| 374 (15200) |
310 (34900) |
||||
| 1b | DMSO/TFA | 378 (sh, 17000) |
1b+·3H | 313 (35100) |
−6.48 |
| 364 (18800) |
1b·2H | 337 (43900) |
−6.87 | ||
| 331 (18300) |
274 (46400) |
||||
| DMSO | 452 (7200) | 1b−·H | 404 (52400) |
−2.21 | |
| 430 (sh, 5400) | 342 (29200) |
||||
| 367 (20500) |
|||||
| DMSO/DBU | 451 (35600) |
1b2− | 428 (61500) |
−0.29 | |
| 430 (sh, 26400) |
313 (56400) |
||||
| 367 (22400) |
|||||
| 2a | DMSO/TFA | 402 (19000) |
2a·2H | 395(42500) |
−5.49 |
| 350 (sh, 10700) |
|||||
| DMSO | 527 (20600) |
2a−·H | 463 (42400) |
0.06 | |
| 391 (10900) |
349 (10500) |
||||
| 336 (18900) |
|||||
| DMSO/DBU | 529 (33000) |
2a2− | 420 (52500) |
3.60 | |
| 336 (12300) |
296 (11200) |
||||
| 228 (3400) | |||||
| 2b | DMSO/TFA | 452 (16000) |
2b·2H | 409 (67000) |
−3.80 |
| 398 (21900) |
340 (32100) |
||||
| DMSO | 617 (7400) | 2b−·H | 546 (75500) |
1.80 | |
| 452 (17100) |
383 (29900) |
||||
| 400 (18200) |
328 (22300) |
||||
| DMSO/DBU | 674 (25400) |
2b2− | 571 (51800) |
4.80 | |
| 534 (7000) | 371 (27900) |
||||
| 421 (19700) |
336 (20766) |
||||
| 3a | DMSO/TFA | 459 (40800) |
3a·2H | 429 (65600)c |
−7.40c |
| 288 (21200) |
|||||
| DMSO | 755 (1700) | 3a−·H | 512 (84600)c |
−1.17c | |
| 600 (17000) |
297 (32600)c |
||||
| 453 (12400) |
|||||
| DMSO/DBU | 595 (8300) | 3a2− | 484 (69700)c |
2.96c | |
| 420 (7200) | |||||
| 3b | DMSO/TFA | 523 (17300) |
3b·2H | 479 (47000)c |
−6.20 |
| 441 (20000) |
409 (93300)c |
||||
| 349 (25200)c |
|||||
| DMSO | 754 (25500) |
3b−·H | 697 (68700)c |
0.50 | |
| 660 (sh, 9100) | 451 (68800)c |
||||
| 521 (17000) |
335 (23400)c |
||||
| DMSO/DBU | 690 (sh, 5000) | 3b2− | 765 (71800)c |
4.40 | |
| 638 (7800) | 403 (52300)c |
||||
| 591 (6800) | |||||
| sp−3b2− | 556 (91100)c |
−1.37 | |||
| 417 (21700)c |
|||||
| dsp−3b2− | 296 (55100) |
−7.65 | |||
First, in the case of the neutral mono-condensed adduct of benzothiazole (1a·2H), the absorption maximum in acidic conditions appears in the UV region at 355 nm with a molar absorptivity of ca. 1.5 × 104 M−1 cm−1. In neutral and basic DMSO, a bathochromic shift occurs towards the blue region at 435 nm, with a nearly doubled ε reaching 2.8 × 104 M−1 cm−1. This shift likely corresponds to the formation of the deprotonated species (1a−·H or 1a2−). Theoretical calculations predict a vertical transition at 413 nm for 1a2−, which is underestimated but generally aligns with the experimental data. In contrast, assigning the experimental 435 nm band to 1a−·H would come with a 0.63 eV error from theory, deemed as large, even for TD-DFT.
Similarly, the mono-indandione derivative (2a·2H) follows a comparable trend, exhibiting a bathochromic shift from 402 nm in acidified DMSO to 527 nm in neutral and basic conditions. This experimental redshift upon deprotonation is also reproduced by theoretical calculations, which predict an absorption maximum at 463 nm for 2a−·H. As the solution transitions from neutral to basic DMSO, ε527 increases from ca. 2.1 × 104 M−1 cm−1 to 3.3 × 104 M−1 cm−1, corresponding to the complete conversion to anion 2a−·H. In such basic condition, the dianion species 2a2− is not observed, since its formation should induce a small (non-observed) redshift compared to 2a·2H, according to theory.
In contrast, the mono-condensed tricyanofuran adduct (3a·2H) displays a distinct behavior. The protonated form 3a·2H absorbs at 459 nm, but in neutral DMSO, deprotonation presumably leads to 3a−·H, which shows a redshifted absorption at 600 nm (ε600 = 1.7 × 104 M−1 cm−1), accompanied with a weaker low-intensity band at 755 nm (ε755 = 0.2 × 104 M−1 cm−1). In basic DMSO, a blueshifted band at 595 nm emerges, suggesting the formation of the dianionic species 3a2−. This is corroborated by the calculations, which also predict a noticeable blueshift (from 512 to 484 nm) when going from the anionic species 3a−·H to the dianionic species 3a2−.
The study of bis-condensed adducts provides striking results, with their symmetrical structures favoring the formation of highly delocalized charged species, and with the establishment of coupled polymethine systems inducing pronounced spectral shifts. The bis-benzothiazole derivative (1b·2H) appears colorless in acidic solution, with absorption in the UV region at 365 nm. Titration of 1b with TFA did not evidenced the formation of the cationic species 1b+·3H (Fig. S46†), for which an absorption maximum is predicted at 313 nm. In neutral DMSO, a band emerges at ca. 450 nm, intensifying in basic conditions to reach ε451 = 3.6 × 104 M−1 cm−1. As observed for the mono-benzothiazole analogue (1a·2H), this evolution suggests the formation of the deprotonated species (1b−·H or 1b2−) in neutral and basic solutions, with theoretical calculations predicting absorption maxima at 404 and 428 nm for 1b−·H and 1b2−, respectively. It is noteworthy that the experimental wavelengths are comparable to the anionic unrigidified benzothiazole-based analogue A, which presents an absorption maximum at 475 nm (Fig. 1).15
For the bis-indandione derivative 2b·2H in acidic solution, the absorption spectrum exhibits two maxima at 452 (ε = 1.60 × 104 M−1 cm−1) and 396 nm (ε = 2.19 × 104 M−1 cm−1). In neutral DMSO, these bands persist, but an additional transition emerges at 617 nm, which we attribute to the presence of deprotonated species 2b−·H, in agreement with theoretical predictions, anticipating a transition at 546 nm for this form. In basic conditions, an intense bathochromic shift occurs, with a new band at 674 nm (ε = 2.5 × 104 M−1 cm−1), a wavelength comparable to that of the bis-indandione anionic pentamethine analogue B, which absorbs at 664 nm (Fig. 1).17 Theoretical calculations suggest that further deprotonation leads to increased redshifts, supporting the assignment of this band to the dianionic species (2b2−). Interestingly, in basified methanol, the absorption is blueshifted to 622 nm (Fig. S44, ESI†), likely due to the hydrogen bonding between the solvent and the multiple oxygen atoms of the dye promoting the monoanionic species 2b−·H.
Finally, the bis-tricyanofuran derivative 3b·2H shows two broad absorption bands at 523 (ε = 1.73 × 104 M−1 cm−1) and 441 nm (ε = 20.0 × 104 M−1 cm−1) in acidic conditions. In neutral DMSO, a strong band develops in the NIR range, peaking at 754 nm (ε = 2.6 × 104 M−1 cm−1). This transition likely corresponds to either the anionic species 3b−·H or its dianionic counterpart 3b2−, for which theoretical absorption maxima are predicted ca. 700 and 760 nm, respectively. The absorption wavelength (754 nm) is notably smaller than the 900 nm absorption maximum of the anionic heptamethine analogue D (Fig. 1). This difference is likely due to the destabilizing effect of the oxonol subunit on the negatively charged polymethine system, combined with the s-cis conformation adopted by the heptamethine bridge, which may hinder linear charge delocalization. In basic conditions, a noticeable blueshift of absorption occurs for 3b2−, a phenomenon discussed in detail in a subsequent section.
Computational study
A conformational analysis of the possible isomers and tautomers of dyes 1b, 2b, and 3b, including their corresponding neutral, mono- and dianionic forms, was carried out using DFT calculations to identify the most thermodynamically stable structures at room temperature (see Fig. S50, ESI†). In few cases, the analysis revealed multiple conformers with relative Gibbs free energies (ΔG) within 2.0 kcal mol−1 of the most stable geometry; these alternatives structures are reported in Fig. S51–S58, ESI.† For all neutral species of 1 and 2 series and their corresponding monoanionic counterparts, the analysis of the tautomers indicates that the hydrogen atoms are preferentially bonded to the oxygen atoms of the central six-membered ring instead of the benzothiazole and indandione moieties.Both 1b·2H and 2b·2H exhibit a single, stable, and planar structure in which the phenolic hydrogen atoms engage in intramolecular hydrogen bonding, either with the nitrogen atoms of the benzothiazole (1b·2H) or with the oxygen atoms of the 1,3-indandione fragments (2b·2H), as shown in Fig. S50 (ESI).† Under basic conditions, deprotonation disrupts one of these hydrogen bonds, leading to a 180° rotation of the corresponding functional group in 2b−·H, whereas 1b−·H retains its initial geometry. In the dianionic forms, the loss of both hydrogen bonds induces a rotation of both peripheral units in 1b2− and 2b2−.
In contrast, the structure of 3b·2H displays notable conformational flexibility at room temperature, with eight conformers lying within 1.22 kcal mol−1 of the global minimum. These arise from rotations of the two tricyanofuran moieties around the C–C bonds of the polymethine chain connecting them to the central core (see Fig. S55, ESI†). Although these rotations have minimal impact on the nature of the principal electronic transitions, they cause minor shifts in the calculated absorption maxima. To account for this, the relevant absorption wavelengths are reported as room-temperature Boltzmann-weighted averages. A similar degree of conformational freedom is observed for both the monoanionic and dianionic forms of 3b, which respectively exhibit seven and four stable conformers within a 2 kcal mol−1 ΔG window (see Fig. S42 and S43, ESI†).
To assess the aromatic or quinoidal nature of the central core of the chromophores depending on their protonation state, nucleus independent chemical shifts (NICS) were computed (Table 1). As anticipated for the symmetrical dyes, the neutral forms exhibit negative NICS(0) values of −6.87, −3.80 and −6.20 ppm for 1b·2H, 2b·2H, and 3b·2H, respectively, indicating a predominantly aromatic character. Upon single deprotonation, the NICS increase significantly for 1b−·H, 2b−·H, 3b−·H, reaching −2.21, 1.80 and 0.50 ppm, respectively, consistent with a progressive loss of aromaticity in favor of a more quinoidal structure. In the fully deprotonated dianionic forms, this trend continues: 1b2− exhibits a nearly non-aromatic character (NICS = −0.29 ppm), whereas both 2b2− and 3b2− display positive NICS of 4.80 and 4.20 ppm, respectively, hinting at a moderately antiaromatic character of the central ring.
Theoretical calculations predict a progressive bathochromic shift in the absorption energies of all three symmetrical dyes upon sequential deprotonation, from the neutral to anionic and then to the dianionic forms. This trend is consistent with the increasing negative charge, which promotes a quinoidal electronic structure, enhancing conjugation along the two coupled polymethine backbones (Fig. 3). In these species, the two polymethine subunits are connected via single bonds, facilitating charge delocalization across the entire system. This delocalization gives rise to cyanine-like electronic transitions, in line with the coupling principle discussed earlier.
Excited-state calculations attribute the long-wavelength absorption bands of the dyes to HOMO → LUMO transitions, each characterized by high oscillator strengths (see Tables S1, ESI†). Electron density difference (EDD) maps between the ground (S0) and first excited (S1) states for neutral 1b, 2b, 3b, and their corresponding anionic and dianionic forms provide further insight into the nature of these electronic excitations (Fig. 5). In the neutral species, the EDD maps reveal pronounced intramolecular charge transfer (ICT) predominantly from the phenolic functions towards the electron-withdrawing moieties. The density redistribution is centered on the chromophoric core and extends along the substituted polymethine chain, with partial involvement of the oxygen atoms attached to the upper methine bridges. In particular, for 3b·2H, the extended conjugation and the stronger electron-withdrawing groups enhance delocalization, resulting in a more extensive ICT character.
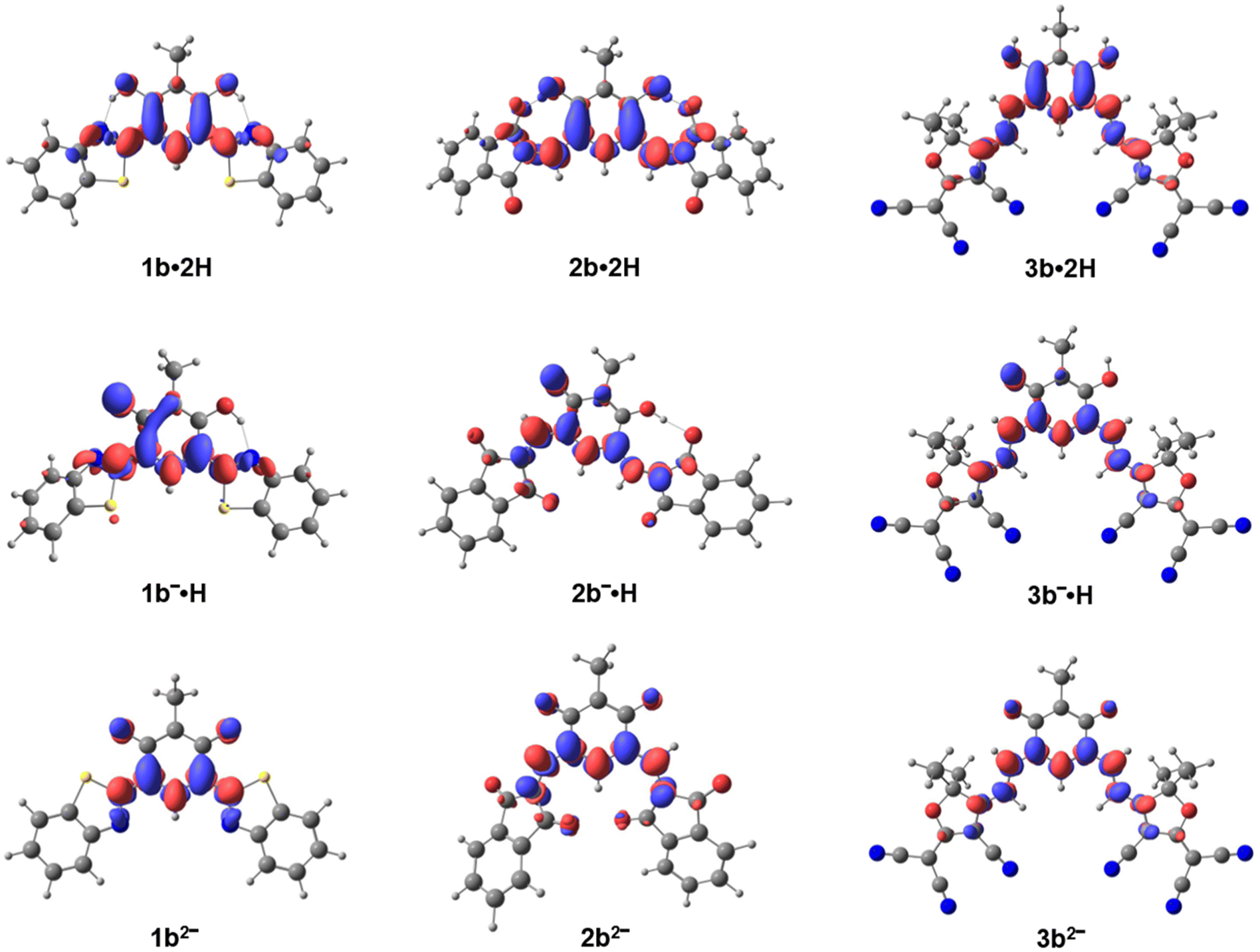 | ||
| Fig. 5 Electronic density differences (EDD) between the ground (S0) and the first excited state (S1) for molecules 1b, 2b and 3b (most stable isomers) under different protonation states. Regions in red indicate an increase in electronic density from S0 to S1, while regions in blue indicate a decrease. The isosurfaces are visualized with an isovalue of 0.003. | ||
In the anionic forms, the EDD maps for the lowest excited state indicate a broader charge delocalization across the molecules compared to the neutral species. However, the presence of a negative charge on the oxygen of the oxonol subunit imparts a quinoidal character on the central core, which localizes the donor group involved in the charge transfer primarily around the oxygen and diminishes the contribution from other parts of the molecule. In the dianionic species, the aromatic character is lost, and the EDD becomes predominantly localized on the lower polymethine chain with only minor contributions from the oxygen atoms of the oxonol subunit.
Spirocyclization of dye 3
Unexpectedly, when 3b2− is dissolved in basic DMSO, a significant hypsochromic effect occurs, shifting the absorption to the 600–700 nm range. This suggests that the dianionic species undergoes structural reorganization, likely evolving into a less-conjugated spirocyclic chromophore sp-3b2− and dsp-3b2−, depicted in Scheme 2. Similar behavior has been previously observed in simpler tricyanofuran-based systems.56–60 Theoretical calculations reveal that the cyclization of one tricyanofuran group leads to two stable isomers, depending on whether the second tricyanofuran unit is rotated 0° or 180° relative to the central ring plane. These sp-3b2− isomers are 7.1 and 8.4 kcal mol−1 more stable than the open form 3b2−, respectively (see Fig. S58, ESI†). Dual cyclization involving both tricyanofuran units leads to the putative dsp-3b2− structure, which is even more stable, with an energy gain of 13.6 kcal mol−1 compared to the open form. These data indicate that the spirocyclic forms are thermodynamically favored. However, their presence in solution may be limited by a high activation barrier associated with the cyclization process from sp-3b2− to dsp-3b2−. The sp-3b2− isomer exhibits a bright electronic transition at 556 nm (see Table 1), whereas for dsp-3b2−, the lowest-energy transition is predicted around 300 nm, consistent with a loss of extended π-conjugation. Interestingly, the spirocyclic forms sp-3b2− and dsp-3b2− show contrasting NICS values of −1.37 and −7.65 ppm, reflecting their quinoidal and aromatic central cores. | ||
| Scheme 2 Spirocyclization of 3b occurring in basic conditions. | ||
The formation of sp-3b2− was also supported by 1H NMR spectroscopy upon addition of Cs2CO3, which revealed the appearance of new doublet signals corresponding to the heptamethine protons (Fig. S29, ESI†). These consist of two distinct pairs of doublets, with coupling constants (3J) of 10.0 Hz and 16.1 Hz, attributed to the cis and trans configurations of the protons on the closed and open sides of the molecule, respectively.
Emission properties
The emission properties of the six chromophores (1a,b, 2a,b, and 3a,b) were investigated in DMSO, with TFA or DBU added as needed to generate either the neutral or charged species. Fluorescence was observed for the bis-benzothiazole dye (1b) and bis-indandione dye (2b) only, whose emission properties are summarized in Table 2.| Experimental | Theoretical | |||||||
|---|---|---|---|---|---|---|---|---|
| Dyes | Conditionsa |
λ
max
[nm] |
λ em [nm] | Stokes shift [cm−1] | Φ [%] | T [ns] | Species | λ theoem [nm] |
| a Pure DMSO or mixture of DMSO + 0.1 M TFA or DBU. b Absorption value extracted from the maximum of the recorded excitation spectra (Fig. S47, ESI†). | ||||||||
| 1b | DMSO/TFA | 365 | 546 | 9100 | 0.2 | 3.2 | EE−1b·2H | 404 |
| KE−1b·2H | 486 (526) | |||||||
| DMSO/DBU | 454 | 498 | 1900 | 11 | 2.3 | 1b−·H | 415 | |
| 1b2− | 481 | |||||||
| 2b | DMSO/TFA | 460 | 704 | 7500 | 2 | 1.1 | 2b·2H | 623 |
| DMSO | 633 | 705 | 1600 | 2 | 1.0 | 2b−·H | 577 (681) | |
| 2b2− | 596 | |||||||
The neutral dye 1b·2H exhibits a yellow fluorescence in acidic DMSO, peaking at 546 nm with a low quantum yield of 0.2% and a notably large Stokes shift of 9100 cm−1 (Fig. 6). The origin of this emission was confirmed by its excitation spectrum, which closely matches the absorption profile of 1b·2H (Fig. S47, ESI†). Benzothiazole-substituted o-phenols are known to undergo excited-state intramolecular proton transfer (ESIPT), leading to significant Stokes shifts.61–65 Thus, we hypothesized that 1b·2H undergoes ESIPT and computed its emission from both the enol (EE-1b·2H, local state excitation) and keto forms (KE-1b·2H, mono-ESIPT process). These excited-state calculations indicate a favorable ESIPT, with a ΔG of −0.27 eV between the excited enol and keto tautomers (Fig. 7). Additionally, the predicted fluorescence maximum for KE-1b·2H at 523 nm aligns well with the experimental value (λexpem = 546 nm), whereas the emission from the enol tautomer (EE-1b·2H) theoretically expected at 404 nm does not fit the experiments.
 | ||
| Fig. 6 Experimental (top) and theoretical (bottom) absorption (plain lines) and emission (dotted lines) spectra of 1b. | ||
 | ||
| Fig. 7 Computed differences of free energies (ΔG) between tautomers of 1b·2H (1st and 2nd ESIPT) at the first excited state and computed emissions. | ||
The possibility of double proton transfer was also investigated, and calculations revealed a favorable ΔG of −0.29 eV for the transition from the excited (mono) keto form (KE-1b·2H) to the doubly proton-transferred form (KK-1b·2H). This species is predicted to emit at 860 nm with a negligible oscillator strength, indicating an emission in the NIR range with an intensity (radiative rate) too low to be detected with our current setup. Note that the presence of a second ESIPT process is consistent with the very low quantum yield observed (0.2%).
Upon deprotonation in DMSO in the presence of DBU, dye 1b displays a blue emission at 498 nm, with a significantly reduced Stokes shift of 1900 cm−1 and a markedly higher fluorescence quantum yield of 11%. This emission aligns well with theoretical predictions, which estimate fluorescence maxima at 415 nm for the monoanionic species (1b−·H) and 481 nm for the dianionic species (1b2−) with a large oscillator strength of 1.09 and 0.60, respectively.
Regarding bis-indandione derivative 2b·2H, fluorescence is observed in the red-NIR region, peaking at 704 nm with a rather large Stokes shift of 7500 cm−1 (Fig. 8). Surprisingly, the deprotonated species 2b−·H exhibits an almost identical emission profile, with comparable fluorescence quantum yield of 2% and lifetime of ca. 1 ns (Fig. S48, ESI†). The excitation spectra of both 2b·2H and 2b−·H closely match their respective absorption profiles (Fig. S47, ESI†), demonstrating that both species relax to the same emissive state.
 | ||
| Fig. 8 Experimental (top) and theoretical (bottom) absorption (plain lines) and emission (dotted lines) spectra of 2b. | ||
This observation suggests that 2b·2H behaves as a photoacid (Fig. 9); upon excitation, its acidity is significantly enhanced, resulting in a proton release and the formation of the excited anionic species 2b−·H, which subsequently relaxes via red light emission. This behavior is reminiscent of known ESIPT fluorophores, such as benzazole-based dyes, which act as photoacids in neutral DMSO.64 Theoretical calculations support this interpretation, indicating that the 2b·2H form undergoes a barrierless ESIPT process, as the excited-state geometry yields a spontaneous proton transfer, likely preceding the proton dissociation described above. The emission spectrum of the keto tautomers closely resembles those of the anionic and dianionic species, with all three peaks appearing between ca. 600–700 nm (see Fig. 9 and Table 2). However, because these spectral differences lie within the typical error bar of TD-DFT calculations, the experimental emission near 700 nm cannot be unambiguously assigned to one specific structure.
 | ||
| Fig. 9 Proposed photoacidic process occurring upon irradiation of 2b·2H. | ||
Conclusions
In this study, we reported the single-step synthesis of six chromophores built around a tetra-substituted six-membered ring incorporating benzothiazole, indandione, or tricyanofuran moieties. The dyes containing indandione and tricyanofuran moieties exhibit visible coloration in solution, attributed to their tendency to spontaneously deprotonate and form anionic species. We systematically explored their optical properties, with a particular focus on their absorption, emission, and protonation-dependent behavior. Experimental and theoretical investigations revealed that deprotonation of the neutral symmetrical dyes, initially featuring aromatic cores, induces pronounced bathochromic shifts, stemming from the formation of highly delocalized oxonol-polymethine structures with a quinoidal six-membered core. Notably, the bis-tricyanofuran derivative (3b) reached NIR absorption in solution and underwent further transformation via spirocyclization under basic conditions, yielding a distinct optical signature.Fluorescence studies revealed that the bis-benzothiazole (1b) and bis-indandione (2b) derivatives exhibit emission. The neutral bis-benzothiazole dye undergoes an ESIPT process, resulting in an exceptionally large Stokes shift, which was further validated through theoretical modeling. Additionally, the bis-indandione dye exhibits a photoacidic behavior, undergoing excited-state deprotonation to generate an emissive anionic species. These findings provide deeper insights into the complex electronic properties of coupled polymethines and anionic dyes while contributing to the development of functional chromophores with tunable absorption and well-defined fluorescence properties suited for specific applications.
Author contributions
Conceptualization: S. P.; investigations: B. M., C. N., S. P.; D. J. supervision: S. P., D. J., O. S.; writing – original draft: B. M., S. P., C. N.; writing – review & editing: B. M., C. N., D. J., O. S., S. P.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the Agence Nationale de la Recherche, in the frame of the SOCOOL (ANR-20-CE07-0024) and CONDOR (ANR-21-CE07-0058) projects. The authors thank the CNRS for continuous support, and the Spectropole (Fédération Sciences Chimiques Marseille) for HRMS analyses. The authors also thank the GLiCID Computing Facility (Ligerien Group for Intensive Distributed Computing, 10.60487/glicid, Pays de la Loire, France) for the computational resources.References
- L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Near-Infrared Heptamethine Cyanines (Cy7): From Structure, Property to Application, Org. Biomol. Chem., 2020, 18, 9385–9397 RSC.
- X. Zhao, F. Zhang and Z. Lei, The Pursuit of Polymethine Fluorophores with NIR-II Emission and High Brightness for in Vivo Applications, Chem. Sci., 2022, 13, 11280–11293 RSC.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Recent Development of Chemosensors Based on Cyanine Platforms, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- H. Janeková, M. Russo and P. Štacko, Cyanine Renaissance: Tailoring the Properties to Applications, Chimia, 2022, 76, 763 CrossRef PubMed.
- D. Saccone, S. Galliano, N. Barbero, P. Quagliotto, G. Viscardi and C. Barolo, Polymethine Dyes in Hybrid Photovoltaics: Structure–Properties Relationships, Eur. J. Org. Chem., 2016, 2244–2259 CrossRef CAS.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Cyanines during the 1990s: A Review, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS.
- M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Syntheses of Cyanines: A Review, Tetrahedron, 2012, 68, 781–805 CrossRef CAS.
- J. L. Bricks, A. D. Kachkovskii, Y. L. Slominskii, A. O. Gerasov and S. V. Popov, Molecular Design of near Infrared Polymethine Dyes: A Review, Dyes Pigm., 2015, 121, 238–255 CrossRef CAS.
- H. Mustroph, Bring Back Order in the Polymethine Dye Medley: Classification, Structure and Spectra, Dyes Pigm., 2022, 208, 110783 CrossRef CAS.
- A. V. Kulinich and A. A. Ishchenko, Design and Photonics of Merocyanine Dyes, Chem. Rec., 2024, 24, e202300262 CrossRef.
- A. V. Kulinich and A. A. Ishchenko, Merocyanines: Electronic Structure and Spectroscopy in Solutions, Solid State, and Gas Phase, Chem. Rev., 2024, 124, 12086–12144 CrossRef CAS.
- C. Elian, B. Mourot, C. Benbouziyane, J.-P. Malval, S. Lajnef, F. Peyrot, F. Massuyeau, O. Siri, D. Jacquemin, S. Pascal and D.-L. Versace, Tris-Benzo[cd]Indole Cyanine Enables the NIR-Photosensitized Radical and Thiol-Ene Polymerizations at 940 nm, Angew. Chem., 2023, 135, e202305963 CrossRef.
- A. Levitz, F. Marmarchi and M. Henary, Introduction of Various Substitutions to the Methine Bridge of Heptamethine Cyanine Dyes Via Substituted Dianil Linkers, Photochem. Photobiol. Sci., 2018, 17, 1409–1416 CrossRef CAS.
- S. Pascal, Y. A. Getmanenko, Y. Zhang, I. Davydenko, M. H. Ngo, G. Pilet, S. Redon, Y. Bretonnière, O. Maury, I. Ledoux-Rak, S. Barlow, S. R. Marder and C. Andraud, Design of Near-Infrared-Absorbing Unsymmetrical Polymethine Dyes with Large Quadratic Hyperpolarizabilities, Chem. Mater., 2018, 30, 3410–3418 CrossRef CAS.
- K. I. Petko, T. M. Sokolenko and L. M. Yagupol'skii, Difluoromethylation of Heterocyclic Compounds Containing an N = C-C Ambident Nucleophilic System, Russ. J. Org. Chem., 2005, 41, 429–433 CrossRef CAS.
- Zh. A. Krasnaya and Yu. V. Smirnova, The Interaction of Dimethylaminomethylenemalonaldehyde Aminal-Acetal with Indandione, Russ. Chem. Bull., 1997, 46, 2086–2088 CrossRef CAS.
- A. V. Kulinich, N. A. Derevyanko, E. K. Mikitenko and A. A. Ishchenko, Merocyanines Based on 1,3-Indanedione: Electronic Structure and Solvatochromism, J. Phys. Org. Chem., 2011, 24, 732–742 CrossRef CAS.
- F. Azamifar, M. Reza Naimi-Jamal and O. M. Demchuk, Design, Synthesis and Characterization of New Trimethine Oxonol Dyes from 1,3-Indandione and 2-Substituted Vinamidinium Salts, Dyes Pigm., 2019, 161, 438–447 CrossRef CAS.
- N. A. Derevyanko, A. A. Ishchenko and A. V. Kulinich, Deeply Coloured and Highly Fluorescent Dipolar Merocyanines Based on Tricyanofuran, Phys. Chem. Chem. Phys., 2020, 22, 2748–2762 RSC.
- M. P. Shandura, V. P. Yakubovskyi, Y. V. Zatsikha, O. D. Kachkovsky, Y. M. Poronik and Y. P. Kovtun, Anionic, Cationic and Merocyanine Polymethine Dyes Based on Dipyrromethene Core, Dyes Pigm., 2013, 98, 113–118 CrossRef CAS.
- V. Polishchuk, A. Kulinich, E. Rusanov and M. Shandura, Highly Fluorescent Dianionic Polymethines with a 1,3,2-Dioxaborine Core, J. Org. Chem., 2021, 86, 5227–5233 CrossRef CAS PubMed.
- V. Polishchuk, M. Filatova, E. Rusanov and M. Shandura, Trianionic 1,3,2-Dioxaborine-Containing Polymethines: Bright Near-Infrared Fluorophores, Chem. – Eur. J., 2022, 28, e202202168 CrossRef CAS PubMed.
- P.-A. Bouit, E. Di Piazza, S. Rigaut, B. Le Guennic, C. Aronica, L. Toupet, C. Andraud and O. Maury, Stable Near-Infrared Anionic Polymethine Dyes: Structure, Photophysical, and Redox Properties, Org. Lett., 2008, 10, 4159–4162 CrossRef CAS.
- P.-A. Bouit, D. Rauh, S. Neugebauer, J. L. Delgado, E. D. Piazza, S. Rigaut, O. Maury, C. Andraud, V. Dyakonov and N. Martin, “Cyanine–Cyanine” Salt Exhibiting Photovoltaic Properties, Org. Lett., 2009, 11, 4806–4809 CrossRef CAS PubMed.
- C. R. Nieto, J. Guilleme, C. Villegas, J. L. Delgado, D. González-Rodríguez, N. Martin, T. Torres and D. M. Guldi, Subphthalocyanine-Polymethine Cyanine Conjugate: An All Organic Panchromatic Light Harvester That Reveals Charge Transfer, J. Mater. Chem., 2011, 21, 15914–15918 RSC.
- Q. Bellier, N. S. Makarov, P.-A. Bouit, S. Rigaut, K. Kamada, P. Feneyrou, G. Berginc, O. Maury, J. W. Perry and C. Andraud, Excited State Absorption: A Key Phenomenon for the Improvement of Biphotonic Based Optical Limiting at Telecommunication Wavelengths, Phys. Chem. Chem. Phys., 2012, 14, 15299–15307 RSC.
- Z. Li, Y. Liu, H. Kim, J. M. Hales, S.-H. Jang, J. Luo, T. Baehr-Jones, M. Hochberg, S. R. Marder, J. W. Perry and A. K. Y. Jen, High-Optical-Quality Blends of Anionic Polymethine Salts and Polycarbonate with Enhanced Third-Order Non-Linearities for Silicon-Organic Hybrid Devices, Adv. Mater., 2012, 24, OP326–OP330 CAS.
- Z. Li, P. Zhao, S. Tofighi, R. Sharma, T. R. Ensley, S.-H. Jang, D. J. Hagan, E. W. Van Stryland and A. K. Y. Jen, Zwitterionic Cyanine–Cyanine Salt: Structure and Optical Properties, J. Phys. Chem. C, 2016, 120, 15378–15384 CrossRef CAS.
- Z. Li, A. A. Syed, P. Zhao, J. C. Yang, R. Sharma, T. R. Ensley, J. D. Matichak, I. Davydenko, S.-H. Jang, D. J. Hagan, S. R. Marder, E. W. Van Stryland and A. K.-Y. Jen, Cationic Polyelectrolyte for Anionic Cyanines: An Efficient Way To Translate Molecular Properties into Material Properties, J. Am. Chem. Soc., 2019, 141, 17331–17336 CrossRef CAS.
- S. Pascal, S. Denis-Quanquin, F. Appaix, A. Duperray, A. Grichine, B. Le Guennic, D. Jacquemin, J. Cuny, S.-H. Chi, J. W. Perry, B. van der Sanden, C. Monnereau, C. Andraud and O. Maury, Keto-Polymethines: A Versatile Class of Dyes with Outstanding Spectroscopic Properties for in Cellulo and in Vivo Two-Photon Microscopy Imaging, Chem. Sci., 2017, 8, 381–394 RSC.
- Y. Arisawa, Y. Kubota, T. Inuzuka and K. Funabiki, Photostability and Halochromic Properties of Near-Infrared Absorbing Anionic Heptamethine Cyanine Dyes, ChemistrySelect, 2022, 7, e202104213 CrossRef CAS.
- S. Dähne and D. Leupold, Coupling Principles in Organic Dyes, Angew. Chem., Int. Ed. Engl., 1966, 5, 984–993 CrossRef.
- B. Mourot, D. Jacquemin, O. Siri and S. Pascal, Coupled Polymethine Dyes: Six Decades of Discoveries, Chem. Rec., 2024, 24, e202400183 CrossRef CAS PubMed.
- K. Elbl, C. Krieger and H. A. Staab, 1,2,4,5-Tetrakis(Dimethylamino)Benzene, a New Electron Donor with Unusual Properties, Angew. Chem., Int. Ed. Engl., 1986, 25, 1023–1024 CrossRef.
- C. J. Adams, R. C. da Costa, R. Edge, D. H. Evans and M. F. Hood, On the Causes of Potential Inversion in 1,2,4,5-Tetrakis(Amino)Benzenes, J. Org. Chem., 2010, 75, 1168–1178 CrossRef CAS PubMed.
- O. Siri, P. Braunstein, M.-M. Rohmer, M. Bénard and R. Welter, Novel “Potentially Antiaromatic”, Acidichromic Quinonediimines with Tunable Delocalization of Their 6π-Electron Subunits, J. Am. Chem. Soc., 2003, 125, 13793–13803 CrossRef CAS.
- S. Pascal and O. Siri, Benzoquinonediimine Ligands: Synthesis, Coordination Chemistry and Properties, Coord. Chem. Rev., 2017, 350, 178–195 CrossRef CAS.
- T. Munteanu, C. Naim, G. Canard, D. Jacquemin, O. Siri and S. Pascal, Small Far-Red Cationic Benzoquinone Diimine Dyes, Org. Biomol. Chem., 2025, 23, 2836–2844 Search PubMed.
- T. Munteanu, V. Mazan, M. Elhabiri, C. Benbouziyane, G. Canard, D. Jacquemin, O. Siri and S. Pascal, A Strategy to Design Substituted Tetraamino-Phenazine Dyes and Access to an NIR-Absorbing Benzoquinonediimine-Fused Quinoxaline, Org. Lett., 2023, 25, 3886–3891 Search PubMed.
- F. Wudl, P. A. Koutentis, A. Weitz, B. Ma, T. Strassner, K. N. Houk and S. I. Khan, Polyazaacenes: New Tricks for Old Dogs, Pure Appl. Chem., 1999, 71, 295–302 CrossRef CAS.
- P. A. Koutentis, Regiospecific Synthesis of 5,7-Disubstituted Quinoxalino[2,3-b]Phenazines, ARKIVOC, 2002, 175–191 Search PubMed.
- K. Hutchison, G. Srdanov, R. Hicks, H. Yu, F. Wudl, T. Strassner, M. Nendel and K. N. Houk, Tetraphenylhexaazaanthracene: A Case for Dominance of Cyanine Ion Stabilization Overwhelming 16π Antiaromaticity, J. Am. Chem. Soc., 1998, 120, 2989–2990 Search PubMed.
- P. Langer, A. Bodtke, N. N. R. Saleh, H. Görls and P. R. Schreiner, 3,5,7,9-Tetraphenylhexaazaacridine: A Highly Stable, Weakly Antiaromatic Species with 16 π Electrons, Angew. Chem., Int. Ed., 2005, 44, 5255–5259 CrossRef CAS.
- C. P. Constantinides, G. A. Zissimou, A. A. Berezin, T. A. Ioannou, M. Manoli, D. Tsokkou, E. Theodorou, S. C. Hayes and P. A. Koutentis, Tetraphenylhexaazaanthracenes: 16π Weakly Antiaromatic Species with Singlet Ground States, Org. Lett., 2015, 17, 4026–4029 CrossRef CAS PubMed.
- S. Pascal, L. Lavaud, C. Azarias, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Controlling the Canonical/Zwitterionic Balance through Intramolecular Proton Transfer: A Strategy for Vapochromism, Mater. Chem. Front., 2018, 2, 1618–1625 RSC.
- S. Pascal, L. Lavaud, C. Azarias, A. Varlot, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Azacalixquinarenes: From Canonical to (Poly-)Zwitterionic Macrocycles, J. Org. Chem., 2019, 84, 1387–1397 Search PubMed.
- T. Horáčková, M. H. E. Bousquet, A. Morice, U. Triballier, G. Canard, P. Lhoták, D. Jacquemin, S. Pascal and O. Siri, Fully Zwitterionic Diaminobenzoquinonediimines Promoted by Cyanoaromatic N-Substituents, Dyes Pigm., 2022, 206, 110681 CrossRef.
- O. Siri and P. Braunstein, Unprecedented Zwitterion in Quinonoid Chemistry, Chem. Commun., 2002, 208–209 RSC.
- A. T. Ruiz, M. H. E. Bousquet, S. Pascal, G. Canard, V. Mazan, M. Elhabiri, D. Jacquemin and O. Siri, Small Panchromatic and NIR Absorbers from Quinoid Zwitterions, Org. Lett., 2020, 22, 7997–8001 CrossRef CAS.
- B. Mourot, V. Mazan, M. Elhabiri, R. Sarkar, D. Jacquemin, O. Siri and S. Pascal, Insights into Extended Coupled Polymethines through the Investigation of Dual UV-to-NIR Acidochromic Switches Based on Heptamethine-Oxonol Dyes, Chem. Sci., 2024, 15, 1248–1259 RSC.
- L. Horner and K.-H. Weber, Zur Kenntnis der o-Chinone, XXVI: Neue Mono- und Dihydroxynaphthochinone, Chem. Ber., 1965, 98, 1246–1251 CrossRef CAS.
- P. Boldt, Darstellung neuer Hydroxy-anthrachlnone und Anthradichinone, Naturwissenschaften, 1964, 51, 137–138 CrossRef CAS.
- V. Khodorkovsky, E. Arkady and N. Ojars, A New Strong Electron Acceptor. The Resurrection of 2,2′-Biindanylidene-1,3,1′,3′-Tetraone (BIT), Tetrahedron Lett., 1994, 35, 2955–2958 Search PubMed.
- C. A. Guido, A. Chrayteh, G. Scalmani, B. Mennucci and D. Jacquemin, Simple Protocol for Capturing Both Linear-Response and State-Specific Effects in Excited-State Calculations with Continuum Solvation Models, J. Chem. Theory Comput., 2021, 17, 5155–5164 CrossRef CAS PubMed.
- B. Le Guennic and D. Jacquemin, Taking Up the Cyanine Challenge with Quantum Tools, Acc. Chem. Res., 2015, 48, 530–537 CrossRef CAS PubMed.
- M. M. Wootten, S. Tshepelevitsh, I. Leito and J. Clayden, Using Color to Control Conformation in a Chemical System Containing Multiple Tricyanofuran Photoacids, Angew. Chem., Int. Ed., 2025, 64, e202502437 CrossRef CAS.
- M. Yu. Belikov, M. Yu. Ievlev, S. V. Fedoseev and O. V. Ershov, Tuning the Photochromic Properties of Chromophores Containing a Nitrile-Rich Acceptor: A Novel Branch in the Investigation of Negative Photochromes, New J. Chem., 2019, 43, 8414–8417 RSC.
- M. Yu. Belikov, M. Yu. Ievlev, S. V. Fedoseev and O. V. Ershov, The First Example of “Turn-off” Red Fluorescence Photoswitching for the Representatives of Nitrile-Rich Negative Photochromes, New J. Chem., 2020, 44, 6121–6124 RSC.
- M. Yu. Belikov and M. Yu. Ievlev, Thermal Studies on Visible-Light-Switchable Negative T-Type Photochromes of a Nitrile-Rich Series, RSC Adv., 2021, 11, 21097–21103 RSC.
- M. Yu. Belikov, M. Yu. Ievlev and I. N. Bardasov, A Novel Water-Soluble Multicolor Halo- and Photochromic Switching System Based on a Nitrile-Rich Acceptor, New J. Chem., 2021, 45, 10287–10295 RSC.
- S. Goswami, S. Das, K. Aich, B. Pakhira, S. Panja, S. K. Mukherjee and S. Sarkar, Chemodosimeter for the Ratiometric Detection of Hydrazine Based on Return of ESIPT and Its Application in Live-Cell Imaging, Org. Lett., 2013, 15, 5412–5415 CrossRef CAS.
- T. Gao, P. Xu, M. Liu, A. Bi, P. Hu, B. Ye, W. Wang and W. Zeng, A Water-Soluble ESIPT Fluorescent Probe with High Quantum Yield and Red Emission for Ratiometric Detection of Inorganic and Organic Palladium, Chem. – Asian J., 2015, 10, 1142–1145 CrossRef CAS PubMed.
- Y. Chen, Y. Fang, H. Gu, J. Qiang, H. Li, J. Fan, J. Cao, F. Wang, S. Lu and X. Chen, Color-Tunable and ESIPT-Inspired Solid Fluorophores Based on Benzothiazole Derivatives: Aggregation-Induced Emission, Strong Solvatochromic Effect, and White Light Emission, ACS Appl. Mater. Interfaces, 2020, 12, 55094–55106 CrossRef CAS.
- T. Pariat, M. Munch, M. Durko-Maciag, J. Mysliwiec, P. Retailleau, P. M. Vérité, D. Jacquemin, J. Massue and G. Ulrich, Impact of Heteroatom Substitution on Dual-State Emissive Rigidified 2-(2′-Hydroxyphenyl)Benzazole Dyes: Towards Ultra-Bright ESIPT Fluorophores, Chem. – Eur. J., 2021, 27, 3483–3495 CrossRef CAS.
- P. Chonlateeroj, S. Arunlimsawat, P. Janthakit, R. Wannapakdee, W. Waengdongbung, T. Sudyoadsuk, P. Kumnorkaew and V. Promarak, Excited-State Intramolecular Proton Transfer Derivatives as Self-Absorption Free Luminophores for Luminescent Solar Concentrators, Mater. Chem. Front., 2025, 9, 695–709 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qo00801h |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |